
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent extraction of rare earths elements from nitrate media in DMDOHEMA/ionic liquid systems: performance and mechanism studies†
Cesar L. Usma*,
S. Dourdain *,
G. Arrachart and
S. Pellet-Rostaing
*,
G. Arrachart and
S. Pellet-Rostaing
ICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: sandrine.dourdain@cea.fr; ceslop33@gmail.com
First published on 22nd September 2021
Abstract
Extraction of La(III), Eu(III) and Fe(III) was compared in n-dodecane and two ionic liquids (ILs) (1-ethyl-1-butylpiperidinium bis (trifluoromethylsulfonyl)imide [EBPip+] [NTf2−] and 1-ethyl-1-octylpiperidinium bis (trifluoromethylsulfonyl)imide [EOPip+] [NTf2−]). Using the extractant N,N′-dimethyl-N,N′-dioctylhexylethoxymalonamide (DMDOHEMA), the effect of pH was investigated in detail to recover extraction mechanisms. The use of ILs as the organic solvent instead of n-dodecane, greatly enhances extraction efficiency, and an ionic liquid with a shorter alkyl chain [EBPip+] [NTf2−] provides higher extraction than [EOPip+] [NTf2−]. The mechanistic study points out that for low nitric acid concentrations ([HNO3] ≤ 0.01 M), metal is extracted via a cation of the ionic liquids, while for higher nitric acid concentrations ([HNO3] ≥ 1.0 M), extraction occurs through pure solvation mechanism of DMDOHEMA as in conventional diluents. This latter case is of high interest for applications, as higher extraction can be obtained without any loss of ILs by ion exchange mechanisms.
1. Introduction
Rare earth elements (REEs) are widely used due to their unique chemical, electrical, magnetic, optical and catalytic properties. They are applied for a large number of essential technological applications such as in hard disc drives, LCD panels, hybrid vehicles, wind turbines, surgical robots, etc.1 Their high economic importance together with their uncertain supply (caused by a fast market evolution), have led to considering REEs as critical metals, generating great interest in the development of new efficient processes for their recycling from electrical and electronic wastes.1 Whatever the source, extraction of rare earth elements involves dissolution and solvent extraction processes that imply the use of acids and organic solvents, generating large amounts of chemical wastes, volatile organic compound release in the atmosphere, water pollution and heat discharge.2In this context, ionic liquids (ILs) are more and more considered as the diluent or solvent in REEs extraction processes.3 Thanks to their ionic nature, ILs present negligible vapor pressure, non-flammability, a high degree of solubility for organic and inorganic substances, and high thermal and chemical stability. For these reasons, ionic liquids arise as a promising alternative to common organic solvents. Their favorable properties are moreover tunable with the adjustment of their cation/anion pair or by the grafting of functional groups on the ILs.
In the field of solvent extraction, ILs were used thirty years ago for the extraction of substituted-benzene derivatives.4 One of the first studies concerning metal extraction in ionic liquids was carried out by Dai et al. in 1999,5 who observed that crown ethers extract strontium with a higher extraction efficiency in ionic liquids than in classical solvents. From this study, the use of ionic liquids as organic phase for metal extraction has notably increased.6–8 Many references report ILs systems for which metal extraction is either higher or lower than in conventional diluents. However, they often don't provide further understanding and prediction of this however essential result.
Extraction of metals in ILs can occur through different mechanisms: (i) the ILs exchange its anion or cation with the metal,9,10 (ii) similar mechanisms than those observed with extractants in classical diluents,11,12 (iii) combinations of these two possibilities.13 However, the description of these mechanisms are not always sufficient to explain and predict the higher or lower extraction efficiency obtained in ILs. In several studies, ion exchange with the IL was shown to induce higher extraction efficiency than in conventional diluents.14,15 This apparently interesting case is however not optimal for application, as the diluent IL is gradually consumed during the extraction. In other cases, higher extraction is obtained when no ion exchange is involved, or on the contrary lower extraction may be achieved while ion exchange is involved.
In all cases, influence of the extraction system parameters on extraction mechanisms and efficiency is essential. For instance, the acidity of the aqueous phase has in some cases no effect on metal extraction,16 while it may drastically enhance or vanish metal extraction for other systems.17,18 Similarly, the alkyl chain length carried by the IL cation has often a strong influence on extraction.19,20
These two essential parameters were therefore studied in details in this study for REE extraction with a malonamide extractant solubilized in two ionic liquids [EBPip+] [NTf2−] and [EOPip+] [NTf2−]. Piperidinuim ionic liquids were chosen because they are known to be good diluents for solvent extraction: depending on their alkyl chain length they are sufficiently hydrophobic to be poorly soluble in aqueous phase, while maintaining a sufficiently low viscosity.21 They were moreover shown to allow a possible electro-deposition of the metal after extraction.22
Malonamides have been used in the DIAMEX solvent extraction process for the recovery of Ln(III) and An(III) in nuclear fuel reprocessing with a high efficiency and stability in nitric acid media.23 Among the malonamides studied for the DIAMEX process, N,N′-dimethyl-N,N′-dioctylhexylethoxymalonamide (DMDOHEMA) has shown a high extraction ability for REEs in organic solvents as benzene,24 n-heptane25 and n-dodecane.26 DMDOHEMA exhibits a polar head and hydrophobic tails, which provide amphiphilic properties and allow the complexation of the ligands with specific cations at the interface and the structuration of the organic phase through the formation of reverse micelle like aggregates.27–29 While several studies have been carried out for REEs extraction in ILs with various extractant molecules (as CMPO,30 DODGAA,31 D2EHPA and HDEHDGA,32 DEHPA33 or mixture of extractants34), to our knowledge, no investigation is reported for REEs extraction with DMDOHEMA in ILs.
In this paper, we report DMDOHEMA extraction of two REEs, La and Eu (chosen as typical REEs), and their separation toward the competitor Fe, in dodecane and in the ionic liquids [EBPip+] [NTf2−] or [EOPip+] [NTf2−]. To account for the ion exchange mechanisms, the equilibrium concentrations of H2O, H+ and NO3− in the organic phase, and of ILs cations and anions in the aqueous phase were determined for various nitric acid concentrations. From these results, some insights on the extraction mechanisms involved in the studied ILs have been proposed and compared to those in dodecane.
2. Experimental
2.1. Materials
High purity grade ILs (≥99.5%), 1-ethyl-1-butylpiperidinium bis (trifluoromethylsulfonyl)imide [EBPip+] [NTf2−] and 1-ethyl-1-octylpiperidinium bis (trifluoromethylsulfonyl)imide [EOPip+] [NTf2−] was obtained from Solvionic. N,N′-Dimethyl-N,N′-dioctylhexylethoxymalonamide (DMDOHEMA) was provided by Pharmasynthèse. Fig. 1 displays the chemical structure of the ionic liquids and of the extractant used in this study. Dodecane and iron(III) nitrate nonahydrate (99%) were purchased from Acros Organics, lanthanum(III) nitrate hexahydrate (99.99%) and europium(III) nitrate pentahydrate (99.99%) from Sigma Aldrich, and nitric acid from Carlo Erba. All chemicals were used without further purification. Deionized water (Milli-Q) was employed for the aqueous phase preparation.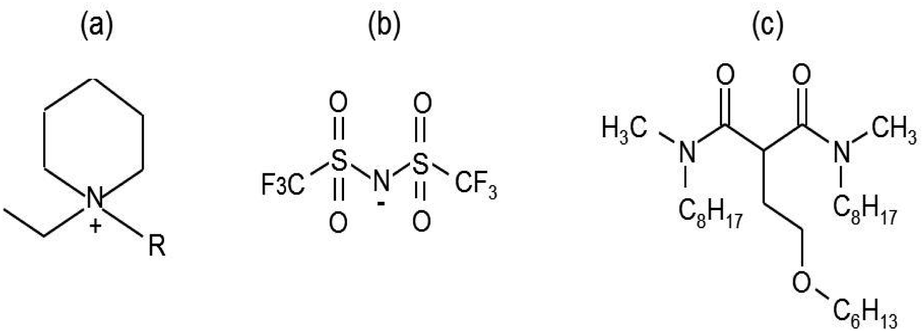 | ||
| Fig. 1 Chemical structures of the ILs and the extractant used in this work. (a) 1-ethyl-1-alkyl piperidinium cation R = butyl (C4H9) or octyl (C8H17) alkyl chain, (b) bistriflimide anion (NTf2−), (c) DMDOHEMA. | ||
2.2. Methods
Aqueous stock solutions containing 250 mg L−1 of each metallic ion were prepared by dissolving the metal nitrates, La(NO3)3, Eu(NO3)3 and Fe(NO3)3, in a nitric acid solution. Organic solutions of DMDOHEMA in ILs or dodecane were prepared and contacted to an equal volume of the aqueous phase (typically, 3 mL of each solutions were introduced in a centrifuge tube). The biphasic system was shaken mechanically in a tube rotator for 1.5 hours at 20 rpm and then separated after centrifugation at 4500 rpm for 30 minutes. All extraction experiments have been carried out at room temperature (25 ± 1 °C).2.3. Quantification
Metal and sulphur (from bistriflimide anion) concentrations in the aqueous phase were determined before and after extraction, by Inductively Coupled Plasma Atomic Emission Spectrophotometry (ICP-AES, Spectro ARCOS spectrometer). The distribution coefficients of the metals (D) and the selectivity (S) towards iron were calculated as follows:
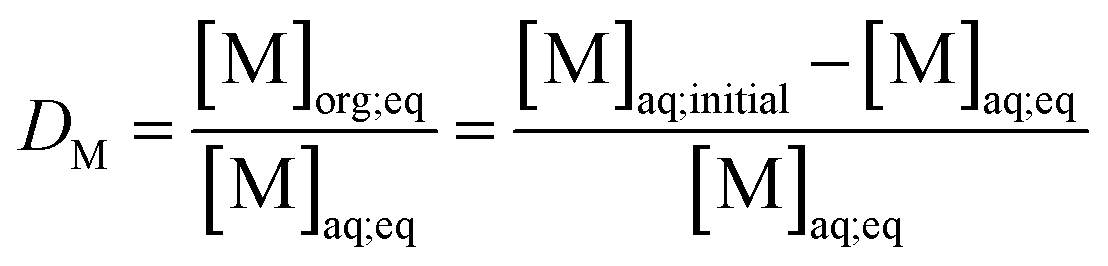 | (1) |
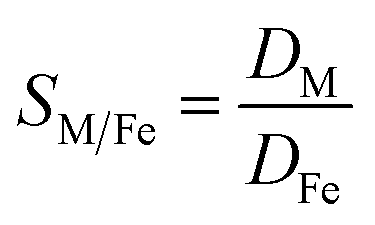 | (2) |
The total carbon concentration (TOC) in the aqueous phase was determined using a Shimadzu TOC-V CSH analyzer based on a 680 °C combustion catalytic oxidation/NDIR method.
The absorbance of NO3− for the aqueous phase before and after contact with the organic phase, were determined using UV-visible spectrophotometer (Varian Cary 50) at 220 nm, and their corresponding concentrations were calculated with Beer's law obtained from standard concentrations of KNO3.
Water extraction was evaluated by Karl Fischer titration of the organic phase using a Metrohm Titrando 809 device, titrations were performed in triplicate, to reduce the uncertainty of the determination. Acid concentration in the organic phase was determined by titration using the same device with 0.001 M NaOH aqueous solution.
3. Results
3.1. Metal extraction
REEs extraction was preliminary studied in the pure dodecane and ionic liquids to distinguish their extraction ability from the one of the extractant. For 3.0 M nitric acid solutions, no metal extraction occurred in absence of extractant in agreement with a study previously reported.35Effect of DMDOHEMA concentration was further investigated. Fig. 2 shows that for all the diluent studied, the distribution coefficients of La3+, Eu3+ and Fe3+ increase with the concentration of extractant. Extraction ability of DMDOHEMA in the various diluents respects the following order for the two REEs tested: dodecane < [EOPip+] [NTf2−] < [EBPip+] [NTf2−]. As already observed by Kubota and co-workers in a different system,36 a higher extraction efficiency of REEs is obtained in ionic liquids with a shorter alkyl chain. It is also important to notice that a good selectivity toward iron was obtained in both ionic liquids, while in dodecane, REEs were less selectively extracted (Table S1†).
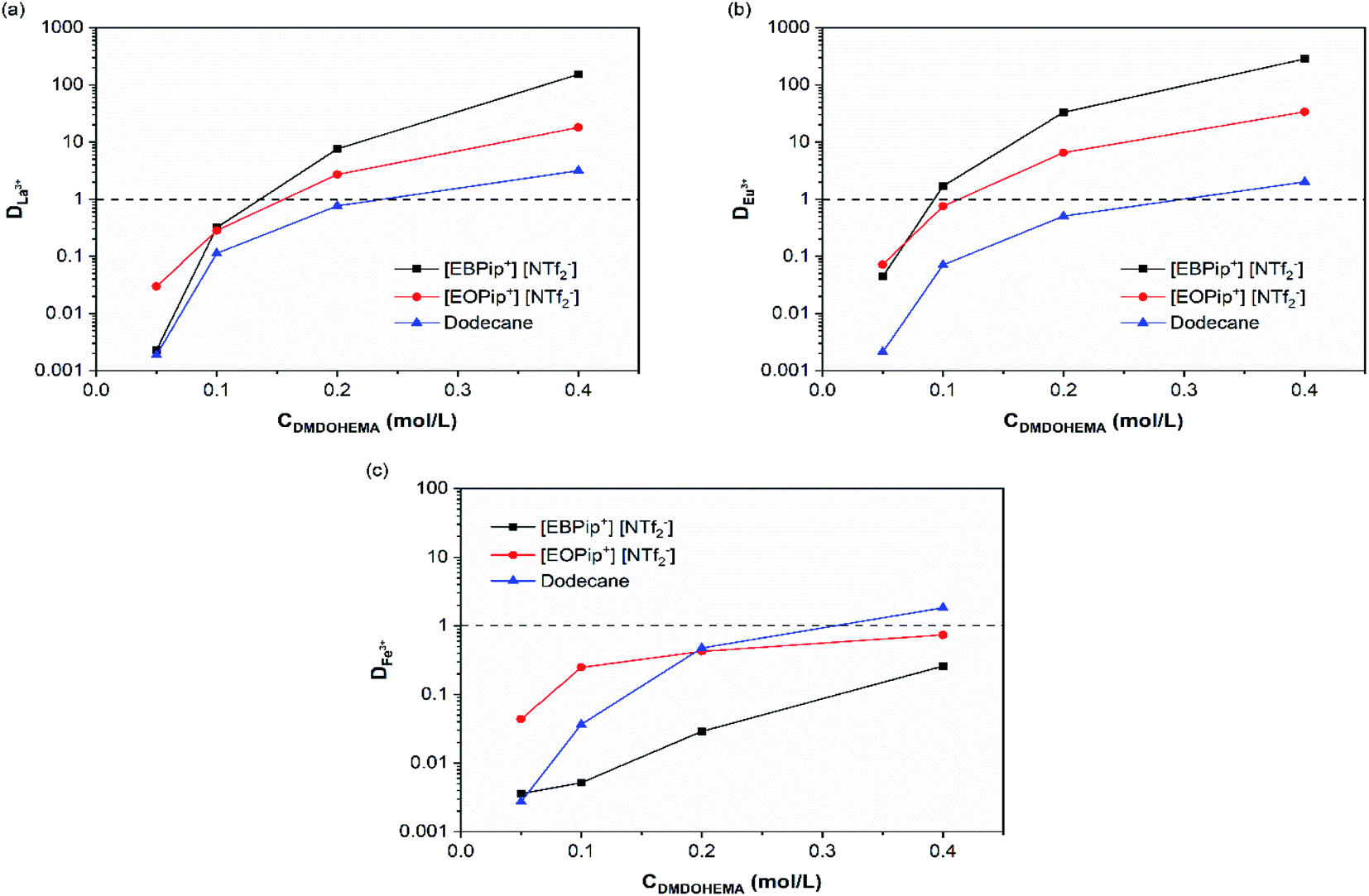 | ||
| Fig. 2 Distribution coefficients as a function of the extractant concentration for [HNO3] = 3.0 M: (a) La3+, (b) Eu+3 and (c) Fe3+. | ||
To evaluate the effect of acidity, distribution coefficients of La, Eu and Fe were determined for DMDOHEMA concentration of 0.05 and 0.4 M at several nitric acid concentrations. In dodecane (Fig. 3c), DMDOHEMA improves its extraction ability for high HNO3 concentration, which is consistent with literature: as a solvating extractant, metal extraction by DMDOHEMA requires anion co-extraction to neutralize the charge of the metallic cation.37 A large amount of nitrate ions is therefore necessary in the aqueous phase to obtain a high metal extraction efficiency. In the two ILs, distribution coefficients of La3+ and Eu3+ decrease significantly with HNO3 concentration but remains higher than 50 for [DMDOHEMA] = 0.4 M. La3+ and Eu3+ extraction is therefore complete on the whole range of nitric acid concentrations and remains 10 times higher than in dodecane (Fig. 3a and b).
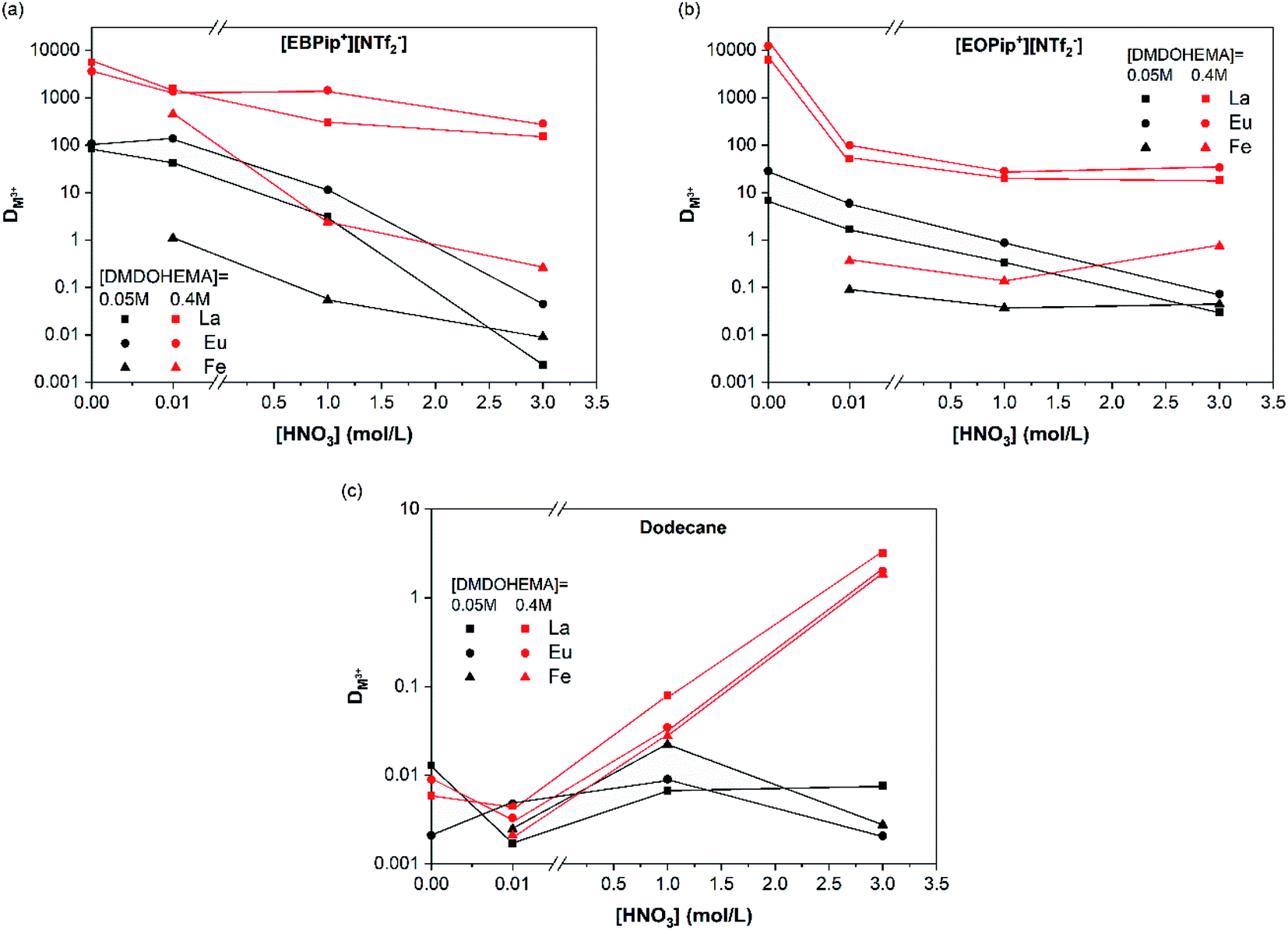 | ||
| Fig. 3 Distribution coefficients as a function of the nitric acid concentration for [DMDOHEMA] = 0.05 M (black symbols) and 0.4 M (red symbols), in: (a) [EBPip+] [NTf2−], (b) [EOPip+] [NTf2−] and (c) dodecane. | ||
In order to determine the stoichiometry of the DMDOHEMA–M3+ complexes, a graphical slope analysis was conducted for each nitric acid concentration. Logarithm of distribution coefficients are plotted as a function of the logarithm of DMDOHEMA concentrations for [HNO3] = 1 and 3.0 M in Fig. S1.† For [EBPip+] [NTf2−] and [EOPip+] [NTf2−], a change in the slope appears at [HNO3] = 1.0 M. Below this concentration, DMDOHEMA forms 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with La3+ and Eu3+ in [EBPip+] [NTf2−] and 2:1 complexes in [EOPip+] [NTf2−]. For higher nitric acid concentration, the stoichiometry of the complexes is 4:1 and 3:1 for [EBPip+] [NTf2−] and [EOPip+] [NTf2−] respectively. These results show that more extractant molecules take part to the complexes when the alkyl chain length of the ionic liquid is smaller. This behaviour was already noticed by Shen et al. with imidazoliun ILs.38 In dodecane, the obtained slope values suggest the formation of 2:1 DMDOHEMA–M3+ complexes on the whole range of nitric acid concentration, which is consistent with previous studies in this diluent.37
:1 complexes with La3+ and Eu3+ in [EBPip+] [NTf2−] and 2:1 complexes in [EOPip+] [NTf2−]. For higher nitric acid concentration, the stoichiometry of the complexes is 4:1 and 3:1 for [EBPip+] [NTf2−] and [EOPip+] [NTf2−] respectively. These results show that more extractant molecules take part to the complexes when the alkyl chain length of the ionic liquid is smaller. This behaviour was already noticed by Shen et al. with imidazoliun ILs.38 In dodecane, the obtained slope values suggest the formation of 2:1 DMDOHEMA–M3+ complexes on the whole range of nitric acid concentration, which is consistent with previous studies in this diluent.37
3.2. Water and acid extraction
The amount of water and H+ uptake in the organic phase were determined by Karl Fischer and acido–basic titration after contact with the aqueous phase. Fig. 4 shows the results obtained as a function of the DMDOHEMA concentration for a nitric acid concentration of 3.0 M in the aqueous phase.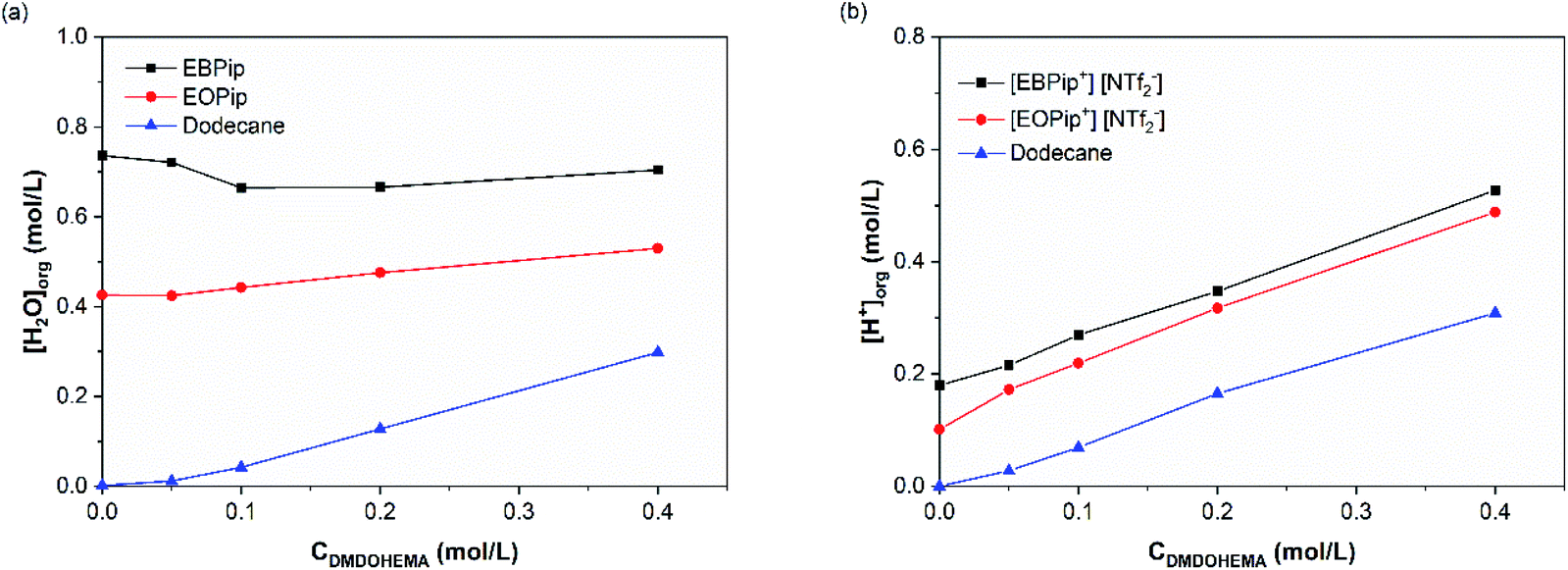 | ||
| Fig. 4 Water (a) and H+ (b) content in the organic phase, after metal extraction, as a function of DMDOHEMA concentration for [HNO3] = 3.0 M. | ||
In dodecane, water extraction starts increasing for a DMDOHEMA concentration of ca. 0.05–0.1 M which is consistent with an extraction induced by the extractants aggregation at the so called Critical Aggregation Concentration (CAC).39 It has indeed been demonstrated in conventional organic solvents, that malonamides extract water in the organic phase thanks to the self-assembly of the extractant molecule into reverse micelle-like aggregates, that are able to solubilize water, acid and polar species in their polar core.40,41 It was also shown that the amounts extracted depend on the initial nitric acid concentration in the aqueous phase which has the property to nucleate the aggregates.42 Being located in the core of the extractant aggregates, extracted water plays therefore an important role, as it contributes to the solvation of the extracted ionic species.24,42
For a constant DMDOHEMA concentration, it can also be noticed that water and acid contents remain practically constant in the ionic liquids and dodecane up to [HNO3] = 1.0 M, and increase for higher nitric acid concentrations (Fig. S2†). This result was already observed by Visser et al. with [C4mim] [NTf2−] contacted to a nitric acid solution.43 Owing to their basic character, malonamides can be protonated by the acid extracted during the solvent extraction process. When nitric acid is present in the aqueous phase, nitrates ions are co-extracted together with H+ forming several HNO3-malonamide adducts, such as: HNO3-L, (HNO3)2-L and HNO3-L2.44,45
As shown in Fig. S2,† a noticeable increase in the acid extraction occurs when 1.0 M HNO3 concentration is reached, coinciding with a higher water concentration in the organic phase. This result confirms that aggregates are nucleated by the acid, and that the formed aggregates further enhance acid extraction.
Looking at the slope values obtained for increased DMDOHEMA concentration in dodecane at 3.0 M HNO3 (Fig. S3†), presence of 1:1 acid–DMDOHEMA complexes is evidenced in the organic phase. In order to discuss all these behaviours, extraction mechanisms are evaluated in more details in the following part.
3.3. Extraction mechanism
To elucidate the extraction mechanism in the studied ionic liquids, we examined the ion and cation exchange of the ionic liquids with the metal extracted as a function of nitric acid concentration. For this, sulfur and total carbon concentrations were determined in the aqueous phase, by ICP and TOC, to evaluate the concentrations of bistriflimide anion (NTf2−) and of EBPip+ or EOPip+ cations transferred to the aqueous phase. Concomitantly, H+ and NO3− concentrations where measured in the organic phases by titration and UV-Vis spectrophotometry. To understand independently the role of each parameter, we evaluated individually the three following cases: ion transfer in absence of extractant and metal; ion transfer with extractant and without metal; ion transfer with extractant and metal extracted.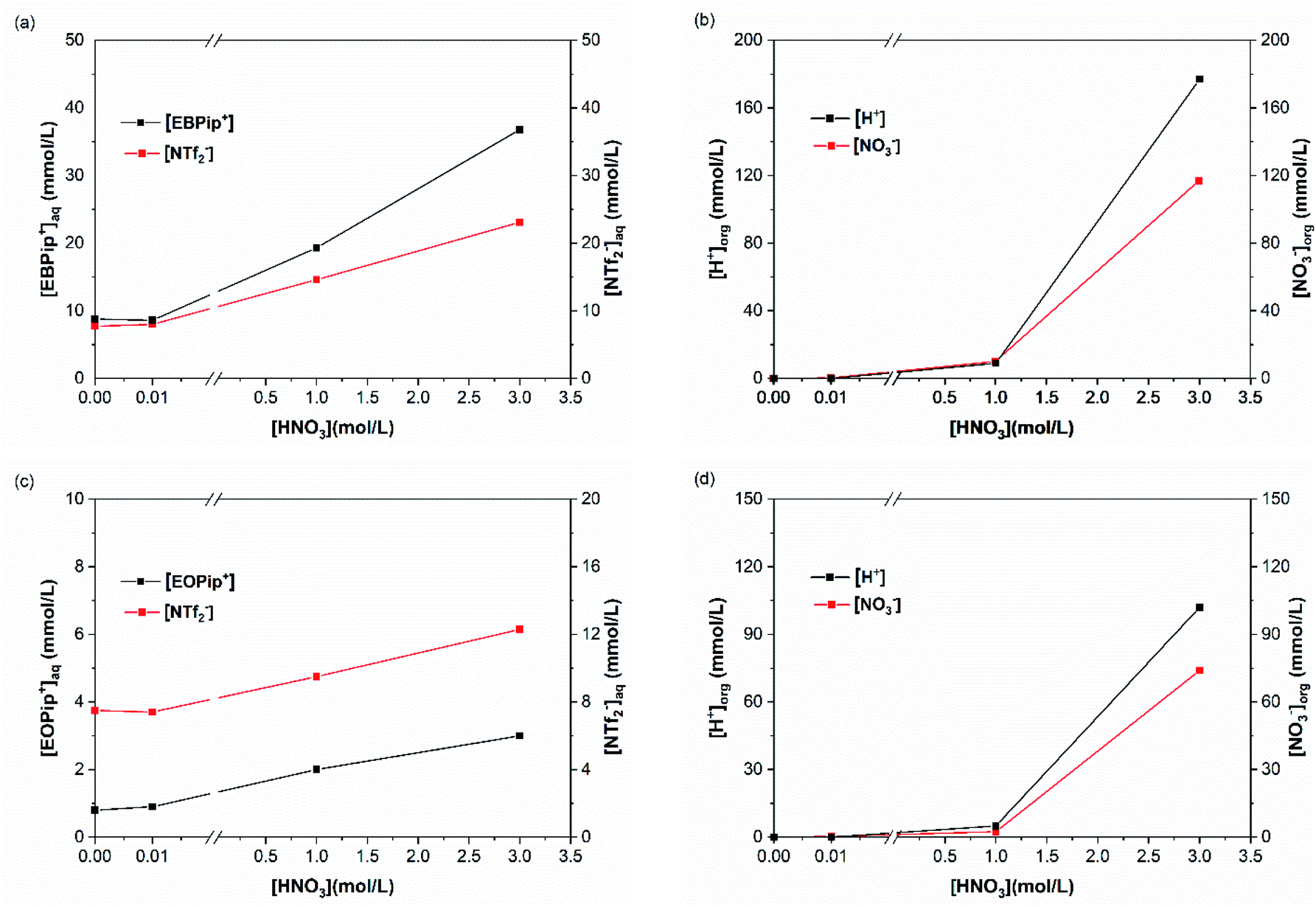 | ||
| Fig. 5 (a) Pip+ and NTf2− concentrations in the aqueous phase, (b) H+ and NO3− concentrations in the organic phase for [EBPip+] [NTf2−]; (c) Pip+ and NTf2− concentrations in the aqueous phase, (d) H+ and NO3− concentrations in the organic phase for [EOPip+] [NTf2−], as a function of the nitric acid concentration, in absence of extractant and metallic cations. | ||
In agreement with a previous study,50 our results show that below a nitric acid concentration of 0.01 M, a negligible amount of IL is either transferred or dissolved in the aqueous phase. Above this concentration, the amount of anions and cations transferred to the aqueous phase increases drastically. This phenomenon can be attributed to an ionic exchange between the ionic liquid and the nitric acid.
However, it has to be noticed that information obtained from the sulfur concentration allowed to determine the overall bistriflimide concentration in the aqueous phase, but not to distinguish between the protonated (HNTf2) and ionic (NTf2−) forms. Another mechanism has therefore to be considered to account for the increase of bistriflimide anions in the aqueous phase. As already proposed in literature,51,52 formation of protonated species (HNTf2) may become significant in the aqueous phase for high acidic conditions:
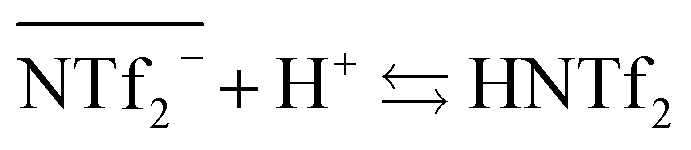 | (3) |
For increased nitric acid concentrations, equilibrium (3) shifts toward the right side of the equation, increasing the ionic liquid solubility in the aqueous phase. This explains the greater solubility of ILs for high HNO3 concentrations. However, the amount of H+ and NO3− transferred to the ionic liquid are higher than the Pip+ and NTf2− transferred in the aqueous phase. We should therefore not only consider the ionic exchange, but also take into account the capacity of the ionic liquids to introduce acid species in their structure.53 It has been reported elsewhere that when contacting [C4mim+] [NTf2−] to HNO3 aqueous solution of concentrations higher than 2 M, the transfer of H+ is of 6% of the initial acid concentration.49,54 In our case, when [HNO3] = 3.0 M we observe a H+ transfer of 6% and 3.5% of the initial acid concentration for [EBPip+] [NTf2−] and [EOPip+] [NTf2−] respectively. For [HNO3] = 1.0 M, quantity of acid extracted in the ILs is within the experimental uncertainty, and below this nitric acid concentration, no H+ extraction was detected.
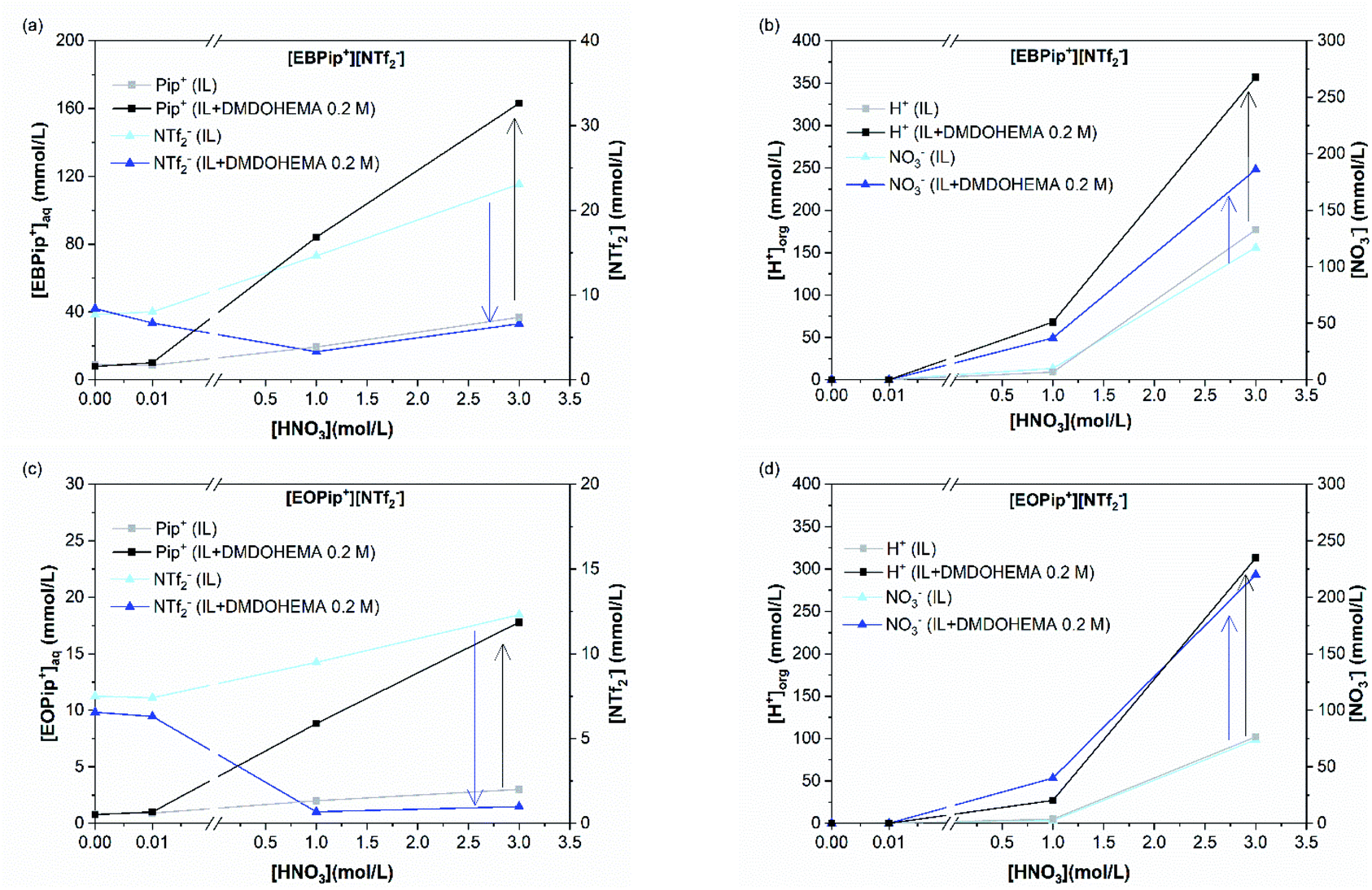 | ||
| Fig. 6 (a) Pip+ and NTf2− concentrations in the aqueous phase, (b) H+ and NO3− concentrations in the organic phase for [EBPip+] [NTf2−]; (c) Pip+ and NTf2− concentrations in the aqueous phase, (d) H+ and NO3− concentrations in the organic phase for [EOPip+] [NTf2−], as a function of the nitric acid concentration, without and with DMDOHEMA 0.2 M. | ||
H+ and NO3− concentrations in ILs show that addition of DMDOHEMA further increases these ions transfer in the ionic liquids which becomes significant from a [HNO3] concentration of 1.0 M. Such an additional transfer of H+ in presence of the extractant has also been reported with N,N′-dimethyl-N,N′-dibutylmalonamide (DMDBMA).54
Considering that malonamides extractants can behave as acidic extractants,44,45 and the increase of piperidinium cations in the aqueous phase in presence of DMDOHEMA, the following chemical model can be proposed involving the extractant protonation and a cationic exchange between protonated DMDOHEMA and IL cations (eqn (4)):
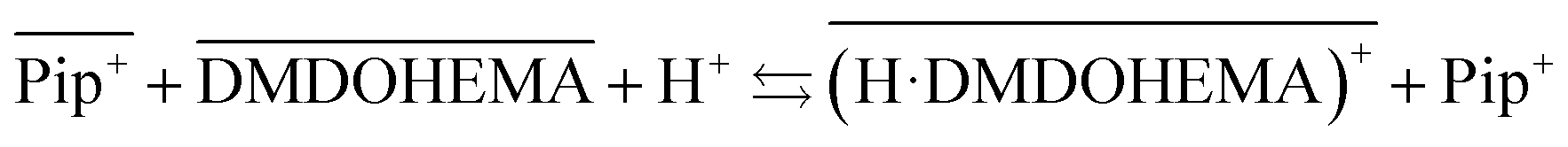 | (4) |
Fig. 6 shows also that amount of H+ is higher in [EBPip+] [NTf2−] than in [EOPip+] [NTf2−]. Transfer of H+ from the acidic aqueous phase to the ILs may therefore be carried out through a cationic exchange mechanism with Pip+, which depends on the solubility of the IL cation in the aqueous phase. In our case, due to its shorter alkyl chain, solubility of EBPip+ cation in the aqueous phase is indeed higher than the one of EOPip+.
When HNO3 concentration is higher than 0.01 M, introduction of DMDOHEMA causes a decrease of bistriflimide concentration in the aqueous phase (Fig. 6 and S4†), which may be due to a protonation competition between NTf2− and DMDOHEMA (eqn (3) and (4)). For higher DMDOHEMA concentrations, the mechanism described by eqn (4) becomes dominant, reducing the HNTf2 form and therefore the total amount of bistriflimide in the aqueous phase.
Taking into account all the results obtained, it can be considered that two main mechanisms are involved in the ion exchange, which strongly depend on the initial nitric acid concentration in the aqueous phase. These mechanisms are studied in presence of metal and as a function of nitric acid concentration in the following section.
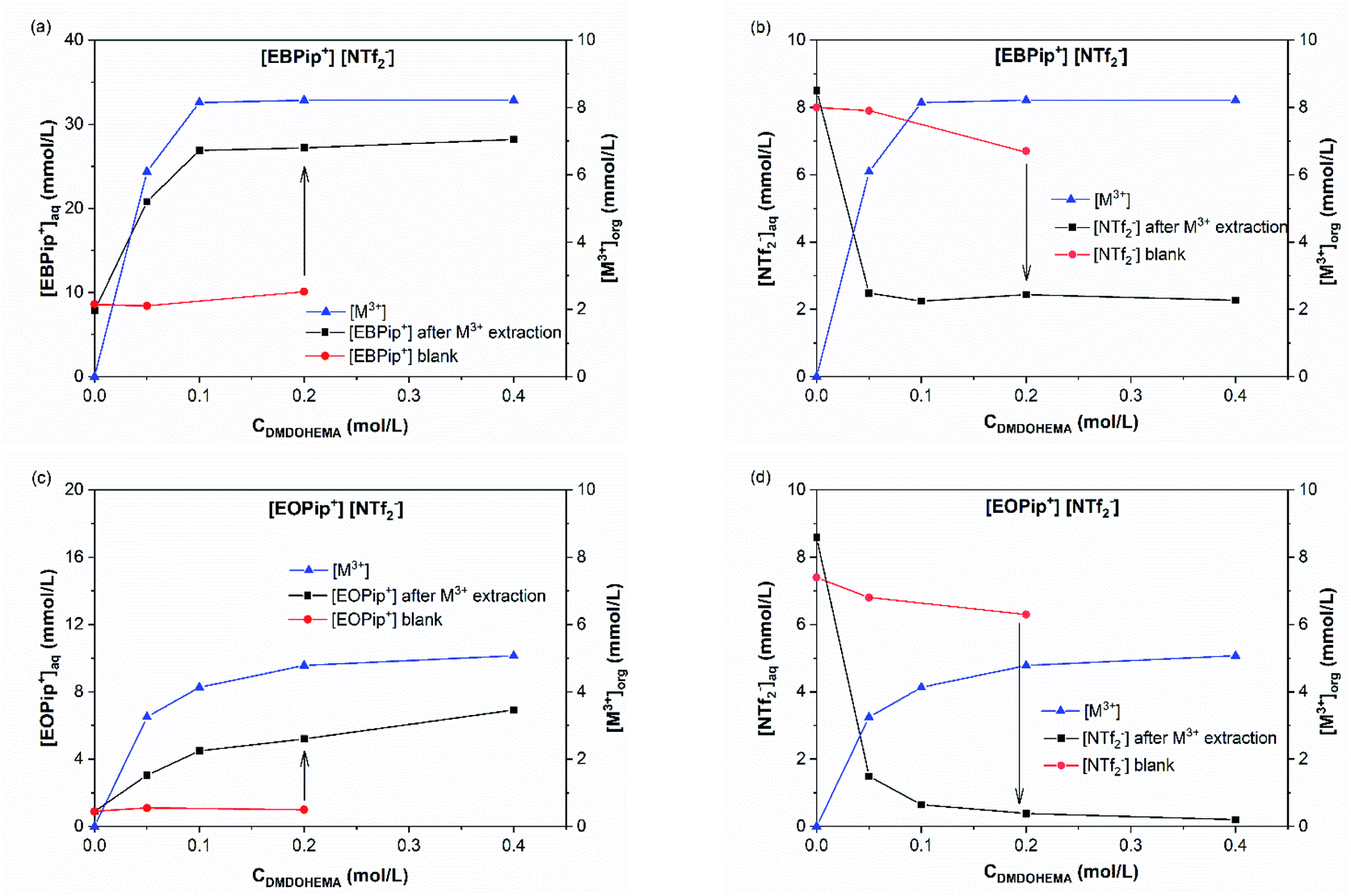 | ||
| Fig. 7 (a) Pip+ and (b) NTf2− concentrations in the aqueous phase for [EBPip+] [NTf2−]; (c) Pip+ and (d) NTf2− concentrations in the aqueous phase for [EOPip+] [NTf2−], as a function of the DMDOHEMA concentration and for an initial nitric acid concentration of 0.01 M in the aqueous phase. M3+ is shown on each graph and refers to the total concentration of extracted metal in the organic phase (La, Eu and Fe). | ||
Results obtained in absence of metal are also shown in this figure (blank) to highlight the effect of metal addition on the various ion exchanges. Fig. 7a and c show that piperidinium cation concentration remains constant with DMDOHEMA when no metal is extracted (red points), while it increases markedly when metal is extracted (black points). This suggests a cationic exchange between the piperidinium cation and the metal extracted. As shown in Fig. 7b and d, a notable decrease in the NTf2− concentration is also observed with DMDOHEMA concentration when metals are extracted (black points). This decrease is concomitant and follows the exact opposite trend of the metal extraction. This single behaviour allows rejecting the possibility of an extraction mechanism involving the exchange of NTf2− anions. In addition, as [NTf2−] is barely affected by the extractant concentration when no metal is extracted (red points), it indicates that the change in the concentration of bistriflimide anion is induced by the extraction process, suggesting that this anion acts in the organic phase as a counter ion in an ion-pair mechanism. As shown in Fig. S5,† a similar behaviour is observed with the aqueous concentrations of piperidinium cation and bistriflimide anion of both ionic liquids, when the nitric acid concentration is 0 M.
In order to quantify the concentration of piperidinium cation(Pip+) and bistriflimide anion (NTf2−) in the aqueous phase related to the metal extraction. Process, we calculated the difference between the concentrations in the blank samples and in the metallic extraction performance samples. Results calculated for both nitric acid concentrations 0 and 0.01 M are summarized in Table 1. For a nitric acid concentration of 0.01 M and at metal extraction saturation (0.2 M DMDOHEMA), it can be estimated that for each mole of metal extracted M3+, two moles of piperidinium cation are transferred to the aqueous phase when [EBPip+] [NTf2−] is used as organic solvent. For [EOPip+] [NTf2−] approximately one mole of EOPip+ is additionally transferred in the aqueous phase for each mole of M3+ extracted in the organic phase. Conversely, there is a reduction in the NTf2− concentration in the aqueous phase when the metal is extracted. The molar relation between metal extracted and this loss of NTf2− is about 2:1 and 1:1 for [EBPip+] [NTf2−] and [EOPip+] [NTf2−].
| Ionic liquid | [HNO3] (M) | [DMDOHEMA] (M) | Δ[M3+] (mM) | Δ[Pip+] (mM) | Δ[NTf2−] (mM) |
|---|---|---|---|---|---|
| [EBPip+] [NTf2−] | 0 | 0.05 | 3.2 | 6.2 | 4.1 |
| 0.2 | 3.2 | 5.3 | 3.8 | ||
| 0.01 | 0.05 | 6.1 | 12.4 | 5.4 | |
| 0.2 | 8.2 | 17.1 | 4.3 | ||
| [EOPip+] [NTf2−] | 0 | 0.05 | 3.0 | 1.6 | 5.4 |
| 0.2 | 3.2 | 2.2 | 5.9 | ||
| 0.01 | 0.05 | 3.2 | 1.9 | 5.3 | |
| 0.2 | 4.8 | 4.2 | 6.2 |
The results obtained indicate therefore that metals La, Eu and Fe are extracted through two simultaneous mechanisms, coordination with cationic exchange and ion pair formation, that can respectively be described as in eqn (5) and (6):
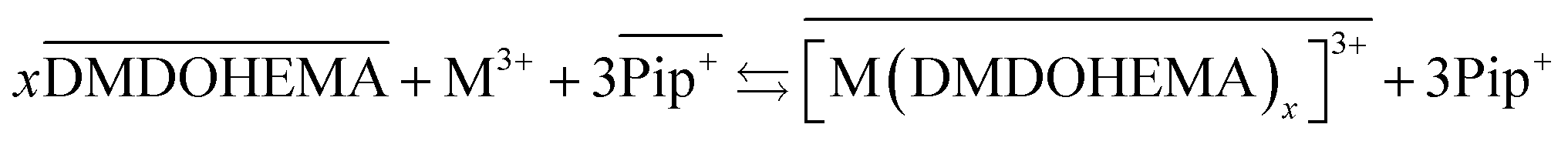 | (5) |
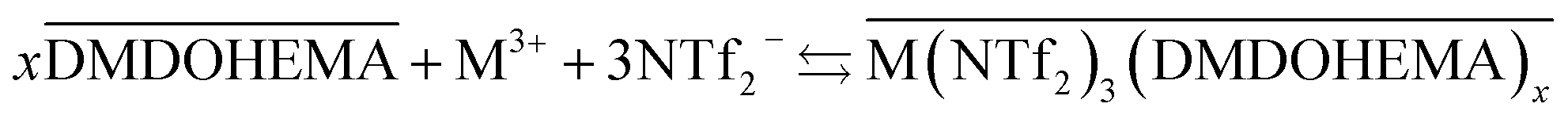 | (6) |
It is important to note that the so-called ion-pair mechanism involves the ionic liquid anion participation as counter ion for the metal extraction while in the solvation mechanism nitrate anion acts as counter ion.
As indicated in the first part of this study from the slope method analysis, the stoichiometric coefficient DMDOHEMA-metal noted “x” in eqn (5) and (6), is of 3 for [EBPip+] [NTf2−] and 2 for [EOPip+] [NTf2−] when [HNO3] is below 1.0 M. The quantity of metal extracted through a cationic mechanism can therefore be determined by taking into account the piperidinum cation concentration in the aqueous phase due to the metal extraction (Δ[Pip+]) and the stoichiometry indicated in eqn (5). By comparing this quantity with the total metal extracted (Δ[M3+]), it can be estimated for [EBPip+] [NTf2−] that around 65% of the metal extraction is carried out by a cationic exchange mechanism (as in eqn (5)), while the remaining 35% of metal extracted are transferred through an ion pair mechanism (as in eqn (6)). For [EOPip+] [NTf2−] the percentage of metal extracted by cationic exchange is only of 25%. A similar mixed mechanisms involving both cation exchange and ion pairing has been reported by Messadi et al. for the extraction of Cu(II) in ammonium based ionic liquids.55 With a similar approach, the proportion of these two mechanisms is estimated for higher nitric concentration in the following section.
(b) Metallic extraction mechanism for high nitric acid concentration ([HNO3] = 1.0 and 3.0 M). Following the same strategy, the transferred concentrations of [M3+]org, [NTf2−] and [Pip+] were measured for 1.0 and 3.0 M nitric acid concentration. The case [HNO3] = 3.0 M is illustrated in Fig. 8 (for [HNO3] = 1.0 M see ESI material Fig. S6†).
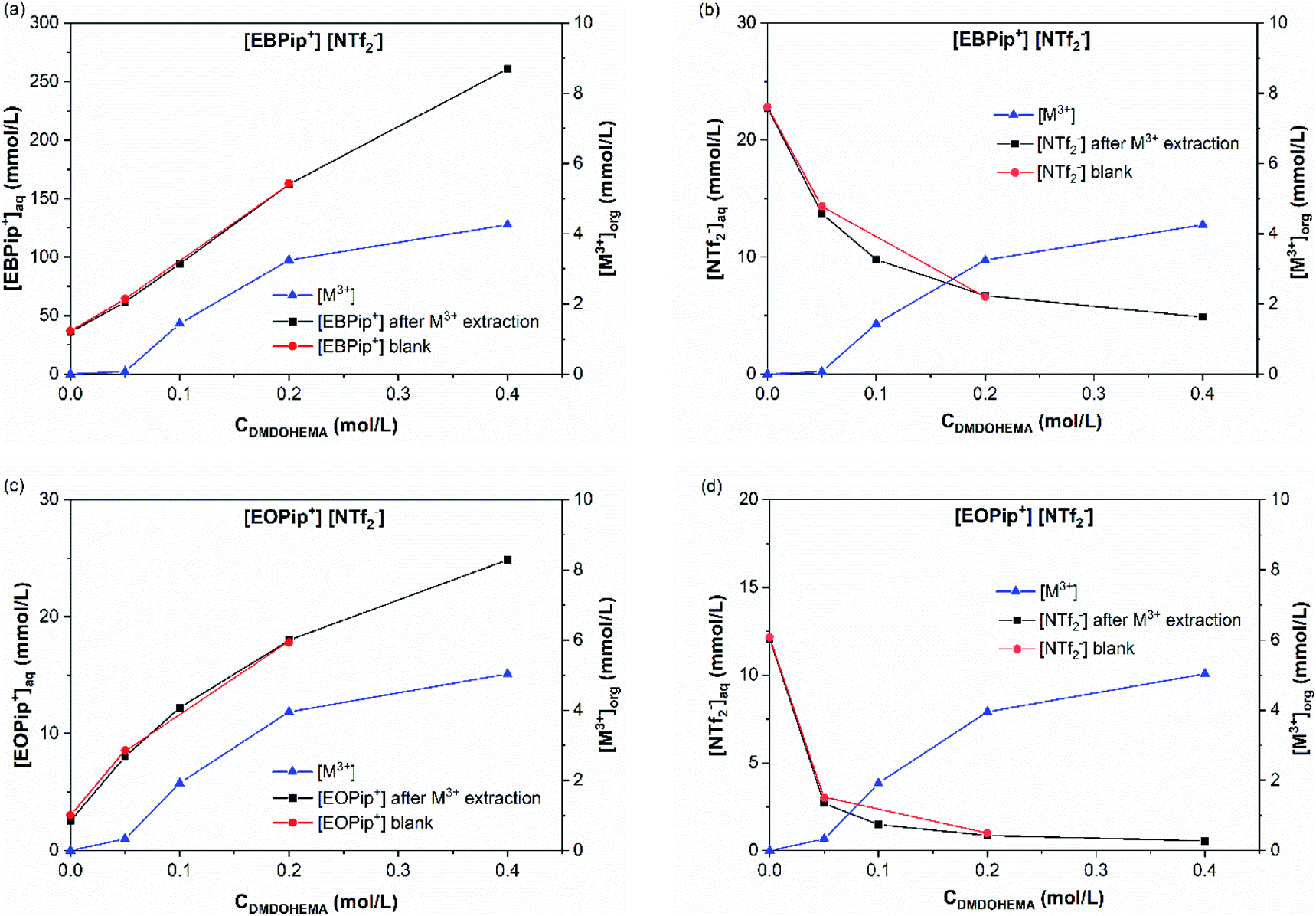 | ||
| Fig. 8 (a) Pip+ and (b) NTf2− concentrations in the aqueous phase for [EBPip+] [NTf2−]; (c) Pip+ and (d) NTf2− concentrations in the aqueous phase for [EOPip+] [NTf2−], as a function of the DMDOHEMA concentration and for an initial nitric acid concentration of 3.0 M in the aqueous phase. M3+ is shown on each graph and refers to the total concentration of extracted metal in the organic phase (La, Eu and Fe). | ||
Fig. 8a and c show that piperidinium cation concentration in the aqueous phase increases with the DMDOHEMA concentration when metal is extracted (black points). However, exactly the same increment is observed with DMDOHEMA concentration when no metal is extracted (red points). As explained in the previous section, piperidinium cations are here exchanged with the acid extracted, but are not further transferred to the aqueous phase to extract the metal.
As previously, Fig. 8b and d show a reduction of the NTf2− concentration when the metal is extracted (black points), which indicates that the extraction process is not carried out through an anionic exchange with the anion of the ILs. As for piperidinium cations, there are no differences between the [NTf2−] transfer before (red points) and after metal extraction. This indicates that the decrease in NTf2− anions concentration in the aqueous phase is only induced by the extractant content and not related to metal extraction. Unlike the systems with [HNO3] ≤ 0.01 M, NTf2− anions do not play an active role in the metal extraction mechanism.
As in the previous section, the concentration variations of piperidinium cation (Pip+) and bistriflimide anion during metal extraction (NTf2−) are compared with the extracted metal concentration in Table 2. It shows that for [HNO3] = 1.0 M, only a small quantity of piperidinium cation is transferred to the aqueous phase when metal is extracted. It suggests that most of the metal is in this case extracted by another mechanism than ion exchange. For [HNO3] = 3.0 M no changes in the [Pip+] is detected, which is also not consistent with a cationic exchange mechanism. It can globally be observed that the quantity of metal extracted through a cationic exchange is gradually decreased with the HNO3 concentration in the aqueous phase.
| Ionic liquid | [HNO3] (M) | [DMDOHEMA] (M) | Δ[M3+] (mM) | Δ[Pip+] (mM) | Δ[NTf2−] (mM) |
|---|---|---|---|---|---|
| [EBPip+] [NTf2−] | 1.0 | 0.05 | 3.3 | 0.5 | 0.1 |
| 0.2 | 6.4 | 5.9 | 0.5 | ||
| 3.0 | 0.05 | 0.1 | 2.5 | 0.6 | |
| 0.2 | 3.2 | 0.9 | 0.1 | ||
| [EOPip+] [NTf2−] | 1.0 | 0.05 | 1.4 | 0.3 | 0.3 |
| 0.2 | 3.5 | 1.7 | 0.2 | ||
| 3.0 | 0.05 | 0.3 | 0.6 | 0.3 | |
| 0.2 | 4.0 | 0.2 | 0.1 |
To account for La, Eu and Fe transfer for [HNO3] ≥ 1.0 M, it has to be considered that the extraction is carried out by a pure solvation mechanism of DMDOHEMA as it is known to operate in conventional diluent as dodecane. This mechanism is described in eqn (7), as it has been reported for lanthanides extraction in DMOHEMA/dodecane system.26
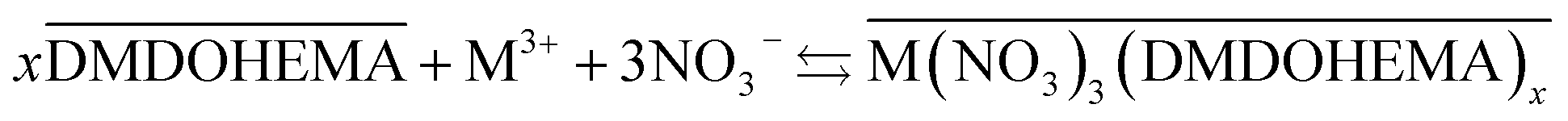 | (7) |
This result is in agreement with a study reported on actinides extraction from nitric acid medium into ionic liquids, where the extraction mechanism was also observed to shift from cationic exchange to solvation mechanism for higher HNO3 concentration.56,57
It is worth highlighting that H+ and metal extraction mechanisms described in eqn (4) and (7), are carried out simultaneously, suggesting that there is a competition between extraction of H+ and M3+ by DMDOHEMA molecules.
The contribution of each mechanism to the metal extraction was evaluated for all the acidic conditions tested. Results are summarized in Table 3. To clarify our point, it is important to note that the so-called ion-pair mechanism involves the ionic liquid anion participation as counter ion for the metal extraction while in the solvation mechanism nitrate anion acts as counter ion.
| [HNO3] (mol L−1) | [EBPip+] [NTf2−] | [EOPip+] [NTf2−] | Dodecane | |||
|---|---|---|---|---|---|---|
| Cat. exch. (%) | Ion pair/solv. (%) | Cat. exch. (%) | Ion pair/solv. (%) | Cat. exch. (%) | Ion pair/solv. (%) | |
| 0 | 65 | 35 | 25 | 75 | — | — |
| 0.01 | 65 | 35 | 25 | 75 | — | — |
| 1.0 | 30 | 70 | 12 | 88 | 0 | 100 |
| 3.0 | 0 | 100 | 0 | 100 | 0 | 100 |
As already suggested, the extraction mechanisms appear to clearly shift from cation exchange to ion pair mechanism when the nitric acid concentration is increased. Ion pair mechanism is reached faster for the most hydrophobic IL [EOPip+] [NTf2−] and is the main mechanism for the conventional dodecane solvent.
4. Conclusions
Our results show that the REEs extraction efficiency is enhanced when DMDOHEMA is used in [EBPip+] [NTf2−] and [EOPip+] [NTf2−] ILs as organic solvent instead of a conventional organic solvent as n-dodecane. A complete metal extraction is achieved at low extractant concentration for low HNO3 concentration, and on the whole range of HNO3 concentration for higher extractant concentration. It allows considering this system in a wide range of processes. Compared to extraction performed in dodecane, La and Eu extraction provide moreover a very interesting selectivity in the ILs, toward the competing element Fe.A mechanistic study has been carried out to unravel these very interesting features. Looking at the role of ion exchange in the extraction mechanisms, effect of extractant addition and of metal extraction were individually investigated. Addition of DMDOHEMA does not affect the water content in the ILs, but increases HNO3 extraction.
When metals are extracted, it was shown that an anionic extraction mechanism can be rejected. La, Eu and Fe were found to be extracted through two simultaneous mechanisms for low nitric acid concentration, i.e. cationic exchange and ion pair formation, while a solvation mechanism similar to the one described in dodecane, involving no ion exchange, was found to be the dominant mechanism for [HNO3] ≥ 1.0 M.
Overall, the DMDOHEMA extractant have shown a higher REEs extraction efficiency in ILs than in dodecane without involving any ion exchange for 3.0 M nitric acid concentration in the aqueous phase, which is of high interest for application. This essential result arises also some fundamental questions on the origin of the higher extraction efficiency in ILs that cannot be related to the exchange of the IL ions. Other hypothesis has therefore to be considered to explain such important results for ILs application, as considering structural aspects as the extractant aggregation as it is nowadays commonly considered in extraction mechanisms in conventional diluents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the LabEx project CheMISyst (ANR-10-LABX-05-01) for financial support.References
- V. Zepf, in Rare Earths Industry, Elsevier, 2016, p. 3 Search PubMed.
- H. Omorodion and R. J. Baker, Irish Chem. News, 2016,(2), 12–14 Search PubMed.
- E. Quijada-Maldonado and J. Romero, Curr. Opin. Green Sustain. Chem., 2021, 27, 100428 CrossRef.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765 RSC.
- S. Dai, Y. H. Ju and C. E. Barnes, J. Chem. Soc., Dalton Trans., 1999, 1201 RSC.
- Ionic Liquids for Better Separation Processes, ed. H. Rodríguez, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016 Search PubMed.
- T. Makanyire, S. Sanchez-Segado and A. Jha, Adv. Manuf., 2016, 4, 33 CrossRef CAS.
- M. Regel-Rosocka and M. Wisniewski, in Applications of Ionic Liquids in Science and Technology, ed. S. Handy, InTech, 2011 Search PubMed.
- S. A. Ansari, P. K. Mohapatra, V. Mazan and I. Billard, RSC Adv., 2015, 5, 35821 RSC.
- M. L. Dietz and J. A. Dzielawa, Chem. Commun., 2001, 2124 RSC.
- V. A. Cocalia, M. P. Jensen, J. D. Holbrey, S. K. Spear, D. C. Stepinski and R. D. Rogers, Dalton Trans., 2005, 1966 RSC.
- V. A. Cocalia, J. D. Holbrey, K. E. Gutowski, N. J. Bridges and R. D. Roqers, Tinshhua Sci. Technol., 2006, 11, 188 CAS.
- M. L. Dietz and D. C. Stepinski, Green Chem., 2005, 7, 747 RSC.
- K. V. Lohithakshan and S. K. Aggarwal, Radiochim. Acta, 2008, 96, 93 CAS.
- H. Luo, S. Dai, P. V. Bonnesen, A. C. Buchanan, J. D. Holbrey, N. J. Bridges and R. D. Rogers, Anal. Chem., 2004, 76, 3078 CrossRef CAS PubMed.
- K. Nakashima, F. Kubota, T. Maruyama and M. Goto, Ind. Eng. Chem. Res., 2005, 44, 4368 CrossRef CAS.
- M. L. Dietz and D. C. Stepinski, Talanta, 2008, 75, 598 CrossRef CAS PubMed.
- I. Billard, A. Ouadi, E. Jobin, J. Champion, C. Gaillard and S. Georg, Solvent Extr. Ion Exch., 2011, 29, 577 CrossRef CAS.
- L. Zhao, Z. Dong, G. Ma and W. Yuan, J. Rare Earths, 2015, 33, 1182 CrossRef CAS.
- E. E. Tereshatov, M. Yu. Boltoeva, V. Mazan, M. F. Volia and C. M. Folden, J. Phys. Chem. B, 2016, 120, 2311 CrossRef CAS PubMed.
- C. Micheau, G. Arrachart, R. Turgis, M. Lejeune, M. Draye, S. Michel, S. Legeai and S. Pellet-Rostaing, ACS Sustainable Chem. Eng., 2020, 8, 1954 CrossRef CAS.
- Y. Traore, S. Legeai, S. Diliberto, G. Arrachart, S. Pellet-Rostaing and M. Draye, Electrochim. Acta, 2011, 58, 532 CrossRef CAS.
- V. Manchanda, Sep. Purif. Technol., 2004, 35, 85 CrossRef CAS.
- E. A. Mowafy and H. F. Aly, Solvent Extr. Ion Exch., 2002, 20, 177 CrossRef CAS.
- R. J. Ellis, Y. Meridiano, J. Muller, L. Berthon, P. Guilbaud, N. Zorz, M. R. Antonio, T. Demars and T. Zemb, Chem. - Eur. J., 2014, 20, 12796 CrossRef CAS PubMed.
- B. Gannaz, R. Chiarizia, M. R. Antonio, C. Hill and G. Cote, Solvent Extr. Ion Exch., 2007, 25, 313 CrossRef CAS.
- E. Scoppola, E. B. Watkins, R. A. Campbell, O. Konovalov, L. Girard, J. Dufrêche, G. Ferru, G. Fragneto and O. Diat, Angew. Chem., Int. Ed., 2016, 55, 9326 CrossRef CAS PubMed.
- C. Dejugnat, L. Berthon, V. Dubois, Y. Meridiano, S. Dourdain, D. Guillaumont, S. Pellet-Rostaing and T. Zemb, Solvent Extr. Ion Exch., 2014, 32, 601 CrossRef CAS.
- S. Dourdain, C. Dejugnat, L. Berthon, V. Dubois, S. Pellet-Rostaing, J.-F. Dufreche and T. Zemb, Solvent Extr. Ion Exch., 2014, 32, 620 CrossRef CAS.
- K. Nakashima, F. Kubota, T. Maruyama and M. Goto, Anal. Sci., 2003, 19, 1097 CrossRef CAS PubMed.
- H. Naganawa, K. Shimojo, H. Mitamura, Y. Sugo, J. Noro and M. Goto, Solvent Extr. Res. Dev., Jpn., 2007, 14, 151 CAS.
- A. Rout, S. Karmakar, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Purif. Technol., 2011, 81, 109 CrossRef CAS.
- S. J. Yoon, J. G. Lee, H. Tajima, A. Yamasaki, F. Kiyono, T. Nakazato and H. Tao, J. Ind. Eng. Chem., 2010, 16, 350 CrossRef CAS.
- H. Okamura, M. Mizuno, N. Hirayama, K. Shimojo, H. Naganawa and H. Imura, Ind. Eng. Chem. Res., 2020, 59, 329 CrossRef CAS.
- R. Turgis, G. Arrachart, V. Dubois, S. Dourdain, D. Virieux, S. Michel, S. Legeai, M. Lejeune, M. Draye and S. Pellet-Rostaing, Dalton Trans., 2016, 45, 1259 RSC.
- F. Kubota, Y. Konagi, K. Nakashima, K. Shimojo, N. Kamiya and M. Goto, Solvent Extr. Res. Dev., Jpn., 2008, 15, 81 CAS.
- M. R. Antonio, R. Chiarizia, B. Gannaz, L. Berthon, N. Zorz, C. Hill and G. Cote, Sep. Sci. Technol., 2008, 43, 2572 CrossRef CAS.
- Y. Shen, S. Wang, L. Zhu, J. Wang and W. Wu, Ind. Eng. Chem. Res., 2011, 50, 13990 CrossRef CAS.
- L. Bosland, Thèse de l'Ecole Centrale Paris. Report CEA-R-6099, 2006 Search PubMed.
- L. Nigond, C. Musikas and C. Cuillerdier, Solvent Extr. Ion Exch., 1994, 12, 261 CrossRef CAS.
- Y. Meridiano, L. Berthon, X. Crozes, C. Sorel, P. Dannus, M. R. Antonio, R. Chiarizia and T. Zemb, Solvent Extr. Ion Exch., 2009, 27, 607 CrossRef CAS.
- C. Dejugnat, S. Dourdain, V. Dubois, L. Berthon, S. Pellet-Rostaing, J. F. Dufrêche and T. Zemb, Phys. Chem. Chem. Phys., 2014, 16, 7339–7349 RSC.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, S. T. Griffin and R. D. Rogers, Ind. Eng. Chem. Res., 2000, 39, 3596 CrossRef CAS.
- L. Spjuth, J. O. Liljenzin, M. J. Hudson, M. G. B. Drew, P. B. Iveson and C. Madic, Solvent Extr. Ion Exch., 2000, 18, 1 CrossRef CAS.
- C. Musikas and H. Hubert, Solvent Extr. Ion Exch., 1987, 5, 151 CrossRef CAS.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2012, 51, 7468 CrossRef CAS PubMed.
- O. Borodin, D. L. Price, B. Aoun, M. A. González, J. B. Hooper, M. Kofu, S. Kohara, O. Yamamuro and M.-L. Saboungi, Phys. Chem. Chem. Phys., 2016, 18, 23474 RSC.
- V. Mazan, I. Billard and N. Papaiconomou, RSC Adv., 2014, 4, 13371 RSC.
- C. Gaillard, V. Mazan, S. Georg, O. Klimchuk, M. Sypula, I. Billard, R. Schurhammer and G. Wipff, Phys. Chem. Chem. Phys., 2012, 14, 5187 RSC.
- D. C. Gaillard, M. Boltoeva, I. Billard, S. Georg, V. Mazan, A. Ouadi, D. Ternova and C. Hennig, ChemPhysChem, 2015, 16, 2653 CrossRef PubMed.
- J. Foropoulos and D. D. DesMarteau, Inorg. Chem., 1984, 23, 3720 CrossRef CAS.
- V. Mazan, M. Yu. Boltoeva, E. E. Tereshatov and C. M. Folden III, RSC Adv., 2016, 6, 56260 RSC.
- K. Matuszek, E. Pankalla, A. Grymel, P. Latos and A. Chrobok, Molecules, 2019, 25, 80 CrossRef PubMed.
- M. Bonnaffé-Moity, A. Ouadi, V. Mazan, S. Miroshnichenko, D. Ternova, S. Georg, M. Sypula, C. Gaillard and I. Billard, Dalton Trans., 2012, 41, 7526 RSC.
- A. Messadi, A. Mohamadou, S. Boudesocque, L. Dupont and E. Guillon, Sep. Purif. Technol., 2013, 107, 172 CrossRef CAS.
- A. Rao and B. S. Tomar, Sep. Purif. Technol., 2016, 161, 159 CrossRef CAS.
- C. Gaillard, M. Boltoeva, I. Billard, S. Georg, V. Mazan and A. Ouadi, RSC Adv., 2016, 6, 70141 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Table S1. See DOI: 10.1039/d1ra05359k |
| This journal is © The Royal Society of Chemistry 2021 |