
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the nucleophilicity of aluminyl anions: targeting selective C–H activation†
Fabian
Kallmeier
 a,
Gareth R.
Nelmes
a,
Claire L.
McMullin
b,
Alison J.
Edwards
c and
Jamie
Hicks
*a
a,
Gareth R.
Nelmes
a,
Claire L.
McMullin
b,
Alison J.
Edwards
c and
Jamie
Hicks
*a
aResearch School of Chemistry, Australian National University, ACT 2601, Australia. E-mail: jamie.hicks@anu.edu.au
bAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, New Illawarra Road, Lucas Heights, NSW 2234, Australia
cDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 13th May 2025
Abstract
Anionic aluminium(I) complexes, or aluminyl anions, are a recently discovered class of main group compounds that can C–H activate simple aromatic molecules. However, functional group tolerance remains an issue, with the activation of functionalised arenes often favouring more kinetically accessible side reactions (e.g. C–O/C–F activation) over the desired C–H activation. Here, we report a new, electron-rich potassium aluminyl complex, which by DFT has been calculated to be the most nucleophilic diamido aluminyl anion reported to date. The anion shows unprecedented rates of reaction towards the C–H activation of arenes, achieving the C–H activation of stoichiometric benzene in seconds at room temperature. Furthermore, the C–H activation is selective even in a range of functionalised arenes, including those with C–O and C–F bonds.
Introduction
The activation of carbon–hydrogen (C–H) bonds is one of the most transformative and challenging areas in modern chemistry.1–3 C–H bonds are ubiquitous in organic molecules, yet their inherent strength and low reactivity have historically made them difficult to functionalise selectively.4,5 Over the past few decades, significant advances in catalysis and synthetic methodology have enabled the direct transformation of C–H bonds into more chemically active functional groups, revolutionising the way chemists approach organic synthesis.2,6,7 However, the field is currently dominated by transition metal complexes, mainly noble, which are rare and expensive.7–11Main group compounds are increasingly being investigated as cheaper, “greener” alternatives to transition metal complexes for various transformations, including C–H activations.12–15 A recently discovered class of compounds that has shown great promise in this area is the aluminyl anions.16–18 These anionic compounds feature an aluminium centre in the +1 oxidation state and are isoelectronic to a carbene.19 Due to aluminium's greater electropositivity and the formal negative charge, aluminyl anions are significantly more nucleophilic than carbenes, and act principally as aluminium-centred nucleophiles.16–18
The ability of aluminyl anions to perform selective C–H activations of challenging substrates has been known since the initial report by Goicoechea and Aldridge in 2018.20 In this landmark publication, the C–H activation of benzene by the diamido potassium aluminium complex K2[Al(DippNON)]2 (I) was reported, leading to the formal oxidative addition product K2[AlH(Ph)(DippNON)]2 (Fig. 1). The reaction occurred under relatively mild reaction conditions (60 °C, 4 days) and represented the first example of a single main group centre to oxidatively add a C–H bond of benzene. In a follow-up publication, the aromatic C–H activation was shown to proceed through a nucleophilic aromatic substitution (SNAr) mechanism, and that the selective meta C–H activation of monoalkylated arenes was also possible with I.21
 | ||
| Fig. 1 (Top) Aluminyl complexes that have been reported to perform the C–H activation of arenes. (Bottom) Reaction conditions and products resulting from the C–H activation of benzene. Dipp = 2,6-diisopropylphenyl; tol = η6-toluene. | ||
Several other aluminyl anions have since been reported that can also achieve the C–H activation of benzene under relatively mild reaction conditions. In 2020, Harder and coworkers reported that the planar, six-membered potassium aluminyl anion II could achieve the double, 1,4 C–H activation of benzene at 35 °C over 7 days (Fig. 1).22 The reaction was also calculated to proceed by an SNAr-type mechanism, with an initial nucleophilic attack on the ring, followed by hydride transfer to the aluminium centre. Teams led by Coles, McMullin and Mulvey, and Hill, Mahon and McMullin have also shown that the more flexible six- (III)23 and seven- (IV)24 membered diamido aluminyl anions can C–H activate benzene.25,26 Both studies found that the accompanying group 1 cation can significantly influence the rate of benzene activation. However, both studies required harsher reaction conditions for benzene activation than I and II (≥80 °C), independent of the cation used. Currently, the most active aluminyl anion towards arene C–H activation is also the most nucleophilic, the dialkyl aluminyl anion V reported by Yamashita.16,27,28 When dissolved in benzene, V performs the C–H activation of the solvent at room temperature over 2.5 hours. This is the only reported aluminyl anion to achieve C–H activation of an unactivated arene at room temperature, but it still requires the complex to be dissolved in the substrate.
In many cases, the selective C–H activation of benzene by aluminyl anions is possible due to benzene's simplicity and lack of alternative sites of reactivity.29 Substituted arenes (for example, toluene, xylenes, aryl ethers or fluorinated arenes) introduce alternative sites of reactivity, which often lead to side reactions or mixtures of products.16,18 For example, the reaction between I and toluene yields a mixture of arene and methyl C–H activation products, whereas the reaction with anisole (PhOMe) shuts down the C–H activation, leading to selective C–O activation.21
The selective C–H activation of aromatic molecules is a desirable reaction. Therefore, designing an aluminyl complex that can selectively activate C–H bonds with some functional group tolerance would be beneficial. Herein, we report [Al(TIPSNON)]−, a modified version of I,20 where the flanking aryl groups of the ligand have been replaced silyl groups, resulting in a more σ-donating ligand. This subtle ligand modification dramatically increases the nucleophilicity of the anion, making it much more selective to C–H activation than I.
Results and discussion
Optimised synthesis of H2(TIPSNON)
The xanthene-based disilylamido proligand H2(TIPSNON) used in this work has previously been reported by Tilley and co-workers through the synthesis of the 4,5-diaminoxanthene, H2(HNON) (Scheme 1).30 However, the two reported routes to this compound are problematic, either low-yielding (13%) or requiring an expensive, specialised chiral phosphine and anhydrous ammonia. We report here a cheaper, multigram preparation for this compound, obtained through the Buchwald–Hartwig coupling between 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene and benzylamine, giving H2(BnNON), followed by a Pd/C catalysed hydrogen reduction of the compound (Scheme 1). This gives 4,5-diaminoxanthene, H2(HNON), in a 60% yield over two steps at >5 g scale. Alternately, the 4,5-diaminoxanthene can also be synthesised by heating the di-tert-butyl proligand, H2(tBuNON),31 in ortho-phosphoric acid for 18 hours at 145 °C, giving the diamine in 97% yield (see ESI for further details†). The diamine can then be converted to the proligand H2(TIPSNON) following a modified procedure to Tilley,30 through the double deprotonation of the diamine with nBuLi followed by quenching with triisopropylsilyl chloride (TIPSCl) giving H2(TIPSNON) in 91% yield (Scheme 1). | ||
| Scheme 1 Optimised synthesis of proligand H2(TIPSNON) originally reported by Tilley and coworkers.30 | ||
Reduction of AlI(TIPSNON) in benzene
Following optimisation of the proligand, our efforts turned to synthesising the desired aluminyl anion [Al(TIPSNON)]−. We recently reported the aluminium iodide complex AlI(TIPSNON) as a precursor to the heterobimetallic aluminium–iron complex (TIPSNON)AlFeCp(CO)2.31 The same AlI(TIPSNON) starting material was used in this work. Following a similar procedure to I,20 a benzene solution of AlI(TIPSNON) was stirred over an excess of KC8 at room temperature for two days, giving a near-colourless solution. Analysis of the reaction mixture by 1H NMR spectroscopy revealed a mixture of three TIPSNON-containing products, K2(TIPSNON) and two new products in an approximate 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio (Scheme 2). Workup of the reaction mixture allowed the separation and isolation of both TIPSNON-containing products as clean, crystalline samples, which were analysed by X-ray crystallography (Fig. 2).
:2:1 ratio (Scheme 2). Workup of the reaction mixture allowed the separation and isolation of both TIPSNON-containing products as clean, crystalline samples, which were analysed by X-ray crystallography (Fig. 2).
 | ||
| Scheme 2 Reduction of AlI(TIPSNON) with excess KC8 at room temperature to give benzene C–H activated products 2 and 3. | ||
 | ||
| Fig. 2 Structures of 2 and 3 as determined by X-ray crystallography. Displacement ellipsoids set at the 50% probability level. Most hydrogen atoms have been omitted and selected organic groups have been shown in wireframe for clarity. | ||
Neither of the two new reaction products were the targeted TIPSNON aluminyl anion, instead, both are benzene C–H activation products. The major product, K[AlH(Ph)(TIPSNON)] (2), is a benzene mono activation product and the minor, K2[{AlH(TIPSNON)}2-1,4-C6H4] (3), a doubly activated product. Both products are thought to be formed from the transiently generated aluminyl anion K[Al(TIPSNON)] reacting with benzene, with 2 being the formal C–H oxidative addition product. The formation of 3 can be explained by a reaction between 2 and a second equivalent of the aluminyl anion, giving the two-fold 1,4 C–H activation product. Compound 2 crystallises as a 1D coordination polymer, with the potassium cations bridging between [AlH(Ph)(TIPSNON)]− anions. This is in contrast to the DippNON analogue, K2[Al(H)(Ph)(DippNON)]2,20 which is dimeric in the solid state due to the potassium cations forming interactions with the Dipp groups of the ligand. Compound 3 is monomeric in the solid state, with the two potassium cations interacting with both faces of the bridging [C6H4]2− ligand in an inverse sandwich-type arrangement. In both structures, the coordinated arene lies perpendicular to the plane of the tridentate TIPSNON ligand and the hydride, leading to aluminium occupying a distorted square pyramidal geometry.
The formation of 2 and 3 was initially surprising as aluminyl anions do not typically C–H activate benzene at room temperature. For example, the activation of benzene by the related aluminyl complex I takes four days at 60 °C,20 whereas the diamido aluminyl anion IV by Hill, Mahon and McMullin takes between 12 hours (Cs) and 14 days (K) at 110 °C, varying according to cation.26 The only aluminyl anion that has been reported to activate benzene at room temperature is the dialkyl aluminyl anion reported by Yamashita and coworkers, which has been calculated to be the most nucleophilic aluminyl anion with the highest energy HOMO.16 The formation of 2 and 3 at room temperature suggests that the nucleophilicity of the transiently generated K[Al(TIPSNON)] complex may be comparable to that of the dialkyl aluminyl anion, which is unexpected for a diamido aluminyl anion due to the weaker σ-donors.
Calculated reaction mechanism for the formation of 2 and 3
To further support that the formation of 2 and 3 proceeds through a transiently generated aluminyl anion, the reaction mechanism was investigated computationally. Two possible mechanisms were calculated using density functional theory at the PBE0-D3BJ-CPCM(benzene)/Def2-TZVPP//PBE0-D3BJ/BS1 (see ESI for details†) level of theory and are shown in Fig. 3. The calculated mechanism was found to occur by two independent C–H activation events, with 2 being an intermediate on the way to 3. McMullin, Fulton and Coles have reported a similar stepwise mechanism for the 1,4-activation of benzene with Na/KIII.32 | ||
| Fig. 3 Gibbs free energy profile (PBE0-D3BJ-CPCM(benzene)/Def2-TZVPP//PBE0-D3BJ/BS1) for the reaction between the transiently generated Kn[Al(TIPSNON)]n (n = 1, 2) aluminyl complex and benzene to give C–H activation complexes 2 and 3. Relative Gibbs free energies are presented in kcal mol−1. Monomeric pathway highlighted in blue. | ||
To begin the mechanism, a number of solution-state conformations of the transiently generated potassium aluminyl were investigated. The lowest energy of these was found to be the dimeric, benzene-free complex, K2[Al(TIPSNON)]2, where the potassium cations bridge the two aluminium centres (K2[Al]2, Fig. 3). Accordingly, all energies quoted are in reference to this complex. However, the corresponding monomeric complex, K[Al(TIPSNON)] (K[Al], Fig. 3), was found to be only +5.5 kcal mol−1 higher in relative energy, suggesting that the first C–H activation (to give 2) could be proceeding through either a monomeric or dimeric mechanism. As such, both reaction pathways were calculated.
The calculated monomeric and dimeric pathways were found to be similar, both starting with the coordination of benzene to the potassium aluminyl. In the dimeric pathway, two different structures of essentially the same Gibbs free energy (+1.6 kcal mol−1) were found: one where the benzene coordinates to a single potassium cation of the potassium-bridged dimer and a second where the benzene inserts into the centre of the dimer, forming an inverse sandwich complex (K2[Al]2C6H6, Fig. 3). Only the second was found to lead to a productive pathway. Nucleophilic attack on the coordinated benzene by the aluminyl anion occurs through TS0-1 (TS0-1mono for the monomeric mechanism and TS0-1di for the dimeric mechanism), leading to the Meisenheimer intermediates INT1mono and INT1di. Consistent with other reported studies,21,26,28,32 this step was found to be rate-determining, with a calculated barrier of +31.7 kcal mol−1 for the dimeric pathway. The transition state for the monomeric pathway (TS0-1mono) was calculated to be slightly lower at +29.9 kcal mol−1 in reference to the solvent-free potassium aluminyl dimer K2[Al]2, but only 24.4 kcal mol−1 when referenced to the monomeric aluminyl anion K[Al]. We propose that this latter value is more indicative of the true activation barrier, as when monitoring the reaction spectroscopically, the aluminyl anion was not observed, suggesting that the aluminyl anion is truly transient and activates benzene before dimerising.
From the Meisenheimer intermediates INT1mono/di, both pathways undergo a facile hydride transfer from the [C6H6]− to the aluminium centre through transition states TS1-2mono/di (with a barrier of <2 kcal mol−1 in both cases), leading to the formation of 2 (or INT2 in the dimeric pathway, which can release an equivalent of K[Al] to give 2). This step is highly exergonic in both pathways (monomeric −47 kcal mol−1; dimeric −43 kcal mol−1).
From 2, the calculated mechanism for the second C–H activation (giving 3) is very similar to that of the first, but now only one pathway is available. This involves the coordination of a second equivalent of potassium aluminyl to 2 (INT2), the nucleophilic attack at the 4-position of the metallated arene (TS2-3; ΔG‡ = 10.4 kcal mol−1), yields a second Meisenheimer intermediate (INT3), and finally, a hydride transfer (TS3-4; ΔG‡ = 6.9 kcal mol−1) gives 3. As with the first C–H activation, the rate-determining step was found to be the nucleophilic attack on the arene, but interestingly, this second C–H activation from 2 has a much lower barrier of only 11.4 kcal mol−1. This suggests the reaction between 2 and a second equivalent of potassium aluminyl is facile at room temperature. As 3 is only formed as a minor product of the reaction, this also supports the transient nature of the aluminyl anion, which preferentially reacts with the benzene solvent over 2, presumably due to its much higher abundance.
Synthesis of [K(18-crown-6)][Al(TIPSNON)] (4)
As the activation of benzene by the TIPSNON aluminyl anion is facile at room temperature, it was clear that isolation of the anion required avoidance of aromatic solvents. Accordingly, the synthesis of the aluminyl anion was repeated but in pentane. A solution of AlI(TIPSNON) was stirred over an excess of KC8 for two days at room temperature, but no reaction was observed. It was only when one equivalent of the potassium sequestering reagent 18-crown-6 was added that a relatively rapid colour change was observed, turning the reaction mixture from colourless to bright yellow/orange. Workup of the reaction mixture after two hours of stirring at room temperature gave the targeted aluminyl anion, [K(18-crown-6)][Al(TIPSNON)] (4), as bright yellow crystals (Scheme 3). | ||
| Scheme 3 Reduction of 1 with excess KC8 and 18-crown-6 in pentane giving the aluminyl anion [K(18-crown-6)][Al(TIPSNON)] (4). | ||
[K(18-crown-6)][Al(TIPSNON)] was characterised by X-ray crystallography, and the structure is shown in Fig. 4. The complex is monomeric in the solid state, crystallising as a contact ion pair, with the aluminium centre of the anion interacting with the 18-crown-6-complexed potassium cation. The structure resembles that of the 18-crown-6 sequestered potassium aluminyl complex IV reported by Hill, Mahon, McMullin and coworkers,24 but with a shorter Al⋯K interaction (Al⋯K: 4 = 3.557(1) Å, IV 3.9133(6) Å) and a much more acute L–Al–K angle, where L is the plane of the diamido ligand.
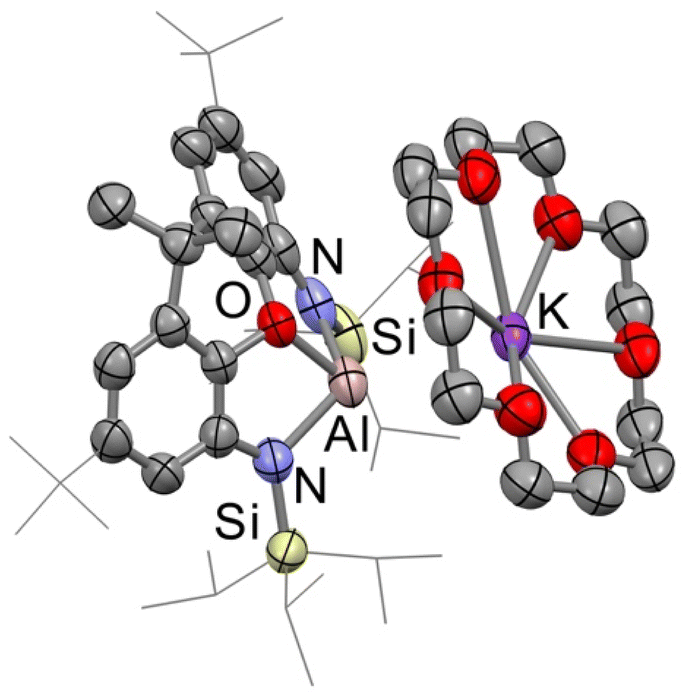 | ||
| Fig. 4 Structure of 4 as determined by X-ray crystallography. Displacement ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted and selected organic groups have been shown in wireframe for clarity. | ||
Structural and electronic comparison between the [Al(TIPSNON)]− and [Al(TIPSNON)]− anions
With the structure of the [Al(TIPSNON)]− anion acquired, a comparison between it and the original [Al(DippNON)]− was carried out. As no [K(18-crown-6)] salt of the [Al(DippNON)]− anion has been reported, comparisons have been made with the anion of the charged-separated [K(222-cryptand)][Al(DippNON)] complex.29 The solid-state structures of the two anions, excluding cations, are shown in Fig. 5, with selected bond lengths and angles in Table 1. | ||
| Fig. 5 A side-by-side comparison of the anions [Al(DippNON)]− (left)29 and [Al(TIPSNON)]− (right) as determined by X-ray crystallography. Anions and hydrogen atoms have been omitted, and selected organic groups have been shown in wireframe for clarity. | ||
| [Al(DippNON)]− (X-ray)29 | [Al(TIPSNON)]− (X-ray) | [Al(TIPSNON)]− (calc) | |
|---|---|---|---|
| Al–O (Å) | 2.1752(9) | 2.006(3) | 2.096 |
| Al–N (Å) | 2.0219(10) | 2.063(3) | 2.103 |
| 2.0489(10) | 2.071(4) | 2.091 | |
| N–Al–N (°) | 126.74(4) | 116.54(13) | 113.2 |
At first glance, the two aluminyl anions appear very similar; both NON-ligands coordinate to the aluminium centre in a tridentate fashion, causing the xanthene backbone to hinge. However, close inspection of some bond lengths and angles (Table 1) reveals some notable differences. In [Al(TIPSNON)]−, the Al–N bonds are slightly elongated compared with [Al(DippNON)] (by approx. 0.03 Å, Table 1), likely due to higher π-acidity of the N-bound TIPS groups lowering the π-donation of the anilides to the aluminium. The more striking difference is the Al–O bond length, which is 0.17 Å shorter in [Al(TIPSNON)]− than in [Al(DippNON)]− (2.006(3) vs. 2.1752(9) Å). This stronger coordination and greater xanthene hinging, decreases the N–Al–N angle by 10° compared to [Al(DippNON)]−.
To assess how these structural changes influence the electronic structure, the [Al(TIPSNON)]− anion was investigated by density functional theory (DFT). The anion was optimised at the PBE0-D3BJ/Def2-TZVP level of theory, with the optimised structure closely matching that obtained from crystallography, including the significant shortening of the Al–O bond length (Table 1). The level of theory is the same as that we recently used to investigate the electronic structure of five reported aluminyl anions (including [Al(DippNON)]−).16
Examination of the frontier molecular orbitals of [Al(TIPSNON)]− reveals an interesting picture (Fig. 6). As is common in aluminyl anions, the HOMO is calculated to be the Al-centred lone pair.16 However, at an energy of −0.63 eV, this is significantly higher than all other reported diamido aluminyl anions, suggesting that [Al(TIPSNON)]− should be the most nucleophilic of all reported diamido aluminyl anions. For comparison, the HOMO of [Al(DippNON)]− is calculated to be −0.97 eV at the same level of theory.16 This increased nucleophilicity has also been observed experimentally, with the transient, non-sequestered TIPSNON potassium aluminyl anion being the only reported diamido aluminyl anion to activate benzene at room temperature. Only the two alkyl-substituted aluminyl anions have calculated higher energy HOMOs,16 with Yamashita's dialkyl aluminyl anion V the only other aluminyl anion to report the activation of benzene at room temperature.27
 | ||
| Fig. 6 (Left) Calculated HOMO and LUMO+n energies of [Al(DippNON)]− and [Al(TIPSNON)]− at the PBE0-D3BJ/Def2-TZVP level of theory.16 LUMO+n indicates the orbital associated with the aluminium empty 3p-orbital. Orbital energies displayed in eV. (Right) Calculated HOMO and LUMO+7 orbitals of [Al(TIPSNON)]− shown at isovalue 0.100 e− Å−3. | ||
The energy of the unoccupied 3p-orbital on the Al-centre is also worthy of note (Fig. 6). In the previously reported [Al(DippNON)]−, this can be found as the LUMO+3 at +2.61 eV. However, in [Al(TIPSNON)]−, the corresponding orbital is significantly higher in energy due to the much stronger O–Al coordination and appears as the LUMO+7 at +3.41 eV – an increase of 0.8 eV compared to [Al(DippNON)]−. This has two major consequences: (1) significantly lowering the electrophilicity of the Al centre in the aluminyl anion and (2) increasing the energy gap between the Al-centred Frontier orbitals (i.e. the HOMO–LUMO+n gap). As the HOMO–LUMO energy separation is critical for explaining the reactivity of numerous low-oxidation state main group compounds,12,13 increasing this energy separation by ∼0.5 eV in comparison to [Al(DippNON)]− is expected to have a significant impact on the aluminyl's reactivity. A side-by-side comparison between the Al-centred frontier molecular orbitals of [Al(TIPSNON)]− and those previously reported for [Al(DippNON)]− is shown in Fig. 6.
Reactivity of [K(18-crown-6)][Al(TIPSNON)] (4)
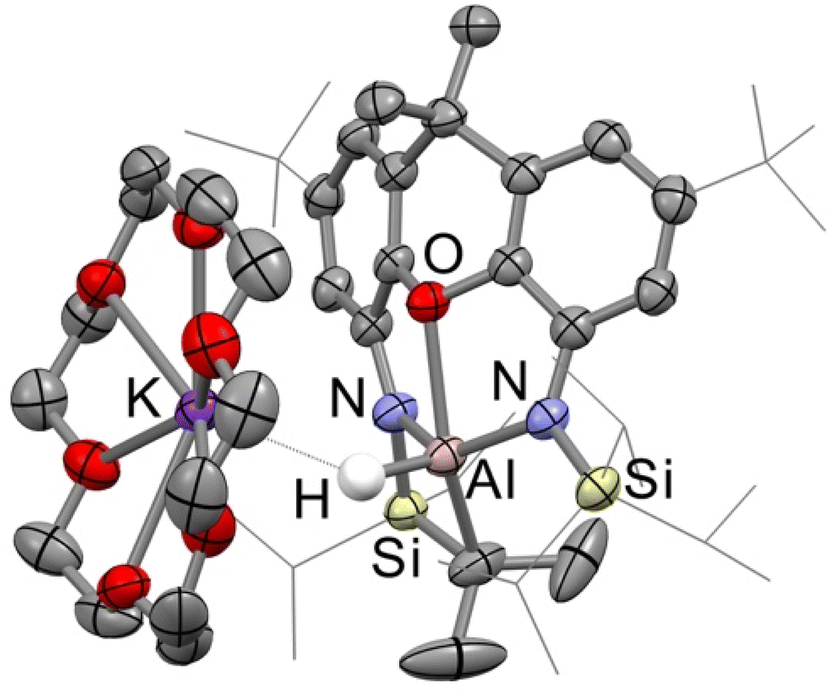
The calculations predict that the TIPSNON aluminyl anon should be more nucleophilic and less electrophilic than the DippNON equivalent. Furthermore, with a larger energy separation between the Al-centred frontier molecular orbitals, the anion should disfavour reactions that occur through more typical oxidative addition-type mechanisms (reactions that require both an occupied and unoccupied orbital in a single step).12 As the rate-determining step in the C–H activation of benzene is calculated to be a nucleophilic attack on the carbon atom (Fig. 3), [K(18-crown-6)][Al(TIPSNON)] (4) may have both increased reactivity and selectivity towards arene C–H activation than other aluminyl anions. As such, the reactivity of 4 was initially investigated towards benzene. 2.5 Equivalents of benzene were added to a solution of 4 in pentane at room temperature. After less than 1 minute, the solution had changed colour from bright yellow/orange to colourless. Workup of the reaction mixture after 10 minutes gave the mono C–H activation product [K(18-crown-6)][AlH(Ph)(TIPSNON)] (5) as a colourless solid (Scheme 4), which was additionally characterised by X-ray crystallography (Fig. 7). Unlike the transient, non-sequestered complex, which gave a mixture of both mono- and di-activation products (Scheme 1), only mono-activation of benzene was observed in this case. This is likely due to the sequestered cation leading to increasingly charge-separated anions in the solution, enhancing coulombic repulsion between the mono-activation (aluminate) product and the aluminyl anion. | ||
| Scheme 4 Reactions between [K(18-crown-6)][Al(TIPSNON)] 4 and benzene, toluene, diphenyl ether and fluorobenzene. All reactions show selectivity towards C–H activation. Outcomes of reactions between K2[Al(DippNON)]2I and the same substrates are shown in boxes for comparison.20,21 | ||
 | ||
| Fig. 7 Structures of 5 (top-left), 6 (top-right), 7 (bottom-left) and 9-H (bottom-right) as determined by X-ray crystallography. [K(18-crown-6)] cations and most hydrogen atoms have been omitted for clarity. Displacement ellipsoids set at the 50% probability level and selected organic groups have been shown in wireframe. | ||
The rate of benzene C–H activation by 4 is unprecedented for aluminyl anions. All other diamido aluminyl anions require heat and significantly longer reaction times to C–H activate benzene.20,22,25,26,32 Only the most nucleophilic aluminyl anion, the dialkyl aluminyl anion V, has been reported to C–H activate benzene at room temperature.27 However, even here, the reaction is reported to take 2.5 hours when dissolved in benzene, not <1 minute with near stoichiometric amounts as in the case of 4.
With the [Al(TIPSNON)]− anion showing enhanced reactivity rates towards arene C–H activation, the selectivity of the C–H activation was explored. The previously reported K2[Al(DippNON)]2 (I) shows little selectivity towards arene C–H activation,16,21 with reactions between I and monofunctionalised arenes (e.g. PhX, where X = a halide, methyl or OR) leading to activation of the X group. With this in mind, the reactivity of 4 towards the functionalised aromatic molecules, toluene, diphenyl ether and fluorobenzene, was investigated (Scheme 4).
Starting with toluene, 2.5 equivalents of toluene were added to a pentane solution of 4 at room temperature. As for the benzene reaction, the mixture turned colourless in less than one minute. Analysis of the reaction mixture by 1H NMR spectroscopy showed selective and complete conversion to the meta C–H activation product [K(18-crown-6)][AlH(3-Me-C6H4)(TIPSNON)] (6), with no evidence of methyl C–H activation observed. The product was isolated in an 88% yield and further characterised by X-ray crystallography (Fig. 7). Tolerance towards the aromatic methyl group is novel for diamido aluminyl anions. For example, the reaction between I and toluene gives a mixture of aromatic and methyl C–H activation products,21 whereas the reaction between the seven-membered diamido aluminyl anion RbIV and toluene gives only the methyl activation product. Only the most nucleophilic dialkyl aluminyl anion V has been reported to selectively activate the meta C–H bond of toluene.28
Extending the functional group tolerance investigations, the reactivity of 4 towards diphenyl ether was investigated. Using similar reaction conditions to those above, the reaction between 4 and diphenyl ether (1.4 equiv.) gave exclusively the meta C–H activation product [K(18-crown-6)][AlH(3-OPh-C6H4)(TIPSNON)] (7) by 1H NMR spectroscopy. The product was isolated in a 52% crystalline yield and additionally characterised by X-ray crystallography (Fig. 7). The reaction is selective towards C–H activation over C–O activation, which is unprecedented for diamido aluminyl anions. As the direct comparison reaction between I and diphenyl ether had not been reported, the reaction was carried out for comparison. In C6D6, no reaction between I and diphenyl ether was observed at room temperature. However, heating the reaction solution to 50 °C saw a gradual discolouration of the solution. Analysis by 1H NMR spectroscopy saw a clean conversion of I to the C–O activation product K2[AlOPh(Ph)(DippNON)]2 (8), which was additionally characterised by X-ray crystallography (see ESI†). This striking contrast in reactivity (C–H vs. C–O activation) between these closely related aluminyl anions shows how these remarkable reagents can be easily tailored to target specific bonds.
Finally, tolerance towards C–F bonds was investigated. The reaction between I and fluorobenzene at room temperature gives a complex mixture of products (>10 DippNON-containing products by 1H and 19F NMR spectroscopy). The reaction with 4 is, however, much more selective, giving only two products in a ∼5:2 ratio by 1H NMR spectroscopy. The major product was found to be the meta C–H activation product [K(18-crown-6)][AlH(3-F-C6H4)(TIPSNON)] 9-H and the minor the C–F activation product [K(18-crown-6)][AlF(Ph)(TIPSNON)] 9-F (Scheme 4). Analysis by X-ray diffraction found the two compounds to cocrystallise in a ∼2:1 ratio, with the structure of 9-H, the major C–H activation product, shown in Fig. 7. Even though the reaction is not completely selective, the reaction is the first example of an aluminyl anion favouring C–H activation over C–F activation.
Single crystal neutron diffraction
In each of the structures of compounds 2–3 and 5–9 studied by single-crystal X-ray diffraction, electron density consistent with an aluminium-bound hydride was observed in the difference map and could be refined isotropically. X-ray diffraction arises from the electron distribution (no proton is necessarily associated with such observed electron density) and deletion of the hydrides from these models and refining to convergence did not significantly perturb the structures. To better understand the structure of the C–H activation products, as well as confirm the presence of an aluminium-bound hydride, compound 6 was additionally characterised by single-crystal neutron diffraction. A crystal of size 3.0 × 1.7 × 1.0 mm was mounted on the KOALA2 instrument at ANSTO (Sydney). 13 images of 6000 second exposures were obtained, rotating the crystal 17° between frames (see ESI for full details†), yielding neutron diffraction data from which the hydride position could be clearly located in the difference map and refined anisotropically, subject only to the constraints implicit in the chosen model (Fig. 8).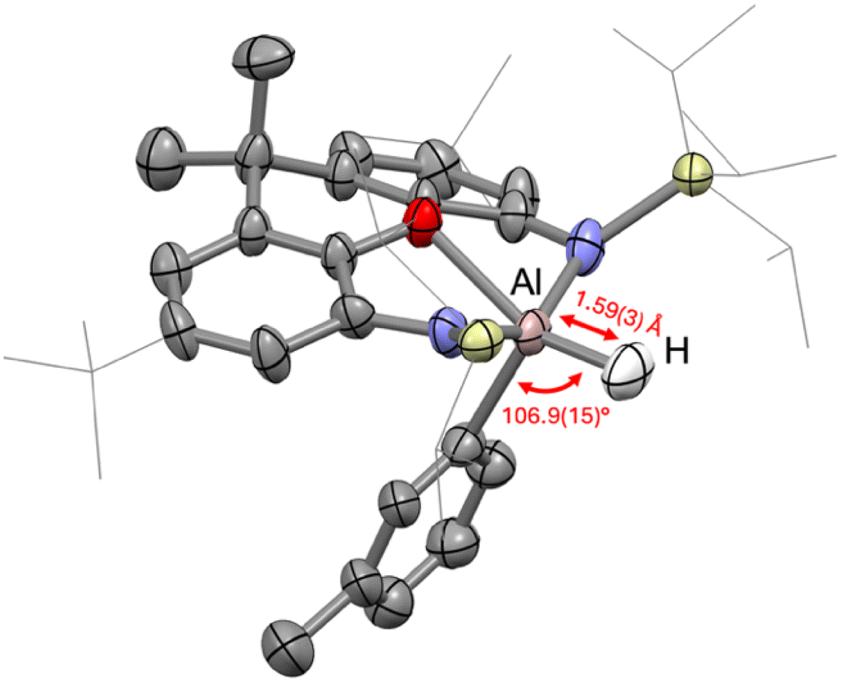 | ||
| Fig. 8 Structure of 6 as determined by single crystal neutron diffraction. Most hydrogen atoms and the [K(18-crown-6)] cation have been omitted for clarity. The TIPS and tolyl groups have been modelled as two-fold disordered, with only one part shown above for clarity. Displacement ellipsoids set at the 50% probability level and selected organic groups have been shown in wireframe for clarity. The Al–H bond length and H–Al–C bond angle are shown in red. | ||
The Al–H bond converged to 1.59(3) Å and lies coplanar with the ligand oxygen and the meta-bound carbon atom of the activated toluene, with an H–Al–C angle of 106.9(15)° and an H–Al–O angle of 153.8(15)°. The τ5 parameter33 for compound 6 is 0.19, meaning that the aluminium centre is best described as occupying a distorted square pyramidal geometry.
sp3 C–H activation with 4
The ease with which 4 can activate aromatic C–H bonds suggests that the aluminyl anion may also be able to activate the sp3 C–H bonds of alkanes. This is not the case as it is clear that 4 does not immediately react with pentane at room temperature, as this is the solvent in which the aromatic C–H activation studies have been performed. Leaving a solution of 4 in pentane for a matter of days at room temperature leads to an intramolecular sp3 C–H activation of a TIPS isopropyl group, giving 10 (Scheme 5). The compound has been characterised by X-ray crystallography, with its structure shown in Fig. 9. Interestingly, 10 can also be synthesised in the solid state by heating a powdered sample of 4 to 100 °C for 2 hours. The fact that this intramolecular activation occurs when dissolved in pentane shows that it is preferred to alkane activation, so intermolecular activation of alkanes with 4 is unlikely. | ||
| Scheme 5 Intramolecular C–H activation of 4 both in solution and in the solid state. | ||
 | ||
| Fig. 9 Solid state structures of 10 as determined by X-ray crystallography. Most hydrogen atoms have been omitted for clarity. Displacement ellipsoids set at the 50% probability level and selected organic groups have been shown in wireframe for clarity. | ||
Conclusions
Since their first report in 2018, aluminyl anions have shown the ability to activate strong C–H bonds under mild reaction conditions. However, selectivity and functional group tolerance continue to be known limitations. The [Al(TIPSNON)]− anion reported here is an extremely nucleophilic aluminyl anion which shows enhanced reactivity towards aromatic C–H bond activation, for the first time allowing for near stoichiometric activation of aromatic substrates at room temperature. Moreover, functional group tolerance has been significantly increased compared to the previously reported aluminyl anions, allowing for selective C–H activation in the presence of aromatic C–O and C–F bonds. The reactivity of [Al(TIPSNON)]− has been compared with the related aluminyl anion, K2[Al(TIPSNON)]2 (I), which typically shows C–X activation (X = N, O, F) over C–H activation, demonstrating how these reagents can be tailored to target specific bonds in substrates.Data availability
The data supporting this article have been included as part of the ESI.† Finalised CIFs for all X-ray diffraction structures have been deposited at the Cambridge Crystallographic Data Centre. These can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/data_request/cif or by emailing E-mail: data_request@ccdc.cam.ac.uk.Author contributions
F. K. performed all the synthesis and characterisation. G. R. N. performed the computational work under the supervision of C. L. M. A. J. E. supervised the collection and refinement of the neutron diffraction data. J. H. was responsible for project management, funding acquisition, and providing resources. All authors were involved in reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JH would like to thank the Australian Research Council (FT240100229) for the funding of this work. We acknowledge the support of the Australian Centre for Neutron Scattering, ANSTO and the Australian Government through the National Collaborative Research Infrastructure Strategy supporting the neutron research infrastructure in this work via ACNS proposal 18886. This research was undertaken with the assistance of resources from the National Computational Infrastructure (NCI Australia), an NCRIS enabled capability supported by the Australian Government, and the University of Bath's Research Computing Group (https://doi.org/10.15125/b6cd-s854) for their support in this work.Notes and references
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- K. M. Altus and J. A. Love, Commun. Chem., 2021, 4, 173 CrossRef.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- H. M. L. Davies, J. D. Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC.
- U. Dhawa, N. Kaplaneris and L. Ackermann, Org. Chem. Front., 2021, 8, 4886–4913 RSC.
- S. Barranco, J. Zhang, S. López-Resano, A. Casnati and M. H. Pérez-Temprano, Nat. Synth., 2022, 1, 841–853 CrossRef CAS.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CAS.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- N. R. Judge, A. Logallo and E. Hevia, Chem. Sci., 2023, 14, 11617–11628 RSC.
- C. I. Raţ, A. Soran, R. A. Varga and C. Silvestru, in Advances in Organometallic Chemistry, ed. P. J. Pérez, F. G. A. Stone and R. West, Academic Press, 2018, vol. 70, pp. 233–311 Search PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS.
- K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942–6956 RSC.
- M. P. Coles and M. J. Evans, Chem. Commun., 2023, 59, 503–519 RSC.
- C. Boehme and G. Frenking, J. Am. Chem. Soc., 1996, 118, 2039–2046 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- R. J. Schwamm, M. S. Hill, H.-Y. Liu, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Chem.–Eur. J., 2021, 27, 14971–14980 CrossRef CAS.
- T. X. Gentner, M. J. Evans, A. R. Kennedy, S. E. Neale, C. L. McMullin, M. P. Coles and R. E. Mulvey, Chem. Commun., 2022, 58, 1390–1393 RSC.
- H.-Y. Liu, M. S. Hill, M. F. Mahon, C. L. McMullin and R. J. Schwamm, Organometallics, 2023, 42, 2881–2892 CrossRef CAS.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- S. Kurumada, K. Sugita, R. Nakano and M. Yamashita, Angew. Chem., Int. Ed., 2020, 59, 20381–20384 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS.
- A. Nicolay, M. S. Ziegler, L. Rochlitz and T. D. Tilley, Polyhedron, 2020, 180, 114420 CrossRef CAS.
- F. Kallmeier, A. J. R. Matthews, G. R. Nelmes, N. R. Lawson and J. Hicks, Dalton Trans., 2024, 53, 12450–12454 RSC.
- A. O'Reilly, A. M. S. Booth, C. L. McMullin, J. R. Fulton and M. P. Coles, Polyhedron, 2024, 264, 117226 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and characterisation data, computational and crystallographic details. CCDC 2432683–2432692. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02682b |
| This journal is © The Royal Society of Chemistry 2025 |