
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights for controlling plutonium behavior in hydrochloric acid solutions†
Yufei
Wang‡
 ,
Natalie T.
Rice‡
,
Julia G.
Knapp‡
,
Sara L.
Adelman
,
Kelly E.
Aldrich
,
Brian T.
Arko
,
Manuel L.
Besmer
,
J. Connor
Gilhula
,
Christopher J.
Godt
,
Jan
Klouda
,
Stosh A.
Kozimor
*,
Brian N.
Long
,
Molly M.
MacInnes
,
Travis
Marshall-Roth
,
Alexandra L.
Nagelski
and
Ida D.
Piedmonte
,
Natalie T.
Rice‡
,
Julia G.
Knapp‡
,
Sara L.
Adelman
,
Kelly E.
Aldrich
,
Brian T.
Arko
,
Manuel L.
Besmer
,
J. Connor
Gilhula
,
Christopher J.
Godt
,
Jan
Klouda
,
Stosh A.
Kozimor
*,
Brian N.
Long
,
Molly M.
MacInnes
,
Travis
Marshall-Roth
,
Alexandra L.
Nagelski
and
Ida D.
Piedmonte
Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: stosh@lanl.gov
First published on 14th April 2025
Abstract
Advancing understanding of aqueous chemistry for plutonium is important because it impacts energy production, environmental management, and national security. Unfortunately, plutonium's aqueous chemistry remains poorly characterized. We addressed this problem by characterizing Pu(IV) redox and coordination chemistry in aqueous solutions as a function of hydrochloric acid concentration using X-ray absorption spectroscopy, ultraviolet-visible near-infrared spectroscopy, and electrochemistry. The impact of Pu–Cl vs. Pu–OH2O bonding was correlated with the stability of different plutonium oxidation states. We discovered that anionic Cl1− ligands stabilized electron-deficient Pu(IV) over Pu(III) and neutral H2O ligands stabilized Pu(III) over Pu(IV). These findings offer a way to control plutonium electron transfer chemistry and imply that selective stabilization of Pu(IV) or Pu(III) may be achieved through tuning the electron donating ability of the ligand. Overall, this work advances predictive capabilities for aqueous plutonium chemistry, particularly within nuclear application spaces.
Introduction
Controlling the aqueous chemistry of plutonium (Pu) is critical for solving technical problems that are important for energy production, environmental management, and national security. On the energy front, there are aqueous processing challenges facing the advancement of nuclear power for clean energy production.1,2 In terms of the environment, the aqueous chemistry of plutonium impacts safely storing nuclear waste,3 reprocessing of spent nuclear fuel,2,3 and remediating sites contaminated by historic activities at nuclear facilities.4 For national security, large-scale aqueous processing and small-scale aqueous radioanalytical separations of plutonium are important for two main reasons: firstly, to ensure the safety, security, and effectiveness of nuclear weapon stockpiles,5,6 and secondly to provide a technical basis for mounting calculated responses to domestic and international nuclear emergencies.7 These needs have inspired researchers to study aqueous plutonium chemistry and substantial advances have been made since the discovery of plutonium in the 1940s.8–13 Unfortunately, many fundamental aspects associated with plutonium chemistry in aqueous matrixes are still poorly defined. This lack of understanding makes plutonium's aqueous chemistry seem unruly, especially in comparison to transition metals,14–17 lanthanides,18–21 and other actinides (uranium22–27 and thorium26–28) that have been studied to a larger extent. Researchers, engineers, and technical staff working in the nuclear sector would be better suited to solve plutonium challenges if this knowledge gap was reduced, predictive capabilities were improved, and the ability to control plutonium reactivity in aqueous solutions was advanced.One aqueous matrix ubiquitously used in the nuclear industry is hydrochloric acid, HCl(aq). Surprisingly, many fundamental aspects about plutonium in this matrix are not well characterized. It is known that plutonium's coordination chemistry in HCl(aq) is defined by the plutonium oxidation state. However, plutonium can adopt four different oxidation states in aqueous media at the same time: Pu(III), Pu(IV), Pu(V) and Pu(VI).13,29 It is also unclear what chemical factors influence plutonium oxidation state stability within HCl(aq) solutions. These attributes engender plutonium with diverse reaction chemistry in HCl(aq) solutions that can be difficult to control and predict, e.g., disproportionation reactions, comproportionation reactions, hydrolysis, colloid formation, to name a few. As a result, there is dogma associated with HCl(aq) solutions that contain plutonium. When things go awry, many variables get blamed blindly (such as temperature, the concentration of plutonium in solution, radiolysis, photolysis, etc.). Better correlating chemical attributes with plutonium electron transfer chemistry would provide clarity and enable avoidance of deleterious side reactions accessible to plutonium in HCl(aq) solutions.
Motivated by the need to reduce the aforementioned knowledge gap, we set out to better define how Pu(IV) coordination chemistry and electron transfer reactivity varied in HCl(aq) solutions as a function of one important chemical factor, ionic strength [H1+(aq) and Cl1−(aq) concentrations]. The complexity of this system was outlined in Scheme 1. The Pu4+(aq) cation can undergo reduction to generate Pu3+(aq). Oxidation and Pu–O bond formation reactions can also occur to generate PuO2n+(aq) (n = 1, 2). In HCl(aq) solutions, plutonium can undergo complexation by neutral H2O and anionic Cl1− ligands. There are also numerous coordination numbers and geometries accessible to Pu4+(aq), Pu3+(aq), and PuO2n+(aq) in HCl(aq).30,31 To provide insight on this topic, we turned to four complimentary analytical methods that are exceptionally suited for speciation studies in aqueous media, namely cyclic voltammetry, X-ray absorption near edge spectroscopy (XANES), extended X-ray absorption fine structure (EXAFS), and Ultraviolet–Visible-Near Infrared (UV-Vis-NIR) spectroscopy. This technique quartet enabled electron transfer reactions, metal oxidation states, ligand identities, coordination numbers, and bond distances accessible to plutonium in HCl(aq) solutions to be rigorously defined.
 | ||
| Scheme 1 Overview of some reactions that impact plutonium speciation in HCl(aq). | ||
Our results suggested that Pu(IV)–aquo ions [Pu(H2O)x4+] dominated the speciation profile in dilute HCl(aq) concentrations (≤3 M). The data also showed that plutonium speciation shifted to anionic chloride complexes (PuCly4−y) when HCl(aq) concentrations were high (>8 M). It was surprising to discover that conversion of the homoleptic Pu(H2O)x4+ aquo complex to the homoleptic PuCly4−y chloride complex occurred rather abruptly and over a relatively small HCl(aq) concentration range (∼4.5 to 8 M). Heteroleptic Pu(IV)-aquochloride complexes [PuCly(H2O)x4−y] were the dominant species present in solution over this HCl(aq) concentration regime. Evaluating the X-ray absorption speciation results alongside the electrochemical data provided additional insight regarding the Cl1− for H2O ligand substitution reaction. This analysis suggested that the plutonium speciation profile shifted suddenly from being dominated by Cl1− to dominated by H2O when the HCl(aq) content crossed the 5.5 M concentration value. Changing the plutonium ligand environment from being homoleptic in Cl1− to homoleptic in H2O also impacted the half-wave potential (E½) for the reversible Pu(IV) + e1− ⇌ Pu(III) (Pu4+/3+) one-electron transfer reaction. For example, the E½ potential was 0.572(2) V for PuCly4−y, 0.744(2) V for Pu(H2O)x4+, and at intermediate values for heteroleptic PuCly(H2O)x4−y species. No additional redox events, e.g., oxidation to PuO2n+(aq) or reduction of Pu2+(aq), were observed electrochemically over the surveyed potential window (ca. 0.4 to 1 V). We interpreted these data as suggesting that anionic Cl1− ligands stabilized more electron-deficient Pu4+(aq) over Pu3+(aq) and neutral H2O ligands stabilized more electron-rich Pu3+(aq) over Pu4+(aq). The data indicated that Pu4+(aq) was a reasonably strong oxidant and able to generate Pu3+(aq) in solutions over the sampled HCl(aq) concentration range. It also highlighted Pu4+(aq) stability toward losing electrons and generating PuO2n+(aq). These observations have general implications on plutonium behavior within aqueous solutions and may help explain unexpected plutonium fractionation that occurs during industrial-scale processing (e.g., using solvent extraction or ion exchange chromatography), while conducting small-scale radioanalytical separations, and in regard to the fate and transport of plutonium through the environment.
Results and discussion
Sample preparation
A series of aqueous solutions were prepared to study how the plutonium speciation varied as a function of HCl(aq) concentration. Toward this end, we first prepared a chemically pure and oxidation state pure stock solution of Pu4+(aq) in HCl(aq) (5.5 or 6 M). Aliquots from this stock solution were gently heated on a hot block until the solution evaporated. The resulting residues were subsequently dissolved in aqueous solutions whose HCl(aq) content systematically varied. For X-ray absorption (XANES and EXAFS) and UV-Vis-NIR measurements the HCl(aq) concentrations were 1, 3, 5.5, 8, and 11 M. For electrochemical measurements the HCl(aq) concentrations were 1, 3, 4.5, 6, 8, and 11 M. The HCl content in the commercially available source of HCl(aq) used to make the more dilute HCl(aq) samples varied from batch-to-batch and ranged 10.8 to 11.1 M. Hence, we estimated the HCl content in the concentrated HCl(aq) solutions as being 11 M and used this value to calculate the HCl(aq) content in all subsequently made serial dilutions. No attempt was made to rigorously characterize the HCl(aq) content in the solutions for our experiments because the HCl(aq) concentration variations were small in comparison to the experimentally observed plutonium speciation changes.Pu L3-edge X-ray absorption near edge spectroscopy (XANES)
The dependence of Pu(IV) oxidation state stability on HCl(aq) concentration (1, 3, 5.5, 8, 11 M) in aqueous solutions was evaluated using Pu L3-edge XANES. Background subtracted and normalized Pu L3-edge XANES data from Pu4+(aq) (∼0.02 M) dissolved in aqueous solutions were compared in Fig. 1 as a function of HCl(aq) concentration. The spectra were dominated by an absorption peak that was superimposed on a step-like absorption threshold. This main absorption peak is often attributed to the electric dipole allowed 2p → 6d transition,32–35 and the inflection point for the rising absorption edge (near 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 063 eV) is routinely correlated with formal plutonium oxidation state values.33 Inflection point energies were determined graphically as the point at which the second derivative of these data equaled zero and these data were used to characterize the oxidation state of plutonium in these samples (Fig. 1). Those energy values ranged from 18060.3 to 18063.1 eV (calibrated to the Zr K-edge from zirconium metal; 17998 eV) with an estimated uncertainty of ±0.1 eV (at 1σ), based on our ability to reproduce XANES spectra on other actinide samples. The inflection energy values were consistent with those determined for other Pu(IV) samples that had Cl1− and/or H2O ligands. For example, the plutonium inflection point energy in dilute HCl(aq) (1 M) was 18063.0(1) eV and similar to that reported previously for the plutonium aquo ion (18063.2 eV).32 Increasing the HCl(aq) concentration to 8 M had essentially no detectable impact on the inflection point energies and the resulting values were equivalent within the estimated uncertainty of our measurement. Pushing the HCl(aq) content even higher, to 11 M, caused the inflection point energy to drop by ca. 3 eV. This 18060.3(1) eV value was slightly higher than the inflection point energy from our Pu(IV) standard Cs2PuCl6 [18059.9(1) eV] and slightly lower than that reported previously for Pu4+(aq) dissolved in 10.8 M HCl (18061.7 eV).33 Hence, we concluded that the dominant plutonium species present in all of these plutonium samples had an oxidation state of +4. Overall, these data indicated that Pu4+(aq) was stable in concentrated and dilute HCl(aq) solutions under conditions associated with shipping the samples to the synchrotron and making the X-ray absorption measurements.
063 eV) is routinely correlated with formal plutonium oxidation state values.33 Inflection point energies were determined graphically as the point at which the second derivative of these data equaled zero and these data were used to characterize the oxidation state of plutonium in these samples (Fig. 1). Those energy values ranged from 18060.3 to 18063.1 eV (calibrated to the Zr K-edge from zirconium metal; 17998 eV) with an estimated uncertainty of ±0.1 eV (at 1σ), based on our ability to reproduce XANES spectra on other actinide samples. The inflection energy values were consistent with those determined for other Pu(IV) samples that had Cl1− and/or H2O ligands. For example, the plutonium inflection point energy in dilute HCl(aq) (1 M) was 18063.0(1) eV and similar to that reported previously for the plutonium aquo ion (18063.2 eV).32 Increasing the HCl(aq) concentration to 8 M had essentially no detectable impact on the inflection point energies and the resulting values were equivalent within the estimated uncertainty of our measurement. Pushing the HCl(aq) content even higher, to 11 M, caused the inflection point energy to drop by ca. 3 eV. This 18060.3(1) eV value was slightly higher than the inflection point energy from our Pu(IV) standard Cs2PuCl6 [18059.9(1) eV] and slightly lower than that reported previously for Pu4+(aq) dissolved in 10.8 M HCl (18061.7 eV).33 Hence, we concluded that the dominant plutonium species present in all of these plutonium samples had an oxidation state of +4. Overall, these data indicated that Pu4+(aq) was stable in concentrated and dilute HCl(aq) solutions under conditions associated with shipping the samples to the synchrotron and making the X-ray absorption measurements.
 | ||
| Fig. 1 Room temperature, background-subtracted, and normalized Pu L3-edge XANES spectra from Pu4+(aq) in HCl(aq) solutions (1–11 M from top to bottom). The orange trace is the low temperature (77 K) background-subtracted and normalized Pu L3-edge XANES spectrum from the solid Pu(IV) standard (Cs2PuCl6).60 Inflection point energies are included. | ||
Pu Ultraviolet–Visible-near infrared spectroscopy (UV-Vis-NIR)
Conclusions regarding the dependence of plutonium oxidation states on HCl(aq) concentrations derived from the Pu L3-edge XANES data were validated by analogous UV-Vis-NIR measurements. Optical spectroscopy samples were prepared in the same way as the XANES samples. Spectra were measured at room temperature and collected every few days over the course of one week. This period was equivalent to the time required to package, ship (from LANL to SSRL), and analyze samples using Pu L3-edge XANES. Because no change in the optical data was observed over the course of one week, we concluded that the plutonium oxidation states were stable in HCl(aq) (1 to 11 M) solutions for the duration of the XANES experiments. This conclusion alleviated concerns of spontaneous redox events that could occur during the shipping process to the synchrotron, which was of concern. Representative spectra have been provided in Fig. 2 (see ESI† also). | ||
| Fig. 2 Room temperature UV-Vis-NIR spectra from Pu4+(aq) after dissolution in HCl(aq) solutions (1–11 M from bottom to top) collected immediately after sample preparation. Relevant peaks were labeled with their associated plutonium oxidation states. | ||
The UV-Vis-NIR spectrum collected in dilute HCl(aq) (1 M) was consistent with other spectra from Pu(IV) collected in dilute acid and reported previously.36,37 Most notable were the intense cluster of peaks centered at 15385 cm−1 (650 nm) and the sharp absorbance peak at 21277 cm−1 (470 nm). Our interpretation of these data agreed with the aforementioned Pu L3-edge XANES spectrum and we concluded that Pu(IV) was the major species present in solution. In addition to these Pu(IV) absorption peaks were small features at 12048 cm−1 (830 nm) and 16639 cm−1 (601 nm) that indicated PuO22+ and Pu(III) were also present in small quantities. Reproducing the sample preparation procedure repeatedly generated PuO22+(aq) and Pu3+(aq) in the dilute HCl(aq) matrix. We estimated the amount of PuO22+ and Pu(III) to be at 5% and 10% of the total plutonium in the samples, respectively. Note, there was a high uncertainty associated with these estimates because the extinction coefficients (ε) used to quantify the relative plutonium concentrations were matrix mismatched, meaning that ε values were determined in HClO4(aq) (2 M)8 and the measurements were made in HCl(aq) (1 M). It was unclear what caused PuO22+(aq) and Pu3+(aq) to form in the dilute HCl(aq) (1 M) solution. We speculated that the redox reaction occurred during sample preparation and assume that the combination of heating the parent Pu4+(aq) stock solution to dryness for 12 hours, followed by dissolution in dilute HCl(aq) (1 M), forced disproportionation of Pu4+(aq) to make PuO22+(aq) and Pu3+(aq).
Increasing the HCl(aq) concentration to be >1 M had marked impact on the plutonium spectra. Most importantly, features associated with PuO22+ and Pu(III) were not detected. We also observed evidence for Cl1− complexation of Pu(IV) when the HCl(aq) was ≥5.5 M in three notable ways. The first change was that increasing the HCl(aq) concentration beyond 3 M caused the cluster of peaks centered at 15385 cm−1 (650 nm) to spread out. Their intensities also changed, such that the low energy absorbance peaks gained intensity relative to the high energy peaks. The second notable change was that a shoulder emerged near 14500 cm−1 (670 nm); see the 5.5 M HCl(aq) spectrum. This feature gained intensity and shifted bathochromically (red) with increasing HCl(aq) content. The third notable observation was that the sharp peak at 21277 cm−1 (470 nm) decreased in intensity and was swallowed by a broad absorption feature. This absorption band commenced near 19000 cm−1 (526 nm) and gained intensity with increased HCl(aq) concentration. Our observations align well with previously reported UV-Vis data in HCl(aq) matrices.37–40
Taken as a whole, the optical and XANES measurements complemented one another. Both characterization techniques confirmed Pu(IV) was the dominant species present in each sample. The UV-Vis-NIR experiments enabled identification of PuO2n+(aq) (n = 1, 2) and Pu3+(aq) contaminants present in solution at quantities below the XANES detection limit (typically estimated at <10% of the total plutonium content in the sample). The Pu L3-edge XANES studies enabled us to conclude that the UV-Vis-NIR spectra obtained in 5.5, 8, and 11 M were characteristic of Pu(IV) dissolved in these higher concentration HCl(aq) solutions. It can be difficult to assign plutonium oxidation states based on UV-Vis-NIR data in concentrated HCl(aq) solutions because Cl1− ligands bind Pu4+(aq), displace neutral H2O, and alter the UV-Vis-NIR spectra. In this regard, the optical data in Fig. 2 provide Pu4+(aq) UV-Vis-NIR reference spectra paired with precise speciation profiles that can be used by those manipulating plutonium in HCl(aq) solutions in the future. This X-ray absorption and UV-Vis-NIR spectroscopy combination is absent from the literature at this time, to the best of our knowledge.
Pu L3-edge extended X-ray absorption fine structure (EXAFS) data
Changes in Pu4+(aq) speciation in aqueous solution were evaluated as a function of HCl(aq) concentration (1, 3, 5.5, 8, 11 M) using Pu L3-edge EXAFS spectroscopy. These room temperature and solution phase k3-weighted Pu L3-edge EXAFS spectra were compared in Fig. 3. The data were best described as having a single wave with five resolved crests that oscillated in intensity from 3 to 11 Å−1. The amplitude for the wave observed in dilute HCl(aq) (1 M) decreased in intensity with increasing energy. Increasing the HCl(aq) concentration from 1 to 11 M did not impact the wave amplitudes, which remained qualitatively constant across the surveyed energy space. However, changing the HCl(aq) concentration caused the wavelength to increase and frequency to become longer in Å−1. We indicated this change in Fig. 3 using black brackets and a green vertical line. | ||
| Fig. 3 The k3-weighted room temperature Pu L3-edge EXAFS spectra displaying the χ(k) function (points) with non-linear curve fitting (lines) from aqueous Pu4+(aq) solutions in HCl(aq) solutions (1–11 M). The gray bracket and the vertical green line were included to help visualize that the wavelength changed with changing HCl(aq) concentration. | ||
The impact of increasing the HCl(aq) concentration (1 to 11 M) on the Pu L3-edge EXAFS data was also evident in the Fourier transforms for the k3-weighted spectra (Fig. 4). In dilute HCl(aq), there was a single symmetric feature just below 2 Å. Increasing the HCl(aq) concentration to 5.5 M caused the feature to change: it broadened, became asymmetric, and its intensity decreased. In 11 M HCl(aq), the feature gained intensity, sharpened, and became symmetric again. Crossing the entire series from 1 to 11 M shifted the peak maximum to longer distances and increased R + δ from ca. 1.9 Å in 1 M HCl(aq) to 2.2 Å in 11 M HCl(aq) (R is the scattering pathway distance and δ is the phase-shift).
 | ||
| Fig. 4 Fourier transforms of room temperature k3-EXAFS spectra for aqueous Pu4+(aq) solutions in HCl(aq) at varied concentrations (1 to 11 M; black circles). Fits for the data are shown as pink traces. The Pu → Cl and Pu → O scattering pathways used to generate the fits are shown as yellow and blue traces, respectively. | ||
Qualitatively, we interpreted these data as follows. Plutonium speciation was dominated by homoleptic aquo tetracations, Pu(H2O)x4+, at low HCl(aq) concentrations (e.g., 1 M). The resulting single shell of H2O ligands manifested as a single scattering peak in the Fourier transform for the k3-weighted spectrum. Heteroleptic aquochloride species, PuCly(H2O)x4−y, existed at intermediate HCl(aq) concentrations (e.g., 5.5 M). Here, the plutonium primary coordination sphere contained H2O ligands with short Pu–OH2O distances and Cl1− ligands with longer Pu–Cl bond distances. The presence of these two coordination environments enabled two distinct scattering pathways (Pu → OH2O and Pu → Cl) that manifested as a broad asymmetric feature in the Fourier transform for the k3-weighted spectra. Moving to concentrated HCl(aq) (11 M) solutions increased the Cl1−(aq) content to the point that Cl1−(aq) outcompeted H2O for Pu4+(aq) complexation. Hence, under these conditions, homoleptic Cl1−(aq) coordination compounds of the general formula PuCly4−y dominated the Pu4+(aq) speciation profile. Having only Cl1−(aq) ligands in the primary coordination sphere manifested as a sharp single peak in the Fourier transform of the k3-weighted spectra that was reminiscent of the Pu(H2O)x4+ spectrum obtained at low HCl(aq) concentrations (e.g., 1 M). The major difference between the spectra obtained from Pu(H2O)x4+vs. PuCly4−y was associated with the intensity (peak area) and R + δ value. The spectrum from PuCly4−y had a smaller intensity and larger R + δ value. This difference suggested that (1) the overall coordination number for PuCly4−y was smaller than that of Pu(H2O)x4+ – fewer Cl1− ligands bound to PuCly4−y than H2O ligands bound to Pu(H2O)x4+ – and (2) the bond distance between Pu(IV) and anionic Cl1− was longer than Pu(IV) bound to neutral H2O.
A more quantitative evaluation of the Pu L3-edge Fourier transforms of the k3-weighted spectra employed conventional shell-by-shell fitting methods.41–43 Those models were consistent with the qualitative interpretation described above. The energy ranges over which usable data were obtained spanned from ca. 3 to 11 Å−1, which limited the resolution in R-space to 0.2 Å (resolution = π/2Δk). Care was taken to constrain our models so that a reasonable number of variables were used, which protected against overparameterizing the fit (typically 75% of the maximum number of free variables allowed for the fitted region). Atomic coordinates from Pu(H2O)93+ and PuCl62− were used to calculate scattering pathways using FEFF8.44–46
The dilute HCl(aq) (1 M) data were fitted using a single scattering pathway associated with a shell of H2O ligands bound by Pu(IV). The obtained model returned 9(1) H2O ligands in the Pu(IV) inner coordination sphere at a Pu–OH2O distance of 2.37(11) Å. The parameters associated with this fit converged to reasonable values with acceptable error bars (Table 1). The measured Pu–O bond distance was consistent with our interpretation of the Pu L3-edge XANES data and suggested that the plutonium oxidation state was +4. For example, subtracting the 8-coordinate Pu(IV) ionic radius (0.96 Å)47 from our Pu–OH2O distance gave a value of 1.41 Å. This value was bracketed by the calculated (1.67 Å) and crystallographically measured H2O ionic radii (1.38 Å).48 The Pu–OH2O distance was shorter than the 2.476(2) and 2.574(3) Å bond distances determined for trivalent Pu(H2O)93+ aquo ion using single crystal X-ray diffraction, which was consistent with the 0.14 Å difference in Pu(IV) vs. Pu(III) ionic radii.45 Including a Pu → Cl1− scattering pathway did not significantly improve our model; hence, it was not included in our final fit. Our coordination numbers – and interpretation that Pu(H2O)9±14+ was the dominant species in solution – were in good agreement with other EXAFS analyses conducted in dilute HCl(aq) solutions. For example, Conradson and coworkers used EXAFS to determine that in dilute HCl(aq) (1.4 M) solutions Pu4+(aq) had a small number of Cl1− ligands (0.9) and that the rest of the coordination sphere was occupied by H2O ligands [Pu–OH2O = 2.39 Å].33
| Pu HCl(aq) Solutions | |||||
|---|---|---|---|---|---|
| 1 M HCl(aq) | 3 M HCl(aq) | 5.5 M HCl(aq) | 8 M HCl(aq) | 11 M HCl(aq) | |
| O coordination number | 9(1) | 10(1) | 6(1) | 3(1) | — |
| σ 2 (O) | 0.009(2) | 0.007(2) | 0.007 | 0.007 | — |
| An–O | 2.37(11) | 2.37(10) | 2.33(14) | 2.30(18) | — |
| Cl coordination number | — | — | 3(1) | 5(1) | 6(1) |
| σ 2 (Cl) | — | — | 0.006 | 0.006 | 0.006(1) |
| An–Cl | — | — | 2.598(6) | 2.588(4) | 2.594(3) |
| Pu coordination number | — | — | — | — | — |
| σ 2 (Pu) | — | — | — | — | — |
| S 0 2 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 |
| E° | 0.21 ± 0.97 | 0.3 ± 1.0 | −2.73 | −3.5 | 2.9 ± 0.9 |
| Total variables used | 4 | 4 | 4 | 4 | 4 |
| Independent variables available for the fitting range | 7.97 | 7.42 | 7.72 | 8.04 | 8.81 |
| Reduced χ2 | 6049.24 | 5626.01 | 2418.60 | 2228.83 | 637.6129 |
| R-Factor | 0.012 | 0.0092 | 0.0072 | 0.011 | 0.010 |
Our data from 3 M HCl(aq) solutions were also best modeled as having only H2O ligands in the inner Pu(IV) coordination sphere. The fit showed 10(1) H2O ligands with a Pu–OH2O distance of 2.37(10) Å. Incorporating a Pu → Cl scattering pathway did not provide a suitable model. The H2O coordination number and Pu–OH2O bond distance were statistically equivalent to that observed in 1 M HCl(aq) and discussed above. We interpreted these data as suggesting that the dominant Pu4+(aq) species present in dilute HCl(aq) (3 M) had an average stoichiometry of Pu(H2O)10±14+ and that the Pu(IV) aquo ion persisted in solution when HCl(aq) concentrations were between 1 and 3 M.
The Pu L3-edge EXAFS data from concentrated HCl(aq) (11 M) solutions were modeled using a Pu → Cl single scattering pathway. The obtained model returned 6(1) chlorides and a 2.594(3) Å Pu–Cl distance. Attempts to include a Pu → H2O scattering pathway into the model failed to converge. This Pu–Cl distance matched the average bond distance determined for PuCl62− using single crystal X-ray diffractometry (2.59 Å).46 Both of these distances were slightly shorter than the sum (2.67 Å) of the Pu(IV) 6-coordinate (0.86 Å) and the Cl1− (1.81 Å) ionic radii. Our Pu–Cl distance in HCl(aq) (11 M) was also in good agreement with other Pu–Cl bond distances determined in the solution phase by EXAFS, e.g., solution phase Pu–Cl distances of 2.63 Å and 2.61 Å have been reported previously.33 Hence, we concluded the dominant Pu4+(aq) species present in concentrated HCl(aq) (11 M) had an average stoichiometry of PuCl6±1(2±1)−.
Fitting the Pu L3-edge EXAFS from plutonium samples dissolved in intermediate concentrations of HCl(aq) (5.5 and 8 M) suggested both Cl1− and H2O ligands were bound to Pu(IV). Modeling data obtained from the 5.5 M HCl(aq) solution showed Pu(IV) was complexed by 6(1) H2O ligands [Pu–OH2O = 2.33(14) Å] and 3(1) Cl1− ligands [Pu–Cl = 2.598(6) Å]. Increasing the HCl(aq) concentration to 8 M decreased the H2O coordination number and increased the Cl1− coordination number: 3(1) H2O ligands [Pu–OH2O = 2.30(18) Å] and 5(1) Cl1− ligands [Pu–Cl = 2.588(4) Å]. We interpreted these data as showing that the dominant species present in 5.5 M HCl(aq) had an average chemical formula of PuCl3±1(H2O)6±1(1±1)+ and a charge that ranged from being neutral to dicationic, based on the uncertainty associated with the Cl1− coordination number. In 8 M HCl(aq), the dominant species in solution had an average chemical formula of PuCl5±1(H2O)3±1(1±1)− and a charge that ranged from being neutral to anionic.
In summary, the Pu L3-edge EXAFS experiments demonstrated that complexation of Pu4+(aq) in aqueous solutions could be controlled by altering the HCl(aq) concentration. The Pu4+(aq) speciation was dominated by the Pu4+-aquo ion in dilute HCl(aq) (between 1 and 3 M). Heteroleptic aquochloride species, PuCly(H2O)x4−y, were detected at intermediate HCl(aq) concentrations (between 4.5 and 8 M). Homoleptic Pu(IV)-chloride complexes, corresponding to PuCl6±1(2±1)−, were observed in concentrated HCl(aq) solutions (11 M). All recorded XANES, UV-Vis-NIR, and EXAFS data were self-consistent with the reduction potentials and Cl1− coordination numbers determined using electrochemical methods (vide infra).
Aqueous cyclic voltammetry
Plutonium redox chemistry was investigated in aqueous media as a function of HCl(aq) (1–11 M) concentration using cyclic voltammetry (Fig. 5). These studies provided additional insight and validation for the conclusions drawn from the X-ray absorption and UV-Vis-NIR measurements described above. In general, the cyclic voltammograms were similar across the sampled HCl(aq) concentrations. They contained a reversible single wave that we attributed to the Pu4+/3+(aq) one-electron transfer reaction (eqn (1)).30| Pu4+(aq) + e1− ⇌ Pu3+(aq) | (1) |
 | ||
| Fig. 5 Cyclic voltammograms from Pu(IV) (16.7 mM) dissolved in HCl(aq) (1–11 M) at a scan rate of 0.01 V s−1 for the Pu4+/3+(aq) redox reaction. Data from HCl(aq) solutions at 1 to 11 M are represented by red, brown, green, orange, blue, and purple traces, respectively. The arrows represent the direction of the potential sweep. The dot with the dashed line indicates the half-wave potential (E½), which are also provided and referenced to Ag/AgCl. Errors are the standard deviation from three replicates. | ||
Closer inspection of the voltammograms obtained in HCl(aq) revealed how peak current (ip), anodic and cathodic peak separation (ΔEp), and the half-wave potentials (E½) varied as a function of scan rate and HCl(aq) concentration. These variations, in turn, informed on how changing the HCl(aq) concentration impacted plutonium speciation.
The influence of scan rate on peak currents (ip) was depicted in Fig. 6. This figure showed that ip varied with scan rate and ranged from ∼0.03 mA at a scan rate of 0.01 V s−1 to 0.6 mA at a scan rate of 10 V s−1. Current passed for the anodic wave (ipa) was nearly equivalent in magnitude to that for the cathodic wave (ipc) for a given HCl(aq) concentration and scan rate, i.e., the ratio of ipa/ipc was near one (see ESI†). We interpreted these data as indicating reversibility of the anodic, Pu(III) → e1− + Pu(IV), and cathodic, Pu(IV) + e1− → Pu(III), electron transfer events. This suggested that the number of electrons associated with the Pu4+(aq) reduction was equivalent to that of the Pu3+(aq) oxidation under all examined experimental conditions and that the primary coordination environment for Pu(IV) was similar to Pu(III) on the electrochemical time scale in HCl(aq).
 | ||
| Fig. 6 Peak current (ip) from cyclic voltametric measurements made on plutonium (16.7 mM) in HCl(aq) (1–11 M) solutions plotted as a function of scan rate (0.01–1 V s−1). Error bars are the standard deviation from three replicates. | ||
Electrochemical reversibility suggested that the Pu(III) coordination environment was equivalent to that of Pu(IV) on the cyclicvoltammographic timescale in all HCl(aq) solutions tested. This observation was anticipated in dilute HCl(aq) solutions, where Pu(IV)-aquo and Pu(III)-aquo ions were both expected to exist. Reversibility was unexpected – however – in more concentrated HCl(aq) for the following reasons. Our Pu L3-edge EXAFS data showed that homoleptic PuCly4−y chlorides exist for Pu(IV) when HCl(aq) concentrations were high. It is also well established that Pu3+(aq) prefers binding H2O over Cl1− in aqueous media when the Cl1−(aq) concentration are similarly high.30 Hence, we expected electrochemical irreversibility for the Pu4+(aq) to Pu3+(aq) redox couple at high HCl(aq) concentrations because of the anticipated H2O for Cl1− ligand substitution reaction: e.g., Pu(III)-homoleptic chloride to Pu(III) heteroletpic aquochloride. Surprisingly, this was not observed. The ensuing electrochemical reversibility showed that the Cl1− for H2O ligand exchange was slow on the cyclicvoltammographic timescale and that electrochemical methods can be effectively used to probe species that contain Pu(III)–Cl bonds in aqueous media, even the illusive PuIIICl63− trianion.
The impact of scan rate on the anodic and cathodic peak separations (ΔEp) was depicted in Fig. 7A. The plot showed ΔEp was weakly dependent on the scan rate when the scan rate was relatively slow, ca. 0.06–0.08 V between 0.01 and 0.2 V s−1. We interpreted this near-independence on scan rate as indicating that a reversible and diffusion-controlled electron transfer processes occurred on the time scale of the experiment, a designation that was consistent with convention49 despite being slightly larger than the theoretical value for a reversible process: 0.057 V at 25 °C.50 At higher scan rates (>0.2 V s−1), more extreme potentials (increased ΔEp values) were needed to drive the Pu4+/3+(aq) redox reactions. This result indicated that the redox process became kinetically controlled, meaning that electron transfer to plutonium was faster than diffusion of plutonium away from the electrode.
 | ||
| Fig. 7 (A) Cathodic and anodic peak separation potential (ΔEp) and (B) half-wave potential (E1/2) as a function of the scan rate from plutonium (16.7 mM) dissolved in HCl(aq) (1–11 M) solutions. Error bars are the standard deviation from three replicates. | ||
Fig. 7A also documented impact of scan rate on ΔEp as a function of HCl(aq) concentration. The anodic and cathodic peak separations were essentially independent of the HCl(aq) content. For example, the ΔEp values were equivalent within measurement uncertainty for each given HCl(aq) concentration. However, we acknowledge that the ΔEp uncertainty increased with the scan rate and the error bars were large (as high as ±0.067 V) at fast scan rates (>1 V s−1).
Fig. 5 showed that the half-wave potentials between the cathodic and anodic peaks (E½) were positive, ranging from 0.57(1) V to 0.74(1) V as the HCl(aq) concentration decreased from 11 to 1 M. The E½ values were also independent of scan rate at each HCl(aq) concentration (Fig. 7b). Utilizing the E½ values in Fig. 5 as estimates for formal Pu4+/3+(aq) potential,51 we estimated Gibbs free energy values (ΔGest) for the reversible Pu4+/3+(aq) electron transfer reaction as a function of HCl(aq) concentration using Eq. 2. These calculations showed that decreasing the HCl(aq) concentration from 11 to 1 M decreased the ΔGest values from −55.2 to −71.8 kJ mol−1, respectively. Uncertainty was estimated to be ±0.1 kJ mol−1 at 1σ. We proposed that the major contributor for the E½ dependence on HCl(aq) was associated with a plutonium speciation change from Pu(H2O)x4+ to PuCly(H2O)x4−y to PuCly4−y that accompanied decreasing the Cl1− content in solution. These calculations also indicated that the Pu(H2O)x4+ aquo cation was more strongly oxidizing than the PuCly4−y anion and that Cl1− complexation stabilized substantially the plutonium +4 oxidation state over the plutonium +3 oxidation state.
| ΔGest = −nFE½ | (2) |
Using half-wave potential (E½) to evaluate Pu4+(aq) coordination number
Electrochemical methods were used to further evaluate how plutonium speciation varied as a function of HCl(aq) concentration. These experiments validated the conclusions described above that were based on the Pu L3-edge EXAFS studies. As an introduction to these experiments, we remind the reader that it is difficult to use EXAFS as a standalone technique for identifying speciation changes. This issue was rooted in the high uncertainties associated with the EXAFS coordination number measurements. A more robust understanding of chemical speciation changes can be obtained if interpretation of the EXAFS data is informed by a complimentary physical method. In this case, we selected electrochemical measurements for two reasons. First, cyclic voltammetry measurements are quite sensitive to chemical speciation changes. Second, it is well established that half-wave potentials (E½) can be used to determine the number of ligands (in this case Cl1−) bound to a redox active metal (in this case plutonium), provided that the measured electron transfer process and the metal–ligand complexation process are both reversible. As stated above, both conditions were met for plutonium dissolved in HCl(aq) (eqn (1) and Scheme 1): the Pu4+/3+(aq) redox reaction was electrochemically reversible and plutonium underwent chemically reversible complexation reactions with Cl1− and H2O ligands (Scheme 1).The mathematical expression that relates the number of Cl1− ligands bound by plutonium to the E½ for the Pu4+/3+(aq) redox reaction is shown in eqn (3).
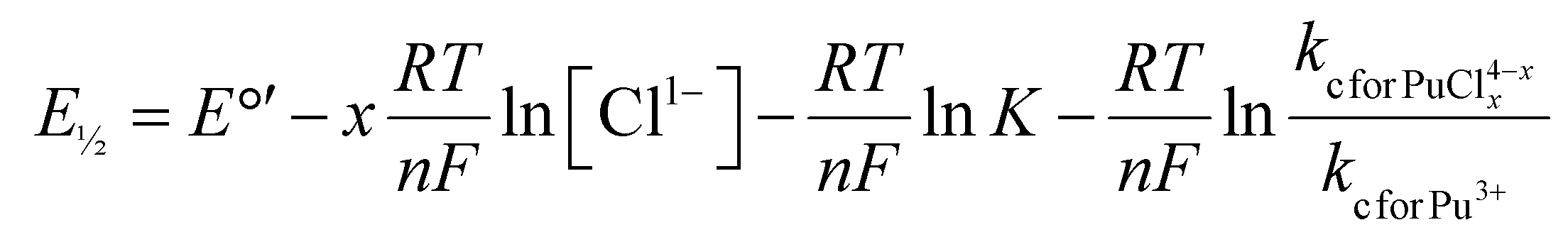 | (3) |
485 C mol−1; kc for Pu(IV) = mass transfer coefficient for Pu4+(aq).
The derivation of this equation is based on the Nernst equation, the steady-state mass transport equation, and the reaction equilibrium equation. For more details, we direct interested readers to the ESI† and the textbook “Electrochemical Methods: Fundamentals and Applications, second edition”, by Bard and Faulkner (pages 36–37 and 186–188) where this relationship was derived.50 Included here is a high-level description that shows how to relate E½ to coordination number within the context of Pu4+/3+(aq) redox reaction and Pu–Cl complexation reactions.
For this scenario, the Cl1− coordination number can be determined by plotting E½vs. the natural logarithm of the chloride concentration, ln[Cl1−(aq)]. The E½ values should be linearly dependent on ln[Cl1−(aq)] and the slope of that relationship equal to x(RT/nF) when a single species is present as the Cl1−(aq) concentration changes (variables defined in eqn (2)). Fig. 8 showed two linear regimes when E½ and ln[Cl1−] were plotted accordingly. The first linear regime occurred at low HCl(aq) concentrations. Here, E½ was highest for the Pu4+/3+(aq) redox reaction in dilute HCl(aq) (1 M) at 0.744(3) V. This value dropped slightly and monotonically to 0.736(1) V when the HCl(aq) concentration was raised to 4.5 M, such that a near horizontal relationship existed between E½ and ln[Cl1−] as the HCl(aq) concentration was increased from 1 to 4.5 M. The slight decrease in E½ suggested that increasing the HCl(aq) concentration to 4.5 M slightly increased the propensity of Cl1−(aq) to complex Pu4+(aq); however, the speciation profile strongly favored Pu(H2O)x4+ over PuCly(H2O)x4−y. Consistent with that notion, we calculated the average Cl1− coordination number for Pu4+(aq) over this HCl(aq) concentration range using eqn (4) (variables defined in eqn (3)). Slope analysis returned a value of −0.0046(9) V, which corresponded to an average Cl1− coordination number of 0.18(3). This result was consistent with the Pu L3-edge X-ray absorption data (vide supra), which showed Cl1−(aq) was not bound by Pu(IV) in dilute HCl(aq). Instead, the homoleptic Pu4+-aquo ion, Pu(H2O)x4+, existed when HCl(aq) concentrations were ≤3(1).
 | (4) |
 | ||
| Fig. 8 Half-wave potentials (E1/2) from cyclic voltammetry measurements made on plutonium dissolved in HCl(aq) (1–11 M) solutions plotted as a function of the natural log of the HCl(aq) concentration. Error bars are the standard deviation of E½ measured at eight scan rates from 0.01 to 10 V s−1. | ||
Linearity also occurred when HCl(aq) concentrations were high (4.5 to 11 M). Here, E½ dropped from 0.736(1) [4.5 M HCl(aq)] to 0.572(3) [11 M HCl(aq)]. Slope analysis of the concentrated HCl(aq) regime returned a value of −0.19(3) V, which corresponded to an average Cl1− coordination number of 7(1) (eqn (3)) over the 4.5 to 11 M HCl(aq) concentration range. This result was consistent with the Pu L3-edge X-ray absorption data (vide supra), which showed Cl1− coordination numbers ≥5(1) when HCl(aq) concentrations were high (≥8 M) and Cl1− coordination numbers ≤3(1) when HCl(aq) concentrations decreased to ≤5.5 M. Other important information extracted from Fig. 8 was related to the domain over which heteroleptic PuCly(H2O)x4−y complexes existed. The graph highlighted a clear and abrupt Pu4+(aq) speciation change when the HCl(aq) concentration was between 4.5 and 6 M.
Evaluating Pu4+(aq) and Pu3+(aq) diffusion coefficients and hydrodynamic radii via peak current (ip)
The diffusion coefficients and hydrodynamic radii for Pu4+(aq) and Pu3+(aq) were determined from the cyclic voltammetry measurements. This was possible because the cyclic voltammetry mass transfer was purely diffusion-limited. Hence, the Randles–Ševčík equation (eqn (5)) could be readily employed to obtain the diffusion coefficient. | (5) |
 ; n = number of electrons, one for Pu4+/3+(aq); T = temperature, 25 K; A = Electrode surface area, cm2; CPu = bulk Pu concentration, M; R = ideal gas constant, 8.314 J mol−1 K−1; F = Faraday constant, 96485 C mol−1; v = scan rate, V s−1.
; n = number of electrons, one for Pu4+/3+(aq); T = temperature, 25 K; A = Electrode surface area, cm2; CPu = bulk Pu concentration, M; R = ideal gas constant, 8.314 J mol−1 K−1; F = Faraday constant, 96485 C mol−1; v = scan rate, V s−1.
In Fig. 6, the peak current (ip) was plotted against the scan rate (v½) for each HCl(aq) concentration value. In all cases, there was a linear relationship between ip and v½ for both anodic Pu(III) → e1− + Pu(IV) and cathodic Pu(IV) + e1− → Pu(III) electron transfer events at scan rates < 1 V s−1. Hence, diffusion coefficients for Pu4+(aq) and Pu3+(aq)
and Pu3+(aq) could be extracted from the slope analyses in this regime using least squares fitting. The goodness of fit was assessed by the coefficient of determination, R2, which ranged from 0.987 to 0.998 for plutonium in HCl(aq) solutions (1–11 M) (see ESI†).
could be extracted from the slope analyses in this regime using least squares fitting. The goodness of fit was assessed by the coefficient of determination, R2, which ranged from 0.987 to 0.998 for plutonium in HCl(aq) solutions (1–11 M) (see ESI†).
The diffusion coefficients were plotted as a function of HCl(aq) concentration in Fig. 9A. All diffusion coefficients were similar in magnitude and subtly varied from 3.5(2) × 10−6 to 5.5(1) × 10−6 cm2 s−1. These values agreed well with estimates we calculated using the Nernst–Haskell equation (eqn (6)). We note that the values determined from eqn (5) should be taken as a rough estimation, as this equation is only valid for ion-pair analytes dissolved in infinitely dilute solutions.
 | (6) |
 | ||
| Fig. 9 (A) Dependence of the diffusion coefficients of Pu4+(aq) (blue) and Pu3+(aq) (red), calculated by both the Randles–Ševčík and Nernst–Haskell equations, on the concentration of the HCl(aq) (1–11 M) solution. (B) Dependence of the hydrodynamic radii of Pu4+(aq) (blue) and Pu3+(aq) (red), calculated from Stokes–Einstein equation and the group contribution method, on the concentration of the HCl(aq) (1–11 M) solutions. | ||
The mathematical expression in eqn (6) related the diffusion coefficient  for both Pu4+(aq) and Pu3+(aq) to the oxidation states of plutonium (m′ = +4 or +3) and chloride (m′′ = –1) and the limiting molar ionic conductivities of plutonium
for both Pu4+(aq) and Pu3+(aq) to the oxidation states of plutonium (m′ = +4 or +3) and chloride (m′′ = –1) and the limiting molar ionic conductivities of plutonium  and chloride
and chloride  : such that
: such that  and 66.6 S·cm2 mol−1 for Pu4+(aq) and Pu3+(aq), respectively52 and
and 66.6 S·cm2 mol−1 for Pu4+(aq) and Pu3+(aq), respectively52 and 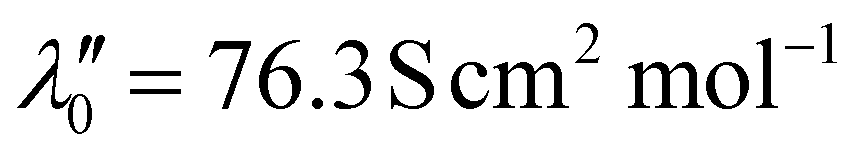 .46 The R, T, and F variables were defined above in eqn (5). This expression enabled the Pu4+(aq) and Pu3+(aq) diffusion coefficients to be estimated based on oxidation state and limiting molar ionic conductivity.
.46 The R, T, and F variables were defined above in eqn (5). This expression enabled the Pu4+(aq) and Pu3+(aq) diffusion coefficients to be estimated based on oxidation state and limiting molar ionic conductivity.
As the HCl(aq) concentration was increased from 1 to 11 M, plutonium species transitioned from Pu(H2O)xn+ (n = 4 or 3), to PuCly(H2O)x4−y, and PuCly4−y. Accompanying this speciation change was a slight decrease in the plutonium diffusion coefficients, such that PuCly4−y moved through the aqueous solution slower than Pu(H2O)x4+. The marginal decrease was attributed to the increased molecular weight of the plutonium species and the increased viscosity of the solution matrixes. Only minor differences between the diffusion coefficients for Pu3+(aq)vs. Pu4+(aq) species were observed.
The diffusion coefficients were used to calculate how the hydrodynamic radii of the Pu4+(aq) and Pu3+(aq) species varied as a function of increasing HCl(aq) concentrations (1 to 11 M) using the Stokes–Einstein equation (eqn (7)).
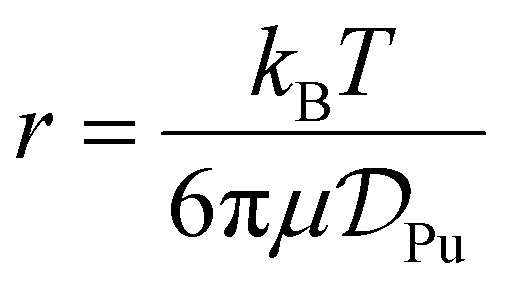 | (7) |
 ; T = temperature, K.
; T = temperature, K.
This analysis assumed the plutonium species were rigid spheres and the experimentally determined hydrodynamic radii for the Pu4+(aq) and Pu3+(aq) species were plotted as a function of HCl(aq) concentration in Fig. 9B. Our experimentally determined hydrodynamic radii tended toward slightly smaller radii [5.1(2) to 2.6(1) Å] as the HCl(aq) concentration increased from 1 to 11 M. These values agreed well with the 3.7 Å theoretical hydrodynamic radius calculated by the group contribution method,53 which summed the atomic volume of each element (Pu and Cl1−). The experimental and theoretical radii were also consistent with the reported hydrodynamic radius of 4.6 Å for Pu(IV) in nitric acid solutions.54,55
These results did not follow the expected inverse relationship between hydrodynamic radii and diffusion coefficients. We suspected the origin of this deviation was related to significant changes in viscosity of the aqueous solutions that occurred when the HCl(aq) concentrations were increased from 1 to 11 M.56 Regarding the size difference between the Pu3+(aq) and Pu4+(aq) species at the same HCl(aq) concentration, there was no significant difference in hydrodynamic radius (t test, two-tailed p > 0.05), indicating the Pu(IV) chloride and aquo complexes were similar in size in solution.
Outlook
Herein, we utilized a combination of XANES and EXAFS, UV-Vis-NIR, and electrochemical methods to advance understanding of how plutonium speciation in aqueous solutions varied as a function of HCl(aq) concentration. Collectively, the data showed how changing the HCl(aq) content from concentrated (11 M) to dilute (1 M) provided control over the electron transfer processes accessible to plutonium. The plutonium speciation profile was dominated by anionic PuCly4−y complexes at high HCl(aq) concentrations (>8 M) and cationic Pu(H2O)x4+ complexes at low HCl(aq) concentrations (≤3 M). Heteroleptic aquochloride complexes, PuCly(H2O)x4−y, dominated the speciation profile over a relatively small HCl(aq) concentration range (∼4.5 to 8 M). Evaluating the X-ray absorption data alongside the electrochemical results provided additional fidelity in terms of the Cl1− for H2O ligand substitution reaction. This analysis showed that an abrupt speciation change occurred when the HCl(aq) content passed over the 5.5 M concentration value, suggesting the plutonium coordination environment was dominated by Cl1− ligands when HCl(aq) concentration was >5.5 M and dominated by H2O ligands when the HCl(aq) concentration was <5.5 M. Despite Cl1− and H2O both being relatively weak field ligands (on the spectrochemical series),57 the ligand environment substantially impacted the half-wave potential (E½) for the reversible Pu4+/3+(aq) one-electron transfer reaction. For instance, the E½ potential was 0.572(2) V for the homoleptic PuCly4−y complex and 0.744(2) V for the homoleptic Pu(H2O)x4+ complex. We interpreted the E½ difference (∼170 mV) as suggesting that anionic Cl1− ligands stabilized electron-deficient Pu4+(aq) over Pu3+(aq) and neutral H2O ligands stabilized electron-rich Pu3+(aq) over Pu4+(aq). These data also showcased the stability of Pu4+(aq) over Pu3+(aq) and the unlikeliness of Pu4+(aq) and Pu3+(aq) to generate PuO2n+(aq) (n = 1, 2) within the surveyed potential window (ca. 0.4 to 1 V).Overall, these findings offer a simple way to control the electron transfer chemistry of plutonium in aqueous solutions. Changing the HCl(aq) concentration altered the coordination chemistry accessible to plutonium, which in turn changed potentials required to access the one-electron Pu(IV) + e1− ⇌ Pu(III) redox reaction. Based on these results, it seemed likely that increasing the electron donating ability of the complexing agent (stronger field than Cl1−) would shift E½ more negative and stabilize electron-deficient Pu4+(aq) further. Conversely, increasing the electron withdrawing properties of the complexing agent should shift E½ more positive and stabilize electron-rich Pu3+(aq) further. It is our hope that these results will help scientists working with plutonium in aqueous solutions by demystifying some of the unexpected electron transfer chemistry plutonium experiences under those conditions. This insight may also aid researchers in better controlling plutonium when conducteing separations for energy-related systems, evaluating the fate and transport of plutonium through aqueous solutions in the environment, and processing aqueous plutonium solutions relevant to national nuclear security missions.
Materials and methods
General consideration
¡Caution! The 238Pu [half-life (t½) = 87.7(1) years], 239Pu [t½ = 24110(30) years], 240Pu [t½ = 6561(7) years], 241Pu [t½ = 14.325(6) years], and 242Pu [t½ = 3.75(2) × 105 years] isotopes – and their radioactive progeny products – present serious health threats because of their α-, β-, and γ-emission.58 Hence, all studies with plutonium were conducted in a laboratory dedicated to studies on radioactive elements. This laboratory is equipped with HEPA-filtered hoods, continuous air monitors, negative pressure gloveboxes, and radiation monitoring equipment. All experiments were carried out with approved safety operating procedures. All free-flowing plutonium solids were handled in negative-pressure gloveboxes equipped with HEPA filters.
Plutonium (a mixture of 238Pu, 0.02 wt%; 239Pu, 93.7 wt%; 240Pu, 6.1 wt%; 241Pu, 0.22 wt%; and 242Pu, 0.03 wt%) used in this study was obtained as either residues from previous experiments, plutonium oxide, or in metallic form dissolved as previously described.59 The NaClO2(s) reagent was obtained commercially (Sigma Aldrich) as a mixture of NaClO2 (80%) and NaCl (20%) and used as received. All water used in these experiments was deionized and passed through a Thermoscientific Barnstead Micropure water purification system until a resistivity of 18.2 MΩ cm was achieved.
The experiments described in this study relied on making aqueous solutions that contained varied amounts of hydrochloric acid, HCl(aq). These solutions were made from serial dilutions of commercially available concentrated HCl(aq) (OPTIMA Grade; Fisher Scientific). The HCl concentration for the concentrated HCl(aq) varied from batch-to-batch, ranging 10.8 to 11.1 M. Hence, we assigned the HCl content in the concentrated solutions as being 11 M and used this value to calculate the HCl(aq) content in all subsequently made serial dilutions.
UV-Vis-NIR absorption measurements of plutonium samples dissolved in HCl(aq)
A Cary 6000i UV-Vis-NIR spectrophotometer was used for all measurements. Data were collected from 10000 cm−1 (1000 nm) to 25000 cm−1 (400 nm). An aliquot (464 μL, 2.1 mg) of the Pu4+(aq) reagent stock solution in HCl(aq) (6 M) was added to a Falcon cone (50 mL). The cone was gently heated on a hot plate (∼70 °C) overnight. The resulting residue was dissolved in a small amount of HCl(aq) (945 μL; 11 M). The solution was loaded into a quartz cuvette equipped with a screw top lid. The lid was closed and sealed with electrical tape. This procedure was used to prepare four additional samples in HCl(aq) at varied concentrations by substituting the HCl(aq) (11 M) used to dissolve the Pu4+(aq) residue for 8, 5.5, 3, and 1 M HCl(aq) solutions. The more concentrated HCl(aq) solutions (11, 8, and 5.5 M) were orange in color, while the more dilute HCl(aq) solutions (3 and 1 M) were peach in color. After data collection was finished, the cuvettes were stored in a room-temperature, dark place. Measurements were taken immediately after sample preparation: 24 hours later, 5 days later, and 7 days later. No colloidal precipitate, solution loss, or color change was observed during this time.
Cyclic voltammetry from Pu4+(aq) dissolved in aqueous solutions
A CH Instruments 620C potentiostat was used for all voltammetric measurements. Experiments were carried out within a HEPA-filtered fume hood using a standard three-electrode cell. An aliquot of a Pu4+(aq) stock solution (16 mg Pu, 1 mL, 67 mM) in HCl(aq) (5.5 M) was transferred to a plastic Falcon cone (50 mL). The solution was heated (near 80 °C) on a hot plate under a stream of filtered air until a soft dryness was achieved. The term “soft dryness” is used to caution against overheating, which can complicate subsequent dissolution efforts. The resulting residue was dissolved in concentrated HCl(aq) (4 mL, 11 M), which generated a solution that was 4 mg L−1 (16.7 mM) in Pu4+(aq). This orange solution was transferred in its entirety (4 mL) to an electrochemical cell. Three electrodes were then submerged into the solution: a platinum disk (2.0 mm diameter) working electrode that had been polished with an alumina (0.05 μm) aqueous slurry on a felt polishing pad, a platinum wire (1.3 mm diameter and 92.4 mm length) counter electrode, and fritted Ag/AgCl reference electrode in a saturated KCl(aq) solution (CH Instruments, Inc.). Voltammograms were collected as a function of scan rates (0.01–10 V s−1) and iR compensation was applied when the scan rate was >1 V s−1 using the CHI630E Electrochemical Analyzer software.The solutions used for the electrochemical assay in concentrated HCl(aq) (11 M) was recovered into a plastic Falcon cone (50 mL). The cone was heated (near 80 °C) on a hot plate under a stream of filtered air to evaporate the solution. The resulting residue was dissolved in HCl(aq) (4 mL; 8 M) and the electrochemical measurements were repeated in analogy to that described above in 11 M HCl(aq). This process was repeated with a series of HCl(aq) solutions: 6, 4.5, 3, and 1 M. Voltammetric measurements were collected in triplicate. This was achieved on three separate days by evaporating a Pu(aq) solution to dryness, dissolving the residue in fresh HCl(aq), and then recovering the Pu(aq) samples. The obtained voltammograms were corrected for the Ag/AgCl drift at non-standard conditions across different chloride concentrations using the Nernst equation.
Preparing plutonium samples dissolved in HCl(aq) for X-ray absorption measurements
An aliquot (29 μL, 2 mg) of the Pu4+(aq) reagent stock solution in HCl(aq) (6 M) was added to a Falcon cone (50 mL). The cone was gently heated on a hot plate (∼65 °C) overnight. The resulting residue was dissolved in a small amount of HCl(aq) (900 μL; 11 M). An aliquot (450 μL, ∼1 mg Pu, ∼0.02 M in Pu) of that solution was loaded into the X-ray absorption sample holder. This procedure was used to prepare four additional samples in HCl(aq) at varied concentrations by substituting the HCl(aq) (11 M) used to dissolve the Pu4+(aq) residue for 8, 5.5, 3, and 1 M HCl(aq) solutions.The X-ray absorption sample holder was custom-made and designed for assaying solutions that contained radiological samples. These holders – and the associated handling procedures – provided adequate containment (three layers) for radiological samples. We additionally deployed appropriate administrative and engineering controls that guarded against the release of radiological material during shipment and data acquisition. The holder consisted of a Teflon body with a 5 mm well. It was equipped with a set of Teflon windows (1 mil thickness) and an additional Kapton window (1 mil thickness). These windows were secured by stainless-steel brackets. Solutions were introduced into the holder through an injection hole that was sealed with a Teflon plug. The plug was secured with an aluminum plate. This primary holder was held within a secondary container, which in turn was nested within the tertiary container. The secondary and tertiary containers were best described as a set of aluminum holders equipped with Kapton windows (2 mil thickness) and rubber gaskets. The assembly was shipped to the Stanford Synchrotron Radiation Lightsource (SSRL). Upon arrival, the samples were surveyed for contamination at the beamline within a radiological tent, placed in a cryostat (which provided tertiary containment), and attached to the SSRL Beamline's 11-2 rail.
Preparing dicesium hexachloroplutonate, Cs2PuCl6, for X-ray absorption measurements
The Cs2PuCl6 compound was made using a modified procedure from the original synthesis.60 In a negative-pressure, air-filled glovebox, a plutonium sample (4.0 g; 16.7 mmol) was suspended in water (11.4 mL). Concentrated HCl(aq) (70.7 mL; 11 M) was then added to the mixture, forming a solution whose total volume was 82.1 mL and that was 0.2 M in plutonium. Analysis by UV-Vis-NIR spectroscopy showed a mixture of Pu(IV) and Pu(III) were present. Separately, NaClO2(s) reagent (80% NaClO2/20% NaCl by weight, 2.24 g; 19.8 mmol) was dissolved in water (3 mL) to make a solution that was 6.6 M in NaClO2(aq). This NaClO2(aq) solution was slowly added to the plutonium solution. After mixing for 20 min, an aliquot (3 mL) was taken from the solution for characterization. Analysis by UV-Vis-NIR spectroscopy showed that the oxidation state for plutonium was +4 and that Pu(III) contaminants had oxidized (see ESI†). The radiochemical analyses (described previously61) confirmed the plutonium concentration was 1.97(1) × 10−4 M. Acid–base titration with a Thermo Scientific Orion Star A121 pH meter using a NaOH solution (0.1 M) showed the HCl(aq) concentration was 9.3 M, and this value was confirmed using a Metrohm chloride ion-selective electrode.Approximately 1 hour after oxidation, a solution of CsCl(aq) [30.1 mL; 3 M in 9.3 M HCl(aq)] was added to the Pu4+(aq) solution [82.1 mL; 1.97(1) × 10−4 M in 9.3 M HCl(aq)] described above. Upon combining these two solutions, a pale-yellow powder immediately precipitated. The slurry was mixed (30 min) and the precipitate was allowed to settle for 12 hours. This powder was isolated by vacuum filtration, washed with HCl(aq) (100 mL, 11 M), and dried in an oven (∼120 °C, 8 hours). Analysis of the filtrate by UV-Vis-NIR absorption spectroscopy showed no detectible plutonium dissolved in solution (see ESI†). Radiochemical analyses showed a precipitation yield of 99.9%. The compound was analyzed by powder X-ray diffraction (see ESI†), and its identity was confirmed as Cs2PuCl6(s) by comparison with previously published data.62
The Cs2PuCl6(s) sample was transferred to a negative-pressure, argon-filled glovebox. A small portion of the sample (3.7 mg, 5.3 μmol) was mixed with boron nitride powder (170.0 mg) in a polystyrene canister (0.5 × 1 inch) that contained a Teflon ball (1/8-inch diameter). The mixture was ground (5 min) using a Wig-L-Bug grinder. The resulting homogeneous powder contained 0.7 wt% Pu(IV), which was calculated to have a plutonium edge jump of 0.15 absorption length in transmission 500 eV above the Pu L3-edge. The fine white powder was loaded into a Teflon NMR tube liner that had been cut to a length of ∼1 inch. This powder was quite flocculant and the potential for spreading unwanted contamination throughout the glovebox was high. The dispersion hazard was mitigated by using a small funnel made from an automated pipette tip (5 mL) to aid in the sample transfer. Additionally, disposable Latex gloves were worn over the glovebox gloves (inside the box) during the solid transfer, and the glovebox floor was covered with aluminum foil to catch any straggling plutonium powder. The powder was packed with a stainless-steel shaft into the Teflon NMR tube liner to generate a tight cylindrical plug (ca. height = 20 mm; diameter = 3 mm). A Teflon stopper was inserted into the tube until it pressed against the plug, which in turn sealed the tube. The tube was transferred to a HEPA-filtered hood and loaded into a slotted aluminum plate. This plate was equipped with two windows made from Kapton tape (1 mil). This primary holder was nested within an additional aluminum sample holder that was equipped with Kapton windows (2 mil). These windows were sealed with indium wire and held in place by stainless-steel brackets and bolts. Care was taken to ensure that the outside of the primary and secondary holders was not contaminated. The sample holder was shipped to SSRL. Upon arrival, the samples were unpackaged at the beamline within a tented workstation and attached to the cold finger of a liquid N2(liq) cryostat, which served as a third layer of containment during the measurements. The cryostat was attached to the SSRL's Beamline 11-2 rail, such that the samples were held at 45° to the incident radiation. The cryostat was attached to a turbo pump and then placed under vacuum (10−6 torr) and cooled with N2(liq) prior to X-ray measurement.
Pu L3-edge X-ray absorption data acquisition
The room-temperature solution-phase Pu L3-edge X-ray measurements were collected under dedicated operating conditions (3.0 GeV, 5%, 500 mA using continuous top-off injections) on end station 11-2. At the time of these measurements, this beamline was equipped with a 26-pole and a 2.0 tesla wiggler. Using a liquid nitrogen-cooled double-crystal Si[220] (ϕ = 90°) monochromator that employed collimating and mirrors, a single energy was selected from the incident white beam. Vertical acceptance was controlled by slits positioned before the monochromator. All measurements were conducted with the monochromator crystals fully tuned. High-energy harmonics were rejected using a 370 mm Rh-coated harmonic rejection mirror. The Rh coating was 50 nm with 20 nm seed coating and the substrate was Zerodur. The harmonic rejection cut-off was set by the mirror angle to 23.5 keV to control which photons experience total external reflection.Beamline 11-2 XAS rail (SSRL) was equipped with three ionization chambers, through which nitrogen gas continually flowed. One chamber was positioned before the sample (10 cm) to monitor the incident radiation (I0). A second chamber was positioned after the sample (30 cm) so that sample transmission (I1) could be evaluated against I0 and so that the absorption coefficient (μ) could be calculated as ln(I0/I1). A third chamber (I2; 30 cm) was positioned downstream from I1 so that the XANES of a calibration foil could be measured against I1. A potential of 1600 V was applied in series to the ionization chambers. The samples were positioned 45° to the incident radiation. Samples were measured in fluorescence. A Lytle detector was used for the HCl(aq) that had concentrations of 1, 3, 5.5, and 8 M. A 100-element Ge detector was used for the 11 M HCl(aq)-containing samples. A Sr (3 mm) filter mounted to Soller slits that were positioned in front of the detector to help remove unwanted X-ray scattering from the samples. All data were collected with vertical slits set at 0.5 mm and horizontal slits set between 7 to 9 mm. A zirconium foil (Zr) was placed between the I1 and I2 ion chambers so that plutonium samples could be calibrated in situ to the energy of the first inflection point of the Zr K-edge (17998 eV).
Pu L3-edge X-ray absorption data analyses
To correct the HCl (11 M) Pu L3-edge data for detector dead time, nonlinear response curves were defined from 0 to ∼70% dead (windowed counts of the emission line versus the total incoming counts into the detector) using a Sr 3 mm filter (300 eV above the Sr K-edge). Each channel was manually surveyed for outliers, which were omitted. The deadtime correction was applied before averaging the individual channels. Then, the six individual scans for each HCl(aq) concentration were calibrated in energy to the first inflection point of the Zr K-edge from a metallic foil (17998 eV), measured in situ. The individual scans were then averaged using IFEFFIT38 within the Athena software package. Within the same program, the data were analyzed by fitting a line to the pre-edge region, which removed the experimental background from the data. Then, a third-order polynomial fit was chosen for the post-edge region of the spectra. The difference between pre- and post-edge functions were set to unity at the first inflection point of the X-ray absorption data. This normalized the absorption jump to 1.0. This normalization procedure gave spectra that were normalized to a single Pu atom. The EXAFS data were analyzed by shell-by-shell fitting methods using the Artemis program and FEFF8.38 The spectra were k3-weighted and Fourier transformed prior to nonlinear least squares curve fitting. The amplitude reduction factor (S02) was set to 0.9 in all solutions. The actinides coordination number (N), scattering path length (R), and mean-squared displacements (σ2) were used as variables and refined as free unless otherwise noted. The FEFF8 calculations were obtained from published SC-XRD data from Pu(H2O)93+, and [PuCl6]2−.39,40
Author contributions
All authors contributed to writing, reviewing, and editing the document. Yufei Wang, Natalie T. Rice, Julia G. Knapp, Stosh A. Kozimor, and Molly M. MacInnes conceived the study and wrote the original draft of the paper. Yufei Wang, Molly M. MacInnes, and Jan Klouda contributed to the cyclic voltammetry measurements. Julia G. Knapp, Natalie T. Rice, and Manuel L. Besmer contributed to the UV-Vis-NIR measurements. Yufei Wang, Molly M. MacInnes, Sara L. Adelman, and Stosh A. Kozimor analyzed the cyclic voltammetry data. Natalie T. Rice, J. Connor Gilhula, Ida D. Piedmonte, Stosh A. Kozimor, Travis Marshall-Roth, and Alexandra L. Nagelski contributed to XAS sample preparation and measurements. Natalie T. Rice and Stosh A. Kozimor analyzed the XAS data. Stosh A. Kozimor and Brian N. Long collected the PXRD measurements. All samples were prepared by Natalie T. Rice, Yufei Wang, Molly M. MacInnes, Julia G. Knapp, Christopher J. Godt, Kelly E. Aldrich, and Brian T. Arko. All authors contributed to writing the manuscript.Data availability
Data associated with this study are available from the corresponding author upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372) for funding the majority of this work. Additional support came from LANL's LDRD projects (20220054DR), the US Department of Energy, National Nuclear Security Association (NNSA), Plutonium Modernization Program (NA-191), the Los Alamos National Laboratory Directors (Rice) and Harold Agnew National Security (Piedmonte) fellowships, and the Glenn T. Seaborg Institute (Gilhula, Klouda, Marshall-Roth). LANL is an affirmative action/equal opportunity employer managed by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, was supported by the US DOE, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515.References
- F. N. von Hippel, Plutonium and Reprocessing of Spent Nuclear Fuel, Science, 2001, 293(5539), 2397–2398, DOI:10.1126/science.1064667%U.
- Y. Wang, Application of a hydroxypyridinone chelator to f-element separations for aqueous reprocessing of used nuclear fuel, University of California, Berkeley, 2021 Search PubMed.
- A. B. Kersting, Plutonium Transport in the Environment, Inorg. Chem., 2013, 52(7), 3533–3546, DOI:10.1021/ic3018908.
- T. E. Payne, J. J. Harrison, C. E. Hughes, M. P. Johansen, S. Thiruvoth, K. L. Wilsher, D. I. Cendón, S. I. Hankin, B. Rowling and A. Zawadzki, Trench ‘Bathtubbing’ and Surface Plutonium Contamination at a Legacy Radioactive Waste Site, Environ. Sci. Technol., 2013, 47(23), 13284–13293, DOI:10.1021/es403278r.
- G. S. Jones, Reactor-Grade Plutonium and Nuclear Weapons: Ending the Debate, Nonproliferation Rev., 2019, 26(1–2), 61–81, DOI:10.1080/10736700.2019.1603497.
- M. Bunn and E. P. Maslin, All Stocks of Weapons-Usable Nuclear Materials Worldwide Must be Protected Against Global Terrorist Threats, J. Nucl. Mater. Manage., 2011, 39, 21–27 Search PubMed.
- S. F. Ashley, G. J. Vaughan, W. J. Nuttall and P. J. Thomas, Considerations in relation to off-site emergency procedures and response for nuclear accidents, Process Saf. Environ. Prot., 2017, 112, 77–95, DOI:10.1016/j.psep.2017.08.031.
- D. L. Clark, S. S. Hecker, G. D. Jarvinen and M. P. Neu, Plutonium, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Netherlands, 2006, pp. 813–1264 Search PubMed.
- C. F. Metz, Analytical Chemistry of Plutonium, Anal. Chem., 1957, 29(12), 1748–1756, DOI:10.1021/ac60132a023.
- B. G. Harvey, H. G. Heal, A. G. Maddock and E. L. Rowley, 188. The chemistry of plutonium, J. Chem. Soc. (Resumed), 1947,(0), 1010–1021, 10.1039/JR9470001010.
- B. Allard, H. Kipatsi and J. O. Liljenzin, Expected species of uranium, neptunium and plutonium in neutral aqueous solutions, J. Inorg. Nucl. Chem., 1980, 42(7), 1015–1027, DOI:10.1016/0022-1902(80)80394-0.
- R. J. Lemire and P. R. Tremaine, Uranium and plutonium equilibriums in aqueous solutions to 200.degree.C, J. Chem. Eng. Data, 1980, 25(4), 361–370, DOI:10.1021/je60087a026.
- G. R. Choppin, A. H. Bond and P. M. Hromadka, Redox speciation of plutonium, J. Radioanal. Nucl. Chem., 1997, 219(2), 203–210, DOI:10.1007/BF02038501.
- I. Zilbermann, E. Maimon, H. Cohen and D. Meyerstein, Redox Chemistry of Nickel Complexes in Aqueous Solutions, Chem. Rev., 2005, 105(6), 2609–2626, DOI:10.1021/cr030717f.
- M. Uudsemaa and T. Tamm, Density-Functional Theory Calculations of Aqueous Redox Potentials of Fourth-Period Transition Metals, J. Phys. Chem. A, 2003, 107(46), 9997–10003, DOI:10.1021/jp0362741.
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Redox and spectral properties of monooxo polypyridyl complexes of ruthenium and osmium in aqueous media, Inorg. Chem., 1984, 23(13), 1845–1851, DOI:10.1021/ic00181a014.
- S. J. Slattery, J. K. Blaho, J. Lehnes and K. A. Goldsby, pH-Dependent metal-based redox couples as models for proton-coupled electron transfer reactions, Coord. Chem. Rev., 1998, 174(1), 391–416, DOI:10.1016/S0010-8545(98)00143-X.
- K. L. Nash and J. C. Sullivan, Chapter 102 Kinetics of complexation and redox reactions of the lanthanides in aqueous solutions, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1991, vol. 15, pp. 347–391 Search PubMed.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Being Excited by Lanthanide Coordination Complexes: Aqua Species, Chirality, Excited-State Chemistry, and Exchange Dynamics, Chem. Rev., 2002, 102(6), 1977–2010, DOI:10.1021/cr010452+.
- L. G. Nielsen and T. J. Sørensen, Including and Declaring Structural Fluctuations in the Study of Lanthanide(III) Coordination Chemistry in Solution, Inorg. Chem., 2020, 59(1), 94–105, DOI:10.1021/acs.inorgchem.9b01571.
- J. A. Rard, Chemistry and thermodynamics of europium and some of its simpler inorganic compounds and aqueous species, Chem. Rev., 1985, 85(6), 555–582, DOI:10.1021/cr00070a003.
- D. E. Morris, Redox Energetics and Kinetics of Uranyl Coordination Complexes in Aqueous Solution, Inorg. Chem., 2002, 41(13), 3542–3547, DOI:10.1021/ic0201708.
- A. Ikeda, C. Hennig, S. Tsushima, K. Takao, Y. Ikeda, A. C. Scheinost and G. Bernhard, Comparative Study of Uranyl(VI) and -(V) Carbonato Complexes in an Aqueous Solution, Inorg. Chem., 2007, 46(10), 4212–4219, DOI:10.1021/ic070051y.
- H. Moll, A. Rossberg, R. Steudtner, B. Drobot, K. Müller and S. Tsushima, Uranium(VI) Chemistry in Strong Alkaline Solution: Speciation and Oxygen Exchange Mechanism, Inorg. Chem., 2014, 53(3), 1585–1593, DOI:10.1021/ic402664n.
- J. P. Austin, M. Sundararajan, M. A. Vincent and I. H. Hillier, The geometric structures, vibrational frequencies and redox properties of the actinyl coordination complexes ([AnO2(L)n]m; An = U, Pu, Np; L = H2O, Cl−, CO32−, CH3CO2−, OH−) in aqueous solution, studied by density functional theory methods, Dalton Trans., 2009,(30), 5902–5909, 10.1039/B901724K.
- Z. Szabó, T. Toraishi, V. Vallet and I. Grenthe, Solution coordination chemistry of actinides: Thermodynamics, structure and reaction mechanisms, Coord. Chem. Rev., 2006, 250(7), 784–815, DOI:10.1016/j.ccr.2005.10.005.
- A. Dash, R. Agarwal and S. K. Mukerjee, Electrochemical behaviour of uranium and thorium aqueous solutions at different temperatures, J. Radioanal. Nucl. Chem., 2017, 311(1), 733–747, DOI:10.1007/s10967-016-5088-7.
- S. Skanthakumar, G. B. Jin, J. Lin, V. Vallet and L. Soderholm, Linking Solution Structures and Energetics: Thorium Nitrate Complexes, J. Phys. Chem. B, 2017, 121(36), 8577–8584, DOI:10.1021/acs.jpcb.7b06567.
- J. M. Cleveland, Aqueous coordination complexes of plutonium, Coord. Chem. Rev., 1970, 5(1), 101–137, DOI:10.1016/S0010-8545(00)80076-4.
- B. W. Stein, S. A. Kozimor and V. Mocko, The Plutonium Handbook, American Nuclear Society, 2019 Search PubMed.
- P. G. Allen, J. J. Bucher, D. K. Shuh, N. M. Edelstein and T. Reich, Investigation of Aquo and Chloro Complexes of UO22+, NpO2+, Np4+, and Pu3 + by X-ray Absorption Fine Structure Spectroscopy, Inorg. Chem., 1997, 36(21), 4676–4683, DOI:10.1021/ic970502m.
- C. H. Booth, S. A. Medling, Y. Jiang, E. D. Bauer, P. H. Tobash, J. N. Mitchell, D. K. Veirs, M. A. Wall, P. G. Allen and J. J. Kas, et al. Delocalization and occupancy effects of 5f orbitals in plutonium intermetallics using L3-edge resonant X-ray emission spectroscopy, J. Electron Spectrosc. Relat. Phenom., 2014, 194, 57–65, DOI:10.1016/j.elspec.2014.03.004.
- S. D. Conradson, K. D. Abney, B. D. Begg, E. D. Brady, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller and P. L. Gordon, et al. Higher Order Speciation Effects on Plutonium L3 X-ray Absorption Near Edge Spectra, Inorg. Chem., 2004, 43(1), 116–131, DOI:10.1021/ic0346477.
- S. D. Conradson, I. Al Mahamid, D. L. Clark, N. J. Hess, E. A. Hudson, M. P. Neu, P. D. Palmer, W. H. Runde and C. Drew Tait, Oxidation state determination of plutonium aquo ions using X-ray absorption spectroscopy, Polyhedron, 1998, 17(4), 599–602, DOI:10.1016/S0277-5387(97)00287-8.
- K. O. Kvashnina, A. Y. Romanchuk, I. Pidchenko, L. Amidani, E. Gerber, A. Trigub, A. Rossberg, S. Weiss, K. Popa and O. Walter, et al., A Novel Metastable Pentavalent Plutonium Solid Phase on the Pathway from Aqueous Plutonium(VI) to PuO2 Nanoparticles, Angew. Chem., Int. Ed., 2019, 58(49), 17558–17562, DOI:10.1002/anie.201911637 , (acccessed 2024/12/18).
- C. Walther, H. R. Cho, C. M. Marquardt, V. Neck, A. Seibert, J. I. Yun and T. Fanghänel, Hydrolysis of plutonium(IV) in acidic solutions: no effect of hydrolysis on absorption-spectra of mononuclear hydroxide complexes, Radiochim. Acta, 2007, 95(1), 7–16, DOI:10.1524/ract.2007.95.1.7 , (acccessed 2024-12-19).
- M. H. Lee, Y. J. Park and W. H. Kim, Absorption spectroscopic properties for Pu(III, IV and VI) in nitric and hydrochloric acid media, J. Radioanal. Nucl. Chem., 2007, 273(2), 375–382, DOI:10.1007/s10967-007-6848-1.
- J. L. Ryan, Actinide(IV) Chloride Species Absorbed by Anion Exchange Resins from Chloride Solutions1, J. Phys. Chem., 1961, 65(10), 1856–1859, DOI:10.1021/j100827a040.
- G. Bailly, D. Maloubier and G. Legay, Plutonium(IV) quantification in acidic process solutions using partial least-squares regression applied to UV–Vis spectrophotometry, J. Radioanal. Nucl. Chem., 2022, 331(3), 1267–1273, DOI:10.1007/s10967-022-08205-4.
- M. H. Lee, M. Y. Suh, K. K. Park, Y. J. Park and W. H. Kim, Determination of Pu Oxidation states in the HCl Media Using with UV-Visible Absorption Spectroscopic Techniques, J. Nucl. Fuel Cycle Waste Technol., 2006, 4, 1–7 Search PubMed.
- E. Gerber, A. Y. Romanchuk, I. Pidchenko, L. Amidani, A. Rossberg, C. Hennig, G. B. M. Vaughan, A. Trigub, T. Egorova and S. Bauters, et al., The missing pieces of the PuO2 nanoparticle puzzle, Nanoscale, 2020, 12(35), 18039–18048, 10.1039/D0NR03767B.
- C. Ekberg, K. Larsson, G. Skarnemark, A. Ödegaard-Jensen and I. Persson, The structure of plutonium(IV) oxide as hydrolysed clusters in aqueous suspensions, Dalton Trans., 2013, 42(6), 2035–2040, 10.1039/C2DT32185H.
- P. Estevenon, T. Dumas, P. L. Solari, E. Welcomme, S. Szenknect, A. Mesbah, K. O. Kvashnina, P. Moisy, C. Poinssot and N. Dacheux, Formation of plutonium(IV) silicate species in very alkaline reactive media, Dalton Trans., 2021, 50(36), 12528–12536, 10.1039/D1DT02248B.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 58(12), 7565–7576, DOI:10.1103/PhysRevB.58.7565.
- J. H. Matonic, B. L. Scott and M. P. Neu, High-Yield Synthesis and Single-Crystal X-ray Structure of a Plutonium(III) Aquo Complex: [Pu(H2O)9][CF3SO3]3, Inorg. Chem., 2001, 40(12), 2638–2639, DOI:10.1021/ic015509p.
- S. D. Reilly, B. L. Scott and A. J. Gaunt, [N(n-Bu)4]2[Pu(NO3)6] and [N(n-Bu)4]2[PuCl6]: Starting Materials To Facilitate Nonaqueous Plutonium(IV) Chemistry, Inorg. Chem., 2012, 51(17), 9165–9167, DOI:10.1021/ic301518g.
- R. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A, 1976, 32(5), 751–767, DOI:10.1107/S0567739476001551.
- C. W. Kammeyer and D. R. Whitman, Quantum Mechanical Calculation of Molecular Radii. I. Hydrides of Elements of Periodic Groups IV through VII, J. Chem. Phys., 1972, 56(9), 4419–4421 CrossRef CAS.
- C. Batchelor-McAuley, M. Yang, E. M. Hall and R. G. Compton, Correction factors for the analysis of voltammetric peak currents measured using staircase voltammetry, J. Electroanal. Chem., 2015, 758, 1–6, DOI:10.1016/j.jelechem.2015.10.004.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2022 Search PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95(2), 197–206, DOI:10.1021/acs.jchemed.7b00361.
- F. H. David and V. Vokhmin, Thermodynamic properties of some tri- and tetravalent actinide aquo ions, New J. Chem., 2003, 27(11), 1627–1632, 10.1039/B301272G.
- J. R. Welty, G. L. Rorrer and D. G. Foster, Fundamentals of Momentum, Heat, and Mass Transfer, John Wiley & Sons, 2015 Search PubMed.
- A. Fallet, N. Larabi-Gruet, S. Jakab-Costenoble and P. Moisy, Electrochemical behavior of plutonium in nitric acid media, J. Radioanal. Nucl. Chem., 2016, 308(2), 587–598, DOI:10.1007/s10967-015-4423-8.
- F. H. David, Structure of trivalent lanthanide and actinide aquo ions, New J. Chem., 1997, 21(2), 167–176 CAS.
- E. Nishikata, T. Ishii and T. Ohta, Viscosities of aqueous hydrochloric acid solutions, and densities and viscosities of aqueous hydroiodic acid solutions, J. Chem. Eng. Data, 1981, 26(3), 254–256, DOI:10.1021/je00025a008.
- J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhi, Inorganic Chemistry: Principles of Structure and Reactivity, Pearson Education, 2006 Search PubMed.
- National Nuclear Data Center, https://www.nndc.bnl.gov/nudat2/(accessed 2024 December 18).
- S. K. Cary, K. S. Boland, J. N. Cross, S. A. Kozimor and B. L. Scott, Advances in containment methods and plutonium recovery strategies that led to the structural characterization of plutonium(IV) tetrachloride tris-diphenylsulfoxide, PuCl4(OSPh2)3, Polyhedron, 2017, 126, 220–226, DOI:10.1016/j.poly.2017.01.013.
- F. J. Miner, R. P. DeGrazio and J. T. Byrne, Dicesium Plutonium Hexachloride, a Proposed Primary Standard for Plutonium, Anal. Chem., 1963, 35(9), 1218–1223, DOI:10.1021/ac60202a003.
- J. M. Blackader, A. S. Wong, N. D. Stalnaker and J. R. Willerton, Radiochemistry Calculations of Plutonium and 241Am Using the Rad Calc III Program, Los Alamos National Laboratory, Los Alamos, NM, 2001 Search PubMed.
- W. Zachariasen, Crystal chemical studies of the 5f-series of elements. II. The crystal structure of Cs2PuCl6, Acta Crystallogr., 1948, 1(5), 268–269, DOI:10.1107/S0365110X48000715.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional UV-Vis spectra, PXRD pattern, and equation derivation. See DOI: https://doi.org/10.1039/d5qi00409h |
| ‡ These are co-first authors because they contributed equally to this manuscript. |
| This journal is © the Partner Organisations 2025 |