
Recent trends in CO2 electroreduction over metal–organic framework-derived materials: a comprehensive review
Nadia
Gholampour
*a,
Chizoba I.
Ezugwu
b,
Hussein A.
Younus
 *ce,
Damien P.
Debecker
a,
Mohamed
Al Abri
cd,
Rashid
Al hajri
d,
Chih-Ming
Kao
f and
Francis
Verpoort
*ghi
*ce,
Damien P.
Debecker
a,
Mohamed
Al Abri
cd,
Rashid
Al hajri
d,
Chih-Ming
Kao
f and
Francis
Verpoort
*ghi
aInstitute of Condensed Matter and Nanosciences (IMCN), Université Catholique de Louvain (UCLouvain), Place Louis Pasteur, 1, Louvain-la-Neuve, Belgium. E-mail: nadia.gh6183@gmail.com
bCentre of Water Technology (WATEC) and Department of Biological and Chemical Engineering, Aarhus University, Universitetsbyen 36, 8000 Aarhus C, Denmark
cNanotechnology Research Center, Sultan Qaboos University, P.O Box 17, Al Khoud, Muscat, PC 123, Oman. E-mail: hay00@fayoum.edu.eg
dPetroleum and Chemical Engineering Department, Sultan Qaboos University, P.O Box 17, Al Khoud, Muscat, PC 123, Oman
eChemistry Department, Faculty of Science, Fayoum University, 63514, Egypt
fInstitute of Environmental Engineering, National Sun Yat-Sen University, Kaohsiung, Taiwan
gJoint Institute of Chemical Research (FFMiEN), Peoples Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya Str., 117198 Moscow, Russia
hNational Research Tomsk Polytechnic University, Lenin Avenue 30, 634050 Tomsk, Russia
iState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, PR China. E-mail: francis@whut.edu.cn
First published on 25th August 2024
Abstract
Carbon dioxide reduction through electrochemical energy is an emerging and appealing approach towards CO2 mitigation, and it is a potential technique in which the current and faradaic efficiencies can be optimized for the efficient/effective conversion of CO2 to solar fuel (storable high-density chemical energy). However, a challenge associated with the current state-of-the-art electrocatalytic systems is developing efficient, selective, and cost-effective heterogeneous catalysts. In this case, materials derived from metal–organic frameworks (MOFs) are promising electrocatalysts that not only possess porous structures similar to their parent MOFs but are also endowed with improved stability and conductivity, which are required in the CO2 reduction reaction (CO2RR). This review surveys the updated strategies to rationally design efficient MOF-based electrocatalysts and MOF-derived materials for CO2 reduction. Various MOF-derived materials are comprehensively discussed, together with the strategies aimed at improving product selectivity. Furthermore, active sites and detailed underlying mechanisms of CO2 reduction are discussed to gain better insights into the future development of electrocatalysts. This investigation aims to highlight the recent advances in the CO2RR to inspire the development of new techniques in designing electrocatalysts based on MOF structures with high performance and high stability.
1. Introduction
Despite the development of various types of renewable energy sources, fossil fuels remain one of the widely used energy sources, resulting in the emission of a massive amount of CO2 into the atmosphere annually.1,2 One of the leading causes of climate change is CO2 emissions, resulting in a record high CO2 concentration of 420.2 ppm in the atmosphere in 2023. It is known that this increase in atmospheric CO2 has led to an enhancement in Earth's average temperature, which is associated with various forms of environmental disasters, including severe hurricanes and ecosystem destruction. There are two main approaches to decrease the levels of atmospheric CO2: (1) restricting the combustion of fossil resources while curbing the energy demand and using cleaner energy sources, including solar and wind energy (however, their storage is a severe concern in this approach) and (2) capturing and utilizing CO2 to produce other valuable chemicals. The latter approach has drawn the attention of numerous researchers for applying several techniques to fix CO2 into fine chemicals, including photochemical reduction,3–6 biological transformation,7 chemical reduction,8–10 and electrochemical reduction.11–13In this regard, the electrochemical CO2 reduction reaction (CO2RR) is among the potential techniques in which renewable energy is converted CO2 into valuable chemicals and fuels such as methanol, methane, formic acid, CO, and ethanol.14–18 It is worth mentioning that many different catalysts, including homogeneous13,19,20 and heterogeneous21 catalysts, have been developed to achieve this goal, and undeniably, heterogeneous catalysts are prominent over homogeneous catalysts due to their easy recycling, high stability, and low toxicity. The products of the CO2RR depend on the electrolyte medium, applied potential, and, remarkably, the type of applied electrocatalyst. For instance, it has been proven that Ag and Au are suitable electrocatalysts to produce CO, while the formation of hydrocarbons and oxygenate products (ethanol, methanol, and methane) is achieved over Cu catalysts.22 Furthermore, numerous metal-free catalysts composed of carbon materials (graphene, carbon nanotubes, nanoporous carbon, and graphene dots) have been shown to be active in the CO2RR.23–25 In the last two decades, porous frameworks, covalent organic frameworks (COFs),26–28 metal–organic frameworks (MOFs),29–31 and materials derived from them32 have been highlighted as highly promising catalysts for CO2RR.
Metal–organic frameworks (MOFs) are a class of porous polymers that coordinate metal nodes with organic ligands to form a 1D–3D structure. Owing to their large surface area, tunable porosity, and large pore volume by adjusting their metal species or organic linkers, MOFs are recognized as promising materials for many applications, including gas storage/separation, adsorption, water treatment, solar cells, sensing, energy storage (batteries and supercapacitors), catalysis,33–39 and in particular, (electro)catalysis.37,38,40–43 However, although several MOFs have been applied in electrochemical CO2RR, issues such as their low conductivity44,45 and their poor chemical stability in water46,47 hinder their application for this purpose. In contrast, MOF-derived materials can remarkably overcome these issues, and thus considered more promising candidates. MOF-derived materials have attracted attention due to the dual roles of their metal nodes, which on the one hand, can create metal-active sites, and organic linkers, on the other hand, creating heteroatom (N, S, P, B)-doped carbonaceous materials after pyrolysis. Here, the MOF structure plays an essential role as a self-template for the homogeneous dispersion of heteroatoms and metals through the carbon matrix, the formation of single-atom sites, and also the emergence of porosity inherited from the pristine MOF.48–52
This review addresses the recent reported developments on MOF-derived materials employed for electrochemical CO2 reduction. Electrochemical CO2 reduction and the applications of MOF materials are topics of significant interest in recent chemical and material research worldwide. Thus, the record of publications on CO2RR over MOFs and MOF-derived materials is astonishing. Therefore, several excellent reviews were recently published, offering diverse perspectives on this matter.53–55 However, to present a thorough study of the design and synthesis of MOF-derived materials for enhanced electrochemical CO2 reduction performance, a comprehensive investigation is still required. This review will also discuss the role of active sites and involved mechanisms to gain a better understanding into the design of new electrocatalysts in CO2RR based on MOF materials. This review selectively presents some of the recent advances and pertinent challenges in this field, focusing on MOF-derived materials applied for electrochemical CO2 reduction. Initially, we present an overview of the electrochemical reduction of CO2. In the second section, the pristine MOFs used in CO2RR are briefly discussed, focusing on their role in tuning the micro-environment during CO2RR and their stability. In Section 3, the MOF-based composites employed in electrochemical CO2RR are explored. Finally, in Section 4, the active sites and mechanisms of MOF-derived materials in CO2RR are discussed in detail.
1.1 Electrochemical CO2RR
| The type of reaction | The number of electrons | The applied potentiala (E0 (V)) | The possible product |
|---|---|---|---|
| a vs. Standard hydrogen electrode (SHE) at PH 7 at 25 °C. | |||
| 2H+ + 2e− → H2 | 2 | −0.41 | Hydrogen |
| CO2 + 2H+ + 2e− → HCOOH | 2 | −0.61 | Formic acid |
| CO2 + 2H+ + 2e− → CO + H2O | 2 | −0.52 | Carbon monoxide |
| CO2 + 4H+ + 4e− → HCHO + H2O | 4 | −0.51 | Formaldehyde |
| CO2 + 6H+ + 6e− → CH3OH + H2O | 6 | −0.38 | Methanol |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | 8 | −0.24 | Methane |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | 12 | −0.34 | Ethylene |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | 12 | −0.33 | Ethanol |
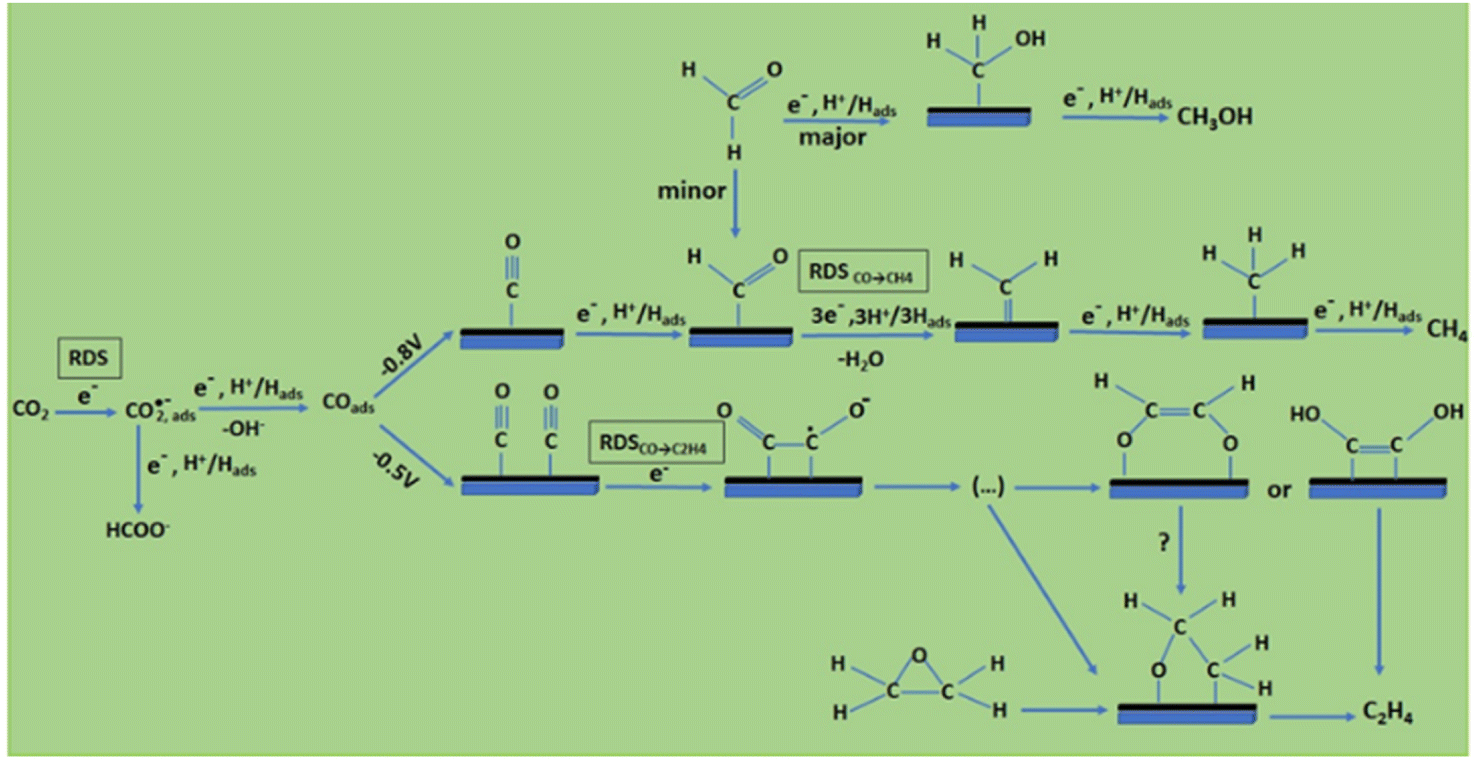
Understanding the mechanism of CO2 electroreduction has been proven to be challenging due to the need for advanced methods to characterize the intermediates formed during the CO2RR. Moreover, given that CO2RR is a multiple electron/proton transfer reaction, controlling the reaction pathways and product selectivity is difficult. According to the applied potential, various C1 and C2 compounds are formed during this reaction process (Table 1). Owing to the high chemical stability of the CO2 molecule, cleaving the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and altering the orientation of the linear CO2 to a bent *CO2δ− molecular structure, which has a lower LUMO energy level, requires a significant energy input of about 750 kJ mol−1. According to the proposed mechanism of electrochemical reduction of CO2 on the Cu electrode surface (Fig. 1),56 a high energy barrier is involved in initial adsorption and transferring one electron to CO2 to generate the *CO2˙− intermediate; this step is identified as the rate-determining step (RDS). Although the exact geometry of the chemisorbed *CO2δ− species on the catalyst surface is still unknown, the three possible orientations are illustrated in Fig. 2.57 The reactivity of the *CO2˙− intermediate on the metal surface plays a significant role in the arrangement of the final products. It was shown that the behavior of the *CO2˙− intermediate on the surface of various metals is different, prompting the formation of different products. For example, on the surface of Sn or In, the *CO2˙− intermediate is bound to the metal via an oxygen atom, which upon further conversion, will lead to the formation of formate (HCOO−). In contrast, the *CO2˙− intermediate is bound via the C atom on the surface of Ag and Au. In this case, *COOH will be formed, which can be reduced to CO.58 Among the metals studied by Hori et al.,58 Cu was found to be the only metal that can still catalyze CO2 conversion to HCOOH, CO, CH4, and C2H4. It has been discovered that the main limiting step for hydrocarbon generation is the protonation of CO* to CHO*.59
O bonds and altering the orientation of the linear CO2 to a bent *CO2δ− molecular structure, which has a lower LUMO energy level, requires a significant energy input of about 750 kJ mol−1. According to the proposed mechanism of electrochemical reduction of CO2 on the Cu electrode surface (Fig. 1),56 a high energy barrier is involved in initial adsorption and transferring one electron to CO2 to generate the *CO2˙− intermediate; this step is identified as the rate-determining step (RDS). Although the exact geometry of the chemisorbed *CO2δ− species on the catalyst surface is still unknown, the three possible orientations are illustrated in Fig. 2.57 The reactivity of the *CO2˙− intermediate on the metal surface plays a significant role in the arrangement of the final products. It was shown that the behavior of the *CO2˙− intermediate on the surface of various metals is different, prompting the formation of different products. For example, on the surface of Sn or In, the *CO2˙− intermediate is bound to the metal via an oxygen atom, which upon further conversion, will lead to the formation of formate (HCOO−). In contrast, the *CO2˙− intermediate is bound via the C atom on the surface of Ag and Au. In this case, *COOH will be formed, which can be reduced to CO.58 Among the metals studied by Hori et al.,58 Cu was found to be the only metal that can still catalyze CO2 conversion to HCOOH, CO, CH4, and C2H4. It has been discovered that the main limiting step for hydrocarbon generation is the protonation of CO* to CHO*.59
 | ||
| Fig. 1 Suggested mechanism of CO2 electrochemical reduction on copper. Reproduced with permission from ref. 56 Copyright 2011, the Royal Society of Chemistry. | ||
 | ||
| Fig. 2 Illustration of the possible orientation of CO2 on the surface of the electrocatalyst. (a) Oxygen coordination, (b) carbon coordination, (c) mixed coordination. Reproduced with permission.57 Copyright 2016, the Royal Society of Chemistry. | ||
Considering the structure and pH effects on the reduction products as well as density functional theory (DFT) results, Kortlever et al.15 proposed the mechanism for CO2 reduction on Cu (Fig. 3). The C1 pathway exhibits methane production, where formyl (*CHO) or *COH species is formed as the reduction product of the CO intermediate, which is subsequently reduced to methane. It has been reported that at high overpotentials, intermediate dimerization may lead to ethylene production in the C1 pathway. The dimerization of CO occurs at low overpotentials, which is the C2 pathway and is the rate-determining step for CO reduction by generating a *C2O2− intermediate through the transfer of an electron, explaining the preference for an alkaline medium for reducing CO.
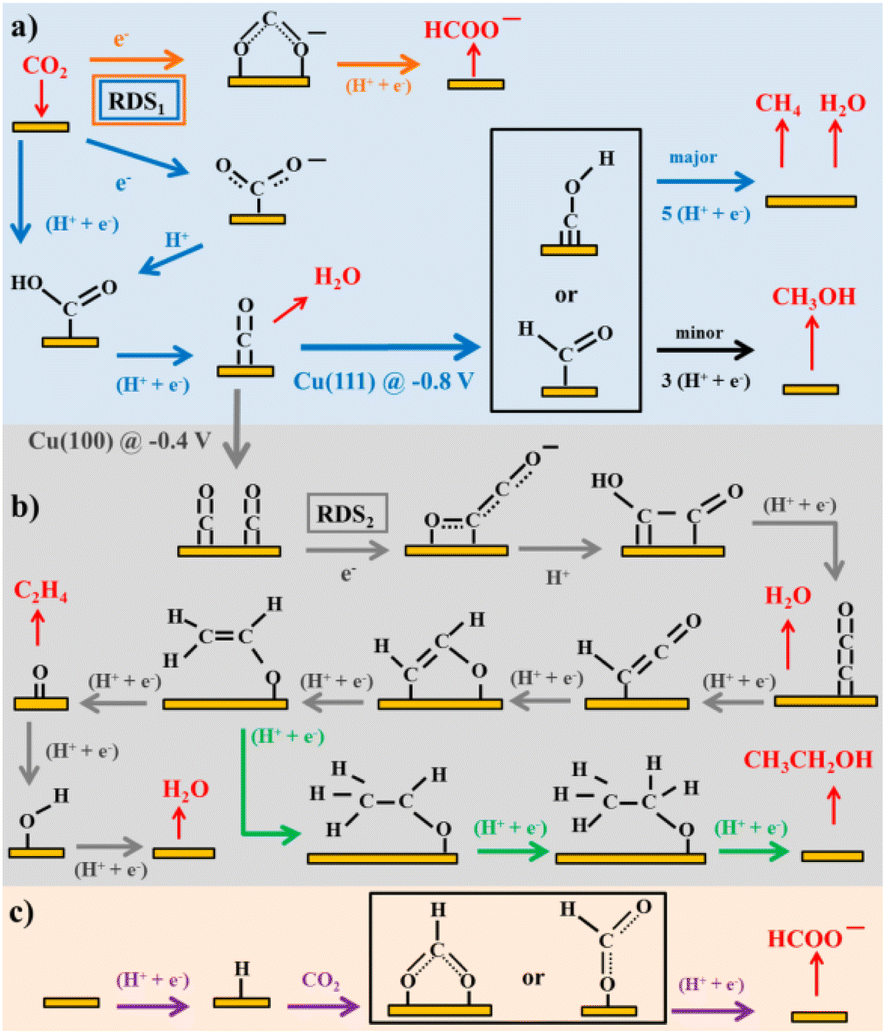 | ||
| Fig. 3 Plausible CO2 electrocatalytic reduction reaction pathways using transition metals and molecular catalysts: (a) blue arrows show the pathways from CO2 to CO and CH4, black arrows to CH3OH, and orange arrows to HCOO−; (b) grey arrows display the pathways from CO2 to ethylene and green arrows to ethanol; and (c) purple arrows demonstrate the pathway of CO2 insertion into a metal–H bond resulting formate. Adsorbates species are in black, while reactants and products in the solution are in red. Potentials are determined vs. RHE. RDS stands for the rate-determining steps and the steps in which either coordinated or separated proton–electron occurs is indicated by H+, e−. Reproduced with permission.15 Copyright 2015, the American Chemical Society. | ||
 | ||
| Fig. 4 Schematic of the cell applied in the electrochemical reduction of CO2. | ||
Practically, there are two values of pH in an H-type cell, the bulk electrolyte pH and the local pH at the surface of the working electrode. The selectivity and overpotentials in CO2RR are affected by the electrolyte pH. For instance, unlike C2H4 formation, the production of CH4 is pH-dependent.66 Therefore, enhanced selectivity for C2H4 in an alkaline solution and a reduced overpotential could be achieved.67,68 It has been realized that larger cations such as Cs+ and K+ prompt an increment in the total current density and selectivity towards H2 and C2H4 production.64,69 However, the effects of anions have not been studied as much as cations. It was discovered that strongly solvated anions such as OH− stabilize the rate-limiting CO2− intermediate, leading to higher selectivity and lower overpotentials for CO production in comparison with anions such as Cl− that are less solvated.66 Alternatively, non-aqueous electrolytes, such as N,N-dimethylformamide (DMF) and acetonitrile, can also be used for CO2RR, offering the advantages of higher CO2 solubility compared to aqueous conditions and low hydrogen concentration to enhance CO2RR and minimize the competitive HER, respectively.70,71 However, these electrolytes may interact with the CO2RR pathways differently.65 In non-aqueous electrolytes, the reaction pathways and reduction products will vary from aprotic to protic solvents, where HER can still be achieved in protic electrolytes, and the electrodes can achieve the formation of hydrocarbon products. Therefore, the choice of electrolyte significantly impacts the product selectivity depending on the capability of the electrolyte to act as a proton source.71,72
In real electrolyzers, CO2RR takes place within a gas–liquid–solid triple-phase boundary. However, achieving economic viability is challenging due to the limited mass transfer, product selectivity issues, and high cell voltages at high current densities, which result in significant ohmic losses and electrode overpotentials.73 Thus, ensuring an adequate supply of gas reactants to the catalyst surface is crucial at higher current densities to sustain high reaction rates.74,75 Nevertheless, most of the current research emphasizes catalyst materials rather than mass transfer or the microenvironment. Often, the solubility and diffusion of CO2 in the electrolyte limit the rate of CO2 mass transfer, and consequently the overall reaction rate. Increasing the solubility of CO2 can be achieved by operation at high pressure or using a more costly, and often more toxic or corrosive solvent as the electrolyte. Alternatively, a more promising approach to overcoming both the CO2 solubility and diffusion limitations is the use of gas diffusion electrodes (GDEs), which are porous electrodes with a high surface area. By using GDEs in CO2RR, researchers have achieved high current densities exceeding 100 mA cm2, which are an order of magnitude greater than that obtained with traditional aqueous systems under similar conditions.76,77
In recent advancements in CO2RR technologies, GDEs and membrane electrode assemblies (MEAs) have been pivotal in achieving higher performances.78,79 GDEs are assembled into MEAs by first depositing a catalyst layer onto a porous gas diffusion layer (GDL), and then hot-pressing this assembly onto a pre-treated proton exchange membrane (PEM) to ensure proper adhesion and optimal contact. Subsequently, this integrated structure is incorporated into the electrochemical cell, facilitating efficient gas transport, catalyst utilization, ion conductivity for enhanced CO2 reduction performance, and efficient removal of products. This design is beneficial to maintain an optimal chemical environment at the catalyst surface, which is crucial for enhancing the CO2RR rate and selectivity. The use of GDEs is particularly effective in suppressing HER by ensuring a higher local concentration of CO2 and reducing the CO2 diffusion distance.80–82 MEA cells incorporating GDEs have demonstrated significant potential in scaling up CO2RR for industrial applications.83 For instance, the highest reported current densities and faradaic efficiencies (FE) in CO2RR to date have been achieved in flow cells utilizing GDEs.84–87
2. MOFs for electrochemical CO2 reduction
One of the first examples of the application of MOF in CO2RR was reported by Kumar et al.88 in 2012, where they studied the catalytic performance of Cu3(BTC)2 immobilized on glassy carbon (GC) in a non-aqueous medium (DMF, comprising tetrabutylammonium tetrafluoroborate, as the supporting electrolyte, saturated with CO2). Oxalic acid (the product) was detected with a faradaic efficiency (FE) of 51%. The highly reproducible reversible redox reactions of Cu(II)/Cu(I) and Cu(I)/Cu(0) were revealed by cyclic voltammetry (CV) measurements in a KCl solution of 0.1 M, indicating the mechanical and electrochemical stability of MOF film@GCE under the experimental conditions. These redox peaks were not observed in copper foil or electrochemically deposited copper metal. In the same year, Hinogami et al.89 described a copper rubeanate metal–organic framework (CR-MOF) coated on carbon paper (CP) as the working electrode for CO2RR in 0.5 M KHCO3 aqueous solution. Cyclic voltammetry revealed that this CR-MOF was more active compared to the Cu electrode, with a selectivity of more than 98% towards the formation of HCOOH. The remaining was attributed to hydrogen selectivity. However, the Cu electrode produced a range of products, including HCOOH, CO, and hydrocarbons. This variation in product selectivity obtained by CR-MOF and metallic Cu was assigned to the different electronic environments of Cu in the two structures, where in CR-MOF, Cu is coordinated to organic ligands, and thus it is ionic and the density of electrons is lower than that in metallic Cu. The decreased electron density resulted in weak CO2 adsorption on the reaction site on CR-MOF, leading to the preferential formation of HCOOH. Achieving different products in these two studies over Cu-based MOFs as electrocatalysts can be explained as follows: Kumar et al.88 applied 1,3,5-benzenetricarboxylic acid (BTC) as the ligand, which contains oxygen atoms (hard donors) coordinated to Cu, while in CR-MOF, the ligand contains N and S donor atoms (soft donors), resulting in the formation of a different electronic environment of metal ions, and accordingly different catalytic activity. Furthermore, the reaction medium in these two studies was different, where in the research performed by Kumar et al., the electrolyte was organic (less protons) compared to the work carried out by Hinogami et al.,89 where an aqueous solution (abundant protons) was employed. Alternatively, Kang et al.90 investigated the effect of different types of ionic liquids (IL) as electrolytes on the product of electrochemical reduction of CO2 over Zn-1,3,5-benzene tricarboxylic acid metal–organic frameworks (Zn-BTC MOFs). The morphology of Zn-MOFs significantly influenced the electrochemical reduction of CO2, with sheet-like Zn-MOFs displaying the greatest activity due to their expansive electroactive surface areas. The effectiveness of the electrolytes is enhanced by using imidazolium-based ILs with fluorine, which interact more strongly with CO2, leading to a higher performance. This combination resulted in a CH4 selectivity exceeding 80% under the optimal conditions, demonstrating the critical role of both the MOF structure and composition of the ILs in the reaction efficiency.2.1 Selectivity
At this stage, the low selectivity for CO2RR has been a challenge that is worth considering. When an aqueous electrolyte is employed in CO2RR, HER is a competitive process due to its lower kinetic barrier in comparison to CO2RR. In addition, unlike HER, which is a single-product reaction, in CO2RR, different carbon-based products, including CO, formate, alcohols, and hydrocarbons may be produced. In this case, multiple MOFs have been created with different metal centers and organic linkers to be applied in CO2RR to produce CO, alcohols, hydrocarbons, or formate with high selectivity. Electrochemical CO2RR is a sustainable pathway to produce syngas, where a stream of CO can be generated from CO2, H2O, and electricity at a high yield under near ambient conditions. Given that CO is a more valuable syngas product,91 efforts have been devoted to driving the CO2RR to the highest possible selectivity for CO.92 In this aspect, several MOFs have been applied in CO2RR to produce CO as the significant product, including ZIF-based catalysts,93–95 1,10-phenanthroline-doped ZIF-8,96 porphyrin-based MOFs,45,47,97–100 nanoparticles embedded in porphyrin-based MOFs,101 and phthalocyanine-based MOFs.102,103In an interesting example, Hod et al.97 developed a catalyst with an FE of almost 100% via the deposition of MOF-525, which contained meso-tetra(4-carboxyphenyl)porphyrin (H4TCPP) as a linker and Zr as the metal node on glass substrates doped with tin oxide fluoride (FTO), followed by post-treatment of the MOF thin film with iron chloride to metalate H4TCPP by iron (Fig. 5a). Three distinct redox waves attributed to Fe(III/II) (Ef = −0.32 V vs. NHE), Fe(II/I) (Ef = −0.87 V vs. NHE), and Fe(I/0) (Ef = −1.4 V vs. NHE) were observed in the CV measurements, signifying the charge transfer by redox hopping between the Fe-TCPP adjacent sites enabled by the Fe-MOF-525 film (Fig. 5b). CO (15.3 μmol cm−2) and H2 (14.9 μmol cm−2) were determined to be the main products. It was demonstrated that the addition of 1 M 2,2,2-trifluoroethanol (TFE) to the catalytic reaction as a weak Brønsted acid resulted in an increase in current density to 5.9 mA cm−2 (Fig. 5c), and also higher catalyst stability, which is in accordance with the results obtained for the homogeneous catalyst (Fe-TPP) by adding TFE.105 Achieving high selectivity toward formate in the electrolysis process is energetically inefficient and is attainable at high cathodic potentials. Interestingly, among the various materials applied in the production of formate via CO2 electrolysis, MOF-based catalysts show potential as excellent platforms for determining the design characteristics of electrocatalysts106 and bimetallic catalysts.107 This can be ascribed to the molecular nature of MOFs and their well-defined tunable structure, enabling detailed studies of the reaction mechanism and reactive intermediates. CO2-derived methane could be effectively utilized in energy infrastructure. To realize the production of potential solar fuel,9,108 MOFs can be promising candidates to convert CO2 to hydrocarbons in electrocatalysis reactions.104,109 In the research carried out by Weng et al.,104 the electrocatalytic activity of the HKUST-1 MOF (comprised of copper(II) and benzene-1,3,5-tricarboxylate (BTC)) together with copper(II) 1,4,8,11-tetraazacyclotetradecane chloride ([Cu(cyclam)]Cl2) and copper(II) phthalocyanine (CuPc) towards CH4 production was investigated, in which Cu(II) was located in a different electronic structure (Fig. 5d). The CuPc catalyst exhibited the highest activity (FE of 66%) and selectivity to produce CH4 compared to [Cu(cyclam)]Cl2 and HKUST-1 (Fig. 5e and f, respectively). Alcohols, including methanol and ethanol, are other targets that can be produced via the electrochemical reduction of CO2. Thus, methanol will be directly produced without applying fossil fuels, and interestingly the synthesized methanol can be converted to gasoline.110 In the discussion of applied materials in CO2RR to produce alcohols, Cu is one of the most promising transition metals applied in CO2 reduction to obtain alcohol and hydrocarbons; nonetheless, the achieved selectivity of the products is low.111 In this regard, several Cu-based MOFs have been successfully utilized in CO2RR to produce alcohols.112–114 In a study performed by Perfecto-Irigaray et al.,115 a faradaic efficiency of 47.2% associated with methanol and ethanol was achieved over a Cu-based MOF. They applied doped metals, including Ru(III), Zn(II), and Pd(II), to partially replace the Cu(II) atoms in the structure of HKUST-1. It was demonstrated that the Ru-doped samples had a noticeable positive impact on the alcohol yield, while Pd(II) hindered the electroreduction of CO2 to liquid products. In a recent study, Zhang et al. demonstrating that manipulating the coordination environment of copper as the metal active site in a copper-based metal–organic framework (Cu-MOF) with various halogen atoms resulted in an improved selectivity for CH4 production in the CO2 reduction reaction (CO2RR).116 A list of the MOFs employed in the CO2RR to produce a range of products is presented in Table 2.
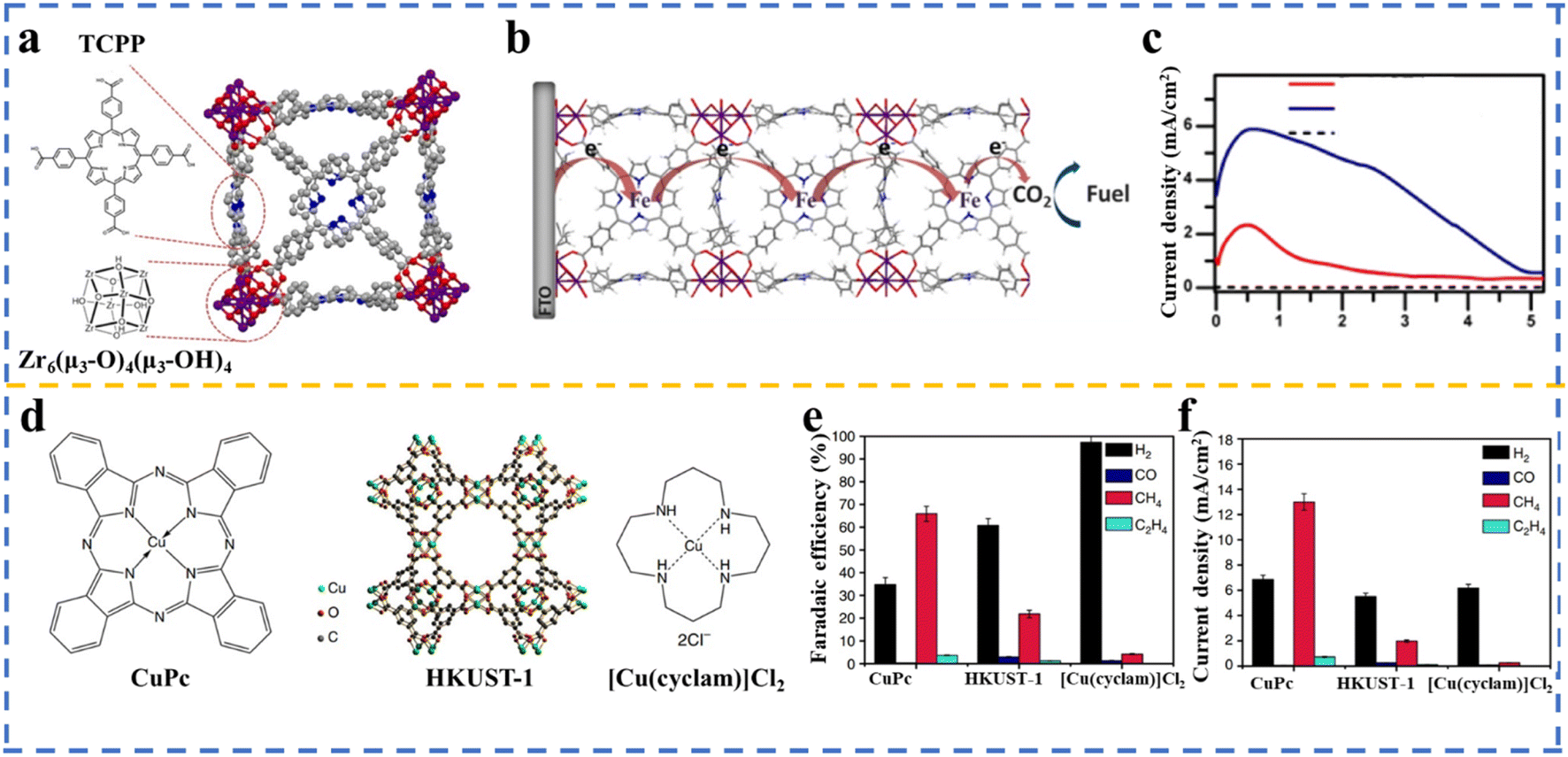 | ||
| Fig. 5 (a) Illustration of a part of MOF-525 and the structure of the TCPP ligand and the Zr6-based node. (b) Transfer charge by redox hopping between neighboring Fe-TCPP sites. (c) Current density of Fe-MOF-525 obtained under various conditions. Reproduced with permission.97 Copyright 2015, the American Chemical Society. (d) Structure of HKUST-1 (copper(II) benzene-1,3,5-tricarboxylate (BTC) MOF), copper(II) phthalocyanine (CuPc), and copper(II) 1,4,8,11-tetraazacyclotetradecane chloride [Cu(cyclam)]Cl2. (e) Faradaic efficiency (%) and (f) current density over three Cu-based catalysts at −1.06 V vs. RHE. Adopted with permission.104 Copyright 2018, Nature Publishing Group. | ||
| Electrocatalyst | Applied potential | Electrolyte | Active site | Catalytic efficiency (%) | Ref. |
|---|---|---|---|---|---|
| a Reversible hydrogen electrode. b TCPP-H2 = 4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate. c Tetrabutylammonium tetrafluoroborate/dimethylformamide. d Standard hydrogen electrode. e 1-Butyl-3-methylimidazolium perchlorate. f Saturated calomel reference electrode. g Tetrabutylammonium hexafluorophosphate. h p-Sulfamidobenzoic acid. i Polyoxometalatemetalloporphyrin organic frameworks. j Al-PMOF [Al2(OH)2(TCPP)], TCPP = tetrakis(4-carboxyphenyl)porphyrin. k Gas diffusion electrode. l Chronopotentiometry. m L = 2,2′-bipyridine-5,5′-dicarboxylic acid. n Normal hydrogen electrode. o Tetrabutylammonium hydroxide. p 4,4′-Dicarboxylic-2,2′-bipyridine. | |||||
| NNU-15 | −0.6 V vs. RHEa | 0.5 M KHCO3 | Ligand (TIPP) | FECO = 99.2 | 45 |
| Co porphyrinb MOF(Al2(OH)2TCPP-Co) | −0.7 V vs. RHE | 0.5 M K2CO3 | Ligand (Co-TCPP) | FECO = 76 | 47 |
| Cu-based MOF | −2.5 V vs. Ag/AgCl | 0.01 M TBATFB/DMFc | Metal cluster (Cu–O) | FEoxalate = 51 | 88 |
| Copper rubeanate MOF | −1.2 V vs. SHEd | 0.5 M KHCO3 | Metal cluster (Cu–O) | FEHCOOH = 42 | 89 |
| Zn-BTC MOFs | −2.2 V vs. Ag/Ag+ | BmimClO4 | Metal cluster (Zn–O) | FECH4 = 88.3 | 90 |
| ZIF-8 | −1.8 V vs. SCEf | 0.5 M NaCl | Metal node (Zn–O) | FECO = 65 | 93 |
| ZIF-8 | −1.1 V vs. RHE | 0.25 M K2SO4 | Metal cluster (Zn–O) | FECO = 81 | 94 |
| Ni(Im)2 nanosheets | −0.85 V vs. RHE | 0.5 M KHCO3 | Metal nodes (Ni2+) | FECO = 78.8 | 95 |
| 1,10-Phenenthroline doped ZIF | −1.1 V vs. RHE | 0.1 M KHCO3 | Ligand(2-methylimidazole) and 1,10-phenanthroline | FECO = 90.6 | 96 |
| MOF-525 | −1.3 V vs. NHE | 1 M TBAPF6 | Ligand (Fe-TCPP) | FECO = almost 55 | 97 |
| TCPP(Co)/Zr-BTB-PSABAh | −0.769 V vs. RHE | 0.5 M KHCO3 | Co | FECO = 85.1 | 98 |
| PCN-222(Fe) | −0.6 V vs. RHE | 0.5 M KHCO3 | Ligand (Fe-TCPP) | FECO = 91 | 99 |
| Co-PMOFi | −0.8 V vs. RHE | 0.5 M KHCO3 | Metal cluster (Zn-ε-Keggin cluster) | FECO = 98.7 | 100 |
| Ag@Al-PMOFj | −1.1 V vs. RHE | 0.1 M KHCO3 | Metal (Ag) | FECO = 55.8 | 101 |
| MOF-1992 | −0.52 V vs. RHE | 0.1 M KHCO3 | Ligand (CoPc) | FECO = 80 | 102 |
| PcCu–O8–Zn | −0.7 V vs. RHE | 0.1 M KHCO3 | Metal cluster (ZnO4) | FECO = 88 | 103 |
| GDEk-Cu(BTC) | −2.3 to −2.5 V vs. SCE | 0.5 M NaHCO3 | Metal cluster (Cu–O) | FECH4 = 11 | 109 |
| HKUST-1doped Zn(II), Ru(III) and Pd(II) | CPl mA cm−2 | 0.5 M KHCO3 | Ru(III) | FECH3OH and C2H5OH = 47.2 | 112 |
| Cu and Bi-based MOF (HKUST-1/CAU-17 blends) | −0.1 to −0.7 V vs. RHE | 0.5 M KHCO3 | (CAU-17) | FECH3OH = 8.6, FEC2H5OH = 28.3 | 113 |
| HKUST-1 | −1.16 V vs. Ag/AgCl | 0.5 M KHCO3 | Metal cluster (Cu–O) | FECH3OH = 5.6, FEC2H5OH = 10.3 | 114 |
| Cu-MOF with Cu–Cl, –Br, –I coordination | −1.08 vs. RHE | 1 M KOH | Cu–halogen | FECH4 = 57.2 for Cu–I | 116 |
| Cu nanoparticles@NU1000 | −0.82 V vs. RHE | 0.1 M NaClO4 | Metal (Cu) | FEformate = 31 | 117 |
| ReLm(CO)3Cl | −1.6 V vs. NHEn | 0.1 M TBAHo/MeCN | Ligandp (ReI(CO)3 (dcbpy)Cl) | FECO = 93 | 118 |
| HKUST-1 | −1.06 V vs. RHE | 0.5 M KHCO3 | Metallic Cu nanoclusters | FECH4 = 27 | 119 |
| ZIF-8 doped with Fe, Ni, Cu | −1.0 vs. RHE | 0.5 M KHCO3 | Imidazolate ligand | FECO = 88.5 | 120 |
2.2 Role of MOFs in tuning the micro-environment during CO2RR
Compared with conventional catalysts, MOFs are unique in that they are comprised of three distinct sites for catalytic functions, i.e., a metal component, organic linker, and pore space. This structure provides ordered architectures and customizable chemical functionalities, which are advantageous for establishing accurate structure–activity relationships for CO2RR. This highlights the potential utility of MOFs, offering a fundamental understanding of how different structural features at the molecular level contribute to selectivity and catalytic efficiency. Recent studies have demonstrated the substantial modular tunability of CO2RR activity and selectivity by carefully tailoring the coordination microenvironment of the metal centres within MOF structures.121 For instance, Meng and colleagues achieved tunable control of the catalytic performance by precisely modulating the chemical and structural features at the atomic level. They used four structural analogues of conductive two-dimensional (2D) MOFs made of metallophthalocyanine (MPc) ligands linked by Cu nodes. The catalytic performance, including activity and selectivity, was found to be governed by two key structural factors, i.e., the metal within the MPc (Co vs. Ni) catalytic subunit and the identity of the heteroatomic cross-linkers (O vs. NH) between these subunits. Among the MOFs, CoPc–Cu–O showed the highest selectivity toward CO with an FE of 85% and high current densities of up to −17.3 mA cm−2 at a low overpotential of −0.63 V. The mechanistic studies supported by DFT calculations indicated that the CoPc-based and O-linked MOFs have lower activation energies for the formation of carboxyl intermediates, resulting in higher activity and selectivity compared to their NiPc-based and NH-linked analogues. These findings present a novel approach for designing high-performance CO2 reduction catalysts by strategically combining various structural factors, such as active metal sites, peripheral groups, and secondary sites.122It was also observed that a catalyst featuring dual-copper sites anchored on ultrathin boron imidazolate layers (BIF-102NSs) could significantly enhance the FE and selectivity for C2H4 production. The catalyst design leveraged the modular tunability of MOFs, allowing precise control of the local chemical environment around the active sites. The dual-copper sites within the MOF structure were found to create a cooperative interaction, which enhanced the adsorption and activation of CO2 molecules. The Cl− ions bridging the Cu2 units in BIF-102NSs played a crucial role in modulating the electronic states and adsorption energies, facilitating the C–C coupling necessary for the formation of multi-carbon products. Electrocatalytic tests showed that BIF-102NSs achieved an FE of 11.3% for C2H4 production, which was significantly higher than its iso-reticular counterpart BIF-103 (7.15%) and single-metal counterpart BIF-104 (3.55%). The enhanced performance was attributed to the cooperative nature of the dual-metal sites, which resulted in charge enrichment at the surrounding Cu centres. This charge enrichment is crucial for stabilizing the reaction intermediates and lowering the activation energy barriers, highlighting the importance of the local micro-environment in determining the CO2RR outcomes.123 Likewise, Cu-HITP (HITP = 2,3,6,7,10,11-hexaiminotriphenylene), a 2D MOF featuring CuN4 nodes, effectively stabilized the key intermediates for C–C coupling. In situ infrared spectroscopy and DFT calculations demonstrated that a polydopamine (PDA) coating on Cu-HITP creates a CO2 reduction-favourable local environment, enriched with proton sources and hydrogen-bond donors around the bi-copper active sites. This combination promoted *CO hydrogenation and stabilized the crucial intermediates (*COH and *OCCOH), leading to high selectivity for C2+ reduced products, achieving FEs of 75% for C2+ products and 51% for C2H4 in KHCO3 electrolyte.124
Sun and co-workers developed a series of Cu4X cluster-based MOFs ([Cu4X(TIPE)3]·3X, [X = Cl, Br, I], TIPE = 1,1,2,2-tetrakis(4-(imidazole-1-yl)phenyl)ethene) to investigate the influence of different halogen atoms in the nodes on the selectivity of CO2RR products. The results showed that the Cu–I MOF achieved the highest FE for CO2RR of up to 83.2% at −1.08 V vs. RHE and a current density of 88 mA cm−2. The FE for CH4 (FECH4) was above 50% over a wide potential range, with a maximum FECH4 of 57.2% at −1.08 V and a partial current density of 60.7 mA cm−2. In comparison, the highest FECH4 for Cu–Cl and Cu–Br was only 32.9% and 40.2% at −1.28 V, respectively. The improved performance of the Cu–I MOF was attributed to the larger radius of the iodine atom, which modulated the electronic properties of the Cu active sites. DFT calculations indicated that the formation energy of the intermediates in the potential-determining steps decreased with an increase in the radius of the halogen atom, explaining the superior catalytic activity of the Cu–I MOF for CO2-to-CH4 conversion. The halogen atoms in the Cu4X clusters altered the d-band centre of the Cu atoms, facilitating the adsorption and activation of CO2 molecules and improving the C–C coupling process, which is crucial for producing multi-carbon products like CH4.125
A nitrile-modified MOF (UiO-66-CN) assembled on the surface of Bi-foil significantly enhanced the local concentration of CO2 near the catalyst surface, increasing it by approximately 27 times compared to the bulk electrolyte, reaching 0.82 M. This concentration boost was crucial for improving the CO2RR reaction rate, given the typically low solubility of CO2 in aqueous solutions. Additionally, the MOF stabilized the reaction intermediates, as revealed by operando infrared spectroscopy and supported by DFT simulations, effectively lowering the energy barriers for their conversion into HCOOH. This stabilization fine-tuned the reaction pathways, enhancing both the selectivity and efficiency of CO2RR. By increasing the local CO2 concentration and stabilizing the reaction intermediates, the MOF also reduced the competition from the HER, allowing CO2RR to proceed more selectively and efficiently. In a conventional H-cell setup, the catalyst achieved a faradaic efficiency of up to 93% for HCOOH, with seven times faster kinetics at −0.75 V vs. RHE. When used in the GDE configuration, the current density for HCOOH production reached levels suitable for technical applications at 166 mA cm−2.126
2.3 MOF stability in CO2RR
Some MOFs can encounter stability challenges due to their relatively weak metal coordination bonds, which make them susceptible to water. In the presence of water, hydrolysis of these metal-linker bonds may occur, leading to the irreversible degradation of the MOF framework and the formation of metal hydroxides or hydrated metal species. This potential instability in water, as well as acidic or basic environments, can limit their practical applications in electrocatalysis, particularly under aqueous conditions.127 The stability of MOFs is influenced by the properties of their linker and inorganic cluster, where higher pKa coordination sites and hydrophobic ligands can enhance their stability by forming stronger bonds and protecting their metal centers.128,129 To address the above-mentioned challenges, researchers are focusing on understanding and improving the stability of MOFs under various conditions. This includes exploring the alkalinity of organic ligands, strengthening metal coordination bonds, and shielding functional groups. Stability considerations also extend to the attachment of MOFs to electrode surfaces and their structural integrity under the reaction conditions. Ensuring strong chemical bonding between the MOF and the electrode surface is crucial for long-term stability, with direct growth of MOFs on the electrode forming robust films that prevent their detachment. However, their structural stability can still be compromised by interactions with the electrolyte, reaction intermediates, or applied current, making the metal–ligand bonds particularly vulnerable to hydrolysis.Under aqueous conditions, MOFs built from high-valence metals such as Zr4+ and Ti4+ with carboxylate ligands often show good stability in aqueous media, although they may be unstable at certain pH levels.130 Low-valent metal MOFs with azolate ligands tend to hydrolyse under acidic conditions, while high-valent metal MOFs with carboxylate ligands can decompose in alkaline media.131 Certain anions in solution, such as hydroxide, carbonate, and phosphate, can also destabilize MOFs by competing with their carboxylate ligands for metal coordination. This issue becomes especially concerning during CO2RR, given that it is commonly conducted in aqueous neutral electrolytes, such as CO2-saturated bicarbonate and phosphate buffer, which can exacerbate the instability of MOFs.132
There are numerous reports on MOFs demonstrating stability in CO2RR across both aqueous and non-aqueous electrolytes. These MOFs are capable of producing a range of products, from simple 2-electron reduction products such as CO and formic acid to more complex products such as ethylene and acetic acid, showcasing their versatility and robustness under various reaction conditions.94,95,133–142 However, some studies have demonstrated that MOFs can undergo structural evolution during the catalytic process, leading to their potential deactivation, loss of their surface area, or the formation of new active phases. For example, Yang et al. reported the formation of Cu nanoparticles functionalized by nitrogen-containing ligands during CO2 electrolysis on a Cu–adenine MOF. This transformation was crucial to the high efficiency of CO2 conversion into hydrocarbons, achieving FEs of over 73% for hydrocarbons, with notable selectivity for ethylene and methane.143 Similarly, the HKUST-1 MOF underwent structural decomposition during CO2RR, leading to the formation of Cu clusters, which act as active sites for catalysis. This structural evolution was confirmed through in situ XAS and EPR studies.144 In the case of CuHHTP MOF (HHTP = hexahydroxytriphenylene), its decomposition during CO2RR resulted in the formation of Cu2O quantum dots. These quantum dots served as highly active catalytic sites, demonstrating a significant FE of 73% for the production of CH4.145 These examples underscore the importance of thorough material characterization before and after the catalytic process to identify the active species and understand the structural evolution of MOFs under the reaction conditions.
Given the potential for pristine MOFs to decompose during CO2RR, rigorous characterization techniques are essential to confirm their stability. In the case of MOF decomposition during electrocatalysis, it will be crucial to confirm that the observed carbon products originate from CO2 rather than from the decomposition of the MOF structure during electrocatalysis. Several characterization techniques can be used to either prove the stability or demonstrate the decomposition of MOFs under the operation conditions. For this purpose, both post-catalysis and in situ/operando characterization methods can be employed to ensure their structure stability and the source of the reduction products. Post-catalysis characterization techniques such as XRD, TEM, and XPS are crucial for analyzing the structural and compositional changes in MOFs after CO2RR. XRD can identify any loss of crystallinity or the emergence of new structures/phases within the MOF material, while TEM can visualize the formation of metal nanoparticles or other decomposition products. XPS can offer detailed information about changes in the chemical states and bonding environments of the metal centers, providing further evidence of whether the MOF has maintained its structural integrity. In situ/operando spectroscopic techniques, such as in situ ATR-FTIR, Raman spectroscopy, and XAS (involving extended X-ray absorption fine structure [EXAFS] and X-ray absorption near edge structure [XANES]) provide real-time insight into the structural integrity of MOFs during the CO2RR process. These techniques can monitor the changes in the metal oxidation states, the formation of the reaction intermediates, and any structural transformations within the MOF. By continuously tracking these parameters, researchers can detect whether the MOF remains intact or begins to decompose under the electrochemical conditions, thereby identifying the actual active species involved in the catalytic process. Isotope labeling with 13CO2 is also one of the most effective techniques to directly trace the carbon atoms involved in the reaction. Using 13CO2 as a feedstock allows tracking of the carbon atoms in the resulting products, thereby confirming whether they originate from CO2 or the MOF framework. This approach, often combined with mass spectrometry or NMR spectroscopy, offers a robust way to validate the source of the carbon in the reaction products, ensuring that the observed products are genuinely the result of CO2 reduction.146–150
2.4 Limitations of pristine MOF-based electrocatalysts
Although MOF materials have an ideal porous structure for CO2 adsorption, they face several drawbacks that hinder their commercial/industrial application in CO2RR. In addition to their low hydrolytic stability over a wide range of pH, as discussed in the previous section,151 the poor electrical conductivity of many MOFs leads to a low charge transfer efficiency, resulting in significant ohmic losses during catalysis. This electrically insulating nature of bulk MOFs affects the overall charge transport rate within the framework. Furthermore, the lack of mesoporosity in MOF materials is a limitation given that mesoporosity facilitates liquid mass transfer.47 Given these limitations, researchers were propelled to convert MOFs into more durable and long-lasting materials. Interestingly, inorganic nanomaterials (metal sulfides151 or metal oxides152,153) and organic–inorganic hybrid nanomaterials (carbon composites154,155) can be fabricated via the electrochemical or thermal treatment of MOFs in the air or inert atmosphere. These materials demonstrate higher stability, and in the case of carbon-based MOFs, higher conductivity compared to their parent MOFs. In addition, the inherent properties of the parent MOFs, such as porosity features and high surface areas. Therefore, the use of MOF-derived materials for CO2 electrochemical reduction has attracted growing attention, which is the crucial point of this study and will be discussed further in the following sections.3. MOF-derived composites for electrochemical CO2RR
Carbon-based nanoporous materials are attractive catalysts in electrochemical reactions due to their chemical stability, high electrical conductivity, and the presence of a significant number of mesopores, which can improve the mass transfer of the liquid phase.156–158 As alternatives to metals such as Ag and Au-based catalysts in electrocatalysis,159,160 carbon-supported transition metal catalysts doped with nitrogen (denoted as NC) display unique electrical and chemical characteristics. Moreover, these materials are less susceptible to poisoning, making them more robust in catalytic applications.161 Interestingly, the arrangement of transition metals within the carbon framework significantly influences the product selectivity and catalytic activity. For instance, when iron atoms are bonded with heteroatoms such as nitrogen and/or oxygen in a carbon matrix, the reaction tends to produce CO.162 In contrast, iron nanoparticles anchored to a nitrogen-doped carbon (NC) support primarily lead to the generation of H2.163Regarding this matter, the unique characteristics of MOFs offer a promising approach to providing well-dispersed active sites throughout their networks. This highlights the fact that MOFs are excellent catalyst precursors for creating hybrid catalysts incorporating metal (oxides) nanoparticles and porous carbon materials.164 The resulting product inherits the porous structure from its parent MOF and has the stability required in CO2RR. Moreover, there is a possibility to introduce a variety of heteroatoms, including N, S, and P, into the MOF-derived carbon structure by choosing diverse organic linkers and modulators. In this case, the properties of the catalyst, such as chemical, electrical and functional characteristics, can be tuned.165
Among the carbon-based electrocatalytic materials produced via MOF templates, zeolitic imidazolate framework (ZIF)-derived carbons are considerably investigated, specifically those originating from ZIF-8. This group of MOFs has attracted attention in CO2RR due to their high surface area, microporous structures, facile synthesis, and presence of nitrogen atoms in the imidazolate linker (given that it creates the possibility to distribute the heteroatom N in the carbon matrix after carbonization). Moreover, during the carbonization process, zinc (the metal node in ZIF-8) will evaporate from the final carbon structure because of the high temperature and there is the possibility to subsequently incorporate active metal sites through various approaches; this strategy has been applied for the design of catalytic materials dedicated to electrochemical CO2RR, as addressed in the following section.
3.1 Supported MOF-based materials
The pyrolysis of multiwall carbon nanotube (MWCNT)-supported Fe-ZIF-8 promoted the generation of N-doped porous carbon (NPC) (ZIF-CNT-FA) (Fig. 6a and b), which exhibited an FE of almost 100% towards CO production and a current density up to 7.7 mA cm−2 at an overpotential of 740 mV.166 It was concluded that the efficient mass and electron transport provided by the CNT support and the significant content of Fe–Nx and pyridinic-N as active sites enhanced the catalytic performance. The superior selectivity for CO was achieved through MWCNT, which accelerated the electron and CO2 transport. Moreover, Huang and coworkers48 employed an oxygen-rich MOF (Zn-MOF-74) to prepare N-doped porous carbon (NPC) (Fig. 6c), which featured a high percentage of active nitrogen (pyridinic and graphitic N) sites and possessed a highly porous structure. Through the optimization of the calcination time and temperature, the amount of the active N species could be regulated. The resultant electrocatalyst was efficient for CO2RR with a high CO FE of 98.4% at −0.55 V vs. RHE (Fig. 6d and e). | ||
| Fig. 6 (a) Schematic of enhanced CO FE obtained over MWCN-supported pyrolyzed ZIF for the CO2RR as a result of enhanced electron conductivity and mass transport in these catalysts compared to pyrolyzed ZIF-FA. (b) TEM image of the MWCN-supported pyrolyzed ZIF catalysts. Reproduced with permission.166 Copyright 2017, the Royal Society of Chemistry. (c) Schematic representation of the synthesis of nitrogen-doped porous carbon (NPC) derived from Zn-MOF-74. (d and e) Faradaic efficiency obtained over NPC and porous carbon (PC), respectively. Reproduced with permission.48 Copyright 2020, Wiley-VCH. (f) Schematic demonstrating the approach of embedding Bi nanoparticles in MOF-derived nitrogen-doped porous carbon Bi@NPC and (g) comparison of faradaic efficiency obtained over Bi-NP and Bi@NPC. Reproduced with permission.167 Copyright 2020, Elsevier. (h) Illustration of the preparation process for the Cu-MOF film and its derivatives on Cu-gauze. (i) SEM image of Cu-MOF-1 derivative formed on Cu-gauze at electrodeposition without CTAB. (j) SEM image of the Cu-MOF-1 derivative formed on Cu-gauze via electrodeposition with CTAB, Reproduced with permission.168 Copyright 2020, Wiley-VCH. | ||
Embedding nanoparticles in MOF-based materials is another reliable means of obtaining an efficient CO2RR electrocatalyst. MOF-derived NPC-doped Bi nanoparticles (Bi@NPC) (Fig. 6f) with a unique microstructure achieved a higher CO2 adsorption capacity and faster electrochemical CO2 reduction to formate than the conventional Bi nanoparticles (Fig. 6g).167 At the low potential of 1.5 V (vs. SCE) in 0.1 M KHCO3 solution, Bi@NPC demonstrated a high formate selectivity of 92.0% and excellent formate current density of 14.4 mA cm−2. On continuous electrolysis, Bi@NPC showed no significant degradation over 12 h.
A potential method to take advantage of the intrinsic and distinguishing characteristics of MOF-derived materials as thin films is the direct growth of active materials on three-dimensional (3D) porous conductive substrates. This technique can enhance the active catalytic sites and rates169–171 due to the enhanced mass transport capacity and the diminished contact resistance, which will lead to smooth electron transfer. Therefore, hollow Cu-MOF thin films were developed on 3D Cu gauze (Cu-G) through a facile and controllable strategy, as reported by Zhu et al.168 By applying an electrochemically assisted self-assembly technique, hollow Cu-MOF was formed on the anode (Cu-gauze substrate), which acted as a source of copper ions in an electrolyte of ethanol/water containing surfactant as a structure-directing agent. During the reduction of CO2, the dendritic Cu0 was realized because of the reduction of the hollow Cu-MOF. At the same time, Cu-MOF (the MOF formed without using surfactant) resulted in the formation of Cu0 nanowires (Fig. 6h–j). Linear sweep voltammetry (LSV) was carried out to assess the catalytic activity of the electrocatalysts, employing CO2-saturated 1-butyl-3-methylimidazolium tetrafluoroborate (BmimBF4, 0.5 M)/acetonitrile (MeCN)/H2O (1.0 M) electrolyte. At −1.85 V versus Ag/Ag+, the highest current density and FE (102.1 mA cm−2 and 98.2%, respectively) were obtained over dendritic Cu0 in comparison to Cu0 nanowires, Cu NPs, and Cu2O. In addition, the electrochemical active surface area (ECSA) analysis revealed that the dendritic Cu0 sample had the largest ECSA compared to Cu0 nanowires, Cu NPs, and Cu2O. Interestingly, it was demonstrated that the generation of formate was a potential function, given that both FE and current density for formate increased quickly with an increase in the negative potential and at −1.85 V vs. Ag/Ag+, reached the maximum of 98.2% before dropping. Furthermore, it was indicated that the FE and the current density of formate were higher in IL/MeCN/H2O than in the aqueous electrolyte at all the applied potentials.
3.2 MOF-derived single-atom/site catalysts
Different methods such as metal oxidation, sulfiding, phosphiding, and metal alloying have been demonstrated to successfully engineer transition metal (TM) electronic states for improved CO2RR activities. However, these processes often result in complex atomic structures and coordination, complicating the study and understanding of the possible catalytic active sites. Alternatively, introducing TM atoms in a well-established material matrix can create significant opportunities to tune the electronic properties of TMs as CO2RR active sites, while maintaining a relatively simple atomic coordination for fundamental mechanism studies. Moreover, TM atoms trapped in a confined environment are less likely to move during catalysis, preventing the nucleation or surface reconstruction commonly observed in catalysis and electrocatalysis. This approach can be critical in developing efficient and stable catalysts for electrochemical CO2 reduction. Building on this concept, single-atom transition metals coordinated with nitrogen (M–Nx) in carbon-based materials have attracted significant attention due to their superior electrocatalytic properties. The unique structure and coordination environment of the M–Nx units confer favorable kinetics, resulting in excellent activity. Additionally, single-atom catalysts (SACs) maximize the metal utilization, given that each atom serves as an active site, enhancing the efficiency and reducing the need for precious metals. Their precise coordination environments offer high selectivity for the desired products, while their strong anchoring prevents their agglomeration and maintains their stability under the reaction conditions. Furthermore, the tunable electronic properties and simple structure of SACs facilitate mechanism studies and optimization. This versatility extends their application to various electrochemical processes, including hydrogen evolution, nitrogen reduction, and oxygen reduction, making SACs a promising pathway for developing efficient, stable, and cost-effective catalysts.172–177 MOFs are ideal for constructing SACs due to their high surface area, uniform and tunable pore structures, and strong coordination bonds, ensuring the stable dispersion and isolation of single metal atoms. Additionally, their versatile chemistry and adjustable coordination environment allows for precise tuning of their electronic properties to optimize the catalytic performance.178–180 However, the commonly reported MOF-based SACs are limited to those with N atoms in their structure, such as ZIFs181,182 and porphyrinic MOFs.183,184 | ||
| Fig. 7 (a) Schematic of the formation NiSAs/N–C and (b) EXAFS spectra for Ni SAs/N–C. (Inset): proposed structure of Ni–Ns. (c) FEs of CO obtained over NiSAs/N–C and Ni NPs/N–C. Reproduced with permission.49 Copyright 2017, the American Chemical Society. (d) Representative process for synthesizing NiSA–Nx–C using bimetallic Mg–Ni-MOF-74. (e) FT-EXAFS spectra of NiSA–Nx–C and Ni foil, and (f) faradaic efficiencies of CO. Reproduced with permission.157 Copyright 2020, Wiley-VCH. (g) Representative process for fabricating Ni–N3–C using ZIF-8 and post-synthetic metal substitution, (h) EXAFS data fitting for Ni–N3–C together with the optimized coordination environment of Ni atoms (inset), and (i) faradaic efficiencies of CO achieved over Ni–N3–C, Ni–N4–C, and N–C. Reproduced with permission.185,186 Copyright 2021, Wiley-VCH. | ||
Gong et al.157 prepared Ni single-atom (SA) embedded in N-doped carbon (NiSA–Nx–C) by applying a non-nitrogenous MOF. In their strategy, bimetallic MgNi-MOF-74 was synthesized, in which a large amount of Mg2+ assisted in realizing the isolation of Ni2+ in the framework, and the pyrrole monomer was filled in the channels of MOF as the source of nitrogen, producing polypyrrole@MgNi-MOF-74 after in situ oxidative polymerization in the presence of I2 (Fig. 7d). Single-atom Ni–N–C was formed upon pyrolysis and MgO was removed by etching. The sample was treated at three different temperatures of 600 °C, 800 °C, and 900 °C, and named NiSA–N2–C, NiSA–N3–C, and NiSA–N4–C, depending on their actual nitrogen ratio, respectively. To elucidate the valence state and coordination environment of NiSA–Nx–C (x = 2, 3, 4), X-ray absorption spectroscopy (XAS) was employed. It was found that the positive valence of the Ni atom located between Ni0 and Ni2+ was also consistent with the XPS results. Moreover, according to the Fourier transform-extended X-ray absorption fine structure (FTEXAFS) spectra, the dominant peaks were assigned to the Ni–N (∼1.36 Å) and Ni–C (∼1.87 Å) scattering paths, and no peak related to Ni–Ni was observed for all three samples, confirming the formation of single atoms (Fig. 7e). The results of FT-EXAFS revealed the Ni–N coordination numbers of 4.0, 3.4, and 2.0 for NiSA–N4–C, NiSA–N3–C, and NiSA–N2–C, respectively, which are consistent with the N ratio determined through the elemental analysis. Subsequently, to assess the function of the Ni–N coordination environment on the performance of the catalyst, electrocatalytic CO2 reduction over NiSA–Nx–C in 0.5 M KHCO3 saturated with CO2 was studied. Noticeably, the current density of NiSA–N2–C was higher than that of its counterparts with an N ratio of 3 or 4. As can be observed in Fig. 7f, this sample afforded the maximum CO FE of 98% at −0.8 V, demonstrating the best electrocatalytic activity among the single-atom Ni–N–C catalysts, revealing that the Ni site coordinated by two nitrogen atoms had the best coordination environment for CO2RR. Also, CO and H2 were identified as the products, and no other carbonaceous products were generated. The much lower current density and FECO for the Ni–N–C catalyst containing Ni NPs compared to that of NiSA–N2–C demonstrated the privilege of single atom sites. Furthermore, according to DFT calculation, NiSA–N2–C resulted in the lowest free energy barrier for the rate-limiting step (given that CO2RR to CO generally involves a two-proton and two-electron transfer process) and very low CO* desorption energy, which is assumed to affect the catalytic performance of this sample compared to its counterparts.
Although single-atom catalysts (SACs) are known for their exceptional catalytic activity and selectivity, the primary challenge with these catalysts is the rational control of their coordination microenvironment. Thus, to overcome this, Hai-Long Jiang and co-workers presented a general approach for the rational fabrication of low-coordinate single-atom Ni electrocatalysts using MOFs for highly selective CO2 electroreduction. The catalysts were synthesized through a post-synthetic metal substitution (PSMS) strategy, which involved replacing Zn atoms in a Zn-based MOF with Ni atoms to create Ni–N3–C, where each Ni atom is coordinated by three nitrogen atoms (Fig. 7g and h). The PSMS strategy allows precise control of the coordination environment of SACs, overcoming the limitations of the traditional one-step pyrolysis methods. The Ni–N3–C catalyst exhibited an impressive CO FE of up to 95.6% at −0.65 V, which was significantly higher than that of Ni–N4–C (89.2%) and N–C (76.1%) (Fig. 7i). Theoretical calculations revealed that the lower coordination number in Ni–N3–C facilitates the formation of COOH* intermediates, thus accelerating the CO2 reduction process. Furthermore, Ni–N3–C demonstrated robust stability, maintaining high CO selectivity and a current density over 10 h of electrolysis. The Ni–N3–C catalyst was further tested in a Zn–CO2 battery, showing excellent CO selectivity (over 90% at 2 mA) and stability across 100 discharge–charge cycles. This work emphasizes the critical role of the coordination microenvironment in enhancing the catalytic performance of SACs and offers a promising approach for the development of efficient electrocatalysts for CO2 reduction.185
Ye et al.194 prepared single sites of Fe–N on a carbon matrix via the pyrolysis of post-modified ZIF-8. In this study, they modified the surface of the as-synthesized ZIF-8 with ammonium ferric citrate (AFC) (as an Fe3+ source), and after pyrolysis under an inert atmosphere, C-AFC©ZIF-8 was formed (Fig. 8a). This catalyst exhibited an FE of 89.1% in CO2RR towards CO production, which is attributed to the isolated Fe–N active sites generated on the carbon matrix surface after the pyrolysis, as confirmed by Fourier transformation of the EXAFS analysis.
 | ||
| Fig. 8 (a) Schematic of Fe–N doped porous carbon synthesized by adding ammonium ferric citrate (AFC) to as-synthesized ZIF-8, followed by pyrolysis. Reproduced with permission.194 Copyright 2017, Elsevier. (b) Synthesis process of Co–N4 and Co–N2 active sites using Co/Zn ZIF followed by pyrolysis. (c) EXAFS spectra demonstrating the atomic dispersion of Co atoms in Co–N2, Co–N3, and Co–N4, confirming the lowest N coordination number in Co–N2. Reproduced with permission.50 Copyright 2018, Wiley-VCH. (d) Fourier transform (FT) at R space showing Cu–N. (e) EXAFS data fitted of CuSAs/TCNFs. Reproduced with permission.173 Copyright 2019, the American Chemical Society. Electrochemical performance comparison: faradaic efficiency at different applied potentials for (f) MnSA/NC and (g) MnSA/SNC. Reproduced with permission.195 Copyright 2021, the American Chemical Society. (h) Schematic of the formation of Fe–N–C and Co–N–C using Fe- and Co-doped ZIF-8. (i) Comparative analysis of CO faradaic efficiency and partial current densities across N-doped carbon, Co–N-doped carbon, and Fe–N-doped carbon. Reproduced with permission.162 Copyright 2018, the American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h−1, surpassing many reported metal-based catalysts under similar conditions. Its high catalytic activity was attributed to its lower coordination number, which facilitated the activation of CO2 to the radical anion intermediate, enhancing the CO2 electroreduction process. Mechanistic insights were gained through experimental and theoretical studies, including DFT calculation and X-ray absorption spectroscopy (XAS). These analyses revealed that reducing the coordination number of Co atoms increases the number of unoccupied 3d orbitals, improving CO2 adsorption and reducing the energy barrier for the formation of the CO2˙− intermediate. This study also found that Co–N2 catalysts exhibit lower charge-transfer resistance and more effective CO2 adsorption compared to Co–N4 catalysts.
200 h−1, surpassing many reported metal-based catalysts under similar conditions. Its high catalytic activity was attributed to its lower coordination number, which facilitated the activation of CO2 to the radical anion intermediate, enhancing the CO2 electroreduction process. Mechanistic insights were gained through experimental and theoretical studies, including DFT calculation and X-ray absorption spectroscopy (XAS). These analyses revealed that reducing the coordination number of Co atoms increases the number of unoccupied 3d orbitals, improving CO2 adsorption and reducing the energy barrier for the formation of the CO2˙− intermediate. This study also found that Co–N2 catalysts exhibit lower charge-transfer resistance and more effective CO2 adsorption compared to Co–N4 catalysts.
Jiao et al.199 presented a general approach for the synthesis of single-atom metal embedded in N-doped carbon (M1–N–C; M = Fe, Co, Ni, and Cu) by pyrolyzing porphyrinic multivariate metal–organic frameworks (MTV-MOFs) named M-PCN-222 (M = Fe, Co, Ni, and Cu), which was assembled through the mixed ligand approach by employing M-TCPP (M = Fe, Co, Ni, and Cu) and H2-TCPP, as shown in Fig. 9a. As can be observed in Fig. 9b, single atoms were formed given that no Ni–Ni peak was detected. Interestingly, in the electrochemical reduction of CO2, a very high CO FE of 96.8% at −0.8 V was achieved over Ni1–N–C, outperforming Fe-, Co-, and Cu-based M1–N–C (Fig. 9c). Searching for the reason for the high catalytic performance of the Ni1–N–C catalyst, DFT calculations presented that the rate-determining step for all the M1–N–C catalysts is the formation of *COOH and Ni1–N–C and Fe1–N–C exhibited much lower energy barriers for *COOH compared the other two catalysts. Moreover, a much lower energy barrier for CO desorption was calculated for the Ni1–N–C catalyst. A more positive value of UL(CO2) − UL(H2) limiting potential difference between CO2RR and HER (UL(CO2) − UL(H2); UL = −ΔG0/e) was measured for Ni1–N–C, suggesting its higher CO2RR selectivity than hydrogen evolution.201
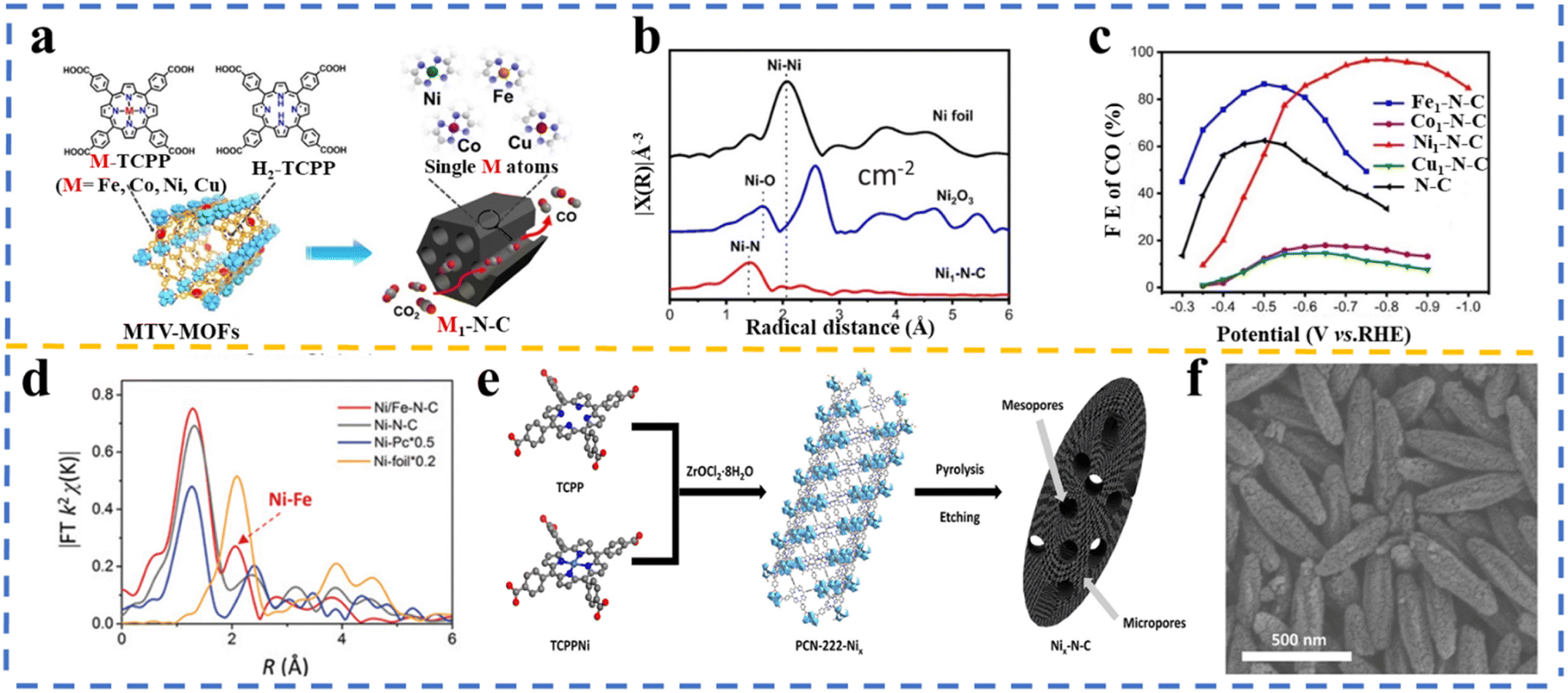 | ||
| Fig. 9 (a) Schematic of the synthesis Fe-, Co-, Ni-, Cu–N–C based on multivariate metal–organic frameworks (MTV-MOFs); (b) k2-weighted FT-EXAFS spectrum and (c) faradaic efficiency of CO obtained over Ni1–N–C, Fe1–N–C, Co1–N–C, Cu1–N–C. Reproduced with permission.200 Copyright 2020, Wiley-VCH. (d) Fourier transform (FT) k2-weighted χ(k) function of the extended X-ray absorption fine structure (EXAFS) spectra. Reproduced with permission.201 Copyright 2019, Wiley-VCH. (e) Schematic of Nix–N–C using Zr-based MOF (PCN-222) and (f) SEM image of Ni20–N–C. Reproduced with permission.202 Copyright 2020, the American Chemical Society. | ||
The synthesis of Fe1–Ni1–N–C was achieved by pyrolyzing ZIF-8 with Fe and Ni-doped ZnO nanoparticles. This process revealed neighboring Fe and Ni single sites, showcasing an impressive faradaic efficiency of 96.2% for CO production in the electroreduction of CO2.52 Subsequently, Zhang et al.203 devised Ni–N–C through the in situ addition of an Ni precursor to the precursor solution of ZIF-8, with the addition of cetyltrimethylammonium bromide (CTAB), followed by the pyrolysis of ZIF-8@CTAB@Ni2+. In the electrochemical reduction of CO2, Ni–N–C demonstrated an outstanding faradaic efficiency of 98% towards CO. It was inferred that the Ni–Nx sites favored CO2 activation, enabling the adsorption of *CO and the establishment of *COOH intermediates.
3.3 MOF-derived nitrogen- and metal–nitrogen-doped porous carbon catalysts
Metal–nitrogen (M–N)204 and metal–nitrogen-grafted porous carbon (M–N–C)205 derived from MOF materials have attracted substantial attention in CO2RR. These M–N and M–N–C materials contain coordinatively unsaturated transition metal–nitrogen sites known as single-atom electrocatalysts.176,177,206–208 The catalytic performance of these sites is highly affected by the coordination environments and the types of metal centers.Wang et al.165 prepared N-doped carbon catalysts employing ZIF-8 as a sacrificial template for CO2RR after etching with acid. In this study, ZIF-8 was carbonized at three different temperatures (700 °C, 800 °C, and 900 °C) denoted as NC-T, with T representing the temperature. Subsequently, the characteristics and electrocatalytic activity of these three samples were investigated. The evaluation revealed that the sample pyrolyzed at the highest temperature exhibited the greatest FE toward CO production, with NC-900 achieving the highest CO FE of approximately 78% at −0.93 V vs. RHE. In addition, the partial current density for CO (jCO) and the highest total current density (jtotal) were also recorded for the NC-900 sample. Raman spectroscopy revealed that the carbon matrix in both NC-800 and NC-900 has equivalent degrees of graphitization, while NC-700 demonstrated an overestimated ratio between the G and D bands of carbon, which was ascribed to the non-pyrolyzed imidazolate.209 It was noted that the surface area of the samples increased by increasing the pyrolysis temperature, suggesting that more residuals were removed from the pores at high temperatures; however, it also led to more agglomeration of the carbon nanoparticles. Furthermore, the study of the N 1s XPS region manifested the presence of four types of nitrogen species, pyridinic-N, pyrrolic-N, quaternary-N, and oxidized-N, respectively.210 Interestingly, as the pyrolysis temperature increased, the nitrogen content decreased, and in all three samples, the dominant N-species was pyridinic-N. When iron was introduced through the ammonia treatment of ammonium ferric citrate post-modified ZIF-7, a significant improvement in catalytic performance was achieved. The optimized catalyst demonstrated a higher faradaic efficiency (∼85% at −0.43 V vs. RHE) and an increased partial current density for CO production (17.8 mA cm−2 at −0.83 V vs. RHE). The XPS results exhibited the presence of five nitrogen configurations, including pyridinic-N, metal–N, pyrrolic-N, graphitic-N, and N-oxidized in the N 1s peak. Interestingly, the amount of pyridinic-N was suppressed in the catalyst showing the highest performance. It was demonstrated that employing NH3 gas augmented the surface and mesoporous areas by promoting the removal of Zn and minimizing the unstable carbon moieties. Altering the NH3 flow and pyrolysis time influenced the current density and CO faradaic efficiency.211
Ren et al.201 synthesized nitrogenated carbon embedded with isolated bimetallic Ni–Fe sites (Ni/Fe–N–C) employing a Zn/Ni/Fe zeolitic imidazolate framework as the precursor, which was investigated as an electrocatalyst for CO2RR. Fe was chemically bonded to the organic ligand in the original MOF, and Ni was capsulated in the framework, as shown in Fig. 9d. A faradaic efficiency of >90% and high selectivity towards CO within a broad potential range of −0.5 to −0.9 V vs. RHE were achieved. DFT calculation demonstrated that by applying the Ni/Fe–N–C catalyst, the energy barrier for the formation of the COOH* intermediate and CO desorption was diminished in comparison to the Ni–N–C and Fe–N–C catalysts, showing the synergistic effect of the bimetal nitrogen sites.
Ni–N–C, Co–N–C, and Fe–N–C as non-noble metal–nitrogen-doped carbon catalysts are relatively inexpensive and have excellent selectivity towards CO production.178,212,213 However, Ni–N–C requires higher overpotentials, and its CO selectivity is more prominent compared to the other two catalysts.212,214 According to these findings, the stable porphyrin-based porous Zr-MOF (PCN-222) was exploited as a sacrificial template to prepare a hierarchical nanoporous Ni–C–N catalyst (Fig. 9e and f).202 This catalyst contained microporous structures originating from its interconnected mesopores and the nano-MOF precursor produced during pyrolysis. To verify the nature of the main active species (in some cases, Ni nanoparticles,215,216 and in other cases Ni–N–C178,213,217 were reported as the active catalytic sites), DFT simulation212 was utilized and Ni nanoparticles were proposed as the most favorable active sites for the HER process. Moreover, the measured Tafel slope for jCO over the Ni–N–C catalyst confirmed that the rate-determining step (RDS) should be the first electron transfer, signifying the Ni–N–C species as the active sites. Although the RDS assigned to the Ni NPs led to a much higher Tafel slope for the CO2 reduction reaction than Ni–N–C active site, the evaluation of the Ni–N–C catalyst synthesized with different molar ratios of TCPP (meso-tetra(4-carboxyphenyl)porphyrin) and TCPPNi indicated that a small content of Ni NPs is favorable for improving the catalytic performance given that it was reported in the literature that Ni–N–C was the main active site.
In the search for catalysts for the selective reduction of CO2 to hydrocarbons and alcohols, Cu metal is considered an ideal catalyst.218 Usually, the hybridization of Cu with carbon materials such as carbon black219 and MOF-derived porous carbon220 can be realized to create more catalytic active sites. However, the local field around Cu is effective in enhancing the CO2RR.221 Cheng et al.222 studied the effects of N-doping on CO2RR by employing a series of N-containing ligand (benzimidazole, BEN) modified Cu-BTC (1,3,5-benzene tricarboxylic acid) MOFs carbonized at three different temperatures. It was shown that the average size of the Cu nanoparticles increased by enhancing the annealing temperature from 400 °C to 800 °C. The high-resolution N 1s spectrum revealed five peaks associated with pyrrolic-N, pyridinic-N, graphitic-N, Cu–N, and oxidized-N, where increasing the annealing temperature decreased the amount of Cu–N and pyrrolic-N. According to the notably high CO2 electrochemical reduction activity and selectivity toward ethylene and ethanol obtained over the sample carbonized at 400 °C, it was deduced that Cu–N and pyrrolic-N play a major role in CO2RR, while for the sample carbonized at 800 °C, HER was substantially suppressed. Although the aggregation of Cu atoms into large-size nanoparticles results in a decrease in the amount of active sites, which is one of the challenges of copper-containing catalysts, Xuan et al.223 prepared bimetal pyrolyzed ZIF of Cu and Zn, in which the atoms did not aggregate. Incorporating Zn atoms in Cu ZIFs helped suppress the aggregation of the atoms and elevated the formation of active sites. By altering the Cu and Zn ratio, the Cu1Zn1–N–C catalyst showed 95.6% CH4 selectivity in the CO2 reduction reaction. It was shown that the Zn atoms played a role in preventing metal aggregation in the carbonization step, which enhanced the vacancy-copper–nitrogen (V-Cu–N) active sites. The results of the X-ray absorption spectroscopy (XAS) measurement unveiled that the catalysts contained many vacancy-metal–nitrogen active sites (V-Cu–N and V-Zn–N sites), and according to DFT simulations, the V-Cu–N sites remarkably increased the selectivity toward CH4.
Multilayer N-doped graphene embedded with Ni–N–C active sites was synthesized using amino-functionalized Ni-based MOF (NH2–Ni-MOF) as a precursor.224 During annealing at 1000 °C and acid treatment, the –NH2 group in the MOF precursors induced more structural defects and edge plane exposure on the graphene layers,225 thus generating more abundant Ni–N–C sites. The catalyst demonstrated remarkable CO2 electrochemical activity towards CO with the maximum FE of 97% at a low overpotential of 0.79 V, which is assigned to the combined effect of the abundant Ni–N–C sites and slight amount of encapsulated Ni cores.224
Using an NH2-containing Ni/Zn bimetallic MOF precursor (NH2–ZnNi1/x-MOF) with varying ratios of Ni/Zn, Wang et al.226 prepared Ni nanoclusters highly dispersed on N-doped carbon. The Ni/Zn ratio and the –NH2 group of the ligand in the MOF structure influenced the size of the Ni catalyst. After pyrolysis of the MOF precursor at 1000 °C, the NH2–ZnNi1/150-MOF sample (Ni/Zn ratio of 1:150) contained an average Ni nanocluster of 1.9 nm grafted on pyridinic N-rich carbon. During the electrochemical conversion of CO2 to CO, the catalyst exhibited an FE of 98.7% and current density of −40.4 mA cm−2. The exceptional long-term stability of the material/catalyst was attributed to the synergistic effect of small Ni clusters and their optimum interaction with the carbon support.
To date, MOFs have been widely explored as hard templates to form N-doped porous carbons employed as heterogeneous catalysts in various reactions,10,240–242 including the electrochemical reduction of CO2. To gain insights into the active sites, Ye et al.48 proposed that pyridinic nitrogen is the active site according to XPS results for N-doped carbon prepared from Zn-MOF-74 and DFT calculations. Four types of nitrogen including pyridinic, pyrrolic, graphitic, and nitrogen oxide were recognized in the high-resolution N 1s XPS on their samples. The amount of nitrogen species was controlled by the calcination temperature. It was shown that there is a relationship between the total pyridinic and graphitic N and the maximum FE for CO. Furthermore, DFT calculations based on the potential limiting steps (PLS) of CO2RR on the surface of the catalyst and the Gibbs free energy changes demonstrated the minor energy barrier for pyridinic N (Fig. 10a). Wang et al.165 also proposed the high catalytic performance of an N-doped carbon catalyst prepared via the pyrolysis of ZIF-8, which was attributed to the domain of quaternary and pyridinic nitrogen species in the carbon structure. Based on DFT calculation, Yang et al.173 suggested that Ni–N4 and Cu–N4 demonstrate lower energy to convert CO2 molecules to *COOH (intermediate) in the rate-determining step (RDS) than pyridinic N (Fig. 10b). In contrast, Cheng et al.222 proposed that Cu–N and pyrrolic N in the carbon matrix are active sites, which enhance the adsorption of CO2 and favor C–C coupling to generate ethylene and ethanol on the surface of Cu. Several studies, relying on both experimental and DFT calculations, demonstrated that metal–N in the carbon structure formed from the pyrolysis of MOFs are active sites in CO2 electrochemical reduction rather than other nitrogen species in the carbon structure.186,223,224
 | ||
| Fig. 10 (a) Production of CO from CO2 reduction in a two-proton and coupled electron pathway, * + CO2 + H+ + e− → *COOH, *COOH + H+ + e− → *CO + H2O, *CO + H2O → * + CO + H2O. Reproduced with permission.48 Copyright 2020, Wiley-VCH. (b) Reduction of CO2 to CO comparing the Gibbs free energy on pyridinic N, Cu–N4, and Ni–N4. Reproduced with permission.173 Copyright 2019, the American Chemical Society. | ||
3.4 MOF-derived metals, metal oxides and metal composites
Given that MOFs exhibit low thermodynamic stability under the reduction potential, additional processes have been applied to convert MOFs to functional materials, including metal and metal oxide nanoparticles, which both inherit some structural properties of MOFs and exhibit electrochemical stability.243 Recent studies demonstrated that the particle size of Cu nanoparticles can highly affect the results of CO2 reduction for methane production.244,245 In search of novel catalysts to convert CO2 to alcohols electrochemically combining both high efficiency and high stability, Zhao et al.246 carbonized a Cu-based MOF (HKUST-1) to achieve an oxide-derived Cu/carbon catalyst (OD Cu/C). Carbonization of HKUST-1 at three different temperatures of 900 °C, 1000 °C, and 1100 °C resulted in a porous carbon matrix embedded with the oxide-derived Cu nanoparticles, denoted as OD Cu/C-900, OD Cu/C-1000, and OD Cu/C-1100, respectively. Cu2O was identified in the PXRD pattern of the three samples (given that Cu2O was determined to be an essential contributor in the production of methanol at a low overpotential),247,248 and SEM analysis showed that with an increase in the carbonization temperature, the size of the Cu nanoparticles increased. Among them, the OD Cu/C-1000 catalyst showed the highest current density 1.0 mA cm−2 at −0.5 V (vs. RHE) compared to the samples carbonized at 900 °C and 1100 °C. Moreover, a relatively lower charge transfer resistance was obtained for the OD Cu/C-1000 catalyst through electrochemical impedance spectroscopy (ESI) tests. Notably, the highest production rate of alcohol was obtained over the OD Cu/C-1000 catalyst. The higher activity of the OD Cu/C-1000 catalyst compared to the two other catalysts was ascribed to the smaller charge transfer residence, the higher Cu2O content, and the well-dispersed copper in the porous carbon matrix.An Ag/Co3O4 nanocomposite was prepared via the pyrolysis of an Ag/Co-based mixed-metal MOF [Ag4Co2(pyz)PDC4][Ag2Co(pyz)2PDC2] at high temperature in air, which exhibited excellent activity in CO2RR.249 The FE for CO reached 55.6% over this catalyst in a KHCO3 aqueous solution of 0.1 M, which was much higher than that obtained over Co3O4. This superior catalytic performance was assigned to the Ag species, which increased the electrical conductivity, accelerated the electron transport rate, and enhanced the selectivity of the electrode materials toward CO production.
A Cu-based MOF (MOF-74) was converted into isolated Cu NP clusters to obtain efficient electrocatalysts for methane production.250 Cu NPs are known to agglomerate, and their high loading usually increases the FE for C2 and C3 products.245 When the porous structure of Cu-MOF-74 was used as a template to generate highly isolated Cu NP clusters, these MOF-derived Cu NPs exhibited outstanding electrocatalytic performances with a high CH4 faradaic efficiency (>50%), which was 2.3 times higher than the commercial Cu NPs.
Liu et al.251 designed copper nanoparticles derived from a Cu-metal organic framework (Cu-MOF/NP) containing Cu as the metal center and 1,3,5-benzenetricarboxylic acid (H3BTC) as the organic linker for CO2 electrochemical reduction to CO in a new flow electrochemical reactor integrated with a membrane electrode assembly (MEA). This Cu-MOF/NP catalyst contained Cu/Cu2O particles with a porous octahedral structure, including Cu0 and Cu+ catalytic active sites. The XPS and XRD results revealed that an increase in the heating rate favored the formation of the Cu+ chemical state, which further resulted in higher catalytic activity.
Porous Cu nanoribbons, obtained through the in situ electrochemical reduction of Cu-MOF with two distinct organic ligands in their structure, exhibited an impressive FE of 82.3% for C2+ production with a partial current density of 347.9 mA cm−2. This surpasses the performance of Cu nanorods and Cu nanoleaves. The mesoporous structure of the Cu nanoribbons enhanced the electric field on their surface, promoting the concentration of K+ and OH− ions at the active sites. This ion concentration facilitated the formation of CO intermediates and C–C coupling, thereby lowering the thermodynamic barriers and improving the selectivity for C2+ products during CO2 electroreduction. These findings highlight the potential of tuning the selectivity of C2+ products through the introduction of mesoporosity in copper catalysts.51
It is known that metal oxides of earth-abundant elements such as In2O3, SnO2, and Bi2O3 are not only cheap but also have high overpotentials for H2 generation, which may be advantageous for CO2 reduction.252–254 In this case, combining these metal oxides with other active metals can enhance the selectivity and efficiency of the product formation in CO2RR. A series of In–Cu bimetallic oxides derived from an MOF was prepared by introducing In2O3 in the structure of a Cu-based MOF.255 By tuning the ratio of Cu/In in the MOF precursor, the production ratio of CO/H2 could be controlled. In the synthesis procedure, after calcination at 400 °C in air, the resulting In–Cu bimetallic oxide was electrochemically reduced to convert Cu2+ to Cu1+. The presence of Cu1+, which plays a vital role in the formation of CO from CO2, was supported by XRD, XPS, and HR-TEM. It was shown that with an increment in the Cu ratio, the morphology changed from needle-like to cubic, and the surface area also increased. By testing the CO2 electrochemical activity of the In–Cu bimetallic oxides in a 0.5 M KHCO3 aqueous solution saturated with CO2, CO and H2 were recognized in the gas phase. Simultaneously, no liquid product was found in the applied potential range (−0.5 V to −1.0 V vs. RHE). The In–Cu bimetallic oxide with a Cu/In ratio of 0.92 displayed the highest current density of 11.2 mA cm−2 and FECO of 92.1% due to its higher electrochemical surface area, porous structure for mass diffusion, lower charge transfer resistance, stronger CO2 adsorption, and synergistic effect between In oxides and Cu oxides.
A copper phosphide/carbon (Cu3P/C) hybrid was synthesized via the pyrolysis of NaH2PO2 and HKUST-1 under Ar at 350 °C.256 These Cu3P/C nanocomposites exhibited an FE of 47% CO at a relatively low potential (−0.3 V). Further, when the catalyst was explored as a cathode in an asymmetrical-electrolyte Zn–CO2 battery, it showed a good performance with an open-circuit voltage of 1.5 V and a power density of 2.6 mW cm−2 at 10 mA cm−2. These promising catalytic properties are likely due to the synergistic interaction between copper and phosphorus, together with the unique structure of Cu3P.
By carbonizing Cu-BTC MOF at controlled temperatures (700 °C, 800 °C, and 900 °C), nitrogen-doped porous carbon frameworks anchored with Cu2O/Cu nanoparticles (Cu2O/Cu@NC) were prepared.257 CV scanning in CO2-purged KHCO3 (0.1 M) electrolyte showed the superior performance of the catalyst carbonized at 800 °C, Cu2O/Cu@NC-800, which exhibited the best catalytic activity and formate selectivity with FE of 70.5% (at −0.68 V vs. RHE). It was found that increasing the carbonization temperature enhanced the Cu content, specific surface area, and pore volume. The Cu content was enhanced by 36.3% as the carbonizing temperature increased from 700 °C to 800 °C, and it was noteworthy that the amount of Cu0, known as the active site in generating formate, also increased. The higher temperature treatment removed more organic residue from the pores and precursor, leading to an enhancement in the specific surface area and pore volume. Furthermore, studying the effect of N-doping in the catalyst in the selectivity, it was recognized that the higher content of Cu–N–Cu in Cu2O/Cu@NC-800 was beneficial for formate production. In comparison, the selectivity towards ethanol was enhanced over Cu2O/Cu@NC-900 due to its lower content of Cu–N–Cu. It was proposed that there are two main intermediates in CO2 electroreduction, as follows (1) *OCHO and (2) *COOH, which will be reduced to formate or CO, respectively. Here, it appears that N-doping assists formate production by lowering the binding energy of *OCHO.258 The Cu2O/Cu@NC-800 catalyst exhibited long-term stability of 30 h, which was ascribed to its well-dispersed Cu2O/Cu nanoparticles, higher Cu content, and higher content of N doped in the Cu2O/Cu lattice.
As the first example of applying a 2D MOF in CO2RR, copper(II)-5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin–Cu(II) (Cu2(CuTCPP)) nanosheets46 were utilized, in which there were two different copper chemical environments for selective and efficient CO2 electroreduction to generate acetate and formate. One Cu was the Cu paddle wheel, which was the cluster in the structure of HKUST-1 (effective in CH4 and C2H4 production),119 and the other was porphyrinic Cu, known as an electrocatalyst for CH4 formation.119 The CO2 electrochemical reduction performance of Cu2(CuTCPP) nanosheets on an FTO electrode was analyzed in a solution of CH3CN containing H2O and 1-ethyl-3-methylimidazolium tetra-fluoroborate (EMIMBF4) ionic liquid as the supporting electrolyte. To control the concentration of protons and increase the solubility of CO2, an organic electrolyte with water and the ionic liquid were employed,62,259 and compared to CuO, Cu2O, Cu, and CuTCPP, the cathodized Cu-MOF nanosheets demonstrated remarkable activity toward the production of formate with an FE of 68.4% at a potential of −1.55 V vs. Ag/Ag+. The Cu catalyst revealed negligible CO2RR activity, producing CO and CH4 (FE of 5%) with no formate and acetate detected. Applying CuTCPP as an electrocatalyst generated CO with an FE of 20% at potentials in the range of −1.50 V to −1.65 V, and H2 was identified as the major product. In contrast, HCOOH (with FE of 14.7% at −1.5 V) and CH3COOH (with FE of 5.8% at −1.45 V) were produced over CuO, while only HCOOH was detected with Cu2O with an FE of up to 21.5%. Furthermore, the structural changes in the Cu(II) paddle-wheel nodes during cathodized reconstruction to CuO, Cu2O, and Cu4O3 were confirmed by ex situ XRD, SEM, HR-TEM, and XPS.
A Bi-based MOF was studied in the electrochemical reduction of CO2 to formate by Lamagni et al.260 It was demonstrated that Bi(1,3,5-tris(4-carboxyphenyl)benzene) Bi(btb), known as CAU-7,261 works as a pre-catalyst and at the reducing potentials, where it undergoes structural rearrangement to form the highly active and selective catalyst of porous organic matrix dispersed with Bi-based nanoparticles. Interestingly, this rearrangement happening in situ during the electrocatalytic reaction enabled the poor conductivity and instability of the MOF to be overcome. Cyclic voltammetry of Bi(btb) deposited on GC recorded under Ar and CO2 atmospheres in an aqueous solution of 0.5 M KHCO3 indicated the structural transformation of Bi(btb) to a steady state of different Bi species for formate production with an FE of 68.4% at a potential of −1.55 V vs. Ag/Ag+. The Cu catalyst revealed negligible CO2RR activity, producing CO and CH4 (FE of 5%) with formate and acetate detected. Applying CuTCPP as an electrocatalyst generated CO with an FE of 20% at potentials in the range of −1.50 V to −1.65 V, and H2 was identified as the major product. Alternatively, HCOOH (with FE of 14.7% at −1.5 V) and CH3COOH (with FE of 5.8% at −1.45 V) were produced over CuO, while only HCOOH was detected with Cu2O with an FE of up to 21.5%. Furthermore, the structural changes in the Cu(II) paddle-wheel nodes during cathodized reconstruction to CuO, Cu2O, and Cu4O3 were confirmed by ex situ XRD, SEM, HR-TEM, and XPS.
A Bi-based MOF was studied in the electrochemical reduction of CO2 to formate by Lamagni et al.260 It was demonstrated that Bi(1,3,5-tris(4-carboxyphenyl)benzene) Bi(btb), known as CAU-7,261 works as a pre-catalyst and at reducing potentials, it undergoes a structural rearrangement to form the highly active and selective catalyst of porous organic matrix dispersed with Bi-based nanoparticles. Interestingly, this rearrangement happening in situ under electrocatalytic reaction resulted in overcoming the poor conductivity and instability of the MOF. Cyclic voltammetry of Bi(btb) deposited on GC recorded under Ar, and CO2 atmosphere in an aqueous solution of 0.5 M KHCO3 indicated the structural transformation of Bi(btb) to a steady state of a different Bi species upon multiple cycling of the Bi3+/Bi0 redox couple (Fig. 11a)262 which then leads to the Bi-based nanoparticle formation as indicated by grazing-incidence X-ray diffraction, HR-TEM, and XPS. In bulk electrolysis experiments, formate was recognized as the main product with a faradaic efficiency of 95% at an overpotential of 770 mV (Fig. 11b). The discussed MOF-derived materials applied in CO2RR are summarized in Table 3.
 | ||
| Fig. 11 (a) Multiple cycling of the Bi3+/Bi0 redox couple under Ar and CO2. (b) Faradaic efficiency of the electrocatalysis products at different potentials. Reproduced with permission.260 Copyright 2020, Wiley-VCH. | ||
| Electrocatalyst | Applied MOF | Potential | Electrolyte | Catalytic efficiency (%) | Ref. |
|---|---|---|---|---|---|
| a Multiwall carbon nanotubes. b Cu single atoms through carbon nanofibers. c Mn single-atom-sulfurized-nitrogen-doped carbon. d Bi(1,3,5-tris(4-carboxyphenyl)benzene) Bi(btb). | |||||
| N-doped porous carbon | ZIF-8-supported MWCNa | −0.56 V vs. RHE | 0.1 M NaHCO3 | FECO = 100 | 166 |
| N-doped porous carbon | Zn-MOF-74 | −0.55 V vs. RHE | 0.5 M KHCO3 | FECO = 98.4 | 48 |
| Bi@N-doped porous carbon | ZIF-67 | −1.5 V vs. SCE | 0.1 M KHCO3 | FEformate = 92 | 167 |
| Dendritic Cu0 and nanowires Cu0 | Cu-MOF | −1.85 vs. Ag/AgCl | (0.5 M) BmimBF4/(1.0 M) acetonitrile/H2O | FEformate = 98.2 | 168 |
| Ni/N-doped porous carbon | ZIF-8 | −0.9 V vs. RHE | 0.5 M KHCO3 | FECO = 71.9 | 49 |
| Fe–N-doped porous carbon | ZIF-8 | −0.33 V vs. RHE | 1 M KHCO3 | FECO = 89.1 | 194 |
| Co–N-doped porous carbon | Co/Zn ZIF | −0.3 V to −0.9 V vs. RHE | 0.5 M KHCO3 | FECO = 94 | 50 |
| N-doped porous carbon | ZIF-8 | −0.93 V vs. RHE | 0.1 M KHCO3 | FECO = 78 | 165 |
| Fe–N-doped porous carbon | ZIF-8 | −0.47 V vs. RHE | 0.1 M KHCO3 | FECO = 93 | 162 |
| Fe–N-doped porous carbon | ZIF-7 | −0.33 V to −0.83 V vs. RHE | 1.0 M KHCO3 | FECO = 86 | 211 |
| Meso N–C–Fe | Fe-ZIF-8@SiO2 | −0.73 V vs. RHE | 0.1 M KHCO3 | FECO = 85 | 189 |
| Ni–N–C | Zn–Ni MOF | −0.54 V vs. RHE | 0.5 M KHCO3 | FECO = 95.1 | 263 |
| Ni/Fe–N–C | Zn/Ni/Fe ZIF | −0.7 V vs. RHE | 0.5 M KHCO3 | FECO = 98 | 201 |
| CuSAs/TCNFsb | Cu/ZIF-8 | −0.9 V vs. RHE | 0.1 M KHCO3 | FECH3OH = 44 | 173 |
| FECO = 56 | |||||
| Ni–C–N | Zr-MOF (PCN-222) | −0.30 V vs. RHE | 0.5 M KHCO3 | FECO = 98.7 | 202 |
| Cu–N | Cu-MOF | −1.01 V vs. RHE | 0.1 M KHCO3 | FEC2H2 = 11.2 | 222 |
| FECH3CH2OH = 18.4 | |||||
| Cu–Zn–N–C | Cu/Zn ZIF | Overall potential difference = 1.5 V | 0.5 M NaHCO3 | FECH4 = 95.6 | 223 |
| NiSA–N–C | Mg–Ni-MOF-74 | −0.8 V vs. RHE | 0.5 M KHCO3 | FECO = 98 | 157 |
| Fe–, Co–, Ni–, Cu–N–C | Porphyrinic MOF | −0.8 V vs. RHE | 0.5 M KHCO3 | FECO = 96.8 | 200 |
| Ni–N3–C | Ni–Zn-MOF | −0.65 V vs. RHE | 0.5 M KHCO3 | FECO = 95.6 | 186 |
| Ni–C–N | NH2–Ni-MOF | −0.79 V vs. RHE | 0.5 M KHCO3 | FECO = 97 | 224 |
| Fe–Ni–N–C | Fe/ZnO, Ni/ZnO doped ZIF-8 | −0.5 V vs. RHE | FECO = 96.2 | 52 | |
| Ni–N–C | ZIF-8@Ni2+ | −0.7 V vs. RHE | 0.1 M KHCO3 | FECO = 98 | 203 |
| MnSA-SNCc | ZIF-8 | −0.45 V vs. RHE | 0.5 M KHCO3 | FECO = 70 | 195 |
| Ni nano cluster on N-doped carbon | NH2–Zn/Ni-MOF | −0.88 V vs. RHE | 0.5 M KHCO3 | FECO = 98.7 | 226 |
| Oxide derived Cu/C | HKUST-1 | −0.7 V vs. RHE | 0.1 M KHCO3 | FEmethanol = 13.8–43.2 | 246 |
| FEethanol = 24–34.8 | |||||
| Ag/Co3O4 | Ag/Co-mixed metal MOF | −1.8 V vs. SCE | 0.1 M KHCO3 | FECO = 55.6 | 249 |
| Cu NPs clusters | MOF-74 | −1.3 V vs. RHE | 0.1 M KHCO3 | FECH4 = 50 | 250 |
| Cu/Cu2O | Cu-MOF | −0.76 V vs. RHE | 0.5 M KHCO3 | FECO = 43.8 | 251 |
| Cu nanoribbons | Cu-MOF | 1 M KOH | FEC2+ = 82.3 | 51 | |
| In–Cu bimetallic oxides | In2O3@Cu-MOF | −0.8 V vs. RHE | 0.5 M KHCO3 | FECO = 92.1 | 255 |
| Cu3P/C | HKUST | −0.3 V vs. RHE | 0.1 M NaHCO3 | FECO = 47 | 256 |
| Cu2O/Cu@N-doped porous carbon | Cu-MOF | −0.68 V vs. RHE | 0.1 M NaHCO3 | FEHCOO− = 70.5 | 257 |
| Bi-based nanoparticles | CAU-7d | −0.97 vs. RHE | 0.5 M NaHCO3 | FEHCOO− = 95 | 260 |
4. Active sites and mechanism of MOF-based materials for electrochemical CO2RR
As discussed earlier, MOFs,39 which are highly ordered coordination polymers, are incredibly appealing materials for several catalytic reactions due to their unique features, combining that of homogenous and heterogeneous catalysts. MOFs have a well-defined structure and highly active catalytic sites incorporated into a stable scaffold, enabling excellent catalytic activity and selectivity, which are crucial parameters in catalysis. Further, their porous structure enables excellent and tunable mass transfer to and from the active sites. Unlike traditional heterogeneous catalysts, the environment around the active sites can be easily tuned, exactly as in the case of small molecular homogenous systems, which enables the fine-tuning of the active sites and the fundamental understanding of the catalytic mechanism. It is well known that the poor electrical conductivity of bulk MOFs is a serious limitation for their applications not only in CO2RR but in most electrocatalytic applications. However, this issue can be partially solved by constructing highly conductive 2D chains or so-called coordination nanosheets (CONASHs).264,265 However, this is limited to a particular type of ligand structure. Another approach to mitigate the low electrical conductivity of MOFs is to utilize them as precursors for creating conductive carbonaceous materials that are doped with some transition metals derived from the starting structure. In this case, the original structure collapses and the molecular nature of MOFs does not exist. Thus, mechanistic studies will be more difficult in this case given that we do not know the exact structure of the active sites. This can explain the poor mechanistic investigations related to MOF-based electrocatalysts in CO2RR.CO2RR has numerous reaction pathways via 2-, 4-, 6-, and 8-electron reduction steps, with multiple possible reduction products. The binding strength of the reaction intermediates as *H, *COOH, *OCOH, and *CO (asterisk (*) indicates the adsorbed species) to the catalyst surface is the critical factor that controls the product selectivity.59,266,267 The binding energy of the catalyst with a particular intermediate may favor the related reaction pathway over other possible paths. Thus, it is necessary to know and accordingly tune the electrocatalyst surface properties to further control their CO2RR performances. The nonselective reduction of CO2 is mainly due to the competition between H+ and CO2 for the key intermediates that produce diverse reduction products through distinct pathways (Table 1). The most common reduction products are carbon monoxide (CO), oxalic acid (C2O4H2), formic acid (HCOOH), formaldehyde (CH2O), and methanol (CH3OH). However, the formation of other reduction products such as methane (CH4), ethylene (CH2CH2), and ethanol (CH3CH2OH) is becoming feasible, particularly with copper-based electrocatalysts.
Scheme 1 outlines one of the plausible mechanisms for the catalytic reduction of CO2 into HCO2− and CO, together with H+ into H2, the most competitive reaction for CO2RR in molecular catalytic systems. Upon the reduction of the transition metal ion, the reduced intermediate can undergo either protonation to form a metal hydride intermediate or directly activate CO2. In the protonation pathway, upon the generation of a metal hydride, it can react either with a second proton to form H2 or with CO2 to produce formate. Conversely, the CO2-activation pathway involves CO2 activation to surpass protonation at the reduced metal center. CO is the specific product; however, in this case, the competition between both pathways is quite complex. For instance, reaching high negative potentials to activate CO2 make the metal sites more Brønsted basic, which favors both pathways, thus negatively affecting the overall selectivity and efficiency of the catalyst. Further, the reaction product, i.e., CO, is often a better ligand than CO2; consequently, enhancing the electron density around the metal center often leads to a more stable M–CO complex, which inhibits the catalyst turnover. Thus, it is clear that for optimal CO2 reduction to CO, an in-depth comprehension of how CO2, CO, and H+ interact with the reduced metal centers is crucial in catalyst design.268
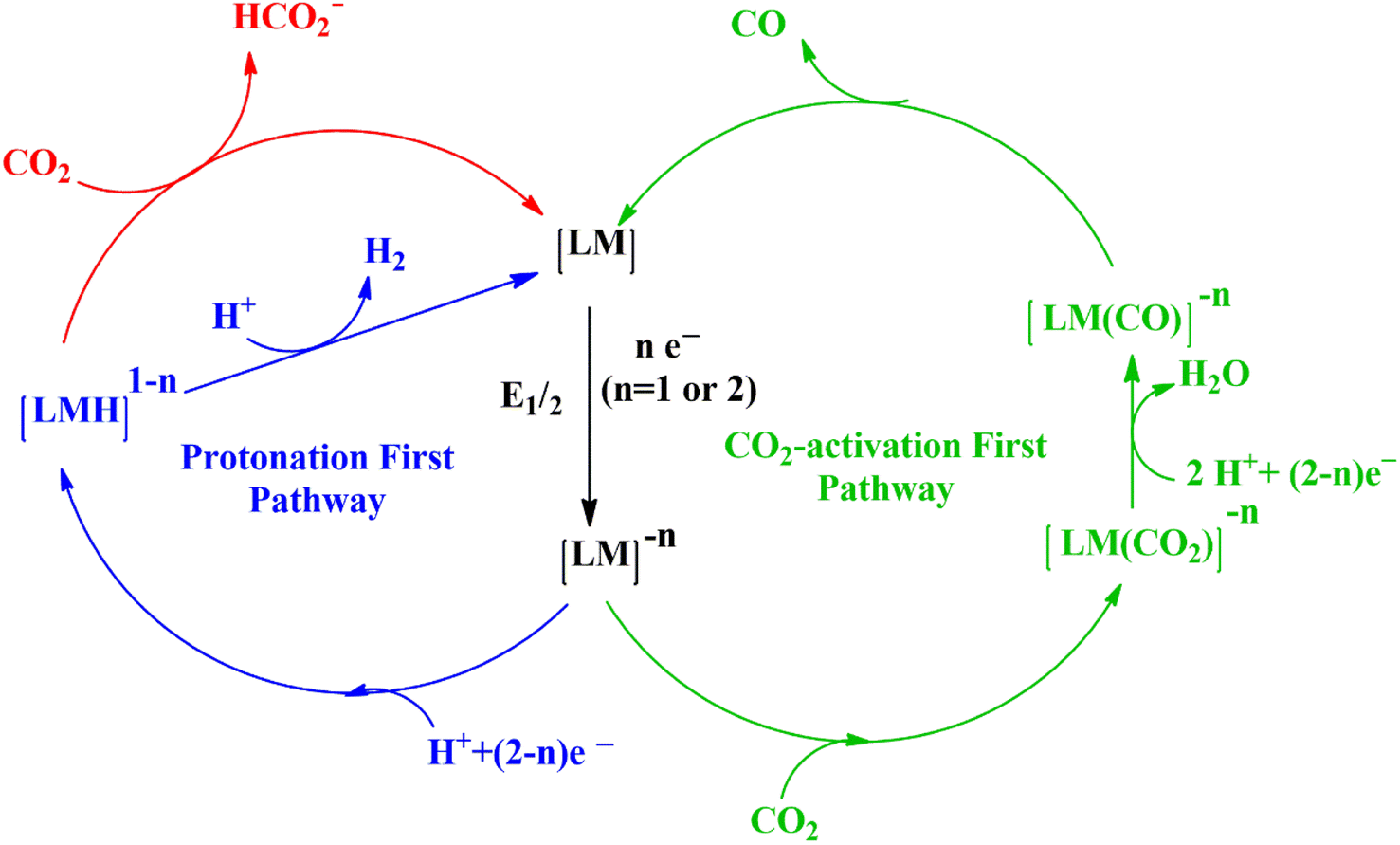 | ||
| Scheme 1 Generalized scheme for CO2 activation using molecular catalysts (M = metal, L = ligand, and n = charge).268 | ||
In heterogeneous systems, it is generally accepted that CO2RR involves several steps, including CO2-adsorption, intermediate formation, production of reaction products, and desorption of the final products.269 Accordingly, to understand the CO2RR mechanism for a particular catalytic system, in situ characterization techniques, together with theoretical/computational studies, are being widely used. For instance, to gain insights into the superior catalytic activity and excellent selectivity of Co-PMOF (cobalt-based polyoxometalate metalloporphyrin organic frameworks), Wang et al. performed thorough DFT calculations. They suggested that CO2 electrochemical reduction to CO includes firstly a proton-coupled electron transfer process to generate a carboxyl intermediate (*COOH), and eventually, for the formation of the *CO intermediate, a second charge transfer (one electron and one proton), followed by CO desorption for the final CO product (Fig. 12a). Based on the calculated free energy diagrams, the RDSs for CO2RR on zinc polyoxometalate (Zn-POM) and Co-TCPP, independently, are the formation of adsorbed intermediates *COOH and *CO with relatively high free energies of ΔG1 = 0.96 eV and ΔG2 = 0.53 eV, respectively. However, when Zn-POM and Co-TCPP were assembled into one polymeric structure, the final compound of Co-PMOF possessed considerably reduced free energies, particularly the RDS of *COOH production (ΔG1 = 0.34 eV), which is consistent with the high activity and selectivity of this bimetallic MOF. Calculations indicated that the Co centers are the favorite sites for CO2 coordination instead of POM, and they also demonstrated the synergistic electron modulation of POM and the porphyrin metal center. It also highlighted that upon the replacement of Co in the porphyrin center with Ni or Zn, the RDS remains the formation of *COOH; however, it has much higher free energies, making its formation more sluggish. In the case of Fe-PMOF, although the energy for the formation of *COOH and *CO decreased as compared to that of Co-PMOF, the desorption of CO becomes more challenging, which can be ascribed to the higher affinity of Fe for the CO ligand.270 Similarly, DFT calculations for a model system of cobalt phthalocyanine (CoPc), with a metal–N4 coordination structure, showed that by compromising on the crucial reaction steps of *COOH production and *CO desorption, it improves the overall reaction thermodynamics, which is consistent with the experimental results, where it achieved an excellent FE of 99% for CO2RR to CO with a mild potential of −0.8 V vs. RHE compared to other metal phthalocyanines (MPcs).272 The in situ spectro-electrochemical characterization of cobalt-metallated [Al2(OH)2TCPPCo] MOF@FTO-electrode in a CO2-saturated electrolyte revealed the formation of Co(I) species under the operating conditions with a Tafel slope of 165 mV per decade, suggesting that the RDS in this catalytic cycle may be either a CO2 binding to a Co(I) porphyrin coupled with a one-electron reduction or a one-electron reduction of a Co(I)–CO2 adduct. A fraction of Co(II) is reduced to Co(I) even at potentials more positive than −0.4 V vs. RHE, which is likely to participate in the catalytic reduction of CO2 to CO.273
 | ||
| Fig. 12 (a) Suggested reaction pathway for the CO2RR on Co-PMOF. Adopted with permission.270 Copyright 2018, Nature Publishing Group. (b and c) Gibbs free energy diagram of the CO2RR on M1O4 units in the structure of PCM-O8-M1. Reproduced with permission.103 Copyright 2020, Nature Publishing Group. (d) Gibbs free energy profile of the CO2RR, (e) proposed mechanism on the BiN4/C surface, (f) difference in the limiting potentials in the reduction of CO2 and H2 evolution. Reproduced with permission.271 Copyright 2019, the American Chemical Society. | ||
In an analogous structure, a 2D conjugated MOF (2D c-MOF) with metal-phthalocyanine as the linker (MN4) and metal-bis(dihydroxy) complex (M1O4) as the metal node (PcM-O8-M1), DFT calculations revealed that the formation of *COOH via protonation is the rate-determining step. Based on the binding energies, it was found that the *COOH intermediate has a more robust interaction. In contrast, *H has a weaker interplay with the linkages (M1O4 complexes) in comparison to that of the phthalocyanine macrocycles. Thus, it was proposed that CO2 activation mainly happens at the M1O4 sites, while the MN4 complex serves as the active site for HER. Further, Gibbs free energy calculations demonstrated that the ZnO4 complex ion of PcCu–O8 has the lowest values for *COOH formation, and also the lowest overpotential compared to other M1O4 structures in PcM-O8-M1 (Fig. 12b), which agreed with the experimental results. In addition, the overpotential for CO2RR at M1O4 was found to be affected by different MN4 complexes in the Pc ligand. Exploring the energy profile for HER on MN4 and M1O4 moieties indicates that the CuN4 moiety in PcCu–O8–Zn has the lowest HER energy barrier and the fastest proton/electron transfer kinetics among the different metal centers (Fig. 12c), suggesting the synergistic effect between the CuN4 and ZnO4 moieties given that the CuN4 complex facilitates the protonation of adsorbed *CO2 on the ZnO4 complexes, and thus accelerates the overall CO2RR kinetics.274 Similarly, computational studies of CO2RR on the surface of a bimetallic Ni–Fe-based 2D MOF suggested that CO2 is activated on the surface of Ni, Fe, and H atom (from the ligand) with proton/electron pair transfer, allowing the intermediate (*COOH) to bind with the metal atoms via a carbon atom. Calculations indicated that Ni-ions are the preferable catalytic sites for CO2RR with a low overpotential of 0.19 V vs. RHE, and they can stabilize *COOH more than the Fe-sites, resulting in a low activation barrier. Additionally, the interaction between *CO and Ni-site is weaker than *CO and Fe-site. Finally, the mechanism for the CO2RR to CO over MOF Ni–Fe was proposed. It was proposed that after the adsorption and attachment of CO2 to the surface of the electrode, it captures an electron to form CO2˙−, a transitional product, which is known to be the rate-determining step. Afterwards, the CO2˙− intermediate will capture another electron for the adsorbed CO (COads), which is then desorbed and released into the electrolyte.275
MOF-derived materials, particularly metal–N–C, metal–N4 structures, and M-SACs, have also received considerable attention in CO2RR, and interesting findings have been attained. However, the actual active sites and reaction mechanisms still need to be clarified due to the uncertainty about the exact atomic structure of the investigated materials. Although advanced characterization techniques such as X-ray absorption spectroscopy (XAS) can provide substantial information about the atomic structure of different materials, it is not easily available. Bismuth single atoms were obtained via the thermal decomposition of a bismuth-based metal–organic framework (Bi-MOF), denoted as Bi–N4 sites on porous carbon networks, which had high intrinsic activity for CO2R to CO with a high FE (FECO up to 97%) and high TOF of 5535 h−1 at a low overpotential of 0.39 V vs. RHE. In the DFT calculations, BiN4/C BiC4 and Bi (110) were considered to understand the effect of the coordination environment on the free energy of the reaction intermediates. It was found that the formation of the COOH* intermediate via CO2 activation is the RDS in all three materials because of the uphill energy barrier for the first proton-coupled electron-transfer step (Fig. 12d–f). BiN4/C had a lower Gibbs free energy for the generation of the *COOH intermediate, matching its better performance and low onset potential. Further, BiN4/C has a more positive UL(CO2) − UL(H2) value (−0.02 V) than Bi(110) (−0.1 V) and BiC4 (−0.2 V), implying its higher selectivity for CO2 conversion to CO, where UL(CO2) − UL(H2) refers to the difference between the limiting potentials for CO2 reduction and H2 evolution. It is noteworthy that previous studies suggested that the value of UL(CO2) − UL(H2) reflects the selectivity of the CO2RR, where a more positive value means a better selectivity for CO2RR over HER.241
5. Conclusions and outlooks
This review comprehensively explored the recent advancements in the design and application of MOFs and MOF-derived materials for electrochemical CO2 reduction reactions (CO2RR). The discussion highlighted the unique properties of MOFs, such as their porous structures, tunable active sites, and the ability to be converted into highly efficient electrocatalysts through pyrolysis and other treatments. Significant progress has been made in enhancing the stability, conductivity, and catalytic activity of these materials, making them promising candidates for CO2RR.Pristine MOFs have shown considerable potential in CO2RR due to their well-defined active sites and the ability to modify their microenvironment, which are crucial for optimizing their catalytic activity and selectivity. MOFs containing transition metals such as Cu, Zn, Ni, Zr, Co, and Fe have demonstrated significant activity in CO2RR. Copper-based MOFs are generally inclined towards the production of hydrocarbons (methane), alcohols (ethanol and methanol), and oxalates. Alternatively, Zn-, Ni-, Co-, Zr-, and Fe-based MOFs generally exhibit high selectivity towards CO. However, the poor stability and electrical conductivity and lack of mesoporosity of these pristine MOFs remain a significant challenge, especially in aqueous environments.
Thus, MOF-derived materials present several distinct advantages over pristine MOFs in the context of CO2RR. One of the most significant benefits is their enhanced stability, especially under electrochemical conditions. Although pristine MOFs often suffer from hydrolytic instability and poor electrical conductivity, MOF-derived materials, typically obtained through processes such as pyrolysis, exhibit improved durability and conductivity. These materials retain the advantageous porous structure of the parent MOFs, allowing for high surface areas and accessible active sites, but with the added benefit of increased conductivity due to the formation of conductive carbon frameworks. Additionally, the thermal treatment involved in the conversion of MOFs to MOF-derived materials generally leads to the incorporation of heteroatoms (e.g., nitrogen, oxygen, and sulfur) in the carbon matrix, which can further enhance the catalytic activity and selectivity by modulating the electronic properties of the active sites.
MOF-derived materials exhibit a variety of catalytic behaviours depending on the type of transition metal and the structural modifications applied to the MOF. N-doped porous carbon materials derived from MOFs such as ZIF-8, Zn-MOF-74, and Co/Zn ZIF show high selectivity towards CO production, with their FE reaching up to 100% under the optimal conditions. Bismuth- and copper-derived materials typically favor the production of formate (HCOO−) and hydrocarbons (such as CH4 and C2H2) as well as oxygenated products (methanol and ethanol), with efficiencies reaching up to 95% for formate in Bi-based catalysts and as high as 82.3% for C2+ products in Cu-derived catalysts. Nickel and iron-doped MOF-derived carbons also display strong selectivity towards CO, often exceeding 90% FE. Moreover, multi-metallic and single-atom catalysts (SACs) derived from MOFs further enhance the selectivity and efficiency, with NiSA–N–C achieving an FE of 98% for CO production, and Cu–Zn–N–C exhibiting a remarkable FE of 95.6% for CH4 production. These results underline the versatility of MOF-derived materials in tailoring the product selectivity, where the choice of metal, doping, and structural modification directly influence the catalytic outcomes.
Mechanistically, the coordination environment around the metal centres, such as M–N4 sites, plays a crucial role in determining the reaction pathways and product selectivity. The catalytic performance is often governed by the adsorption and activation of CO2 on the metal sites, followed by proton-coupled electron transfer steps. The presence of dopants or additional heteroatoms (such as N and S) within MOF-derived materials further modulates the electronic environment of the active sites, enhancing their ability to stabilize the reaction intermediates and lower the energy barriers for the rate-limiting steps towards a particular product.
The stability and durability of MOF-based materials under electrochemical conditions, particularly in aqueous environments, represent a significant challenge in CO2RR applications. Pristine MOFs often undergo hydrolysis and structural degradation, leading to a decline in their catalytic activity over time, which hinders their practical application. Additionally, the poor electrical conductivity of many MOFs presents another obstacle, given that it limits efficient charge transfer during CO2RR, resulting in substantial ohmic losses and reduced catalytic efficiency. Furthermore, the lack of mesoporosity in MOF materials is a limitation given that mesoporosity facilitates liquid mass transfer. Identifying and controlling the active sites within MOF-derived materials are also complex, especially in systems with multiple potential active sites, which complicates the determination of the specific sites responsible for catalysis. Furthermore, the scalability and cost of synthesizing MOFs and their derivatives pose challenges for their widespread industrial application, necessitating the development of more cost-effective and scalable synthesis methods.
Moving forward, enhancing the stability of MOF-based materials in aqueous and electrochemical environments is a critical focus for future research. This can involve designing MOFs with stronger metal–ligand bonds, incorporating hydrophobic ligands, or developing hybrid materials that combine the advantages of MOFs with more stable substances. To address their conductivity issue, efforts can focus on using conductive coordination nanosheets or 2D MOFs, integrating conductive polymers, carbon-based materials, or metallic nanoparticles into MOF structures. Additionally, using MOFs as precursors for synthesizing metal or metal-heteroatom-doped, highly conductive carbonaceous materials or single-atom catalysts (SACs) can be a promising approach to further expand the research in this direction. Advanced in situ and operando characterization techniques will be essential in better understanding the active sites and mechanisms involved in CO2RR, helping to guide the rational design of more effective catalysts. Finally, efforts to bridge the gap between laboratory research and industrial application should prioritize the development of catalysts that not only offer high selectivity and efficiency but also demonstrate robustness and scalability in real devices.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
D. P. D. thanks the Francqui Foundation for the Francqui Research Professor chair. H. A. Y. and M. A. A acknowledge the support from His Majesty Trust Fund (HMTF), Oman (Project ID: SR/DVC/NRC/24/01).Notes and references
- T. Boden, R. Andres and G. Marland, Global, Regional, and National Fossil-Fuel CO2 Emissions (1751–2014), Environmental System Science Data Infrastructure for a Virtual Ecosystem, 2017 Search PubMed.
- O. Edenhofer, Climate Change 2014: Mitigation of Climate Change, Cambridge University Press, 2015 Search PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 Search PubMed.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 Search PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, Chem. Rev., 2015, 115, 12888–12935 Search PubMed.
- C. I. Ezugwu, S. Liu, C. Li, S. Zhuiykov, S. Roy and F. Verpoort, Coord. Chem. Rev., 2022, 450, 214245 Search PubMed.
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981–6000 Search PubMed.
- A. Taheri Najafabadi, Int. J. Energy Res., 2013, 37, 485–499 Search PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 Search PubMed.
- N. Gholampour, Y. Zhao, F. Devred, C. Sassoye, S. Casale and D. P. Debecker, ChemCatChem, 2023, 15, e202201338 Search PubMed.
- J. Durst, A. Rudnev, A. Dutta, Y. Fu, J. Herranz, V. Kaliginedi, A. Kuzume, A. A. Permyakova, Y. Paratcha and P. Broekmann, Chimia Int J. Chem., 2015, 69, 769–776 Search PubMed.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 Search PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 Search PubMed.
- J. P. Jones, G. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 Search PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 Search PubMed.
- U. Nwabara, S. Verma, A. A. Gewirth and P. J. Kenis, ECS Meeting Abstracts, 2018, 01(28), 1630 Search PubMed.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 Search PubMed.
- M. F. Hasan, L. M. Rossi, D. P. Debecker, K. C. Leonard, Z. Li, B. C. Makhubela, C. Zhao and A. Kleij, ACS Sustain. Chem. Eng., 2021, 9, 12427–12430 Search PubMed.
- R.-Z. Zhang, B.-Y. Wu, Q. Li, L.-L. Lu, W. Shi and P. Cheng, Coord. Chem. Rev., 2020, 422, 213436 Search PubMed.
- H. A. Younus, N. Ahmad, W. Ni, X. Wang, M. Al-Abri, Y. Zhang, F. Verpoort and S. Zhang, Coord. Chem. Rev., 2023, 493, 215318 Search PubMed.
- C. Long, X. Li, J. Guo, Y. Shi, S. Liu and Z. Tang, Small Methods, 2019, 3, 1800369 Search PubMed.
- Y. i. Hori, in Modern Aspects of Electrochemistry, Springer, 2008, pp. 89–189 Search PubMed.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 Search PubMed.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 Search PubMed.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 Search PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 Search PubMed.
- M. Lu, M. Zhang, J. Liu, Y. Chen, J.-P. Liao, M.-Y. Yang, Y.-P. Cai, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202200003 Search PubMed.
- J. Francis Kurisingal, H. Kim, J. Hyeak Choe and C. Seop Hong, Coord. Chem. Rev., 2022, 473, 214835 Search PubMed.
- P. Shao, L. Yi, S. Chen, T. Zhou and J. Zhang, J. Energy Chem., 2020, 40, 156–170 Search PubMed.
- X. Li and Q.-L. Zhu, EnergyChem, 2020, 2, 100033 Search PubMed.
- S. Gulati, S. Vijayan, S. Kumar, B. Harikumar, M. Trivedi and R. S. Varma, Coord. Chem. Rev., 2023, 474, 214853 Search PubMed.
- X. Li and Q.-L. Zhu, EnergyChem, 2020, 2, 100033 Search PubMed.
- L. Valenzuela, G. Amariei, C. I. Ezugwu, M. Faraldos, A. Bahamonde, M. E. Mosquera and R. Rosal, Sep. Purif. Technol., 2022, 285, 120351 Search PubMed.
- C. I. Ezugwu, S. Ghosh, M. E. Mosquera and R. Rosal, in Nanomaterials for Water Treatment and Remediation, CRC Press, 2021, pp. 371–408 Search PubMed.
- D. Skoda, T. Kazda, L. Munster, B. Hanulikova, A. Styskalik, P. Eloy, D. P. Debecker, P. Vyroubal, L. Simonikova and I. Kuritka, J. Mater. Sci., 2019, 54, 14102–14122 Search PubMed.
- O. Shekhah, J. Liu, R. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 Search PubMed.
- N. Gholampour, C. I. Ezugwu, S. Rahmdele, A. G. Gilanie and F. Verpoort, Resour. Chem. Mater., 2022, 1(3–4), 201–210 Search PubMed.
- C. I. Ezugwu, J. M. Sonawane and R. Rosal, Sep. Purif. Technol., 2021, 120246 Search PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 Search PubMed.
- V. Remya and M. Kurian, Int. Nano Lett., 2019, 9, 17–29 Search PubMed.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, TrAC, Trends Anal. Chem., 2019, 118, 401–425 Search PubMed.
- M.-R. Ahmadian-Yazdi, N. Gholampour and M. Eslamian, ACS Appl. Energy Mater., 2020, 3, 3134–3143 Search PubMed.
- M. Hao, M. Qiu, H. Yang, B. Hu and X. Wang, Sci. Total Environ., 2021, 760, 143333 Search PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen and D. Tan, J. Am. Chem. Soc., 2020, 142, 13606–13613 Search PubMed.
- Q. Huang, Q. Li, J. Liu, R. Wang and Y.-Q. Lan, Matter, 2019, 1, 1656–1668 Search PubMed.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 Search PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 Search PubMed.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 Search PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 Search PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 Search PubMed.
- H. Huo, J. Wang, Q. Fan, Y. Hu and J. Yang, Adv. Energy Mater., 2021, 11, 2102447 Search PubMed.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou and S.-H. Yu, J. Am. Chem. Soc., 2021, 143, 19417–19424 Search PubMed.
- V. K. Abdelkader-Fernandez, D. M. Fernandes and C. Freire, J. CO2 Util., 2020, 42, 101350 Search PubMed.
- Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi, J. Ge and J. Tang, Small, 2021, 17, 2006590 Search PubMed.
- S. S. A. Shah, T. Najam, M. Wen, S.-Q. Zang, A. Waseem and H.-L. Jiang, Small Struct., 2022, 3, 2100090 Search PubMed.
- K. Schouten, Y. Kwon, C. Van der Ham, Z. Qin and M. Koper, Chem. Sci., 2011, 2, 1902–1909 Search PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 Search PubMed.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 Search PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 Search PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 Search PubMed.
- F. Song, M. M. Busch, B. Lassalle-Kaiser, C.-S. Hsu, E. Petkucheva, M. l. Bensimon, H. M. Chen, C. Corminboeuf and X. Hu, ACS Cent. Sci., 2019, 5, 558–568 Search PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. Kenis and R. I. Masel, Science, 2011, 334, 643–644 Search PubMed.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 Search PubMed.
- S. Kaneco, K. Iiba, H. Katsumata, T. Suzuki and K. Ohta, Electrochim. Acta, 2006, 51, 4880–4885 Search PubMed.
- M. Moura de Salles Pupo and R. Kortlever, ChemPhysChem, 2019, 20, 2926–2935 Search PubMed.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 Search PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. Kenis, J. Power Sources, 2016, 301, 219–228 Search PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna and O. S. Bushuyev, Science, 2018, 360, 783–787 Search PubMed.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 Search PubMed.
- R. M. Arán-Ais, D. Gao and B. Roldan Cuenya, Acc. Chem. Res., 2018, 51, 2906–2917 Search PubMed.
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, ACS Catal., 2016, 6, 2382–2392 Search PubMed.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2022, 12, 331–362 Search PubMed.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 Search PubMed.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2018, 4, 317–324 Search PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 Search PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 Search PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave and E. B. Duoss, Nat. Energy, 2022, 7, 130–143 Search PubMed.
- E. W. Lees, B. A. Mowbray, F. G. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 Search PubMed.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 Search PubMed.
- L. Li, X. Zhang, C. Liu, V. S. S. Mosali, J. Chen, A. M. Bond, Q. Gu and J. Zhang, Appl. Catal., B, 2023, 331, 122597 Search PubMed.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 Search PubMed.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 Search PubMed.
- A. Al Harthi, M. A. Abri, H. A. Younus and R. A. Hajri, J. CO2 Utiliz., 2024, 83, 102819 Search PubMed.
- J. Y. Zhao, K. Huang, C. Liu, X. Wu, Y. N. Xu, J. Li, M. Zhu, S. Dai, C. Lian and P. F. Liu, Adv. Funct. Mater., 2024, 2316167 Search PubMed.
- X. Yu, Y. Xu, L. Li, M. Zhang, W. Qin, F. Che and M. Zhong, Nat. Commun., 2024, 15, 1711 Search PubMed.
- T. Liu, K. Ohashi, K. Nagita, T. Harada, S. Nakanishi and K. Kamiya, Small, 2022, 18, 2205323 Search PubMed.
- S. Yan, C. Peng, C. Yang, Y. Chen, J. Zhang, A. Guan, X. Lv, H. Wang, Z. Wang, T.-K. Sham, Q. Han and G. Zheng, Angew. Chem., Int. Ed., 2021, 60, 25741–25745 Search PubMed.
- R. Senthil Kumar, S. Senthil Kumar and M. Anbu Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 Search PubMed.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, H17–H19 Search PubMed.
- X. Kang, Q. Zhu, X. Sun, J. Hu, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 266–273 Search PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 Search PubMed.
- M. B. Ross, C. T. Dinh, Y. Li, D. Kim, P. De Luna, E. H. Sargent and P. Yang, J. Am. Chem. Soc., 2017, 139, 9359–9363 Search PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 Search PubMed.
- X. Jiang, H. Li, J. Xiao, D. Gao, R. Si, F. Yang, Y. Li, G. Wang and X. Bao, Nano Energy, 2018, 52, 345–350 Search PubMed.
- J.-X. Wu, W.-W. Yuan, M. Xu and Z.-Y. Gu, Chem. Commun., 2019, 55, 11634–11637 Search PubMed.
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z. F. Huang, Z. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 Search PubMed.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 Search PubMed.
- X. D. Zhang, S. Z. Hou, J. X. Wu and Z. Y. Gu, Chemistry, 2020, 26, 1604–1611 Search PubMed.
- B.-X. Dong, S.-L. Qian, F.-Y. Bu, Y.-C. Wu, L.-G. Feng, Y.-L. Teng, W.-L. Liu and Z.-W. Li, ACS Appl. Energy Mater., 2018, 1, 4662–4669 Search PubMed.
- Y.-R. Wang, Q. Huang, C.-T. He, Y. Chen, J. Liu, F.-C. Shen and Y.-Q. Lan, Nat. Commun., 2018, 9, 1–8 Search PubMed.
- Y. T. Guntern, J. R. Pankhurst, J. Vavra, M. Mensi, V. Mantella, P. Schouwink and R. Buonsanti, Angew. Chem., Int. Ed., 2019, 58, 12632–12639 Search PubMed.
- R. Matheu, E. Gutierrez-Puebla, M. Á. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. Pei, N. Hanikel, B. Zhang and P. Yang, J. Am. Chem. Soc., 2019, 141, 17081–17085 Search PubMed.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech and E. Brunner, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 Search PubMed.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 Search PubMed.
- D. Yang, Q. Zhu, X. Sun, C. Chen, L. Lu, W. Guo, Z. Liu and B. Han, Green Chem., 2018, 20, 3705–3710 Search PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 Search PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. Garcia, J. Am. Chem. Soc., 2014, 136, 6798–6801 Search PubMed.
- Y. L. Qiu, H. X. Zhong, T. T. Zhang, W. B. Xu, P. P. Su, X. F. Li and H. M. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 2480–2489 Search PubMed.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 Search PubMed.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 Search PubMed.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 Search PubMed.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castano and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 Search PubMed.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 Search PubMed.
- Y. Zhang, Q. Zhou, Z. F. Qiu, X. Y. Zhang, J. Q. Chen, Y. Zhao, F. Gong and W. Y. Sun, Adv. Funct. Mater., 2022, 32, 2203677 Search PubMed.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 Search PubMed.
- L. Ye, J. Liu, Y. Gao, C. Gong, M. Addicoat, T. Heine, C. Wöll and L. Sun, J. Mater. Chem. A, 2016, 4, 15320–15326 Search PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig and V. S. Batista, Nat. Commun., 2018, 9, 1–9 Search PubMed.
- J. H. Cho, C. Lee, S. H. Hong, H. Y. Jang, S. Back, M. g. Seo, M. Lee, H. K. Min, Y. Choi and Y. J. Jang, Adv. Mater., 2023, 35, 2208224 Search PubMed.
- C. Li, Y. Ji, Y. Wang, C. Liu, Z. Chen, J. Tang, Y. Hong, X. Li, T. Zheng, Q. Jiang and C. Xia, Nano–Micro Lett., 2023, 15, 113 Search PubMed.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 Search PubMed.
- P. Shao, W. Zhou, Q.-L. Hong, L. Yi, L. Zheng, W. Wang, H.-X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 Search PubMed.
- Z.-H. Zhao, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 7986–7993 Search PubMed.
- Y. Zhang, Q. Zhou, Z.-F. Qiu, X.-Y. Zhang, J.-Q. Chen, Y. Zhao, F. Gong and W.-Y. Sun, Adv. Funct. Mater., 2022, 32, 2203677 Search PubMed.
- S. Mukhopadhyay, M. S. Naeem, G. Shiva Shanker, A. Ghatak, A. R. Kottaichamy, R. Shimoni, L. Avram, I. Liberman, R. Balilty, R. Ifraemov, I. Rozenberg, M. Shalom, N. López and I. Hod, Nat. Commun., 2024, 15, 3397 Search PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 Search PubMed.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 Search PubMed.
- J. Duan, W. Jin and S. Kitagawa, Coord. Chem. Rev., 2017, 332, 48–74 Search PubMed.
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 Search PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang and P. Zhang, Adv. Mater., 2018, 30, 1704303 Search PubMed.
- D. Narváez-Celada and A. S. Varela, J. Mater. Chem. A, 2022, 10, 5899–5917 Search PubMed.
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z. F. Huang, Z. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 Search PubMed.
- Y. Wang, P. Hou, Z. Wang and P. Kang, ChemPhysChem, 2017, 18, 3142–3147 Search PubMed.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu and D. D. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202204967 Search PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 Search PubMed.
- Y. Liu, S. Li, L. Dai, J. Li, J. Lv, Z. Zhu, A. Yin, P. Li and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 16409–16415 Search PubMed.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 Search PubMed.
- Y. Zhou, S. Chen, S. Xi, Z. Wang, P. Deng, F. Yang, Y. Han, Y. Pang and B. Y. Xia, Cell Rep. Phys. Sci., 2020, 1(9), 100182 Search PubMed.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech and E. Brunner, Nat. Commun., 2020, 11, 1409 Search PubMed.
- H. Zhong, M. Wang, G. Chen, R. Dong and X. Feng, ACS Nano, 2022, 16, 1759–1780 Search PubMed.
- Y.-H. Xiao, Y.-X. Zhang, R. Zhai, Z.-G. Gu and J. Zhang, Sci. China Mater., 2022, 65, 1269–1275 Search PubMed.
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 Search PubMed.
- D.-H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C.-T. Dinh, F. P. García de Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorović, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M.-J. Choi, S.-W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 Search PubMed.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 Search PubMed.
- X. Cao, D. Tan, B. Wulan, K. S. Hui, K. N. Hui and J. Zhang, Small Methods, 2021, 5, 2100700 Search PubMed.
- Z. Chen and Y. Wang, Fundam. Res., 2023 DOI:10.1016/j.fmre.2023.03.019.
- A. Wuttig, C. Liu, Q. Peng, M. Yaguchi, C. H. Hendon, K. Motobayashi, S. Ye, M. Osawa and Y. Surendranath, ACS Cent. Sci., 2016, 2, 522–528 Search PubMed.
- Y. Gong and T. He, Small Methods, 2023, 7, 2300702 Search PubMed.
- Y. Yang, Y. Yang, Y. Liu, S. Zhao and Z. Tang, Small Sci., 2021, 1, 2100015 Search PubMed.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1701759 Search PubMed.
- W. Chen, B. Han, C. Tian, X. Liu, S. Liang, H. Deng and Z. Lin, Appl. Catal., B, 2019, 244, 996–1003 Search PubMed.
- D. Yu, L. Ge, B. Wu, L. Wu, H. Wang and T. Xu, J. Mater. Chem. A, 2015, 3, 16688–16694 Search PubMed.
- C. Wang, Y. V. Kaneti, Y. Bando, J. Lin, C. Liu, J. Li and Y. Yamauchi, Mater. Horiz., 2018, 5, 394–407 Search PubMed.
- N. Zaman, N. Iqbal and T. Noor, Arabian J. Chem., 2022, 103906 Search PubMed.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H.-L. Jiang, J. Mater. Chem. A, 2019, 7, 26371–26377 Search PubMed.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew Chem. Int. Ed. Engl., 2020, 59, 2705–2709 Search PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 Search PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 Search PubMed.
- S. Back, J.-H. Kim, Y.-T. Kim and Y. Jung, ACS Appl. Mater. Interfaces, 2016, 8, 23022–23027 Search PubMed.
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 Search PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 Search PubMed.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed.
- R. Wang, X. Sun, S. Ould-Chikh, D. Osadchii, F. Bai, F. Kapteijn and J. Gascon, ACS Appl. Mater. Interfaces, 2018, 10, 14751–14758 Search PubMed.
- Y. Guo, H. Yang, X. Zhou, K. Liu, C. Zhang, Z. Zhou, C. Wang and W. Lin, J. Mater. Chem. A, 2017, 5, 24867–24873 Search PubMed.
- D. Zhang, Z. Tao, F. Feng, B. He, W. Zhou, J. Sun, J. Xu, Q. Wang and L. Zhao, Electrochim. Acta, 2020, 334, 135563 Search PubMed.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew Chem. Int. Ed. Engl., 2020, 59(23), 8896–8901 Search PubMed.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C. W. Kung, M. J. Katz, M. So, B. Klahr, D. Jin and Y. W. Chung, Adv. Mater., 2014, 26, 6295–6300 Search PubMed.
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Adv. Energy Mater., 2017, 7, 1602114 Search PubMed.
- S. Alizadeh and D. Nematollahi, J. Am. Chem. Soc., 2017, 139, 4753–4761 Search PubMed.
- J. Li, S. Chen, N. Yang, M. Deng, S. Ibraheem, J. Deng, J. Li, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2019, 58, 7035–7039 Search PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 Search PubMed.
- Y. Ren, J. Wang, M. Zhang, Y. Wang, Y. Cao, D. H. Kim and Z. Lin, Angew. Chem., Int. Ed., 2024, 63, e202315003 Search PubMed.
- J. Li, C. Chen, L. Xu, Y. Zhang, W. Wei, E. Zhao, Y. Wu and C. Chen, JACS Au, 2023, 3, 736–755 Search PubMed.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 Search PubMed.
- Y. Li, S. Wang, X.-s. Wang, Y. He, Q. Wang, Y. Li, M. Li, G. Yang, J. Yi, H. Lin, D. Huang, L. Li, H. Chen and J. Ye, J. Am. Chem. Soc., 2020, 142, 19259–19267 Search PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai and R. Chen, Nat. Energy, 2018, 3, 140–147 Search PubMed.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang and C. Liu, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 Search PubMed.
- H. Huang, K. Shen, F. Chen and Y. Li, ACS Catal., 2020, 10, 6579–6586 Search PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang and D. Su, J. Am. Chem. Soc., 2017, 139, 14143–14149 Search PubMed.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 Search PubMed.
- T. He, S. Chen, B. Ni, Y. Gong, Z. Wu, L. Song, L. Gu, W. Hu and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 3493–3498 Search PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 Search PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 Search PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 Search PubMed.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 Search PubMed.
- X. Sun, R. Wang, S. Ould-Chikh, D. Osadchii, G. Li, A. Aguilar, J.-l. Hazemann, F. Kapteijn and J. Gascon, J. Catal., 2019, 378, 320–330 Search PubMed.
- L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590–6602 Search PubMed.
- N. L. Torad, M. Hu, Y. Kamachi, K. Takai, M. Imura, M. Naito and Y. Yamauchi, Chem. Commun., 2013, 49, 2521–2523 Search PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 Search PubMed.
- Z. Chen, K. Mou, S. Yao and L. Liu, ChemSusChem, 2018, 11, 2944–2952 Search PubMed.
- Y. Ye, F. Cai, H. Li, H. Wu, G. Wang, Y. Li, S. Miao, S. Xie, R. Si, J. Wang and X. Bao, Nano Energy, 2017, 38, 281–289 Search PubMed.
- H.-Y. Tan, S.-C. Lin, J. Wang, C.-J. Chang, S.-C. Haw, K.-H. Lin, L. D. Tsai, H.-C. Chen and H. M. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 52134–52143 Search PubMed.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54(37), 10758–10762 Search PubMed.
- K. Liu, G. Wu and G. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 Search PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 Search PubMed.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 Search PubMed.
- F. O. Ochedi, D. Liu, J. Yu, A. Hussain and Y. Liu, Environ. Chem. Lett., 2020, 19, 941–967 Search PubMed.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 Search PubMed.
- J.-H. Guo, X.-Y. Zhang, X.-Y. Dao and W.-Y. Sun, ACS Appl. Nano Mater., 2020, 3, 2625–2635 Search PubMed.
- Y. Zhang, T. Sun, P. Zhang, K. Liu, F. Li and L. Xu, J. Colloid Interface Sci., 2023, 631, 96–101 Search PubMed.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 Search PubMed.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 Search PubMed.
- G. Chen, H. Zhong and X. Feng, Chem. Sci., 2021, 12, 15802–15820 Search PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 Search PubMed.
- Y. Li, S. L. Zhang, W. Cheng, Y. Chen, D. Luan, S. Gao and X. W. Lou, Adv. Mater., 2022, 34, 2105204 Search PubMed.
- D. A. Carter and J. E. Pemberton, J. Raman Spectrosc., 1997, 28, 939–946 Search PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai and B. I. Yakobson, Nano Lett., 2016, 16, 466–470 Search PubMed.
- C. Yan, Y. Ye, L. Lin, H. Wu, Q. Jiang, G. Wang and X. Bao, Catal. Today, 2019, 330, 252–258 Search PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 Search PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 Search PubMed.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. N. Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 Search PubMed.
- C. F. Wen, F. Mao, Y. Liu, X. Y. Zhang, H. Q. Fu, L. Zheng, P. F. Liu and H. G. Yang, ACS Catal., 2020, 10(2), 1086–1093 Search PubMed.
- R. Daiyan, X. Lu, X. Tan, X. Zhu, R. Chen, S. C. Smith and R. Amal, ACS Appl. Energy Mater., 2019, 2, 8002–8009 Search PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie and F. Yang, Energy Environ. Sci., 2018, 11, 1204–1210 Search PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 Search PubMed.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu and A. Epshteyn, ACS Catal., 2014, 4, 3682–3695 Search PubMed.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 Search PubMed.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 Search PubMed.
- Y.-S. Cheng, X.-P. Chu, M. Ling, N. Li, K.-L. Wu, F.-H. Wu, H. Li, G. Yuan and X.-W. Wei, Catal. Sci. Technol., 2019, 9, 5668–5675 Search PubMed.
- X. Xuan, J. Cheng, X. Yang and J. Zhou, ACS Sustain. Chem. Eng., 2020, 8, 1679–1686 Search PubMed.
- S. Zhang, C. Chen, K. Li, H. Yu and F. Li, J. Mater. Chem. A, 2021, 9, 18785–18792 Search PubMed.
- D. Geng, S. Yang, Y. Zhang, J. Yang, J. Liu, R. Li, T.-K. Sham, X. Sun, S. Ye and S. Knights, Appl. Surf. Sci., 2011, 257, 9193–9198 Search PubMed.
- H. Wang, X. Wu, G. Liu, S. Wu and R. Xu, Nano Res., 2022, 1–8 Search PubMed.
- H. Kiuchi, R. Shibuya, T. Kondo, J. Nakamura, H. Niwa, J. Miyawaki, M. Kawai, M. Oshima and Y. Harada, Nanoscale Res. Lett., 2016, 11, 1–7 Search PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson and J. Lou, ACS Nano, 2015, 9, 5364–5371 Search PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li and C. Ma, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 Search PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 Search PubMed.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K. H. Wu, Y. Lin, X. Sun, Q. Li and G. Centi, ChemSusChem, 2016, 9, 1085–1089 Search PubMed.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 Search PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 Search PubMed.
- G.-L. Chai and Z.-X. Guo, Chem. Sci., 2016, 7, 1268–1275 Search PubMed.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- H. R. M. Jhong, C. E. Tornow, B. Smid, A. A. Gewirth, S. M. Lyth and P. J. Kenis, ChemSusChem, 2017, 10, 1094–1099 Search PubMed.
- X. Cui, Z. Pan, L. Zhang, H. Peng and G. Zheng, Adv. Energy Mater., 2017, 7, 1701456 Search PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 Search PubMed.
- M. Jiang, X. Cao, D. Zhu, Y. Duan and J. Zhang, Electrochim. Acta, 2016, 196, 699–707 Search PubMed.
- L. Chai, L. Zhang, X. Wang, L. Xu, C. Han, T.-T. Li, Y. Hu, J. Qian and S. Huang, Carbon, 2019, 146, 248–256 Search PubMed.
- F. Liu, S. Liu, J. Meng, F. Xia, Z. Xiao, Z. Liu, Q. Li, J. Wu and L. Mai, Nano Energy, 2020, 73, 104758 Search PubMed.
- Y. Xu, Q. Li, H. Xue and H. Pang, Coord. Chem. Rev., 2018, 376, 292–318 Search PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 Search PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 10560–10565 Search PubMed.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 Search PubMed.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 Search PubMed.
- K. W. Frese, J. Electrochem. Soc., 1991, 138, 3338–3344 Search PubMed.
- S.-Y. Zhang, Y.-Y. Yang, Y.-Q. Zheng and H.-L. Zhu, J. Solid State Chem., 2018, 263, 44–51 Search PubMed.
- M. K. Kim, H. J. Kim, H. Lim, Y. Kwon and H. M. Jeong, Electrochim. Acta, 2019, 306, 28–34 Search PubMed.
- J. Liu, L. Peng, Y. Zhou, L. Lv, J. Fu, J. Lin, D. Guay and J. Qiao, ACS Sustain. Chem. Eng., 2019, 7, 15739–15746 Search PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 Search PubMed.
- C. I. Shaughnessy, D. T. Jantz and K. C. Leonard, J. Mater. Chem. A, 2017, 5, 22743–22749 Search PubMed.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798–8801 Search PubMed.
- W. Guo, X. Sun, C. Chen, D. Yang, L. Lu, Y. Yang and B. Han, Green Chem., 2019, 21, 503–508 Search PubMed.
- M. Peng, S. Ci, P. Shao, P. Cai and Z. Wen, J. Nanosci. Nanotechnol., 2019, 19, 3232–3236 Search PubMed.
- D. Li, T. Liu, Z. Yan, L. Zhen, J. Liu, J. Wu and Y. Feng, ACS Appl. Mater. Interfaces, 2020, 12, 7030–7037 Search PubMed.
- Z. Zhao, X. He, J. Yi, C. Ma, Y. Cao and J. Qiu, RSC Adv., 2013, 3, 84–90 Search PubMed.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012–9016 Search PubMed.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu and K. Daasbjerg, Adv. Funct. Mater., 2020, 30, 1910408 Search PubMed.
- M. Feyand, E. Mugnaioli, F. Vermoortele, B. Bueken, J. M. Dieterich, T. Reimer, U. Kolb, D. De Vos and N. Stock, Angew. Chem., Int. Ed., 2012, 51, 10373–10376 Search PubMed.
- V. Vivier, A. Regis, G. Sagon, J.-Y. Nedelec, L. Yu and C. Cachet-Vivier, Electrochim. Acta, 2001, 46, 907–914 Search PubMed.
- Z. Ma, D. Wu, X. Han, H. Wang, L. Zhang, Z. Gao, F. Xu and K. Jiang, J. CO2 Util., 2019, 32, 251–258 Search PubMed.
- W.-H. Li, W.-H. Deng, G.-E. Wang and G. Xu, EnergyChem, 2020, 2, 100029 Search PubMed.
- R. Sakamoto, K. Takada, T. Pal, H. Maeda, T. Kambe and H. Nishihara, Chem. Commun., 2017, 53, 5781–5801 Search PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 Search PubMed.
- J. S. Yoo, R. Christensen, T. Vegge, J. K. Nørskov and F. Studt, ChemSusChem, 2016, 9, 358–363 Search PubMed.
- J. M. Barlow and J. Y. Yang, ACS Cent. Sci., 2019, 5, 580–588 Search PubMed.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 Search PubMed.
- Y. R. Wang, Q. Huang, C. T. He, Y. Chen, J. Liu, F. C. Shen and Y. Q. Lan, Nat. Commun., 2018, 9, 4466 Search PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji and X. Zheng, J. Am. Chem. Soc., 2019, 141, 16569–16573 Search PubMed.
- Z. Zhang, J. Xiao, X. J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 16339–16342 Search PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 Search PubMed.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 Search PubMed.
- R. Iqbal, M. B. Akbar, A. Ahmad, A. Hussain, N. Altaf, S. Ibraheem, G. Yasin, M. A. Khan, M. Tabish and A. Kumar, Adv. Mater. Interfaces, 2022, 9, 2101505 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |