
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methane steam reforming over Ni/YSZ cermet anode materials synthesized by different methods
S.
Bassil†
,
T.
Caillot
,
F. J.
Cadete Santos Aires
,
F. C.
Meunier
 and
A.
Kaddouri
*
and
A.
Kaddouri
*
Université Lyon 1 – CNRS, UMR 5256, IRCELYON, Institut de Recherches sur la Catalyse et l'Environnement de Lyon, 2 Avenue Albert Einstein, F-69626 Villeurbanne, France. E-mail: akim.kaddouri@ircelyon.univ-lyon1.fr
First published on 27th January 2024
Abstract
Ni/YSZ is considered as a benchmark material for solid oxide fuel cells (SOFCs) but suffers from both Ni agglomeration and carbon deposition when hydrocarbons are used as fuels. In the present study, samples of Ni/yttria-stabilized zirconia (Ni/YSZ) with 1.25 < molar ratio Ni/(Y + Zr) < 1.5 were prepared by a sol–gel method and by impregnation of YSZ with nickel, in both aqueous and organic media. The Ni/YSZ materials were characterized by dynamic TG analysis, FTIR, XRD, UV-vis, TPR, TPO and TEM techniques. The properties of the Ni/YSZ materials were discussed in relation with the corresponding catalytic activities for methane steam reforming, which were also compared to that of a commercial material. The superior catalytic performance of the sol–gel-prepared catalyst and its resistance to poisoning despite noticeable (though comparable to those measured on the other samples) carbon deposits were attributed to the greater homogeneity of the mixture of nickel and YSZ phases.
1 Introduction
Porous Ni/YSZ cermet is currently the most common anode material for solid oxide fuel cell (SOFC) applications because it is chemically stable in reducing atmospheres at high temperatures and its thermal expansion coefficient is close to that of the YSZ electrolyte. However, multiple problems hinder the effective implementation of Ni/YSZ, among which are nickel agglomeration and redox instability.1,2In previous decades significant advancements have been made in developing novel anode materials for direct oxidation of hydrocarbon fuels in SOFCs. Systems such as perovskites, pyrochlores, tungsten bronzes and rare-earth-doped ceria materials were investigated as alternatives to Ni/YSZ cermet but with limited success.3 Yet, Ni–YSZ remains the benchmark material for SOFCs due to a lack of alternative high-performance anodes. Despite the outstanding electrocatalytic properties of Ni/YSZ cermet materials for operations using H2 as fuel, Ni/YSZ-based anodes suffer from a number of drawbacks when hydrocarbons are used, notably carbon deposition.
In spite of efforts to produce hydrogen by different methods,4–8 steam reforming of hydrocarbons has remained as a topic of intense research interest because this technique is the most cost-effective syngas (H2/CO) production method.9 Methane can be transformed ex situ into synthesis gas,10 which can be used as a fuel in solid oxide fuel cells (SOFCs)11 or converted in situ directly on the Ni–YSZ anode within the fuel cell system12 to produce electrical and heat energy. The following equilibria have been widely reported for the two strongly endothermic methane steam reforming reactions (1) and (3) and for the moderately exothermic water-gas shift (WGS) reaction (2)
| CH4 + H2O ⇔ CO + 3H2 ΔH = +206 kJ mol−1 | (1) |
| CO + H2O ⇔ CO2 + H2 ΔH = −41 kJ mol−1 | (2) |
| CH4 + 2H2O ⇔ CO2 + 4H2 ΔH = +165 kJ mol−1 | (3) |
The carbon formed can cause blockage of the reactive sites for electro-oxidation and lead to a volumetric expansion of the cell components and deterioration of the microstructure.13,14 Three main kinds of carbon are generally observed on nickel-based catalysts: pyrolytic carbon, encapsulating carbon and filamentous carbon. A number of investigations have shown that the rate of carbon formation is largely dependent on the chemical composition of the catalyst as well as its preparation procedure.15–17
Several preparation techniques have been employed in previous studies to synthesize Ni/YSZ cermet, among which are mechanical mixing,18 sonochemical process,19 electroless technique,20 gel precipitation,21 precipitation22 and combustion.23 However, for all these methods a number of factors vary markedly during the preparation and can affect to some extent the homogeneity of powder composition and the uniformity of particle size. To overcome these drawbacks, an improved sol–gel process, where propionic acid is used as both the chelating agent and the solvent, is proposed to be used here.24 It has already been used successfully to prepare various oxides such as Co3O4,25 NiO/Al2O3, Ni/SiO226 or La0.8Sr0.2TiO3+δ27 and LaNiO3 perovskite or CuO–CuCrO428 and NiCo2O4 spinel.29
This study presents the synthesis of Ni/YSZ by using both impregnation techniques (either in aqueous medium or in organic solvent) and the sol–gel method. The impregnation methods initially involve the interaction between the surface of particles of solid YSZ and a liquid solution containing nickel ions. Subsequent heat treatments lead to precursor decomposition and solid-state reactions. The sol–gel method initially involves the formation of a homogeneous gel containing Ni2+, Y3+ and Zr4+ cations, which then leads to an organic resin before final calcination. Sol–gel techniques typically lead to solids more homogeneous than those prepared by impregnation because of the better initial mixing of the reagents.30,31
Various physicochemical characterisation techniques and the catalytic activity of the Ni/YSZ samples for methane steam reforming were investigated and compared to that of a commercial catalyst.
2 Experimental
2.1 Sample synthesis
The synthesis of Ni/YSZ catalysts has been performed using the following reagents: nickel nitrate (Merck, >97%), nickel acetylacetonate (Alfa Aesar, >95%), yttrium acetylacetonate (Aldrich 99.9%), zirconium acetylacetonate (Merck, >98%), YSZ (TOSOH) and propionic acid (Alfa Aesar, 99%). Prior to any synthesis the metal nitrates, which are typically hygroscopic, and the metal acetylacetonates were decomposed at a temperature of up to 800 °C in a TG/DTA apparatus (Perkin-Elmer, mod. Pyris Diamond) to determine their exact composition. The commercial catalyst nickel oxide–yttria-stabilized zirconia was obtained from Sigma Aldrich Chemie Gmbh (Product reference 704229, batch number 85097MJ).2.2 Characterization
Thermogravimetric (TG) studies were carried out in a thermal analyzer (Perkin Elmer-Pyris Diamond) under atmospheric pressure in the temperature range of 25–800 °C. 20–30 mg of the samples was placed in a platinum crucible and heated using a helium flow rate of 6 L h−1. Elemental analysis of the solids was performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES). X-ray diffraction (XRD) patterns were recorded by a Siemens D5000 diffractometer between 2θ = 5 and 80°, employing filtered CuKα radiation with a count time of 1 s.The general morphology of the catalysts and local chemical and structural information as well as eventual carbon poisoning were assessed by transmission electron microscopy (TEM) using a JEOL JEM 2010 TEM operated at 200 kV and equipped with a LaB6 thermoionic electron gun, a high-resolution polepiece (0.196 nm point resolution) and an energy-dispersive X-ray (EDX) spectrometer (pentafet from Oxford Instruments). Fourier transform infrared (FTIR) study was carried out using an FTIR NEXUS spectrometer (over the region 400–4000 cm−1) and samples mixed with spectroscopic grade KBr pellets. Diffuse reflectance ultraviolet-visible spectroscopy (DR-UV-vis) measurements were performed using a Lambda 950 UV-vis spectrophotometer (Perkin Elmer) equipped with an integrating sphere detector. Temperature-programmed reduction (TPR) of fresh catalysts was performed in the presence of 5% H2 in N2 at a temperature interval of 25–800 °C with a heating rate of 10 °C min−1. Temperature-programmed oxidation (TPO) experiments were performed, at the end of the catalytic tests, in the presence of 5% O2 in N2 at a temperature interval of 25–800 °C with a heating rate of 10 °C min−1.
2.3 Catalytic activity evaluation
The continuous flow reaction was carried out within a fixed bed tubular quartz reactor (0.025 g of catalysts, particle size around 40 μm) at atmospheric pressure and in the 700–800 °C temperature interval using 5% CH4, 5% H2O, N2 balance and a total flow rate of 6.5 L h−1. Before testing, each catalyst was pretreated in situ at 800 °C for 1 h in a H2/N2 flow of 30 mL min−1. The reactor effluents were analyzed on-line by an Agilent gas micro-chromatograph equipped with high-sensitivity thermal conductivity detectors (TCDs).3 Results and discussion
3.1 Elemental analysis and specific surface area of the samples
Elemental analysis of samples (ICP) is reported in Table 1. Nickel-based cermet materials used as anodes in solid oxide fuel cells fed with hydrocarbons or hydrogen typically exhibit high nickel concentrations to allow sufficient electronic and ionic conductivity.32–34 The chemical compositions of the AQ, ORG and SG NiO/YSZ catalysts were similar. The molar ratio of Ni to Zr of SG was ca. 1.74 compared to 1.84 for the Ni/YSZ commercial catalyst. The AQ, ORG and SG samples, which were calcined at 1200 °C, presented lower BET surface areas (between 0.8 and 1.5 m2 g−1) as compared to the commercial catalyst (3 m2 g−1). The final calcination temperature (1200 °C) was significantly higher than the maximal operating temperature (800 °C). This is a common practice and enables activation for use in high-temperature fuel cell applications.35,363.2 Thermal analysis
Fig. 1 shows the TG curves of the precursor decomposition upon heating in a helium atmosphere at 5 °C min−1. The final step, which is completed at about 600 °C, brings the total weight loss up to 51.4% for AQ, 53.4% for ORG and 65.5% for SG precursors. The profiles of the decomposition of AQ and ORG precursors were similar to those obtained for nickel nitrate and nickel acetylacetonate, respectively.37,38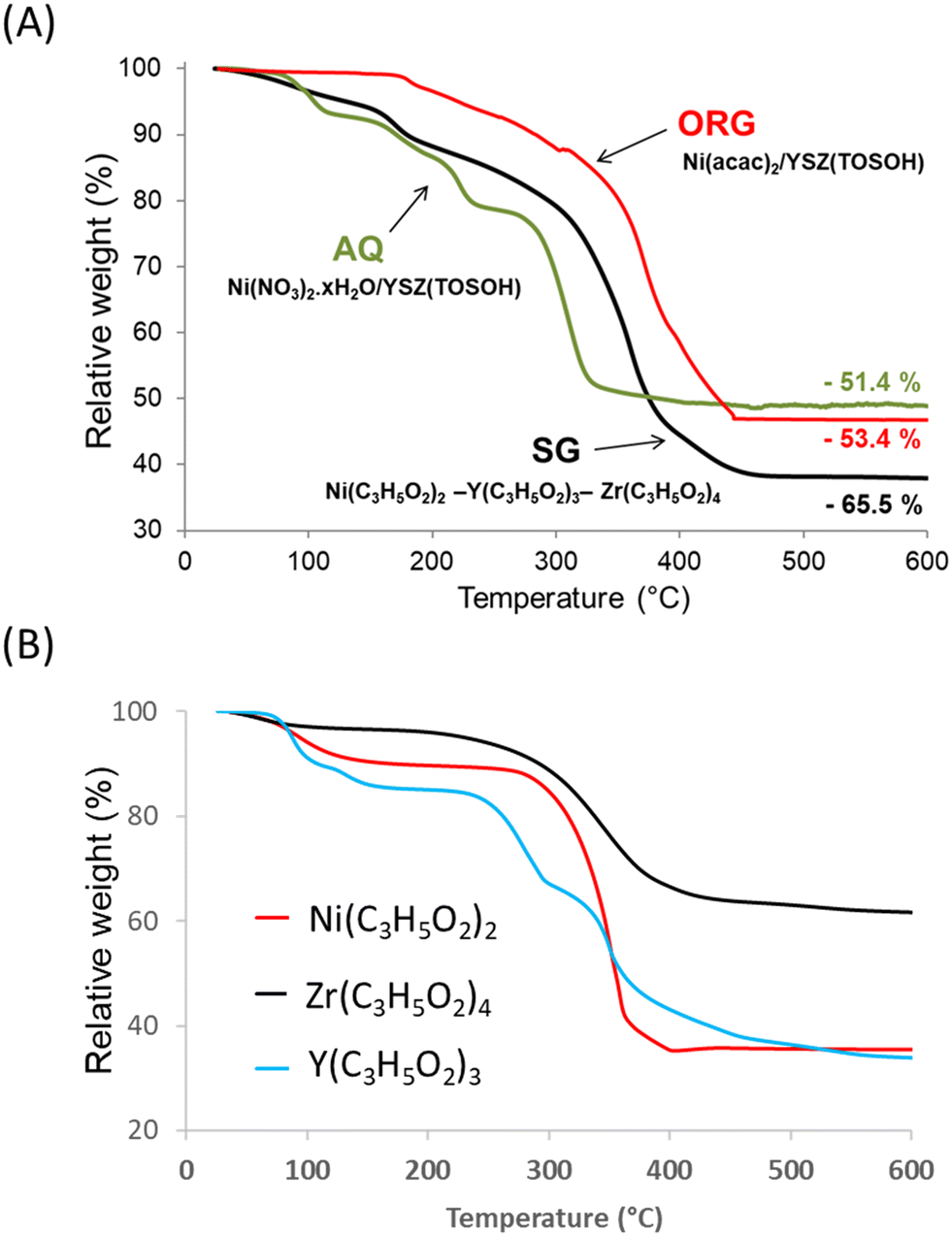 | ||
| Fig. 1 TG curves of the (A) AQ, ORG and SG precursors and (B) Ni2+, Zr4+ and Y3+ propionates. | ||
The TG result obtained with the SG sample was found to be a combination of thermal decomposition of individual propionates of Ni, Y and Zr intimately mixed, prepared with the same sol–gel procedure (Fig. 1B). The weight loss originated predominantly from the evolution of water (desorption of physisorbed water), which was already present in the gel, and from the combustion of organic species present in the precursor. For the SG sample the evolution of H2O began at room temperature and reached a maximum at ca. 100 °C.
After the removal of water, the percentage of weight loss in the 100–600 °C temperature interval registered during the decomposition of the SG precursor was ca. 63.5% with respect to the theoretical value of ca. 63.8%. Taking into account the elemental analysis of the samples (Table 1) the overall process of the decomposition of the SG precursor can be characterized by the following equation:
| 0.7Ni(C3H5O2)2 − 0.04Y(C3H5O2)3 − 0.26Zr(C3H5O2)4 → 0.7NiO − 0.02Y2O3 − 0.26ZrO2 |
3.3 FTIR analysis
The FTIR spectra of the AQ, ORG and SG precursors are shown in Fig. 2A. In AQ a wide peak is observed in the range of 3200–3700 cm−1, corresponding to hydroxyl groups of physisorbed water. The absorption band at around 1650 cm−1 is assigned to the bending mode of H–O–H (ref. 39) and the bands at ca. 830 and 1380 cm−1 are attributed to nitrate ions.37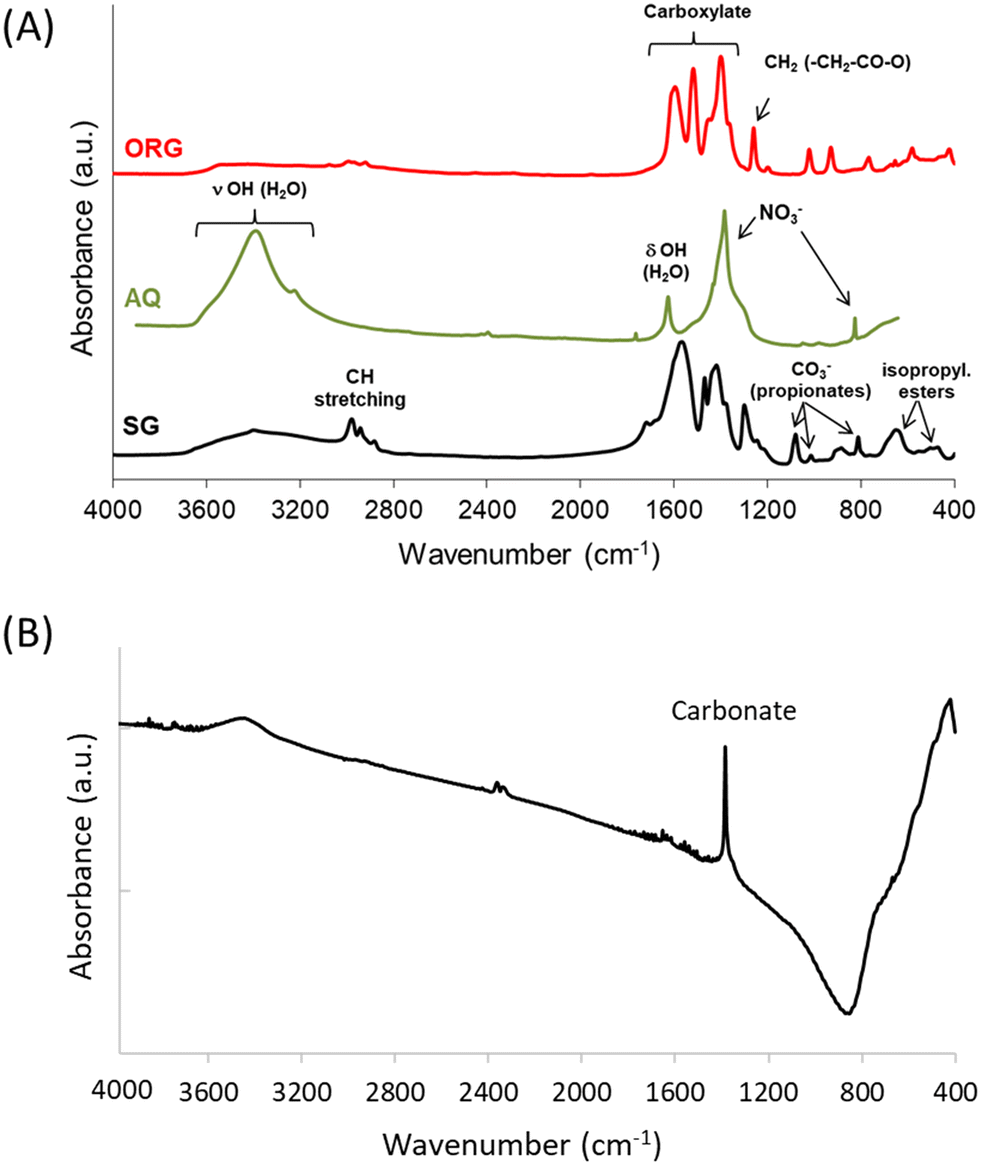 | ||
| Fig. 2 FTIR spectra of (A) the AQ, ORG and SG precursors and (B) the SG sample calcined at 1200 °C. | ||
The spectra of the ORG and SG precursors showed bands in the 1100–1800 cm−1 region, which are due to oxygenated species, and also bands at ca. 1450 cm−1 and 1570 cm−1 due, respectively, to symmetric and asymmetric vibrations of carboxylate groups CO2−.40
The FTIR spectrum of the SG precursor showed several bands in the 400–3800 cm−1 region. The bands in the 2900–2950 cm−1 region are attributed to the stretching modes of CH groups of carboxylic acid, and those at ca.1450 cm−1, 1410 cm−1 and around 1380 cm−1 correspond to the bending modes δasCH3, δCH2 and δsCH3 associated with propionates and propionic acid, while the strong adsorption around 1540 cm−1 is assigned to νasCOO.41,42 Upon increasing the temperature to 1200 °C, the absorption of organic residues is no longer detected (i.e. no bands present around 3000 cm−1), and the band typical of physisorbed water disappears while the weak absorption band of nickel oxide at ca. 500 cm−1 is broadened, with a strong carbonate band also being observed around 1380 cm−1 (Fig. 2B).37
3.4 XRD data
The X-ray diffraction patterns of the SG precursor calcined at 600 and 1200 °C are shown in Fig. 3. The SG calcined at 600 °C is in a poorly crystalline state. With an increase in calcination temperature from 600 °C to 1200 °C an increase in the intensity of the lines was observed due to further improvement in the crystallinity. The solid presents several peaks characteristic of NiO (JCPDF 01-089-5881) and YSZ (JCPDF 01-070-4431). Most importantly, no change in the NiO/YSZ biphasic system was noted even after 5 h at 1200 °C. NiO and YSZ do not form solid solutions, even at high temperatures.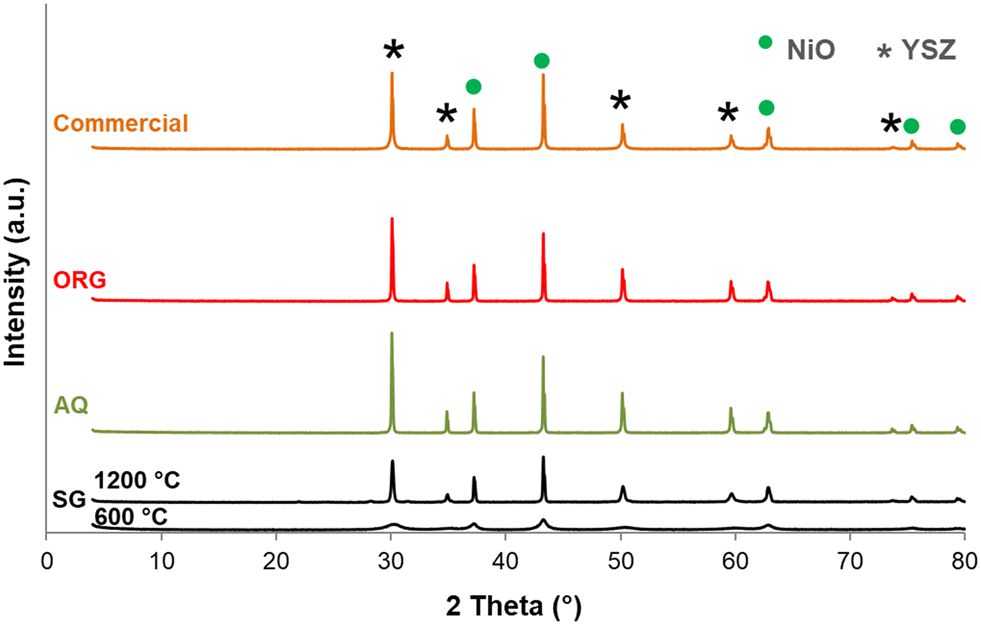 | ||
| Fig. 3 X-ray diffraction patterns of SG (calcined at 600 and 1200 °C for 5 h) and AQ, ORG and the NiO–YSZ commercial samples calcined at 1200 °C for 5 h (JCPDF files: NiO: 01-089-5881; YSZ: 01-070-4431). | ||
Another interesting point to mention from Fig. 3 is that there are no XRD lines due to free ZrO2 and Y2O3 even up to the highest calcination temperature. These results are in agreement with reports on YSZ samples prepared by other methods.43,44
For comparison XRD diffractograms of AQ, ORG and the corresponding commercial NiO/YSZ are also shown in Fig. 3. All compounds calcined at 1200 °C are crystalline and show similar XRD patterns to that of SG. Generally, this green solid is first sintered and then reduced under hydrogen to form a porous Ni/YSZ cermet before use.45
3.5 UV-vis data
UV-vis spectra of AQ, ORG and SG are shown in Fig. 4A. The spectra show an absorption at approximately 320 nm which is attributed to band gap adsorption in NiO.44 Two additional peaks can be seen at approximately 720 and 1170 nm that are characteristic of the presence of β-Ni(OH)2.46 The UV-vis absorption signal around 212 nm corresponds to the interband transition in monoclinic zirconia.47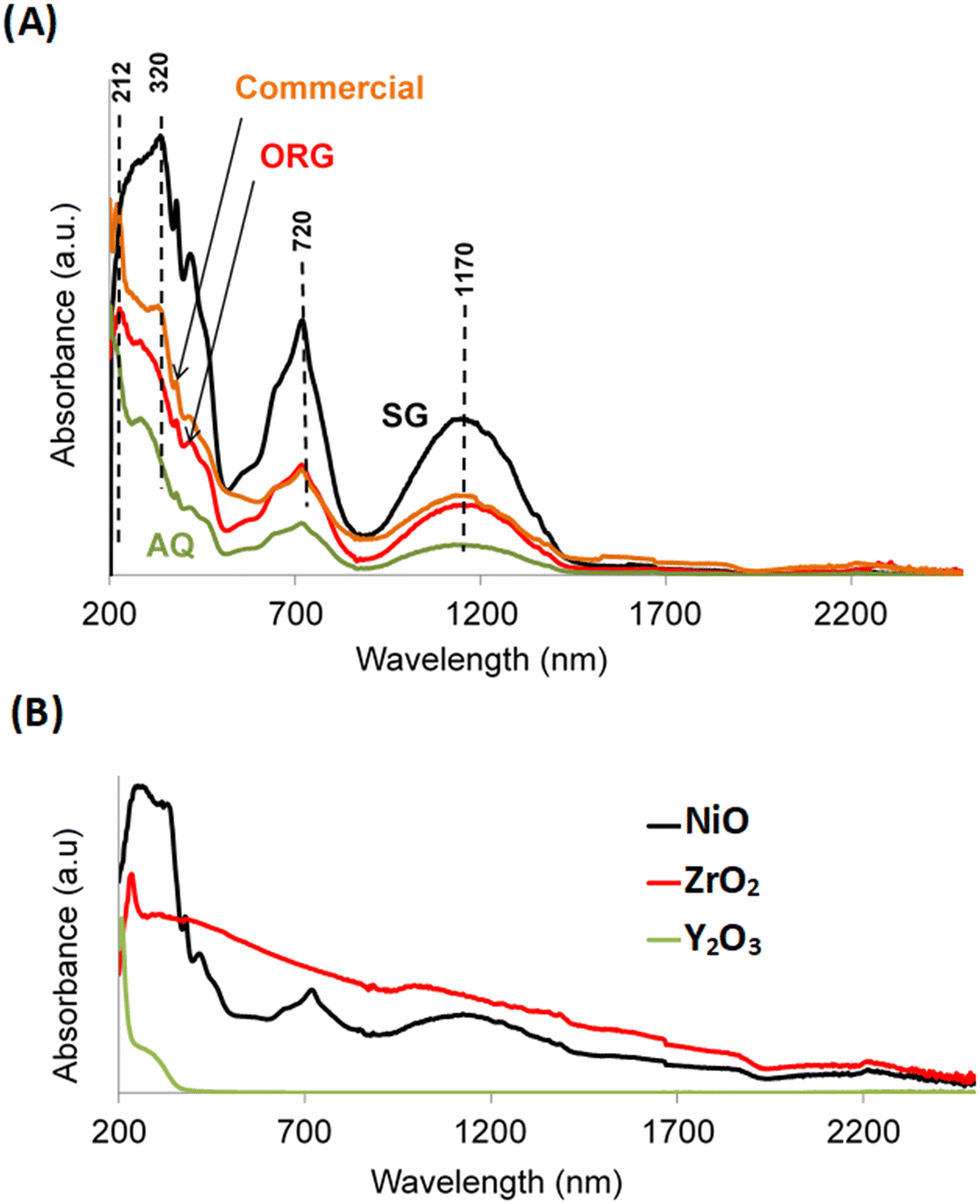 | ||
| Fig. 4 UV-visible absorption spectra of (A) AQ, ORG, SG and the commercial NiO–YSZ and (B) reference materials NiO, Y2O3 and ZrO2. | ||
The presence of this phase in the sample is in agreement with literature reports that showed a relationship between an increase in the calcination temperature of YSZ and a gradual transition from the metastable tetragonal phase of ZrO2 to the stable monoclinic one.48 For the sake of completeness, Fig. 4B shows the optical absorption spectra of the NiO, ZrO2 and Y2O3 solids prepared by the sol–gel process, using propionic acid, under the same conditions as for SG.
3.6 Temperature-programmed reduction (TPR) measurements
Results of the TPR data (Fig. 5) showed that the peak maximum temperature of all catalysts is located in the 380–500 °C temperature range, in agreement with literature reports.49 The degree of reduction of NiO from the AQ, ORG and SG catalysts, in TPR experiments, was 36.2% (500 °C), 36.4% (410 °C) and 37.6% (440 °C), respectively (Table 1). The degree of reduction is, however, increased appreciably (43.3% at 380 °C) for commercial NiO/YSZ. For the SG catalyst, there is a small additional TPR peak centered at 530 °C. This reduction peak is, however, shifted towards the higher temperature side with respect to the parent reduction peak observed at 440 °C. This is probably due to the strong interaction of NiO with YSZ in the SG sample.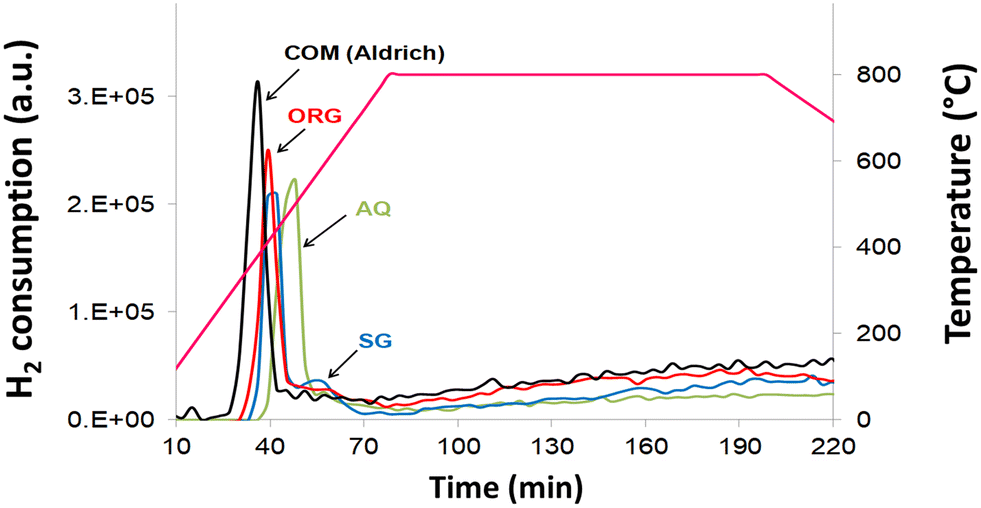 | ||
| Fig. 5 Temperature-programmed reduction plots of AQ, ORG, SG and the commercial NiO–YSZ. | ||
3.7 Catalytic testing
In this investigation the activity of the differently prepared AQ, ORG, SG and the corresponding commercial material was probed by the methane steam reforming reaction at high temperature (700–800 °C). Results showing the influence of the preparation method on the catalytic activity of the NiO/YSZ catalysts, after pre-reduction (5% H2 in N2) at 800 °C for 1 h, are presented in Table 2. The catalytic data reported here are taken after running the reaction for 7 h (for each temperature) when steady state has essentially been reached. The results are presented as methane conversion and hydrogen and carbon oxide concentrations.| Temperature/sample | CH4 conversion (%) | H2 concentration (%) | CO concentration (%) | CO2 concentration (%) |
|---|---|---|---|---|
| 700 °C | ||||
| AQ | 63.70 | 8.50 | 2.40 | 0.33 |
| ORG | 62.70 | 8.20 | 2.26 | 0.36 |
| SG | 75.70 | 10.01 | 2.97 | 0.27 |
| Commercial Ni/YSZ | 57.90 | 7.60 | 2.02 | 0.40 |
| 750 °C | ||||
| AQ | 69.20 | 9.20 | 2.70 | 0.24 |
| ORG | 67.20 | 8.60 | 2.60 | 0.23 |
| SG | 77.50 | 10.20 | 3.20 | 0.12 |
| Commercial Ni/YSZ | 65.10 | 8.40 | 2.46 | 0.28 |
| 800 °C | ||||
| AQ | 77.40 | 9.91 | 3.09 | 0.17 |
| ORG | 76.00 | 9.86 | 3.07 | 0.16 |
| SG | 85.50 | 11.10 | 3.52 | 0.05 |
| Commercial Ni/YSZ | 75.30 | 9.67 | 3.03 | 0.15 |
Methane conversion of all samples increased with temperature. Interestingly the corresponding selectivity profiles for H2 and CO follow an identical pattern to that of methane conversion. In contrast, there is a sharp fall in selectivity for CO2 (especially for Ni/YSZ-SG; only CO2 traces at 800 °C). The comparison between the prepared Ni/YSZ catalysts and the corresponding commercial sample reveals distinct catalytic performances. The difference in reactivity with methane and the product selectivity is clearly apparent at all temperatures. The activity ranking was SG > AQ > ORG > commercial Ni/YSZ.
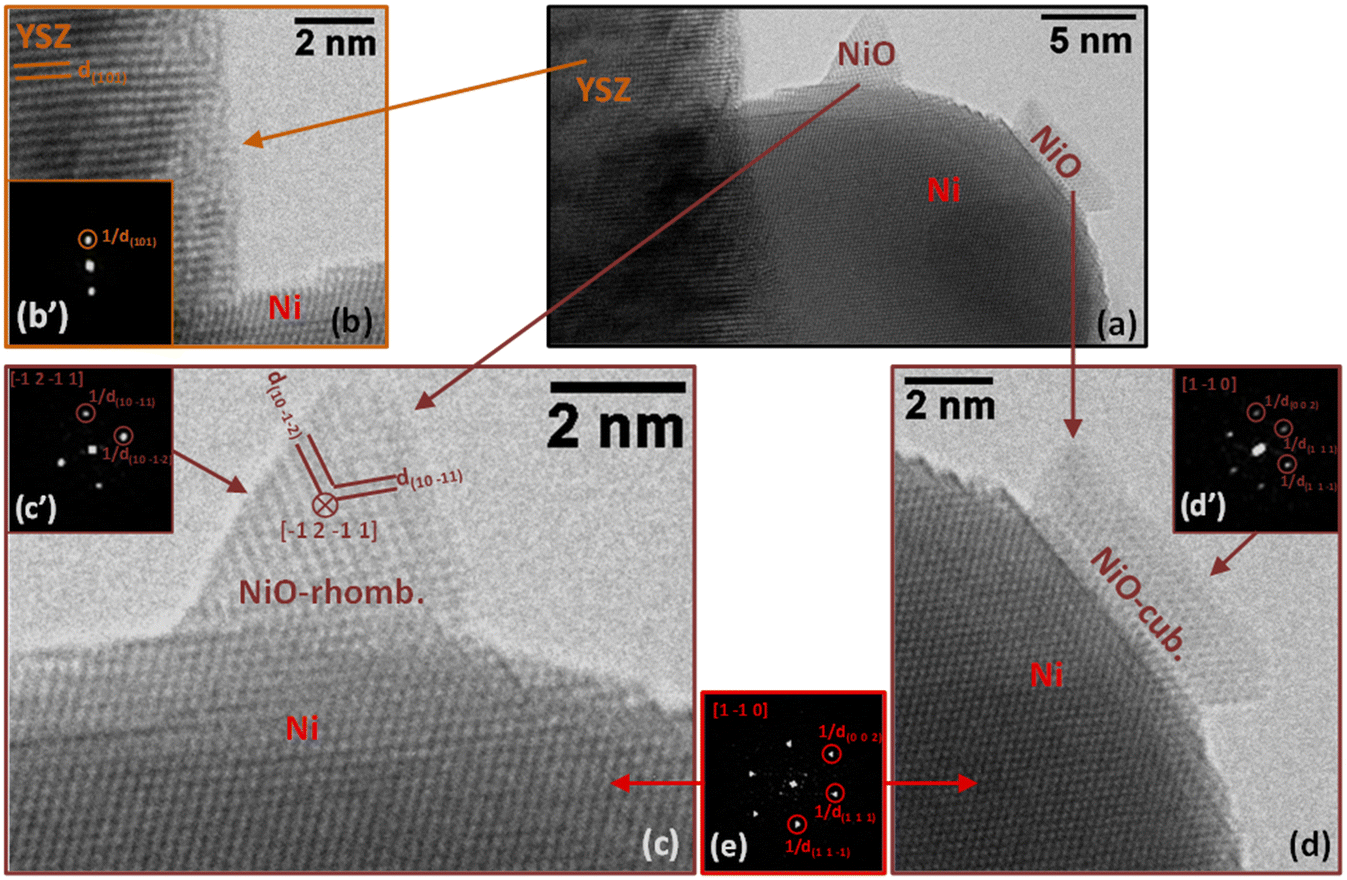
The superior catalytic performance of the SG sample was likely associated to the intimate and homogeneous mixture of nickel and YSZ and their interaction. A comparison of the TEM characterization of the commercial Ni/YSZ (Fig. 6 and 8) and SG (Fig. 7 and 9) reveals subtle differences. The most common regions of the sample Ni/YSZ-Comm (Fig. 6a) show the presence of rather large Ni particles (up to several tens of nm). A close look at those particles by high-resolution TEM clearly reveals that those are metallic Ni with the characteristic face-centered cubic structure. Enlarged regions of the HR image in Fig. 6b show the structures of both the Ni particle (Fig. 6c and c′) and the YSZ support (Fig. 6d and d′). The 2D-FFT of the Ni particle (Fig. 6c′) reveals a [1 −1 0] zone axis of cubic Ni (space group 225: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with a = 0.35238 nm; PDF 00-004-0850) with two visible type {1 1 1} planes (through typical spatial frequencies, arbitrarily taken as 1/d(1 1 −1) and 1/d(1 1 1)) together with a type {0 0 1} plane (through typical spatial frequency 1/d(0 0 2), since the structure factor is 0 for all {hkl} spots with mixed parity in a fcc structure and thus no {0 0 1} spot can be observed) (see Table 3 for measurement details).
m with a = 0.35238 nm; PDF 00-004-0850) with two visible type {1 1 1} planes (through typical spatial frequencies, arbitrarily taken as 1/d(1 1 −1) and 1/d(1 1 1)) together with a type {0 0 1} plane (through typical spatial frequency 1/d(0 0 2), since the structure factor is 0 for all {hkl} spots with mixed parity in a fcc structure and thus no {0 0 1} spot can be observed) (see Table 3 for measurement details).
 | ||
| Fig. 6 TEM images of particles on commercial Ni–YSZ: (a) region with several large Ni particles supported on YSZ. (b) Detailed view of one of those particles. (c) High-resolution image of the surface of the Ni particle in (b) and corresponding 2D-FFT (c′). (d) High-resolution image of the YSZ support surface in (b) and corresponding 2D-FFT (d′). | ||
 | ||
| Fig. 7 TEM images of particles on SG: (a) detailed view of one Ni particle supported on YSZ revealing the presence of two NiO regions at its surface. (b) High-resolution image of the YSZ support and corresponding 2D-FFT (b′). (c) Higher-resolution image of the pyramidal-shaped NiO particle in (a) and corresponding 2D-FFT (c′). (d) Higher-resolution image of the raft-shaped NiO particle in (a) and corresponding 2D-FFT (d′). (e) 2D-FFT of Ni particle in (a) (also seen at higher resolution in (c) and (d)). | ||
| Fig. 6c and c′: Ni particle | ||
|---|---|---|
| Measured | Expected value (from PDF files) | |
| d (1 1 −1) | 0.203 ± 0.010 nm | 0.20340 nm |
| d (1 1 1) | 0.206 ± 0.010 nm | 0.20340 nm |
| d (0 0 2) | 0.179 ± 0.010 nm | 0.17620 nm |
| Angle between (1 1 −1) and (1 1 1) | 70.2 ± 0.5° | 70.53° |
| Angle between (1 1 1) and (0 0 2) | 55.1 ± 0.5° | 54.74° |
| Fig. 6d and d′: YSZ support | ||
|---|---|---|
| Measured | Expected value (from PDF files) | |
| d (0 −1 1) | 0.291 ± 0.010 nm | 0.296514 nm |
| d (1 0 1) | 0.297 ± 0.010 nm | 0.296514 nm |
| d (1 1 0) | 0.252 ± 0.010 nm | 0.256609 nm |
| Angle between (1 1 −1) and (1 1 1) | 70.9 ± 0.5° | 70.59° |
| Angle between (1 1 1) and (0 0 2) | 54.8 ± 0.5° | 54.71° |
The 2D-FFT of the YSZ support (Fig. 6d′) reveals a [−1 1 1] zone axis of tetragonal YSZ (space group 137: P42/nmc with a = 0.36290 nm and c = 0.51430 nm; PDF 01-070-4431) corresponding to 8% yttria in zirconia (((ZrO2)0.92(Y2O3)0.08)0.926). The visible planes are two planes of the {1 0 1} type and one plane of the {1 1 0} type (see details in Table 3). It must be stressed that d(1 1 0) and d(0 0 2) in this structure are very similar to each other (0.256609 nm and 0.257150 nm, respectively); only the measurement of the angles between the three planes allowed us to validate the (1 1 0) plane and dismiss the presence of the (0 0 2) plane.
In sample SG, most of the particles supported on YSZ are in the range of several nm to several tens of nm in size as is the case for the particle in Fig. 7a. In this region (Fig. 7a), we can detect the presence of two small islands of NiO (Fig. 7c and d) on the surface of a larger Ni particle supported on YSZ. It is interesting to note that the Ni particle is clearly metallic while the two islands, both NiO, have different structures: the pyramidal-shaped one (Fig. 7c) has a trigonal (rhombohedral) structure (space group 166: Rm with a = 0.29552 nm and c = 0.72275 nm expressed in a hexagonal system; PDF 00-044-1159), while the raft-like one (Fig. 7d) has a classical cubic bunsenite NiO structure (space group 225: Fmm with a = 0.41771 nm; PDF 00-047-1049).
In both images in Fig. 7c and d, the 2D-FFT of the image of the Ni particle (Fig. 7e) gives a [1 −1 0] zone axis (it is the same particle in both cases; see Table 4 for details). In the image and corresponding 2D-FFT in the inset (Fig. 7c′) of the pyramidal-shaped NiO particle in Fig. 7c only two planes are visible, corresponding to planes (1 0 −1 1) and planes (1 0 −1 2) separated by an angle of 78.95°, revealing a [−1 2 −1 1] zone axis (see Table 4 for details). In the image and corresponding 2D-FFT in the inset (Fig. 7c′) of the raft-shaped NiO particle in Fig. 7d three planes are visible, revealing a [1 −1 0] zone axis (see Table 4 for details). In Fig. 7b, only one plane of the YSZ support is visible (type {1 0 1}, measured d(1 0 1) is 0.292 ± 0.010 nm), preventing the identification of the zone axis of the support. The presence of oxidized regions at the nm-level at the surface of Ni particles indicates that either some Ni particles were not fully reduced during the pre-reduction step or, most probably, partial oxidation of the metal particles occurred under reaction conditions. This was not at all observed in the case of the commercial Ni/YSZ.
| Fig. 7c, d and e: Ni particle | ||
|---|---|---|
| Measured | Expected value (from PDF files) | |
| d (1 1 −1) | 0.207 ± 0.010 nm | 0.20340 nm |
| d (1 1 1) | 0.202 ± 0.010 nm | 0.20340 nm |
| d (0 0 2) | 0.174 ± 0.010 nm | 0.17620 nm |
| Angle between (1 1 −1) and (1 1 1) | 70.7 ± 0.5° | 70.53° |
| Angle between (1 1 1) and (0 0 2) | 54.3 ± 0.5° | 54.74° |
| Fig. 7c and c′: NiO particle (rhombohedral) | ||
|---|---|---|
| Measured | Expected value (from PDF files) | |
| d (1 0 −1 1) | 0.238 ± 0.010 nm | 0.241197 nm |
| d (0 1 −1 2) | 0.212 ± 0.010 nm | 0.208849 nm |
| Angle between (1 0 1 −1) and (0 1 −1 2) | 78.5 ± 0.5° | 78.95° |
| Fig. 7d and d′: NiO particle (rhombohedral) | ||
|---|---|---|
| d (1 1 −1) | 0.238 ± 0.010 nm | 0.24120 nm |
| d (1 1 1) | 0.241 ± 0.010 nm | 0.24120 nm |
| d (0 0 2) | 0.212 ± 0.010 nm | 0.20890 nm |
| Angle between (1 1 −1) and (1 1 1) | 70.1 ± 0.5° | 70.53° |
| Angle between (1 1 1) and (0 0 2) | 54.8 ± 0.5° | 54.74° |
The amounts of carbon deposited over Ni/YSZ-SG and Ni/YSZ-AQ were found to be higher (1.4 to 1.7 wt%) than that observed with the corresponding commercial catalyst (0.4 wt%). This can be attributed to the higher reforming reactivity of methane over these catalysts since the reaction is first order with methane which is consistent with rate-determining methane adsorption.50
3.8 Temperature-programmed oxidation (TPO) measurements
Steam reforming of hydrocarbons suffers from carbon formation, which deactivates the catalyst.50–55 A number of investigations have shown that the rate of carbon formation is largely dependent on the chemical composition of the catalyst as well as its preparation procedure.15,56–58 Coke formation on metals is considerably more complex. In studies of coke formation on nickel, the reaction sequence has been suggested to involve hydrocarbons adsorbing on the surface of nickel and undergoing reactions to produce carbon atoms. These carbonaceous species may stay on the surface to encapsulate the metal and deactivate the catalyst or may dissolve in the nickel and migrate to growth centers such as grain boundaries. Catalyst deactivation may be irreversible or reversible. Generally, deactivation by coke formation can be reversed by gasification of the coke.In order to investigate the extent of catalyst coverage following carbon deposition, the Ni/YSZ catalysts were subjected to temperature-programmed oxidation (TPO) after exposure to the reaction mixture at high temperature. Although the amount of carbon deposited was higher over the Ni/YSZ-SG catalyst with respect to the corresponding commercial one (1.4 vs. 0.4 wt%), the resistance to deactivation was found to be higher in the case of the sample prepared by the sol–gel technique, which remained the most active and selective for all temperatures used. TEM images show that the carbon formed over the catalyst Ni/YSZ-Comm is essentially composed of amorphous carbon and (rather amorphous) filamentous carbon (Fig. 8); the carbon formed on the catalyst Ni/YSZ-SG is mainly graphitic carbon (Fig. 9) around large Ni particles (Fig. 9a, c, d and e), whereas smaller Ni particles remain un-coked (Fig. 9b), although some tubular and filamentous carbon can also be detected (Fig. 9g). For both samples the YSZ surface remains free of any carbon deposits (Fig. 8 and 9, in particular 9f). The amount of carbon dioxide produced through TPO experiments indicated that the carbon formed was generally very limited (<2%) and does not deactivate the catalysts most of which are free of carbon. Higher resistance to deactivation observed for the Ni/YSZ-SG catalyst can be attributed to the higher carbon oxidation by the YSZ phase in this catalyst delaying the poisoning of the Ni particles. Indeed, this is consistent with the ETEM observation of carbonaceous species oxidation at the soot/YSZ interface.59
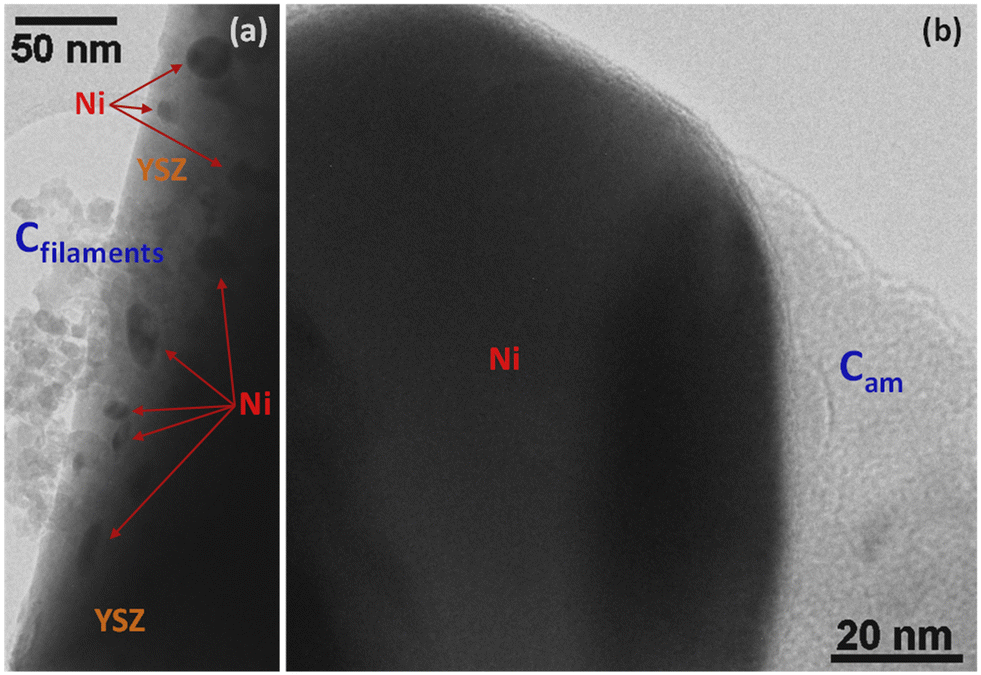 | ||
| Fig. 8 TEM images of carbon deposits on commercial Ni–YSZ: (a) region revealing the presence of filamentous carbon (Cfilaments) near a group of Ni particles supported on YSZ; (b) a thick amorphous carbon (Cam) layer partially covers a large Ni particle. | ||
 | ||
| Fig. 9 TEM images of carbon deposits on SG: (a) general region showing large Ni particles covered by thick layers of graphitic carbon (Cgr) while smaller Ni particles and the YSZ surface remain carbon-free; (b) magnified view of a small carbon-free Ni particle present in (a); (c) detail of one of the graphitic carbon-covered Ni particles present in (a). (d) Region showing a large Ni particle covered by several layers of graphite and carbon-free YSZ support; (e) enhanced view of the rectangle drawn on the Ni particle in (d) showing the graphitic nature of the carbon layers with corresponding 2D-FFT (e′); (f) enhanced view of the rectangle drawn on the YSZ support in (d) with corresponding 2D-FFT (f′). (g) Detail of carbon filaments showing the graphitic nature of their walls and 2D-FFT corresponding to the drawn square (g′). | ||
4 Conclusions
The present work has addressed the use of a variety of preparation routes, such as an impregnation technique in aqueous or organic medium and the sol gel process, for the synthesis of Ni/YSZ anode material. The sol gel method appears to be the most effective method to facilitate intimate mixing and interaction between Ni and YSZ phases. It results in a Ni/YSZ composite, which is both highly active in methane steam reforming into syngas and stable at high temperatures without segregation of the components. The improved catalytic activity is likely associated to the uniform distribution of the porous metallic nickel in Ni–YSZ rather than to specific surface areas, which were rather low and similar for all samples. The results suggest that Ni/YSZ prepared by the sol–gel route has a great potential, in terms of activity and resistance to poisoning by carbon deposits, compared to the corresponding commercial catalyst.Author contributions
All authors conducted experimental work and analyses, read and agreed with the content of the manuscript.Conflicts of interest
The authors declare they have no conflict of interests.Acknowledgements
The authors acknowledge the Region Rhone Alpes Auvergne for financial support through the PhD fellowship grant of S. Bassil (BIODOMPAC project).Notes and references
- T. Iwata, J. Electrochem. Soc., 1996, 143, 1521–1525 CrossRef CAS.
- M. Cassidy, G. Lindsay and K. Kendall, J. Power Sources, 1996, 61, 189–192 CrossRef CAS.
- P. I. Cowin, C. T. G. Petit, R. Lan, J. T. S. Irvine and S. Tao, Adv. Energy Mater., 2011, 1, 314–332 CrossRef CAS.
- R. G. Compton and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1986, 1225–1229 RSC.
- C. Königstein, J. Photochem. Photobiol. A, 1995, 90, 141–152 CrossRef.
- I. Willner and B. Steinberger-Willner, Int. J. Hydrogen Energy, 1988, 13, 593–604 CrossRef CAS.
- H. K. Abdel-Aal, Int. J. Hydrogen Energy, 1985, 10, 333–333 CrossRef CAS.
- T. Capurso, M. Stefanizzi, M. Torresi and S. M. Camporeale, Energy Convers. Manage., 2022, 251, 114898 CrossRef CAS.
- J. R. Rostrup-Nielsen and T. Rostrup-Nielsen, CATTECH, 2002, 6, 150–159 CrossRef CAS.
- H. Zhang, Z. Sun and Y. Hanghu, Renewable Sustainable Energy Rev., 2021, 14, 111330 CrossRef.
- I. Rossetti and A. Tripodi, Top. Catal., 2022 DOI:10.1007/s11244-022-01563-z.
- A. Khaliq and F. Karl, Catal. Today, 2000, 63, 479–487 CrossRef.
- M. D. Gross, J. M. Vohs and R. J. Gorte, J. Mater. Chem., 2007, 17, 3071–3077 RSC.
- E. W. Park, H. Moon, M. S. Park and S. H. Hyun, Int. J. Hydrogen Energy, 2009, 34, 5537–5545 CrossRef CAS.
- T. Borowiecki, Appl. Catal., 1987, 31, 207–220 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar, L. Xinmei, L. Qiann and Z.-F. Yan, Energy Fuels, 2007, 21, 581–589 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, Energy Fuels, 2005, 22, 2195–2202 CrossRef.
- J. H. Lee, H. Moon, H. W. Lee, J. Kim, J. D. Kim and K. H. Yoon, Solid State Ionics, 2002, 148, 15–26 CrossRef CAS.
- E. Sominski, A. Gedanken, N. Perkas, H. P. Buchkremer, N. H. Menzler, L. Z. Zhang and J. C. Yu, Microporous Mesoporous Mater., 2003, 60, 91–97 CrossRef CAS.
- S. K. Pratihar, A. Das Sharma, R. N. Basu and H. S. Maiti, J. Power Sources, 2004, 129, 138–142 CrossRef CAS.
- M. Marinsek, K. Zupan and J. Macek, J. Power Sources, 2000, 86, 383–389 CrossRef CAS.
- C. M. Grgicak, R. G. Green, W. F. Du and J. B. Giorgi, J. Am. Ceram. Soc., 2005, 88, 3081–3087 CrossRef CAS.
- U. Anselmi-Tamburini, M. Arimondi, F. Maglia, G. Spinolo and Z. A. Munir, J. Am. Ceram. Soc., 1998, 81, 1765–1772 CrossRef CAS.
- D. D. Dunuwila, C. D. Gagliardi and K. A. Berglund, Chem. Mater., 1994, 6, 1556–1562 CrossRef CAS.
- M. El Baydi, G. Poillerat, J. L. Rehspringer, J. L. Gautier, J. F. Koening and P. Chartier, J. Solid State Chem., 1994, 109, 281–288 CrossRef CAS.
- P. Gronchi, A. Kaddouri, P. Centola and R. Del Rosso, J. Sol-Gel Sci. Technol., 2003, 26, 843–846 CrossRef CAS.
- S. Bassil, A. Kaddouri, B. Béguin and P. Gélin, Thermochim. Acta, 2013, 567, 35–39 CrossRef CAS.
- A. Kaddouri, N. Dupont, P. Gélin and A. Auroux, Catal. Commun., 2011, 15, 32–36 CrossRef CAS.
- M. El Baydi, S. K. Tiwari, R. N. Singh, J. L. Rehspringer, P. Chartier, J. F. Koening and G. Poillerat, J. Solid State Chem., 1995, 116, 157–169 CrossRef CAS.
- S. D. Kim, H. Moon, S. H. Hyun, J. Moon, J. Kim and H. W. Lee, J. Power Sources, 2006, 163, 392–397 CrossRef CAS.
- P. Lenormand, M. Rieu, R. F. Cienfuegos, A. Julbe, S. Castillo and F. Ansart, Surf. Coat. Technol., 2008, 203, 901–904 CrossRef CAS.
- N. Q. Minh and T. Takahashi, Science and Technology of Ceramic Fuel Cells, Elsevier, Amsterdam, 1995 Search PubMed.
- E. Drozdz-Ciesla, J. Wyrwa, J. Bros and M. Rekas, J. Therm. Anal. Calorim., 2012, 108, 1051–1057 CrossRef CAS.
- T. Klemenso, C. Chung, P. H. Larsen and M. Mogensen, J. Electrochem. Soc., 2005, 152, A2186–A2192 CrossRef.
- B. S. Prakash, S. S. Kumar and S. T. Aruna, Renewable Sustainable Energy Rev., 2014, 36, 149–179 CrossRef.
- K. Haberko, M. Jasinski, P. Pasierb, M. Radecka and M. Rekas, J. Power Sources, 2010, 195, 5527–5533 CrossRef CAS.
- S. A. A. Mansour, Thermochim. Acta, 1993, 228, 173–189 CrossRef CAS.
- N. N. Nichio, M. L. Casella, E. N. Ponzi and O. A. Ferretti, Thermochim. Acta, 2003, 400, 101–107 CrossRef CAS.
- Y. B. Khollam, A. S. Deshpande, A. J. Patil, H. S. Potdar, S. B. Despande and S. K. Date, Mater. Chem. Phys., 2001, 71, 235–241 CrossRef CAS.
- H. M. Ismail, J. Anal. Appl. Pyrolysis, 1991, 31, 315–326 CrossRef.
- E. Spinner, P. Yang, P. T. T. Wong and H. H. Mantsch, Aust. J. Chem., 1986, 39, 475–486 CrossRef CAS.
- P. F. R. Ewings and P. G. Harisson, J. Chem. Soc., Dalton Trans., 1975, 1717–1721 RSC.
- L. Ying, X. Yusheng, G. Jianghong, C. Yunfa and Z. Zhongtai, Mater. Sci. Eng., B, 2001, 86, 119–122 CrossRef.
- D. Adler and J. Feinleib, Phys. Rev. B: Solid State, 1970, 2, 3112–3134 CrossRef.
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS.
- S. H. David, J. L. David, B. Christina and R. M. D. Barry, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- S. H. Ranjan and R. G. Ranga, Bull. Mater. Sci., 2000, 23, 349–354 CrossRef.
- N. V. Vlasenko, P. I. Kyriienco, K. V. Valihura, G. R. Kosmambetova, S. O. Soloviev and P. E. Strizhak, ACS Omega, 2019, 4, 21469–21476 CrossRef CAS PubMed.
- F. P. Ribeiro, M. M. V. M. Souza, O. R. M. Neto, S. M. R. Vasconcelos and M. Schmal, Appl. Catal., A, 2009, 353, 305–309 CrossRef.
- A. L. Dicks, K. D. Pointon and A. Siddle, J. Power Sources, 2000, 86, 523–530 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1997, 37, 225–232 CrossRef CAS.
- O. Yamazaki, K. Tomishige and K. Fujimoto, Appl. Catal., A, 1996, 136, 49–56 CrossRef CAS.
- D. L. Trimm, Catal. Today, 1999, 49, 3–10 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- U. Olsbye, O. Moen, A. Slagtern and I. M. Dahl, Appl. Catal., A, 2002, 228, 289–303 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1974, 33, 184–201 CrossRef.
- I. R. Azevedo, A. A. A. Da Silva, Y. T. Xing, R. C. Rabelo-Neto, N. T. J. Luchters, J. C. Q. Fletcher, F. B. Noronha and L. V. Mattos, Int. J. Hydrogen Energy, 2022, 47, 15624–15640 CrossRef CAS.
- M. Torimoto and Y. Sekine, Catal. Sci. Technol., 2022, 12, 3387–3411 RSC.
- A. Serve, T. Epicier, M. Aouine, F. J. Cadete Santos Aires, E. Obeid, M. Tsampas, K. Pajot and P. Vernoux, Appl. Catal., A, 2015, 504, 74–80 CrossRef CAS.
Footnote |
| † Currently on leave in Lebanon. |
| This journal is © The Royal Society of Chemistry 2024 |