
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactivated solid-state self-assembly: a mechanochemistry-free route to high-purity aromatic amine crystals†
Xia
Wang
,
Ming-Hui
Su
,
Jia-Yong
Zhou
,
Shao-Shuai
Liu
,
Jin-Wei
Yuan
 ,
Liang-Ru
Yang
,
Meng
Yan
,
Ya-Xin
Li
and
Yun-Tao
Xia
*
,
Liang-Ru
Yang
,
Meng
Yan
,
Ya-Xin
Li
and
Yun-Tao
Xia
*
College of Chemistry & Chemical Engineering, Henan University of Technology, Academician Workstation for Natural Medicinal Chemistry of Henan Province, Zhengzhou 450001, P. R. China. E-mail: xyt@haut.edu.cn
First published on 16th June 2025
Abstract
The rate-limiting step in solid-state reactions involves the diffusion of atoms, molecules, or ions through the crystalline phases of the reactant, intermediate, and product. This process is slow, often requiring days or even weeks of continuous or intermittent treatment, while consuming a significant amount of energy. This study describes a light-driven spontaneous solid-state synthesis strategy for the preparation of solid aromatic amines. Under ambient conditions (25 °C, 1 atm H2), natural light irradiation (≥100 W) triggers surface plasmon resonance in 12R-Pd-NCs, inducing directional adsorption of solid nitroarenes and facilitating spontaneous ultrafast electron transfer through non-mechanochemical pathways. The system achieves exceptional efficiency with product yields exceeding 99% and chemical selectivity >99% for aromatic amines. Gram-scale experiments (15 g substrate) reveal remarkable catalytic performance, exhibiting a turnover number (TOF) of 1.39 × 105 h−1 while maintaining full catalytic activity through five consecutive cycles. This methodology transcends conventional thermodynamic limitations by establishing a novel “photon-induced electron tunneling-proton-coupled interface” mechanism in solid-state reactions, opening new avenues for sustainable chemical transformations.
Crushed fresh grapes produce wine by fermentation, but dried grapes cannot. While milk tends to go sour with time, powdered milk remains unchanged. Similarly, dried meat can be stored for a long time, whereas broth spoils quickly. By observing these phenomena, one can deduce that the transformation of one material into another occurs in the liquid state, not in the solid state. Aristotle, one of the most famous ancient Greek philosophers, summarized these observations and concluded, “No Corpora nisi Fluida”, meaning “no reaction occurs without a solvent”. This philosophy had a significant impact on the development of modern European science, providing a historical reason why most organic reactions have been studied in solution, even when no special reason for using solvents was apparent.1 As a result, the current pharmaceutical and fine chemical industries are heavily reliant on solvent-based organic synthesis, leading to severe solvent waste issues, as organic solvents typically account for 80–90% of the total mass used in any organic reaction (Scheme 1a).2 Although solvent recovery is a highly effective way to reduce solvent waste, organic chemists should focus on (re)designing organic synthesis to reduce or eliminate solvent use. In this context, solid-state organic transformations have attracted considerable attention as cleaner and more sustainable synthetic alternatives.3
 | ||
| Scheme 1 (a) Synthesis strategies and challenges in different phase states; (b) traditional solid-state synthesis strategies; (c) this work: solar-driven spontaneous solid-state synthesis without external forces. | ||
In solid-state synthesis, reactant molecules are in a constrained state, and their molecular conformations are relatively stable. This limits their ability to participate in reactions in the solid state. Based on topochemistry theory, solid-state synthesis reactions can generally be divided into four stages: first, crystal defects, deformations, and molecular looseness occur within one or several crystal nuclei. The second step involves the breaking of old chemical bonds and the formation of new ones under established conditions. Third, a small amount of the product quickly forms a solid solution within the original crystal. Fourth, product crystallization and separation occur.4 From the above steps, it is not difficult to find that the rate-limiting step in solid-state reactions is the diffusion of atoms, molecules or ions through the crystal phases of the reactants, intermediates, and products.5 This process is slow and can take days or even weeks, often requiring high-temperature treatment and consuming large amounts of energy. Methods such as solid-state grinding, ultrasonic irradiation, mechanical shaking or stirring, spark plasma sintering, and high-temperature melting are effective ways to accelerate this process (Scheme 1b).3b–l,6 Among these, solid-state grinding dominates the field of solid-state synthesis and has been widely applied in drug cocrystal synthesis, material development, electroplating, etc.3c,j,4b,7,11a Mechanical grinding increases the free energy of the solid surface, and the combined shear forces, friction, and elastic tension generated during compression produce instant micro-heating, which activates the reaction system and accelerates the reaction. However, the unavoidable use of external forces leads to what is commonly known as mechanochemistry. In the context of the energy crisis, green chemistry, and sustainable development, the field of solid-state organic synthesis faces new opportunities and challenges: (1) achieving spontaneous solid-state organic synthesis under mild conditions is difficult; (2) most solid-state synthesis strategies are often suitable for only a few specific cases, with activity and selectivity often failing to achieve synchronous regulation control; (3) conventional catalysts tend to exhibit high activity on the contact surface during solid-state reactions, while the uncontacted portion often reacts slowly or not at all, making it difficult to achieve unified control of the conversion rate and selectivity across spatial dimensions.3a,4b,5,7a
Photoactivated synergistic solid-state self-assembly catalysis driven by solar energy may provide an interesting pathway for spontaneous, efficient and highly selective solid-state synthesis under mild conditions. Using the photo effect of solar energy to drive organic synthesis holds promise for replacing traditional thermal catalytic techniques, thus enabling low-energy chemical production.8,10b In fact, the photo effect has been extensively studied in various fields, including energy utilization, biomedicine, catalytic conversion, smart devices, etc.9 It has also been applied in areas such as solar steam generation, phototherapy, photocatalysis, agricultural heaters, photoenergy storage, photo-induced self-healing materials, photo-driven soft robots, and photofunctional materials.10 Considering the current energy crisis, energy consumption is one of the critical factors limiting the development of modern chemical industries. Using solar energy to drive organic synthesis and converting and storing solar energy as chemical energy, offers a new approach to alleviate the current energy dilemma. Metal nanostructures, with their unique plasmonic optical properties, provide opportunities to achieve this technological pathway.11
However, there are two key scientific and technical challenges facing the full utilization of solar energy in solid-state synthesis and chemical production: how to efficiently capture solar energy across a broad spectrum and how to effectively channel the captured photon energy into solid-state chemical reactions.12 Palladium metal is an efficient catalyst for many organic reactions, but the localized surface plasmon resonance properties of palladium nanostructures are often unsatisfactory compared to those of common metals like gold and silver. Palladium has a smaller absorption cross-section and its response spectrum is limited to the ultraviolet range, posing significant difficulties for solar energy capture and utilization.12c,13 The key to addressing these issues lies in optimizing and controlling these processes to meet the needs of organic synthesis. One effective method is to load photosensitive metals or semiconductor materials to enable absorption across a broad visible-light spectrum. The resulting photoelectric effect generates localized high temperatures that can provide the necessary heat for organic reactions, achieving the unification of solar energy utilization and catalytic activity distribution in space.14 Reducing structural symmetry is another effective way to enhance visible-light absorption.12b It is worth noting that these strategies work well for organic reactions in the liquid state, but in solid-state reactions, the penetration of light is reduced, and the confinement of molecules in the solid state makes photocatalytic organic synthesis reactions renders very difficult, especially under spontaneous conditions without external forces. In fact, current photocatalytic technology is mainly limited to liquid-phase reactions, sometimes supplemented by stirring.12b,14b,14c,15 Compared to this, solid-state photocatalytic reactions hold even greater potential in the fields of energy and the environment and should be regarded as a powerful tool for green chemistry, although they are more challenging. Therefore, developing photocatalytic materials for visible light is the ultimate goal of this research, aiming for spontaneous solid-state synthesis driven by solar energy.
Sometimes, interesting discoveries come from unfettered imagination. As McQueen stated, “Most material discoveries in the field of solid-state chemistry have historically been made by accident, not by carefully designed reactions…”.16 Inspired by the aforementioned work, we have developed a straightforward class of palladium nanoclusters (12R-Pd-NCs). The defect structure of the clusters displays remarkable asymmetry, which markedly enhances visible light absorption and provides a continuous supply of energy. The flexible alkyl chains exhibit optimal capture and driving properties. This pre-designed catalyst enables spontaneous, efficient, and highly selective solid-state synthesis of aromatic amines at room temperature (25 °C), requiring only the continuous supply of solar energy (Scheme 1c). Notably, this catalyst is widely applicable to various substrates and can easily be used in the preparation of several products of potential interest from a pharmaceutical perspective, such as benzocaine, butamben or bioactive molecules like p-aminophenyl ether. Even in the preparation of chemical intermediates like p-anisidine, it is possible to achieve up to 15 g production with >99% yield and >99% chemical selectivity, using only a minimal amount of catalyst (75 mg). This is a new discovery in the field of solid-state photocatalysis. The development of this strategy offers opportunities and limitless possibilities for the large-scale application of solar-driven green solid-state synthesis.
Results and discussion
Catalytic evaluation of solid-state photoactivated spontaneous hydrogenation
Herein, we report marked gains in monolayer-protected nanocluster 12R-Pd-NCs, outperforming the classical Pd/C catalyst. The approach involves Pd species bonded to laurylbenzene via metal carbon [M-C(sp2)] from the homogeneous reduction method by sodium borohydride, characterized by strong lipophilicity and high flexibility (see the ESI†). The constitutive model of monolayer organic capping ([M-C(sp2)]) has the great potential to make the dispersion and catalytic properties of nanoscale palladium clusters different from those of conventional Pd/C, homogeneous palladium complexes and some palladium colloids. We focused on nitrobenzene hydrogenation, an important reaction for the production of drugs and dyes, and a model reaction for evaluating hydrogenation catalysts. Previous studies on this reaction were usually performed with diluted nitrobenzene with a large amount of solvents for the fast removal of the aniline product on the metal surface.17 These catalysts are insufficient for this reaction under solvent-free conditions, especially at ambient temperature and hydrogen pressure, even when the reactants are in the liquid form and mechanical stirring is introduced at the same time.18a Here, we found that the 12R-Pd-NCs catalyst was highly efficient for spontaneous photoactivated hydrogenation of solid-state nitrobenzene, achieving a >99% yield of the whole reaction and a turnover frequency of 139, 169 h−1 at ambient temperature (25 °C) and hydrogen pressure (1 atm H2) without any external force, requiring only the continuous supply of solar energy (Fig. 1a). In order to prevent the introduction of mechanical energy into the reaction system, a series of preparatory steps are performed prior to the introduction of hydrogen gas. These include the shaking and standing of solid reactants and catalysts, which are carried out manually. After an interval of standing, hydrogen is introduced. This activity was about 24 times greater than that of the 3–5 nm Pd/C catalyst (5718 h−1) as well as 4 times that of the classical Pd(OAc)2 catalyst (36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 626 h−1, 4 h) under the same conditions, respectively (Fig. 1c, more details can be found in the ESI†), exceeding the performance of the state-of-the-art Pd-, Pt-, and Ru-based catalysts previously reported for nitrobenzene hydrogenation.18Fig. 1 also illustrates the utilisation of solar energy to drive and enhance the hydrogenation reaction in a solid state configuration via photothermic chemical processes. While light is critical to the reaction, its role as a heat source is an important consideration. The results of the reaction at different temperatures under dark conditions, as illustrated in Fig. 1d, indicate that temperature effects are unlikely to be the controlling factor in the process. The hypothesis of a ‘gradient-driven’ phenomenon appears to be a more reasonable assumption. Due to the nature of how light versus heat (in the dark) is applied, the resulting temperature ‘gradients’ are distinctly different. According to thermal measurements, these gradients are larger in the case of light bursts (Scheme 3b). This is significant because it is the gradient, and not uniformity, that drives molecular or particulate motion. The presence of a gradient facilitates the preferential migration of the catalyst relative to the reactant/product, enabling complete transformation. This is something that heat, by itself, cannot achieve in the absence of a gradient that drives motion (Fig. 1d). The data illustrated in Fig. 1 also suggest the significance of gradients. The “step-like” function in yield in Fig. 1b suggests that there is a critical light level above which the dynamics required for high yield are achieved. The gradient needs to exceed a “local motion inhibited” threshold value in order for the yield to change. The change in the physical forms of the reaction is also consistent with the higher yield arising due to motion induced by a temperature gradient (Fig. 6). The physical form of the reaction mixture dramatically changes from a highly aggregated crystal structure to a plastic-like appearance during the reaction. In solid-state reactions, the microscopic alterations in particle morphology have a significant impact on the yield of the reaction. This presents a considerable challenge to the reproducibility of solid-state reactions. It is worth noting that in this system the substrate has almost no effect on the reaction yield within the particle size range of 0.05–5.5 mm (Fig. 2).
626 h−1, 4 h) under the same conditions, respectively (Fig. 1c, more details can be found in the ESI†), exceeding the performance of the state-of-the-art Pd-, Pt-, and Ru-based catalysts previously reported for nitrobenzene hydrogenation.18Fig. 1 also illustrates the utilisation of solar energy to drive and enhance the hydrogenation reaction in a solid state configuration via photothermic chemical processes. While light is critical to the reaction, its role as a heat source is an important consideration. The results of the reaction at different temperatures under dark conditions, as illustrated in Fig. 1d, indicate that temperature effects are unlikely to be the controlling factor in the process. The hypothesis of a ‘gradient-driven’ phenomenon appears to be a more reasonable assumption. Due to the nature of how light versus heat (in the dark) is applied, the resulting temperature ‘gradients’ are distinctly different. According to thermal measurements, these gradients are larger in the case of light bursts (Scheme 3b). This is significant because it is the gradient, and not uniformity, that drives molecular or particulate motion. The presence of a gradient facilitates the preferential migration of the catalyst relative to the reactant/product, enabling complete transformation. This is something that heat, by itself, cannot achieve in the absence of a gradient that drives motion (Fig. 1d). The data illustrated in Fig. 1 also suggest the significance of gradients. The “step-like” function in yield in Fig. 1b suggests that there is a critical light level above which the dynamics required for high yield are achieved. The gradient needs to exceed a “local motion inhibited” threshold value in order for the yield to change. The change in the physical forms of the reaction is also consistent with the higher yield arising due to motion induced by a temperature gradient (Fig. 6). The physical form of the reaction mixture dramatically changes from a highly aggregated crystal structure to a plastic-like appearance during the reaction. In solid-state reactions, the microscopic alterations in particle morphology have a significant impact on the yield of the reaction. This presents a considerable challenge to the reproducibility of solid-state reactions. It is worth noting that in this system the substrate has almost no effect on the reaction yield within the particle size range of 0.05–5.5 mm (Fig. 2).
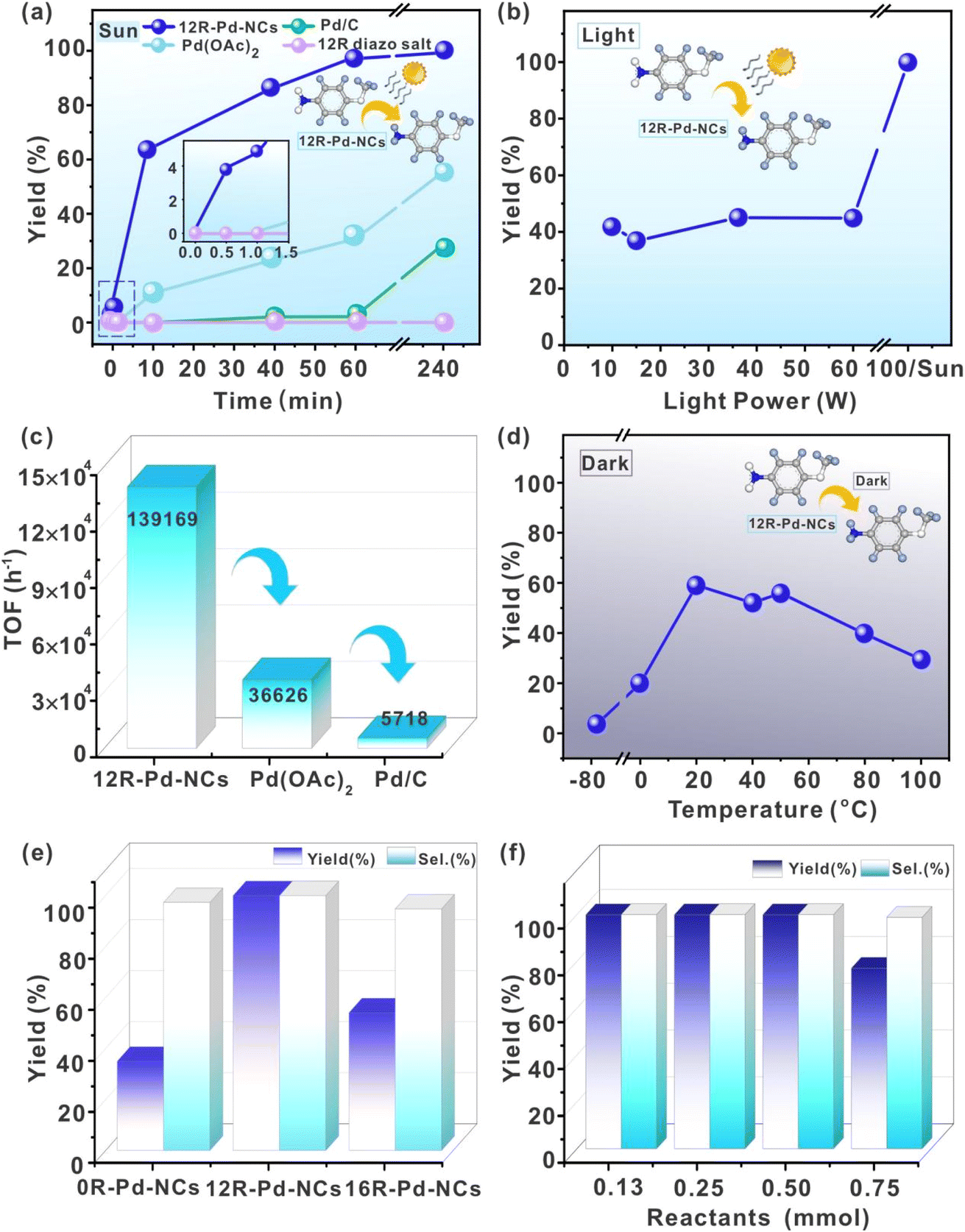 | ||
| Fig. 1 Catalytic evaluation of spontaneous solid-state photocatalytic hydrogenation: catalytic conditions unless otherwise noted: 0.5 mmol of solid-state nitro compound, 12R-Pd-NCs (2 mg), H2 balloon (1 atm), static illumination for 4 hours under sunlight. The yields presented in the article represent the overall HPLC yield of the reaction, rather than the yield of a localised reaction. (a) Investigation of the catalytic performance of 12R-Pd-NCs, Pd(OAc)2, Pd/C and 12R-diazosalt in solar light; (b) catalytic evaluation of 12R-Pd-NCs at different light intensities; (c) the TOF values of the 12R-Pd-NCs, Pd(OAc)2 and Pd/C catalysts; (d) the catalytic evaluation of 12R-Pd-NCs under dark conditions; (e) catalytic performance of 12R-Pd-NCs, 16R-Pd-NCs and 0R-Pd-NCs in sunlight; (f) investigation of the substrate/catalysts ratio. | ||
 | ||
| Fig. 2 Catalytic evaluation of solid reactants under different particle size distributions: aHPLC yield of the whole reaction system; bchemoselectivity. | ||
Initially, solid-state 4-nitroanisole 1a was selected as a model substrate. As shown in Fig. 1a, palladium played a crucial role in the formation of the target solid-state anilines. Successful attempts showed that spontaneous photoactivated hydrogenation yielded aniline 2a in 28% yield after 240 minutes. In contrast, in the absence of palladium, the reaction does not proceed when the precursor, p-dodecyl diazonium salt, is added directly. To our satisfaction, the utilization of palladium acetate as a catalyst led to a higher yield of the target product 2a, reaching 55%. It is highly probable that this significant increase in catalytic performance is due to the fact that a small amount of palladium acetate was in situ reduction under solid conditions in a hydrogen atmosphere and formed a small amount of palladium clusters, which reacted rapidly with solid nitrobenzene on the contact surface. However, since the surface of this metallic palladium cluster lacked flexible, lipophilic alkyl chains, the uncontacted portion did not react sufficiently, likely explaining why the reaction yield was only half. Fig. 1e provides compelling evidence to support the conclusion that the length of the alkyl chain directly impacts the reaction. Chains of insufficient length are ineffective in “trapping” reactants, whereas chains of excessive length reduce flexibility in “trapping” solid reactive objects, simultaneously weakening the departure rate of solid products. Further studies show that 12R-Pd-NCs are the most efficient catalysts for spontaneous catalysis of solid nitrobenzene for photoactivated hydrogenation without external forces. This process has the potential to achieve yields exceeding 99% under sunlight irradiation. It is noteworthy that only 2 mg of 12R-Pd-NCs is sufficient for 0.5 mmol of the solid reactant to achieve the aforementioned outcome, thereby offering a promising avenue for the industrial implementation of solar-driven spontaneous solid-state green synthesis (Fig. 1f). The efficiency of the reaction was investigated at varying light intensities. Interestingly, the findings indicate that irradiation with a 100 W light source (xenon or tungsten lamps) yields reaction outcomes comparable to those observed under solar conditions (Fig. 1b). Fig. 1d shows that the enhanced activity and selectivity of this spontaneous solid-state reaction is the result of a “photothermal temperature gradient driver”. It can be observed that under dark conditions, the catalytic efficiency is significantly lower compared to that under light conditions, regardless of the temperature. Establishing a temperature gradient using only an external heat source is challenging, as heat alone cannot generate a gradient that drives motion. It should be noted that thermal adjustments can result in an initial increase in yield, which subsequently declines. Another reason could be that an excessively high temperature causes the generated water molecules to vaporise, thereby weakening the facilitating role of generated water in solid-state reactions. This constitutes a reverse experimental argument in favour of the facilitating role of generated water in topological chemical theory.5
Synthesis and characterization
The nanocluster can be either synthesized in situ and utilized directly, or isolated and stored for at least six months at ambient temperatures. The lipophilic characteristics of the particles result in their effective dispersion in organic solvents, forming approximately 3.2 ± 0.8 nm spherical particles. As shown in Fig. 3, no considerable formation of aggregates was observed for the catalysts by high resolution transmission electron microscopy (HR-TEM). The length of the fully extended capped ligand was estimated to be 2.0 nm (Spartan software), which to some extent accelerates substrate uptake on the surface of the nanocluster and the synergistic activation of molecular hydrogen. HR-TEM of 0R-Pd-NCs revealed the presence of a minor degree of palladium aggregation, thereby indicating that the flexibility and cross-linking of longer alkyl chains exert a stabilizing influence on the clusters to a certain extent (Fig. S2†). | ||
| Fig. 3 The HR-TEM images of 12R-Pd-NCs. | ||
Energy dispersive X-ray spectroscopy (EDS) results indicate the presence of palladium on the surface of the nanocluster, but this should not be taken as an accurate indication of the palladium metal content (Table S1 and Fig. S1†). The 12R-Pd-NCs were characterized by inductively coupled plasma-optical emission spectrometry (ICP-OES) to reveal a metal content of 73.66 wt% (Table S2†).
X-ray photoelectron spectroscopy (XPS) of 12R-Pd-NPs was then performed, showing two strong Pd signals at 335.60 eV (3d5/2) and 341.06 eV (3d3/2). As illustrated in Fig. 4b, the core level spectral peaks at 335.0 eV (3d5/2) and 336.9 eV (3d5/2) can be attributed to Pd0 and Pd2+, respectively. The O 1s peak at 530.50 eV (Fig. S7†), which resides within the characteristic binding energy range of lattice oxygen in metal oxides (529.5–530.5 eV), is probably assigned to Pd–O coordination.18e This assignment rigorously excludes contributions from surface-adsorbed hydroxyl groups or water molecules (typically observed at 531–533 eV). Concurrently, the C 1s spectrum (Fig. 4a) exhibits a distinct C–O peak at 286.68 eV, corresponding to oxygen-bridged metal–carbon hybrid interfaces (C–O–Pd), as established in metal–carbon hybrid systems.18f Quantitative analysis of the C speciation (Fig. 4a) reveals a near 1:1 ratio between C–O and C–Pd bonding configurations (deducting interference from C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, 51.3% vs. 48.7%, respectively). This stoichiometric correspondence is further corroborated by the Pd 3d spectral deconvolution (Fig. 4b), which demonstrates a comparable distribution between Pd2+ (51.9%, Pd–O–C) and Pd0 (48.1%, Pd–C) species. The remarkable consistency (<1% deviation) in these paired ratios provides compelling evidence for the coexistence of divalent Pd coordination (Pd–O–C) and zero valent Pd–C domains (Pd–C) through oxygen-mediated interfacial bonding and metal carbon bonds, respectively. This distinctive oxidation/reduction state markedly accelerates the solid-phase catalysis of the substrate in comparison to palladium carbon of an equivalent particle size. No peaks corresponding to Pd–N bonds were observed, while distinct peaks corresponding to C–C/CC and Pd–C bonds were observed at 284.78 eV and 283.78 eV, respectively (Fig. 4a). This indicates that the precursor species are bonded to the surface of the nanocluster via metal–carbon bonds. In addition, the FT-IR spectra of 12R-Pd-NPs (Fig. 4d) exhibit significant peaks at 2940 and 729 cm−1, attributed to methylene C–H stretches and CC stretches in the aromatic rings, respectively. Notably, there are no observations of N
C, 51.3% vs. 48.7%, respectively). This stoichiometric correspondence is further corroborated by the Pd 3d spectral deconvolution (Fig. 4b), which demonstrates a comparable distribution between Pd2+ (51.9%, Pd–O–C) and Pd0 (48.1%, Pd–C) species. The remarkable consistency (<1% deviation) in these paired ratios provides compelling evidence for the coexistence of divalent Pd coordination (Pd–O–C) and zero valent Pd–C domains (Pd–C) through oxygen-mediated interfacial bonding and metal carbon bonds, respectively. This distinctive oxidation/reduction state markedly accelerates the solid-phase catalysis of the substrate in comparison to palladium carbon of an equivalent particle size. No peaks corresponding to Pd–N bonds were observed, while distinct peaks corresponding to C–C/CC and Pd–C bonds were observed at 284.78 eV and 283.78 eV, respectively (Fig. 4a). This indicates that the precursor species are bonded to the surface of the nanocluster via metal–carbon bonds. In addition, the FT-IR spectra of 12R-Pd-NPs (Fig. 4d) exhibit significant peaks at 2940 and 729 cm−1, attributed to methylene C–H stretches and CC stretches in the aromatic rings, respectively. Notably, there are no observations of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N signals at 2250 cm−1.
N signals at 2250 cm−1.
 | ||
| Fig. 4 (a) The C 1s XPS core level spectra of 12R-Pd-NPs; (b) Pd 3d XPS core level spectra; (c) XRD spectra of the diazonium salt precursor, 12R-Pd-NCs, 16R-Pd-NCs and 0R-Pd-NCs; (d) FT-IR spectra of the 12R-Pd-NCs; (e) the BET results of 12R-Pd-NPs; (f) results of 12R-Pd-NCs valence bond energies by DFT calculations. | ||
Furthermore, density functional theory (DFT) calculations revealed that the valence state of palladium exerts a significant influence on the bonding of the Pd–C bond, with a tendency towards the Pd0–C bonding form (Fig. 4f, see the computational method, parameters, and more details in the ESI†). The unprecedentedly high activity of 12R-Pd-NCs is attributed to the support, which exhibits a strong interaction with the Pd–NCs, maintaining the oxidation–reduction states of metals and accelerating substrate uptake, while facilitating the rapid shipping of the aniline product from the surface of Pd–NCs to accelerate the reaction. Brunauer–Emmett–Teller (BET) results indicated that the nanocluster exhibited a relatively small specific surface area and lacked a fully porous structure (Fig. 4e). The trace mesopores (10 nm) were likely to be due to inter-particle spatial stacking. This result was found to be in exact alignment with the previously observed nanocluster size result from HR-TEM. X-ray diffraction (XRD) patterns of the 12R-Pd-NCs have been recorded for the as-prepared samples in the form of dry powders. As shown in Fig. 4c, the broad diffraction peak observed at 20–35° may be attributed to the presence of carbon elements within amorphous alkyl carbon chains; peaks at 40.0°, 46.5°, 67.9°, 81.8°, and 86.3° exhibit diffraction lines corresponding to the crystal planes at (111), (200), (220), (311), and (222), respectively, indicating the presence of Pd in 12R-Pd-NCs. The particle size (35–45°) calculated according to the Scherrer formula demonstrates a gradual increase with the lengthening of alkyl chains, a finding that is corroborated by the HR-TEM results (Fig. S2†). On the basis of the above characterization studies, we conclude that the novel nanocluster has been successfully monolayer-protected and decorated with laurylbenzene by metal–carbon bonds. The remarkable stability and high activity observed in this type of nanocluster are likely attributed to a synergistic effect among the flexible alkyl chains, Pd0–C bonds, and Pd2+charges of palladium on the nanocluster.
Monitoring the reaction progress
The reaction progress of the spontaneous solid-state synthesis strategy around 1e (4-nitrobiphenyl, white solid) in the presence of sunlight was monitored by powder X-ray diffraction (PXRD) analysis (Fig. 5). This circumvents the complication of superimposed absorption peaks that frequently arise in conventional solid-state catalytic reactions using infrared detection. After 2 h, new diffraction peaks derived from hydrogenation products 2e (4-aminobiphenyl, yellow solid) appeared, while the peaks associated with the starting materials remained. After 8 h, the diffraction peaks derived from the starting materials had nearly completely disappeared, and large amounts of hydrogenated product 2e and traces of 12R-Pd-NCs were observed, demonstrating a clean solid-to-solid conversion without melting during this transformation.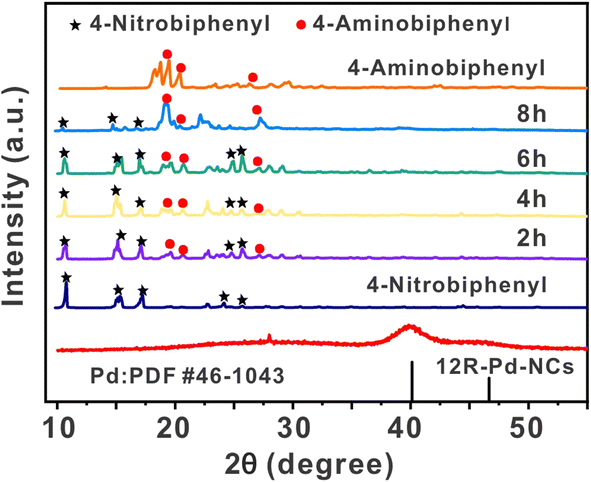 | ||
| Fig. 5 Monitoring the reaction progress by PXRD analysis. | ||
We also discovered unusual sigmoidal kinetics in the non-mechanical chemistry solid-state photoactivated catalytic hydrogenation of nitrobenzene, in which the physical form of the reaction mixture dramatically changes from a highly aggregated crystal structure to a plastic-like appearance during the reaction (Fig. 6). This dramatic change in rheology can result in a rapid increase in the reaction rate.20b The alteration is readily discernible when observed under a 50× microscope. As periodic sampling of the reaction requires opening the reaction bottle and exposing it to air, each data point was obtained from an individual reaction.
 | ||
| Fig. 6 Monitoring the reaction progress by optical microscopy. The physical form of 1e in the reaction dramatically changes from a highly aggregated crystal structure to a plastic-like appearance during the reaction. | ||
Substrate scope of the spontaneous solid-state photoactivated hydrogenation reaction
To explore the scope of the present spontaneous solid-state photoactivated hydrogenation reaction, various monosubstituted and polysubstituted nitrobenzenes were investigated. As shown in Scheme 2, solid-state photoactivated hydrogenation to anilines gave yields ranging from moderate to excellent under the optimized reaction conditions, depending on the spatial potential resistance effect and electron effect of the substituents. Interestingly, if the substituent group in the substrate is located at the para position of the reaction site and has a small spatial site barrier, the electronic effect has little impact on the reaction (yields >90%). For instance, when nitrobenzene substituents are located at the para position, they can donate methoxy- (2a, >99%), methyl- (2c, >99%), and phenolic hydroxyl- (2g, 92%), or possess electron-withdrawing groups p-F (2j, 91%). These substituents resulted in good yields of products. The spatial potential resistance effect dramatically reduced the hydrogenation efficiency. But the target products 2b, 2d, 2e, 2f, 2h, 2m and 2k can be obtained in high yields with extended reaction times. Of these, 2f is not only a raw material for the herbicide oxaziflam and the insecticide fumonisole, but also an important reagent for gold and silver assays. And 2m is a raw material for the industrial synthesis of the medical contrast agent iodophthalamide. In contrast, the catalytic efficiency of iodine-, bromine-, and chlorine-substituted anilines was found to be inadequate (poor yield and selectivity, not listed). As solvent-free solid-state reactions can be regarded as reactions that proceed under extremely-high-concentration conditions, the reagents and catalysts in the solid state interact much more strongly with each other than those in solution. This could tentatively explain the observed low reactivity of the halogenated anilines, which could potentially coordinate strongly with any off-cycle palladium species, leading to catalyst deactivation. This transformation exhibits exceptional substrate generality, enabling precise construction of diverse functionalized aromatics bearing sensitive groups or heteroatom substituents through controllable regioselective pathways (2l–2p). Imidazole-, thiazole-, carbonyl- and cyano-containing arylamines (2n, 2o, 2p, and 2l) were also obtained in good yield (87%, 49%, 88% and 51%, respectively). 2k was also obtained from the corresponding 1-naphthylamine (43%). Scheme 2 also illustrates several products of potential interest from a pharmaceutical perspective, namely the reduction of nitro-containing species that may lead to either bioactive or drug-like fragments. For example, benzocaine (2q), which has a clinical anaesthetic effect, can be obtained with a yield of 90%. Similarly, a long-acting local anaesthetic used in the treatment of chronic pain, butamben (2r), and the potentially biologically active compounds 2e were obtained with yields of 80% and 79%, respectively. Even in the preparation of chemical intermediates like p-anisidine, it is possible to achieve a production of up to 15 g with >99% yield and >99% chemoselectivity, using only a minimal amount of catalyst (75 mg). | ||
| Scheme 2 Solar-driven and spontaneous solid-state synthesis of functionalized nitro compounds over 12R-Pd-NCs. aCatalytic conditions unless otherwise noted: 0.5 mmol of solid-state nitro compound, 12R-Pd-NCs (2 mg), H2 balloon (1 atm), illuminate for 4 hours under sunlight, or a 100 W xenon lamp/tungsten filament lamp; bHPLC yield of the whole reaction system; cchemoselectivity. | ||
Encouraged by these results, this attractive protocol was further applied to the hydrogenation of nitro compounds with structures comprising either aromatic heterocyclic compounds with multisubstituted or double-nitro groups. The N-methyl substitution of nitrobenzene did not demonstrate a spatial site-blocking effect. Conversely, the yields of nitrobenzene aromatic rings with neighbouring substitution of methylamino groups (2s) were markedly higher than those of the para-substituted products (2t). Notably, 2s was obtained with a yield of up to 99%. This phenomenon may be attributed to the presence of intramolecular hydrogen bonding in the neighbour-substituted reactants, which facilitates the reaction. Polysubstituted double-nitro groups, such as 2u, 2v, 2w, and 2x, can undergo effective solid-state photoactivated spontaneous hydrogenation, resulting in the target products with yields of 75%, 63%, 52% and 59%, respectively. However, if the molecular weight of nitrobenzene is large, the reaction proceeds poorly, resulting in only traces of the target product. Understanding the selectivity toward functional groups prone to overreduction is essential for evaluating catalytic performance. To address this, we conducted additional experiments with nitroarenes bearing sensitive alkyne, alkene, and aldehyde moieties. Experimental results revealed that the catalytic system exhibited limited performance in the reduction of nitrobenzene derivatives containing alkynyl groups (3a), achieving only 25% yield with 61% chemoselectivity. This process was accompanied by complete reduction of the alkynyl moiety, generating side product 3a′ as a major byproduct. For substrates bearing alkenyl groups, although improved chemoselectivity was observed (3b), the reaction efficiency remained unsatisfactory with merely 79% yield of the desired product. Notably, the system demonstrated particularly poor reactivity toward aldehyde-containing substrates, where only trace amounts of the product could be detected under standard reaction conditions (3c). Further analysis of potential byproducts generated during the photoactivated hydrogenation of nitrobenzene was conducted – particularly focusing on low-selectivity products 2x and 2v. High resolution mass spectrometry detection results show the generation of intermediate nitroso compounds (Fig. S10 and S11†). The complete absence of coupling byproducts (azo/azoxy compounds) strongly indicates that the reaction proceeds via a direct hydrogenation pathway rather than undergoing condensation processes.
Mechanistic studies
To gain insight into the superior activity of 12R-Pd-NCs on the photoactivated solid-state self-assembly, a series of single-factor controlled experiments were conducted. As shown in Scheme 3, eqn (a)–(f), at −78 °C under light conditions, only traces of the product could be obtained, which indicates that the reaction is thermodynamically controlled. The solid reactant 1a was statically placed under sunlight for 4 h at ambient temperature (25 °C) and the target product 2a was almost quantitatively obtained (>99%, Scheme 3, eqn (a)–(a)). At this time the temperature within the reaction system was measured to be around 40–45 °C (data from multiple parallel tests). Nevertheless, under conditions of light avoidance, with other variables held constant (45 °C), the target product 2a could be obtained in only moderate yields (55%, Scheme 3, eqn (a) and (b)). Even when the reaction temperature was varied continuously in the dark, the catalytic efficiency was simply not comparable to that in the presence of light (Table S3†). The above results suggest that the spontaneous solid-state reaction operates through a photoactivated catalytic mechanism in the presence of palladium nanoclusters. Moreover, the heating effect produced by photogenerated hot electrons (carriers) is much greater than that of an external heat source. This new discovery in the field of photoactivated solid-state self-assembly will provide a new insight into the rational design of photocatalysts for other solid-state catalytic reactions. | ||
| Scheme 3 Controlled experiments and infrared thermal imaging. | ||
The process was visualized using an infrared thermographic display. When the solid mixture was statically placed in sunlight for 2 minutes, the temperature of the catalyst surface increased to 32.9 °C, significantly higher than the ambient temperature of 25.7 °C, causing the ambient temperature inside the reaction flask to increase to 30 °C. This visual contrast became more pronounced as the reaction time increased, with the temperature of the catalyst surface increasing to 46 °C after 4 hours (Scheme 3b). The above findings and single-factor controlled experiments at 0 °C further validate the proposed mechanism (Scheme 3, eqn (a)–(d)). In order to investigate the π–π stacking of the aromatic rings of the substrate and catalyst, we introduced solvents into the reaction system. Notably, solvent introduction significantly attenuates π–π stacking interactions (Table S8†). Also the inhibition brought about by the introduction of radical trappers cannot exclude the effect caused by the isolation of the solid state crystal image by foreign additions (Table S5,† entry 6).19
The electron paramagnetic resonance (EPR) spectrum of 12R-Pd-NPs exhibits a prominent signal at g = 2.003 (Fig. S9†), characteristic of oxygen vacancy defects.21a Previous reports have shown that the defect energy levels introduced by oxygen vacancies can narrow the band gap of materials and reduce the energy required for electron transitions, thereby expanding the light absorption range to the visible light region and enhancing the absorption of visible light.12a,21b,21c The UV-vis diffuse reflectance spectroscopy analysis of 12R-Pd-NPs demonstrated a significant enhancement in visible light absorption (Fig. S8†), thus providing convincing validation for the aforementioned conclusions through distinct absorption band expansion in the 400–600 nm wavelength range. Cryogenic photon-injection experiments show that controlled tests at −10 °C with varying light intensities (15–200 W) yielded constant product yields (Table S6†), demonstrating no light-intensity dependence. Thermodynamic analysis reveals that under dark reaction conditions, the observed activity originates from thermodynamic processes, which demonstrates that the activity stems from local thermal activation rather than photocatalysis. These controlled experiments collectively provide evidence to rule out photocatalysis as a dominant mechanism.
The recovered catalyst exhibited uncompromised hydrogenation efficiency (Fig. 7). Even after five cycles of catalysis, this catalyst still exhibited high stability. This feature is significantly different from that of the Pd/C and resin-based metal catalysts with relatively low stability. Post-cycle characterization of the fifth-generation catalyst through integrated high-resolution transmission electron microscopy (HR-TEM) and inductively coupled plasma (ICP) analyses revealed that nanoparticle agglomeration and structural decomposition collectively drive catalytic deactivation, with HR-TEM imaging (Fig. S12†) demonstrating pronounced Pd nanoparticle coalescence and ICP quantification confirming substantial palladium loss (Table S10†, 73.66 → 56.1 wt%, Δ = 17.6 wt%), mechanistically attributed to progressive cluster disintegration under cyclic operational stresses.
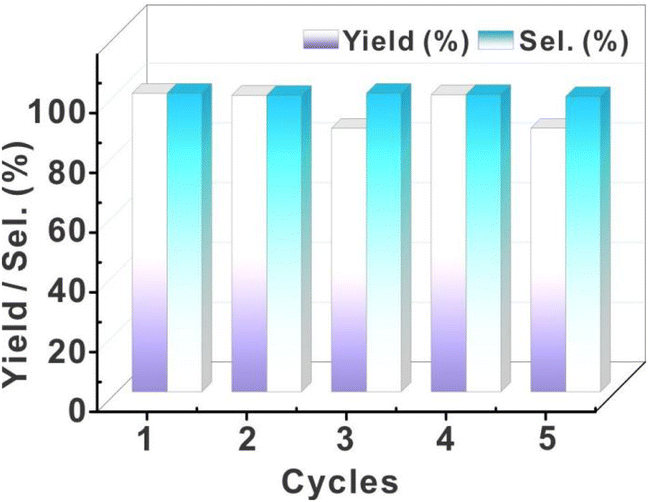 | ||
| Fig. 7 The recycling test of 12R-Pd-NCs. | ||
Conclusions
In summary, we propose a photoactivated solid-state self-assembly strategy to address the challenges encountered in current solid-state catalysis. The reaction can proceed smoothly and spontaneously without the introduction of mechanochemistry, and can be easily scaled up to a gram scale, requiring only sunlight for static irradiation. The “accidental” design of the palladium nanoclusters and the ‘temperature gradient drive’ under the coordinated action of light were fundamental to the fact that their catalytic performance, without the introduction of mechanochemistry, was an order of magnitude higher than that of the state-of-the-art Pd-, Pt-, and Ru-based catalysts previously reported for the nitrobenzene hydrogenation reaction.18a The substrate has almost no effect on the reaction yield within the particle size range of 0.05–5.5 mm. Further studies have shown that the unprecedented high activity of 12R-Pd-NCs is related to the support, which has a strong interaction with 12R-Pd-NCs to maintain the oxidation–reduction states of metals and accelerate the reaction. In addition, the dramatic change in rheology can lead to a rapid increase in reaction rate. The positive facilitation of the photocatalytic spontaneous solid-state reaction by the produced water was demonstrated in reverse. We anticipate that the strategy developed in this study could unlock broad areas of chemical space for palladium-catalyzed solid-state syntheses of valuable synthetic targets in various scientific fields, while also offering opportunities and prospects for applications based on green solid-state organic synthesis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. T. X. supervised and provided guidance to the project. X. W. performed the reaction optimisation. J. Y. Z., M. H. S. and S. S. L. carried out the synthetic experiments and formal analysis. J. W. Y., L. R. Y. and M. Y. conducted the mechanistic investigations. All the authors analysed and discussed the experimental data. Y. X. L. and Y. T. X. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Fundamental Research Funds for the Henan Provincial Colleges and Universities in Henan University of Technology (No. 2021BS010, 2021BS026, 2021BS028, and 2021BS021), Colleges and Universities Key Research Program Foundation of Henan Province (No. 22A150037), the Innovative Funds Plan of Henan University of Technology (No. 2020ZKCJ29), the Natural Science Foundation of Henan Province (No. 242300420542, 252300421284 and 252300420767), the Natural Science Project of Zhengzhou Science and Technology Bureau (No. 22ZZRDZX08), the Science and Technology Foundation of Henan Province (No. 222102230069, 232102320084, and 222102320056) and the National Natural Science Foundation of China (No. 22305068).Notes and references
- For representative books and articles, see
(a)
F. Toda, Organic Solid State Reactions, Springer, Berlin Heidelberg, 2004, pp. 115–245 Search PubMed
; (b) K. Tanaka, Solvent-Free Organic Synthesis, 2nd revised edn, Wiley-VCH, Weinheim, 2009, pp. 5–135 Search PubMed
-
(a) D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11(1), 133–137 CrossRef CAS
- For representative studies, see
(a) F. Toda, Acc. Chem. Res., 1995, 28, 480–486 CrossRef CAS
-
(a) F. Toda, K. Kiyoshige and M. Yagi, Angew. Chem., Int. Ed., 1989, 28, 320–321 CrossRef
-
(a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
-
(a) Y.-C. Zhao, S. V. Rocha and T. M. Swager, J. Am. Chem. Soc., 2016, 138, 13834–13837 CrossRef CAS PubMed
-
(a) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3(1), 13–19 CrossRef CAS PubMed
- C.-A. Wang, Y. Ouyang, Y.-B. Luo, X.-R. Gao, H.-Y. Gao, G. Wang and X.-T. Shu, Chin. J. Catal., 2024, 60, 128–157 CrossRef CAS
- Z.-Q. Wang, Z.-Q. Yang, Z. C. Kadirova, M.-N. Guo, R.-M. Fang, J. He, Y.-F. Yan and J.-Y. Ran, Coord. Chem. Rev., 2022, 473, 214794 CrossRef CAS
-
(a) Y.-T. Chen, M.-P. Zhuo, X.-Y. Wen, W.-B. Chen, K.-Q. Zhang and M.-D. Li, Adv. Sci., 2023, 10, 2206830 CrossRef CAS PubMed
-
(a) Y.-J. Xiong, J.-Y. Chen, B. Wiley, Y.-X. Xia, Y.-D. Yin and Z.-Y. Li, Nano Lett., 2005, 5, 1237–1242 CrossRef CAS PubMed
-
(a) N.-N. Jiang, X.-L. Zhuo and J.-F. Wang, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS PubMed
-
(a) L.-W. Li, Z.-X. Cai, Q.-H. Wu, W.-Y. Lo, N. Zhang, L.-X. Chen and L.-P. Yu, J. Am. Chem. Soc., 2016, 138(24), 7681–7686 CrossRef CAS PubMed
-
(a) R.-H. Zhang, H. Wang, S.-Y. Tang, C.-J. Liu, F. Dong, H.-R. Yue and B. Liang, ACS Catal., 2018, 8(10), 9280–9286 CrossRef CAS
- N. Zhang, A. Jalil, D.-X. Wu, S.-M. Chen, Y.-F. Liu, C. Gao, W. Ye, Z.-M. Qi, H.-X. Ju, C.-M. Wang, X.-J. Wu, L. Song, J.-F. Zhu and Y.-J. Xiong, J. Am. Chem. Soc., 2018, 140(30), 9434–9443 CrossRef CAS PubMed
- J. R. Chamorro and T. M. McQueen, Acc. Chem. Res., 2018, 51, 2918–2925 CrossRef CAS PubMed
-
(a) W. Liu, H.-S. Feng, Y. S. Yang, Y.-M. Niu, L. Wang, P. Yin, S. Hong, B.-S. Zhang, X. Zhang and M. Wei, Nat. Commun., 2022, 13, 3188 CrossRef CAS PubMed
-
(a) Q.-S. Luo, H. Wang, Q. Xiang, Y.-T. Lv, J.-B. Yang, L.-J. Song, X.-M. Cao, L. Wang and F.-S. Xiao, J. Am. Chem. Soc., 2024, 146(38), 26379–26386 CrossRef CAS PubMed
-
(a) J.-S. Chen, A. N. Vasiliev, A. P. Panarello and J. G. Khinast, Appl. Catal., A, 2007, 325, 76–86 CrossRef CAS
-
(a) M. Králik and A. Biffis, J. Mol. Catal. A: Chem., 2001, 177, 113–138 CrossRef
-
(a) N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02185e |
| This journal is © The Royal Society of Chemistry 2025 |