
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtmosphere-directed reconstruction of Cu-based metal–organic frameworks toward efficient CO2 electroreduction†
Jiye
Feng‡
,
Danni
Shi‡
,
Fei
Wang
,
Yiming
Zou
,
Weicheng
Li
,
Wenbiao
Zhang
,
Huaijun
Lin
 ,
Yuying
Meng
and
Qingsheng
Gao
*
,
Yuying
Meng
and
Qingsheng
Gao
*
College of Chemistry and Materials Science, Jinan University, Guangzhou 510632, P. R. China. E-mail: tqsgao@jnu.edu.cn
First published on 2nd June 2025
Abstract
The electrochemical reconstruction of metal–organic frameworks (MOFs) offers a promising approach for in situ fabrication of high-performance electrocatalysts. However, this innovation is often hindered by unpredictable structural transformations due to the complex thermodynamic and kinetic interplay of such multiple electrochemical and chemical processes. Herein, the reaction-atmosphere (Ar or CO2) guided reconstruction of Cu-based MOFs to Cu nanoparticles with mixed-valence surfaces/interfaces was investigated for the first time to unravel the kinetic contribution made by intermediate chemisorption. As shown, Cu-1,3,5-benzenetricarboxylate (HKUST-1) with frangible Cu–O4 nodes undergoes thermodynamically favored reduction quickly upon applying cathodic potentials, followed by varied surface changes kinetically governed by the intermediates of the hydrogen evolution reaction or CO2 reduction reaction (HER or CO2RR). Under an Ar atmosphere, the predominant HER increases the [OH−] in the microenvironment near the cathode and thereby boosts the re-oxidation of in situ formed Cu toward Cu/Cu2O interfaces. Conversely, the CO2RR facilitates the strong adsorption of *CO on Cu surfaces, effectively preserving Cu(0) species. Thanks to the rich Cu/Cu2O interfaces with a lowered energy barrier for *CO–*CO coupling during the subsequent CO2RR test, the electrocatalysts restructured under Ar afford the obviously improved CO2-to-C2H4 conversion as compared with their counterparts restructured under CO2. Such an atmosphere-controlled reconstruction strategy is further validated using CuBDC (BDC = 1,4-benzenedicarboxylate) with labile Cu–O4 nodes, while CuPz2 (Pz = pyrazole), with robust Cu–N4 coordination, remains stable, highlighting the framework-dependent nature. These findings establish atmosphere-controlled reconstruction of metastable MOFs as a powerful tool for rational electrocatalyst design.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) powered by renewable electricity is a promising route toward carbon neutrality.1–3 To overcome the high energy barrier of CO2 activation and rationalize the reaction pathways toward target products, precise control is highly demanded over the microstructure of electrocatalysts.4–6 Thanks to the atomic-level periodicity of metal nodes and the resulting accurate control over active sites, metal–organic frameworks (MOFs) have recently garnered significant attention for CO2RR; they also feature other merits, such as high surface area, rich nanoporosity, and tunable coordination environments that can promote CO2 adsorption and activation.7–9 They reconcile the precision of homogeneous catalysis with the practicality of heterogeneous systems, enabling new opportunities beyond traditional copper catalysts.10,11 In particular, Cu-based MOFs have demonstrated potential for selectively converting CO2 to value-added C2+ products, attributable to their optimum adsorption energy and facilitated coupling of *CO intermediates on coordination-controlled Cu sites.12,13 While the issue of low intrinsic conductivity in pristine MOFs can be addressed by designing conjugated organic linkers or incorporating conductive materials,14 the persistent challenge of framework instability continues to hinder their practical implementation.15 Under the harsh conditions of CO2RR, Cu-based MOFs containing frangible nodes (such as Cu–O4) undergo in situ reconstruction due to the extensively disrupted coordination along with electrochemical and chemical changes of copper ions.11,16,17 Such reconstruction during CO2RR undoubtedly poses a great challenge not only to long-term durability,18,19 but also to the atomic-level understanding of the electrocatalytic mechanism.20,21Very recently, the electrochemical reconstruction of MOFs was introduced to produce active and selective catalysts on site, avoiding the time and energy-consuming preparation of catalysts and working electrodes.22–25 The in situ restructured electrocatalysts inherit the structural merits of MOFs, and more importantly generate accommodative surfaces with highly active and robust sites under controlled electrochemical conditions thanks to quasi-molecular-imprinting effects.21 Efforts have been devoted to boosting the electroreduction of CO2 to C2+ products by controlling the reconstruction of Cu-based MOFs.22,26–28 For instance, Wen et al. employed S doping to direct the in situ reconstruction of Cu-1,3,5-benzenetricarboxylate (HKUST-1) toward Cu/CuxSy interfaces, resulting in efficient ethylene production.22 Peng's group fabricated copper/polyamine hybrid composites by impregnating HKUST-1 with polyamines and then performing in situ electrochemical reconstruction, achieving favorable C2H4 production on polyaniline/HKUST-1 and CH4 production on polypyrrole/HKUST-1.27 However, these studies primarily focused on precursor modification, overlooking the thermodynamic and kinetic effects of reconstruction conditions (such as electrolytes and atmospheres), thereby limiting the general applicability of these strategies. It is noteworthy that HER invariably accompanies CO2RR in aqueous electrolytes, where their interplay critically influences the dynamic evolution of the Cu surface, although the underlying mechanisms remain elusive. Buonsanti and co-workers pointed out that the adsorption of either *H or *CO (* denotes an active-site) eventually alters the thermodynamic order of Cu facets, leading to potential-dependent nanoclustering of Cu electrocatalysts.29 By contrast, Lee et al. proved that continuous reconstruction from polycrystalline Cu to Cu(100) took place only in the presence of CO2, suggesting an aggravated reconstruction by the CO intermediate of CO2RR.30 Therefore, unravelling the directional functionalities of HER and CO2RR is necessary to consolidate the thermodynamic and kinetic fundamentals of electrochemical Cu-MOFs reconstruction,31,32 which is however a research gap to the best of our knowledge.
To decouple the above effects, the reaction-atmosphere (Ar or CO2) guided reconstruction of Cu-based MOFs was investigated herein for the first time. A series of characterization studies, including ex situ X-ray diffraction (XRD) and in situ Raman, showed that HKUST-1 consisting of frangible Cu–O4 nodes underwent thermodynamically favored reduction at cathodic potentials, followed by surface reconfigurations kinetically governed by HER or CO2RR intermediates (Scheme 1). Under an Ar flow, the predominant HER led to excessive OH− in the microenvironment of the working cathode, thereby facilitating the easy transformation to Cu–OH or Cu–Oad.33 Such enhanced re-oxidation of the in situ formed Cu surface ultimately produced rich Cu/Cu2O interfaces, which benefited the CO2-to-C2H4 conversion in the subsequent CO2RR thanks to the lowered energy barrier for *CO–*CO coupling. In sharp contrast, a CO2 atmosphere resulted in the strong adsorption of in situ formed CO from CO2RR on the metallic Cu surface, serving as protection to maintain Cu(0) species that however favors CO generation rather than coupling.34 Furthermore, similar reconstruction in a controlled manner and the correspondingly altered CO2RR were identified on another Cu-MOF with fragile Cu–O4 nodes, i.e., CuBDC (BDC = 1,4-benzenedicarboxylate), but negligible for CuPz2 (Pz = pyrazole) with much stronger Cu–N4. This revealed the coordination-dependent dynamic evolution of MOFs, which can be further modulated to design efficient catalysts simply by varying the atmospheric conditions.
 | ||
| Scheme 1 Schematic illustration of the atmosphere-guided directional reconstruction of HKUST-1. | ||
Experimental section
Chemicals
Potassium hydroxide (KOH, 85%), copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, >99.0%), terephthalic acid (H2BDC, 99%), pyrazole (Pz, 99%), polyvinylpyrrolidone (PVP, M. W. 58000), ammonia solution (NH4OH, 28%) and N,N-dimethylformamide (DMF, 99.8%) were provided by Macklin Co., Ltd (Shanghai, China). 1,3,5-Benzenetricarboxylic acid (H3BTC, 98%) was purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). Nafion (5 wt%) solution was purchased from Sigma-Aldrich. Ethanol (99.7%) was bought from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All aqueous solutions were prepared using ultrapure water (>18 MΩ cm).Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vethanol = 1:1). The two solutions were subsequently combined under continuous stirring for 15 minutes to ensure homogeneity. The mixture was transferred into a 50 mL Teflon-lined stainless-steel autoclave and heated at 100 °C for 10 hours. After the system cooled to room temperature, the obtained product was washed with ethanol three times and finally dried overnight at 60 °C under vacuum.
:Vethanol = 1:1). The two solutions were subsequently combined under continuous stirring for 15 minutes to ensure homogeneity. The mixture was transferred into a 50 mL Teflon-lined stainless-steel autoclave and heated at 100 °C for 10 hours. After the system cooled to room temperature, the obtained product was washed with ethanol three times and finally dried overnight at 60 °C under vacuum.
Physical characterization
Microstructural characterization, including transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDS), and corresponding elemental mapping analysis, was conducted using a JEOL 2100F. Surface morphology examination was performed on a ZEISS ULTRA55 scanning electron microscope. Crystalline structure analysis was performed through XRD measurements using a Bruker D8 diffractometer equipped with Cu Kα radiation (λ = 1.54056 Å). Surface chemical analysis was conducted using X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) on a Thermo Scientific Escalab 250Xi system. Molecular vibrational characteristics were investigated through Fourier transform infrared (FT-IR) spectroscopy (4000–400 cm−1) using a PerkinElmer spectrometer. Raman spectroscopic measurements were performed with a Horiba HR-800 laser confocal microspectrometer employing a 532 nm excitation source.Preparation of working electrodes
The catalyst ink was prepared by dispersing 5 mg of electrocatalyst and 50 μL of Nafion into 450 μL of ethanol. The mixture was ultrasonicated for 30 minutes to ensure homogeneous dispersion. Subsequently, 100 μL of the ink was pipetted onto a carbon paper electrode (dimensions: 0.5 cm × 2 cm), achieving a catalyst loading of 1 mg cm−2. The coated electrode was then dried at room temperature before being used for subsequent electrochemical tests.Electrochemical test
The electrochemical CO2 reduction reaction was performed in a flow cell at room temperature. All electrochemical experiments were conducted in a three-electrode setup utilizing a CHI660E potentiostat. The prepared gas diffusion electrode (GDE) served as the working electrode, while a platinum plate and a solid Ag/AgCl electrode were employed as the counter electrode and reference electrode, respectively. The electrolyte (1.0 M KOH) was circulated at a flow rate of 15 mL min−1 on both the cathode and anode sides using a peristaltic pump. An anion exchange membrane was used to separate the anode and cathode chambers. CO2 gas was fed into the cathode chamber at a flow rate of 20 mL min−1, regulated by a mass flow controller. All potentials relative to the reference electrode were converted to the reversible hydrogen electrode (RHE) scale based on the following equation:| E(vs. RHE) = E(vs. Ag/AgCl) + 0.059 × pH + 0.197 V |
Product analysis
An online gas chromatograph (GC, FULI-9790 II) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD) was used to analyze the gas products. High-purity N2 (99.999%) served as the carrier gas. Based on the peak areas obtained from the GC, the partial current densities (j) and the faradaic efficiency (FE) of gas products were calculated using the following equations:
where x represents one of the products, n denotes the number of electrons transferred for each product, α is the conversion factor for CO, CH4, C2H4 and H2 (based on the calibration of standard samples), F represents the Faraday constant (F = 96
 485C mol−1), and Vm is the molar volume of an ideal gas under standard conditions (Vm = 22.4 L mol−1).
485C mol−1), and Vm is the molar volume of an ideal gas under standard conditions (Vm = 22.4 L mol−1).
The liquid products were analyzed using 1H nuclear magnetic resonance (NMR) spectroscopy. A 100 ppm DMSO solution was employed as the internal standard. For analysis, 800 μL of the electrolyte was mixed with 100 μL of D2O and 100 μL of the internal standard, and the product concentrations were calculated based on a standard calibration curve.
Theoretical calculations
The density functional theory (DFT) calculations were carried out using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional, as implemented in the CASTEP module of Materials Studio software. The total energy calculations were conducted with a plane-wave basis set, employing a kinetic energy cutoff of 450.0 eV to ensure accurate determination of the density of states. Computational parameters included a self-consistent field (SCF) convergence criterion of 1 × 10−6 eV and a 5 × 5 × 1 k-point grid for Brillouin zone integration. The core electrons were treated using ultrasoft pseudopotentials. Initially, a face-centered cubic (fcc) copper model was constructed, upon which the Cu–Cu2O system was further developed. Subsequently, a 3 × 3 relaxed rhombic Cu2O(001) bilayer was selected and positioned atop a relaxed 3 × 3 Cu(001) surface to construct the Cu/Cu2O heterojunction model. In this configuration, oxygen atoms were introduced to saturate the copper atoms on the Cu(001) surface, thereby achieving effective passivation of the surface atoms. Such a heterojunction structure constructed from Cu(001) and Cu2O(001) ensured the exposure of the Cu(100) and Cu2O(100) facets. To investigate the catalytic performance for CO2 reduction, five-atomic-layer slab models were constructed for both Cu(100) and Cu2O(100) crystallographic planes. A vacuum region of 15 Å between repeated slabs was applied to avoid interactions between repeated slabs along the z-direction.The binding energy (BE) of an adsorbate was calculated as:
| BE(adsorbate) = E(slab+adsorbate) − E(slab) − E(adsorbate) |
The Gibbs free energy (G) of a species was calculated as:
| G = E + ZPE − TS |
| ΔG = ΔE + ΔZPE − TΔS |
The transition state search was performed based on the complete LST/QST method, with an RMS convergence criterion of 0.25 eV Å−1, utilizing the optimized geometric configurations of both reactants and products as initial structures. CASTEP employed algorithms including the nudged elastic band (NEB) method and the dimer approach to explore the potential energy surface and determine the transition state configuration. The transition state configuration was accurately resolved between the reactant and product geometries by implementing a 10-step QST energy minimization protocol.
Results and discussion
XRD analysis showed that the diffraction pattern of the as-prepared HKUST-1 was consistent with the simulated one (Fig. 1a), confirming the formation of a framework composed of dimeric Cu units connected by benzene-1,3,5-tricarboxylate (BTC) linkers. Accordingly, FT-IR spectroscopy clearly identified the characteristic vasym(C–O2) and vsym(C–O2) bands of BTC at 1650–1590 cm−1 and 1450–1370 cm−1,16 respectively (Fig. S1, ESI†). SEM analysis further presented uniform octahedral crystals with smooth surfaces in the as-obtained samples (Fig. 1b). Subsequently, HKUST-1 was loaded onto carbon paper as a pre-catalyst for electrochemical reconstruction in 1.0 M KOH under Ar or CO2 atmospheres. Initially, the optimal potential required for such reconstruction was determined by comparing chronoamperometry tests (Fig. S2, ESI†). Under a CO2 flow, the output current at −1.3 V (vs. RHE) was obviously increased as compared with that at −1.2 V (vs. RHE), but comparable to the one at −1.4 V (vs. RHE). Thus, a working potential of −1.3 V (vs. RHE) was adopted for the following measurements. The morphological evolution of HKUST-1 was visually observed by using SEM (Fig. 1c and d). It was evident that HKUST-1 underwent reconstruction from well-defined octahedra to smaller nanoparticles in both Ar and CO2 atmospheres. However, the reconstruction rate was relatively slower under Ar. Despite this, the complete reconstruction into nanoparticles was achieved after 45 minutes. For clarity in discussion, the restructured samples after 1 h under Ar and CO2 atmospheres were denoted as HKUST-Ar and HKUST-CO2, respectively. Considering the inherent instability of the Cu–O4 coordination under alkaline conditions, potential-free reconstruction of HKUST-1 was further performed. Raman spectroscopy clearly identified the characteristic bands of Cu(OH)2 at 292 and 491 cm−1 in the resulting samples (Fig. S3a, ESI†), and SEM demonstrated the collapse of octahedra to nanowires (Fig. S3b, ESI†), rather than nanoparticles observed after electrochemical reconstruction. It's suggested that the framework undergoes collapse in alkaline solution, but the applied negative potentials play a critical role in reducing Cu2+ to Cu nanoparticles. | ||
| Fig. 1 (a) Experimental and simulated XRD patterns, and (b) SEM image of pristine HKUST-1. SEM images of HKUST-1 reconstructed under (c) Ar and (d) CO2 for 10, 30, 45 and 60 min. | ||
The reconfigured HKUST-CO2 and HKUST-Ar were characterized through a series of physical measurements to understand their structural and compositional differences. FT-IR analysis confirmed the collapse of the HKUST-1 framework following reconstruction, as evidenced by the disappearance of characteristic adsorption bands associated with the carboxylate ligand (Fig. S4, ESI†). As shown in the XRD patterns (Fig. 2a), the diffraction peaks of HKUST-1 disappeared, and new ones corresponding to Cu(111) and Cu(200) emerged at 2θ = 43.3° and 50.4°, respectively, confirming the complete transformation of the Cu–O4 framework into dominant metallic Cu. This could be ascribed to the thermodynamically favored reduction at the highly negative potential of −1.3 V (vs. RHE) compared to the Cu(OH)2/Cu redox potential of 0.61 V (vs. RHE). Different from HKUST-CO2, HKUST-Ar presented two additional peaks associated with Cu2O(111) and Cu2O(200), indicating the presence of Cu/Cu2O interfaces. XPS and AES were further utilized to investigate the chemical composition and elemental valence states. In the Cu 2p XPS profile (Fig. 2b), the absence of Cu(II) peaks and the presence of a peak related to Cu(I)/Cu(0) at 932.4 eV indicated the reduction of Cu(II) in HKUST-1 frameworks during electrolysis in Ar or CO2 atmospheres.22 In the AES profiles, HKUST-Ar displayed peaks at 914.3 and 917.3 eV, corresponding to Cu(I) and Cu(0),35 respectively, whereas HKUST-CO2 exhibited only the Cu(0) signal (Fig. 2c), consistent with the XRD results. High-resolution TEM (HR-TEM) images provided further insights into the structural configurations. HKUST-CO2 exclusively exhibited lattice fringes corresponding to Cu(111) (Fig. 2d), whereas HKUST-Ar distinctly revealed Cu/Cu2O interfaces, with clearly resolved lattice spacings of 0.204 nm and 0.245 nm assigned to Cu(111) and Cu2O(111), respectively (Fig. 2e). These comprehensive analyses confirm that under an Ar atmosphere, the Cu/Cu2O interface is restructured, while only metallic Cu is achieved in the presence of CO2.
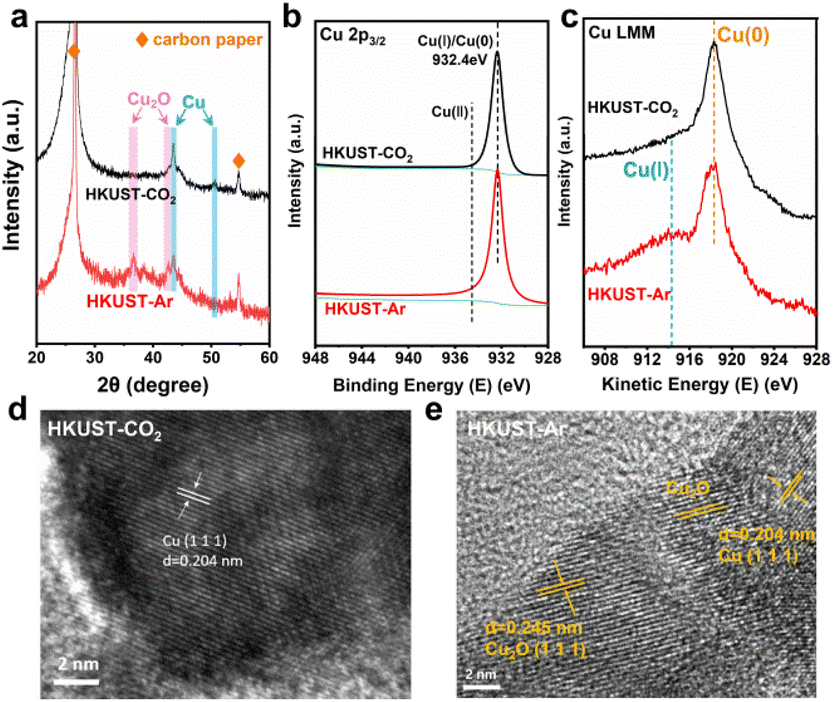 | ||
| Fig. 2 (a) XRD patterns, (b) high-resolution Cu 2p3/2 XPS profiles, (c) Cu LMM AES profiles and (d and e) HR-TEM images of the restructured HKUST-CO2 and HKUST-Ar. | ||
Further attention was given to how the valence states of the restructured samples varied under different atmospheric conditions. At first, the changes in phase structures during reconstruction under CO2 or Ar atmospheres were analyzed by using ex situ XRD (Fig. 3a and b). Only the diffraction peaks of Cu appeared in the presence of CO2 over time. By contrast, the peaks of Cu2O gradually increased alongside a decrease in Cu peaks under Ar, suggesting the re-oxidation of the in situ formed metallic Cu. This evolution was then further examined by in situ Raman spectroscopy (Fig. 3c and d). At a working potential of −1.3 V vs. RHE, the absorption bands corresponding to Cu(II)–O and C–H disappeared rapidly, indicating the collapse of the metal–organic framework.36 Notably, Cu–CO peaks appeared under a CO2 atmosphere (Fig. 3c), attributed to the strong adsorption of *CO on the in situ formed Cu(0) surface.37 In sharp comparison, new bands at 520, 700, and 600 cm−1 emerged within only 5 minutes under Ar (Fig. 3d), corresponding to the characteristic vibrations of Cu2O, Cu–OH, and Cu–Oad, respectively,38,39 which gradually increased in intensity during electrolysis. The adsorbed Cu–Oad species are re-oxidized in the electrolyte, generating CuOx species,40 and thus the signals of Cu2O can be further observed at different reduction potentials by XRD (Fig. S5, ESI†) and Raman (Fig. S6, ESI†). This indicated that the re-oxidation process can prevent the reduction of Cu2O at highly negative potentials.33,41 Moreover, the restructured electrodes, HKUST-Ar and HKUST-CO2, were subjected for cyclic voltammetry (CV) analysis to identify the adsorption of *CO and *OH.42 After reconstruction under Ar for 1 h, the CV profile of HKUST-Ar exhibited visible adsorption/desorption peaks of OH− in 1.0 M KOH (Fig. 3e), which were subsequently weakened when the carrier gas was switched to CO, along with the emergence of a Cu–CO couple. As indicated, the restructured Cu preferentially adsorbs *CO due to their strong interactions. Concertedly, only the Cu–CO peaks could be observed on HKUST-CO2 in 1.0 M KOH initially (Fig. 3f), associated with the predominant *CO adsorption on Cu during HKUST-1 reconstruction. These peaks declined after switching the gas to Ar followed by 30 cycles of CV, and those of Cu–OH reappeared again.
 | ||
| Fig. 3 (a and b) Ex situ XRD patterns and (c and d) in situ Raman spectra of HKUST-1 collected at different times during electrochemical reconstruction under (a and c) CO2 and (b and d) Ar flows. CVs of (e) HKUST-Ar in 1.0 M KOH before and after injecting CO, and (f) HKUST-CO2 in 1.0 M KOH and after 30 CV scans under an Ar flow. | ||
There was still a doubt that whether variation in local [OH−] near electrodes would influence the valence states of the restructured surface. At a fixed potential of −1.3 V (vs. RHE) adopted for electrochemical reconstruction, the j under a CO2 atmosphere was approximately −200 mA cm−2 (Fig. S7, ESI†), lower than that observed under Ar (−300 mA cm−2). Given the synchronous proton consumption along with electron transfer in both the HER and CO2RR,20 we can infer that the local [OH−] formed in situ varies at different j values. To this end, the HKUST-1 pre-catalyst was restructured in a CO2 atmosphere at j = −200 and −300 mA cm−2, but the received HKUST-CO2 samples showed only the diffraction peaks of Cu in XRD (Fig. S8a, ESI†). Analogously, Cu2O was found in HKUST-Ar under an Ar atmosphere, regardless of the j value (−200 or −300 mA cm−2) used during reconstruction (Fig. S8b, ESI†). These results rule out the influence of local [OH−] variations caused by different rates of HER and CO2RR.
According to the above results, it's reasonable to propose that Cu(II) in HKUST-1 pre-catalysts undergoes rapid reduction toward metallic Cu(0), which is then re-oxidized to Cu–OH, Cu–Oad and even Cu2O under an Ar atmosphere because the predominant HER produces excess OH− at the local surface of the electrode and boosts the thermodynamic of re-oxidation according to the Nerst equation.33,43 By contrast, Cu(0) can be stabilized by the *CO intermediate of the CO2RR in a CO2 flow, preventing re-oxidation even in a microenvironment rich in OH−.
The varied valence states of the restructured electrocatalysts would lead to differences in CO2RR performance.44 In particular, mixed-valent Cu species, e.g., Cu(I)–Cu(0) ensembles, have demonstrated high efficiency for C2+ production.45,46 We afterward evaluated the CO2RR performance of HKUST-Ar and HKUST-CO2 in a flow cell with 1.0 M KOH as the electrolyte (Fig. S9, ESI†). The gaseous and liquid products were analyzed by GC (Fig. S10, ESI†) and 1H NMR (Fig. S11, ESI†), respectively. The FE of liquid products was less than 10% based on calculations derived from the standard curves (Fig. S12, ESI and Table S1, ESI†), so we only discussed gaseous products here, and focused on ethylene. Linear sweep voltammetry (LSV) curves revealed that the current density for HKUST-Ar was higher than that for HKUST-CO2 (Fig. 4a), indicating the enhanced activity of HKUST-Ar. Potentiostatic tests were conducted at sequentially decreasing potential from −1.1 to −1.5 V (vs. RHE), and the corresponding product FEs and current densities are depicted in Fig. 4b and c. As shown, the FE for C2H4 production through HKUST-Ar was substantially higher than that with HKUST-CO2. Notably, HKUST-Ar achieved a maximum FE of C2H4 of 47.8% at −1.2 V vs. RHE, compared to only 13.4% on HKUST-CO2 (Fig. S13, ESI†).
 | ||
| Fig. 4 (a) LSV curves and (b and c) product FEs of HKUST-Ar and HKUST-CO2 in 1.0 M KOH at different applied potentials. Chronoamperometric stability tests of (d) HKUST-Ar and (e) HKUST-CO2. (f) Energy profiles of *CO–*CO coupling on Cu and Cu/Cu2O models. | ||
The electrochemically active surface areas of the catalysts were measured by deriving the double-layer capacitance (Cdl) through cyclic voltammetry (Fig. S14, ESI†). The results indicated that HKUST-Ar exhibited a Cdl value of 3.26 mF cm−2, surpassing the 2.04 mF cm−2 observed for HKUST-CO2, which suggested a higher number of active sites in HKUST-Ar. Furthermore, electrochemical impedance spectroscopy (EIS) was used to analyze electron transport dynamics during the CO2RR (Fig. S15, ESI†). The Nyquist plot revealed that HKUST-Ar has a smaller radius, indicative of lower electron transfer resistance (Rct). Meanwhile, HKUST-Ar exhibited a significantly lower Tafel slope compared to HKUST-CO2, indicating faster kinetics and better catalytic performance for CO2 electroreduction (Fig. S16, ESI†). This evidence suggests that in situ electrochemical reconstruction under Ar gas can effectively enhance electron transfer rates and accelerate reaction kinetics, thereby improving catalytic activity for CO2RR.
The long-term stability was evaluated by conducting a chronoamperometric test. The HKUST-Ar sample presented a significantly higher C2H4 FE of approximately 45% (Fig. 4d), which remained above 40% over 300 minutes of reaction. By contrast, the main products on HKUST-CO2 were H2 and CO with the FEs of ∼40% and ∼44%, respectively, and the FE of C2H4 was below 17% (Fig. 4e). Furthermore, we characterized the two catalysts after the long-term CO2RR test. There was no obvious change in the phase or valence on HKUST-CO2 (Fig. S17, ESI†). In contrast, the Cu2O(111) and (200) peaks of HKUST-Ar gradually weakened (Fig. S18a, ESI†), and the AES results confirmed a decrease in the Cu(I) signal (Fig. S18b, ESI†). Meanwhile, TEM images of both materials revealed only the presence of Cu(111), indicating the stable existence of Cu after prolonged reaction (Fig. S19, ESI†). By employing pulse electrolysis methods to stabilize the Cu(I) species, the long-term durability will be further improved.47
According to the abovementioned structural characterization, HKUST-CO2 predominantly consisted of metallic Cu, while HKUST-Ar featured multi-phase Cu/Cu2O interfaces. To gain more insights into the enhancement of C2H4 production, density functional theory (DFT) calculations were performed on the Cu and Cu/Cu2O structural models (Fig. S20, ESI†), which served as the representatives of HKUST-CO2 and HKUST-Ar, respectively. As a critical step toward C2+ products, the coupling of two *CO intermediates was brought into focus. The corresponding reaction models are illustrated in Fig. S21, ESI,† with computational data summarized in Table S2, ESI.† As shown in Fig. 4f, the kinetic barrier (0.36 eV) and free-energy change (−0.14 eV) for *CO coupling on Cu/Cu2O are lower than those on Cu (0.72 and 0.16 eV), suggesting that the Cu(0)–Cu(I) interfaces can effectively boost C2 production.48,49 This conclusion is consistent with previous studies.20,50 Additionally, the CO adsorption energies on both Cu and Cu/Cu2O were quite similar (Fig. S22, ESI†), further suggesting that the key contribution lies in reducing the *CO–*CO coupling energy barrier.
To demonstrate the universality of the proposed strategy, we substituted Ar gas with He for the reconstruction. The XRD analysis revealed the generation of Cu2O (Fig. S23, ESI†), and the SEM image showed a similar morphology to that of HKUST-Ar (Fig. S24, ESI†). The resulting CO2RR performance was essentially consistent (Fig. S25, ESI†). This finding suggests that the strategy is applicable across different inert gases. Subsequently, we expanded our study to include other Cu-based MOFs, first focusing on CuBDC (BDC = 1,4-benzenedicarboxylate), with a similar Cu–O4 coordination but a layered crystalline structure. The XRD pattern of the obtained CuBDC closely matched the simulated structure, and SEM showed that CuBDC exhibited a flake-like morphology (Fig. 5a). Then CuBDC was subjected to reconstruction under Ar or CO2 for 1 hour, followed by XRD analysis. The results mirrored those of HKUST-1, with Cu2O diffraction peaks observed under Ar gas (Fig. 5b). Furthermore, CuBDC-Ar significantly outperformed CuBDC-CO2 for the CO2RR to C2H4 production (Fig. 5d), indicative of the universality for Cu-MOFs with Cu–O4 coordination.
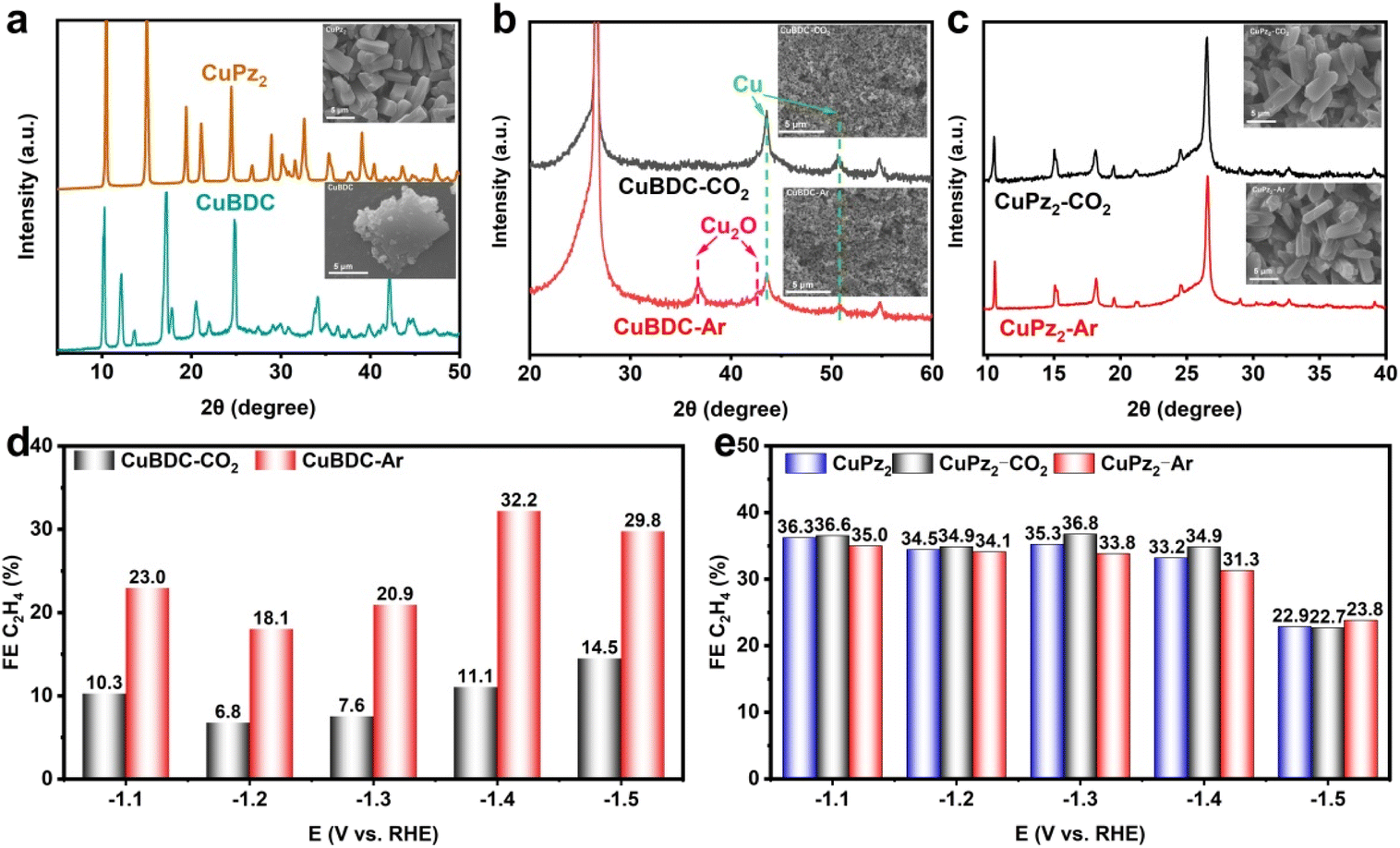 | ||
| Fig. 5 XRD patterns and SEM images of (a) CuBDC and CuPz2, and their products after electrochemical reconstruction under (b) CO2 (i.e., CuBDC-CO2 and CuPz2-CO2) and (c) Ar (i.e., CuBDC-Ar and CuPz2-Ar) atmospheres. FE of C2H4 for the CO2RR on (d) CuBDC-CO2 and CuBDC-Ar, and (e) CuPz2, CuPz2-CO2 and CuPz2-Ar in 1.0 M KOH at different applied potentials. | ||
Next, we investigated CuPz2 (Pz = pyrazole), which possesses a Cu–N4 structure. The XRD results demonstrated that the MOF structure remained intact, and SEM observations corroborated this by showing an unchanged morphology (Fig. 5c). The applied potential was incrementally elevated to −1.5 V and −2.0 V (vs. RHE), with XRD analysis revealing no substantial alterations in the characteristic diffraction peaks corresponding to CuPz2 (Fig. S26, ESI†). This stability is likely due to the strong coordination of Cu–N4, making CuPz2 resistant to reconstruction.51 Subsequent performance testing revealed no significant differences compared to the measurement without reconstruction (Fig. 5e), further substantiating that our strategy is effective only in facilitating the reconstruction process for specific structural configurations and is not applicable to MOFs with inherently stable structures. The frangible Cu–O4 nodes are a prerequisite for reconstruction, while the strong Cu–N4 coordination requires harsher conditions with corrosive ions (e.g., halogen) to drive reconfiguration.51,52
Conclusions
This study investigated the atmosphere-guided reconstruction of HKUST-1, focusing on the key influence of electrocatalysis intermediates and their critical roles in designing reactive surfaces to enhance CO2RR performance. The HKUST-1 pre-catalyst, featuring frangible Cu–O4 nodes, underwent rapid reduction under cathodic potential, leading to atmosphere-dependent restructuring of surfaces and interfaces. Under an Ar atmosphere, the dominant HER process generated excessive OH− in the cathodic microenvironment, promoting the formation of Cu/Cu2O interfaces in HKUST-Ar through the re-oxidation of Cu(0). In contrast, under a CO2 atmosphere, the strongly adsorbed *CO intermediate from CO2RR inhibited Cu(0) re-oxidation, resulting in a restructured HKUST-CO2 surface dominated by metallic Cu(0) sites. Thanks to the rich interfaces of Cu/Cu2O that facilitate *CO coupling, finally, HKUST-Ar achieved improved C2H4 production during CO2RR compared to HKUST-CO2. This highlights the contribution of controlled dynamic reconstruction in optimizing catalytic performance. Further validation using inert gases (e.g., He) and other Cu-MOFs (e.g., CuBDC) demonstrated the general applicability of this strategy. This work presents a novel approach to regulating the reconstruction of Cu-based MOFs, offering a pathway to design highly efficient catalysts for CO2RR and beyond.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Jiye Feng: synthesis, investigation, formal analysis, writing of the original draft. Danni Shi: experimental analysis, image polishing. Fei Wang: proofreading of the original draft. Yiming Zou: data curation. Weicheng Li: experimental analysis. Wenbiao Zhang: DFT calculation, supervision. Huaijun Lin: conceptualization, supervision, funding acquisition. Yuying Meng: conceptualization. Qingsheng Gao: conceptualization, supervision, writing – review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22175077), Innovation Team Project in Guangdong Colleges and Universities (2021KCXTD009), Guangdong Basic and Applied Basic Research Foundation (2023A1515240081), National Natural Science Foundation of Guangdong Province (2024A1515010427) and Fundamental Research Funds for the Central Universities (No. 21623103 and 21624412).Notes and references
- S. Nitopi, E. Bertheussen and S. B. Scott,
et al.
, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
.
- C. P. O'Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124, 3648–3693 CrossRef PubMed
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed
- X. She, Y. Wang, H. Xu, S. Chi Edman Tsang and S. Ping Lau, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed
- J. Han, X. Bai, X. Xu, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC
- Y.-F. Tang, L.-B. Liu, M. Yu, S. Liu, P.-F. Sui, W. Sun, X.-Z. Fu, J.-L. Luo and S. Liu, Chem. Soc. Rev., 2024, 53, 9344–9377 RSC
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC
- M. Yang, N.-N. Zhu, Y.-F. Huang, J.-M. Xiao, Y. Fang, Z.-J. Yi, D.-S. Bin, L. Liu and D. Li, Sci. China Mater., 2025, 68, 724–743 CrossRef CAS
- A. H. Mamaghani, J. Liu, Z. Zhang, R. Gao, Y. Wu, H. Li, M. Feng and Z. Chen, Adv. Energy Mater., 2024, 14, 2402278 CrossRef CAS
- N. Sun, S. S. A. Shah, Z. Lin, Y.-Z. Zheng, L. Jiao and H.-L. Jiang, Chem. Rev., 2025, 125, 2703–2792 CrossRef CAS PubMed
- F. Yang, D. Zhu, C. Xia, Z. Shahid, S. Chen and B. Y. Xia, Coord. Chem. Rev., 2024, 517, 216021 CrossRef CAS
- Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS PubMed
- L. S. Xie, G. Skorupskii and M. Dinca, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed
- Y. Peng, S. Sanati, A. Morsali and H. García, Angew. Chem., Int. Ed., 2023, 62, e202214707 CrossRef CAS PubMed
- J. Feng, W. Zhang, D. Shi, Y. Jia, Y. Tang, Y. Meng and Q. Gao, Chem. Sci., 2024, 15, 9173–9182 RSC
- J. Feng, X. Wang and H. Pan, Adv. Mater., 2024, 36, 2411688 CrossRef CAS PubMed
- X. Tan, H. Zhu, C. He, Z. Zhuang, K. Sun, C. Zhang and C. Chen, Chem. Sci., 2024, 15, 4292–4312 RSC
- L. Xie, Y. Jiang, W. Zhu, S. Ding, Y. Zhou and J.-J. Zhu, Chem. Sci., 2023, 14, 13629–13660 RSC
- W. Zhang, Y. Yang, Y. Tang and Q. Gao, J. Energy Chem., 2022, 70, 414–436 CrossRef CAS
- W. Zhang, C. Huang and J. Zhu,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202112116 CrossRef CAS PubMed
- C. F. Wen, M. Zhou and P. F. Liu,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202111700 CrossRef CAS PubMed
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS PubMed
- Z. Han, D. Han and Z. Chen,
et al.
, Nat. Commun., 2022, 13, 3158 CrossRef CAS PubMed
- K. Yue, R. Lu and M. Gao,
et al.
, Science, 2025, 388, 430–436 CrossRef CAS PubMed
- C. F. Wen, S. Yang, J. J. He, Q. Niu, P. F. Liu and H. G. Yang, Small, 2024, 20, 2405051 CrossRef CAS PubMed
- J. Cheng, L. Chen and X. Xie,
et al.
, Angew. Chem., Int. Ed., 2023, 62, e202312113 CrossRef CAS PubMed
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 RSC
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed
- S. H. Lee, J. C. Lin and M. Farmand,
et al.
, J. Am. Chem. Soc., 2020, 143, 588–592 CrossRef PubMed
- W. Dai, K. Wan and K. Pang,
et al.
, Chem, 2025, 11, 102345 CAS
- X. Ma, T. Yang and D. He,
et al.
, Nat. Synth., 2025, 4, 53–66 CrossRef CAS
- C.-J. Chang, S.-C. Lin, H.-C. Chen, J. Wang, K. J. Zheng, Y. Zhu and H. M. Chen, J. Am. Chem. Soc., 2020, 142, 12119–12132 CrossRef CAS PubMed
- T.-C. Chou, C.-C. Chang and H.-L. Yu,
et al.
, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed
- Y. Guo, C. Feng and S. Wang,
et al.
, J. Mater. Chem. A, 2020, 8, 24477–24485 RSC
- H. Sun, X. Han, K. Liu, B. Shen, J. Liu, D. Wu and X. Shi, Ind. Eng. Chem. Res., 2017, 56, 9541–9550 CrossRef CAS
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. r. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS
- X. Chang, M. He, Q. Lu and B. Xu, Sci. China:Chem., 2023, 66, 96–106 CrossRef CAS
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS PubMed
- Y. Wu, J. Feng, D. Shi, W. Zhang, Y. Tang and Q. Gao, Chem. Commun., 2023, 59, 10428–10431 RSC
- C.-J. Chang, S.-F. Hung, C.-S. Hsu, H.-C. Chen, S.-C. Lin, Y.-F. Liao and H. M. Chen, ACS Cent. Sci., 2019, 5, 1998–2009 CrossRef CAS PubMed
- X. He, L. Lin and X. Li,
et al.
, Nat. Commun., 2024, 15, 9923 CrossRef CAS PubMed
- J. Wang, J. Zhang and C. Chen, Chin. J. Catal., 2025, 68, 83–102 CrossRef
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed
- X.-D. Zhang, T. Liu and C. Liu,
et al.
, J. Am. Chem. Soc., 2023, 145, 2195–2206 CrossRef CAS PubMed
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef
- Y. Zhou, Y. Yao and R. Zhao,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202205832 CrossRef CAS PubMed
- G. Xie, W. Guo and Z. Fang,
et al.
, Angew. Chem., Int. Ed., 2024, 136, e202412568 CrossRef
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and data for CO2RR performance details, and additional Raman, XRD, IR, SEM, CV, and NMR spectra. See DOI: https://doi.org/10.1039/d5sc02601f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |