
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombating multiple aetiologies of Alzheimer's disease to rescue behavioural deficits†
Madhu
Ramesh
 ,
Chenikkayala
Balachandra
,
Ashish
Kumar
,
Sourav
Samanta
and
Thimmaiah
Govindaraju
*
,
Chenikkayala
Balachandra
,
Ashish
Kumar
,
Sourav
Samanta
and
Thimmaiah
Govindaraju
*
Bioorganic Chemistry Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bengaluru, Karnataka 560064, India. E-mail: tgraju@jncasr.ac.in
First published on 6th June 2025
Abstract
Alzheimer's disease (AD) encompasses a range of intricate pathologies characterized by aberrant protein aggregation, atypical accumulation of metal ions, increased levels of reactive oxygen species (ROS), oxidative stress, neuroinflammation, and synaptic dysfunction. These collectively contribute to a decline in learning, memory, and cognitive abilities, broadly classified as dementia. AD accounting for most of the dementia cases remains a significant health challenge. Despite extensive research, therapeutic advancements for AD and other neurodegenerative diseases (NDDs) have achieved modest success. In this context, our study presents a hybrid drug design approach involving strategic and tactical repurposing of the structural and functional pharmacophores of current or failed drugs and biologically active compounds by integrating them into a single structural framework to concurrently target key pathological hallmarks viz., amyloid beta (Aβ), tau, metal ions, ROS, and neuroinflammation (NLRP3 inflammasome). The evaluation of in vitro and cellular models of Aβ, tau, and microglia highlights the efficacy of the fluoro-derivative DM4 in mitigating multiple etiological factors. DM4 exhibits excellent blood–brain barrier (BBB) permeability and biocompatibility. DM4 effectively reduced the amyloid burden, neuroinflammation, synaptic dysfunction, and neurodegeneration in the APP/PSEN1 transgenic AD mouse model. Behavioural assessments corroborated the rescue of learning and memory deficits, thereby presenting a viable strategy for the treatment of neurodegeneration and its associated cognitive decline.
Introduction
Neurodegenerative diseases (NDDs) affect millions of people worldwide with profound socio-economic impact. Some of the major NDDs include Alzheimer's disease (AD), Parkinson's disease (PD), tauopathies, synucleinopathies, frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HTT).1 Decades of research have unveiled shared pathological characteristics among these conditions, such as aberrant protein aggregation, dysregulated metal ion homeostasis, elevated levels of ROS, oxidative stress, biomolecular and mitochondrial damage, neuroinflammation, and synaptic dysfunction, leading to motor and cognitive impairments and onset of dementia.2,3 AD is a complex multifactorial disease that accounts for 70–80% of total dementia cases.4,5 The intricate interplay of various aetiological factors complicates the understanding of disease progression and the development of targeted therapeutics.6 The amyloid hypothesis, which focuses on the accumulation of Aβ, has been a central research theme, yet therapies targeting Aβ have achieved limited success due to a complex interplay with other disease mechanisms.7,8 Metal ion dyshomeostasis, in conjunction with Aβ, contributes to ROS production, leading to oxidative stress and further biomolecular damage.9–11 Tau pathology, another significant aspect of AD and tauopathies, has emerged as a promising therapeutic target, with several tau-focused therapies currently undergoing clinical trials.12–14 Recent studies have highlighted neuroinflammation, particularly the NLRP3 inflammasome pathway, as a critical factor in AD, with microglia playing a central role in mediating neuroinflammation and contributing to neurodegeneration.15,16 Given the multifactorial nature of AD, it is becoming evident that therapies targeting a single disease factor are unlikely to be effective.17 Therefore, there is an imperative need to develop multifunctional molecules targeting multiple etiological factors, offering a promising approach to therapeutic development for AD.18,19 There is an urgent need to develop multifunctional molecules to target multiple aetiological factors for better therapeutic management of AD.The multifunctional approach involves the design of small molecules tailored to address multiple pathological factors, which encompasses three distinct strategies.15,19–21 The hybrid multifunctional method combines two functional entities to synthesize new multifunctional molecules, the linkage strategy connects two established pharmacologically active compounds with chemical linkers, and the fusion technique merges pharmacophore units to form a drug-like molecule with multifunctional properties.22–25 Several dual-targeting molecules have been evaluated in vitro and in preclinical models, showing promising results and superiority over single-target therapies.26–28 This success has spurred efforts to rationally design multifunctional molecules that can concurrently target Aβ, metals, acetylcholine esterase (AChE), ROS, mitochondrial damage, and neuroinflammation.20–29 However, reports of small molecules effectively targeting all these major disease factors in vivo are exceedingly rare.30–33 A significant challenge in developing such molecules is the increased size resulting from the integration of multiple components, which often leads to reduced activity and failure in their in vivo translation.29,30 Consequently, there is a pressing need to develop novel design approaches for small multifunctional molecules that effectively target the major disease factors, thereby advancing the development of improved therapeutics for AD.
In combating AD, we embarked on the rational design of multifunctional small molecules that target Aβ, tau, metals, ROS, and NLRP3 inflammasome, thereby addressing neuroinflammation (Fig. 1). We have employed a hybrid drug design approach involving strategic and tactical repurposing of structural and functional pharmacophores of current or failed drugs and biologically active compounds by integrating them into a single structural framework. Utilizing a diazo-based molecular backbone, we incorporated metal chelation and anti-inflammatory functionalities to target the complex etiology (Fig. 1A). The molecules were subjected to in vitro evaluations, assessing their ability to modulate Aβ aggregation, chelate metals, mitigate metal-associated toxicity, and inhibit tau aggregation. These studies identified fluoro-derivative DM4 as the lead multifunctional molecule. Subsequent cellular investigations demonstrated the efficacy of DM4 in reducing amyloid and tau toxicity, suppressing microglial activation, and preventing NLRP3 inflammasome assembly. Encouraged by the multifunctional activities of DM4, we proceeded to assess its effectiveness in an APP/PSEN1 double transgenic (Tg) mouse model of AD. DM4 exhibited BBB permeability and biocompatibility with an LD50 of 97.8 mg per kg body weight in the mouse model. In vivo toxicity evaluations, including serum biochemical and histopathological analyses, confirmed biocompatibility of DM4 in the murine model. DM4 demonstrated efficacy in reducing the amyloid burden, neuroinflammation, neurodegeneration, and synaptic dysfunction. Behavioural studies further indicated a restoration of learning and memory capabilities (Fig. 1B). Through this integration approach, we successfully developed a multifunctional small molecule that targets the multiple pathological features of AD. The promising results from in vivo preclinical studies underscore the therapeutic potential of this molecule.
 | ||
| Fig. 1 Schematic showing the design and mechanism of action. (A) Rational design of multifunctional diazo-based small molecules aimed at addressing various disease aetiologies of Alzheimer's disease (AD) through a hybrid drug design approach. This involves the strategic and tactical repurposing of structural and functional pharmacophores from existing or failed drugs and biologically active compounds, integrating them into a single structural framework. (B) Schematic representation illustrating the targeting of multiple aetiologies by DM4 and its therapeutic effects in a mouse model. | ||
Results and discussion
Design and synthesis
AD is a complex multifactorial condition that necessitates multifunctional drug candidates to address its various aetiologies. Designing multifunctional drugs presents several challenges. To achieve multifunctionality, multiple pharmacophores of different types must be incorporated, which can lead to increased molecular weight (loss of drug-likeness), heightened toxicity, poor BBB permeability, and reduced multifunctionality, limiting their preclinical and clinical applications. In our novel hybrid drug design, we have introduced a strategy that integrates multiple pharmacophore units within a single molecular framework to achieve drug-likeness, biocompatibility, and BBB permeability while retaining multifunctionality. We have focused on incorporating pharmacophores targeting Aβ, tau protein, metals, and neuroinflammation, which are major pathologies associated with AD. Careful consideration was given to ensure the design results in a small molecule with drug-likeness and the potential to cross the BBB, while being biocompatible and multifunctional. The specific pharmacophores were selected based on their potential to (1) target pathology with functional pharmacophore fragments from molecules demonstrated to be effective in the literature and previous work from our group, (2) be non-toxic, (3) be chemically feasible to integrate into a single compact molecular framework, and (4) balance hydrophilicity and hydrophobicity to ensure BBB permeability. While the novel hybrid molecules are designed to function as multifunctional agents, they may lose some functionality or efficacy compared to the parent pharmacophores due to structural and conformational changes. Our novel design approach significantly reduces drug development efforts by minimizing the need to synthesize, screen, and evaluate a large library of molecules. Instead, it allows for the design and development of a small set of multifunctional molecules to identify leads with potential for clinical translation, followed by further lead optimization.The misfolding and aggregation of Aβ and tau driven by hydrogen bonding and hydrophobic interactions has been extensively studied.34,35 Various dyes have been developed to target these aggregates through similar interactions including Congo red (CR).36,37 The studies indicate that these compounds not only allow detection but also inhibit or dissolve the pre-formed amyloid aggregates over time.38–40 These observations have inspired us to design multifunctional molecules based on the CR core, which is known to bind both amyloid and tau aggregates.35 The aromatic units around two diazo moieties enhance the ability to inhibit and dissolve amyloid aggregates through improved π–π stacking and hydrophobic interactions with the β-sheets.23,41,42 Furthermore, Aβ aggregation, exacerbated by metal ions such as CuII, ZnII, and FeIII, is known to generate ROS and subsequent oxidative stress.10,43 To incorporate the multifunctionality of the diazo core framework, a dicarboxylic acid moiety derived from ethylenediaminetetraacetic acid (EDTA) was integrated. EDTA is a potent metal chelator, and its dicarboxylic acid moiety derived from EDTA can effectively chelate metal ions to inhibit metal-mediated toxicity. Additionally, it imparts hydrophilicity to molecules, balancing other hydrophobic moieties. Recent studies underscore the significant role of neuroinflammation in AD and other neurodegenerative disorders, with the NLRP3 inflammasome emerging as a key therapeutic target.15,44,45 We have incorporated sulphonamide, nitro benzene, carboxy benzene, and trifluoromethoxy benzene groups inspired by established NLRP3 inhibitors such as CY-09, JC-124, tranilast, and IIIM-1269 to impart NLRP3 inhibition properties (Fig. 1A).46 These molecules have been shown to effectively inhibit the NLRP3 inflammasome, but they have limitations such as toxicity. We have derived active pharmacophore units from these inhibitors that potentially inhibit the NLRP3 inflammasome. A major challenge in CNS drug discovery is the lack of BBB permeability, which restricts many promising AD treatments.47 Our molecular design, featuring various functional groups with optimal hydrophilic and hydrophobic balance, is anticipated to cross the BBB. Notably, the fluoro-moiety further enhances bioavailability and biocompatibility. Overall, we have meticulously integrated various moieties to create a multifunctional molecular framework capable of targeting amyloid aggregation, metal toxicity, ROS, and the NLRP3 inflammasome, with the potential for BBB permeability. These multifunctional ligands are expected to effectively address the multifaceted pathology of AD (Fig. 1B). DMs were synthesized as outlined in Scheme S1 of the ESI† and characterized using 1H, 13C, and 19F NMR, HRMS, and LCMS analytical techniques (ESI†).
Modulation of amyloid aggregation and rescue Aβ toxicity
The diazo-based multifunctional molecules were designed to modulate amyloid aggregation and we have assessed them for their ability for Aβ aggregation inhibition by ThT fluorescence assay. Aβ42 (10 μM) was incubated independently and in the presence of DM1–4 (50 μM) in phosphate buffered saline (PBS, 10 mM, pH 7.4). After 48 h incubation, the extent of aggregation inhibition was quantified via ThT fluorescence, which revealed that all molecules exhibited inhibitory properties, with DM4 demonstrating superior efficacy, achieving 86% reduction in aggregation (Fig. S1A†). Further analysis of the inhibitory effect of DM4 with varying concentrations indicated a dose-dependent response, with inhibition rates of 50%, 67%, and 83% at concentrations of 10 μM, 20 μM, and 50 μM, respectively (Fig. 2A). DM4 exhibits maximum absorbance at 480 nm, which overlaps with the emission spectrum of ThT. To validate the inhibition effect of DM4 on Aβ aggregation, we employed a sensitive and specific antibody-based dot blot assay (Table S1†). The results from dot blot and subsequent quantification reinforced these findings, demonstrating a concentration-dependent activity against Aβ aggregation by DM4, with a maximum inhibition of 97.5% at 50 μM (Fig. 2B and S2A†). Transmission electron microscopy (TEM) imaging was employed to visualize the morphological characteristics of fibril formation and its inhibition by DM4. TEM micrographs illustrated that DM4 effectively inhibited the formation of fibrils, resulting in amorphous aggregates, in contrast to the Aβ42 alone (Fig. S3A†). In the dissolution experiment, the preformed fibrillar aggregates, prepared by incubating Aβ42 in PBS for 48 h, were treated with the compounds. DM4 showed dissolution of amyloid aggregates by 77%, while others showed minimal dissolution effects (Fig. S1B†). A concentration-dependent dissolution was observed for DM4, with 33%, 62%, and 85% dissolution at 10 μM, 20 μM, and 50 μM, respectively (Fig. 2C). Further, the dot blot assay and the corresponding quantification data confirmed the dissolution effect of DM4 in a concentration-dependent manner, with a 68% dissolution at 50 μM (Fig. 2D and S2B†). TEM imaging provided further visualization of the dissolution of Aβ fibrillar aggregates by DM4, which led to the formation of amorphous aggregates (Fig. S3B†). To understand the direct interaction of DM4 with Aβ42, we performed a fluorescence titration assay. HiLyte Fluor 555-labeled Aβ42 was titrated with increasing concentrations of DM4 (10 nM to 1 μM), and changes in the fluorescence spectra were monitored. As the concentration of DM4 increased, the fluorescence decreased, suggesting the direct binding of DM4 to Aβ42 and the induction of structural changes (Fig. 2E). The change in fluorescence was plotted and fitted to determine the apparent dissociation constant (Kd) (Fig. 2F). The results show that DM4 strongly binds to Aβ42 with a nanomolar Kd value of 117 ± 24 nM. Furthermore, we employed microscale thermophoresis (MST) using fluorescently labeled Aβ42 (Aβ42 HiLyte Fluor 555) to demonstrate DM4 binding and determine the Kd value. The results show an altered MST response with increasing DM4 concentration, confirming the nanomolar binding affinity (Kd = 732 ± 164 nM) (Fig. 2G). These binding studies confirm the direct and strong interaction of DM4 with Aβ42 and suggest that it induces structural changes that may prevent its aggregation. These comprehensive studies unequivocally demonstrate the ability of DMs to modulate Aβ aggregation, with DM4 exhibiting the highest activity. The modulation of Aβ aggregation is attributed to the diazo-moiety substituted with naphthyl and aryl groups, inspired by known Aβ modulators Congo red and TGR63. The enhanced Aβ modulation of DM4 is possibly due to the trifluoro moiety (CF3) in the aryl substitution, which has higher hydrophobicity compared to other moieties with relatively lower hydrophobicity. This increased hydrophobicity potentially disrupts the hydrophobic and hydrogen bonding interactions involved in Aβ fibrillation.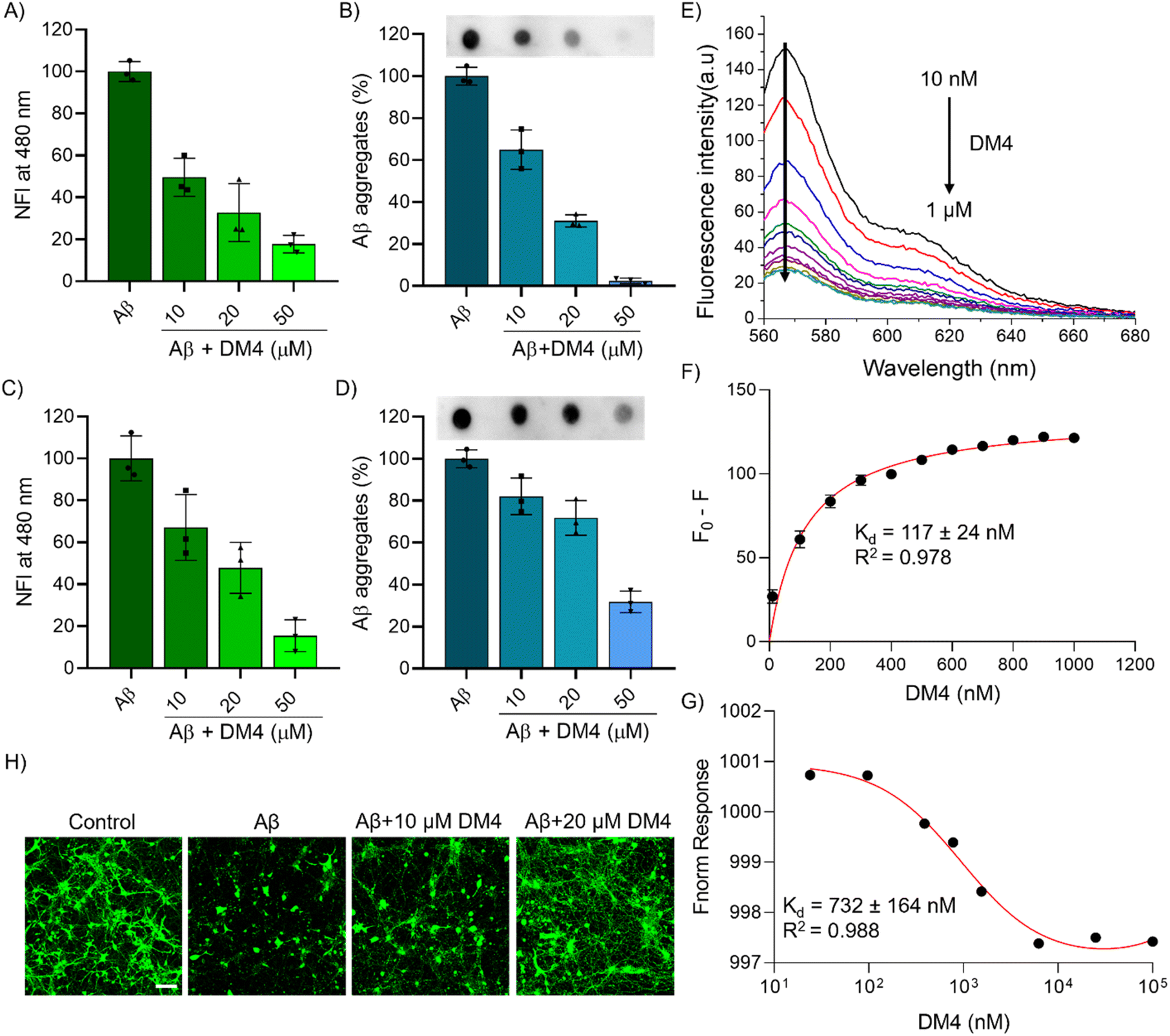 | ||
| Fig. 2 DMs modulate Aβ aggregation. (A) ThT fluorescence assay of Aβ (10 μM) aggregation inhibition by DM4 in a concentration dependent manner. (B) Dot blot assay and its quantification for Aβ (10 μM) aggregation inhibition by DM4 (10, 20 and 50 μM) in a concentration dependent manner. (C) ThT fluorescence assay of Aβ (10 μM) aggregate dissolution by DM4 (10, 20 and 50 μM) in a concentration dependent manner. (D) Dot blot assay and its quantification for Aβ aggregate dissolution by DM4 (10, 20 and 50 μM) in a concentration dependent manner. (E) Fluorescence spectral change of Aβ42-HiLyte Fluor 555 (1 μM) upon titration with DM4 (10 nM to 1 μM) to understand the direct binding interaction of DM4 with Aβ42 and (F) fitting the change in fluorescence to determine the dissociation constant (Kd) of DM4 with Aβ42. (G) Microscale thermophoresis (MST) assay of DM4 interaction and binding with Aβ42 to determine Kd. (H) Representative confocal images showing the rescue of mouse hippocampal primary neuronal cells from Aβ (5 μM) toxicity by DM4 at concentrations of 10 and 20 μM, visualized using calcein staining (scale bar: 150 μm). (NFI: Normalised fluorescence intensity). | ||
The neuroprotective potential of DM4 against Aβ-induced toxicity was assessed in SH-SY5Y neuroblastoma cells. The cell viability of all DMs across a range of concentrations revealed minimal cytotoxicity up to 100 μM, thereby affirming their compatibility for biological applications (Fig. S4A–D†). Furthermore, the cytotoxicity of the lead compound DM4 was evaluated in the normal cell line HEK293 and was found to be minimally toxic up to a concentration of 100 μM (Fig. S4E†). The neuronal rescue efficacy of DM4 was then evaluated through a live-dead cell assay, employing calcein-AM and propidium iodide (PI) staining. Confocal microscopy demonstrated a reduction in neuronal cell death attributable to Aβ toxicity in the presence of DM4, as evidenced by a decrease in PI-positive dead cells relative to those subjected solely to Aβ (Fig. S5A†). This protective effect was substantiated by additional cell viability measurements via an MTT assay, which showed that while Aβ (10 μM) induced cell death led to 50% viability, DM4 treatment improved cell survival to 80% and 92% at concentrations of 20 μM and 50 μM, respectively (Fig. S5B†). We further investigated the ability of DM4 to rescue primary hippocampal neuronal cells from Aβ toxicity. Neurons were isolated from the mouse hippocampus and cultured for 8 days. The neuronal cells were then treated with Aβ (5 μM) alone and in combination with DM4 (10 and 20 μM) for 24 h. Following treatment, the cells were subjected to calcein staining and confocal imaging to assess the toxic effects and the protective efficacy of DM4. The imaging results clearly demonstrate a significant decrease in neuronal cells and neurite length following Aβ treatment (Fig. 2H). However, DM4 effectively rescued the neuronal cells from Aβ toxicity, as evidenced by the preservation of neuronal cells and neurite length. These in vitro and cellular studies corroborate the efficacy of DM4 in modulating Aβ aggregation and mitigating its neurotoxic effects.
Metal chelation, inhibition of metal dependent Aβ aggregation, ROS generation and rescue toxicity
DMs were functionalized with a dicarboxylic acid moiety derived from EDTA to facilitate metal ion sequestration and inhibit metal-dependent Aβ aggregation and ROS generation. UV-vis absorbance spectroscopy was employed to assess the chelation efficacy of DMs towards CuII, ZnII, and FeIII. DMs (10 μM) were titrated with metal ions in the range of 1 to 50 μM, and the resulting absorbance spectra were recorded in a HEPES buffer (10 mM, pH 7.4). The spectral shifts observed upon metal coordination with DMs were analyzed to determine the dissociation constant (Kd), indicating effective interaction between DMs and metal ions, as evidenced by notable hypochromic shifts (Fig. S6 and S7†). All DMs demonstrated efficient chelation with CuII and ZnII, while DM2 and DM4 also showed strong binding affinity for FeIII. The results revealed that DM2 and DM4 bind strongly to the tested metal ions with micromolar Kd values (Fig. 3A). DM4 exhibited the highest binding affinity for CuII, ZnII, and FeIII, with hypochromic and hypsochromic shifts (Fig. 3B). Matrix-assisted laser desorption/ionization (MALDI) spectrometry analysis was performed to elucidate the metal ion binding of DM4. After incubating DM4 with metal ions in PBS for 1 h, samples were subjected to MALDI analysis, which confirmed the formation of DM4-metal complexes (DM4 + Mn+), as indicated by the mass peaks corresponding to the complexes (Fig. S8†).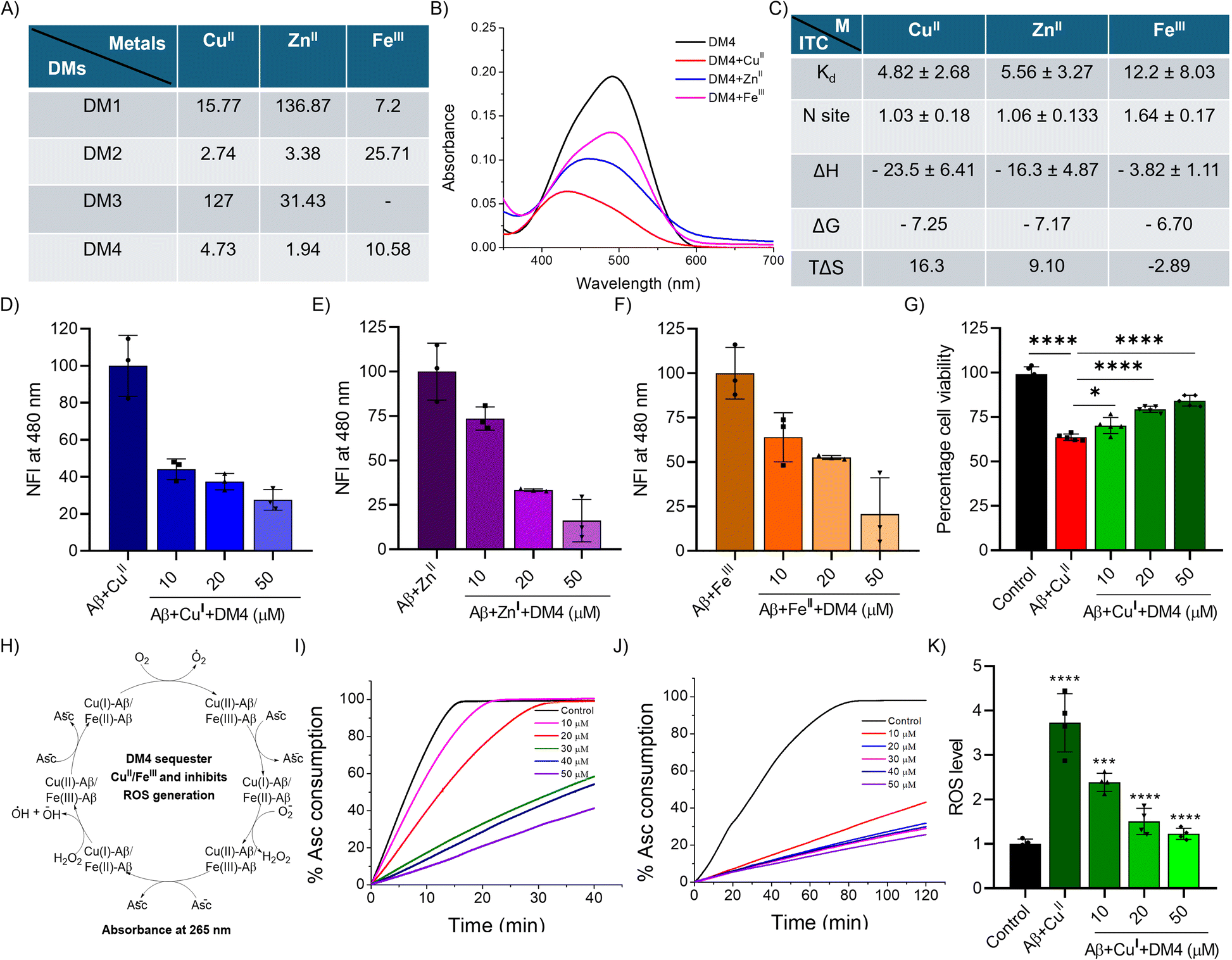 | ||
| Fig. 3 DMs chelate metal ions, inhibit metal dependent Aβ aggregation and toxicity, and inhibit ROS generation. (A) The dissociation constant (Kd in μM) of DMs and metal ions (CuII, ZnII and FeIII) determined from UV spectral changes from metal titration. (B) UV-vis spectra of DM4 (10 μM) alone and with metal ions (50 μM). (C) Isothermal titration calorimetry (ITC) data of DM4 interaction with CuII, ZnII and FeIII. Inhibition of (D) CuII, (E) ZnII and (F) FeIII (20 μM) dependent Aβ (10 μM) aggregation by DM4 in a concentration dependent manner (10, 20 and 50 μM). (G) DM4 rescue of neuronal cells from Aβ-CuII (10 μM) toxicity in SH-SY5Y cells from cell viability measured by MTT assay (n = 5 biologically independent replicates. One-way ANOVA with Tukey's post hoc test *p < 0.05). (H) Schematic representation of Aβ-CuII/FeIII ascorbate consumption assay. Ascorbate (Asc) consumption in the presence of (I) Aβ-CuII and (J) Aβ-FeIII with increasing concentration of DM4 (10, 20, 30, 40 and 50 μM) shows inhibition of ROS production. (K) Intracellular ROS detection by DCFDA assay in SH-SY5Y cells treated with Aβ-CuII alone and in the presence of DM4 (10, 20 and 50 μM) shows reduction in ROS production (n = 4 replicates in well scan mode to calculate mean fluorescence from each well. One-way ANOVA with Tukey's post hoc test *p < 0.05). | ||
To further characterize the metal-binding ability of the lead compound DM4, we performed an isothermal titration calorimetry (ITC) experiment. The ITC data show that DM4 strongly binds to all three metal ions with micromolar Kd values. DM4 displays binding affinity in the order CuII > ZnII > FeIII, with Kd values of 4.82 ± 2.68, 5.56 ± 3.27, and 12.2 ± 8.03 μM, respectively (Fig. 3C and S9†). The interaction between DM4 and CuII, ZnII, and FeIII was found to be spontaneous, with negative ΔG values of −7.25, −7.17, and −6.70 kcal mol−1 respectively. The binding of DM4 with CuII and ZnII was favored by both enthalpy and entropy (Cu-DM4: ΔH = −23.5 ± 6.41 and TΔS = 16.3 kcal mol−1; Zn-DM4: ΔH = −16.3 ± 4.87 and TΔS = 9.10 kcal mol−1). In contrast, the interaction of DM4 with FeIII was primarily favored by enthalpy (ΔH = −23.5 ± 6.41 and TΔS = −2.89 kcal mol−1). The binding site (N) for DM4 and metal ions was found to be ∼1, suggesting a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding ratio between DM4 and the metal ions. These ITC studies indicate the strong and favorable binding of DM4 with all three metal ions. Electron paramagnetic resonance (EPR) spectroscopy was employed to characterize the binding of paramagnetic CuII with DM4. The EPR spectra revealed a strong signal of CuII with a parallel g value of 2.147, indicating coordination with DM4 and suggesting the formation of a CuII-DM4 complex (Fig. S10†). The lower-than-usual g value suggests a moderately covalent coordination environment and a square planar or distorted octahedral geometry with carboxylate donors, similar to EDTA-type coordination. The superior metal binding and Aβ aggregation inhibition properties of DM4 led us to perform metal-dependent Aβ aggregation inhibition assays. DM4 significantly inhibited CuII, ZnII, and FeIII-dependent Aβ aggregation, with inhibition rates of 62%, 84%, and 85%, respectively, achieved at 50 μM (Fig. 3D–F). The neuroprotective potential of DM4 against metal-Aβ induced toxicity was evaluated which revealed concentration-dependent rescue of neuronal cells from metal-Aβ toxicity with complete rescue achieved at 50 μM, attributed to its potent Aβ aggregation inhibition and metal chelation capabilities (Fig. 3G and S11†).
:1 binding ratio between DM4 and the metal ions. These ITC studies indicate the strong and favorable binding of DM4 with all three metal ions. Electron paramagnetic resonance (EPR) spectroscopy was employed to characterize the binding of paramagnetic CuII with DM4. The EPR spectra revealed a strong signal of CuII with a parallel g value of 2.147, indicating coordination with DM4 and suggesting the formation of a CuII-DM4 complex (Fig. S10†). The lower-than-usual g value suggests a moderately covalent coordination environment and a square planar or distorted octahedral geometry with carboxylate donors, similar to EDTA-type coordination. The superior metal binding and Aβ aggregation inhibition properties of DM4 led us to perform metal-dependent Aβ aggregation inhibition assays. DM4 significantly inhibited CuII, ZnII, and FeIII-dependent Aβ aggregation, with inhibition rates of 62%, 84%, and 85%, respectively, achieved at 50 μM (Fig. 3D–F). The neuroprotective potential of DM4 against metal-Aβ induced toxicity was evaluated which revealed concentration-dependent rescue of neuronal cells from metal-Aβ toxicity with complete rescue achieved at 50 μM, attributed to its potent Aβ aggregation inhibition and metal chelation capabilities (Fig. 3G and S11†).
The redox-active metals, CuII and FeIII, coordinate with Aβ and undergo Fenton-type reactions to generate excessive ROS. This process contributes to oxidative stress and biomolecular damage (Fig. 3H). The potent metal-binding properties of DM4 prompted an investigation into its ability to sequester these metal ions in a redox-inactive state, and thereby alleviate ROS production. An ascorbate consumption assay was utilized to measure the inhibition of the redox cycle responsible for ROS generation, monitoring ascorbate absorbance at 267 nm.48 The assay demonstrated a decrease in ascorbate consumption as a function of DM4 concentration, indicating its efficacy in sequestering CuII and FeIII (Fig. S12†). In the presence of Aβ, CuII and FeIII coordinate with peptide and facilitate ROS generation by consuming ascorbate. The sequestration ability of DM4 was further evidenced by the Aβ-CuII/FeIII ascorbate assay, which showed concentration-dependent inhibition of ROS generation, with DM4 reducing ascorbate consumption by 60% and 80% for Aβ-CuII and Aβ-FeIII complexes, respectively at 50 μM (Fig. 3I and J). The superior activity of DM4 towards FeIII is possibly due to its higher ability to maintain the metal in a redox-inactive state. To confirm the decrease in ROS generation in neuronal cells, a dichlorodihydrofluorescein diacetate (DCFDA) assay was performed. Neuronal cells treated with Aβ and CuII to induce ROS generation were co-treated with DM4. The results indicated that Aβ-CuII treatment significantly increased ROS levels, which were substantially decreased by DM4 in a concentration-dependent manner (Fig. 3K). These studies collectively demonstrate the strong chelation of redox-active metal ions, inhibition of metal-dependent Aβ aggregation, and reduction of ROS generation by DM4, which contains a dicarboxylic acid moiety. EDTA is a very strong metal chelator that can sequester metals from essential metalloproteins, potentially causing toxicity (Kd ∼10−19). The dicarboxylic acid moiety in DM4 has reduced the affinity but retained it to an extent (Kd ∼10−6) that effectively inhibits metal-mediated toxicity.
Modulation of tau aggregation and rescue tau toxicity
DMs were designed to potentially interact with tau protein and modulate their aggregation. A ThT fluorescence assay was performed to assess the ability of these DMs (50 μM) to inhibit the aggregation of tau (10 μM) using a heparin-induced aggregation model.26,49 Among the DMs, DM4 exhibited a notable 70% inhibition of tau aggregation (Fig. S13A†). Further analysis of the inhibitory effect of DM4 revealed a concentration-dependent reduction in tau aggregation, with 66%, 68%, and 70% inhibition observed for 10 μM, 20 μM, and 50 μM, respectively (Fig. 4A). This inhibition was validated by sedimentation and SDS-PAGE assays, which showed a concentration-dependent effect of DM4 on the retaining of the tau monomer band, indicating effective inhibition of aggregation (Fig. 4B). TEM imaging was employed to visualize the structural features of the inhibited samples. While tau incubated with heparin alone formed fibrillar aggregates, those samples treated with DM4 exhibited a reduction in fibrils and the formation of amorphous aggregates, suggesting effective inhibition (Fig. 4C). To explore the potential of DMs to disassemble the preformed tau fibrils, a ThT fluorescence assay was performed. The results indicated that DM2 and DM4 could effectively disassemble tau fibrils, with DM4 achieving a superior 70% reduction in fibrils (Fig. S13B†). Concentration-dependent studies with DM4 further revealed a 30%, 63%, and 70% reduction in tau aggregates at concentrations of 10 μM, 20 μM, and 50 μM, respectively (Fig. 4D). TEM imaging of the dissolved species supported the dissolution of tau fibrils by DM4, as evidenced by the reduction of fibrils and the emergence of amorphous structures (Fig. 4E). The ThT assay, sedimentation assay, and TEM imaging demonstrate that DM4 effectively modulates tau aggregation and may offer neuroprotective benefits against toxicity. To understand the direct interaction and binding of DM4 with tau protein, we performed binding studies. We employed a tau intrinsic fluorescence assay by titrating with increasing concentrations of DM4. Tau protein in its native form exhibits fluorescence due to tyrosine in a specific local environment. The binding of any ligand that potentially induces structural changes results in altered intrinsic fluorescence. The intrinsic fluorescence of tau decreased upon the addition of DM4, suggesting the binding of DM4 to tau protein, which potentially alters its structure and conformation to inhibit aggregation (Fig. S14A†). The change in fluorescence was plotted against DM4 concentration to determine the Kd value. The results show that DM4 strongly binds to tau with a Kd value of 1.31 ± 0.12 μM (Fig. S14B†). Furthermore, we performed an MST assay to confirm the binding of DM4 to tau and determine the Kd value. Alexa-647 conjugated tau protein was incubated with different concentrations of DM4, and the MST assay was conducted. The results demonstrate DM4 binding to tau, as evidenced by the change in MST response and a Kd value of 1.54 ± 0.24 μM (Fig. 4F). These binding studies validate the direct binding of DM4 to tau protein, inducing structural changes that inhibit aggregation. The diazo-moiety derived from Congo red, along with other substitutions, has been implicated in the modulation of tau aggregation. DM4 exhibits good binding to tau protein and modulates tau aggregation, although the efficacy is slightly compromised compared to Aβ binding and modulation.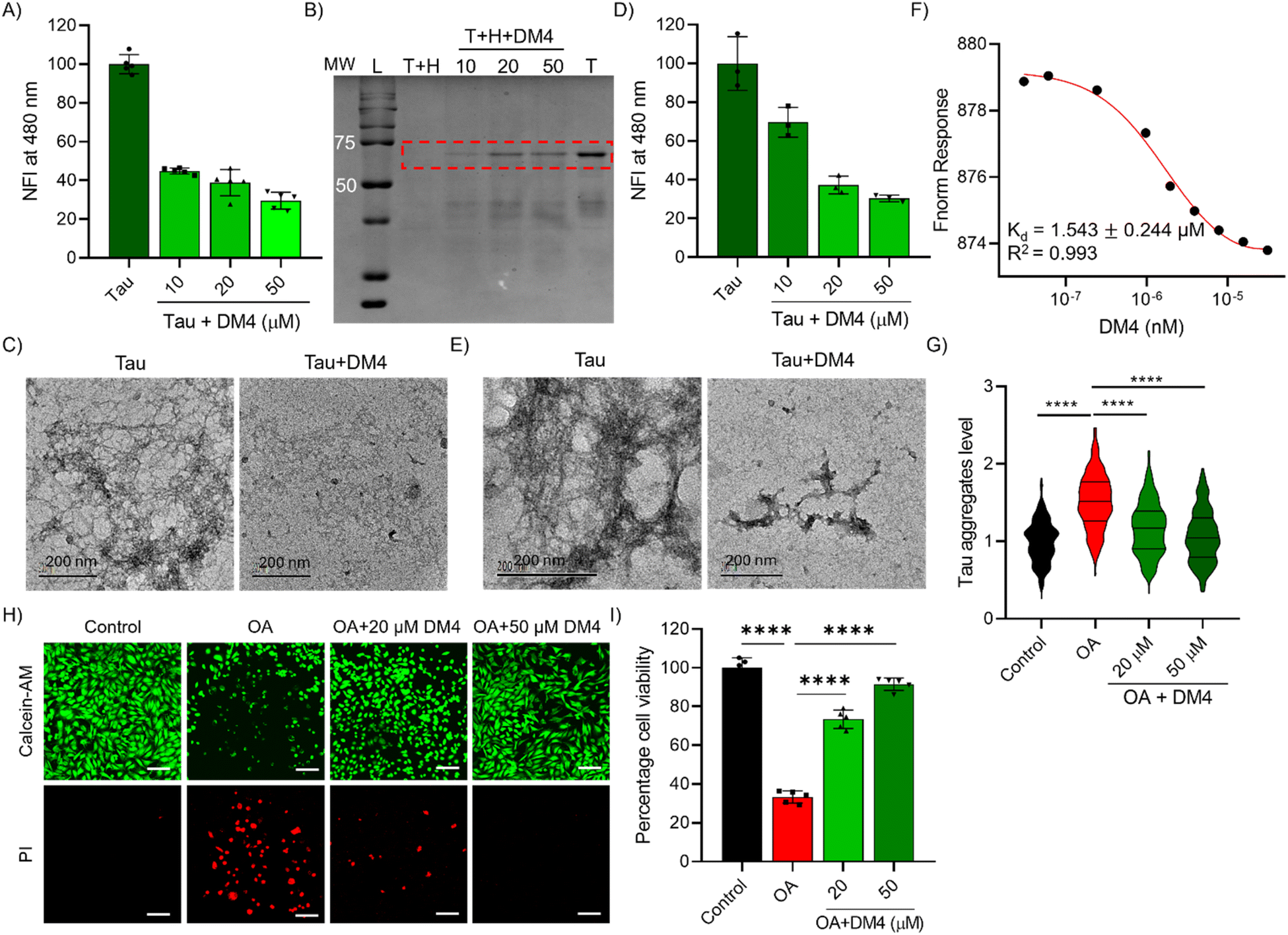 | ||
| Fig. 4 DMs modulates tau aggregation. (A) ThT fluorescence assay of tau aggregation inhibition by DM4 in a concentration dependent manner (10, 20 and 50 μM). (B) SDS-PAGE image of sedimentation assay shows tau aggregation inhibition by DM4 in a concentration dependent manner as evident from the tau monomer band (MW – molecular weight in KDa, L-ladder, T-tau and H-Heparin). (C) TEM images of tau aggregation inhibition by DM4 (50 μM). (D) ThT fluorescence assay of tau aggregate dissolution by DM4 in a concentration dependent manner (10, 20 and 50 μM). (E) TEM images of preformed tau aggregate dissolution by DM4 (50 μM). (F) Microscale thermophoresis (MST) assay of DM4 interaction and binding with tau protein to determine the Kd value. (G) Immunofluorescence assay quantification of tau aggregates (AT8 positive tau aggregates) in okadoic acid (OA)-induced tau toxicity and its reduction by DM4 in a dose-dependent manner in SHSY-5Y cells (normalised to control, n > 350 cells quantified for each group. One-way ANOVA with Tukey's post hoc test *p < 0.05). (H) Representative confocal images of live-dead cell imaging by using calcein-AM and PI for rescue of OA-induced tau toxicity in SH-SY5Y neuronal cells by DM4 (20 and 50 μM) (scale bar 100 μM). (I) Rescue of neuronal cells by DM4 from tau toxicity determined by MTT cell viability assay (n = 5 replicates. One-way ANOVA with Tukey's post hoc test *p < 0.05). | ||
Okadaic acid (OA), a potent inhibitor of serine/threonine protein phosphatases PP1 and PP2A at nanomolar concentrations, induces hyperphosphorylation of tau, leading to its aggregation and subsequent cellular toxicity.50 Using the OA-induced tau toxicity in the SH-SY5Y cell model, we assessed the ability of DM4 to inhibit tau aggregation and pathology. We performed an immunofluorescence assay using the AT8 anti-tau antibody to probe tau aggregates of phospho-tau (Phospho-Tau Ser202 and Thr205) levels. The immunofluorescence results show that OA (40 nM) treatment for 18 hours in SH-SY5Y cells resulted in increased levels of phospho-tau and aggregates (Fig. 4G and S15†). Treatment with DM4 (20 and 50 μM) significantly reduced the pathological tau aggregates, suggesting its ability to mitigate tau pathology in the cell model. Further we have performed a live/dead cell assay to evaluate DM4's ability to rescue from tau toxicity by employing calcein-AM and PI staining. Cells were exposed to 40 nM OA both with and without DM4 co-treatment at concentrations of 20 μM and 50 μM, followed by incubation for 24 h. Post-incubation, cells were stained with calcein-AM and PI for confocal microscopy analysis. The findings indicated that OA exposure elevated the proportion of PI-stained dead cells with a concurrent decrease in calcein stained live cells. However, DM4 treatment resulted in a decreased PI positive dead cell count, indicating its neuroprotective effect against tau-induced cytotoxicity (Fig. 4H). The neuroprotective impact of DM4 on OA-induced tau toxicity was further corroborated through cell viability assessments using the MTT assay. Cell viability assays revealed that OA exposure reduced viability to 33%, whereas DM4 treatment improved viability to 73% and 92% at concentrations of 20 μM and 50 μM respectively (Fig. 4I). We further investigated the ability of DM4 to rescue primary hippocampal neuronal cells from tau toxicity induced by OA. Neuronal cells were treated with OA (20 nM) alone and in combination with DM4 (10 and 20 μM) for 24 h, followed by calcein imaging. The results demonstrated a significant decrease in neuronal cell count and neurite length due to OA-induced tau toxicity. However, DM4 effectively rescued the neuronal cells from this toxicity, as evidenced by the preservation of neuronal cells and neurite length (Fig. S16†). These in vitro and cellular analyses substantiate the ability of DM4 to modulate tau protein aggregation effectively and mitigate its neurotoxic effects.
DM4 inhibits microglial activation and NLRP3 inflammasome formation
Neuroinflammation is a critical factor in the pathology of NDDs, with activated microglia driving inflammatory responses.51 Activation of the NLRP3 inflammasome in AD produces inflammatory mediators such as IL1β and caspase 1, leading to neuroinflammation and subsequent neuronal cell death.52 DMs with anti-inflammatory moieties have been designed to inhibit NLRP3 inflammasome formation. Using the N9 microglial cell model, we investigated the anti-neuroinflammatory effects by inducing NLRP3 inflammasome formation with lipopolysaccharide (LPS) and adenosine triphosphate (ATP) treatment, as well as LPS and AβO treatment for AD-associated neuroinflammation.53,54 Initially, we assessed the inhibition of microglial activation by DM4 through the Iba1 level via immunofluorescence. Cells were cultured in confocal dishes for 24 h, primed with LPS (500 ng mL−1) for 6 h, followed by AβO (5 μM) treatment for another 18 h. Immunofluorescence imaging and quantification indicated that Iba1 levels increased upon AβO exposure. However, DM4 effectively reduced microglial activation in a concentration-dependent manner (Fig. 5A and B). To further evaluate NLRP3 inflammasome inhibition by DM4, ASC speck imaging was performed through immunofluorescence. Induction of NLRP3 inflammasome formation via LPS (500 ng mL−1) and ATP (5 mM) treatment alongside DM4 presence (20 μM) revealed large inflammasome structures comprised of multiple NLRP3-ASC units resulting in speck formation detectable through ASC protein immunofluorescence (Fig. 5C).55 Significantly reduced ASC speck formation following DM4 treatment confirmed the inhibition of NLRP3 inflammasome formation (Fig. 5C and D). The formation of the NLRP3 inflammasome leads to secretion of pro-inflammatory mediators culminating in cell death. We conducted a cell viability assay to assess inflammation-mediated cell death rescue with an AβO-induced model. The assay demonstrated that treatment with AβO (5 μM) in LPS-primed microglial cells reduced cell viability to 38%, whereas DM4 treatment improved viability to 73% and 92% at concentrations of 20 μM and 50 μM respectively (Fig. 5E). These findings demonstrate the ability of DM4 to rescue microglial activation and NLRP3 inflammasome formation to curb neuroinflammation. The trifluoromethoxy benzene moiety on DM4, inspired by CY-09, an effective NLRP3 inhibitor, has contributed to the inhibition of NLRP3 inflammasome activity.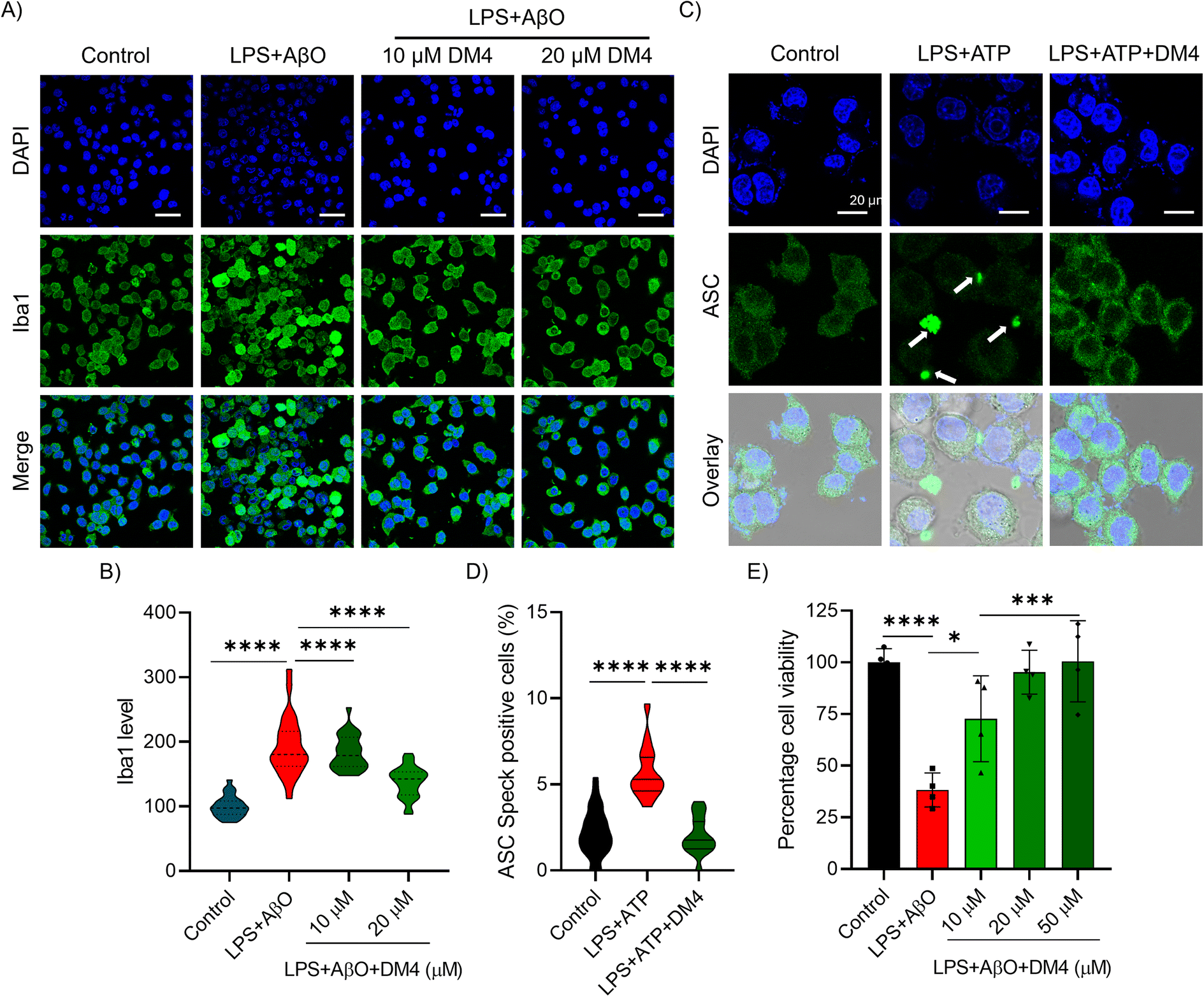 | ||
| Fig. 5 DM4 ameliorates microglial activation and NLRP3 inflammasome formation. (A) Representative confocal images of Iba1 immunofluorescence of N9 microglial cells primed with LPS (500 ng mL−1) and challenged with AβO (5 μM) alone and with DM4 (10 and 20 μM) incubated for 18 h and its (B) quantification normalised against control (n > 500 cells quantified for each group. One-way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 200 μM). (C) Representative confocal images of ASC speck (white arrows) immunofluorescence in N9 microglial cells primed with LPS (500 ng mL−1) and challenged with ATP (5 mM) alone and with DM4 (20 μM) incubated for 1 h and its (D) quantification (n > 30 ROI quantified for each group. One-way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 20 μM). (E) Rescue of AβO induced neuroinflammation mediated cell death by DM4 in a concentration dependent manner from cell viability measured using MTT assay. One-way ANOVA with Tukey's post hoc test *p < 0.05. | ||
BBB permeability and in vivo biocompatibility of DM4
A primary requirement for any CNS therapeutic is permeability across the BBB, a semipermeable physiological barrier. The multifunctional properties of DM4 prompted an investigation into its in vivo efficacy. First, the lipophilicity of DM4 was evaluated by determining its logP value using the shake flask method to ascertain the partition coefficient between octanol and water (Fig. S17A and B†). The logP value of DM4 was found to be 0.964, suggesting its propensity for BBB penetration. Subsequently, a murine model was utilized to assess the BBB permeation and brain accumulation.56 Wild type (WT) mice were administered a vehicle (control) and DM4 at a dosage of 5 mg per kg body weight. Post-injection, the animals were sacrificed at 30 min and 24 h to harvest the brain tissue. The brain samples were homogenized and extracted with methanol, and the extracts were then analysed by MALDI-TOF mass spectrometry to detect the presence of DM4 (Fig. S17C†). The mass results indicated that control samples showed no mass peaks corresponding to DM4 or adducts, whereas brain lysates from DM4-treated mice displayed peaks at 30 min and 24 h indicative of DM4 with a sodium adduct (M + Na), confirming the ability of DM4 to cross the BBB and accumulate within the brain. To further confirm the BBB permeability of DM4, we performed UPLC analysis to determine its presence in brain samples. The UPLC chromatograms clearly show the accumulation of DM4 in the brain within 30 min, with trace amounts still detectable 24 h post intraperitoneal injection (Fig. S18†). DM4, with a balance of hydrophobicity (trifluoro benzene moiety) and hydrophilicity (dicarboxylic acid) in the diazo-core was BBB permeable. This suggests the importance of a hydrophobic-hydrophilic balance in brain-permeant molecules.
The in vivo biocompatibility profile of DM4 was assessed in a WT mouse model aged 12 months. First, lethal dose 50 (LD50) was established to ascertain the lethal threshold of DM4 and to understand the dosing for subsequent toxicity studies. The LD50 was determined by administering increasing doses of DM4 intraperitoneally and monitoring the animals over a 14-day period for changes in physical appearance, activity, and mortality rate. DM4 was tolerated up to a dose of 50 mg per kg BW, exhibited lethal effects beyond 80 mg per kg BW, and was fatal at 180 mg per kg BW, with the LD50 calculated to be 97.8 mg per kg BW (Fig. 6B). For biocompatibility evaluation, a daily intraperitoneal dose of 5 mg per kg BW, which is twentyfold less than that of the LD50, was selected. The dosage investigated for toxicity in 12-month-old WT mice, was divided into control (vehicle) and treatment (DM4) groups over a two-month period. Weekly BW monitoring, post-treatment serum collection, and visceral organ analysis were performed to assess toxicity through serum biochemistry and histopathology (Fig. 6A). BW served as a toxicity indicator, with no significant differences observed between the treated and control groups throughout the study duration (Fig. 6C).57 Serum biomarkers indicative of liver, kidney, heart, and immune function including aspartate aminotransferase (AST), alanine transaminase (ALT), creatinine, blood urea nitrogen (BUN), bilirubin, total protein, triglycerides, and alkaline phosphatase (ALP) were measured. Biochemical assays revealed that these biomarkers remained significantly unaltered in DM4-treated mice compared to controls, except for a slight decrease in ALP levels (Fig. 6D).58,59 Histopathological examinations of liver, kidney, lung, spleen, and heart tissues stained with haematoxylin and eosin (H&E) showed no microscopic pathological lesions in DM4-treated mice relative to controls (Fig. 6E).60 These findings affirm the biocompatibility of DM4 in a preclinical in vivo mouse model, supporting its advancement to in vivo models of AD.
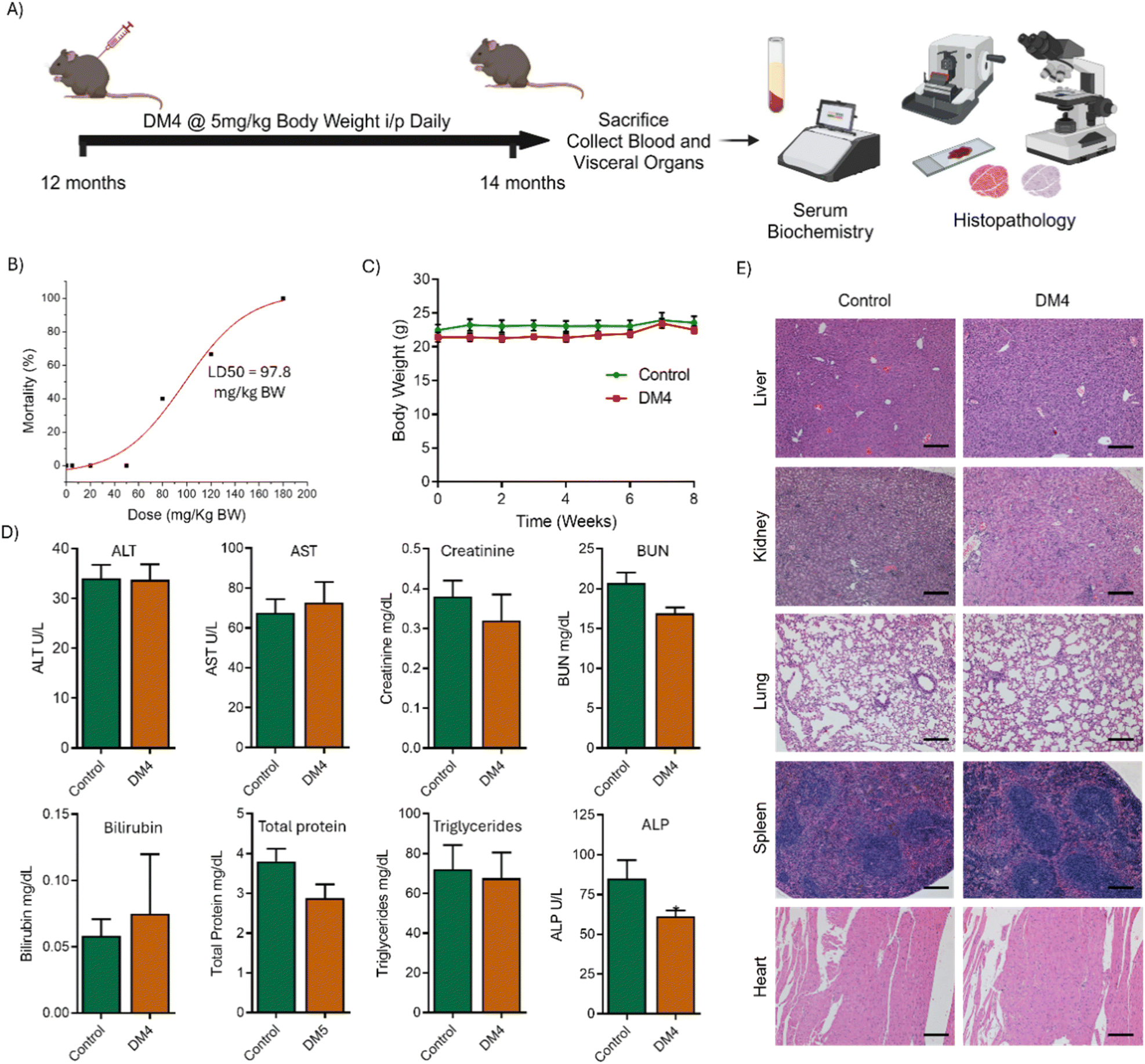 | ||
| Fig. 6 In vivo biocompatibility of DM4 in a mouse model. (A) Schematic showing the experimental timeline and protocol for in vivo biocompatibility studies in mice. (B) Dose response curve of DM4 to determine lethal dose 50 (LD50) in a mouse model revealed that DM4 displayed higher LD50 (n = 5 mice in each dose group). (C) Time dependent body weight of mice treated with DM4 at dose rate of 5 mg per kg BW daily for 8 weeks (n = 6 mice per group). (D) The serum levels of organ function biomarkers in control and DM4 treated mice determined by serum biochemical tests (n = 5 to 6 mice for each group. Unpaired, two-tailed t-test *p < 0.05). (E) H & E staining images of different visceral organs of control and DM4 treated mice show the absence of microscopic pathological lesions indicating biocompatibility of the compound (n = 3 biologically independent animals with 2 sections per animal. Scale bar: 200 μm). | ||
DM4 ameliorates the amyloid burden and neuroinflammation in an APP/PSEN1 Tg mouse model
Encouraged by the promising in vitro results demonstrating the efficacy of DM4 in modulating AD pathologies, its BBB permeability, and favourable biocompatibility profile, we proceeded to assess the in vivo efficacy. We employed an APP/PSEN1 Tg AD mouse model to evaluate the therapeutic effect of DM4 to ameliorate various pathologies of AD. Twelve-month-old animals were categorized into four groups: WT controls receiving vehicle treatment (WTC), WT treated with DM4 (WTT), AD controls with vehicle treatment (ADC), and AD subjects treated with DM4 (ADT) at a dosage of 5 mg per kg BW administered intraperitoneally daily for eight weeks. Following the treatment regimen, the animals were sacrificed, and their brains were harvested to assess the reduction of pathological features (Fig. 7A). Aβ plaque deposition, a hallmark of AD, was prevalent in the APP/PSEN1 Tg AD mouse model. Fluorescence staining with ThT was performed on brain tissue sections to visualize and quantify the amyloid burden. Confocal imaging revealed basal ThT-positive amyloid deposits in WT mice and dense Aβ plaques in the cortex and hippocampus of the AD mouse brains (Fig. 7B, C and S19†). Quantitative analysis of the Aβ burden in both the cortex and hippocampus indicated a significant decrease in amyloid plaques following DM4 treatment (Fig. 7B and C). Remarkably, DM4 treatment led to a complete reduction of amyloid load in AD mice as evident from dot blot analysis of brain lysates (Fig. S20†). To understand the effect of DM4 on the amyloid production pathway, we analyzed the APP protein and its C-terminal cleaved fragments (APP-CTFs), which result from β-secretase cleavage, using western blotting. The results indicated that APP expression remained unchanged upon DM4 treatment (Fig. S21A and B†). As expected, APP-CTF expression significantly increased in AD animals, and DM4 failed to reduce these levels (Fig. S21A and C†). These findings suggest that the amyloid production pathway is unaffected by DM4 treatment. Our design strategy and in vitro results clearly demonstrated that DM4 effectively inhibits fibril formation and promotes the dissolution of preformed fibrils. DM4 directly targets Aβ peptide aggregation, preventing further accumulation and build-up of amyloid load. As demonstrated in vitro, DM4 also disassembles preformed fibrillar aggregates into shorter fibrils and amorphous aggregates. These disassembled products are likely cleared through innate clearance mechanisms in the brain, such as cerebrospinal fluid (CSF) and glymphatic clearance. Another possible mechanism might be microglia-mediated clearance through increased phagocytosis and degradation. This warrants further detailed studies to elucidate the mechanism of amyloid clearance. | ||
| Fig. 7 DM4 ameliorates the amyloid burden and neuroinflammation in an APP/PSEN1 Tg AD mouse model. (A) Schematic showing the experimental timeline and protocol for immunofluorescence studies in AD mice. Representative confocal images of amyloid load stained by Thioflavin T (ThT) in (B) the cortex and (C) hippocampus and its quantification (n = 3 biologically independent animals with 2 to 3 sections per animal. One way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 200 μm). Representative confocal images of Iba1 staining to probe activated microglial cells in (D) the cortex and (E) hippocampus and its quantification (n = 3 biologically independent animals with 2 to 3 sections per animal. One way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 100 μm). (F) Schematic showing the procedure to perform sandwich ELISA for quantification of proinflammatory markers from mouse brain tissue. (G) Quantification of proinflammatory markers of neuroinflammation IL1β, TNFα and IL6 by sandwich ELISA of control and DM4 treated mouse brain tissue (n = 5 biologically independent animals. One way ANOVA with Tukey's post hoc test *p < 0.05). | ||
Neuroinflammation is predominantly driven by activated microglial cells and the proinflammatory mediators they secrete.51 Typically dormant, microglia become activated in AD, leading to neuroinflammation and subsequent neurodegeneration.17In vitro studies using the N9 microglial cell model have shown that DM4 inhibits microglial activation and inflammatory cell death. This was further investigated in vivo, where Iba1 immunofluorescence was used to probe the activated microglial population in the brain sections.61 Confocal imaging revealed a significant increase in Iba1 positive areas in the cortex and hippocampus of ADC brains compared to WTC, indicating heightened microglial activation (Fig. 7D, E and S22†). However, DM4 treatment markedly reduced the Iba1 positive activated microglial population, bringing levels close to those observed in WTC, suggesting potent anti-neuroinflammatory activity. The suppression of AβO-induced microglial activation and NLRP3 inflammasome formation by DM4 was corroborated by cellular studies. Downstream effects were assessed by quantifying proinflammatory mediators in the brain samples using sandwich ELISA (Fig. 7F). The results showed a significant elevation in the levels of IL1β, TNFα, and IL6 in ADC brain samples, which were substantially reduced following DM4 treatment, confirming the mitigation of neuroinflammation (Fig. 7G). The immunofluorescence imaging and ELISA quantification underscore DM4's ability to alleviate neuroinflammation in vivo, supporting its therapeutic potential.
DM4 ameliorates neurodegeneration and synaptic dysfunction
Amyloid aggregates are toxic to neuronal cells, and the activation of microglial cells leads to neuroinflammation and subsequent neuronal death.62 Our in vivo analysis indicates a substantial reduction in amyloid load and neuroinflammation in DM4 treated AD mice. It is expected that DM4 treatment may synergistically mitigate neurodegeneration and synaptic defects. The population of mature neurons in the brain was examined using the neuronal marker NeuN (neuronal nuclear protein) through immunofluorescence, which is specifically expressed in mature neuronal cells. Confocal imaging revealed a decrease in NeuN staining in both the cortex and hippocampus of ADC compared to WTC, indicating neurodegeneration (Fig. 8A, B and S23†). AD mice treated with DM4 exhibited a NeuN staining area akin to WTC, implying the prevention of neurodegeneration. Further, immunofluorescence for microtubule-associated protein 2(MAP2), a dendritic marker, indicated recovery in the DM4-treated AD brains comparable to WTC (Fig. 8C, D and S24†). The rescue of MAP2 levels suggests the preservation of neuronal integrity and the neural network, which is compromised in AD and other NDDs. The protective effect of DM4 against neurodegeneration can be attributed to its multifunctional properties that target multiple pathologies of AD. Synaptic defects and dysfunction, hallmarks of many NDDs including AD, result in neurological impairments. Immunofluorescence was employed to measure postsynaptic density protein 95 (PSD95), a synaptic function and integrity marker. Immunofluorescence imaging and quantification have revealed a substantial decrease in the levels of PSD95 in AD models relative to WT (Fig. 8E, F and S25†). Treatment with DM4 has significantly restored PSD95 levels, suggesting an improvement in synaptic integrity. DM4 appears to synergistically ameliorate synaptic defects by concurrently targeting amyloid deposition, microglial activation, NLRP3 inflammasome activation, and neuroinflammation. | ||
| Fig. 8 DM4 ameliorates neurodegeneration and synaptic abnormalities in an APP/PSEN1 Tg AD mouse model. Representative confocal images of NeuN immunofluorescence, a marker of mature neurons in (A) the cortex and (B) hippocampus and its quantification (n = 3 biologically independent animals with 2 to 3 sections per animal. One way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 100 μm). Representative confocal images of MPA2 immunofluorescence to probe dendrites in (C) the cortex and (D) hippocampus and its quantification (n = 3 biologically independent animals with 2 to 3 sections per animal. One way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 500 μm). Representative confocal images of PSD95 immunofluorescence to stain synaptic density in (E) the cortex and (F) hippocampus and its quantification (n = 3 biologically independent animals with 2 to 3 sections per animal. One way ANOVA with Tukey's post hoc test *p < 0.05. Scale bar 200 μm). | ||
DM4 rescues behavioural deficits
AD is phenotypically characterized by impairments in learning, memory, and cognition, leading to behavioural deficits. DM4 has demonstrated significant mitigation of major pathological phenotypes, as evident from results discussed vide supra. We have further assessed the therapeutic efficacy of DM4 in rescuing behavioural deficits through open field (OF) and Morris water maze (MWM) behavioural tests (Fig. 9A).42 The OF test was performed to evaluate the alleviation of agitation, and locomotor anomalies in AD models. Following a three-day acclimatization period with the experimenter, subjects were introduced into the OF arena, and their movements were captured using a roof-mounted digital camera. Analysis of the movement within the novel OF arena, facilitated by video tracking software, indicated heightened agitation in AD models, characterized by increased total distances travelled and frequency of central region crossings (Fig. 9B). Quantitative analysis revealed a significant increase in the number of entries and duration spent in the central zone for AD models compared to WT controls, which was notably reduced following DM4 treatment (Fig. 9C and D). Further, the total distance travelled, particularly within the central zone, was substantially increased in AD models but was mitigated in those treated with DM4 (Fig. 9E and F). These OF outcomes suggest that DM4 treatment effectively addresses agitation and locomotor issues in AD models.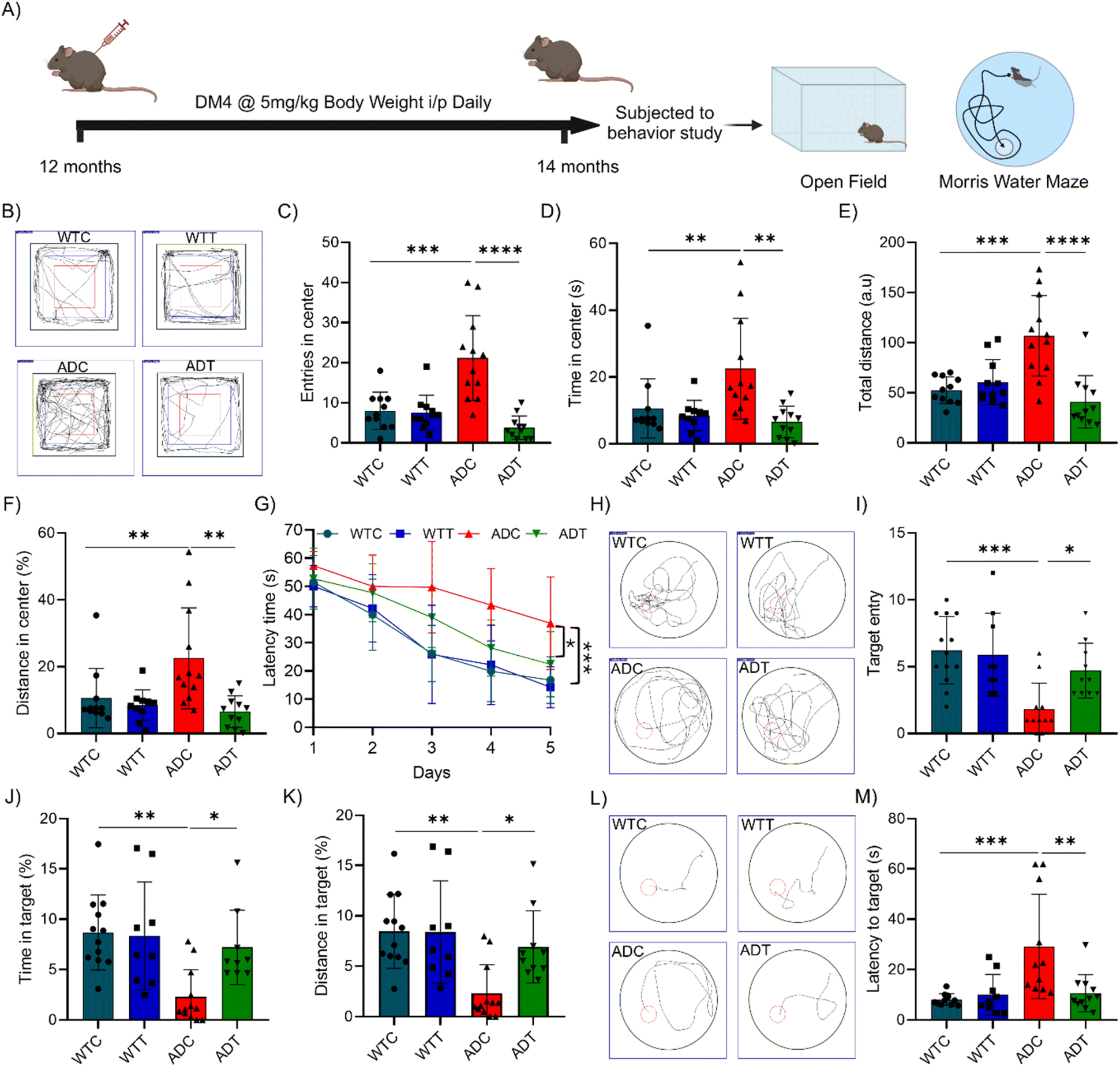 | ||
| Fig. 9 DM4 ameliorates learning and memory deficits in an APP/PSEN1 Tg AD mouse model. (A) Schematic showing experimental timeline and behaviour studies in APP/PSEN1 Tg AD mice. (B) Representative tracking of animal locomotion (WT and AD mice treated with vehicle and DM4) during an open field (OF) test; quantification of (C) entries in the centre zone of the OF arena, (D) percentage time spent in the centre zone, (E) total distance travelled by animals in the OF arena and (F) percentage time spent in the centre zone. (G) Quantification of latency to the platform during the training period of the Morris water maze (MWM) behaviour test. (H) Representative tracking of animal movements (WT and AD mice treated with vehicle and DM4) during the MWM probe trial test. Quantification of (I) target zone (platform) entries during the probe trial, (J) percentage of total time spent in the target zone (platform) and (K) percentage of distance travelled in the target. (L) Representative tracking of latency to the platform zone during the MWM probe trial test and (M) its quantification. (n = 9 to 12 mice per group, one way ANOVA with Tukey's post hoc test *p < 0.05). | ||
AD is primarily characterised by learning and memory deficits. To evaluate the rescue of spatiotemporal learning and memory deficiencies, we employed the MWM test. Subjects were trained over a five-day period to locate and rest on a designated platform within one of the quadrants of the maze, with the latency to reach the platform serving as a measure of spatiotemporal learning. A subsequent probe trial, conducted after a day of rest and involving the removal of the platform, allowed for the assessment of spatiotemporal memory based on subject movement. Latency measurements during the training phase highlighted a pronounced learning impairment in AD models, as evidenced by extended latency periods, which were significantly ameliorated by DM4 treatment on the fourth and fifth days (Fig. 9G). Tracking during the probe trial elucidated a deficit in spatiotemporal memory among AD models, manifesting as erratic exploration within the MWM, in contrast to the targeted exploration exhibited by WT controls (Fig. 9H). However, DM4-treated AD models showed marked improvements in exploratory behavior, aligning more closely with that of WT controls. Quantitative metrics, including the frequency of target platform entries, time spent, and distance covered within the target platform zone, underscored a reduction in AD models indicative of compromised memory, which was significantly rescued by DM4 treatment (Fig. 9I–K). The latency to enter the platform zone during the probe trial was notably increased in ADC relative to controls, that was substantially reduced in DM4 treatment, suggesting an enhancement in spatial memory (Fig. 9L and M). These behavioral studies affirm the ability of DM4 to alleviate behavioral deficits. The therapeutic impact of DM4 is attributed to its multifunctional effect in ameliorating the amyloid burden, neuroinflammation, neurodegeneration, and synaptic dysfunction in vivo. The observed restoration of synaptic integrity, as evidenced by the recovery of PSD95 levels, suggests a possible mechanism underlying the behavioral improvements conferred by DM4 treatment.
In silico ADMET studies
The pharmacokinetic properties of DM4, including absorption, distribution, metabolism, excretion, and toxicity (ADMET), were evaluated by in silico predictions using multiple tools.63–66 These prediction models were developed by considering various structural and functional parameters of chemical compounds that influence biocompatibility and distribution. The models were constructed using algorithmic or machine learning approaches and were trained with extensive, experimentally validated datasets to accurately predict the ADMET parameters of novel drugs based on their chemical structures. The prediction accuracy of these models, tested using experimentally known drugs, ranges from 70% to 80%. These in silico studies provide valuable insights into the suitability of the molecule for detailed in vivo pharmacokinetic studies. The in silico prediction results for DM4 indicate several favorable ADMET properties (Table 1 and S2–S5†). The compound exhibits good solubility and oral bioavailability, as evidenced by favorable logP values, gastrointestinal absorption, and bioavailability metrics from various in silico prediction tools (Table 1). Additionally, the results demonstrate effective distribution and metabolism with moderate retention of DM4. Toxicity parameters, including AME tests and hERG results, suggest that the compound is non-toxic and biocompatible in vivo.
| ADMET | Parameter | Swiss ADME | pkCSM | ADMETlab 3.O | DeepPK |
|---|---|---|---|---|---|
| Absorption | logP |
4.75 | 6.1759 | 4.432 | 3.62 |
| GI absorption | Low | 47.23% | — | Absorbed | |
| Bioavailability | 0.56 | — | >50% | >50% | |
| Caco2 permeability | — | 0.034 | −4.94 | −5.88 | |
| P-Glycoprotein substrate | No | Yes | No | No | |
| P-Glycoprotein inhibitor | — | No | No | No | |
| Distribution | PPB | — | −0.492 | 98.6% | 98.73 |
| VDss | — | — | 1.613 | 0.66 | |
| Metabolism | CYP2D6 substrate | Yes | No | No | No |
| CYP1A2 inhibitor | No | No | No | No | |
| Excretion | Total clearance | — | −0.668 | — | 2.9 |
| Half life | — | — | 1.328 | 3 | |
| Toxicity | AMES test | — | Safe | 0.572 | Safe |
| hERG inhibitor | — | No | 0.538 | Safe | |
| Hepatotoxicity | — | Yes | 0.947 | Toxic | |
| Genotoxicity | — | — | No | Safe |
Conclusions
The pathological complexity of AD necessitates the development of multifunctional therapeutic agents capable of targeting multiple pathological features to enhance treatment efficacy. We designed multifunctional small molecules by integrating functional moieties to address key pathological factors, including Aβ, tau, metal toxicity, ROS, NLRP3 inflammasome activation and synaptic dysfunction. In vitro studies have shown that the lead molecule DM4 chelates metal ions, reduces ROS generation, modulates metal-independent and -dependent Aβ aggregation, and tau aggregation. Cellular studies have further confirmed the potential of DM4 in mitigating Aβ and tau toxicity, preventing ROS generation, attenuating microglial activation, and targeting neuroinflammation via the NLRP3 inflammasome pathway. Subsequent in vivo investigations have demonstrated the ability of DM4 to cross the BBB and exhibit biocompatibility. DM4 effectively alleviates the pathological hallmarks of AD, including amyloid accumulation, neuroinflammation, neurodegeneration, and synaptic dysfunction in an APP/PSEN1 Tg AD mouse model. Behavioural studies employing OF and MWM tests have demonstrated the efficacy of DM4 in reversing behavioural impairments. The strategic design of DM4, targeting multiple pathological processes while ensuring biocompatibility and BBB permeability, has proven promising and lays the groundwork for the development of improved therapeutics for a range of NDDs. The preclinical in vivo performance of DM4 underscores its potential for clinical translation as a therapeutic candidate.Data availability
The data that support the findings of this study are available in the manuscript and the ESI† of this article.Author contributions
T. G. designed the molecules. T. G. and M. R. designed the experiments. C. B. performed the synthesis and characterization. A. K. synthesized and scaled up the lead compound. M. R. performed the experiments, curated the data, and carried out the analysis. S. S. helped with the in vivo experiments. M. R. and T. G. wrote the manuscript, and others gave inputs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank JNCASR, SERB (core grant: CRG/2020/004594), Govt. of India, New Delhi, India, for the funding. We thank the central animal facility, JNCASR for help in animal experiments. We thank Shiva prasad P. S. for help in confocal imaging and UPLC experiments. MR thanks Prathamesh for help in the western blot experiment and Autophagy lab JNCASR for neuronal media. MR and AK thank UGC for their student fellowship.References
- B. N. Dugger and D. W. Dickson, Cold Spring Harbor Perspect. Biol., 2017, 9, a028035 CrossRef PubMed
.
- D. M. Wilson, M. R. Cookson, L. Van Den Bosch, H. Zetterberg, D. M. Holtzman and I. Dewachter, Cell, 2023, 186, 693 CrossRef CAS PubMed
- K. Suri, M. Ramesh, M. Bhandari, V. Gupta, V. Kumar, T. Govindaraju and N. A. Murugan, ChemBioChem, 2024, e202400224 CrossRef CAS PubMed
- J. M. Long and D. M. Holtzman, Cell, 2019, 179, 312 CrossRef CAS PubMed
-
Alzheimer's Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities, ed. T. Govindaraju, The Royal Society of Chemistry, 2022, pp. 1–669 Search PubMed
- R. M. Anderson, C. Hadjichrysanthou, S. Evans and M. M. Wong, Lancet, 2017, 390, 2327 CrossRef PubMed
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184 CrossRef CAS PubMed
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306 CrossRef CAS PubMed
- A. I. Bush and R. E. Tanzi, Neurotherapeutics, 2008, 5, 421 CrossRef CAS PubMed
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Acc. Chem. Res., 2019, 52, 2026 CrossRef CAS PubMed
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252 CrossRef CAS PubMed
- E. E. Congdon and E. M. Sigurdsson, Nat. Rev. Neurol., 2018, 14, 399–415 CrossRef CAS PubMed
-
P. Gopinath, M. Ramesh and T. Govindaraju, Tau-targeting Therapeutic Strategies for Alzheimer's Disease, in Alzheimer's Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities, The Royal Society of Chemistry, 2022, pp. 487–514 Search PubMed
- C.-W. Chang, E. Shao and L. Mucke, Science, 2021, 371, eabb8255 CrossRef CAS PubMed
- M. T. Heneka, M. J. Carson, J. E. Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica, R. M. Ransohoff, K. Herrup, S. A. Frautschy, B. Finsen, G. C. Brown, A. Verkhratsky, K. Yamanaka, J. Koistinaho, E. Latz, A. Halle, G. C. Petzold, T. Town, D. Morgan, M. L. Shinohara, V. H. Perry, C. Holmes, N. G. Bazan, D. J. Brooks, S. Hunot, B. Joseph, N. Deigendesch, O. Garaschuk, E. Boddeke, C. A. Dinarello, J. C. Breitner, G. M. Cole, D. T. Golenbock and M. P. Kummer, Lancet Neurol., 2015, 14, 388 CrossRef CAS PubMed
- M. T. Heneka, R. M. McManus and E. Latz, Nat. Rev. Neurosci., 2018, 19, 610 CrossRef CAS PubMed
- F. Leng and P. Edison, Nat. Rev. Neurol., 2021, 17, 157 CrossRef PubMed
- M. J. Oset-Gasque and J. Marco-Contelles, ACS Chem. Neurosci., 2018, 9, 401 CrossRef CAS PubMed
-
T. Mondal, S. Samanta, A. Kumar and T. GovindarajuMultifunctional Inhibitors of Multifaceted Aβ Toxicity of Alzheimer's Disease, in Alzheimer's Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities, The Royal Society of Chemistry, 2022, pp. 455–486 Search PubMed
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221 CrossRef CAS PubMed
- T. Storr, Can. J. Chem., 2021, 99, 1 CrossRef CAS
- K. Rajasekhar, K. Mehta and T. Govindaraju, ACS Chem. Neurosci., 2018, 9, 1432 CrossRef CAS PubMed
- M. Rana, H. J. Cho, H. Arya, T. K. Bhatt, K. Bhar, S. Bhatt, L. M. Mirica and A. K. Sharma, Inorg. Chem., 2022, 61, 10294 CrossRef CAS PubMed
- D. Padhi, P. Baruah, M. Ramesh, H. Moorthy and T. Govindaraju, Redox Biol., 2024, 71, 103119 CrossRef CAS PubMed
- M. Ramesh, C. Balachandra, P. Andhare and T. Govindaraju, ACS Chem. Neurosci., 2022, 13, 2209 CrossRef CAS PubMed
- M. Ramesh, A. Acharya, N. A. Murugan, H. Ila and T. Govindaraju, ChemBioChem, 2021, 22, 3348 CrossRef CAS PubMed
- A. Gandini, A. E. Gonçalves, S. Strocchi, C. Albertini, J. Janočková, A. Tramarin, D. Grifoni, E. Poeta, O. Soukup, D. Muñoz-Torrero, B. Monti, R. Sabaté, M. Bartolini, G. Legname and M. L. Bolognesi, ACS Chem. Neurosci., 2022, 13, 3314 CrossRef CAS PubMed
- D. Zhao, Y. Tang, X. Suo, C. Zhang, Y. Dou and J. Chang, Innovation, 2021, 2, 100160 CAS
- M. Ramesh and T. Govindaraju, Chem. Sci., 2022, 13, 13657 RSC
- J. M. Kim, J. Kang, M. Lee, J. Han, G. Nam, E. Tak, M. S. Kim, H. J. Lee, E. Nam, J. Park, S. J. Oh, J. Y. Lee, J. Y. Lee, M. H. Baik and M. H. Lim, J. Am. Chem. Soc., 2020, 142, 8183 CrossRef PubMed
- L. Sun, H. J. Cho, S. Sen, A. S. Arango, T. T. Huynh, Y. Huang, N. Bandara, B. E. Rogers, E. Tajkhorshid and L. M. Mirica, J. Am. Chem. Soc., 2021, 143, 10462 CrossRef CAS PubMed
- M. Habashi, S. Vutla, K. Tripathi, S. Senapati, P. S. Chauhan, A. Haviv-Chesner, M. Richman, S. A. Mohand, V. Dumulon-Perreault, R. Mulamreddy, E. Okun, J. H. Chill, B. Guérin, W. D. Lubell and S. Rahimipour, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210766119 CrossRef CAS PubMed
- N. Guzior, A. Wieckowska, D. Panek and B. Malawska, Curr. Med. Chem., 2015, 22, 373 CrossRef CAS PubMed
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed
- E. I. Yakupova, L. G. Bobyleva, I. M. Vikhlyantsev and A. G. Bobylev, Biosci. Rep., 2019, 39, BSR20181415 CrossRef PubMed
- B. Frieg, L. Gremer, H. Heise, D. Willbold and H. Gohlke, Chem. Commun., 2020, 56, 7589 RSC
- M. I. Sulatsky, A. I. Sulatskaya, O. I. Povarova, I. A. Antifeeva, I. M. Kuznetsova and K. K. Turoverov, Prion, 2020, 14, 67 CrossRef CAS PubMed
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405 CrossRef CAS PubMed
- C. Lendel, B. Bolognesi, A. Wahlström, C. M. Dobson and A. Gräslund, Biochemistry, 2010, 49, 1358 CrossRef CAS PubMed
- J. Hatai, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2017, 139, 2136 CrossRef CAS PubMed
- M. Rana, H. J. Cho, T. K. Roy, L. M. Mirica and A. K. Sharma, Inorg. Chim. Acta, 2018, 471, 419 CrossRef CAS PubMed
- S. Samanta, K. Rajasekhar, M. Ramesh, N. A. Murugan, S. Alam, D. Shah, J. P. Clement and T. Govindaraju, Adv. Ther., 2021, 4, 2000225 CrossRef CAS
-
C. Hureau, Role of Metal Ions in Alzheimer's Disease: Mechanistic Aspects Contributing to Neurotoxicity, in Alzheimer's Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities, The Royal Society of Chemistry, 2022, pp. 170–192 Search PubMed
- M. S. Tan, J.-T. Yu, T. Jiang, X. C. Zhu and L. Tan, Mol. Neurobiol., 2013, 48, 875 CrossRef CAS PubMed
- Y. Zhang, Z. Dong and W. Song, Signal Transduct. Targeted Ther., 2020, 5, 37 CrossRef PubMed
- A. Zahid, B. Li, A. J. K. Kombe, T. Jin and J. Tao, Front. Immunol., 2019, 10, 2538 CrossRef PubMed
- W. J. Geldenhuys, A. S. Mohammad, C. E. Adkins and P. R. Lockman, Ther. Deliv., 2015, 6, 961 CrossRef CAS PubMed
- E. Atrián-Blasco, M. del Barrio, P. Faller and C. Hureau, Anal. Chem., 2018, 90, 5909 CrossRef
- A. Paul, G. K. Viswanathan, A. Huber, E. Arad, H. Engel, R. Jelinek, E. Gazit and D. Segal, FEBS J., 2021, 288, 4267 CrossRef CAS PubMed
- C.-H. Chou and C.-R. Yang, Int. J. Mol. Sci., 2021, 22, 5347 CrossRef CAS PubMed
- D. V. Hansen, J. E. Hanson and M. Sheng, J. Cell Biol., 2018, 217, 459 CrossRef CAS PubMed
- M. T. Heneka, M. P. Kummer, A. Stutz, A. Delekate, S. Schwartz, A. Vieira-Saecker, A. Griep, D. Axt, A. Remus, T.-C. Tzeng, E. Gelpi, A. Halle, M. Korte, E. Latz and D. T. Golenbock, Nature, 2013, 493, 674 CrossRef CAS PubMed
- K. V. Swanson, M. Deng and J. P. Ting, Nat. Rev. Immunol., 2019, 19, 477 CrossRef CAS PubMed
- G. Zito, M. Buscetta, M. Cimino, P. Dino, F. Bucchieri and C. Cipollina, Int. J. Mol. Sci., 2020, 21, 4294 CrossRef CAS PubMed
- A. Stutz, G. L. Horvath, B. G. Monks and E. Latz, Methods Mol. Biol., 2013, 1040, 91 CrossRef CAS PubMed
- J. A. Nicolazzo, S. A. Charman and W. N. Charman, J. Pharm. Pharmacol., 2006, 58, 281 CrossRef CAS PubMed
- D. van Berlo, M. Woutersen, A. Muller, M. Pronk, J. Vriend and B. Hakkert, Regul. Toxicol. Pharmacol., 2022, 134, 105235 CrossRef CAS PubMed
- S. Campion, J. Aubrecht, K. Boekelheide, D. W. Brewster, V. S. Vaidya, L. Anderson, D. Burt, E. Dere, K. Hwang, S. Pacheco, J. Saikumar, S. Schomaker, M. Sigman and F. Goodsaid, Expert Opin. Drug Metab. Toxicol., 2013, 9, 1391 CrossRef CAS PubMed
- L. Wang, Z. Yang, F. Lu, J. Liu, Y. Song and D. Li, Molecules, 2014, 19, 12676 CrossRef PubMed
-
P. Greaves, Introduction, in Histopathology of Preclinical Toxicity Studies, ed. P. Greaves, Academic Press, Boston, 4th edn, 2012, pp. 1–10 Search PubMed
- B. Kenkhuis, A. Somarakis, L. R. T. Kleindouwel, W. M. C. van Roon-Mom, T. Höllt and L. van der Weerd, Neurobiol. Dis., 2022, 167, 105684 CrossRef CAS PubMed
- H. S. Kwon and S.-H. Koh, Transl. Neurodegener., 2020, 9, 42 CrossRef PubMed
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066 CrossRef CAS PubMed
- L. Fu, S. Shi, J. Yi, N. Wang, Y. He, Z. Wu, J. Peng, Y. Deng, W. Wang, C. Wu, A. Lyu, X. Zeng, W. Zhao, T. Hou and D. Cao, Nucleic Acids Res., 2024, 52, W422 CrossRef PubMed
- Y. Myung, A. G. C. de Sá and D. B. Ascher, Nucleic Acids Res., 2024, 52, W469 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis, characterisation and supplementary results. See DOI: https://doi.org/10.1039/d4sc08585j |
| This journal is © The Royal Society of Chemistry 2025 |