
Insights into electrolyte flooding in flexible gas diffusion electrodes for CO2 electrolysis: from mechanisms to effective mitigation strategies
Yuming
Wu
 *a,
Hesamoddin
Rabiee
bc,
Xiu Song
Zhao
d,
Geoff
Wang
b and
Yijiao
Jiang
*a
*a,
Hesamoddin
Rabiee
bc,
Xiu Song
Zhao
d,
Geoff
Wang
b and
Yijiao
Jiang
*a
aSchool of Engineering, Macquarie University, Sydney, NSW 2109, Australia. E-mail: yuming.wu@mq.edu.au; yijiao.jiang@mq.edu.au
bSchool of Chemical Engineering, The University of Queensland, St Lucia 4072, Australia
cCentre for Future Materials, University of Southern Queensland, Springfield, QLD 4300, Australia
dInstitute of Materials for Energy and Environment, Qingdao University, 308 Ningxia Road, Qingdao 266071, China
First published on 16th May 2024
Abstract
The advancement of CO2 electrolysis has reached a stage where practical CO2 electrolysers show promise for high conversion rate, low manufacturing cost, and extended system durability. While gas diffusion electrodes (GDEs) as flexible cathodes play a pivotal role in flow cell electrolysers, a prevalent issue arises with the implemented GDEs. Electrolyte flooding refers to the infiltration of bulk liquid electrolyte into the GDEs' gas diffusion channels. This typically occurs when the hydrophobicity of the GDEs towards the electrolyte diminishes, and then lowers the conversion efficiency and hence forecloses the durability of CO2 electrolysis. Compared to a proven track record of reporting substantial advancements in various novel catalysts, there is a scarcity of publications addressing the fundamental challenge of electrolyte flooding. In this review, the recent advancements in flexible GDEs for CO2 electrolysis are summarized, covering the evolution of different GDE types used in CO2 electrolysis and the current design trends in various flow cell electrolysers. In addressing the critical challenge, valuable insights into the fundamental mechanisms of triggering electrolyte flooding and in situ or ex situ approaches to observe flooding are discussed. A key segment of this review covers a comprehensive summary and evaluation of state-of-the-art methods of mitigating electrolyte flooding in flexible GDEs for CO2 electrolysis, considering three distinct perspectives within flow cell electrolysers: each layer of GDEs, properties of the membrane, and operating conditions. Finally, we discuss the remaining challenges and propose prospective research directions for alleviating the persistent issue of electrolyte flooding.
1. Introduction
Reducing CO2 emissions is widely acknowledged as a major research priority to reduce the greenhouse effect, which is one of the most daunting challenges to overcome global warming. Carbon capture, utilisation and storage (CCUS) technologies are acknowledged as among the most promising technologies to achieve the transition goal.1 The nascent carbon utilisation strategy is gaining attraction owing to being able to treat CO2 as a commodity instead of a burden.2 Employing waste CO2 as a feedstock to produce chemicals and fuels would offer a route to a carbon-neutral economy or even negative carbon emissions. CO2 electrolysis, or electrochemical CO2 reduction, is an attractive and sustainable option owing to mild electrolyser operating conditions (i.e., ambient pressure and temperature and neutral pH conditions), tunability towards desired products, and modular reactor designs.3 Electricity for CO2 electrolysis could directly be from renewable energy such as wind and solar if the future grid comprises abundant renewable energy. As a result, this strategy has great potential to achieve a carbon-neutral economy and negative CO2 emissions, ultimately addressing the rising climate change issue. Motivated by the urgent abatement of CO2 emissions, CO2 electrolysis has progressed to the point that efforts can now contribute to translating this knowledge toward the development of practical CO2 electrolysers.4The advancement of CO2 electrolysis, however, is confined to the laboratory scale with an electrode geometric area per unit commonly less than 250 cm2.5 A much larger electrode area is required to process about 50 t day−1 of CO2 to achieve competitive cost compared to petrochemical processes.6 Although the selectivity to desired products such as CO and C2H4 and current density achieved in bench-scale experiments approach likely industrial requirements,7 the present energy efficiency and stability of the lab electrolysers are far from the targets set by Sargent et al.8 The challenges in attaining a scalable performance primarily stem from operational stability of a gas diffusion electrode (GDE) as the cathode. The GDE not only exhibits exceptional mechanical resilience due to its flexibility but, more importantly, effectively addresses the mass transport issue with planar electrodes in static electrolysers.9 It is crucial to acknowledge that the voltage consumption at the anode poses a significant challenge, but this is not the central focus of this review. The most prolonged stable operation for a CO2 electrolysis flow cell is 1200 h at 300 mA cm−2 at a faradaic efficiency of CO of around 60% while it was initially 80%.10 Still, the required continuous reaction time is 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h to minimize the capital-expenditure of a conversion unit to economically compelling levels.11 More gravely, the extensively applied carbon-based GDEs often exhibit decaying after only a few hours of operation in flow cell CO2 electrolysers.8,11
000 h to minimize the capital-expenditure of a conversion unit to economically compelling levels.11 More gravely, the extensively applied carbon-based GDEs often exhibit decaying after only a few hours of operation in flow cell CO2 electrolysers.8,11
The related literature describing the stability of CO2 electrolysis links the degradation to electrolyte flooding. Burdyny et al.12 stated that flooding of the gas diffusion layer (GDL) will typically occur within several hours of operation, resulting in a diminished selectivity towards products of the CO2 reduction reaction. Xu et al.13 observed that the intensity of liquid signals indicating flooded electrolyte increases by roughly 31% in the initial 30 min of electrolysis within the GDE. Flooding is the ingress of bulk liquid electrolyte into the gas diffusion channels of the GDE. When flooding occurs, as illustrated in Fig. 1, the liquid occupies the microporous layer (MPL) and macroporous layer of the GDE that are originally hydrophobic. The infiltration of electrolyte into the GDL not only hinders CO2 access to the catalyst surface's active site by elongating the diffusion pathway but also has the potential to trigger salt precipitation. The desired gas–electrode–electrolyte interfaces are progressively substituted by the catalyst immersed in the electrolyte. The CO2 reduction (CO2R) performance undergoes a shift marked by a change in selectivity towards the hydrogen evolution reaction (HER). The electrolyte flooding ultimately results in critical failure of the CO2 electrolysis system.
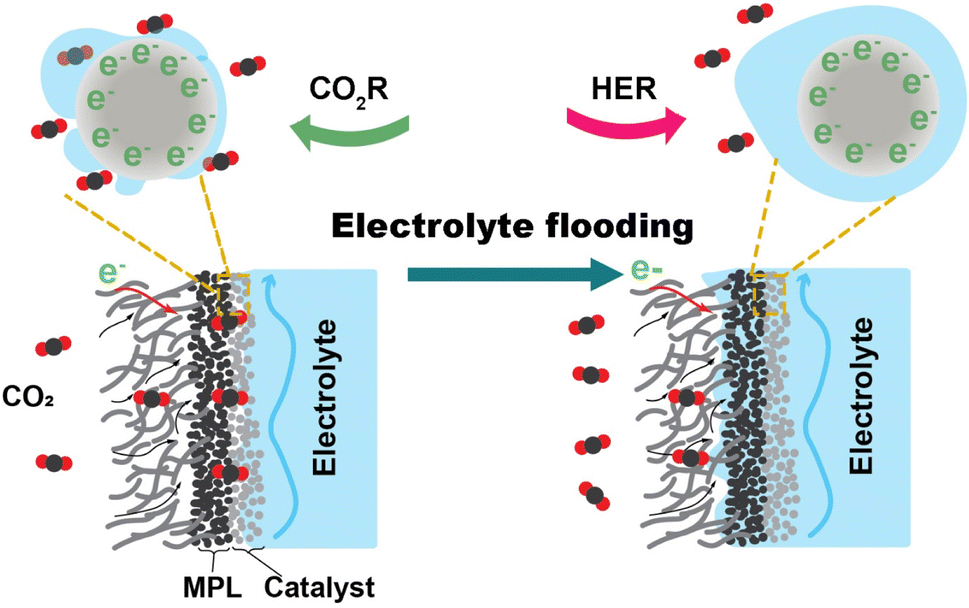 | ||
| Fig. 1 Illustration of electrolyte flooding in a GDE: the left depicts the ideal conditions of gas and liquid streams within a GDE composed of a microporous layer (MPL) and catalyst during CO2 electrolysis, while the right side illustrates the microenvironment of a GDE confronting electrolyte flooding. | ||
In any case, ensuring the resilient three-phase interface at the active sites of the catalyst layer with high-performance is crucial for establishing an efficient and long-lasting reaction cell for CO2 electroreduction. Managing the flooding of liquid electrolytes into the porous structure of the GDL remains a critical practical challenge for GDEs with operational stability in CO2 electrolysers.14 Especially at industrially relevant current densities >200 mA cm−2, flooding commonly becomes a critical issue and decreases the long conducting time of the system by drastically reducing the electroreduction selectivity and reaction rate.15 At present, there is still debate on the exact cause of electrolyte flooding. The complexity of the issue can be attributed to several factors, such as structures and compositions of GDEs, preparation methods of the GDE, pressure drop across the GDE, electrolyte concentration, gas stream humidity, operating temperatures, and other related variables.
In addressing the critical challenge of protecting GDEs from electrolyte flooding, this review paper delves into the comprehensive knowledge of gas-fed CO2 electrolysis and latest achievements in application of GDEs in this context. This perspective aims to offer valuable insights into the fundamental mechanisms of triggering electrolyte flooding and approaches for characterizing electrolyte flooding, and explore potential strategies for mitigating electrolyte flooding when employing the GDEs in electrolysers for CO2 electrolysis.
2. Advancements in flexible GDEs for CO2 electrolysis
The scale-up of CO2 electrolysis relies on the development of GDEs to overcome the substantial mass-transfer limitations observed with traditional planar electrodes. Before delving into the advancements of flexible GDEs in CO2 electrolysis systems, it is essential to review the mechanisms of electrochemical CO2R and the initial benefits of using GDEs in CO2 electrolysers.2.1 Evolution of applying GDEs in CO2 electrolysis
Analogous to electrolysis cells for other applications such as water splitting, CO2 electrolysis is commonly carried out in an electrolyser (also called a reactor or a cell) composed of a cathode, separator, electrolyte, anode and energy source.26 The desired products are produced at the cathode whilst water is oxidized at the anode. The two chambers are divided by a separator, such as a diaphragm membrane or an ion-exchange membrane, such as Nafion® or Sustainion® membranes. Common anolytes include acids (e.g. sulfuric acids), bases (aqueous KOH solutions), or aqueous inorganic salt solutions. Catholytes commonly include aqueous solutions of inorganic salts such as KHCO3,16 ionic liquids (e.g. 1-ethyl-3-methylimidazolium tetrafluoroborate),17 and organic solvents such as acetonitrile.18 Catholyte-free cells (also known as a vapor-fed cell) have also recently emerged as an effective alternative to the conventional reaction set-up.4,19The pursuit of electrochemically reducing CO2 to the desired C1–C3 products has seen considerable improvements in selectivity and the conversion rate, primarily through advancements in catalyst design. Catalysts serve critical roles at the cathode in achieving a sufficient rate of CO2 reduction and desired products at viable overpotentials through optimizing the free energy landscape of the reaction pathways, which makes the CO2 electroreduction application practically viable.20 Since CO2 electrolysis involves multiple electron and proton transfers, the process can proceed via various reaction pathways involving different intermediates. Fig. 2a shows that different catalysts could facilitate different reaction pathways, yielding a diverse range of products, including CO, HCOO−/HCOOH, methane, multiple-carbon products and undesired hydrogen.21–23
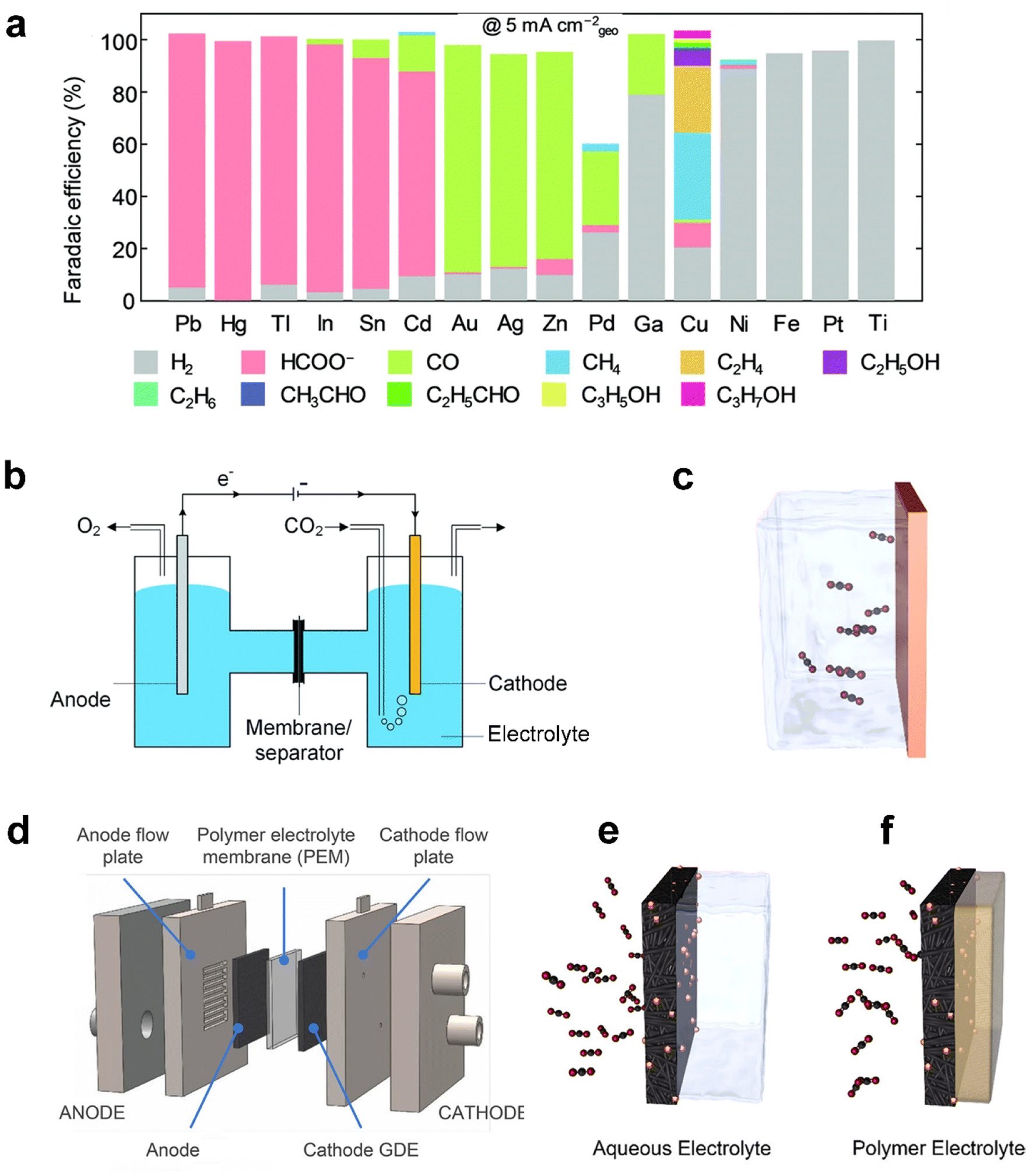 | ||
| Fig. 2 (a) Faradaic efficiency (FE) of various reaction products measured by Hori et al.41 after constant current electrolysis in 0.1 M KHCO3 on single metal electrodes. (b) Schematic of a laboratory electrochemical H-cell reactor and (c) planar electrode as the cathode. (d) Schematic of a typical membrane-based flow cell reactor, (e) gas diffusion electrodes (GDEs) in a flow cell electrolyser, and (f) a GDE in a membrane-electrode assembly without aqueous electrolyte. (a) is reproduced from ref. 42 with permission from The Royal Society of Chemistry, copyright 2020. (b) is reproduced from ref. 9 with permission from The Royal Society of Chemistry, copyright 2020. (c), (e), and (f) are reproduced from ref. 4 with permission of American Chemical Society, copyright 2019. (d) is reproduced from ref. 40 with permission of American Chemical Society, copyright 2018. | ||
Metals are generally employed as catalysts, classified into three major groups according to the products: (i) Sn, In, Hg, Bi, Tl, Cd and Pb favor the production of HCOO−/HCOOH;24 (ii) Au, Ag, Zn, and Pd tend to produce CO;25,26 (iii) Cu-based catalysts that are selective towards alkanes (e.g. CH4 and C2H4) and alcohols (e.g. CH3OH and C2H5OH).27 Engineering the material factors of metal catalysts, such as the particle size, exposed facets, grain boundary, vacancies, edges, and corner sites, has been reported to significantly influence the overpotential and product selectivity since the intermediate binding energies are highly sensitive to the atomic arrangement of active sites.28,29 The electrocatalysts have, furthermore, broadened the spectrum of materials from monometallic to bimetallic catalysts with various combinations of transition metals.30,31 Furthermore, aiming to predict promising electrocatalysts, a machine-learning density functional theory (DFT) framework was developed, and it simulated catalytic activity of hundreds of copper-containing intermetallic catalysts.32
Prior to introducing flexible GDEs in CO2 electrolysis systems, the initial CO2 electroreduction experiments were conducted in a static H-cell electrolyser33 (Fig. 2b). This reactor uses planar or simple porous electrodes as a cathode immersed in an electrolyte saturated with CO2. The planar electrode, typically metal foil or a glassy carbon plate, as shown in Fig. 2c, is useful to screen catalyst materials on the lab scale because of its relatively simple geometry that rules out impacts induced by complex factors such as structures of the electrodes.34–36 In addition, the use of a simple porous electrode allows the catalyst to maximize its surface area and inspection of the catalytic mechanism at an identical location during the electrochemical reactions.37,38
However, the cathode reaction rates within the H-cell reactor are indeed limited by the rate of CO2 transfer across the hydrodynamic layer from the bulk electrolyte to the electrode surface due to the low solubility and diffusivity of CO2 in the aqueous electrolyte,39 as shown in Fig. 2c. The situation becomes worse, especially at a high current density. In such a system, mass-transfer rates could be improved by operating the H-cell reactor at high pressure or low temperature to increase the solubility of CO2 in the electrolyte. Nonetheless, these configurations are far from commercially available conditions, but mass transport in an H-cell also limits testing at current densities of <100 mA cm−2.40
To overcome the mass-transport limitations of H-cells, a flow cell electrolyser has been proven to be highly effective in the development of commercial-scale fuel cells and water electrolysers.43 The schematic of the most widely studied CO2 electrolysis flow cell is displayed in Fig. 2d. Owing to applying a GDE as the cathode, the reactor achieved continuous flows of the reactants and products transferring into and away from the electrodes. As illustrated in Fig. 2e, CO2 is directly fed to the interface between the catalyst and catholyte through the highly-porous structure, instead of being dissolved in the liquid phase of the electrolyte before reaching the catalyst like in Fig. 2c,44 or CO2 in the gas phase mixed with vapor flows to the interface between the catalyst and membrane as shown in Fig. 2f.45 Weber et al.46,47 developed a multiphase model and found that the increased active surface area and decreased mass-transfer resistances are the reasons that a GDE cathode can realize an order of magnitude of current density improvement of CO2 electrolysis compared with the planar cathode. As a result, the current density in such a system is able to reach up to 3.37 A cm−2,48 compared with the planar electrode in the order of 10 mA cm−2.9 The significant enhancement in the reaction rate, consequently, enables flow cells to be commonly built and operated at a laboratory scale. The design of the flow cell units constituting larger stacks made the electrolyser configuration promising to meet the productivity required by commercial CO2 electrolysis.9
Moreover, the oxygen evolution reaction (OER) is driven at the anode. The choice of the GDE anode is beneficial for CO2 electrolysis as well.49 The polymer electrolyte membrane is applied to separate the two chambers and facilitates the flow of ions. The thickness of the electrolyte membrane is a key factor since the thinner membrane can result in less ohmic losses, but increased crossover risk of products and reactants of the two electrodes.47
2.2 Contemporary types of GDEs
Recent advances in GDEs employed in CO2 electrolysis are pushing current density and selectivity into a realm of industrial use. As mentioned before, GDEs overcome the significant mass-transfer resistances because of the large mass-transfer boundary layer near the planar electrodes. In this section, two main categories of GDEs according to different base materials are reviewed: a carbon-based GDE and polytetrafluoroethylene (PTFE)-based GDE. A carbon-based GDE commonly includes one or multiple porous carbon materials as the GDL such as carbon fibres, carbon cloth, and graphene, while the PTFE-based GDE is built up on a PTFE membrane composed of PTFE fibres. Other home-made GDEs were reported as well, such as metal oxide50 and metal gauze-based51 GDEs. Cook et al.51 chose a copper mesh-based GDE using copper acetate monohydrate as the catalyst layer and further achieved a current density 667 mA cm−2 with 53% FE of ethylene at 2 °C. This study opened up an avenue for Cu catalyst-based CO2 electrolysis at higher efficiency. Even so, these home-made GDEs unlike carbon or PTFE-based GDEs have not been commonly validated.Carbon-based GDEs are now generally utilized in flow-cell electrolysers owing to their low-cost (0.10–0.40 US$ per cm2, according to the price on the Fuel Cell Store), flexibility, electronic conductivity, and the catalyst simply being deposited and fixed. It is typically composed of two layers: the catalyst layer (CL) and gas diffusion layer (GDL), as illustrated in Fig. 3a; thus the catalyst-coated GDL is referred to as a GDE. The CL is usually deposited on the GDL by spraying catalyst ink and then evaporating the solvent, which ends with at least two components: active metal particles as catalytic sites and carbon black that is crucial to disperse and support metal particles.52 The GDL is a porous carbon layer which consisted of a microporous layer (MPL) and a carbon fibre substrate (CFS) allowing the reactant CO2 to diffuse to the CL. The MPL is typically carbon black mixed with hydrophobic PTFE to mitigate permeation of electrolyte into the gas chamber. The CFS or substituted carbon cloth serves as a current collector for electrons to flow from an external circuit to the interface between the CL and electrolyte.53 A typical cross-sectional morphology of the GDE with multiple layers is shown in Fig. 3b.
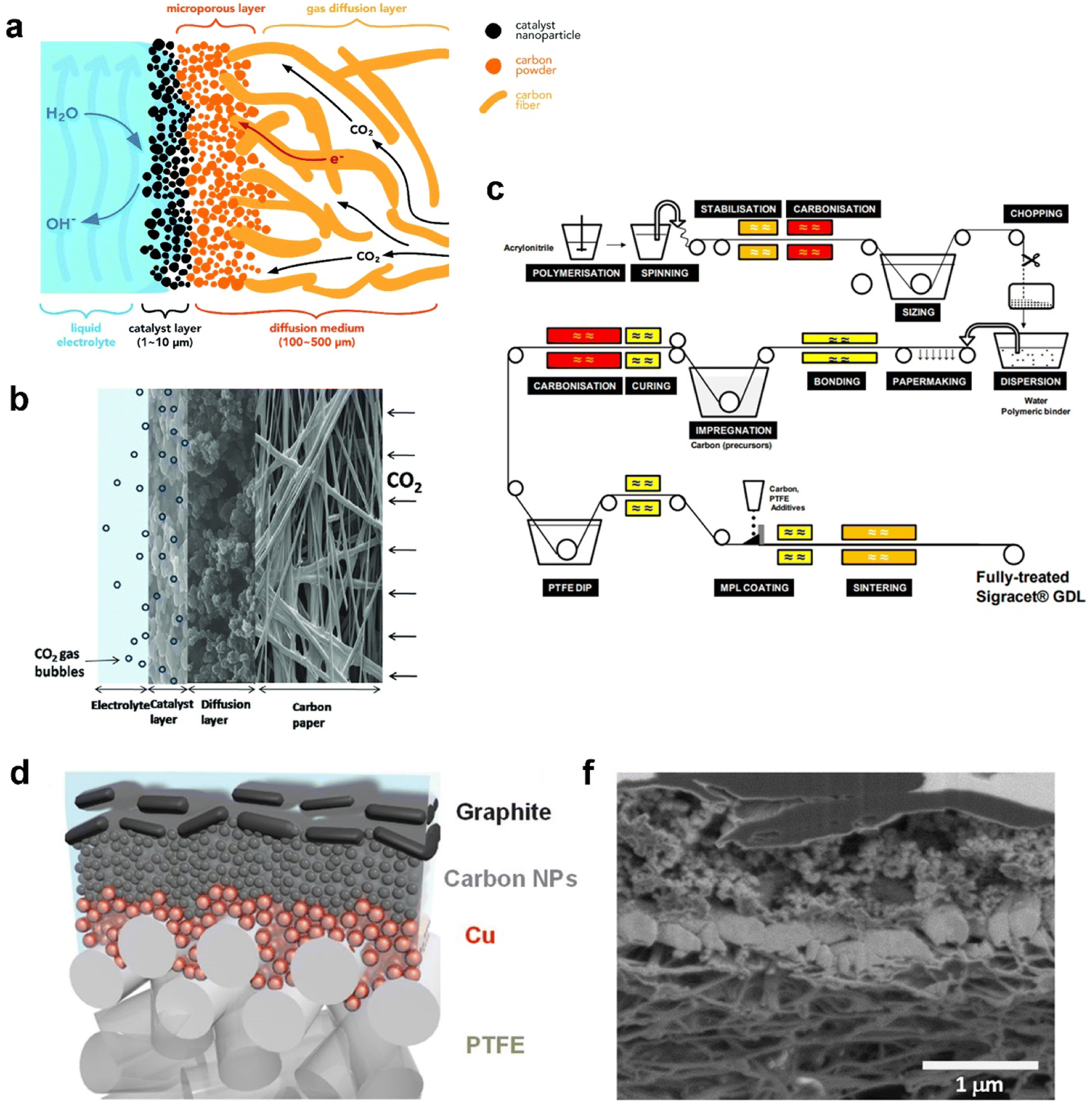 | ||
| Fig. 3 (a) Schematic, (b) cross-sectional morphology of the carbon-based GDE, and (c) manufacturing process of SIGRACET® carbon-based gas diffusion layers; (d) schematic illustration and (f) cross-sectional SEM image of each layer of the PTFE-based GDE. (a) is reproduced from ref. 46 with permission from The Royal Society of Chemistry, copyright 2018. (b) is reproduced from ref. 55 with permission from Elsevier, copyright 2014. (c) is reproduced from ref. 56 with permission from White Paper SGL Group. (d) and (f) are reproduced from ref. 57 with permission from AAAS, copyright 2018. | ||
At the very beginning, a carbon-based GDE was investigated in a proton exchange membrane (PEM) fuel cell nearly 30 years ago. Within PEM fuel cells, effective water management is necessary to meet fast response to requirement of high-power output system. Water deficiency reduces ionic conductivity in the membrane and the CL and induces severe contact resistance between the membrane and the CL, whereas excess water produced by the oxygen reduction reaction reduces catalytic sites for electrochemical reactions and impedes reactant transport through the non-reactive region.54 As a result, the GDE demonstrated great influence on the performance and stability of PEM fuel cells.
Along with the development of PEM fuel cells, GDLs in carbon-based GDEs are commercially available. A typical process of carbon GDL manufacture, SIGRACET® GDL as an example, is based on technologies from the paper and textile industries, as shown in Fig. 3c. For the fabrication of a CFS, continuous carbon fibres are produced from polyacrylonitrile or cellulose fibres pyrolysis. After carbonization, small fibre segments are cut and dispersed with additional binders. The mixture is then transferred to paper-making equipment and the resulting rolls are impregnated with phenolic resin which is then cured in air. Once all the solvents have been removed, the paper can be cut and pressed or moulded into the desired thicknesses or shapes.58 The CFS sheets are then heated to higher temperatures (1750–2700 °C) for graphitization where an amorphous carbon phase transforms into crystalline graphite. Subsequently, the sheets must be hydrophobized with fluoropolymers such as PTFE or fluorinated ethylene propylene (FEP) to create the porous volume. Before the addition of the MPL, carbon black powders are mixed with PTFE, surfactants and light alcohols in aqueous solution. The resulting slurry is then coated onto the CFS by spraying, screen-printing, or manual deposition. Finally, the MPL-coated GDLs undergo a separate three-step heat-treatment process to evaporate the solvents (approximately 120 °C), volatilize the surfactants (above 200 °C), and sinter PTFE (approximately 350–380 °C).58,59
In 1987, Williams et al.60 firstly reported the application of a carbon-based GDE in CO2 electrolysis to conduct in situ Raman spectroscopy for investigating the mechanism at the interface between the GDE and electrolyte, instead of trying to charge high current density for high rate CO2 reduction. The Kenis group placed commercial a carbon-based GDE in a flow cell61 and has thoroughly studied the relative functions in the performance of CO2 electrolysis.62–66 However, the carbon-based GDE tends to be wetted with electrolyte due to electrowetting and salt precipitation. This means the initial hydrophobic surface of the MPL deteriorated, which further allows the electrolyte to penetrate through the MPL and occupy the pores of the carbon substrate. The invaded pores cannot play a role in eliminating mass-transfer resistances; thus the carbon-based GDE hardly works under stable condition for over 1000 h. The operating lifetime of a commercial carbon-based GDE is far from the industry requirement of thousands of hours.8 Vermaas et al.67 prepared GDEs from a series of commercial carbon-based GDLs with a range of structural parameters (carbon fibre structure, thickness, and cracks), and found that there is a trade-off between flooding resistance and mass transfer capabilities that limits the maximum performance of GDEs during CO2 electrolysis. This trade-off depends strongly on the thickness and the structure of the carbon fibre substrate.
A PTFE-based GDE with a PTFE membrane as the GDL has been employed and reported in CO2 electrolysis in recent years.68 A typical structure of a PTFE-based GDE is shown in Fig. 3d. The hydrophobic membrane consists of a backbone made from polymer fibres. The corresponding catalyst is often sputtered on the membrane. Due to the nonconductive properties, an additional current collector is required to append such as a thin layer (∼1 μm) of copper or graphite as shown in Fig. 3f. Sargent's group reported a sequence of CO2 electrolysis studies on PTFE-based GDEs. Dinh et al.57 started to use the PTFE membrane composed of PTFE fibres as the GDL. Various layers were deposited one by one on the PTFE-GDL: Cu, carbon nanoparticles, and graphite. The Cu as catalyst was sputtered onto the porous PTFE GDL, while carbon nanoparticles and graphite played a role in ensuring the uniform distribution of current and overall support and current collector, respectively. The unprecedented GDE contacting alkaline catholyte reduces CO2 to ethylene with 70% faradaic efficiency at −0.55 V vs. RHE, for an initial 150 operating hours. Following this ground-breaking work, the PTFE-based GDE worked well with a Ag catalyst in both alkaline and neural catholytes.69 Furthermore, García de Arquer et al.70 achieved a big improvement in CO2 electrolysis performance achieving a current density of 1.3 A cm−2 at 45% cathodic energy efficiency by optimizing the PTFE-based GDE. Here a hybrid catalyst consisting of ionomer-coated copper was deposited onto the PTFE fibres.
Besides, other groups chose the PTFE-based GDE as a tool to eliminate flooding and boost stable partial current density of the desired products after a series of studies from Sargent's group. Jännsch et al.71 compared the PTFE-based GDE with a carbon-based GDE with a commercial Cu catalyst. The average FE and j for ethylene could be increased from 35% and 106 mA cm−2 to 42.6% and 118 mA cm−2. Shi et al. used a Ag catalyst on a PTFE GDL as the cathode to produce CO when Cl2 was oxidized from aqueous NaCl anolyte.72 Andronescu et al.73 reported that the PTFE membrane thickness used in the GDE plays an essential role in CO2 electrolysis performance. A moderate thickness (75 μm) of the GDL is needed to suppress hydrogen production.
As illustrated in the above literature, the PTFE-based GDE is a promising choice to achieve desirable stability owing to steady hydrophobicity. However, hardly adhering to catalyst ink solvents, catalyst particles must be sputtered or by other tedious and high-cost deposition strategies on the PTFE membrane. This restricts applications of catalysts that cannot be sputtered or have low electrical conductivity. Plus, the current collector itself is additional expenditure. In a nutshell, the PTFE-based GDE provides an opportunity to achieve stable CO2 electrolysis, while the fewer catalyst candidate and higher cost should be seriously considered prior to scaling up.
2.3 Design of flow cell electrolysers
Current flow cell electrolysers are usually classified into two types: microfluidic cells with an aqueous catholyte layer and membrane-electrode assembly (MEA) cells without a catholyte layer.74 First demo of a microfluidic cell was presented by Mahmood et al.75 in 1987, as shown in Fig. 4a. In a semibatch microfluidic cell, gas-phase CO2 was continuously supplied to the horizontal cathode surface, though the electrolytes remained static. At a cathode potential of approximately −1.4 V vs. RHE, the lead-based cathode exhibited a current density larger than 100 mA cm−2 for the formation of formic acid. This ground-breaking work also demonstrated the distinct role of the GDE in promoting the rate of CO2 electrolysis. With the rapid increasing CO2 emissions and the development of GDEs in PEM fuel cells, many efforts have been made to further improve CO2 electrolysis in microfluidic cells based on GDEs. A two-compartment microfluidic electrochemical cell with the simultaneous CO2 gas and electrolyte flowing by GDE was reported by Kenis et al.61 Two-compartment means the cell mainly consists of gas and electrolyte compartments without a membrane to separate the electrolyte chamber. Later on, they improved their cell design by including an ion exchange membrane to prevent the reduction products from being oxidized around the anode.65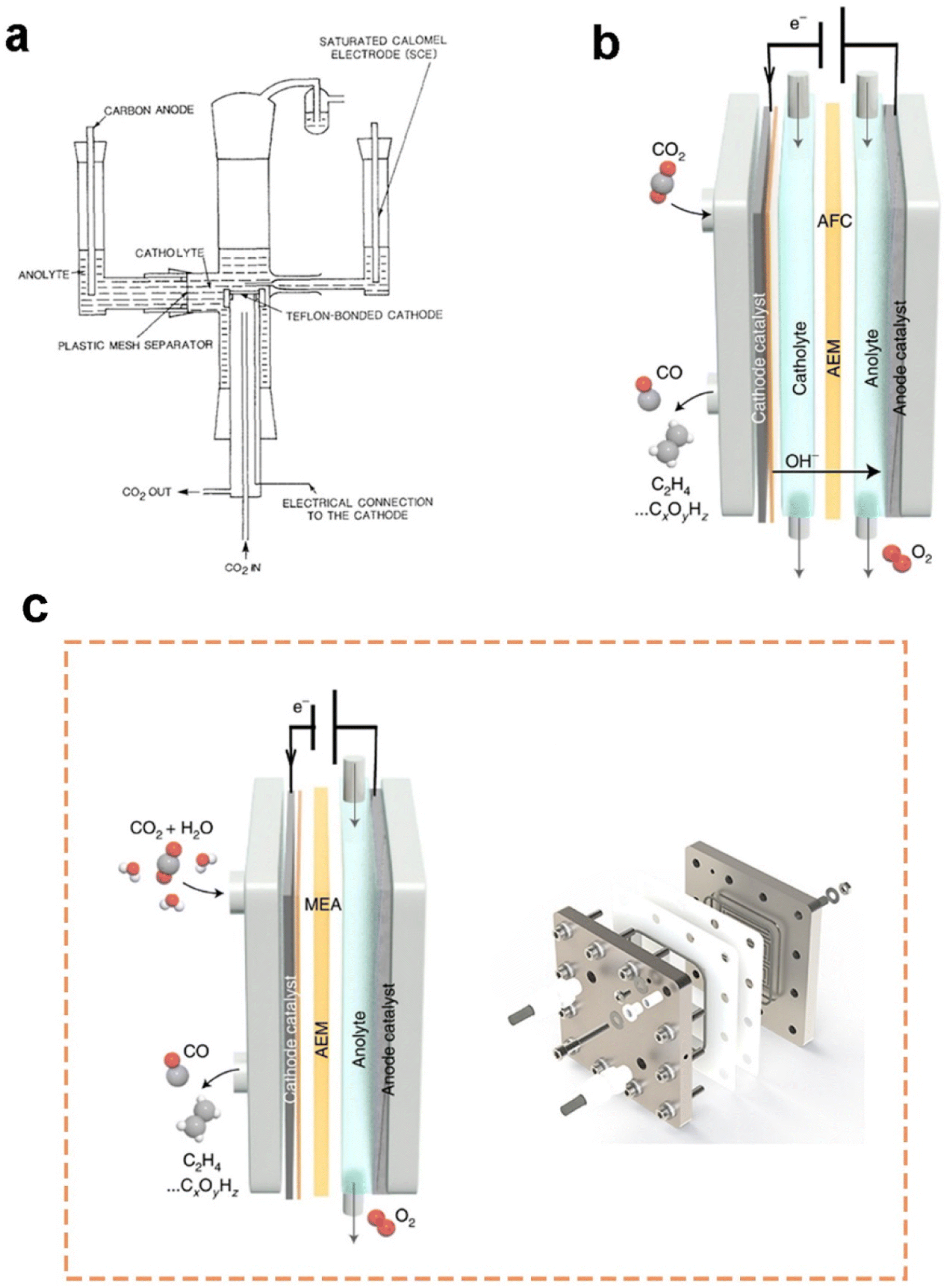 | ||
| Fig. 4 (a) Schematics of the first demo of a microfluidic flow cell and (b) a three-compartment microfluidic flow cell employing a GDE (note that the membrane is variable along with different electrolytes or catalysts); (c) schematics of a typical MEA employing a GDE (please note that the membrane is variable along with different electrolytes or catalysts) and MEA electrolyser product from Dioxide Materials®. (a) is reproduced from ref. 75 with permission from Springer Nature, copyright 1987. (b) and (c) are reproduced from ref. 7 with permission from Springer Nature, copyright 2022. | ||
Until now, the three-compartment design shown in Fig. 4b is the most employed microfluidic cell for CO2 electrolysis measurements owing to the high current density and energy efficiency achieved. A microfluidic cell developed by García de Arquer et al.70 can even reach a current density of 1.3 A cm−2 at 45% cathodic energy efficiency. The superb performance is due to a unique GDE design where a hybrid catalyst structure was developed through decoupling gas, ion, and electron transport by depositing an ionomer-coated copper catalyst onto a PTFE fibre GDL. Nonetheless, the extremely high concentration of 7 M KOH used as the catholyte could pose a significant concern for future applications.
A membrane-electrode assembly (MEA) cell is another flow cell design that exhibits low ohmic loss and high energy efficiency owing to minimized distance between electrodes. Here, the GDE as the cathode is pressed onto a membrane directly, allowing for reducing the ohmic resistance caused by the absence of a catholyte layer, as shown in Fig. 4c. In this configuration, liquid catholyte is not supplied to the cathode side while humidified CO2 gas was provided to the cathode from the macro-porous side. Cook et al.76 first introduced MEA configurations to electrochemically convert CO2 in 1988. Now, the MEA reactor is commercially available (see Fig. 4c). There are two common approaches to design cathodes inside MEA: (1) depositing the catalyst on the membrane prior to assembly, and (2) depositing the catalyst on a GDL followed by hot-pressing to the membrane. It should also be noted that flooding is likely to occur in the cathode GDE of an MEA, as water diffuses from the anode to the cathode.
Owing to the reduced electrolyte contact with the GDE and less inner resistance within the electrolyser, the stability performance of MEA and energy efficiency are generally better than that of the microfluidic cell. However, MEAs still encounter challenges with electrolyte flooding originating from the anode and permeating through the membrane.77,78 The invasive liquid and precipitated salts considerably increase the mass-transport resistance for CO2 and diminish the stability of CO2 electrolysis processes. On the other hand, it is difficult to compare the CO2 electrolysis performance with others reported in the literature due to the different GDL materials, deposition methods of catalysts, exposed areas of catalyst layer electrolytes, membranes, and components in flow cells. Therefore, standard conditions for CO2 electrolysis measurements in a flow cell are necessary to evaluate the behaviour of electroreduction at faradaic efficiency, partial current density, energy efficiency, operating lifetime, and so on.
3. Insights into electrolyte flooding in GDEs
As per the literature, the investigation on electrolyte flooding in GDEs during CO2 electrolysis commenced from 2011 and then has drawn increasing attention with time. Compared to the huge number of publications on catalysts for CO2 electrolysis (1930 papers in 2021 and 1758 papers in 2022, publications searched from the Web of Science™), flooding research is not a hot spot at all, although we must admit its crucial role in scaling up the technology. Therefore, investigation of electrolyte flooding in GDEs for CO2 electrolysis is not just significant but urgent.3.1 Mechanisms of electrolyte flooding
As reported in plenty of literature studies, most emerging carbon-based GDEs applied in flow cell CO2 electrolysers often exhibit limited durability, with performance decay after only a few hours of operation.8,11,79 This is due to the invasion of electrolyte into the gas channel by permeating through the porous GDE. The ineffective pores occupied by the liquid limit the direct transfer of reactant CO2 to CL, which decompose the high mass-transfer merit of GDEs. The performance decay is resulted from the factor that the electrolyte flooding is able to squash CO2 electrolysis especially with long conducting time or at high current densities. Managing flooding of liquid electrolytes into the porous structure remains a critical practical challenge for GDEs with operational stability in CO2 electrolysers.12,14,80–82To date, the flooding pitfall of the carbon-based GDE is mainly on account of three reasons: electrowetting,83 uneven pressure distributions across the GDE,84,85 and salt precipitation.14 Electrowetting means that the liquid generally becomes easily spread over the solid surface under an applied electrical field. The phenomenon has been explained by Lippmann–Young's equation that describes the relations between the contact angle and the applied potential (see eqn (1) and left of Fig. 5).86 Under CO2 electrolysis conditions, the contact angle of the CL is forced to decrease with larger potential applied.87 Gao et al.81 set up a home-made apparatus for measuring real-time contact angle on the GDE charged by currents, see right of Fig. 5, although the influence from oxygen evolution in droplets was not decoupled. Electrowetting can take place in both electrically conductive materials and dielectric materials, but the wettability of conductive materials (such as a carbon-based GDE) is more sensitive to an electric field than that of dielectric materials.83
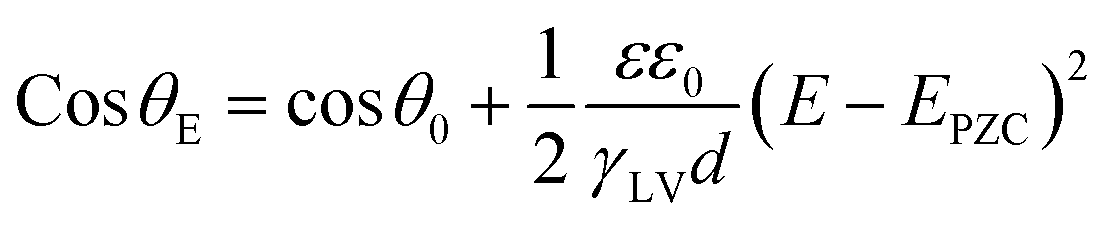 | (1) |
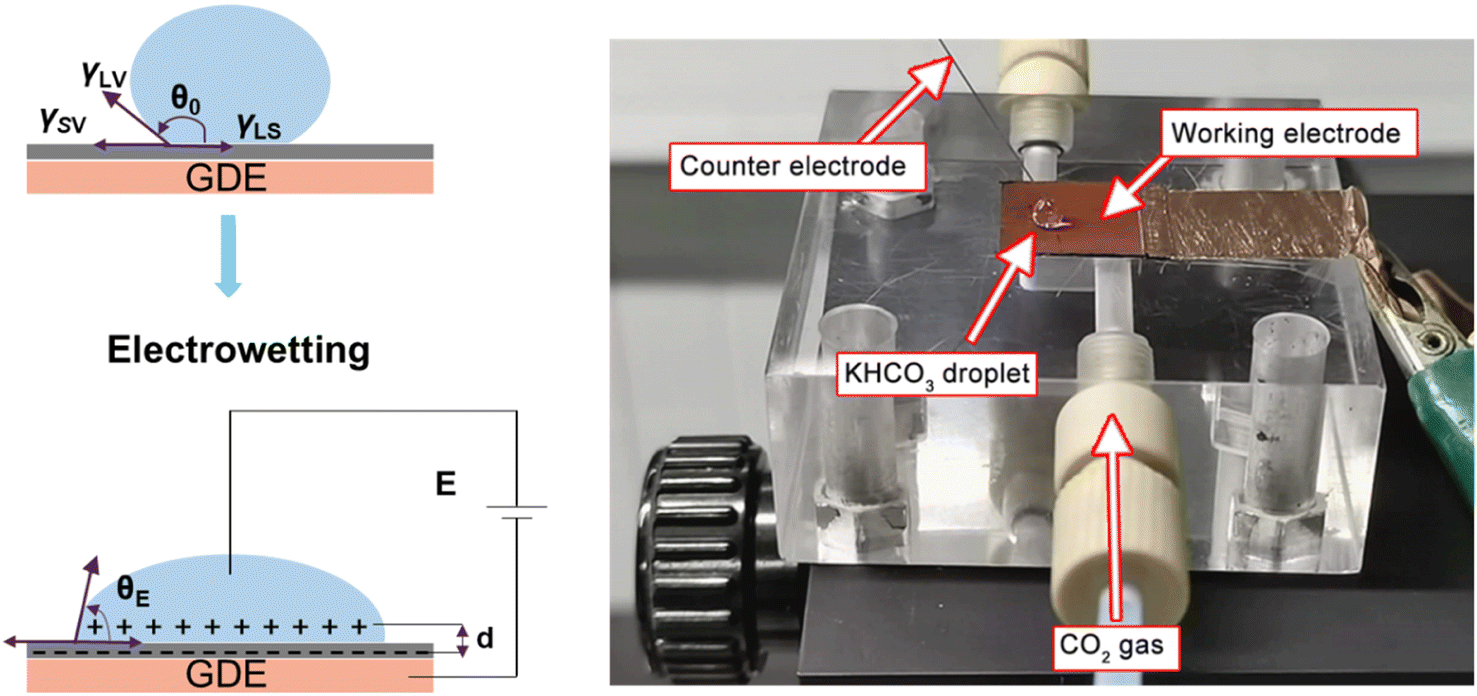 | ||
| Fig. 5 Illustration of electrowetting (left) and photo of the homemade platform that permits the operando observations of electrowetting during CO2 electrolysis developed by Gao et al. Adapted from ref. 81 with permission from American Chemical Society, copyright 2021. | ||
The differential pressure between gas and liquid phases within the GDL should be delicately controlled close to the active interfaces. Even slight overpressures on either the gas or liquid side of the GDL can cause bubbles in the liquid phase or result in flooding of the GDL.88 A maintained differential pressure should be constrained. Unfortunately, an imbalanced differential pressure within the GDE is common in present studies.89 Jeanty et al.85 presented that the position of the three-phase boundary in the pores of the GDE and the degree of electrolyte flooding are dependent on the differential pressure. Breugelmans et al.84 investigated the effect of pressure drop through the GDE on the flooding that was characterized by the electrolyte penetrating flow rate. Breugelmans et al. concluded that none of the differential pressure between catholyte and gas chamber is best for the CO2 electrolysis performance. However, the penetration flow rate is measured by visually analysing the drops on back of the GDE, so the method lacks accuracy.
Salt precipitation (or carbonation) is a critical factor giving rise to deactivation of GDEs in both a microfluid and MEA flow cell.90,91 These hydroxide ions tend to react with dissolved CO2 and then produce bicarbonate ions on route to carbonate ions.92,93 The negative potential on the cathode forms an interfacial electric field that attracts metal cations from the electrolyte to the cathode outer Helmholtz plane,94 which also responds to the electrowetting of absorbing the catholyte. The additional bicarbonate/carbonate salts from the transferred metal cations (potassium normally), plus consumed water during CO2 electrolysis make the salt concentration exceed the solubility limit, resulting in the formation of solid potassium carbonate salts.95 These salts precipitate within the GDE, progressively reducing CO2 mass transport to the catalyst until the pores are completely blocked and CO2 electrolysis is eliminated. The degradation process is briefly shown in Fig. 6. Shortly, the electrolyte flooding allows salts to precipitate in the GDE along with the other inducing factors. The highly interconnected relationship between salt precipitation and electrolyte flooding is crucial but has just begun to be explored.90
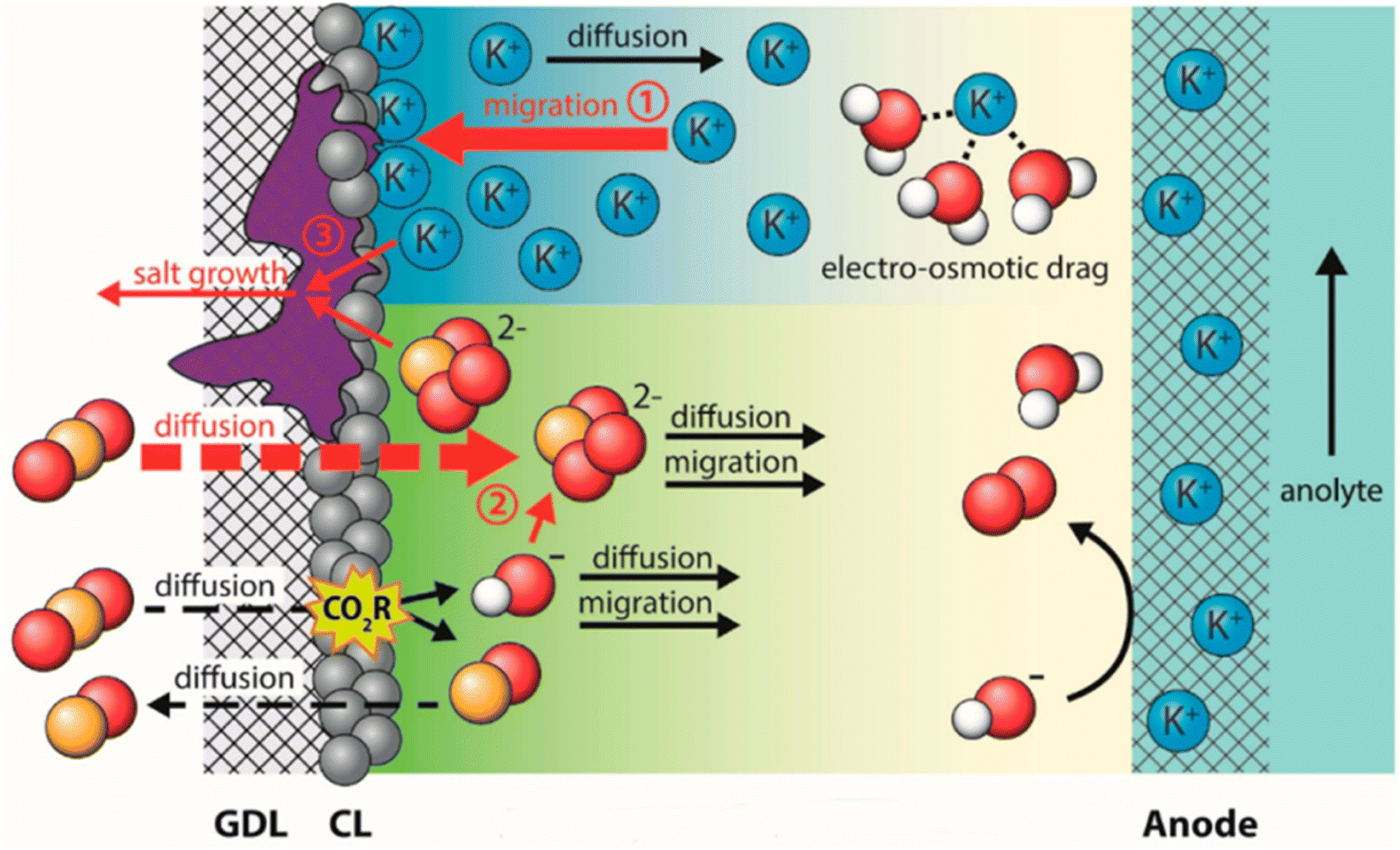 | ||
| Fig. 6 Schematic of the salt precipitation degrading the GDE together with electrolyte flooding. Reproduced from ref. 96 with permission from American Chemical Society, copyright 2023. | ||
3.2 Approaches to observe flooding in GDEs
Prior to delving into the methodologies for observing electrolyte flooding, it is imperative to establish a foundation through general techniques for characterizing GDEs. While assessing mechanical properties such as strength and toughness of GDEs is significant,97,98 the majority of the reported methods rely on microscopies or spectroscopies to analyse physical and chemical properties. Transmission electron microscopy (TEM) and atomic force microscopy (AFM) are able to obtain more detailed morphology on a very tiny scale that provides significant information for explaining the phenomenon during CO2 electrolysis.70,99 X-ray diffraction (XRD) and Fourier-transform infrared spectroscopy (FTIR) are quick and robust characterization methods. Integral structure information of the GDE can be obtained through them, but it becomes cumbersome when targeting a specific layer of the GDE.100,101 The region scanning of X-ray photoelectron spectroscopy (XPS) was applied to check the variation of the chemical state of the catalyst when investigating the electrocatalytic mechanism.102,103 Jovanovic et al.104 used Raman spectra to investigate bonding structure changes on a silver based GDE after CO2 electrolysis. In situ Raman spectroscopy is an effective technique to unveil the pH variation from the cathodic GDE surface to the electrolyte bulk. Lu et al.92 employed it to analyse the concentrations of HCO3− and CO32− and then derived related pH values from the concentrations and equilibrium constants.In a PEM fuel cell, if the water removal rate does not keep up with the generation rate at the cathode, excess water will accumulate, causing water flooding and thus blocking the pores in the porous CL and GDL. Owing to extensive research conducted on flooding of polymer electrolyte membrane fuel cells (PEMFCs),105 techniques and methods for characterizing water flooding have been established, providing measurements, such as the polarization curve,106 electrochemical impedance spectroscopy,107,108 pressure drop,109 membrane resistance measurement,110 and visualization of flooding.111,112 There is a consensus that cathode flooding plays a vital role in affecting the reduction efficiency of CO2 electrolysis by favouring selectivity of the competitive HER.14 Well-developled characterization methods for electrolyte flooding in CO2 electrolysis, however, were explored merely a few years ago. How to characterize flooding is a requisite step to unveil the deterioration mechanism and further overcome the insufficient stability that is limiting the large-scale development of CO2 electrolysis. Here, the current characterization of flooding extent is classified into two categories: ex situ or in situ methods.
Ex situ methods for observing and quantifying flooding are focused on the detection of residual salts post CO2 electrolysis measurement. A few common techniques can be employed to acquire the salt precipitated in the GDE post reaction, such as energy dispersive spectroscopy (EDS) and micro-computed tomography (micro-CT). As the close relationship between flooding and formation of alkali salts in the GDE from catholyte, potassium was mainly used as a tracer for flooding. Thiele et al.113 obtained EDS maps of the GDE to indicate signs of electrolyte infiltration. They observed potassium deposited on the catalyst layer and minor potassium signals were also observed in the GDL likely from the flooding at higher current densities. The EDS mapping has been combined with inductively coupled plasma mass spectrometry (ICP-MS) to try to quantitatively describe the salt concentration dependent on the depth of GDEs (as shown in Fig. 7a), which was introduced by the Broekmann group. Kong et al.114 started to use the combined approach to yield concentration depth profiles after different times of CO2 electrolysis. They then used the methodology to study flooding phenomena in GDEs differing in the abundance of cracks in the MPL, and concluded that cracks play an important role in the electrolyte management of CO2 electrolysers, since the electrolyte penetrating through cracks is paramount in avoiding flooding-related performance drops.115 Recently, they still employed this method to indicate a direct correlation between the break-down of effective electrolyte diffussion and the appearance of flooding.116 However, the depth profiles of potassium were obtained by statistical analysis of many EDS images. Moreover, the potassium concentration determined by ICP-MS is not from the surface observed by EDS, although there are two sides of one GDE sample after cutting.
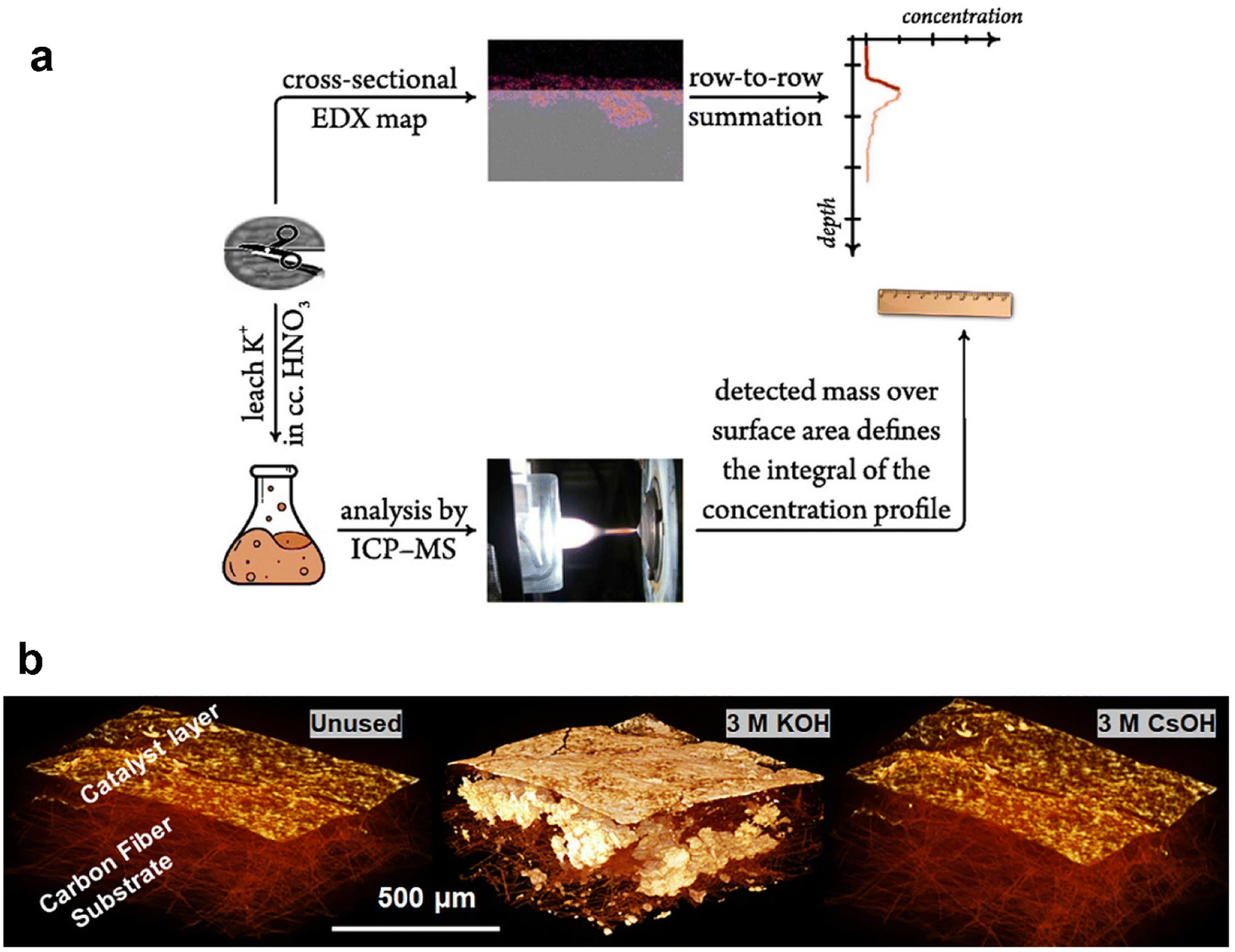 | ||
| Fig. 7 Ex situ methods for characterizing the extent of flooding: (a) a combined EDX/ICP-MS approach by using cross-sectional EDX mapping calculated by using a scaling factor that sets the integral of the K mass determined by ICP-MS; reproduced from ref. 114 with permission from Elsevier, copyright 2022 and (b) micro-CT images of an unused GDE and the precipitated salts within the GDE tested with different catholytes. Reproduced from ref. 118 with permission from American Chemical Society, copyright 2021. | ||
Micro-CT is a robust apparatus to image internal solid deposits of a porous GDE with a non-destructive advantage. X-rays were utilized to scan the GDE completely, which is usually time-consuming. It measures variation in X-ray attenuation upon rotating the samples, and then detects the tomographic images sequentially to reconstruct 3D images (see Fig. 7b) with high spatial resolution. The Kenis group demonstrated competent application of the technique on GDEs. Jhong et al.117 utilized micro-CT to visualize the catalyst layer and investigate the effect of different deposition methods of the catalyst layer. For studying flooding, Cofell et al.118 utilized micro-CT to visualize the presence of residual salts within the GDL from the penetrated electrolyte during CO2 electrolysis, as shown in Fig. 7b. There are other techniques (such as SEM and XRD) to detect alkali salts but they are not quantitative analysis techniques.
Ex situ methods generally cannot precisely characterize and quantitatively assess flooding. In situ strategies are desired to observe the real-time progress of electrolyte flooding. Electrochemical double-layer capacitance (EDLC) measurements have been used to quantify the wetted surface area of GDEs.119 Leonard et al.14 measured the currents from cyclic voltammetry divided by the sweep rate as EDLC. The variation of EDLC indicates the movement of the electrode–electrolyte interface. This means the EDLC can be used to track the condition of the electrolyte within the GDE to a certain degree during electrolysis. The increased current and EDLC is evidence of a flooded GDE. EDLC is commonly measured using an electrochemical workstation and there is no need for additional accessories. Larrazábal et al.120 then utilized EDLC measurements to confirm whether flooding was the factor affecting the performance of two types of GDEs in an MEA electrolyser.
Fluorescence spectroscopy coupled with confocal laser scanning microscopy is a newly developed in situ method to quantitatively analyse the interfacial electrolyte transportation through GDEs during CO2 electrolysis. An appropriate fluorescent agent is requisite for labelling the liquid phase before CO2 electrolysis measurements. Shi et al.121 first introduced an in situ technique to track real-time electrolyte flooding. They prepared a GDE labelled with a pH-responsive dye, allowing them to track the shift of the phase boundary with changes in the hydrophilicity of the GDE, as shown in Fig. 8a. The pioneering work further revealed the fundamental role of interfacial CO2 transportation in determining the stability of CO2 equilibrium concentration during the electrochemical reaction. Kalde et al.122 utilized fluorescence spectroscopy to achieve in-operando visualization of flooding through obtaining high-resolution information at the pore-scale on the liquid distribution and active reaction areas inside a simulated GDE, where structure and pores were designed and printed during operation.
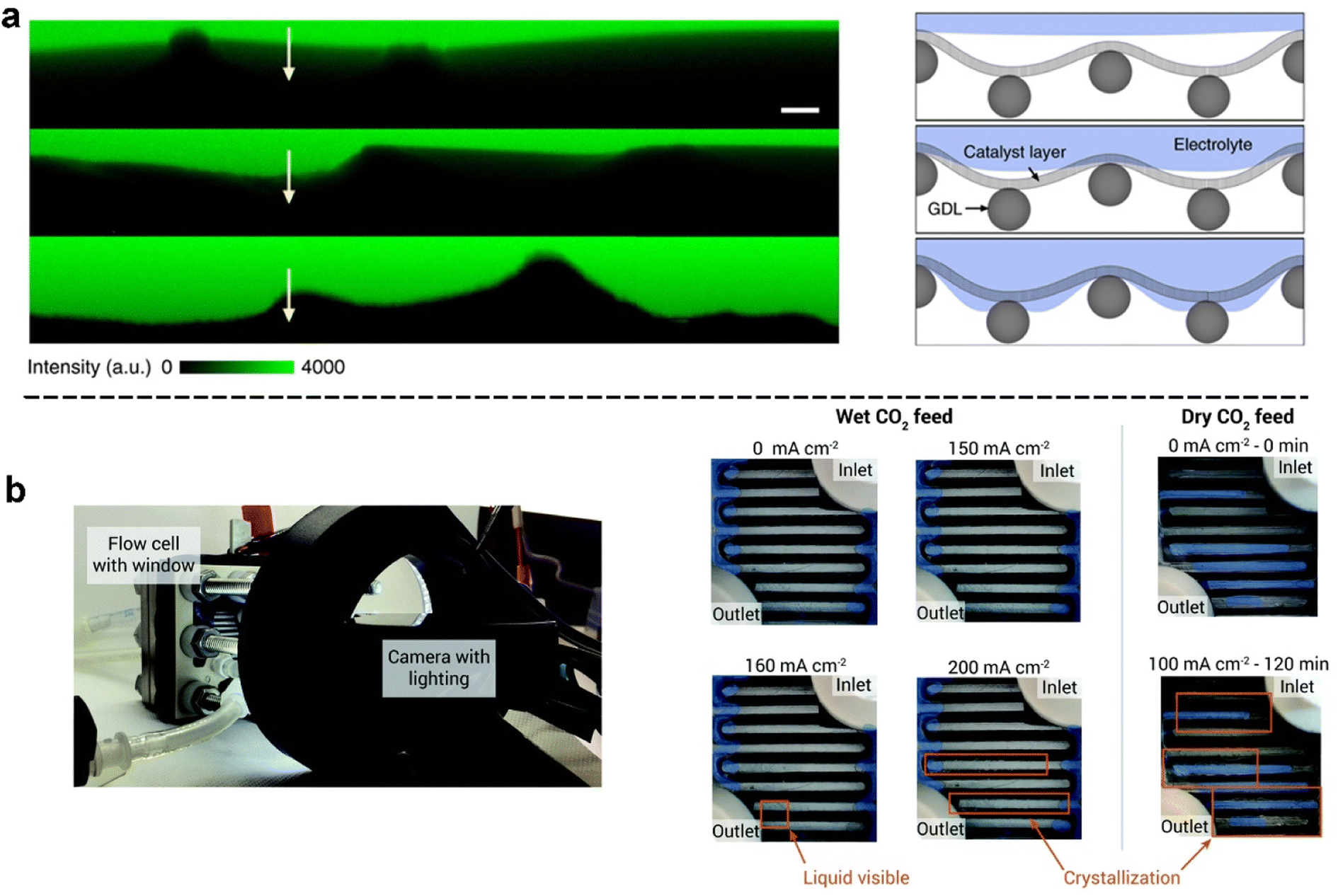 | ||
| Fig. 8 In situ methods for characterizing the extent of flooding: (a) fluorescence intensity line scans of labelled regions on the cross-section of GDEs that has different catalyst wettabilities after plasma treatment (yellow arrows from the CL to the GDL) and (right) schematic illustration of the corresponding interfacial structures; reproduced with permission from ref. 121. Copyright Nature Publishing 2020. (b) Experimental setup of a flow cell electrolyser used to observe flooding: images of the cathode flow field captured during electrolysis (left), and progressive liquid accumulation and crystallization in a flow cell as the current density was increased (right). Orange rectangles indicate areas of observable crystallization or liquid accumulation. Shaded blue regions represent residual adhesive from cell construction. Reproduced from ref. 78 with permission from Royal Society of Chemistry, copyright 2020. | ||
There are also simple but effective in situ approaches reported to observe flooding. Observation of electrolyte accumulation on the substrate side of the GDE facing the gas chamber has been reported by various experimental set-ups to detect flooding. As demonstrated in Fig. 8b, Berlinguette et al.78 designed and built a camera-incorporated electrolyser with embedded relative humidity sensors to monitor water with spatial and temporal resolution in the cathode chamber. The cathode flow plate was modified with a transparent viewing window for real-time monitoring by using a camera. They successfully observed the dynamic evolution of electrolyte flooding at different current densities (see right of Fig. 8b).
Jeanty et al.85 applied a feasible and more straightforward method to monitor electrolyte penetration. Small droplets start to emerge on the carbon support of the GDE driven by electro-wetting after the current was applied. The droplets grow in size over time and flow down the gas side of the GDE. Mot et al.84 used a transparent polymethyl methacrylate (PMMA) plate as the cover of the electrolyser to observe the flooded droplets under various pressure distributions between gas and liquid chambers. They observed and classified electrolyte flooding into four stages. In addition, Reyes et al.15 proposed a water-trap method to quantify real-time flooding. They calculated flooded water mass by measuring the water flux differentiation between the cathode outlet and cathode inlet and then subtracted the water consumed by CO2 electrolysis and the HER, to quantify the extent of cathode flooding in a zero-gap CO2 reactor.
4. Progress in mitigating electrolyte flooding
Commercial carbon-based GDEs were designed and fabricated for PEMFCs that experienced very different operational conditions to CO2 electrolysis.64,79,123,124 Carbon-based GDEs in PEM fuel cells are responsible for facilitating gas, liquid, electron, and heat transport in the presence of reactant gases and water, so material composition and microstructures have been optimized accordingly.58,123 For example, removing water from the cathode CL is crucial to device operation at high currents when the generated liquid water inhibits oxygen flux. Thus, densely packed MPLs serve both as high-surface-area conductive contacts to the CL and as effective media for water management.101,123,125,126 In PEMFC research, the inspection of flooding phenomena has reached a significant degree of maturity.105,127 For CO2 electrolysis, however, the water management is totally different. The cathode is driven by the electric potential to convert CO2 gas into either gas or liquid products, which is drastically different from the reaction scenario for PEMFCs.11,123 The electrolyte flow is desired to be constrained in the liquid channel within the CO2 electrolyser, even though the invasion of electrolyte into the gas channel, flooding, always occurs.Aiming to resolve the flooding issue with the development of GDEs in future scale-up of flow cell electrolysers, a great deal of efforts have been made along with understanding the GDE deterioration mechanism. Considering the vulnerable nature of the commercial GDE, developed strategies have been categorized to target specific layers within the GDE. Additionally, the impacts of membrane selection and operating conditions on mitigating flooding are thoroughly examined and discussed in this chapter.
4.1 Mitigating flooding from the CL
The catalyst layer (CL) provides active sites for CO2 electrolysis when sufficient protons are supplied from electrolyte. Notably, recent developments have identified the influence of overlayers at the CL, the interface facing the electrolyte to enhance CO2 electrolysis performance.128–130 The CO2 concentration in the local electrochemical environment of catalytic sites has also been shown to affect the product distribution of copper catalysts.131 As mentioned before, parameters such as the catalyst layer thickness and porosity, CO2 feed concentration, and feed flow rate are avenues to control the productivity and product distribution.132 Because of facing the electrolyte directly, the CL is penetrated by the electrolyte prior to other layers, which means the water management within the CL not only determines the reactive sites but also the limitation on mass transfer.Functionalization of a catalyst with fluorine-containing agents inside the catalyst layer could notably increase carbon-based GDE stability against flooding from a few hours to slightly more than ten hours.121,133,134 For example, Wang et al.134 reported a fluoroalkyl silane-modified copper catalyst enhancing water activation (mitigating electrolyte flooding), CO adsorption and hydrogenation of adsorbed CO to the CHO intermediate, as illustrated in Fig. 9a. As a result, the functionalized CL exhibits an ultrahigh current density of 1.6 A cm−2 with a C2+ (mainly ethylene and ethanol) faradaic efficiency of 80% for electrocatalytic CO2 reduction in a flow cell; Shi et al.121 modified the structure of gas–liquid–solid interfaces over GDEs by coupling a fluorine-terminated silane to carbon black in the catalyst layer to enhance hydrophobicity. They further achieved wettability modification on the GDE by plasma treatment, as shown in Fig. 9b. Polyvinylidene fluoride (PVDF) has also been identified as a hydrophobic binder used in catalysts on carbon-based GDEs.135
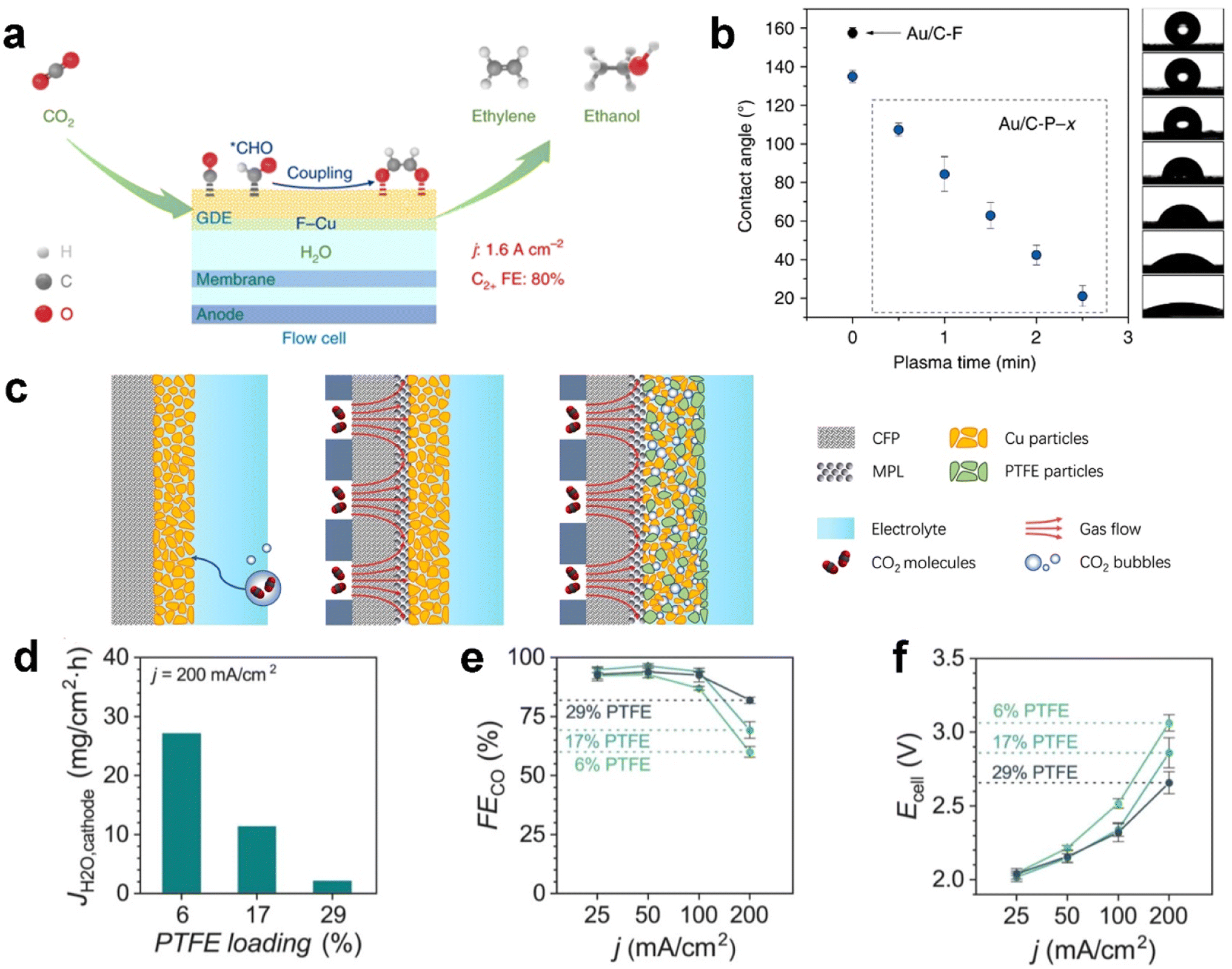 | ||
| Fig. 9 Functionalization of the catalyst layer with a fluorine-containing agent against flooding: (a) addition of a fluoroalkyl silane to mitigate electrolyte flooding; (b) fluorine-terminated silane to carbon black; (c) blending PTFE particles with the Cu catalyst, and effect of PTFE content in the CL on GDE flooding: (d) JH2O,cathode after applying a current density of 200 mA cm−2, (e) FECO, and (f) total cell voltage at 25, 50, 100, and 200 mA cm−2. (a) is reproduced from ref. 134 with permission from Springer Nature, copyright 2020. (b) is reproduced from ref. 121 with permission from Springer Nature, copyright 2020. (c) is reproduced from ref. 133 with permission from Springer Nature, copyright 2021. (d)–(f) are reproduced from ref. 15 with permission of American Chemical Society, copyright 2020. | ||
Polytetrafluoroethylene (PTFE) particles are another popular fluoropolymer added to the CL as a hydrophobic agent.133,136,137 Feng et al.133 proposed hydrophobic microenvironment construction that is able to significantly boost CO2 electrolysis on the GDE through protecting the CL from flooding, as shown in Fig. 9c. They dispersed hydrophobic PTFE nanoparticles inside commercial copper nanoparticles. Consequently, the PTFE-added CL achieves a greatly improved activity and faradaic efficiency for CO2 reduction, with a partial current density >250 mA cm−2 and a single-pass conversion of 14% at moderate potentials, which are around twice that of a regular electrode without adding PTFE. Berlinguette et al.15 also blended PTFE with a silver catalyst on the GDL, and they concluded that higher PTFE loading yielded better CO2 electrolysis performance (see Fig. 9d). The elevated FECO and reduced cell voltage (Fig. 9e and f) are attributed to the improved flooding conditions, as evidenced by a diminished flux of water to the cathode (JH2O,cathode), which is the change in mass of the water trapped in the MEA electrolyser.
Subsequently, Li et al.136 introduced PTFE into the CL that is a new nickel–nitrogen-doped carbon (Ni–N–C) electrocatalyst, and the PTFE-modified CL compared to a conventional electrode without PTFE displayed a substantially outstanding water-flooding-resistant ability, decreased overpotential, and nearly 100% CO selectivity. Besides, there are a few studies demonstrating blending of non-fluorine agents in the CL for mitigating electrolyte flooding, such as urea138 and 1-octadecanethiol.139,140
Tuning the morphology of the catalyst is also effective to prevent carbon-based GDEs from being wetted.81,141 Copper is an ideal catalyst to tune the surface structure. Han et al.142 deposited a mesoporous film on copper foam to enhance the hydrophobicity of the surface facing membrane. The nanoporous catalyst prevents the occurrence of electrolyte flooding without voltage loss. Gao et al.81 electrodeposited the desired hierarchical Cu catalyst with a sharp needle structure on a commercial GDL, as shown in Fig. 10. This hierarchical copper structure allowed the CO2 reduction electrode with sufficient hydrophobicity to build a robust gas–liquid–solid triple-phase boundary, which can not only trap more CO2 close to the active copper surface but also effectively resist electrolyte flooding even under high-rate operation. It consequently achieved a high C2+ production rate of 255 ± 5.7 mA cm−2 with a faradaic efficiency of 64 ± 1.4%, as well as outstanding operational stability at 300 mA cm−2 over 45 h in a flow reactor.
 | ||
| Fig. 10 Tuning the morphology of the catalyst to prevent carbon-based GDEs from being wetted. Reproduced from ref. 81 with permission of American Chemical Society, copyright 2021. | ||
4.2 Mitigating flooding from the GDL
Maintenance of the hydrophobicity in the CL alone is insufficient to prevent the GDE from being over flooded at high current densities.143 The commercial carbon-based GDL ranging from 100–500 μm as a water barrier also plays a vital role in water management, although the thickness of the CL is about 1–10 μm.46 Zhang et al.144 suggested that water management in the GDL remains a priority compared with the CL for efficient gas transport. They compared the influences of treatments on the CL or GDL on flooding respectively, whereas the method they used to adjust hydrophobicity of the GDL was drop casting polydimethylsiloxane (PDMS) on copper particles instead of solely modifying the GDL substrate.Beyond the use in the CL, PTFE is also a popular additive to boost the hydrophobicity of a commercial GDL. Kim et al.64 established that a certain amount of PTFE in a commercial GDL is key to mitigating flooding for CO2 electrolysis to CO (see Fig. 11). 20 wt% of PTFE in the MPL provides sufficient hydrophobicity and the highest partial current density of CO owing to the lowest charge transfer resistance (Fig. 11a and b). They also concluded that a certain minimum substrate thickness is required for the long-term stability of CO2 electrolysis, by observing that a commercial GDL with a thinner substrate exhibited extensive flooding. Zhang et al.145 coated PTFE on a carbon fibre skeleton to enhance the hydrophobicity of the GDL while maintaining an optimal porosity to avoid gas blocking during CO2 electrolysis. An additional PTFE microporous layer, nonetheless, was then attached to the GDL. The PTFE-coated GDE is stable for at least 103 h, which is 16 times more than that achieved by a commercial hydrophobic carbon GDL. Kong et al.115 investigated the effect of cracks within the MPL from commercial carbon-based GDEs on the electrolyte flooding. They demonstrated that electrodes with an appropriate abundance of cracks show high and sustained catalytic activity, since cracks serve as preferential pathways for the electrolyte transport through the MPL. Then the cracks drain excess electrolyte from the catalytic layer, which prevents flooding of micropores and enables them to function as efficient transport channels for gaseous CO2. It is noted that the above studies are focused on hydrophobicity enhancement on the MPL not comprehensively including the carbon substrate.
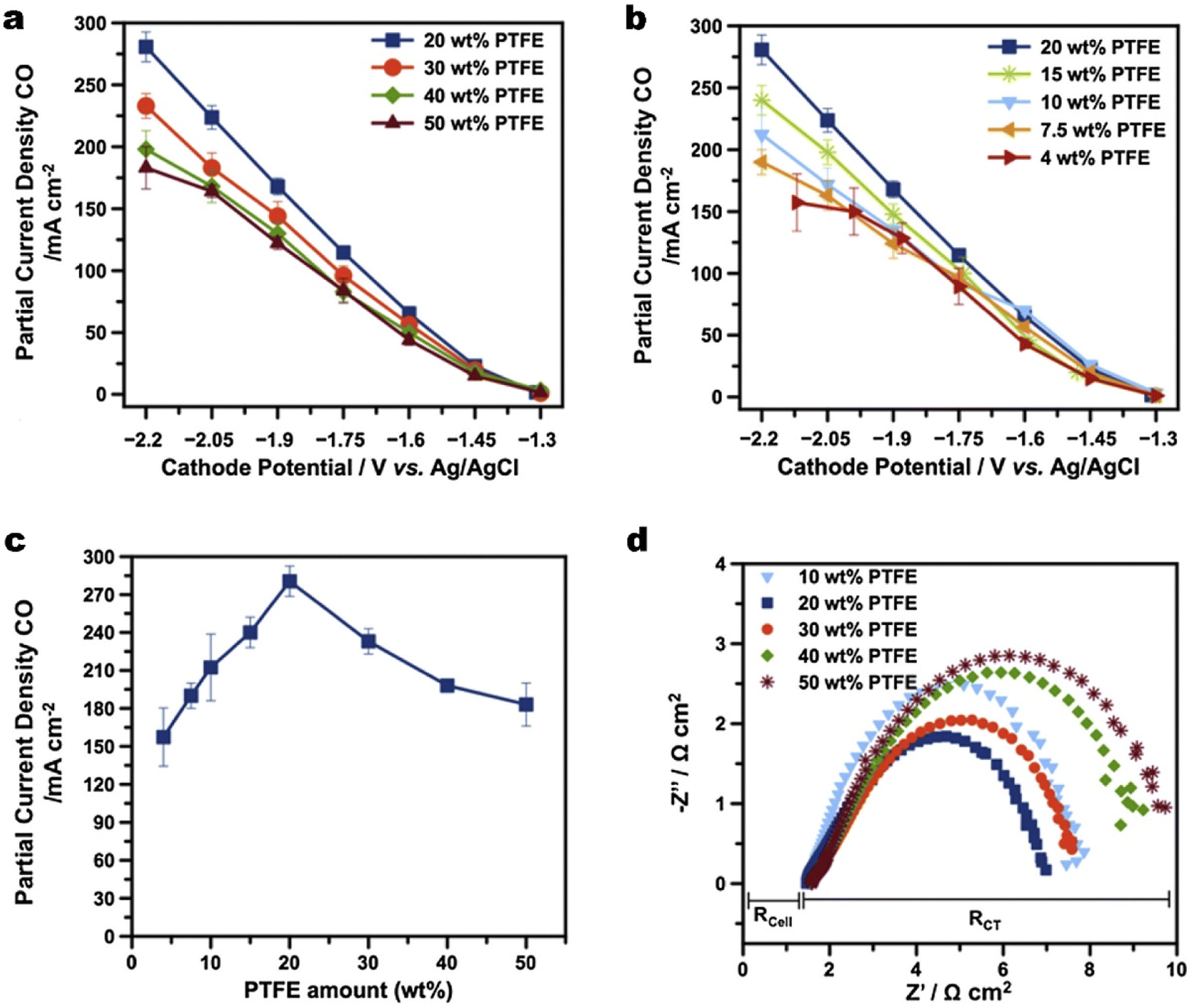 | ||
| Fig. 11 (a) and (b) Partial current density of CO and H2 as a function of different potentials for GDEs composed of MPLs with different amounts of PTFE wt%; (c) a comparison of partial current densities of CO for a Ag-based GDE as a function of PTFE content in the MPLs; and (d) the Nyquist plot of electrochemical impedance spectra for the GDEs with different PTFE contents in MPLs. Reproduced from ref. 64 with permission from Elsevier, copyright 2016. | ||
In our previous work, we reported a simple, vacuum-assisted infiltration method to deposit PTFE particles and carbon black preferentially at the interface between the MPL and the carbon cloth in a commercial GDL.146 As illustrated in Fig. 12a and b, vacuum-assisted infiltration allows some PTFE particles to be transported through existing cracks in the MPL and protrude into the CL to provide additional protection against electrolyte flooding through the MPL cracks. In CO2 electrolysis measurements with a commercial GDL covered by a silver nanoparticle catalyst, the PTFE-embedded GDE achieved a FECO of nearly 80% at 300 mA cm−2. Remarkably, at 100 mA cm−2 the PTFE-embedded GDE operated stably for more than 100 h with a FECO above 80% (see Fig. 12c), which was more than 50 times longer than for the untreated GDE.
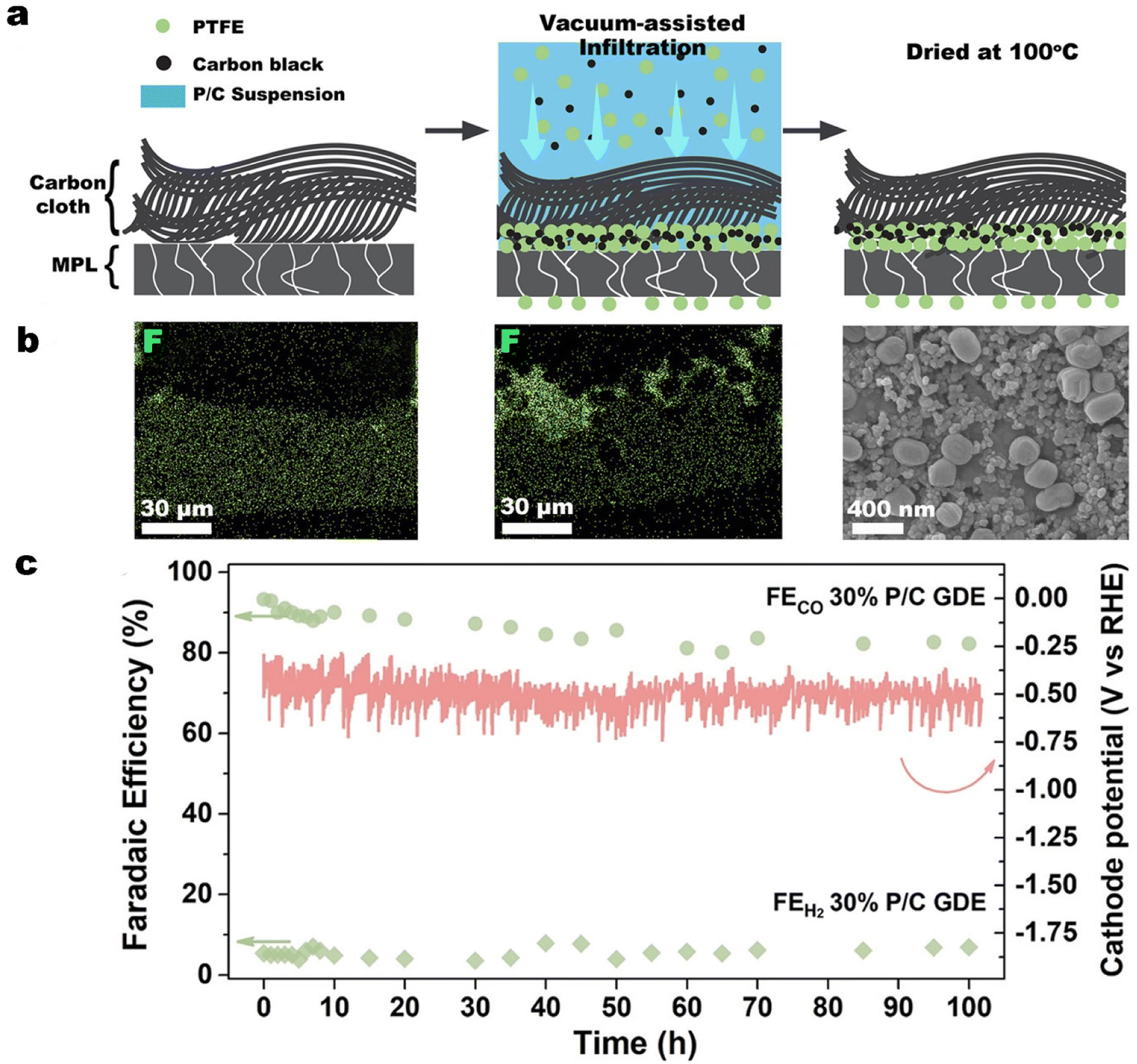 | ||
| Fig. 12 (a) Schematic of the vacuum-assisted infiltration method, the corresponding, (b) energy-dispersive X-ray spectroscopy (EDS) mapping of fluorine in the cross section of the commercial GDL, and (c) faradaic efficiency for CO and H2 and cathode potential of the PTFE-embedded GDE (30% P/C GDE) in an additional 100 h test at a current density of 100 mA cm−2. Reproduced from ref. 146 with permission of American Chemical Society, copyright 2022. | ||
Moreover, other fluoropolymer substrates have been demonstrated to be effective in mitigating electrolyte flooding. Yamaguchi et al.147 employed a PVDF GDL for flooding mitigation. It is noted that a conductive layer of aluminium is incorporated between the CL and the polymer substrate to provide an electron conduction path to the GDEs with the polymer substrate. The choice of aluminium layer was based on its limited activity in the parasitic hydrogen evolution reaction. Meanwhile, GDLs composed solely of fluoropolymers has been tested to isolate the effects of electrolyte flooding. Wicks et al.148 first fabricated a 3D-printed fluoropolymer GDL by photocuring a ternary solution of difunctional perfluoropolyether urethane methacrylate monomer Fluorolink MD700, N-methyl-2-pyrrolidone (NMP), and triethylene glycol (TEG), and then coated a conformal Cu catalyst layer on the structured GDL. Owing to the capability of 3D printing, several structure parameters, such as porosity, microstructure, and macrostructure, can be modulated to investigate the impact on mass transport and product distribution during CO2 electrolysis.
4.3 Mitigating flooding from the membrane
Besides the modification on each layer of commercial GDEs, Seger et al.13 utilized a thicker anion exchange membrane (AEM) to mitigate electrolyte flooding and impurity crossing over, explained by a longer water-ion transfer pathway within the AEM. Nonetheless, Berlinguette et al.15 showed that flooding can be mitigated by using a thinner (≤40 μm) AEM with low water uptake, in tandem with a hydrophobic cathode. They demonstrated that the thinnest membrane (20 μm) renders more efficient CO2 electrolysis at 200 mA cm−2 (as shown in Fig. 13a and b), owing to the lower electrolyte flooding through the thinner membrane. The group additionally found that a wet CO2 feed helps mitigate the flooding in the GDE by maintaining a uniformly hydrated cathode chamber (see Fig. 13c and d). Moreover, when using a dry CO2 feed, a higher flux of electrolyte across the membrane promotes salt precipitation and electrolyte flooding.78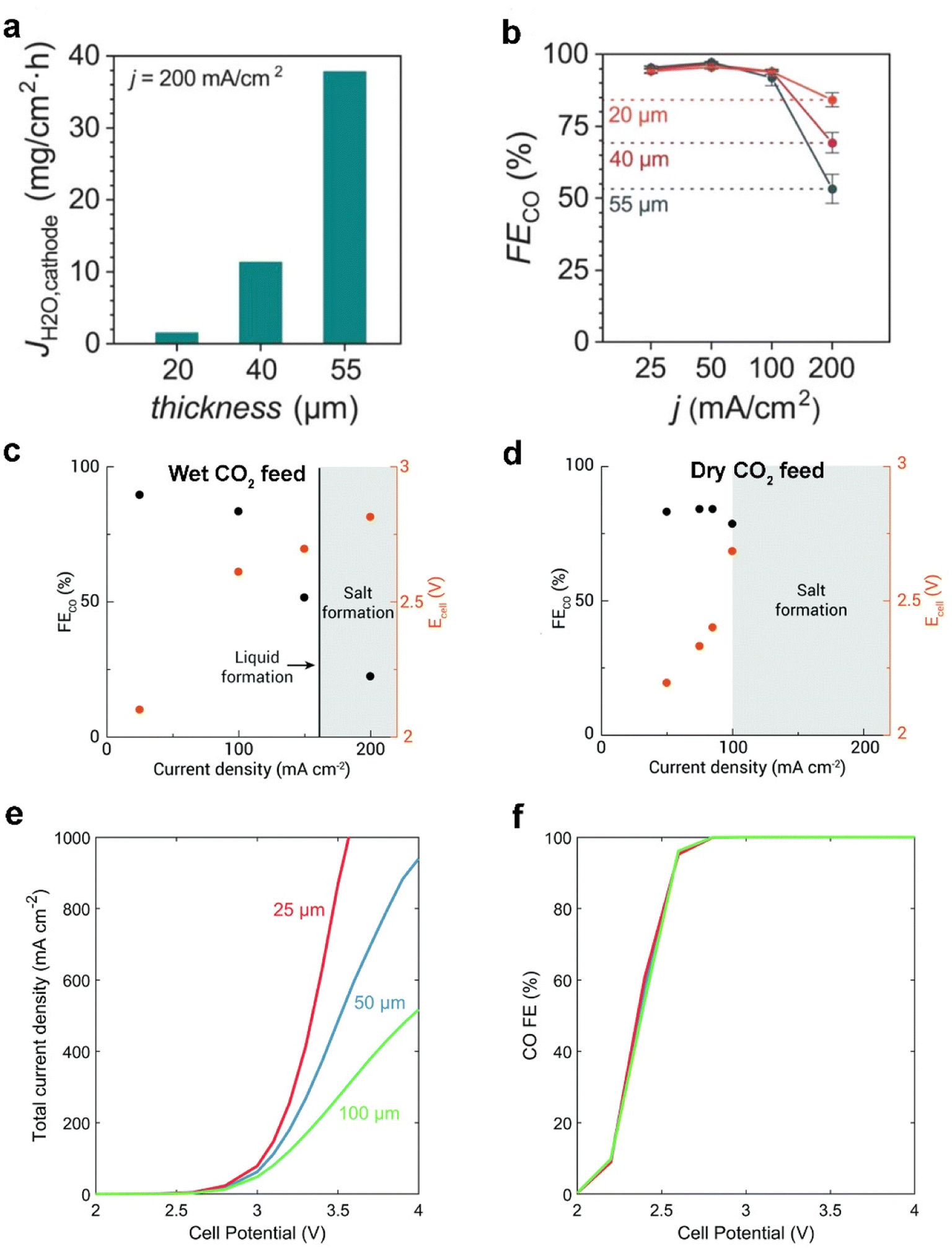 | ||
| Fig. 13 Effect of different thicknesses (20, 40, and 55 μm) of the membrane on (a) the flooding conditions (flux of water to the cathode, JH2O) and (b) CO faradaic efficiencies (FECO) at 200 mA cm−2. Reproduced from ref. 15 with permission from the American Chemical Society, copyright 2020. FECO and Ecell obtained for (c) wet CO2 feed and (d) dry CO2 feed with constant 100 sccm at varied current densities, where shaded areas represent conditions where the cell was not stable and salt precipitation (or liquid formation) was observed. Reproduced from ref. 78 with permission from The Royal Society of Chemistry, copyright 2020. (e) Total current density and (f) CO faradaic efficiency for the full-MEA case simulated with a 25 μm membrane, 50 μm membrane, and 100 μm membrane. Reproduced from ref. 77 with permission from The Royal Society of Chemistry, copyright 2019. | ||
Weng et al.77 also reported a similar modelling result that decreasing the membrane thickness is beneficial for alleviating the dehydration issue in a MEA electrolyser. Fig. 13e shows the effects of membrane thickness on the full-MEA performance. The current density increases with a thinner membrane, with the difference becoming more significant with higher cell potentials due to both enhanced water transport and lower ohmic losses. However, the selectivity to CO is not strongly impacted as shown in Fig. 13f. They suppose that decreasing membrane thickness will result in increased crossover and lower CO2 utilization. This trade-off between CO2 utilization and flooding conditions in a MEA electrolyser may result in an unexpected outcome.
4.4 Operating conditions
Besides the above modification on a commercial GDE and regulations of membrane thickness, different operating conditions in CO2 electrolysis systems play a crucial role in determining the extent of electrolyte flooding. The recommended conditions to constrain flooding are reviewed.As stated above, uneven pressure distributions across GDEs significantly induce electrolyte flooding in the GDE.84,85 Baumgartner et al.149 correlate the electrolyte flooding with a CO2 permeability constant, which is measured by the pressure drop across the GDE between the catholyte and gas compartment. A high permeability constant of the GDE is very promising for scale-up because it determines how well the GDE could maintain the separation of gas and liquid phases at a large scale, but it is merely one of operating conditions. The orientation of the GDE placed in an electrolyser has been considered to relieve electrolyte flooding. Cheng et al.150 reported a reverse-assembled GDE that can mitigate flooding effectively within a flow cell, where the silver nanoparticle catalyst was facing away from the electrolyte and toward the CO2 gas supply. This strategy was validated to operate GDE for over 150 h without degradation.
Operating temperature is critical for the performance of the electrolyser, influencing not only the activity of the catalyst but also the evaporation of the electrolyte. A modelling study by Weng et al.77 suggested that increasing the temperature at the cathode GDE is able to potentially minimize flooding in an exchange-MEA, because more permeated electrolyte is evaporated from the GDE. The simulation results indicate that the water vapor pressure is 0.46 atm at 80 °C, compared to 0.03 atm at 25 °C. Thus, a higher current density can be achieved at higher operating temperature with more rapid kinetics although a lower solubility of CO2 is a negative factor at elevated temperatures.
The electrolyte used plays a crucial role in determining the degree of flooding. Electrolyte composition and concentration also have significant effects on the morphology, distribution, and surface coverage of the carbonate deposits.42,118,151 Cofell et al.118 reported that the concentration of the electrolyte influences the CO2 electrolysis performance. They observed that decreases in selectivity are caused by occlusion of the catalyst layer surface by carbonate deposits, which are introduced by catholyte flooding during a 6 h testing period, and these deposits form more quickly in higher concentration of flooded electrolyte as shown in Fig. 14a and b. Moreover, they employed different alkaline catholytes for conducting CO2 electrolysis and found that a GDE operated in CsOH electrolyte is the most promising for long-term operation (see Fig. 14c), since the CsOH electrolyte results in fewer precipitated salts remaining in the GDE (see Fig. 7b). Qin et al.152 found that the presence of potassium cations in the acid catholyte results in more severe flooding, as evidenced by measuring the mass of electrolyte permeating through the GDE after CO2 electrolysis. Yet, metal cations in the electrolyte play a vital role in stabilizing crucial reaction intermediates and facilitating the reduction of CO2 to CO.153
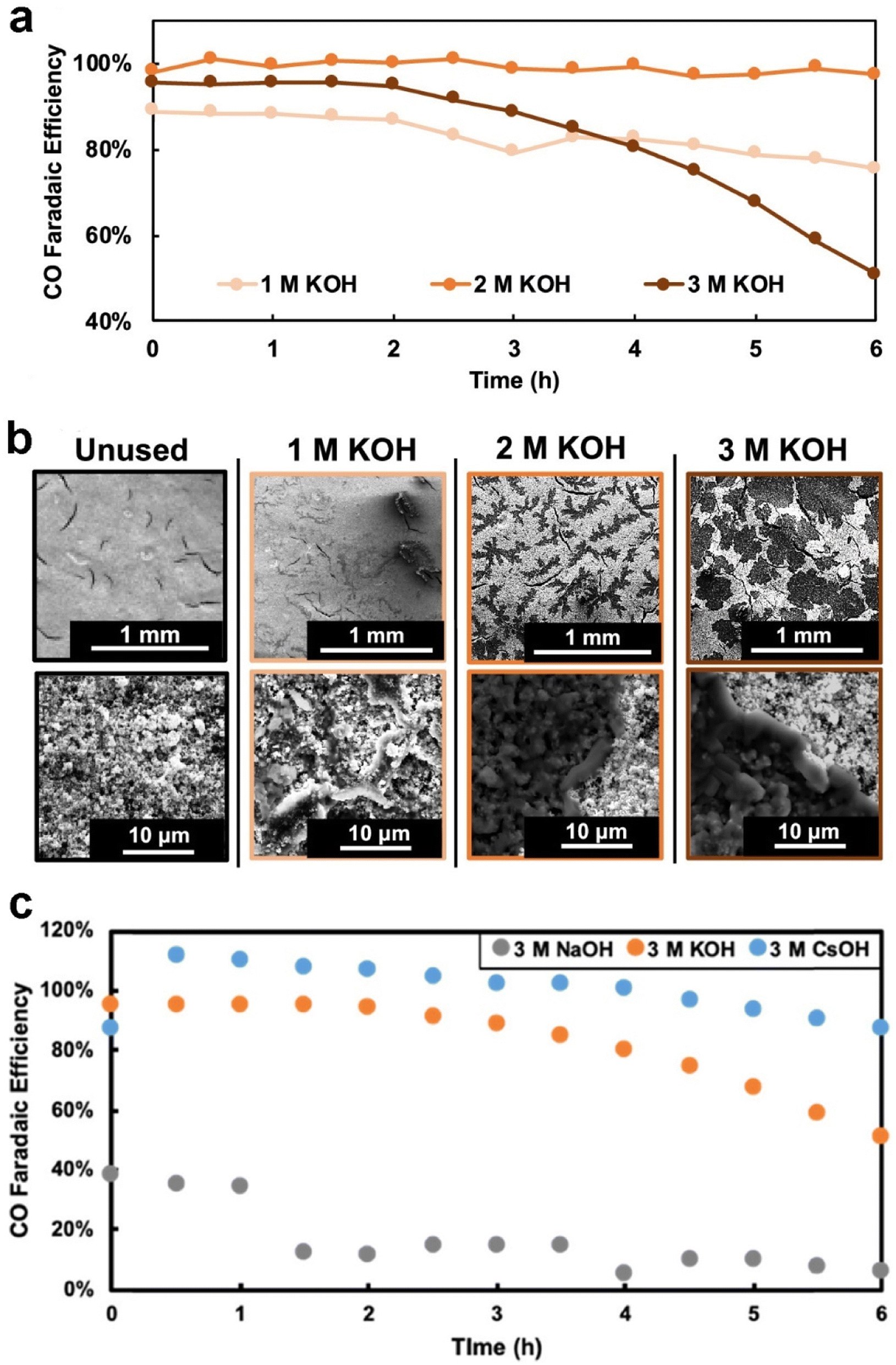 | ||
| Fig. 14 (a) Selectivity to CO over time for each GDE during the electrochemical testing at 200 mA cm−2 constant current; (b) SEM images (80× magnification top row and 500× bottom row) of the surface of each of the four GDEs, one pristine and three that were tested for 6 h in 1, 2, and 3 M KOH, respectively; (c) selectivity to CO over time for each of the GDEs facing different electrolytes during the electrochemical testing at 200 mA cm−2. Reproduced from ref. 118 with permission from American Chemical Society, copyright 2021. | ||
5. Summary and outlook
Ensuring the sustained performance of the GDE amidst the challenge of electrolyte flooding emerges as a critical imperative in advancing practical CO2 electrolysis. This perspective reviewed the CO2 electrolysis system, advancements in using GDEs, the mechanism of electrolyte flooding, and state-of-the-art studies on mitigating electrolyte flooding. The advancements of GDEs within flow cell electrolysers for CO2 electrolysis are summarized in the initial sections. The insight into flooding, subsequently, is discussed based on the underlying mechanisms of flooding approaches to observe it. The latest progress on mitigating flooding in GDEs are eventually discussed and summarized.Despite significant advances in recent years, the advancements in mitigating flooding remain inadequate to operate GDEs perform for a longer period and achieve economically feasible goals. Achieving this goal requires a comprehensive approach, beginning with a fundamental perspective. Understanding the mechanism of wettability declining on the catalyst layer is essential to reveal the primary reason for electrolyte flooding, yet it remains poorly understood. Systematic studies are required to examine the effects of each component of the catalyst layer on the performance degradation. These components include the active catalyst particles, supporting particles, Nafion® resin, and PTFE particles. Besides the methods of retaining the catalyst layer's hydrophobicity, strategies for decoupling the GDE from serious flooding at high current densities should be developed in other GDE layers.
We expect more accurate, robust, and powerful techniques to be explored for characterizing electrolyte flooding to further study the fundamental mechanisms. Advanced in situ characterization studies are essential to monitor and assess electrolyte flooding and the condition of all electrolytic systems under real-time CO2 electrolysis conditions. As discussed above, a few in situ techniques have been employed to investigate GDEs during CO2 electrolysis, such as an in situ fluorescence microscopy,121in situ Raman spectroscopy,92in situ FTIR154 and X-ray absorption spectroscopy155 for detecting catalytic reaction intermediates. However, the accuracy (or resolution) and sensitivity are not enough to monitor real-time mass transfer or distinguish the new chemicals or intermediates produced. In addition, the extensive data from in situ studies could be used for machine-learning-aided calculation and prediction.
With the goal of scalable CO2 electrolysis, unremitting efforts are being made to develop practical strategies for controlling electrolyte flooding, which are valuable and deserving of attention. Besides the lab-scale progress on mitigating flooding, we recommend exploring practical and cost-effective methodologies by modifying commercial GDEs to leverage existing production lines. Utilizing readily available materials as additives for these modifications would be advantageous and should be considered. Simultaneously, fabrication of new types of GDEs should also be further explored with innovative additives or substrate materials to decouple the influence of electrolyte flooding. In MEA electrolysers, the stability of CO2 electrolysis is better than that of a microfluidic cell, owing to the reduced electrolyte contact with the cathode GDE. However, the membrane plays a pivotal role in mitigating flooding by either preventing or restraining liquid electrolyte transfer from the anode.15,78 Thus, an effective combination of an anolyte and membrane could help control the transfer of liquid electrolyte and prevent salt precipitation. Priority should be given to optimize the composition and concentration of the anolyte, as well as the ion selectivity and thickness of the membrane, to move closer to achieving this goal.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support received from the Australian Research Council (DP230102577) which facilitated our research activities pertinent to this review. Special thanks are extended to Associate Professor Thomas Rufford for his invaluable insights and contributions during the preparation of this manuscript.References
- D. L. McCollum, W. Zhou, C. Bertram, H.-S. de Boer, V. Bosetti, S. Busch, J. Després, L. Drouet, J. Emmerling, M. Fay, O. Fricko, S. Fujimori, M. Gidden, M. Harmsen, D. Huppmann, G. Iyer, V. Krey, E. Kriegler, C. Nicolas, S. Pachauri, S. Parkinson, M. Poblete-Cazenave, P. Rafaj, N. Rao, J. Rozenberg, A. Schmitz, W. Schoepp, D. van Vuuren and K. Riahi, Nat. Energy, 2018, 3, 589–599 CrossRef
.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 263 CrossRef PubMed
- S. Garg, M. Li, M. N. Idros, Y. Wu, G. Wang and T. E. Rufford, ChemRxiv, 2021, DOI:10.26434/chemrxiv-2021-7chqx.
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O'Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat Sustainability, 2022, 5, 563–573 CrossRef
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS
- Q. Xu, S. Garg, A. B. Moss, M. Mirolo, I. Chorkendorff, J. Drnec and B. Seger, Nat. Catal., 2023, 6, 1042–1051 CrossRef CAS
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed
- A. Reyes, R. P. Jansonius, B. A. W. Mowbray, Y. Cao, D. G. Wheeler, J. Chau, D. J. Dvorak and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 1612–1618 CrossRef CAS
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS
- D. L. T. Nguyen, Y. Kim, Y. J. Hwang and D. H. Won, Carbon Energy, 2020, 2, 72–98 CrossRef CAS
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- T. Ma, Q. Fan, H. Tao, Z. Han, M. Jia, Y. Gao, W. Ma and Z. Sun, Nanotechnology, 2017, 28, 472001 CrossRef PubMed
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- T. Mizuno, K. Ohta, A. Sasaki, T. Akai, M. Hirano and A. Kawabe, Energy Sources, 1995, 17, 503–508 CrossRef CAS
- R. Daiyan, X. Lu, X. Tan, X. Zhu, R. Chen, S. C. Smith and R. Amal, ACS Appl. Energy Mater., 2019, 2, 8002–8009 CrossRef CAS
- B. Qin, Q. Zhang, Y.-H. Li, G. Yang and F. Peng, ACS Appl. Mater. Interfaces, 2020, 12, 30466–30473 CrossRef CAS
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS
- H. Liu, Y. Zhu, J. Ma, Z. Zhang and W. Hu, Adv. Funct. Mater., 2020, 30, 1910534 CrossRef CAS
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS
- C. W. Lee, K. D. Yang, D.-H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater., 2018, 30, 1704717 CrossRef PubMed
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, Nature, 2020, 581, 178–183 CrossRef CAS
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef CAS
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS PubMed
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC
- P. Millet, R. Ngameni, S. A. Grigoriev, N. Mbemba, F. Brisset, A. Ranjbari and C. Etiévant, Int. J. Hydrogen Energy, 2010, 35, 5043–5052 CrossRef CAS
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC
- G. Wen, B. Ren, X. Wang, D. Luo, H. Dou, Y. Zheng, R. Gao, J. Gostick, A. Yu and Z. Chen, Nat. Energy, 2022, 7, 978–988 CrossRef CAS
- S. Ma, R. Luo, S. Moniri, Y. Lan and P. J. A. Kenis, J. Electrochem. Soc., 2014, 161, F1124 CrossRef CAS
- F. Bienen, A. Löwe, J. Hildebrand, S. Hertle, D. Schonvogel, D. Kopljar, N. Wagner, E. Klemm and K. A. Friedrich, J. Energy Chem., 2021, 62, 367–376 CrossRef CAS
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 607 CrossRef CAS
- C. Lim, W. H. Lee, J. H. Won, Y.-J. Ko, S. Kim, B. K. Min, K.-Y. Lee, W. S. Jung and H.-S. Oh, Adv. Sustainable Syst., 2021, 5, 2100216 CrossRef CAS
- S. M. Moosavi, M. Niffeler, J. Gostick and S. Haussener, Chem. Eng. Sci., 2018, 176, 503–514 CrossRef CAS
- S. Park, J.-W. Lee and B. N. Popov, Int. J. Hydrogen Energy, 2012, 37, 5850–5865 CrossRef CAS
- J. Wu, P. P. Sharma, B. H. Harris and X.-D. Zhou, J. Power Sources, 2014, 258, 189–194 CrossRef CAS
-
R. Schweiss, C. Meiser, T. Damjanovic, I. Galbiati and N. Haak, SIGRACET Gas Diffusion Layers for PEM Fuel Cells, Electrolyzers and Batteries, 2016 Search PubMed
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
-
D. L. Wood and R. L. Borup, in Polymer Electrolyte Fuel Cell Durability, ed. F. N. Büchi, M. Inaba and T. J. Schmidt, Springer New York, New York, NY, 2009, pp. 159–195, DOI:10.1007/978-0-387-85536-3_8
-
W. Mérida, Porous Transport Layer Degradation, CRC Press, Boca Raton, 2011 Search PubMed
- D. Masheder and K. P. J. Williams, J. Raman Spectrosc., 1987, 18, 387–390 CrossRef CAS
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS
- K. Wu, E. Birgersson, B. Kim, P. J. A. Kenis and I. A. Karimi, J. Electrochem. Soc., 2014, 162, F23–F32 CrossRef
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS
- S. Ma, R. Luo, J. I. Gold, Z. Y. Aaron, B. Kim and P. J. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, ACS Sustain. Chem. Eng., 2022, 10, 4683–4693 CrossRef CAS PubMed
- D. Raciti, T. Braun, B. M. Tackett, H. Xu, M. Cruz, B. J. Wiley and T. P. Moffat, ACS Catal., 2021, 11, 11945–11959 CrossRef CAS
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661 CrossRef PubMed
- Y. Jännsch, M. Hämmerle, E. Simon, M. Fleischer and R. Moos, Energy
Technol., 2022, 10, 2200046 CrossRef
- J.-Z. Zhang, S. Wu, F. Shen, W. Song, Y. Hua, Z. Wu, X.-G. Zhang and J. Shi, Ionics, 2022, 28, 4321–4329 CrossRef CAS
- F. Huq, I. Sanjuán, S. Baha, M. Braun, A. Kostka, V. Chanda, J. R. C. Junqueira, N. Sikdar, A. Ludwig and C. Andronescu, ChemElectroChem, 2022, 9, e202101279 CrossRef CAS
- D. Wu, F. Jiao and Q. Lu, ACS Catal., 2022, 12, 12993–13020 CrossRef CAS
- M. N. Mahmood, D. Masheder and C. J. Harty, J. Appl. Electrochem., 1987, 17, 1159–1170 CrossRef CAS
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1988, 135, 1470 CrossRef CAS
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC
- D. G. Wheeler, B. A. W. Mowbray, A. Reyes, F. Habibzadeh, J. He and C. P. Berlinguette, Energy Environ. Sci., 2020, 13, 5126–5134 RSC
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS
- M. E. Leonard, M. J. Orella, N. Aiello, Y. Román-Leshkov, A. Forner-Cuenca and F. R. Brushett, J. Electrochem. Soc., 2020, 167, 124521 CrossRef CAS
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed
- Z. Qi, A. R. Kashi, A. K. Buckley, J. S. Miller, J. Ye, M. M. Biener, A. C. Foucher, E. A. Stach, S. Ma, K. P. Kuhl and J. Biener, J. Phys. Chem. C, 2022, 126(46), 19637–19646 CrossRef CAS
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC
- B. De Mot, J. Hereijgers, M. Duarte and T. Breugelmans, Chem. Eng. J., 2019, 378, 122224 CrossRef CAS
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS
-
L. Yeo and H.-C. Chang, in Encyclopedia of Microfluidics and Nanofluidics, ed. D. Li, Springer US, Boston, MA, 2008, pp. 600–606, DOI:10.1007/978-0-387-48998-8_470
- R. Chai, Y. Liu, J. Wang, Q. Liu and Z. Rui, Appl. Energy, 2022, 323, 119584 CrossRef CAS
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, ACS Appl. Energy Mater., 2022, 5, 15125–15135 CrossRef CAS PubMed
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS
- J. Disch, L. Bohn, L. Metzler and S. Vierrath, J. Mater. Chem. A, 2023, 11(14), 7344–7357 RSC
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, J. Phys. Chem. C, 2015, 119, 55–61 CrossRef CAS
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed
- J. Osiewacz, M. Löffelholz, B. Ellendorff and T. Turek, J. Power Sources, 2024, 603, 234430 CrossRef CAS
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed
- K. Han, B. K. Hong, S. H. Kim, B. K. Ahn and T. W. Lim, Int. J. Hydrogen Energy, 2011, 36, 12452–12464 CrossRef CAS
- I. Nitta, T. Hottinen, O. Himanen and M. Mikkola, J. Power Sources, 2007, 171, 26–36 CrossRef CAS
- N. T. Nesbitt and W. A. Smith, J. Electrochem. Soc., 2021, 168, 044505 CrossRef CAS
- G. Zhang, Z.-J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed
- H. Ungan and A. Bayrakçeken Yurtcan, Int. J. Energy Res., 2019, 43, 5946–5958 CrossRef CAS
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed
- H. Xiang, S. Rasul, K. Scott, J. Portoles, P. Cumpson and E. H. Yu, J. CO2 Util., 2019, 30, 214–221 CrossRef CAS
- S. Jovanovic, R. Krause, A. Lüken, J. Ackermann, S. Merz, P. Jakes, R.-A. Eichel and J. Granwehr, J. Electrochem. Soc., 2020, 167, 086505 CrossRef CAS
- N. Yousfi-Steiner, P. Moçotéguy, D. Candusso, D. Hissel, A. Hernandez and A. Aslanides, J. Power Sources, 2008, 183, 260–274 CrossRef CAS
- S. D. Knights, K. M. Colbow, J. St-Pierre and D. P. Wilkinson, J. Power Sources, 2004, 127, 127–134 CrossRef CAS
- J.-M. Le Canut, R. M. Abouatallah and D. A. Harrington, J. Electrochem. Soc., 2006, 153, A857 CrossRef CAS
- H. Görgün, M. Arcak and F. Barbir, J. Power Sources, 2006, 157, 389–394 CrossRef
- X. Liu, H. Guo and C. Ma, J. Power Sources, 2006, 156, 267–280 CrossRef CAS
- T. Abe, H. Shima, K. Watanabe and Y. Ito, J. Electrochem. Soc., 2004, 151, A101 CrossRef CAS
- M. Afra, M. Nazari, M. H. Kayhani, M. Sharifpur and J. P. Meyer, Energy, 2019, 175, 967–977 CrossRef CAS
- M. N. Islam, U. Shrivastava, M. Atwa, X. Li, V. Birss and K. Karan, ACS Appl. Mater. Interfaces, 2020, 12, 39215–39226 CrossRef CAS PubMed
- D. McLaughlin, M. Bierling, R. Moroni, C. Vogl, G. Schmid and S. Thiele, Adv. Energy Mater., 2020, 10, 2000488 CrossRef CAS
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, J. Catal., 2022, 408, 1–8 CrossRef CAS
- Y. Kong, M. Liu, H. Hu, Y. Hou, S. Vesztergom, M. d. J. Gálvez-Vázquez, I. Zelocualtecatl Montiel, V. Kolivoška and P. Broekmann, Small Methods, 2022, 6, 2200369 CrossRef CAS PubMed
- H. Hu, Y. Kong, M. Liu, V. Kolivoška, A. V. Rudnev, Y. Hou, R. Erni, S. Vesztergom and P. Broekmann, J. Mater. Chem. A, 2023, 11(10), 5083–5094 RSC
- H.-R. M. Jhong, F. R. Brushett and P. J. A. Kenis, Adv. Energy Mater., 2013, 3, 589–599 CrossRef CAS
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, ACS Appl. Mater. Interfaces, 2021, 13, 15132–15142 CrossRef CAS PubMed
- P. K. Sow, Z. Lu, H. Talebian, L. Damron and W. Mérida, J. Phys. Chem. C, 2016, 120, 24794–24802 CrossRef CAS
- G. O. Larrazábal, V. Okatenko, I. Chorkendorff, R. Buonsanti and B. Seger, ACS Appl. Mater. Interfaces, 2022, 14, 7779–7787 CrossRef PubMed
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed
- A. M. Kalde, M. Grosseheide, S. Brosch, S. V. Pape, R. G. Keller, J. Linkhorst and M. Wessling, Small, 2022, 18, 2204012 CrossRef CAS PubMed
- H. Li, Y. Tang, Z. Wang, Z. Shi, S. Wu, D. Song, J. Zhang, K. Fatih, J. Zhang, H. Wang, Z. Liu, R. Abouatallah and A. Mazza, J. Power Sources, 2008, 178, 103–117 CrossRef CAS
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS
- J. T. Gostick, M. A. Ioannidis, M. W. Fowler and M. D. Pritzker, Electrochem. Commun., 2009, 11, 576–579 CrossRef CAS
- J. P. Owejan, J. E. Owejan, W. Gu, T. A. Trabold, T. W. Tighe and M. F. Mathias, J. Electrochem. Soc., 2010, 157, B1456 CrossRef CAS
- L. Cindrella, A. M. Kannan, J. F. Lin, K. Saminathan, Y. Ho, C. W. Lin and J. Wertz, J. Power Sources, 2009, 194, 146–160 CrossRef CAS
- S. C. Perry, S. M. Gateman, R. Malpass-Evans, N. McKeown, M. Wegener, P. Nazarovs, J. Mauzeroll, L. Wang and C. Ponce de León, Chemosphere, 2020, 248, 125993 CrossRef CAS PubMed
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS
- S. Garg, M. Li, Y. Wu, M. Nazmi Idros, H. Wang, A. J. Yago, L. Ge, G. G. X. Wang and T. E. Rufford, ChemSusChem, 2021, 14, 2601–2611 CrossRef CAS PubMed
- F. L. P. Veenstra, N. Ackerl, A. J. Martín and J. Pérez-Ramírez, Chem, 2020, 6, 1707–1722 CAS
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- F. Yu, P. Leung, Q. Xu, S. Mavrikis, P. Nazarovs, A. Shah, L. Wang and C. Ponce de León, J. Power Sources, 2023, 580, 233201 CrossRef CAS
- X. Sheng, W. Ge, H. Jiang and C. Li, Adv. Mater., 2022, 34, 2201295 CrossRef CAS PubMed
- Q. Wang, H. Dong, H. Yu and H. Yu, J. Power Sources, 2015, 279, 1–5 CrossRef CAS
- S. Garg, M. Li, T. Hussain, M. N. Idros, Y. Wu, X. S. Zhao, G. G. X. Wang and T. E. Rufford, ACS Appl. Mater. Interfaces, 2022, 14, 35504–35512 CrossRef CAS PubMed
- L. Xue, X. Wu, Y. Liu, B. Xu, X. Wang, S. Dai, P. Liu and H. Yang, Nano Res., 2022, 15, 1393–1398 CrossRef CAS
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- W. H. Lee, Y.-J. Ko, Y. Choi, S. Y. Lee, C. H. Choi, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Nano Energy, 2020, 76, 105030 CrossRef CAS
- J. Han, B. Tu, P. An, J. Zhang, Z. Yan, X. Zhang, C. Long, Y. Zhu, Y. Yuan, X. Qiu, Z. Yang, X. Huang, S. Yan and Z. Tang, Adv. Mater., 2024, 2313926 CrossRef CAS PubMed
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS
- T. Zhang, Z. Li, X. Lyu, J. Raj, G. Zhang, H. Kim, X. Wang, S. Chae, L. Lemen, V. N. Shanov and J. Wu, J. Electrochem. Soc., 2022, 169, 104506 CrossRef CAS
- L. Li, J. Chen, V. S. S. Mosali, Y. Liang, A. M. Bond, Q. Gu and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208534 CrossRef CAS PubMed
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS
- S. Yamaguchi, H. Ebe, T. Minegishi and M. Sugiyama, ACS Appl. Mater. Interfaces, 2024, 16, 17371–17376 CrossRef CAS PubMed
- J. Wicks, M. L. Jue, V. A. Beck, J. S. Oakdale, N. A. Dudukovic, A. L. Clemens, S. Liang, M. E. Ellis, G. Lee, S. E. Baker, E. B. Duoss and E. H. Sargent, Adv. Mater., 2021, 33, 2003855 CrossRef CAS PubMed
- L. M. Baumgartner, A. Goryachev, C. I. Koopman, D. Franzen, B. Ellendorff, T. Turek and D. A. Vermaas, Energy Adv., 2023, 2, 1893–1904 RSC
- W.-H. Cheng, M. H. Richter, I. Sullivan, D. M. Larson, C. Xiang, B. S. Brunschwig and H. A. Atwater, ACS Energy Lett., 2020, 5, 470–476 CrossRef CAS
- S. Garg, Q. Xu, A. B. Moss, M. Mirolo, W. Deng, I. Chorkendorff, J. Drnec and B. Seger, Energy Environ. Sci., 2023, 16, 1631–1643 RSC
- H.-G. Qin, Y.-F. Du, Y.-Y. Bai, F.-Z. Li, X. Yue, H. Wang, J.-Z. Peng and J. Gu, Nat. Commun., 2023, 14, 5640 CrossRef CAS PubMed
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS
- F. N. Ajjan, M. J. Jafari, T. Rębiś, T. Ederth and O. Inganäs, J. Mater. Chem.
A, 2015, 3, 12927–12937 RSC
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. García de Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |