
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-atom molybdenum doping induces nickel oxide-to-hydroxide transformation for enhanced alkaline hydrogen evolution†
Yuanqi
Liu
 a,
Qiang
Gao
b,
Lei
Shi
b,
Joseph
Kearney
b,
Xue
Han
c,
Zhenhua
Xie
d,
Maoyu
Wang
e,
Hua
Zhou
e and
Huiyuan
Zhu
*ab
a,
Qiang
Gao
b,
Lei
Shi
b,
Joseph
Kearney
b,
Xue
Han
c,
Zhenhua
Xie
d,
Maoyu
Wang
e,
Hua
Zhou
e and
Huiyuan
Zhu
*ab
aDepartment of Chemical Engineering, University of Virginia, Charlottesville, VA 22903, USA. E-mail: kkx8js@virginia.edu
bDepartment of Chemistry, University of Virginia, Charlottesville, VA 22904, USA
cChemistry Division, Brookhaven National Laboratory, Upton, New York 11973, USA
dDepartment of Chemical Engineering, Columbia University, New York, New York 10027, USA
eX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 19th June 2025
Abstract
NiMoOx compounds are widely regarded as among the most efficient non-noble metal catalysts for the hydrogen evolution reaction (HER). Nevertheless, understanding the structural evolution under in situ conditions and further enhancing their performance remain key challenges. Herein, we report that single-atom Mo doping in NiO significantly enhances its HER activity, reducing the overpotential to 131 mV at 10 mA cm−2 compared to undoped NiO. In situ X-ray absorption spectroscopy and Raman spectroscopy reveal that under catalytic conditions, Mo single atoms remain structurally stable, while Ni2+ species in NiO are converted to Ni(OH)2 in alkaline media under the applied working potential for HER. Notably, this transformation is absent in undoped NiO, indicating that Mo doping promotes the formation of active Ni(OH)2 sites, which, in turn, accelerate the rate-limiting water dissociation step. These findings provide critical mechanistic insights into the structural evolution of NiMoOx during alkaline HER and highlight the importance of in situ studies in the development of highly efficient catalysts.
 Huiyuan Zhu | Our first Nanoscale Horizons paper was published in 2022 and was featured in the Emerging Investigator Series, highlighted on the cover, and recognized as one of Nanoscale Horizons' Most Popular Articles of 2022. I was truly honored and grateful for the recognition as the journal's first Emerging Investigator. Now, as I transition from an early-career researcher to a mid-career scientist, Nanoscale Horizons remains our go-to journal—a place we are proud to publish in and hope to continue contributing to in the years ahead. Congratulations on 10 years of leading nanoscience—here's to many more of breaking new ground! |
New conceptsIn this work, we report a colloidal synthesis strategy for fabricating single-atom-doped oxides—specifically, NiO nanoparticles (15–25 nm) incorporating isolated Mo atoms (Mo–NiO). We introduce the concept of single-atom-Mo-induced phase transformation, wherein atomically dispersed Mo acts as an electronic switch that drives in situ lattice evolution during alkaline hydrogen evolution reaction (HER). The resulting Mo–NiO exhibits excellent performance, requiring only 131 mV to reach 10 mA cm−2 with a Tafel slope of 117 mV dec−1—dramatically outperforming undoped NiO (305 mV, 180 mV dec−1) and comparable to state-of-the-art NiMoOx catalysts. These metrics suggest that Mo doping significantly accelerates the water dissociation step, thereby enhancing overall HER kinetics. In situ X-ray absorption and Raman spectroscopy reveal that under applied potential, the oxidation states of Ni and Mo remain unchanged while NiO transforms into Ni(OH)2. This phase transition is likely driven by electron withdrawal at Mo sites, which elongates neighboring Ni–O bonds and promotes the formation of the catalytically active Ni(OH)2, with Mo centers remaining structurally intact. Our study highlights how integrating single-atom electronic modulation with dynamic phase engineering offers a generalizable strategy for designing low-cost, high-performance electrocatalysts under operating conditions. |
Introduction
The world's heavy reliance on fossil fuels has not only accelerated their depletion but also led to severe environmental consequences, including shifts in climate patterns.1 Consequently, advancing sustainable energy schemes is paramount for ensuring future energy security and environment preservation. Especially as many countries aspire to achieve carbon neutrality by 2050, hydrogen is gaining momentum for its strong potential to decarbonize both the industrial and transportation sectors—if it can be produced through clean routes. Nevertheless, the majority of H2 is still produced from fossil fuels, mainly via steam reforming, which emits significant CO2 and requires harsh reaction environment.2–4 This highlights the urgent need to develop efficient electrochemical hydrogen evolution reaction (HER) systems powdered by renewable energy.5,6 Alkaline HER plays a pivotal role in the large-scale production of green hydrogen through water electrolysis, serving as a cornerstone for decarbonizing energy systems and enabling renewable energy storage. Alkaline water electrolysis is a cost-effective technology that allows the use of non-precious metal electrodes, thereby lowering capital costs compared to acidic PEM electrolysis while maintaining competitive efficiency under renewable power input.7,8 However, ensuring long-term stability in alkaline environments remains a major challenge. Transition metal-based catalysts and their oxide/hydroxide derivatives are susceptible to corrosion, dissolution–reprecipitation cycles, phase transformations, and surface reconstruction during prolonged operation, which can result in the loss of active sites and diminished conductivity.9 Precious metals, such as Pt, Pd, and Ru, are the state-of-the-art catalysts for the HER, but their high cost and scarcity limit broad use.10To reduce or replace precious metals, single-atom catalysts (SACs), featuring atomically dispersed, well-defined metal centers anchored on solid supports (e.g., oxides, carbon materials, etc.), have gained attention for their high catalytic performance in critical reactions,11–13 including nitrate reduction,14,15 selective hydrogenation,16,17 CO2 reduction,18,19 HER,20,21 and oxygen evolution reaction (OER).22,23 Many non-precious metal-based SACs, such as Ni,24 Co,25 W,26 and Mo,27 have been developed as viable HER catalysts. However, the precise role of the single-atom site—whether as the primary active center, a promoter through synergistic effects, or a co-catalyst that facilitates favorable hydroxide/water adsorption and decouples the adsorption, activation, and desorption steps—remains unclear, especially when both the single-atom sites and the solid supports are active toward HER. For example, bimetallic NiMo oxides28 like NiMoOx has shown promising activity and stability for alkaline HER,29–31 but the true active sites remain debated, with unconventional Ni4Mo and metastable β-NiMoO4 sites being proposed as key catalytic motifs. The inherent structural heterogeneity was also found to optimize the intermediate binding strengths, thereby reducing the energy barrier of the Volmer step in alkaline HER.32,33 Under reaction conditions, the true nature of the active sites can become even more complex, as they may either remain metallic or transform into surface hydroxides in alkaline environments. This is attributed to the electronegative and nucleophilic nature of hydroxide ions, with metal hydroxides playing a critical role in enhancing HER performance in alkaline media.34,35
Herein, we synthesized single-atom Mo doped NiO nanoparticles (Mo–NiO NPs) and investigated the role of single-atom Mo in NiO, focusing on how Mo dopants modulate the electronic environment to enhance alkaline HER activity. Compared to undoped NiO, which exhibits an overpotential of 305 mV and a Tafel slope of 180 mV dec−1 in 1 M KOH, Mo–NiO exhibits a notably lower overpotential of 131 mV at 10 mA cm−2 and a reduced Tafel slope of 117 mV dec−1. In situ X-ray adsorption spectroscopy (XAS) and Raman spectroscopy reveal that Mo single atoms remain structurally stable during alkaline HER, while Ni2+ species in NiO undergo a dynamic transformation to Ni(OH)2 under negative bias. Significantly, this phase transformation, which is absent in undoped NiO, is driven by localized electronic perturbations induced by the Mo dopant, ultimately stabilizing the active Ni(OH)2 phase. These findings clarify the nature of active sites in Mo–NiO catalysts and highlight the importance of in situ characterization for illustrating dynamic electrocatalytic processes.
Synthesis and characterization of Mo–NiO NPs
Mo–NiO NPs were synthesized via colloidal reduction of a nickel precursor and thermal decomposition of a molybdenum precursor, followed by air annealing to obtain Mo single-atom-doped NiO (ESI:† Experimental section). Inductively coupled plasma optical emission spectrometry (ICP-OES) confirmed an elemental molar ratio of approximately 4% Mo and 96% Ni, with the low Mo content indicating isolated single-atom doping rather than the formation of Mo clusters or NPs. As schematically illustrated in Fig. 1a, this synthesis strategy enables the incorporation of Mo atoms into the NiO lattice while maintaining structural integrity. The representative bright field (BF)-transmission electron microscopy (TEM) image (Fig. 1b) demonstrates that the Mo–NiO NPs predominantly range in size from 15 to 25 nm before air annealing. The Mo–NiO NPs loading on Vulcan carbon after air annealing is shown in Fig. S1 (ESI†). Fig. 1c displays the X-ray diffraction (XRD) patterns of Mo–NiO NPs and pristine NiO NPs, both of which match the typical characteristic face-centered cubic (fcc) NiO phase (JCPDS #47-1049). According to the Debye–Scherrer's equation , the crystallite sizes of Mo–NiO NPs and NiO NPs have been determined to be approximately 20 nm and 70 nm, respectively, consistent with the TEM results (Fig. 1b and Fig. S2, ESI†). Furthermore, the absence of characteristic Mo phase peaks can be attributed to the low Mo concentration resulting from single-atom Mo formation, making them difficult to detect using lab-based XRD. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1d) directly reveals Mo single atoms, visible as bright spots due to the higher Z-contrast of Mo compared to Ni. The HR-TEM image of Mo–NiO NPs also demonstrates the high crystallinity of NiO, displaying (200) lattice fringes with interplanar spacings of 0.208 nm; these values align with direct measurements from the corresponding fast Fourier transform (FFT) pattern (Fig. S3, ESI†).36 Energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 1e) further confirms the homogeneous distribution of Ni, Mo, and O within the Mo–NiO NP matrix. As a comparison, we synthesized Mo-rich, MoO3/NiO NPs (ESI:† Experimental section; Fig. S4a) with a particle size of 5–10 nm, and ∼9% and ∼2% Mo-doped Mo–NiO NPs samples (Fig. S4b and c, ESI†). The XRD pattern displays pronounced peaks from the NiO (200) and MoO3 (021) planes (JCPDS # 05-0508) in an orthorhombic structure, confirming the co-existence of NiO and MoO3 crystalline domains within the NPs (Fig. S4d, ESI†). Moreover, the ICP-OES shows a Mo atomic fraction about 40%, indicating that Mo is hardly present as isolated single atoms in the MoO3/NiO NPs.
, the crystallite sizes of Mo–NiO NPs and NiO NPs have been determined to be approximately 20 nm and 70 nm, respectively, consistent with the TEM results (Fig. 1b and Fig. S2, ESI†). Furthermore, the absence of characteristic Mo phase peaks can be attributed to the low Mo concentration resulting from single-atom Mo formation, making them difficult to detect using lab-based XRD. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1d) directly reveals Mo single atoms, visible as bright spots due to the higher Z-contrast of Mo compared to Ni. The HR-TEM image of Mo–NiO NPs also demonstrates the high crystallinity of NiO, displaying (200) lattice fringes with interplanar spacings of 0.208 nm; these values align with direct measurements from the corresponding fast Fourier transform (FFT) pattern (Fig. S3, ESI†).36 Energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 1e) further confirms the homogeneous distribution of Ni, Mo, and O within the Mo–NiO NP matrix. As a comparison, we synthesized Mo-rich, MoO3/NiO NPs (ESI:† Experimental section; Fig. S4a) with a particle size of 5–10 nm, and ∼9% and ∼2% Mo-doped Mo–NiO NPs samples (Fig. S4b and c, ESI†). The XRD pattern displays pronounced peaks from the NiO (200) and MoO3 (021) planes (JCPDS # 05-0508) in an orthorhombic structure, confirming the co-existence of NiO and MoO3 crystalline domains within the NPs (Fig. S4d, ESI†). Moreover, the ICP-OES shows a Mo atomic fraction about 40%, indicating that Mo is hardly present as isolated single atoms in the MoO3/NiO NPs.
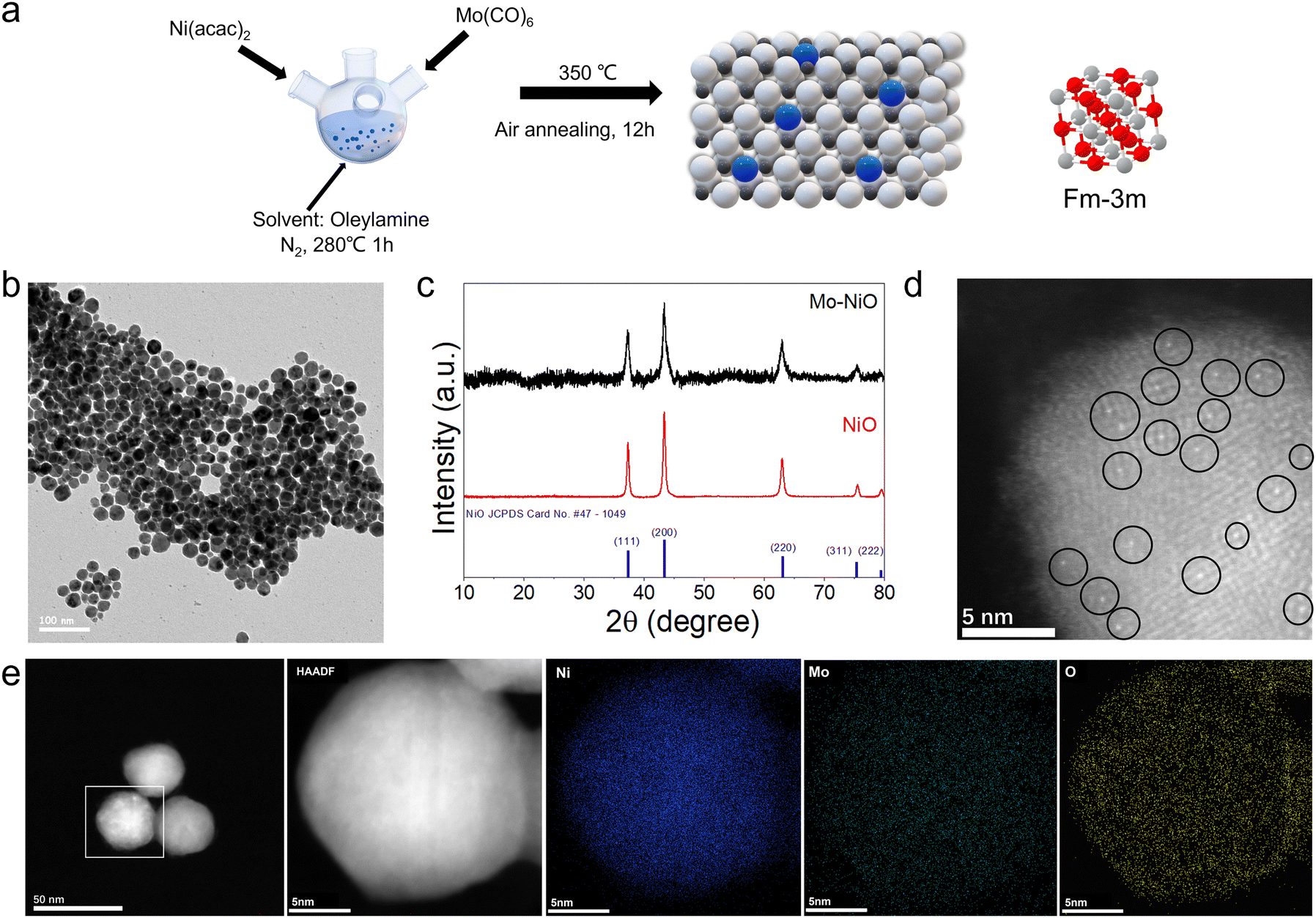 | ||
| Fig. 1 (a) Schematic illustrations of the synthesis of Mo–NiO NPs. Ni: white ball; Mo: blue ball; O: black ball. (b) The BF-TEM image of Mo–NiO as-synthesized NPs. (c) XRD pattern of Mo–NiO NPs/C and NiO. (d) High-resolution HAADF-STEM image and some of the Mo single atoms marked with black circles, (e) HAADF-STEM EDS elemental mapping of Mo–NiO NPs/C. | ||
X-ray photoelectron spectroscopy (XPS) results further reveal the valence states of Ni and Mo in the Mo–NiO NPs (Fig. S5, ESI†). In the Ni 2p spectrum, the characteristic peaks at 854.7 eV (2p3/2) and 872.3 eV (2p1/2), accompanied by satellite features at 861.0 eV and 880.6 eV, confirm the presence of Ni2+, consistent with the NiO phase in the Mo–NiO NPs.37–39 For the Mo 3d region, the observed peaks at 232.6 eV (3d5/2) and 235.8 eV (3d3/2) are assigned to Mo6+ species.40,41 In the O 1s spectrum, the dominant peak at 529.3 eV is attributed to lattice O2− in crystalline NiO, while a weak shoulder at 531.5 eV corresponds to surface hydroxyl groups. The minor intensity of this shoulder indicates limited surface hydroxylation.42 This suggests that the Mo atoms in the Mo–NiO NPs structure likely substitute Ni atoms within the lattice and coordinate with adjacent oxygen atoms, forming a stable integrated oxide framework.
HER performance
The alkaline HER performance of the Mo–NiO NPs, MoO3/NiO, NiO NPs and commercial Pt/C was evaluated in a standard three-electrode single cell using 1.0 M KOH as the electrolyte, at a scan rate of 5 mV s−1 (ESI:† Electrochemical measurements). As shown in Fig. 2a and b, the linear scan voltammetry (LSV) curves of Mo–NiO NPs requires a significantly lower overpotential of 131 mV at 10 mA cm−2 and a smaller Tafel slope (117 mV dec−1) than NiO NPs (305 mV and 180 mV dec−1, respectively). Fig. S6 (ESI†) shows that MoO3/NiO requires an overpotential of 190 mV, significantly higher than Mo–NiO, and it also reveals overpotentials at 10 mA cm−2 of 154 mV for 2% Mo–NiO, and 149 mV for 9% Mo–NiO. These results demonstrate a non-linear relationship between Mo concentration and catalytic performance: even moderate doping enhances activity compared to undoped NiO, with a medium loading (4%) achieving the lowest overpotential. Compared to other reported Ni oxides based electrocatalysts, Mo–NiO NPs exhibit top-tier HER performance (Table S1, ESI†). The Tafel slopes of Mo–NiO NPs and NiO NPs indicate that a Volmer–Heyrovsky mechanism and the Volmer step is the rate-determining step (RDS).43 It also implicates that single-atom Mo doping is a critical promoter of the water dissociation process as a much lower Tafel slope was obtained comparing with the pure NiO case. To quantify the kinetic benefit introduced by Mo, the activation energy at thermodynamic equilibrium (η = 0 mV, denoted Eη=0a) is determined from the temperature dependence of the exchange current density (j) (Fig. S7 and S8, ESI†).44 LSV curves for Mo–NiO NPs and NiO NPs were recorded at five temperatures – 298, 306, 312, 320, and 328 K – to obtain the corresponding LSV curves (Fig. S7a and S8a, ESI†). According to the Arrhenius equation, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j vs. η plots (Fig. S7b and S8b, ESI†) and the resulting Arrhenius plots were plotted (Fig. S7c and S8c, ESI†). The extracted Eη=0a values are 24.08 kJ mol−1 for Mo–NiO NPs and 39.61 kJ mol−1 for NiO NPs (Fig. S7d and S8d, ESI†), which confirms that Mo doping effectively lowers the activation energy barrier and promotes the water-dissociation step in alkaline HER.
j vs. η plots (Fig. S7b and S8b, ESI†) and the resulting Arrhenius plots were plotted (Fig. S7c and S8c, ESI†). The extracted Eη=0a values are 24.08 kJ mol−1 for Mo–NiO NPs and 39.61 kJ mol−1 for NiO NPs (Fig. S7d and S8d, ESI†), which confirms that Mo doping effectively lowers the activation energy barrier and promotes the water-dissociation step in alkaline HER.
 | ||
| Fig. 2 HER performance in 1 M KOH electrolytes. (a) LSV curves, (b) Tafel plots, (c) calculated Cdl values, (d) ECSA-corrected polarization curves, (e) EIS plots of Mo–NiO and NiO NPs. (f) Stability of Mo–NiO NPs at a fixed overpotential of −0.28 V vs. RHE. | ||
Catalytic activity is intrinsically linked to the electrochemical surface area (ECSA), which was quantified using double-layer capacitance (Cdl) measurements derived from cyclic voltammetry (CV) in the non-faradaic potential regime (0.826–0.926 V vs. RHE) across varying scan rates (Fig. S9, ESI†). By linear fitting of the corresponding plots (Fig. 2c), Mo–NiO NPs exhibit a Cdl of 19.9 mF cm−2, whereas NiO NPs demonstrate a value of 7.16 mF cm−2. Assuming a typical specific capacitance (Cs) of 40 μF cm−2 for an atomically smooth surface in alkaline media, the ECSA is given by ECSA = Cdl/Cs.45 Consequently, Mo–NiO NPs have an ECSA of 497.5 cm2, substantially larger than that of NiO NPs (179 cm2). After normalizing the polarization curves by the respective ECSA (Fig. 2d), Mo–NiO NPs still achieve a higher current density at the same overpotential, reflecting their superior intrinsic HER activity.
From the electrochemical impedance spectroscopy (EIS) analysis (Fig. 2e), the high-frequency semicircle diameter – corresponding to charge-transfer resistance (Rct) – was remarkably smaller for Mo–NiO NPs, indicating enhanced charge-transfer kinetics at the Mo–NiO NPs/electrolyte interface. This enhanced electron transfer supports the observed improvements in both overpotential and Tafel slope, thus mechanistically linking Mo doping to optimized interfacial charge dynamics. Beyond activity, the long-term stability of Mo–NiO NPs was assessed through chronoamperometry (CA) at a fixed overpotential corresponding to 100 mA cm−2 under Ar atmosphere (Fig. 2f). The results show no significant decline in current density for Mo–NiO NPs after 100 h, demonstrating its excellent catalytic durability. The durability of the catalysts was also evaluated by potential cycling between 0 and −0.3 V for 3000 cycles at a scan rate of 50 mV s−1. The overpotential required to reach 10 mA cm−2 remains unchanged. At 100 mA cm−2 the overpotential increases by only ∼7 mV. This negligible performance loss confirms that the Mo–NiO NPs catalyst maintains structural and electrochemical stability during prolonged potential cycling (Fig. S10, ESI†).
In situ spectroscopic investigation
To investigate how single-atom Mo doping influences the HER catalytic property of NiO NPs, in situ XAS and Raman spectroscopy were employed. In situ XAS was used to directly probe the local bonding and coordination environments of Ni and Mo centers under the reaction conditions. Fig. 3a presents the X-ray absorption near edge structure (XANES) spectra of the Ni K-edge in Mo–NiO for each applied potential. The adsorption edge energy of the Ni K-edge under open-circuit potential (OCP) closely matches that of the NiO reference and is significantly higher than that of the Ni foil, confirming that the valence state of Ni in Mo–NiO NPs remains close to Ni2+ under OCP conditions. Upon applying the reduction potential from −0.1 V to −0.4 V vs. RHE, both the adsorption edge energy and white line intensity remain essentially unchanged compared to those OCP. This indicates that the oxidation state of the Ni2+ species remains largely unchanged under alkaline HER reaction conditions. Fig. 3b shows the Mo K-edge XANES of Mo–NiO NPs, where no significant shift in absorption energy or change in white line intensity is observed upon applying reduction potentials (from OCP to −0.4 V vs. RHE), indicating that the oxidation state of Mo species also remains stable. Moreover, the Mo K-edge absorption energy of Mo–NiO NPs at OCP is significantly higher than that of Mo foil, suggesting an oxidation state of Moδ+ existed in Mo species. Notably, the XANES of Mo–NiO sample demonstrates a shoulder around 20004 eV in the pre-edge region, indicating the existence of MoOx with distorted structures.46–48
 | ||
| Fig. 3 In situ XAS of Mo–NiO NPs. (a) Ni K-edge XANES spectra. (b) Mo K-edge XANES spectra. (c) The k2-weighted Fourier transformation of the Ni K-edge EXAFS spectra. (d) The k2-weighted Fourier transformation of the Mo K-edge EXAFS spectra. (e) Mo K-edge wavelet transform EXAFS of Mo–NiO and Mo foil reference. The color contour represents the intensity range. | ||
In order to further understand the coordination environment, the fitting results of Ni K-edge and Mo K-edge k2-weight Fourier transforms of the extended X-ray absorption fine structure (EXAFS) spectra for Mo–NiO NPs along with Ni foil, Mo foil and NiO sample were plotted in R space and k space (Fig. 3c, d and Fig. S11–S14, ESI†).49 The corresponding fitting parameters are provided in Tables S2–S5 (ESI†). In the Ni K-edge EXAFS spectra, there are two major peaks of the scattering paths. The first shell at ∼2.05 Å can be assigned to the Ni–O scattering path, while the second shell at ∼3.12 Å can be attributed to the Ni–O–Ni scattering path. Notably, no significant Ni–Ni coordination signal is observed at ∼2.48 Å, in contrast to the characteristic Ni–Ni scattering peak observed in Ni foil, indicating the absence of metallic Ni coordination in the sample. The absence of significant positional shifts for these two shells under applied potentials (from OCP to −0.4 V vs. RHE) suggests that the oxidation states of Ni species remain unchanged. However, due to the resolution limits in EXAFS data fitting analysis (Artemis), it remains challenging to definitively assign the second shell Ni–O–Ni to either NiO or Ni(OH)2, as both contain the Ni–O–Ni scattering path. Consequently, additional characterization is required to determine whether any active site structural changes occur under applied potentials.
Regarding the Mo, in the R space, the K-edge EXAFS spectra display a major peak at ∼1.75 Å, attributed to the Mo–O scattering path from the MoOx species in Mo–NiO NPs. The characteristic Mo–Mo scattering peak (∼2.72 Å for the Mo foil) is nearly absent, confirming that Mo primarily exists as atomically dispersed sites without long-range Mo–Mo coordination. Furthermore, the wavelet transform (WT) EXAFS of the Mo K-edge shows a Mo–O scattering maximum at ∼1.3 Å and ∼4.7 Å−1 in Mo–NiO NPs with negligible scattering contributions at higher shells, in contrast to the Mo–Mo scattering maximum at ∼2.4 Å and ∼8.2 Å−1 observed in Mo foil (Fig. 3e), indicating that Mo exhibits the feature of isolated metal atoms. Taken cumulatively, the above results demonstrate the successful creation of single-atom Mo doped NiO NPs. In addition, compared to the OCP conditions, the intensity and position of the Mo–O scattering peaks did not change during the reaction conditions, indicating that the Mo single atom species in NiO remain stable, with negligible structure or oxidation state changes.
In situ Raman spectroscopy under applied potentials was used to track the dynamic surface chemical bonding evolution of Mo–NiO NPs. The Raman spectra of pure NiO NPs at both ex situ and under OCP conditions exhibits two characteristic peaks at 510 cm−1 and 729 cm−1, which can be assigned to the one-phonon (1P) TO and LO modes and two-phonon (2P) 2TO modes of NiO, respectively (Fig. 4a).50 Upon applying a reductive potential (from OCP to −0.4 V vs. RHE), the characteristic peaks positions of the Ni–O vibrational modes exhibit no significant shifts, and no new peaks, indicating that in the absence of Mo single-atom doping, the NiO active sites undergo minimal structural evolution. In contrast, Mo-doped NiO NPs under ex situ and OCP conditions exhibit a new Raman signal at 822 cm−1, attributed to the stretching vibration of terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (Fig. 4b).51,52 The position of this MoO stretching peak remains unchanged under applied potentials, indicating that the Mo single-atom species undergo negligible structural evolution, consistent with the in situ XAS results. When applied the potential from −0.1 V to −0.4 V vs. RHE, two new peaks emerged at 313 cm−1 and 448 cm−1, corresponding to the E-type vibration of Ni–OH (γNi–OH) and A1g-type stretch of Ni–O (νNi–O) from Ni(OH)2, respectively. This result indicates potential-dependent structural evolution of Mo–NiO NPs toward hydroxide formation.53 Therefore, in situ Raman spectra reveal that Mo single-atom doping in NiO NPs promotes the transformation of Ni2+ species into Ni(OH)2 under applied potentials. Under strongly alkaline conditions (pH > 12), DFT-based Pourbaix diagram indicate that residual chemical potential of NiO is 0.13 eV lower than that of Ni(OH)2.54 Consequently, a cathodic bias (<0 V vs. RHE) initially drives the hydration of NiO into β-Ni(OH)2, well before the potential becomes negative enough to reach the stability region of metallic Ni. While the alkaline environment sets the thermodynamic driving force, Mo tends to withdraw electron density from adjacent Ni–O bonds. This electron withdrawal leads to bond elongation, lowers the bond dissociation energy, and weakens the covalent character within the Ni–O coordination sphere.55 The concomitant distortion of the Mo–O coordination environment may also give rise to oxygen vacancies and electron-rich-regions near the Mo center. Under negative potential, these electron enriched domains could act as charge reservoirs that compensate for the electron depletion associated with OH− adsorption, while the oxygen vacancies provide additional adsorption sites and reduce lattice-diffusion barriers.56 The synergistic interplay of electronic buffering and defect activation could substantially decrease the activation enthalpy for Ni–OH bond formation, enabling Ni2+ to re-coordinate with OH− without changing its oxidation state.57 The generation of Ni(OH)x further promotes the water dissociation step and the subsequent proton-coupled electron transfer in the HER, thereby accounting for the significantly enhanced catalytic performance of Mo–NiO.58
O bonds (Fig. 4b).51,52 The position of this MoO stretching peak remains unchanged under applied potentials, indicating that the Mo single-atom species undergo negligible structural evolution, consistent with the in situ XAS results. When applied the potential from −0.1 V to −0.4 V vs. RHE, two new peaks emerged at 313 cm−1 and 448 cm−1, corresponding to the E-type vibration of Ni–OH (γNi–OH) and A1g-type stretch of Ni–O (νNi–O) from Ni(OH)2, respectively. This result indicates potential-dependent structural evolution of Mo–NiO NPs toward hydroxide formation.53 Therefore, in situ Raman spectra reveal that Mo single-atom doping in NiO NPs promotes the transformation of Ni2+ species into Ni(OH)2 under applied potentials. Under strongly alkaline conditions (pH > 12), DFT-based Pourbaix diagram indicate that residual chemical potential of NiO is 0.13 eV lower than that of Ni(OH)2.54 Consequently, a cathodic bias (<0 V vs. RHE) initially drives the hydration of NiO into β-Ni(OH)2, well before the potential becomes negative enough to reach the stability region of metallic Ni. While the alkaline environment sets the thermodynamic driving force, Mo tends to withdraw electron density from adjacent Ni–O bonds. This electron withdrawal leads to bond elongation, lowers the bond dissociation energy, and weakens the covalent character within the Ni–O coordination sphere.55 The concomitant distortion of the Mo–O coordination environment may also give rise to oxygen vacancies and electron-rich-regions near the Mo center. Under negative potential, these electron enriched domains could act as charge reservoirs that compensate for the electron depletion associated with OH− adsorption, while the oxygen vacancies provide additional adsorption sites and reduce lattice-diffusion barriers.56 The synergistic interplay of electronic buffering and defect activation could substantially decrease the activation enthalpy for Ni–OH bond formation, enabling Ni2+ to re-coordinate with OH− without changing its oxidation state.57 The generation of Ni(OH)x further promotes the water dissociation step and the subsequent proton-coupled electron transfer in the HER, thereby accounting for the significantly enhanced catalytic performance of Mo–NiO.58
 | ||
| Fig. 4 Characterizations of Mo–NiO. (a) In situ Raman spectra of NiO NPs. (b) In situ Raman spectra of Mo–NiO NPs. | ||
Conclusions
In summary, we report the synthesis of the single-atom-doped Mo–NiO NPs and their enhanced performance in alkaline HER, achieving a low overpotential of 131 mV at 10 mA cm−2. Through combined in situ XAS and in situ Raman spectroscopic studies, we demonstrate the critical role of single-atom Mo doping in driving the structural evolution of NiO. While the Mo dopants maintain their coordination environment without structural changes, they play a key role in promoting the transformation of Ni2+ species from NiO to Ni(OH)2 under electrocatalytic conditions. This dynamic evolution optimizes water dissociation kinetics, leading to superior HER activity compared to undoped NiO. This work provides insights into how single-atom Mo doping in NiO operates, thereby advancing the exploration and mechanistic understanding of active site evolution in alkaline HER.Author contributions
Y. L. and H. Z. conceptualized the project. L. S., Q. G., and J. K. helped with the synthesis of the catalysts and collected the data. X. H., Z. X., M. W. and H. Z. helped with the collection and analysis of the XAS data. We thank Dr. Yizhen Chen (University of Virginia) and Dr. Yipeng Zang (University of Virginia) for their valuable discussions on this project.Conflicts of interest
The authors declare no competing interests.Data availability
All data are available in the main text and the ESI.†Acknowledgements
This work was supported by the US Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division (DE-SC00234430). This research was performed on APS beam time award at beamline 12-BM-B from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research used resources of beamline 7-BM (QAS) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- J. Fan, H. Wang, G. Liao, C. Song and J. Zou, Sci. China: Technol. Sci., 2025, 68, 1620206 CrossRef.
- Z. Zhang, C. Song, J. Fan, Z. Fang, H. Wang and J. Zou, J. Mater. Chem. A, 2025, 13, 6385–6396 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- T. Wen, M. Yang, J. Zou and H. Wang, Ind. Eng. Chem. Res., 2025, 64, 9209–9216 CrossRef CAS.
- H. Tüysüz, Acc. Chem. Res., 2024, 57, 558–567 Search PubMed.
- M. Lao, P. Li, Y. Jiang, H. Pan, S. X. Dou and W. Sun, Nano Energy, 2022, 98, 107231 CrossRef CAS.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Q. Gao, X. Han, Y. Liu and H. Zhu, ACS Catal., 2024, 14, 6045–6061 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2017, 8, 1701343 CrossRef.
- H. Wang, X. Li, Y. Deng, J. Jiang, H. Ma and J. Zou, Coord. Chem. Rev., 2025, 529, 216462 CrossRef CAS.
- Q. Gao, B. Yao, H. S. Pillai, W. Zang, X. Han, Y. Liu, S.-W. Yu, Z. Yan, B. Min, S. Zhang, H. Zhou, L. Ma, H. Xin, Q. He and H. Zhu, Nat. Synth., 2023, 2, 624–634 CrossRef CAS.
- Q. Gao, B. Yao, Y. Liu, L. Shi, Z. Yan, L. Xu, Q. He and H. Zhu, Chem Catal., 2025, 5, 101328 CrossRef CAS.
- Q. Gao, Z. Yan, W. Zhang, H. S. Pillai, B. Yao, W. Zang, Y. Liu, X. Han, B. Min, H. Zhou, L. Ma, B. Anaclet, S. Zhang, H. Xin, Q. He and H. Zhu, J. Am. Chem. Soc., 2023, 145, 19961–19968 CrossRef CAS PubMed.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., 2023, 62, e202218167 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., 2021, 60, 6170–6176 CrossRef CAS PubMed.
- Y. Cai, J. Fu, Y. Zhou, Y. C. Chang, Q. Min, J. J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 586 CrossRef CAS PubMed.
- K. Shah, R. Dai, M. Mateen, Z. Hassan, Z. Zhuang, C. Liu, M. Israr, W. C. Cheong, B. Hu, R. Tu, C. Zhang, X. Chen, Q. Peng, C. Chen and Y. Li, Angew. Chem., 2022, 61, e202114951 CrossRef CAS PubMed.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS PubMed.
- Y. Li, Z. S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed.
- Z. Lei, W. Cai, Y. Rao, K. Wang, Y. Jiang, Y. Liu, X. Jin, J. Li, Z. Lv, S. Jiao, W. Zhang, P. Yan, S. Zhang and R. Cao, Nat. Commun., 2022, 13, 24 CrossRef CAS PubMed.
- B. Pattengale, Y. Huang, X. Yan, S. Yang, S. Younan, W. Hu, Z. Li, S. Lee, X. Pan, J. Gu and J. Huang, Nat. Commun., 2020, 11, 4114 CrossRef CAS PubMed.
- M. Wang, K. Sun, W. Mi, C. Feng, Z. Guan, Y. Liu and Y. Pan, ACS Catal., 2022, 12, 10771–10780 CrossRef CAS.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W. C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, e1800396 CrossRef PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W. C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- S. H. Park, D. T. To and N. Myung, Appl. Catal., A, 2023, 651, 119013 CrossRef CAS.
- A. Serban, M.-T. Liu, N. Chen, H. M. Chen and X. Hu, Energy Environ. Sci., 2025, 18, 1533–1543 RSC.
- Z. Wang, J. Chen, E. Song, N. Wang, J. Dong, X. Zhang, P. M. Ajayan, W. Yao, C. Wang, J. Liu, J. Shen and M. Ye, Nat. Commun., 2021, 12, 5960 CrossRef CAS PubMed.
- Y. An, X. Long, M. Ma, J. Hu, H. Lin, D. Zhou, Z. Xing, B. Huang and S. Yang, Adv. Energy Mater., 2019, 9, 1901454 CrossRef CAS.
- H. Zhang, X. Wu, C. Chen, C. Lv, H. Liu, Y. Lv, J. Guo, J. Li, D. Jia and F. Tong, Chem. Eng. J., 2021, 417, 128069 CrossRef CAS.
- M. Guo, Y. Lian, Y. Yuan, T. Yu, Y. Qu and C. Yuan, Appl. Phys. Lett., 2023, 123 Search PubMed.
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., 2021, 60, 18981–19006 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- P. Ahuja, S. K. Ujjain, I. Arora and M. Samim, ACS Omega, 2018, 3, 7846–7855 CrossRef CAS PubMed.
- N. Pauly, F. Yubero, F. J. García-García and S. Tougaard, Surf. Sci., 2016, 644, 46–52 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- A. F. Carley, S. D. Jackson, J. N. O'Shea and M. W. Roberts, Surf. Sci., 1999, 440, L868–L874 CrossRef CAS.
- M. A. Bica de Moraes, B. C. Trasferetti, F. P. Rouxinol, R. Landers, S. F. Durrant, J. Scarmínio and A. Urbano, Chem. Mater., 2003, 16, 513–520 CrossRef.
- F. Xie, W. C. Choy, C. Wang, X. Li, S. Zhang and J. Hou, Adv. Mater., 2013, 25, 2051–2055 CrossRef CAS PubMed.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 159–166 CrossRef CAS.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, e1802880 CrossRef PubMed.
- L. Cheng, L. Zhou, A. Xie, A. Tan, H. Jiang, R. Zhang, J. Miao, J. Liu, P. Wan and Y. Tang, Appl. Surf. Sci., 2025, 703, 163404 CrossRef CAS.
- J. Wang, J. Hu, S. Niu, S. Li, Y. Du and P. Xu, Small, 2022, 18, e2105972 CrossRef PubMed.
- L. He, M. Li, L. Qiu, S. Geng, Y. Liu, F. Tian, M. Luo, H. Liu, Y. Yu, W. Yang and S. Guo, Nat. Commun., 2024, 15, 2290 CrossRef CAS PubMed.
- H. Wang, S. Hamanaka, Y. Nishimoto, S. Irle, T. Yokoyama, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS PubMed.
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- N. Mironova-Ulmane, A. Kuzmin, I. Steins, J. Grabis, I. Sildos and M. Pärs, J. Phys. Conf. Ser., 2007, 93, 012039 CrossRef.
- J. Z. Ou, J. L. Carnpbell, D. Yao, W. Wlodarski and K. Kalantar-zadeh, J. Phys. Chem. C, 2011, 115, 10757–10763 CrossRef CAS.
- X. Guan, Y. Ren, S. Chen, J. Yan, G. Wang, H. Zhao, W. Zhao, Z. Zhang, Z. Deng, Y. Zhang, Y. Dai, L. Zou, R. Chen and C. Liu, J. Mater. Sci., 2020, 55, 5808–5822 CrossRef CAS.
- X. W. Yu, J. Zhao, L. R. Zheng, Y. Tong, M. Zhang, G. C. Xu, C. Li, J. Ma and G. Q. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- L. F. Huang, M. J. Hutchison, R. J. Santucci, Jr., J. R. Scully and J. M. Rondinelli, J. Phys. Chem. C, 2017, 121, 9782–9789 CrossRef CAS.
- H.-C. Chen, A. Shabir and K.-H. Tu, J. Mater. Chem. A, 2025 10.1039/D5TA00935A.
- A. Khalaf, R. Saghir, A. M. Abdallah, M. Noun and R. Awad, Appl. Phys. A: Mater. Sci. Process., 2024, 130, 691 CrossRef CAS.
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z.-L. Wang, Chem. Sci., 2023, 14, 3400–3414 RSC.
- G. S. Wang, Z. X. Yan, M. Xiang, Y. J. Ding, J. Q. Chen and Z. H. Xu, Mater. Today Chem., 2024, 42, 102358 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00302d |
| This journal is © The Royal Society of Chemistry 2025 |