DOI:
10.1039/D4RA01747A
(Review Article)
RSC Adv., 2024,
14, 14919-14933
Advancements in double decarboxylative coupling reactions of carboxylic acids
Received
6th March 2024
, Accepted 30th April 2024
First published on 8th May 2024
Abstract
The double decarboxylative coupling reaction between two (similar or different) molecules of carboxylic acids is an emerging area that has gained considerable attention as a new avenue for forging carbon–carbon bonds. Since this synthetic strategy only utilizes carboxylic acids as easily accessible, non-toxic and stable starting materials, and extrudes carbon dioxide (CO2) as the only waste by-product, it can be considered as an environmentally benign alternative to traditional coupling reactions which mainly rely on the use of toxic organic halides or organometallic reagents. The aim of this review is to highlight the recent advances and developments in this exciting new field that may serve as inspiration for future research to mature it.
1. Introduction
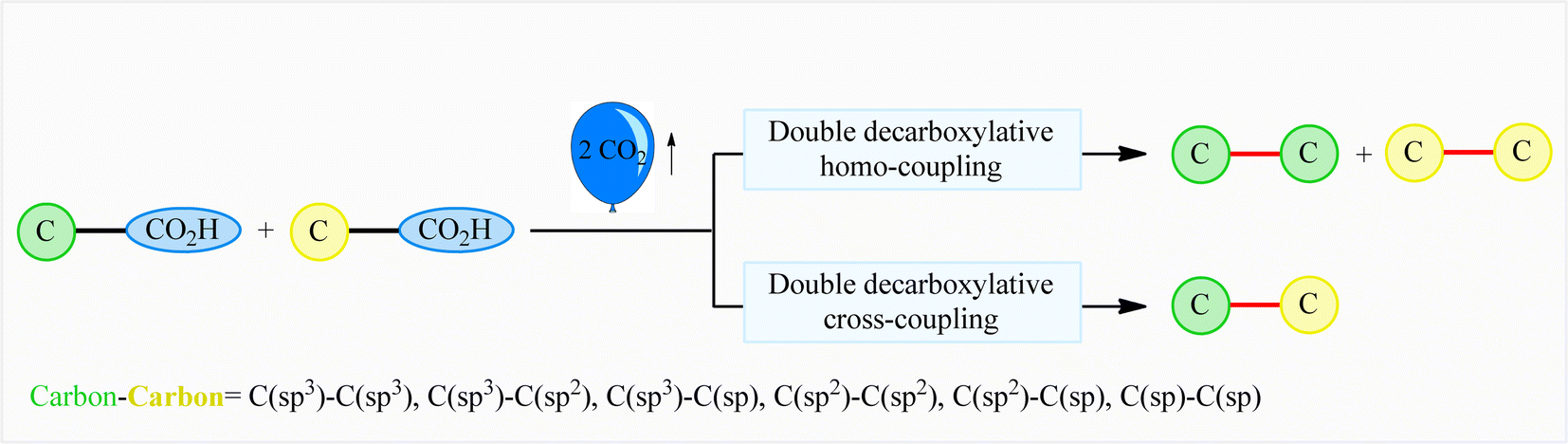
The decarboxylative cross-coupling reactions of carboxylic acids have emerged within recent years as one of the most promising strategies for constructing carbon–carbon1 and carbon–heteroatom bonds,2 not only because carboxylic acids are widely available with significant structural diversity and at low cost, but also because these reactions form small amounts of CO2 as an innocuous by-product. In this field, the double decarboxylative coupling of two molecules of (similar or different) carboxylic acids has recently been developed as a green and attractive strategy to form new carbon–carbon bonds that avoids the use of any toxic organic halides or organometallic reagents which are normally involved in traditional coupling reactions.3 Interestingly, this new methodology has been successfully applied to efficient construction of all six kinds of C–C bonds [i.e., C(sp3)–C(sp3), C(sp3)–C(sp2), C(sp3)–C(sp), C(sp2)–C(sp2), C(sp2)–C(sp), and C(sp)–C(sp)], demonstrating the general applicability of this synthetic approach (Fig. 1). Since many advances have occurred during the past few years on the double decarboxylative C–C coupling reactions, a comprehensive review in this fast growing research field seems to be timely. In continuation of our review articles on decarboxylative4 and modern cross-coupling reactions,5 in the present review we will highlight recent advances and achievements in the fabrication of C–C bonds via double decarboxylative coupling reactions. It should be noted that we will not speak on the double decarboxylative coupling of other substrates than carboxylic acids (e.g., esters, carbonates)6 or multi-component reactions.7
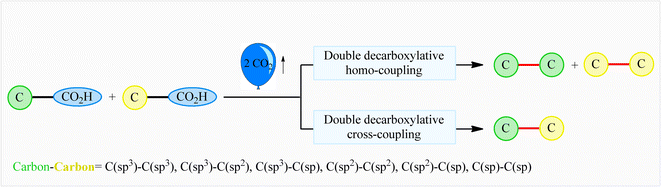 |
| | Fig. 1 Double decarboxylative coupling reactions of carboxylic acids. | |
2. C(sp3)–C(sp3) bond formation
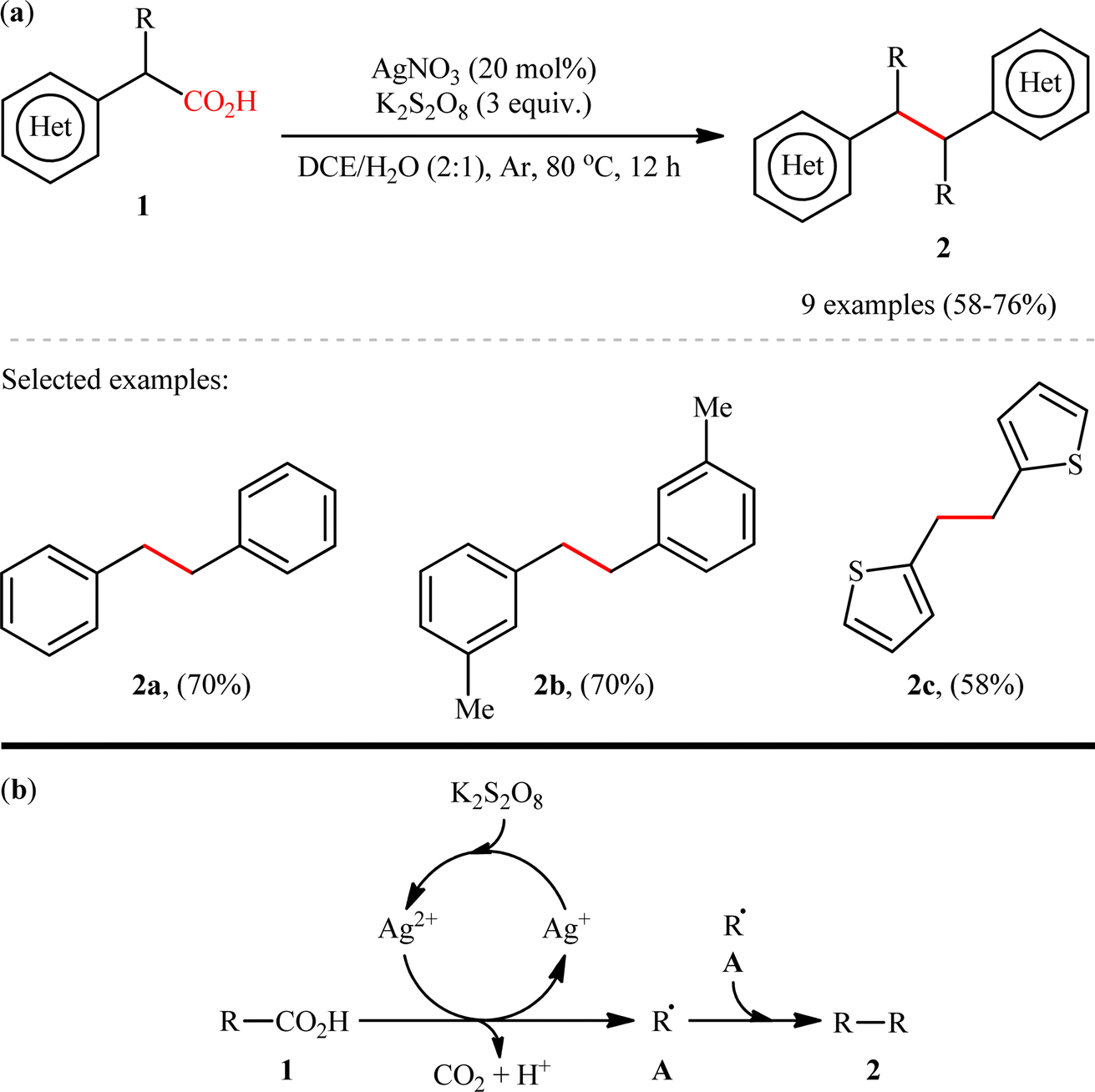
The construction of C(sp3)–C(sp3) bonds through double decarboxylative coupling of aliphatic carboxylic acids has been scarcely investigated; in fact, only two examples of such reactions were reported in literature thus far. In 2018, Li, Xiang, and their co-workers disclosed for the first time the possibility of formation of a new C–C bond at sp3-hybridized centers via a double decarboxylative synthetic protocol.8 They revealed that the treatment of (hetero)benzylic carboxylic acids 1 with 3 equiv. of K2S2O8 in the presence of a catalytic amount of AgNO3 in binary solvent 1,2-dichloroethane (DCE)/H2O with ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 resulted in the corresponding (hetero)aryl-substituted ethanes 2 in moderate to good yields, ranging from 58% to 75% (Scheme 1a). The results showed that both silver catalyst and the oxidant played a pivotal role in this transformation, as no reaction was observed in the absence of either of them. It is worth mentioning that the presence of H2O was crucial for the success of the reaction probably because the presence of H2O promotes the dissolution of K2S2O8. The results indicated that electronic and steric effects of the substituents on the phenyl ring periphery of substrates had no significant impact on the outcome of this homocoupling reaction. Notably, this catalytic platform was also found to be suitable for decarboxylative homocoupling of α-keto acids for producing corresponding 1,2-diketones. Unfortunately, neither simple aliphatic carboxylic acids (e.g., heptanoic acid) nor benzoic acids could react in this system. Based on a series of control experiments, the authors proposed a possible mechanistic pathway for this transformation which is outlined in Scheme 1b. Initially, the oxidation of Ag+ species with persulfate generates the active Ag2+ species. Subsequently, carboxylic acid 1 undergoes single electron oxidative decarboxylation with the active Ag2+ species to produce the alkyl free radical A and regenerate the Ag+ species. Finally, radical–radical homocoupling of two equivalents of intermediate A affords the observed product 2.
:1 resulted in the corresponding (hetero)aryl-substituted ethanes 2 in moderate to good yields, ranging from 58% to 75% (Scheme 1a). The results showed that both silver catalyst and the oxidant played a pivotal role in this transformation, as no reaction was observed in the absence of either of them. It is worth mentioning that the presence of H2O was crucial for the success of the reaction probably because the presence of H2O promotes the dissolution of K2S2O8. The results indicated that electronic and steric effects of the substituents on the phenyl ring periphery of substrates had no significant impact on the outcome of this homocoupling reaction. Notably, this catalytic platform was also found to be suitable for decarboxylative homocoupling of α-keto acids for producing corresponding 1,2-diketones. Unfortunately, neither simple aliphatic carboxylic acids (e.g., heptanoic acid) nor benzoic acids could react in this system. Based on a series of control experiments, the authors proposed a possible mechanistic pathway for this transformation which is outlined in Scheme 1b. Initially, the oxidation of Ag+ species with persulfate generates the active Ag2+ species. Subsequently, carboxylic acid 1 undergoes single electron oxidative decarboxylation with the active Ag2+ species to produce the alkyl free radical A and regenerate the Ag+ species. Finally, radical–radical homocoupling of two equivalents of intermediate A affords the observed product 2.
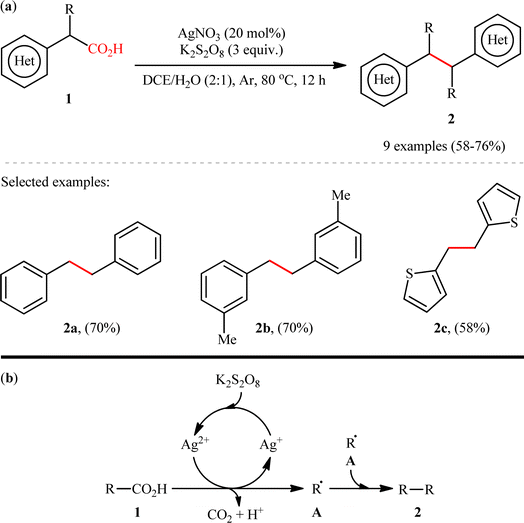 |
| | Scheme 1 (a) Ag-catalyzed double decarboxylative coupling of (hetero)benzylic carboxylic acids 1; (b) a plausible mechanism for the formation of (hetero)aryl-substituted ethanes 2.8 | |
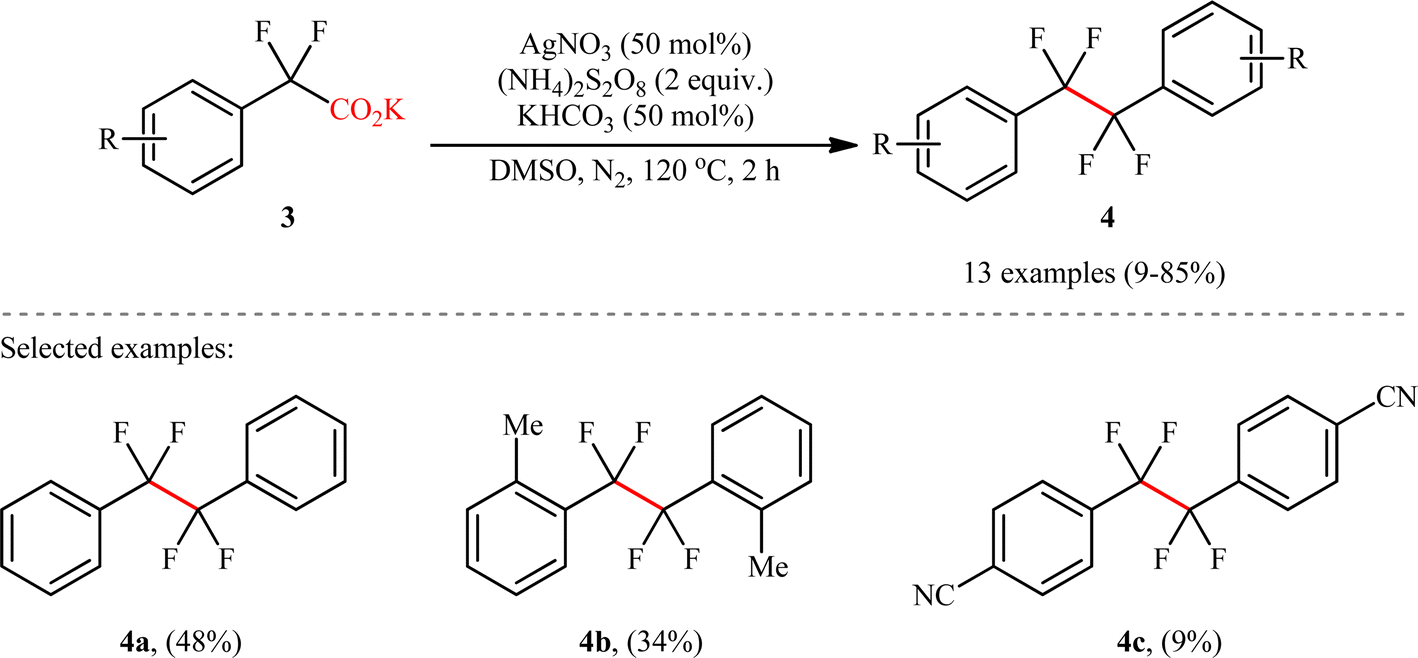
Shortly afterwards, Wan's research group reported a conceptually similar C(sp3)–C(sp3) coupling strategy to synthesis of symmetric CF2–CF2 containing dimers.9 In this study, a library of thirteen 1,1,2,2-tetrafluoro-1,2-diarylethanes 4 was efficiently synthesized through the decarboxylative homocoupling of corresponding potassium 2,2-difluoro-2-phenylacetate derivatives 3 employing AgNO3/(NH4)2S2O8/KHCO3 combination as a catalytic system in dimethyl sulfoxide (DMSO) at 120 °C (Scheme 2). Besides AgNO3, other silver salts such as Ag2CO4, AgBF4, AgOAc and AgSO3CF3 were also found to promote this decarboxylative homocoupling reaction; albeit, in lower yields. However, replacement of silver salts with copper(I) or (II) salts completely suppressed reaction. Unfortunately, no comment was made by the authors regarding these observations. Depending on the electronic effects of substituents on the aromatic ring, substrates with electron-donating groups provided higher yields than those with electron-withdrawing groups. Interestingly, the reaction proved insensitive to steric hindrance. While the dimethylated substrate produced 46% yield, the trimethylated one produced 85% yield. A drawback of the protocol is the requirement for an inert atmosphere and an elevated temperature (120 °C), which may limit its application profile. Unfortunately, 2,2-difluoro-2-thienylacetic acid did not take part in this homocoupling and therefore no other heterobenzylic carboxylic acids were examined in the protocol. According to the authors, the reaction proceeds via a mechanism analogous to that of Li, Xiang, and co-workers for the formation of (hetero)aryl-substituted ethanes 2.
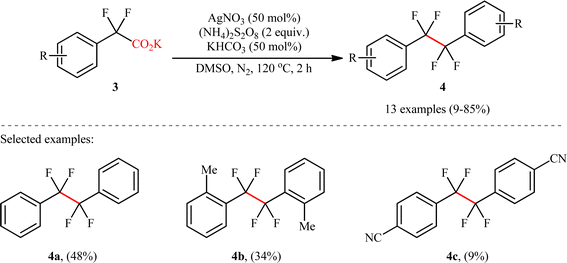 |
| | Scheme 2 Wan's synthesis of 1,1,2,2-tetrafluoro-1,2-diarylethanes 4.9 | |
3. C(sp3)–C(sp2) bond formation
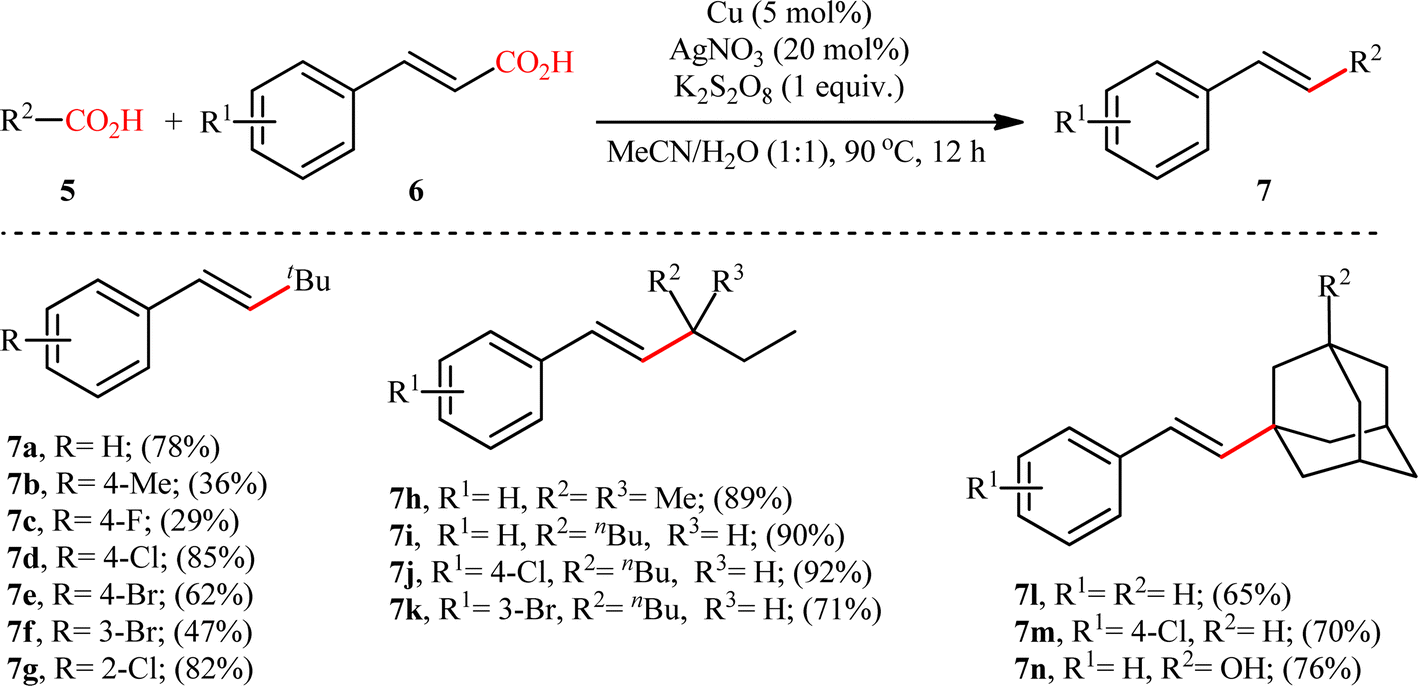
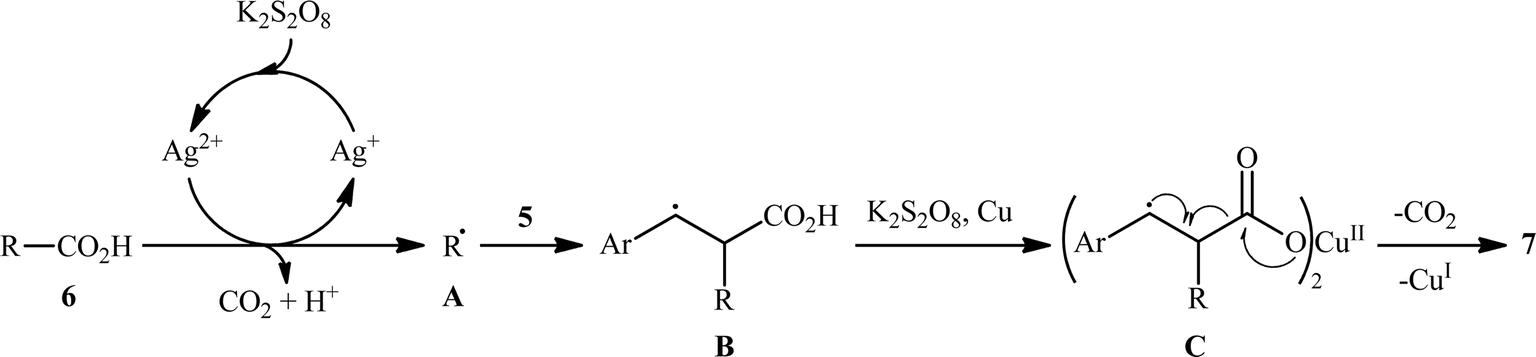
The first and only example on the formation of C(sp3)–C(sp2) bonds through double decarboxylative cross-coupling reaction between two carboxylic acids was published by Mai et al.,10 who revealed that the treatment of various aliphatic carboxylic acids 5 and cinnamic acid derivatives 6 in the presence of a cooperative AgNO3 and copper(0) catalytic system produced the corresponding β-alkyl styrene derivatives 7 in satisfactory yields (Scheme 3). Under the optimized conditions, both cyclic and acyclic (secondary and tertiary) aliphatic carboxylic acids were compatible substrates in this transformation. The reaction, however, appears to be limited to unsubstituted and cinnamic acids bearing weak electron-withdrawing groups. Strangely, the primary carboxylic acids failed to participate in this reaction. Moreover, when cinnamic acid bearing 4-OMe substitution was used as the substrate under the identical conditions, the unwanted 4-methoxybenzaldehyde was isolated in a yield of 82%, without the formation of desired product. The synthetic potential of this double decarboxylative coupling protocol was further demonstrated by alkenylation of gemfibrozil, an oral drug used to lower lipid levels. An important feature of this synthetic strategy is that no homo-coupled products were detected in this transformation since the nucleophilic alkyl radicals were preferentially trapped by the cinnamate acceptor in an exclusive manner. Mechanistically, this C(sp3)–C(sp2) bond forming reaction starts with the oxidation of Ag(I) by persulfate to Ag(II), which decarboxylates the aliphatic carboxylic acid 5 and generates the key alkyl radical A. Afterwards, this alkyl radical attacks α-position of cinnamic acid 6 to produce benzyl radical intermediate B, which after interaction with Cu(0) affords carboxylate species C. Finally, the newly formed intermediate C undergoes a single electron transfer (SET) process to release CO2 and form the observed product 7 (Scheme 4).
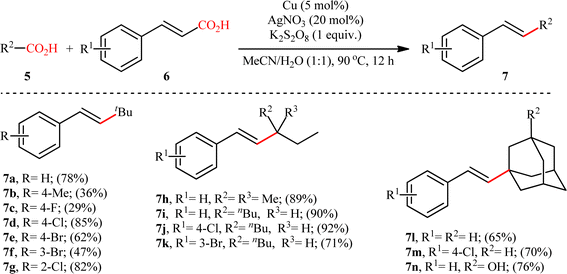 |
| | Scheme 3 Selected examples of the Cu/Ag-catalyzed decarboxylative alkenylation of aliphatic carboxylic acids 5 with cinnamic acids 6.10 | |
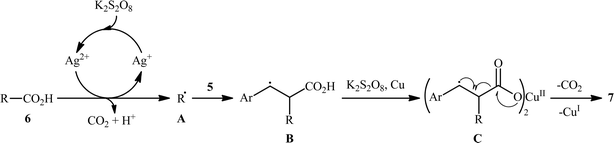 |
| | Scheme 4 Mechanism proposed to explain the formation of β-alkyl styrenes 7. | |
4. C(sp3)–C(sp) bond formation
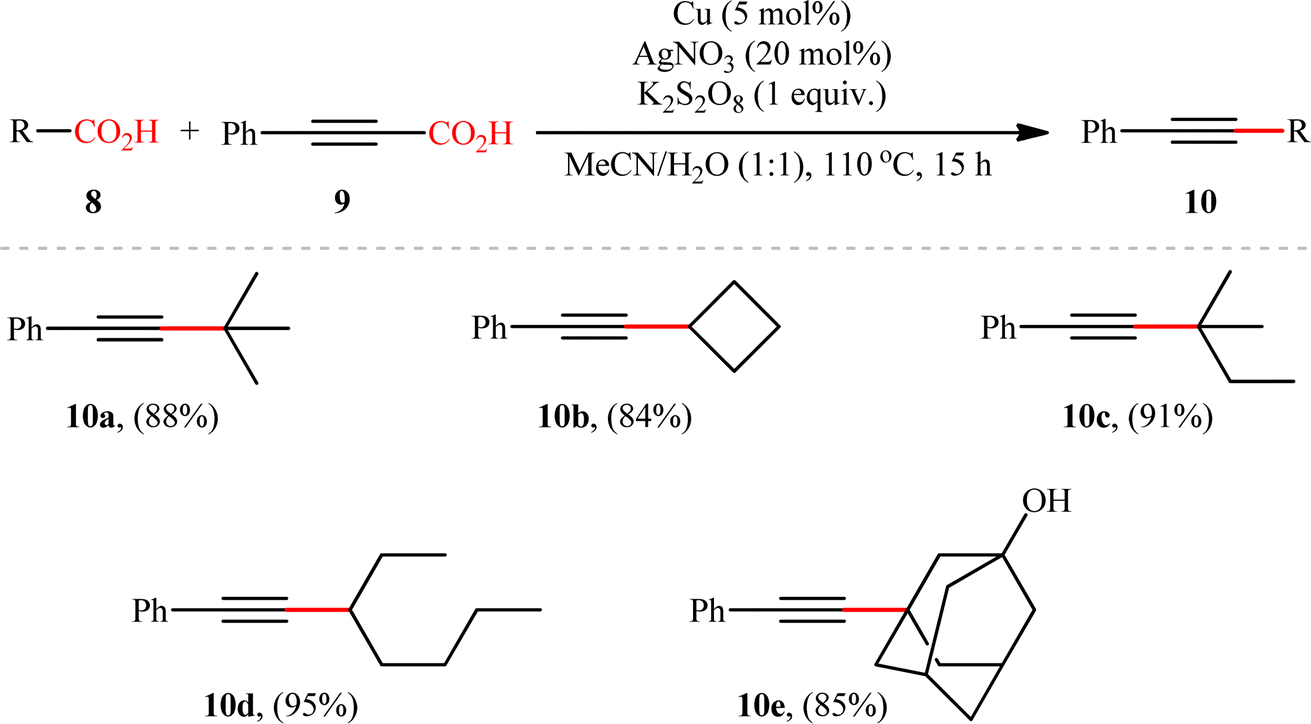
Based on their established strategy, Mai, Sun, and co-workers further developed an alkyl–alkynyl double decarboxylative cross-coupling to forge C(sp3)–C(sp) bonds through the similar Ag/Cu bimetallic tandem catalysis and just by elevating the reaction temperature to 110 °C.10 In this preliminary work, the authors investigated relatively broad scope of aliphatic carboxylic acids 8 (14 substrates), but very limited scope of alkynoic acids (only phenylpropiolic acid 9); however, the yields were good (Scheme 5). Nevertheless, it should be mentioned that primary aliphatic carboxylic acids and secondary aliphatic carboxylic acids containing ether, halide, or ketone groups on the adjacent position were not compatible with the reaction conditions. The author proposed mechanism for this transformation is analogous to the one depicted for alkenylation of aliphatic carboxylic acids.
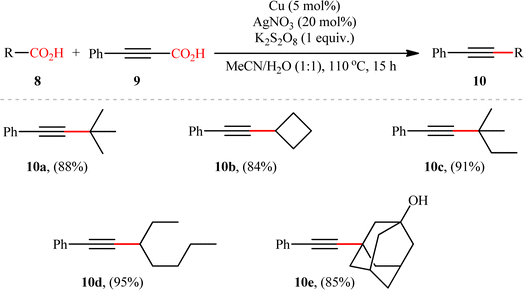 |
| | Scheme 5 Cu/Ag-catalyzed double decarboxylative cross-coupling reaction between aliphatic carboxylic acids 8 and phenylpropiolic acid 9.10 | |
5. C(sp2)–C(sp2) bond formation
In this section, we describe the current literature on the double decarboxylative coupling reactions that form C(sp2)–C(sp2) bonds via cleavage of C(sp2)–CO2H bonds. The section is organized according to the type of product formed in three different sub-sections. Thus, the synthesis of biaryl derivatives through the decarboxylative homo-/hetero-coupling of benzoic acids are discussed first. This is followed by double-decarboxylative cross-coupling between α-keto acids and α,β-unsaturated acids to form α,β-unsaturated carbonyls. Finally, the only reported example for the synthesis of 1,2-diketones via the decarboxylative homocoupling of α-keto acids will be covered at the end of the section.
5.1. Biaryls synthesis
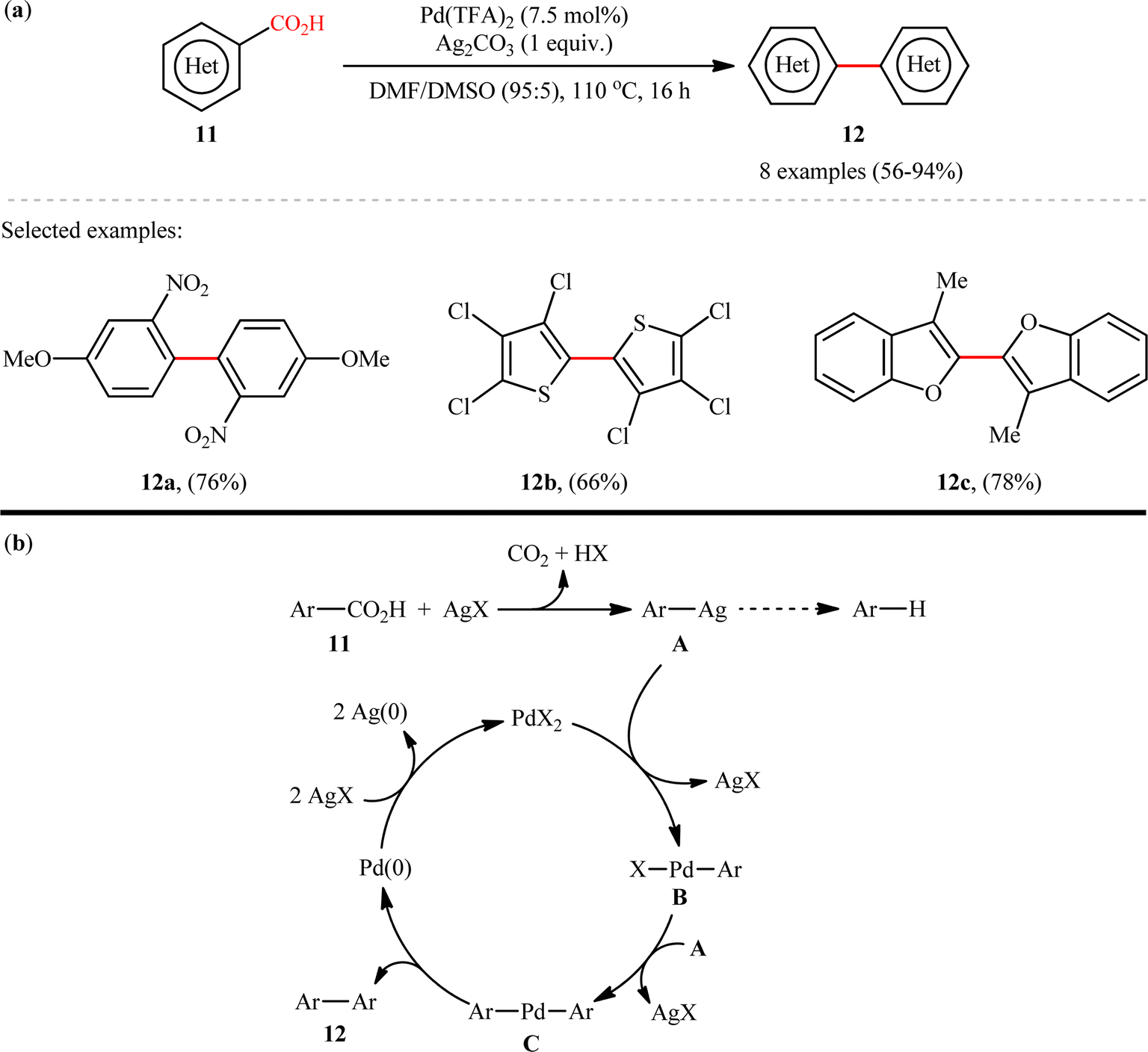
The first example on decarboxylative C(aryl)–CO2H/C(aryl)–CO2H coupling reactions has been reported by the group of Larrosa in 2010,11 when a range of hetero(aromatic) carboxylic acids 11 underwent decarboxylative homocoupling in the presence of a Pd/Ag system in a mixed solvent of dimethylformamide (DMF) and DMSO to form the corresponding symmetrical biaryls 12 in moderate to excellent yields (Scheme 6a). Although only 2-heteroaromatic carboxylic acids or aromatic carboxylic acids with an ortho electron-withdrawing substituent were tolerated, this method could be an inspiration for further researchers. A limitation of this procedure was the competing proto-demetalation of the Ag(I)–arene intermediate which results in the formation of unwanted protodecarboxylated products. Unfortunately, when benzoic acids ortho-substituted with Br, OMe, or F were applied under the identical conditions, decarboxylation to the corresponding arenes were the main products observed, with only small amounts (10–20% NMR yield) of the desired dimer products. These results clearly indicated that the relative rates of protodemetallation versus transmetallation are highly affected by the nature of the group in ortho position. The following mechanistic cycle was proposed by the authors for this decarboxylative homocoupling reaction (Scheme 6b): Initially, Ag(I) salt promotes the decarboxylation of (hetero)arene carboxylic acid 11 to generate the Ag(I)–arene intermediate A, which after transmetalation to the Pd(II) catalyst provides Pd(II)–arene species B. Subsequently, a second transmetalation with another Ag(I)–arene A takes place to form the bisaryl–Pd species C, that after reductive elimination provides the target biaryl products 12 and the Pd(0) species. Finally, two equiv. of Ag(I) oxidizes Pd(0) to regenerate Pd(II) catalyst and complete the catalytic cycle.
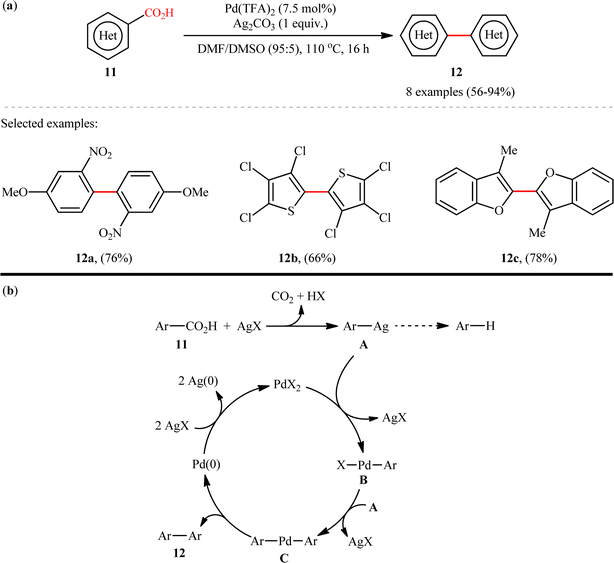 |
| | Scheme 6 (a) Larrosa's synthesis of symmetrical biaryls 12; (b) suggested mechanism for decarboxylative homocoupling of hetero(aromatic) carboxylic acids 11.11 | |
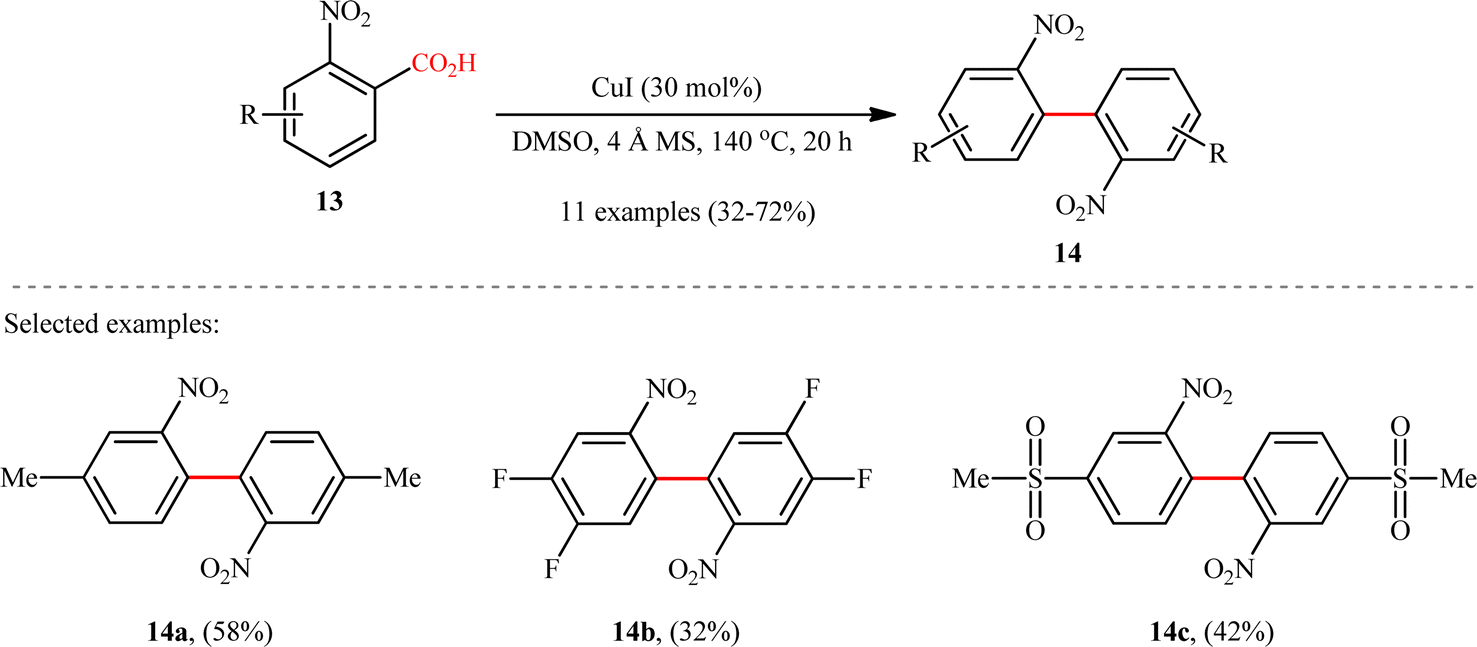
Subsequently, copper-catalyzed version of this homocoupling reaction was disclosed by Cai and co-workers,12 who revealed that the treatment of various ortho-nitrobenzoic acids 13 bearing electron-deficient (fluoro, chloro, trifluoromethyl, and sulfonyl) and electron-donating groups (methyl and methoxy) with a catalytic amount of CuI in the presence of 4 Å molecular sieves (MS) in DMSO, afforded the corresponding 2,2′-dinitrosubstituted biaryls 14 in poor to good yields (Scheme 7). It is worthwhile to note that the 4-methylated substrate produced 58% yield, whereas the 6-methylated one was quite inert under the identical conditions, indicating high sensitivity of this transformation to steric hindrance. In the investigation of the scope of this transformation, it was found that presence of a nitro group at the ortho-position of benzoic acid substrate was vital for the success of the reaction. The authors explained this observation by stabilizing effect of the nitro group for the transition structure of decarboxylation procedure and enhancing the rate of formation of aryl-copper intermediate in the transformation. It should be mentioned that other copper catalysts such as CuBr, CuOAc, and Cu2O were also effective in this homocoupling reaction but gave lower yield of product.
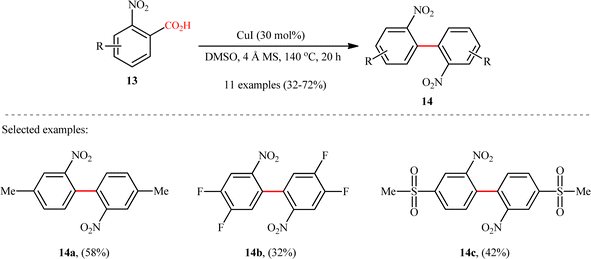 |
| | Scheme 7 Cu-catalyzed decarboxylative homocoupling of ortho-nitrobenzoic acids 13.12 | |
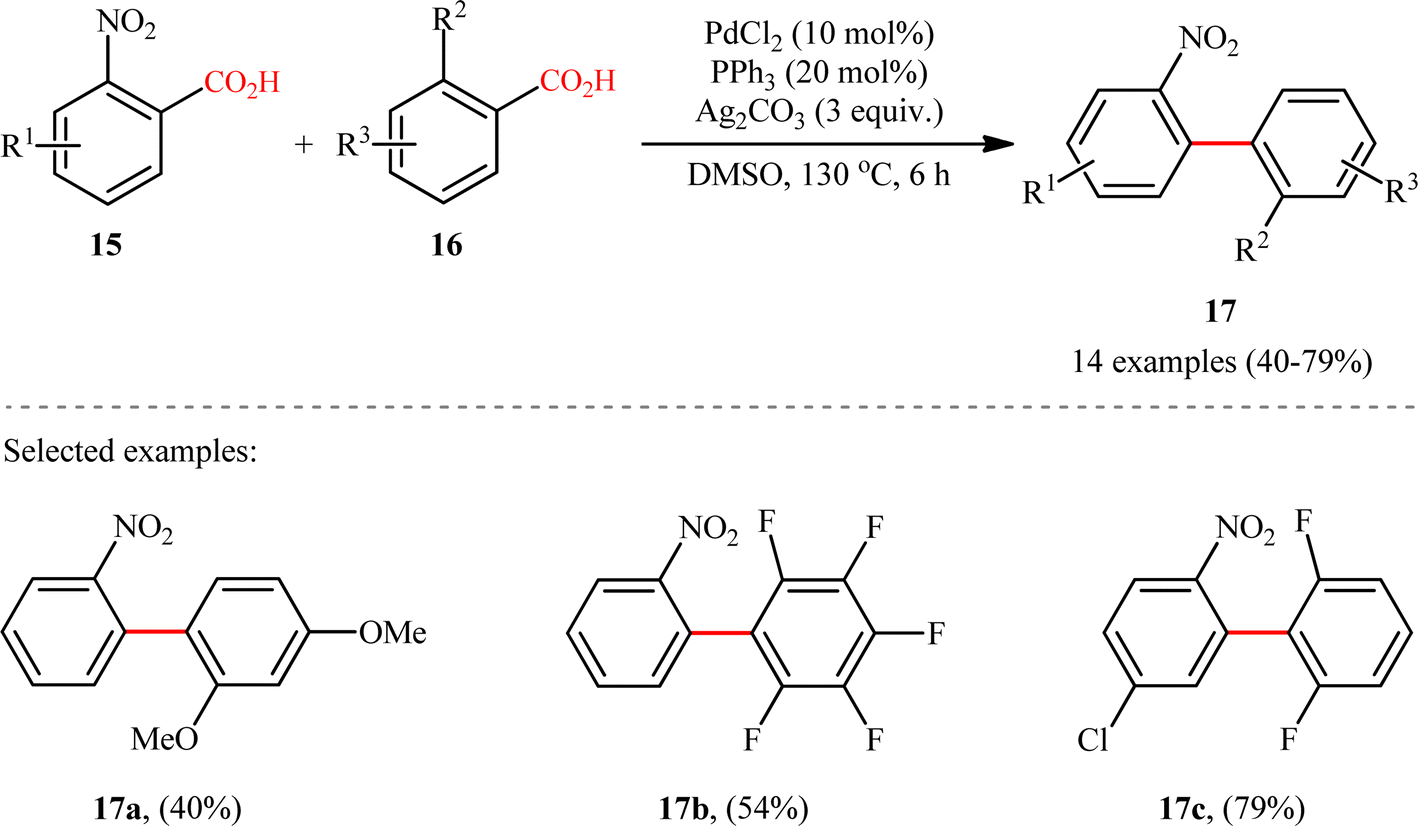
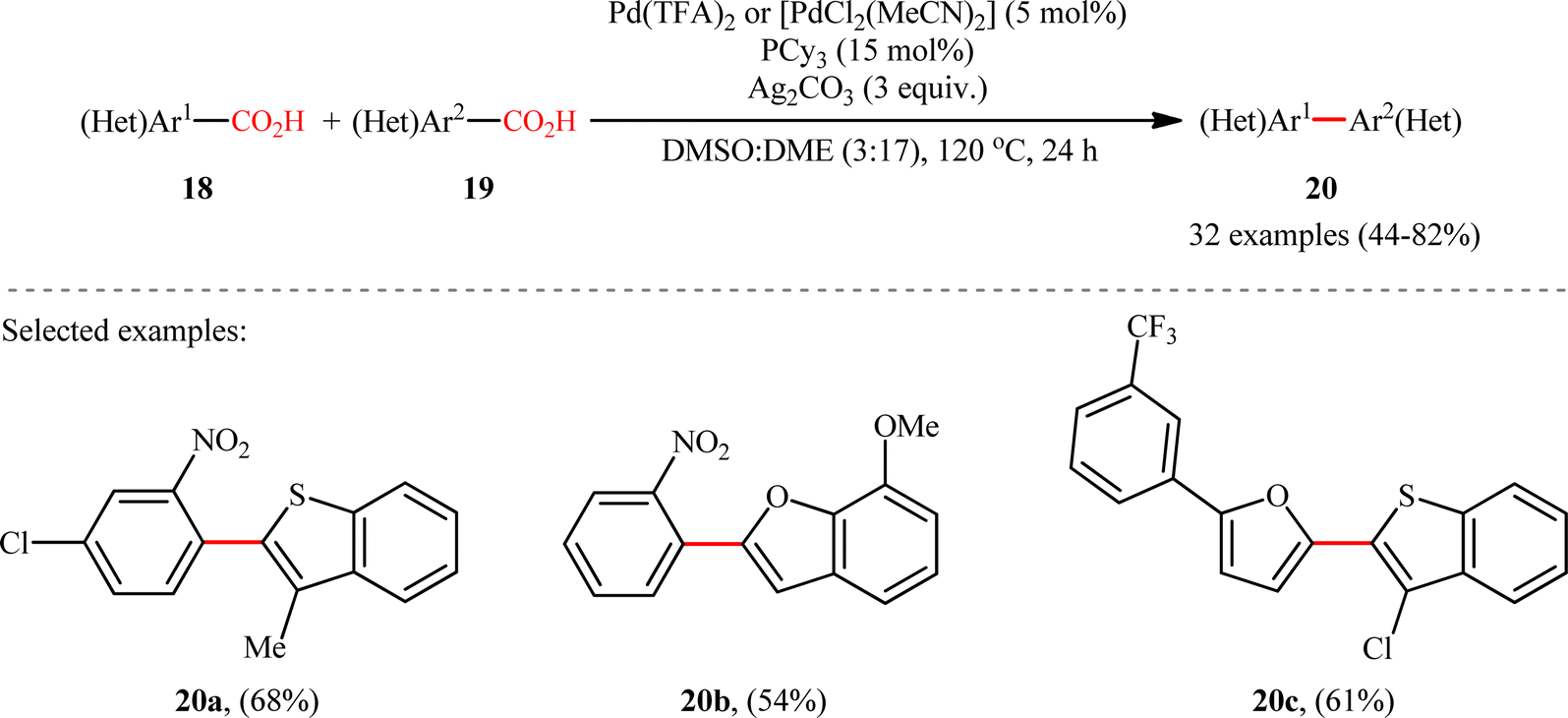
A notable contribution to this field was reported by Tan, Deng, and co-workers in 2011.13 They disclosed that a merge of PdCl2 and PPh3 with Ag2CO3 could enable challenging double decarboxylative cross-coupling between two benzoic acids substrates. On the basis of the optimal conditions, a series of o-nitrobenzoic acid derivatives 15 smoothly coupled with various o-substituted benzoic acids 16 by a double decarboxylation process procedure to generate desired unsymmetrical biaryl products 17 in moderate to good yields (Scheme 8). It should be mentioned that significant amounts of symmetrical biaryl by-products, formed by homocoupling of arenecarboxylic acids, were also observed in all cases. Indeed, the cross-coupling/homocoupling selectivity was less than 3.5:1 at best. In this system, silver salt should play multiple roles:14 (i) the decarboxylating promoter, (ii) the transmetalating agent, and (iii) an external oxidant. In a closely related study, Su and co-workers disclosed that a variety of unsymmetrical bi(hetero)aryl compounds 20 were successfully formed from two different (hetero)aryl carboxylic acids 18 and 19 using a Pd/Ag bimetallic system equipped with a phosphine ligand (Scheme 9).15 It is noteworthy that this protocol not only allows the cross-coupling of arenecarboxylic acids that are electronically different but also those that are electronically similar, and provided good yields. However, like Tan–Deng's work, this process also relies on an elevated temperature (120 °C) and ortho-substituent in benzoic acids, which limited its broad application in practical synthesis. Along this line, the catalytic system involved PdCl2 (20 mol%) in presence of Cu(OH)2 (75 mol%) was demonstrated by Rameau et al. for the same cross decarboxylative coupling, albeit with much lower efficiency (9 examples with average yield of 19%).16
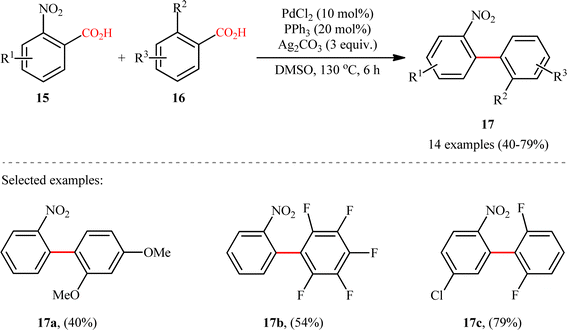 |
| | Scheme 8 Tan–Deng's synthesis of unsymmetrical biaryls 17.13 | |
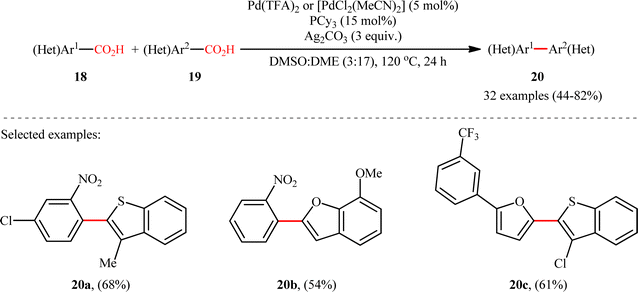 |
| | Scheme 9 Su's synthesis of unsymmetrical biaryls 20.15 | |
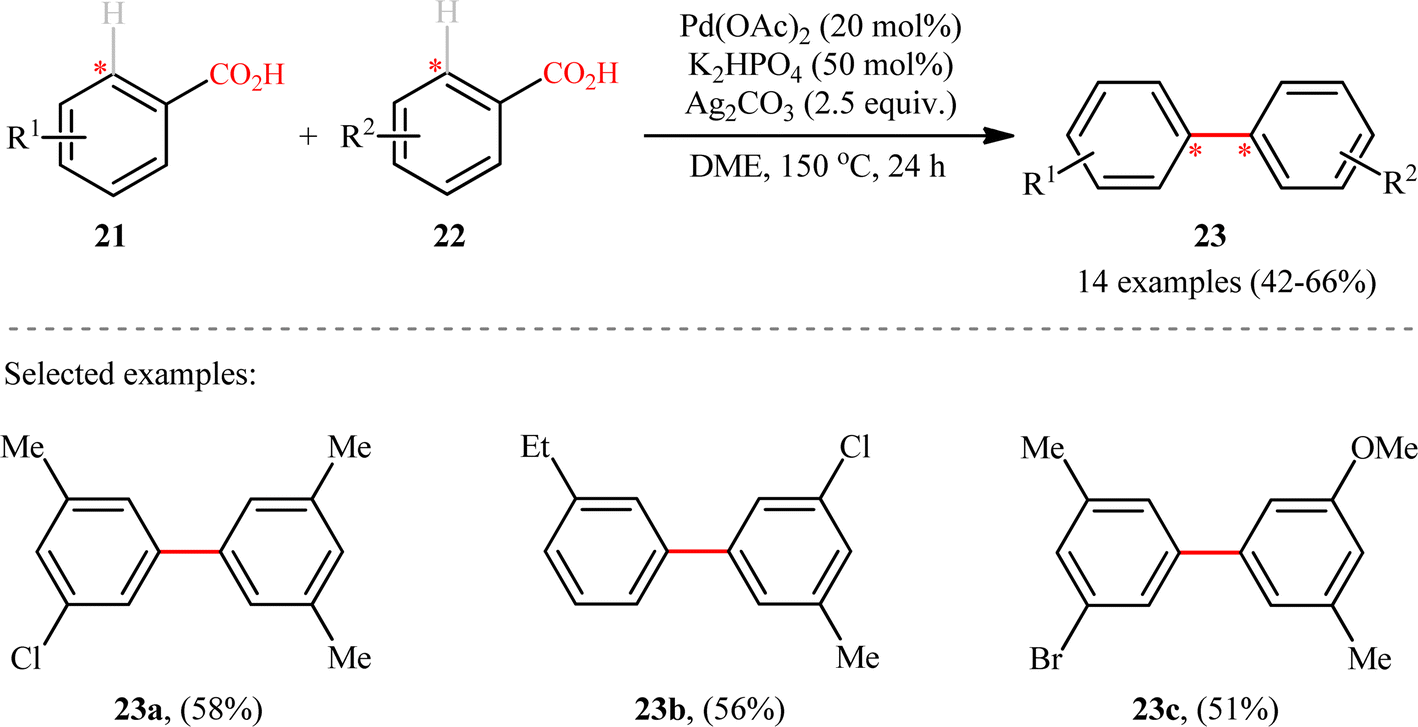
Following these works, Shi and colleagues unraveled an elegant strategy for the synthesis of unsymmetrical meta-substituted biaryls 23 from two different electron-rich benzoic acids 21 and 22 through a Pd-catalyzed carboxyl-directed intermolecular cross-dehydrogenative coupling and subsequent decarboxylation.17 The reaction took place in the presence of Pd(OAc)2/Ag2CO3/K2HPO4 combination as an efficient catalytic system in refluxing DME, and provided the desired products in synthetically useful yields (Scheme 10). It should be mentioned that in these reactions, the carboxyl group served as a traceless directing group that after formation of the desired C–C bond is easily removed via a Pd-catalyzed protodecarboxylation process. Unlike previous double decarboxylative coupling protocols which were limited to the use of o-substituted benzoic acids where at least one of the them was electron deficient, this new strategy tolerated electron-rich acids and the new C–C bonds were formed in the ortho position of carboxyl.
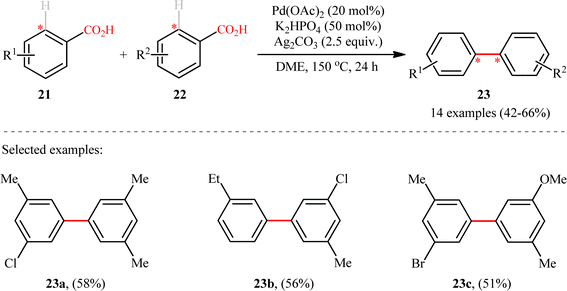 |
| | Scheme 10 Synthesis of biaryls 3 through Pd-catalyzed decarboxylative cross-dehydrogenative coupling between two different benzoic acids 21 and 22.17 | |
5.2. α,β-Unsaturated carbonyls synthesis
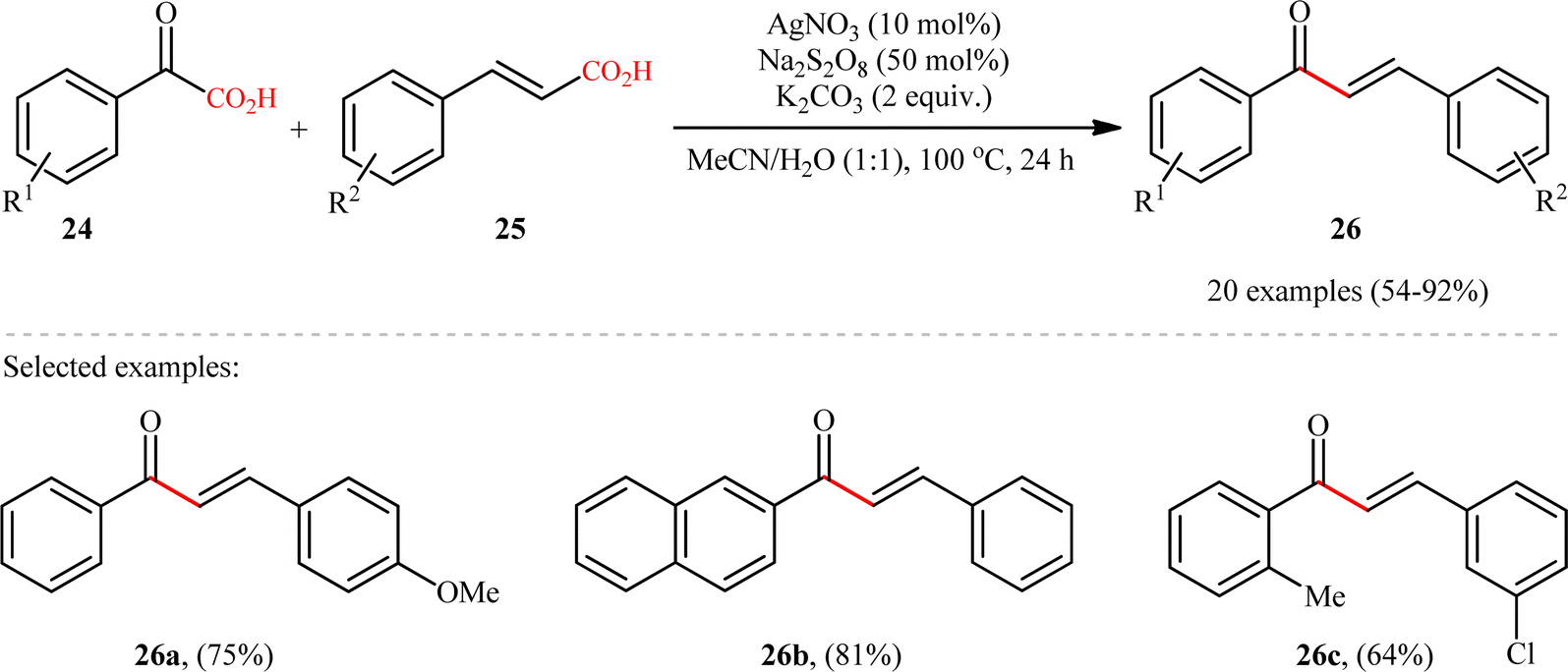
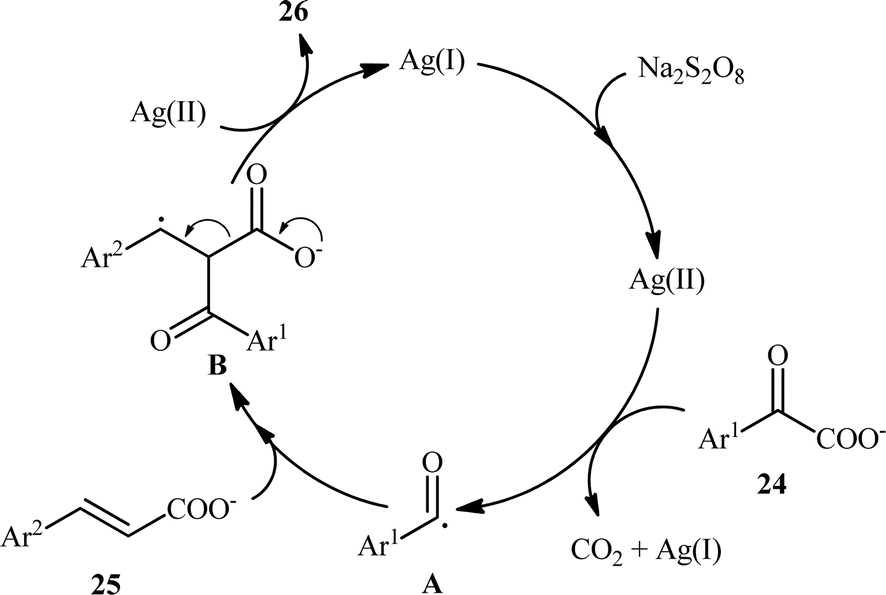
In 2015, Yang and Wang along with their co-workers developed a novel scheme for the synthesis of chalcone derivatives 26 by silver-catalyzed double-decarboxylative cross-coupling of aromatic α-keto acids 24 with cinnamic acids 25.18 In this study, twenty functionalized chalcones 26 have also been obtained with AgNO3 as the catalyst and Na2S2O8 as the oxidant in an aqueous medium with yields up to 92% (Scheme 11). Although a series of cinnamic acids bearing both electron-donating (e.g., Me, OMe) and electron-withdrawing (e.g., F, Cl, Br) functional groups were well tolerated under the reaction conditions, the scope of α-keto acids restricted to electron-rich derivatives. Besides that, neither aliphatic α-keto acids nor aliphatic conjugated acids, such as crotonic acid could react in this system, thus limiting the range of applications this procedure. The proposed mechanism for this transformation is outlined in Scheme 12. Initially, the acyl radical A was generated from α-keto acid 24 by an oxidative radical decarboxylation procedure. This acyl radical attacked to the α-position of the double bond of the cinnamic acid 25, followed by elimination of CO2 to provide the target product 26. There is no need to say that such a radical mechanism is preferred in water because of the inertness of water toward radical species and enhance the reaction rates.
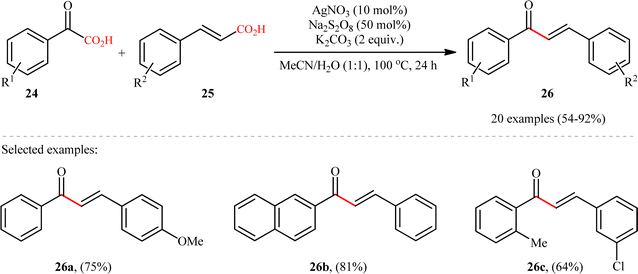 |
| | Scheme 11 Ag-catalyzed double-decarboxylative cross-coupling of aromatic α-keto acids 24 with cinnamic acids 25.18 | |
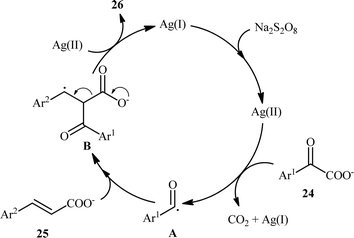 |
| | Scheme 12 Plausible mechanism for the Ag-catalyzed synthesis of chalcones 26. | |
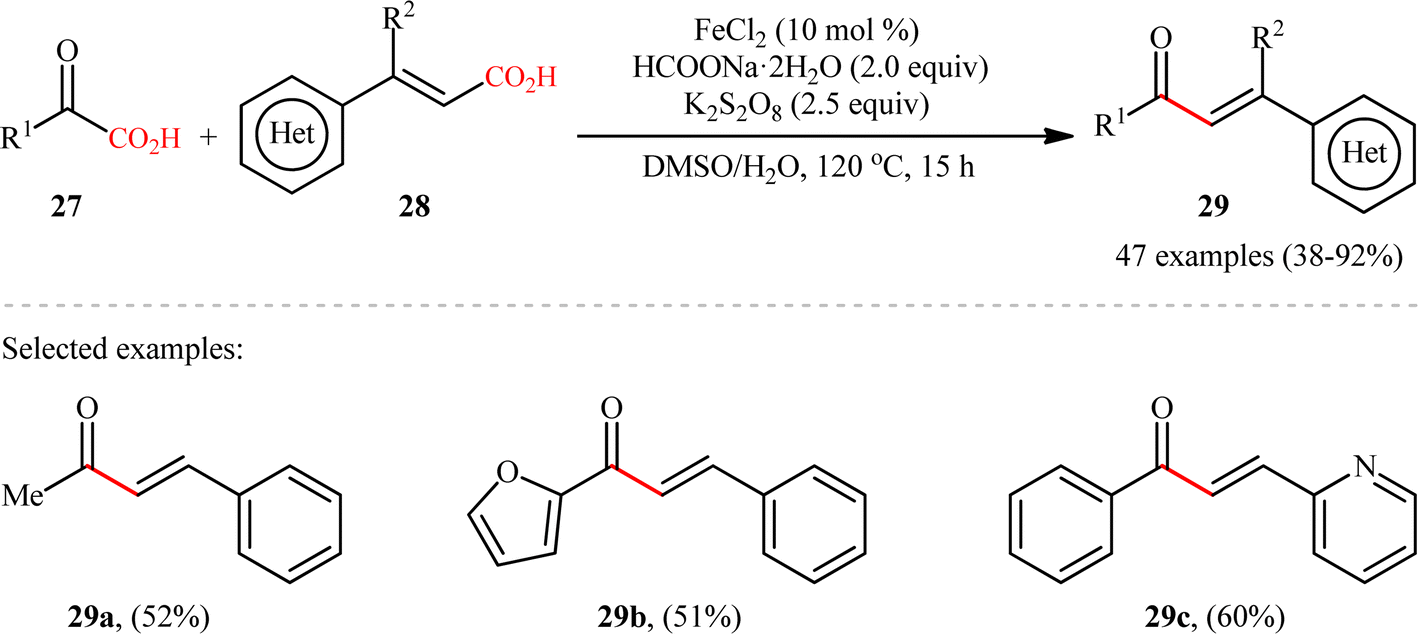
Concurrently, the Guo laboratory developed iron-catalyzed version of the above reaction.19 The reaction was carried out under nitrogen atmosphere in a H2O/DMSO mixture solvent, using 10 mol% inexpensive FeCl2 as the catalyst, 2 equiv. of sodium formate as the base, 2.5 equiv. of potassium persulfate as the oxidant at 120 °C. Under the optimized conditions, decarboxylative coupling of various 2-oxoacetic acid derivatives 27 (including aryl, heterocyclic, aliphatic, amino 2-oxoacetic acids) and cinnamic acids 28 with either electron-donating or -withdrawing groups at the para, meta or ortho position of the phenyl ring, afforded the corresponding α,β-unsaturated carbonyl products 29 in moderate to high yields (Scheme 13). Besides cinnamic acids, conjugated dienoic acids also could be applied as suitable substrates under the identical conditions. The selectivity of this C–C coupling was demonstrated in the reaction using 1,4-phenylenediacrylic acid that afforded exclusively the monoacylated product without any diacylated derivative. Unfortunately, β-alkyl acrylic acid such as crotonic acid was not amenable to this procedure. Noteworthy, the scalability of the reaction was demonstrated through the synthesis of chalcone (1,3-diphenyl-2-propene-1-one) in 1.29 g scale in good yield of 62%. Generally, compared to the Ag-catalyzed variant, this Fe-catalyzed alternative showed the advantage of a broader substrate scope and a higher average yield.
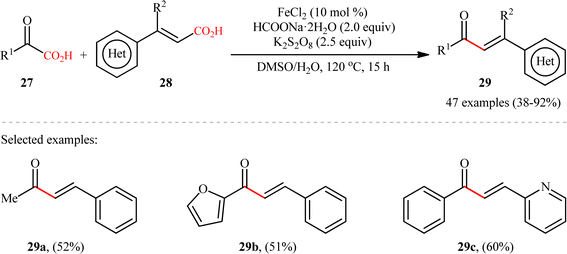 |
| | Scheme 13 Fe-catalyzed double decarboxylative cross-coupling between 2-oxoacetic acids 27 and cinnamic acid derivatives 28.19 | |
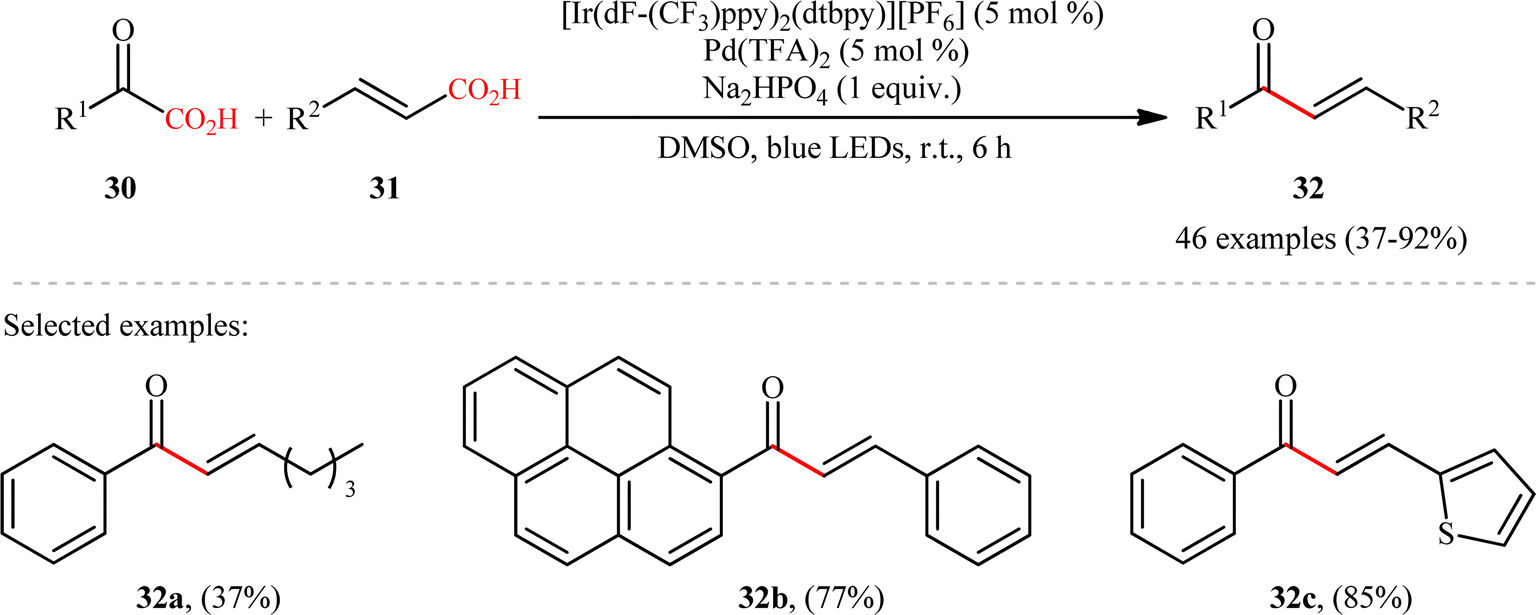
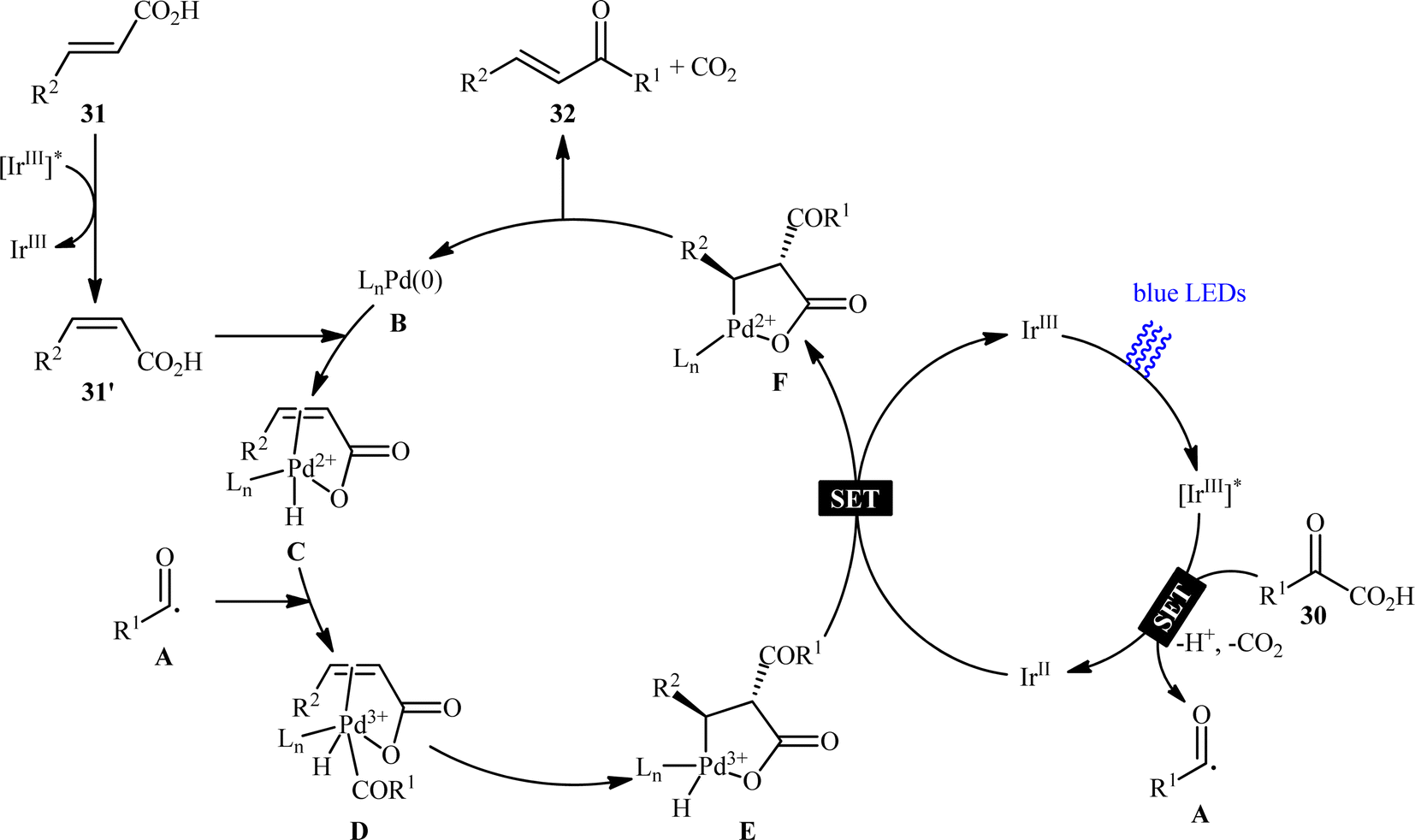
Pursuing in this direction, very recently, Ghosh's research team devised a dual catalytic system comprised of a palladium and an iridium photocatalyst, which enabled double decarboxylative coupling of a wide range of α-keto acid derivatives 30 (including mono and polycyclic aromatic, heteroaromatic, cyclic and open chain aliphatic α-keto acids) with various β-alkyl/aryl/heteroaryl α,β-unsaturated acids 31 to form the corresponding α,β-unsaturated ketones 32 in modest to excellent yields with outstanding (E)-selectivity (Scheme 14).20 The catalytic reaction was found to tolerate the presence of a diverse range of important functional groups (e.g., OMe, OCOMe, OCF3, OH, F, Cl, Br, CF3, CN, NO2, SO2Me) on both partners, thus expanding on the scope of previous reports in this field. Beside broad substrate scope and excellent stereoselectivity, beside good yields, scale ability and mild reaction conditions can be considered as the advantages of this Ir/Pd-catalyzed doubly decarboxylative cross-coupling protocol. According to the authors, the reaction proceeds through two intertwined catalytic cycles as shown in Scheme 15. At first, the ground state photocatalyst IrIII undergoes photoexcitation under visible-light irradiation to produce the excited state [IrIII]*, which oxidizes α-keto acid 30 by a SET process and promote decarboxilative acyl radical A formation. Meanwhile, photoexcited [IrIII]* species converts trans-α,β-unsaturated acid 31 into its cis-form 31′ through energy transformation. Subsequently, coordination of cis-α,β-unsaturated acid 31 to the in situ generated palladium(0) active catalyst B provides PdII-intermediate C. Afterwards, acyl radical A addition to cinnamate coordinated PdII-intermediate C experiences one electron oxidation of palladium species and forms intermediate D. Next, β-migratory insertion of acyl group to the coordinated double bond affords a five member pallado-cycle E, which after taking up one electron from IrII via SET provides PdII-intermediate F. Finally, concerted decarboxylative demetallation of intermediate F affords the target (E)-α,β-unsaturated ketone product 32 and regenerate the active palladium species B.
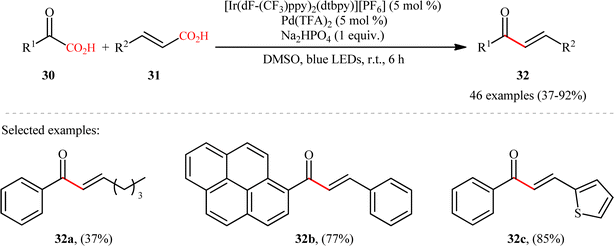 |
| | Scheme 14 Ir/Pd-catalyzed double decarboxylative cross-coupling of α-keto acid derivatives 1 with α,β-unsaturated acids 2.20 | |
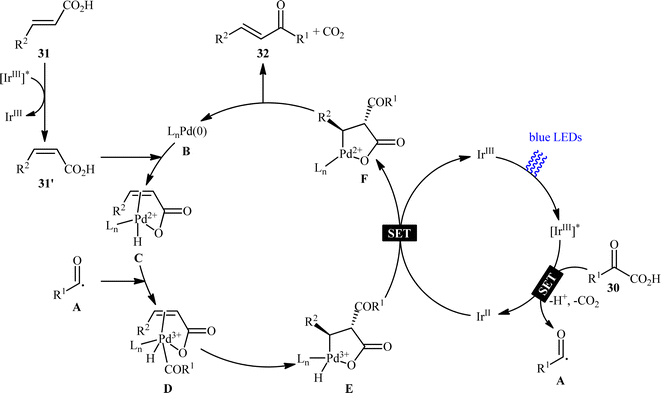 |
| | Scheme 15 Proposed mechanism for the reaction in Scheme 14. | |
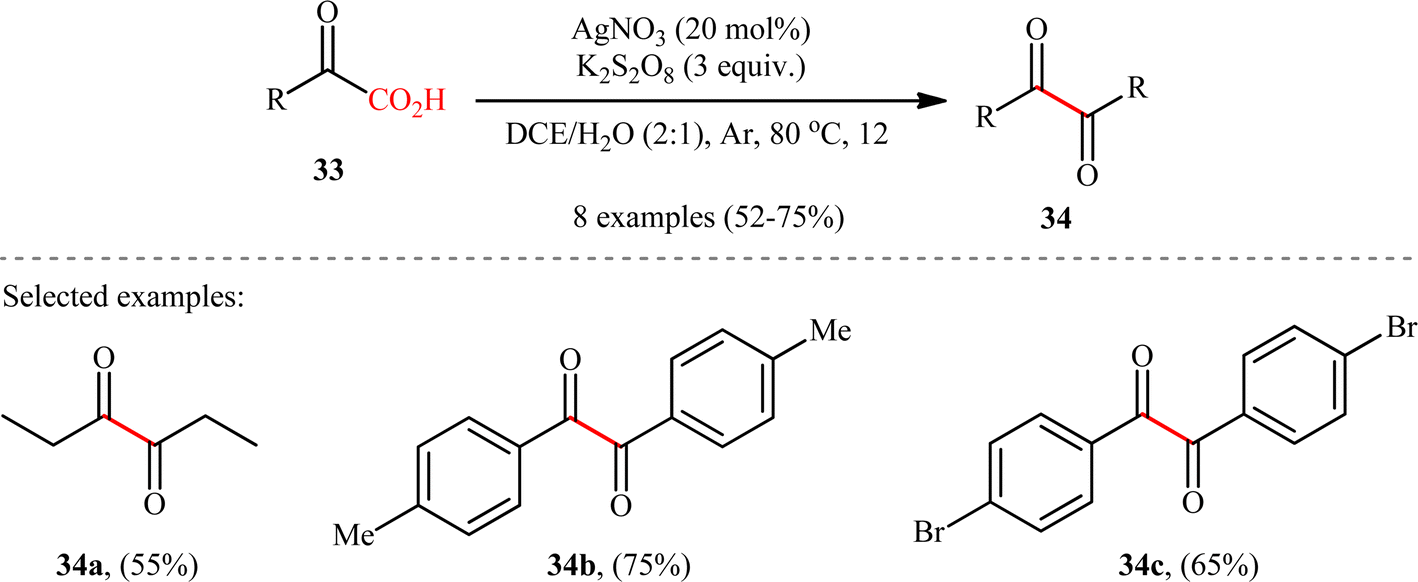
5.3. 1,2-Diketones synthesis
In 2018, in the same paper describing construction of C(sp3)–C(sp3) bonds through the double decarboxylative coupling of aliphatic carboxylic acids,8 Li–Xiang's research group unraveled the preparation of a library of 1,2-diketones 34 in moderate to good yields via the oxidative decarboxylative homocoupling of respective α-keto acids 33 under their standard conditions. As shown in Scheme 16, various aliphatic, aromatic, and heteroaromatic α-keto acids were utilized to establish the general applicability of the protocol. It is worthwhile to mention that the judicious selection of catalyst and oxidant was crucial for the reaction to proceed successfully, otherwise poor yields or no desired product was obtained at all. Next to 1,2-diketones, anhydrides could be also applied in this decarboxylative coupling reaction. Thus, under the similar conditions and just by adding a few drops of H2SO4, an array of symmetrical and unsymmetrical anhydrides underwent decarboxylative coupling to give corresponding 1,2-diketones in synthetically useful yields. Mechanistically, the reaction proceeds along a similar pathway to that described in Scheme 2. To the best our knowledge, this is the only reported example on the direct synthesis of 1,2-diketones from the corresponding α-keto acids.
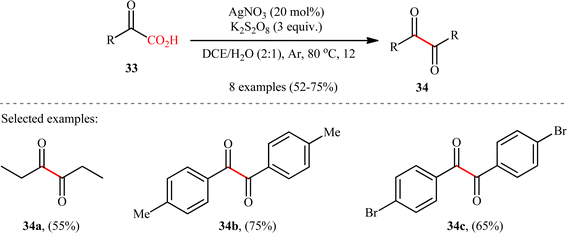 |
| | Scheme 16 Li–Xiang's synthesis of 1,2-diketones 34.8 | |
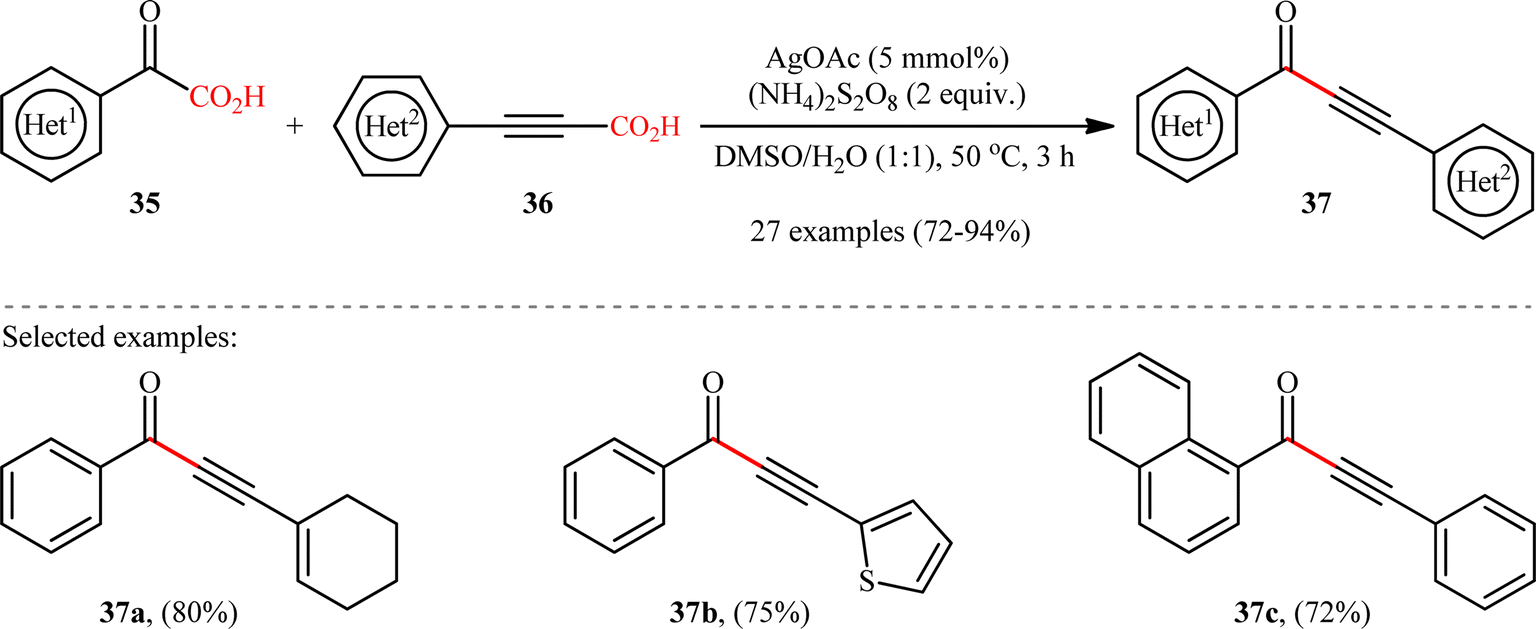
6. C(sp2)–C(sp) bond formation
In 2018, Cheng and co-workers reported an elegant study on the preparation of synthetically and biologically important aryl ynones 37 by the Ag(I)-catalyzed double decarboxylative alkynylation of (hetero)aromatic α-keto acids 35 with (hetero)arylpropiolic acids 36.21 As illustrated in Scheme 17, the reaction proceeded efficiently in aqueous medium (DMSO/H2O 1:1) under relatively mild conditions and provided the expected ynones 37 in high yields (up to 94%) within 3 h. Concerning the substrate scope, the reaction was almost independent of the steric- and electronic-factors of both partners. Therefore, either electron-donating or electron-withdrawing functionalities on both reaction components were well tolerated by this protocol. The strategy was compatible with alkenyl propiolic acid derivatives, as exemplified by high yielding synthesis of 3-(cyclohexen-1-yl)-1-phenylprop-2-yn-1-one from 2-oxo-2-phenylacetic acid and 3-(cyclohexen-1-yl)propionic acid. However, the protocol was unfruitful with both aliphatic α-keto acids and alkyl propiolic acids. Based on some control experiments, the authors have proposed that this C(sp2)–C(sp) bond forming reaction may proceed through a radical addition/decarboxylation sequence. Intriguingly, this methodology was also brilliantly adapted to the synthesis of a panel of nineteen 2-phenyl-4H-chromen-4-one derivatives through the decarboxylative annulation reaction between 2-hydroxyphenyl glyoxylic acids and arylpropiolic acids.
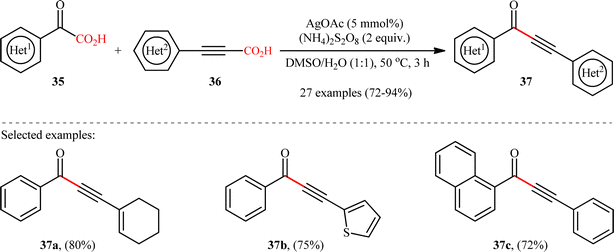 |
| | Scheme 17 Cheng's synthesis of ynones 37.21 | |
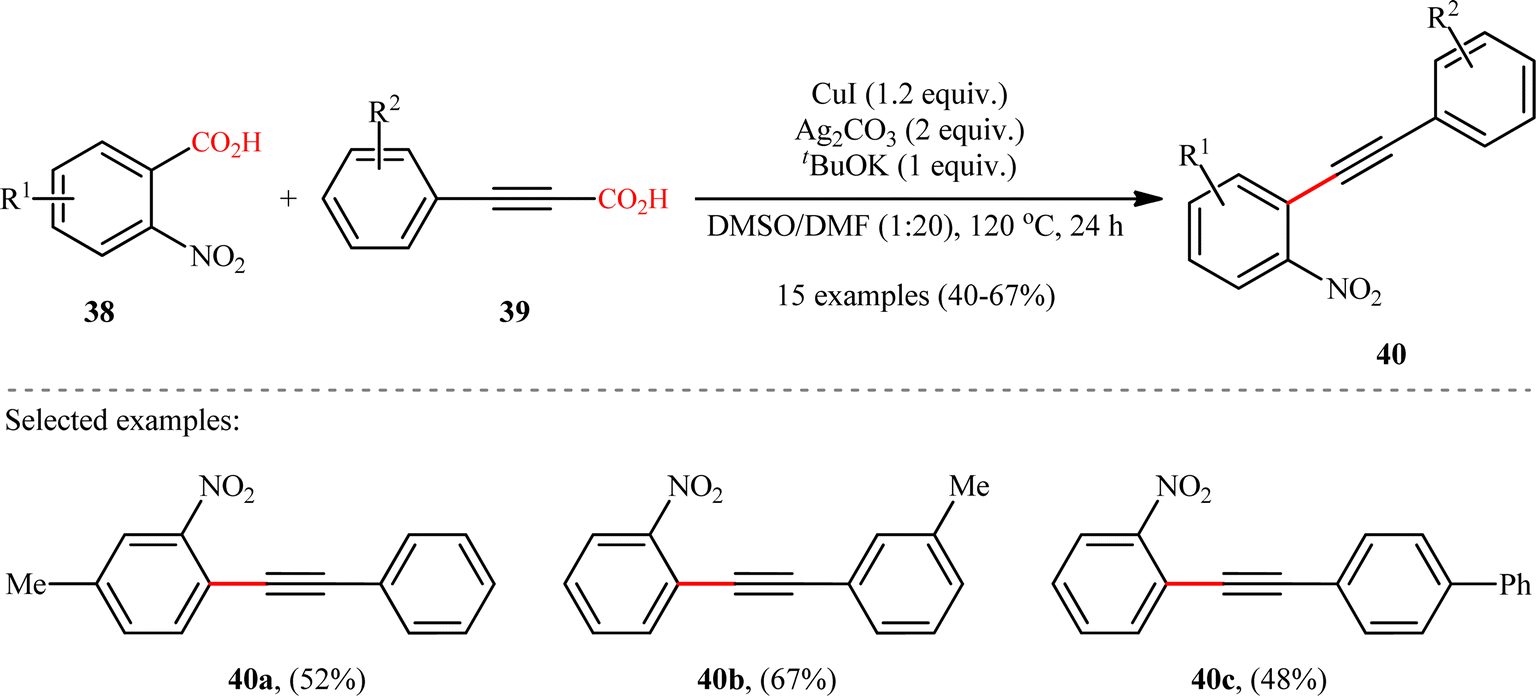
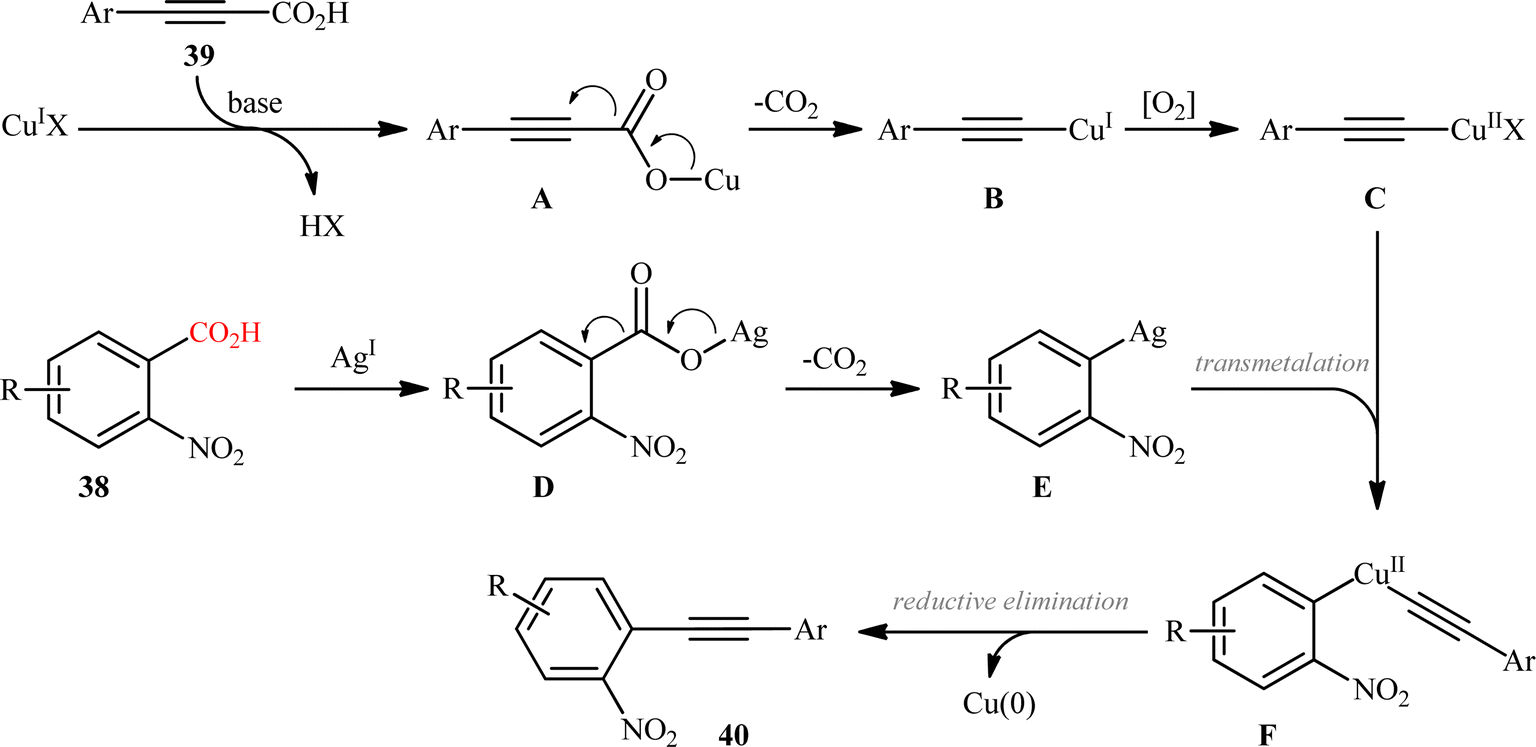
In the same year, Jana and colleagues reported a conceptually analogous C(sp2)–C(sp) bond forming reaction using a silver(I)/copper(I) bimetallic system.22 Thus, a variety of 1-aryl-2-(2-nitrophenyl)acetylenes 40 were synthesized in moderate yields via Ag2CO3/CuI-promoted double decarboxylative cross-coupling between o-nitrobenzoic acid derivatives 38 and arylpropynoic acids 39 (Scheme 18). It is worthwhile to note that presence of nitro group at ortho position of arene carboxylic acids was crucial for the success of this reaction. Therefore, other benzoic acids such as p-nitrobenzoic acid, o-methoxybenzoic acid, pentafluorobenzoic acid, and heteroaryl carboxylic acids were proven not to be adaptable substrates for this reaction. Notably, ortho nitrobenzoic acids with another electron-deficient substituent (e.g., 2,4-dinitro-benzoic acid) resulted in decarboxylative protonation product only. Besides these general limitations, inactivity of alkylated propiolic acids and formation of significant amounts (40–50%) of 1,4-diarylsubstituted 1,3-diyne by-products through homocoupling of arylpropynoic acids were other disadvantages reported by the authors for their methodology. The proposed mechanism for this decarboxylative alkynylation is outlined in Scheme 19. Transmetalation between arylethynylcopper complex C (generated via decarboxylation of arylpropynoic acid 39) and arylsilver complex E (generated via the decarboxylation of o-nitrobenzoic acid 38) affords an aryl(arylethynyl)copper complex F, which after reductive elimination delivers the observed product 40.
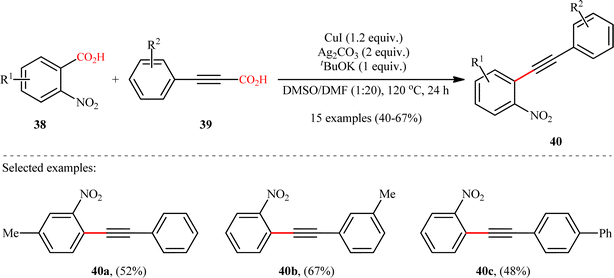 |
| | Scheme 18 Ag/Cu-promoted decarboxylative alkynylation of 2-nitrobenzoic acids 38 with arylpropynoic acids 39.22 | |
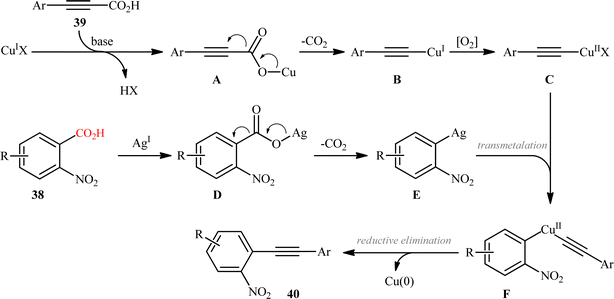 |
| | Scheme 19 Proposed mechanism for the formation of 1-aryl-2-(2-nitrophenyl)acetylenes 40. | |
7. C(sp)–C(sp) bond formation
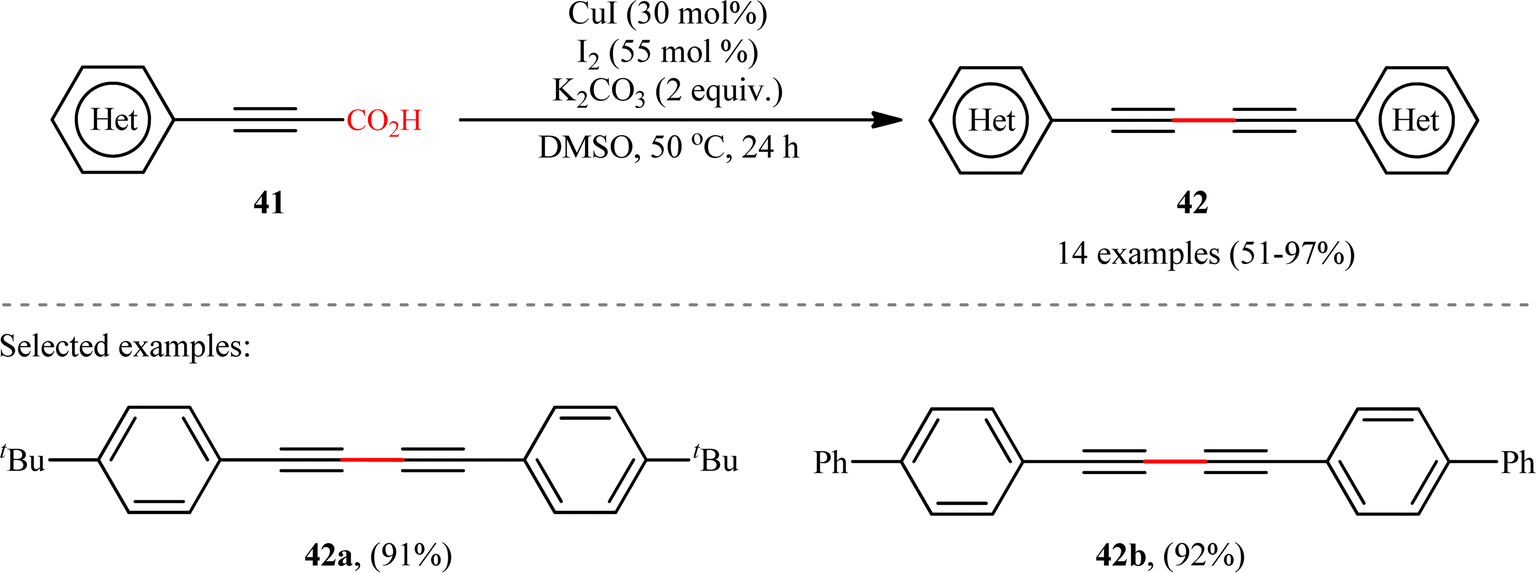
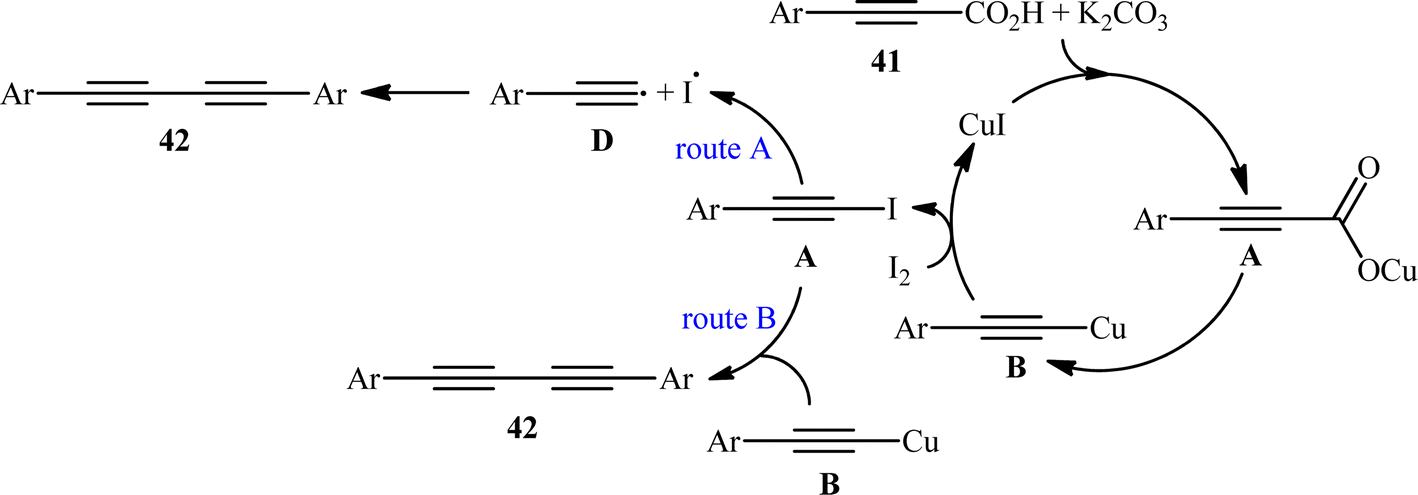
The idea to synthesis of 1,3-diynes through decarboxylative homocoupling of corresponding propiolic acids was exploited in 2014 by Lang and co-workers.23 By employing 3-phenylpropiolic acid as the model substrate, quite some catalysts, oxidants, bases, and solvents were screened, choosing eventually the CuI/I2/K2CO3 combination as the most suitable catalytic system and DMSO as the optimal solvent. Under the optimized conditions, fourteen (hetero)aryl propiolic acids 41 underwent decarboxylative homocoupling and gave the expected symmetrical 1,4-di(hetero)arylsubstituted 1,3-diynes 42 in moderate to almost quantitative yields (Scheme 20). However, no examples of the coupling of alkyl propynoic acids were given in this study. In order to expand the scope of their methodology, the authors applied their catalytic system to the cross-coupling reaction of a series of aryl propiolic acids with 4-ethynylanisole in order to synthesize of unsymmetric 1,3-diynes. Although in all examples a mixture of three possible products were obtained, the reaction showed good selectivity toward cross-coupling versus homocoupling. The results showed that the presence of an electron-donating group on the phenyl ring of aryl propiolic acids enhanced the ratios of the cross-coupling/homocoupling products. The plausible mechanism for this C(sp)–C(sp) bond forming reaction is shown in Scheme 20 and starts with the formation of the carboxylate intermediate A via the reaction of aryl propiolic acid 41 with copper catalyst in the presence of K2CO3, which is followed by its decarboxylation to furnish the Cu(I)-acetylide intermediate B. Subsequently, the oxidative reaction of this intermediate with molecular iodine affords the alkynyliodide intermediate C and regenerates CuI. Finally, intermediate C undergoes decomposition to produce alkyne radical D, which after homocoupling forms the observed 1,3-diyne 2 (Scheme 21, path a). In another possibility, the alkynyliodide intermediate C may react with Cu(I)-acetylide intermediate B to give the target product 42 (Scheme 21, path b).
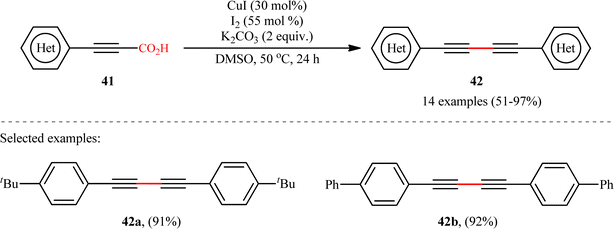 |
| | Scheme 20 Cu-catalyzed oxidative decarboxylative homocoupling of aryl propiolic acids 41, developed by Lang.23 | |
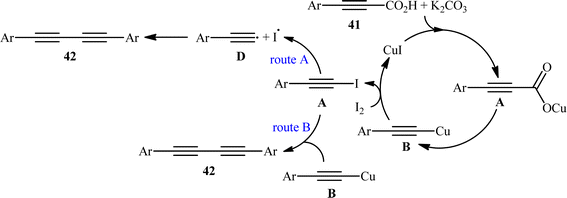 |
| | Scheme 21 Mechanism proposed to explain the 1,4-diarylsubstituted 1,3-diynes 42 synthesis. | |
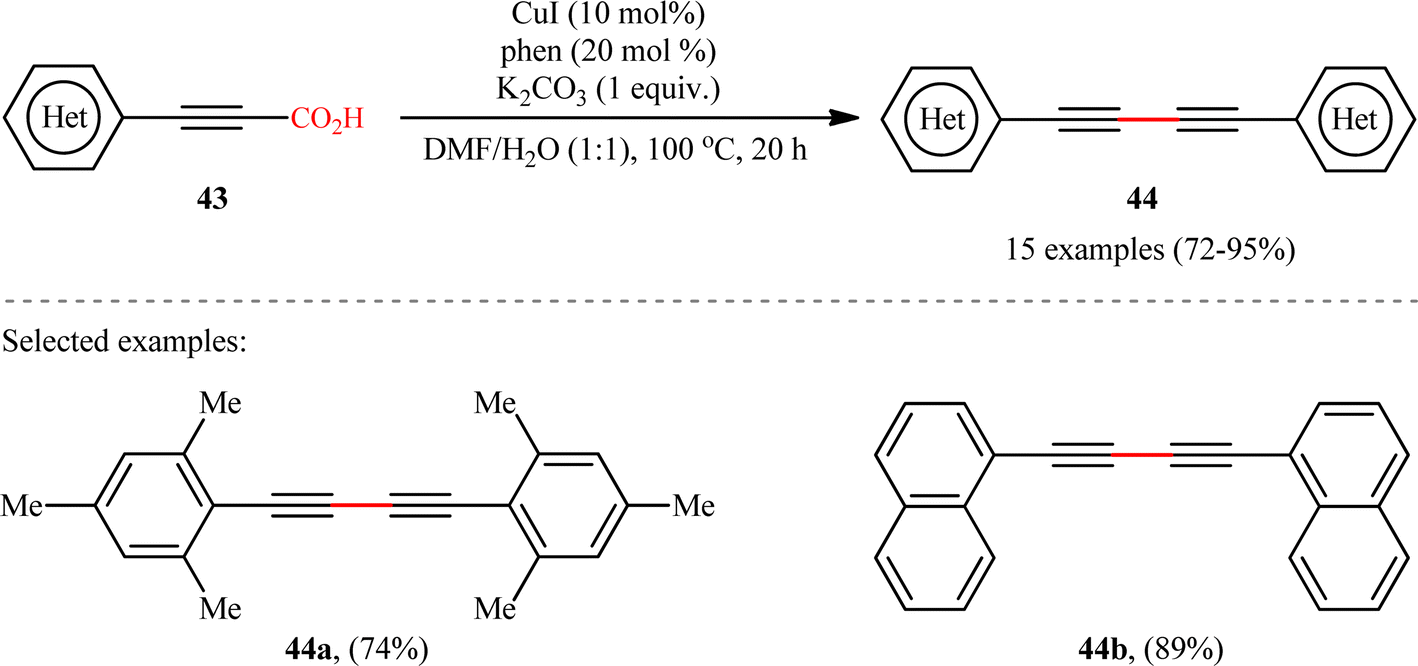
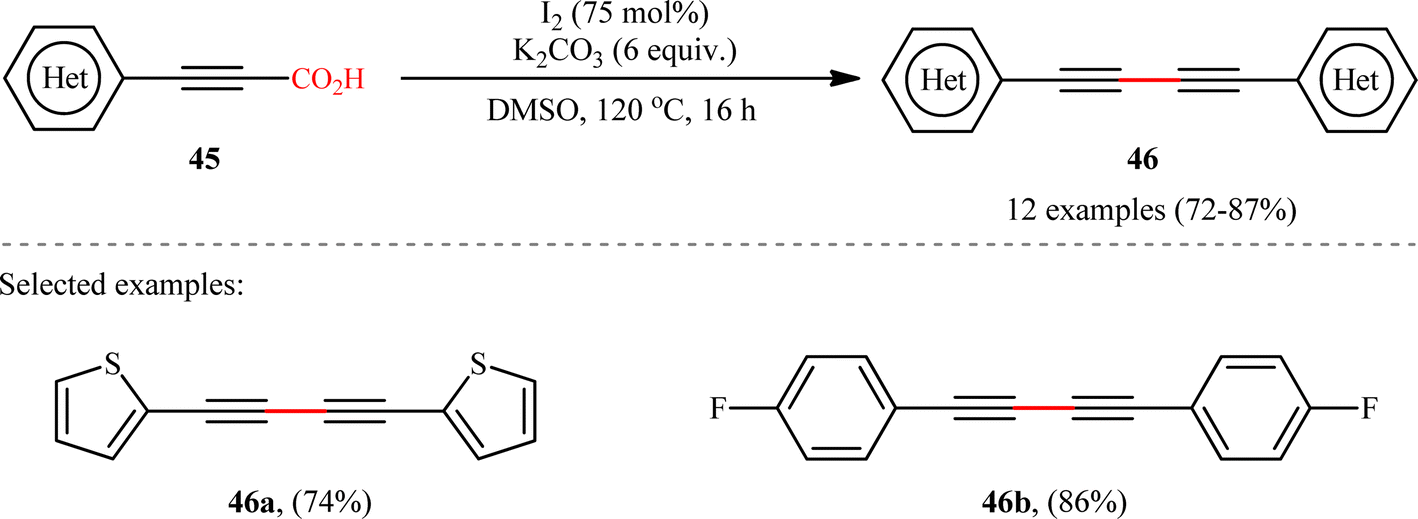
In another study, the same research group reported that a lower loading of copper catalyst (10 mol%) could be employed for similar purposes when combined with 1,10-phenanthroline (phen) ligand (Scheme 22), resulting in a protocol that significantly improved yields for sterically hindered substrates when compared to their previous work (74% versus 51% yield for 2,4,6-trimethyl-substituted substrate).24 Along this line, Ghosh and Chattopadhyay described that a similar transformation can be effected with molecular iodine in the presence of K2CO3, thus avoiding the use of metal catalysts and ligands.25 Thus, a panel of 12 (hetero)aryl propiolic acids 45 reacted well under the standard conditions [I2 (0.75 equiv.), K2CO3 (6 equiv.), DMSO, 120 °C, 16 h] to produce the corresponding symmetrical 1,4-di(hetero)arylsubstituted 1,3-diynes 46 in high yields (Scheme 23). After a series of mechanistic investigations, it was confirmed that this reaction most likely proceeds through a radical pathway with a 1-iodoalkyne intermediate.
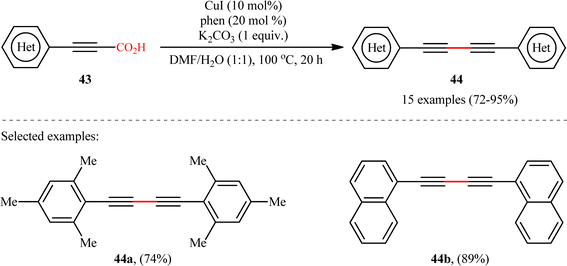 |
| | Scheme 22 CuI/phen-catalyzed decarboxylative homocoupling of (hetero)aryl propiolic acids 43.24 | |
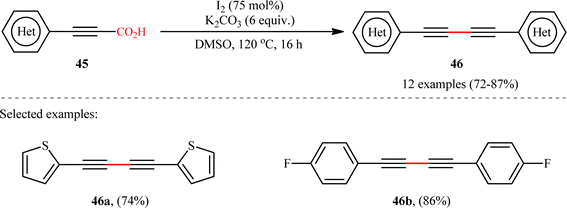 |
| | Scheme 23 Ghosh–Chattopadhyay's synthesis of 1,4-di(hetero)arylsubstituted 1,3-diynes 45.25 | |
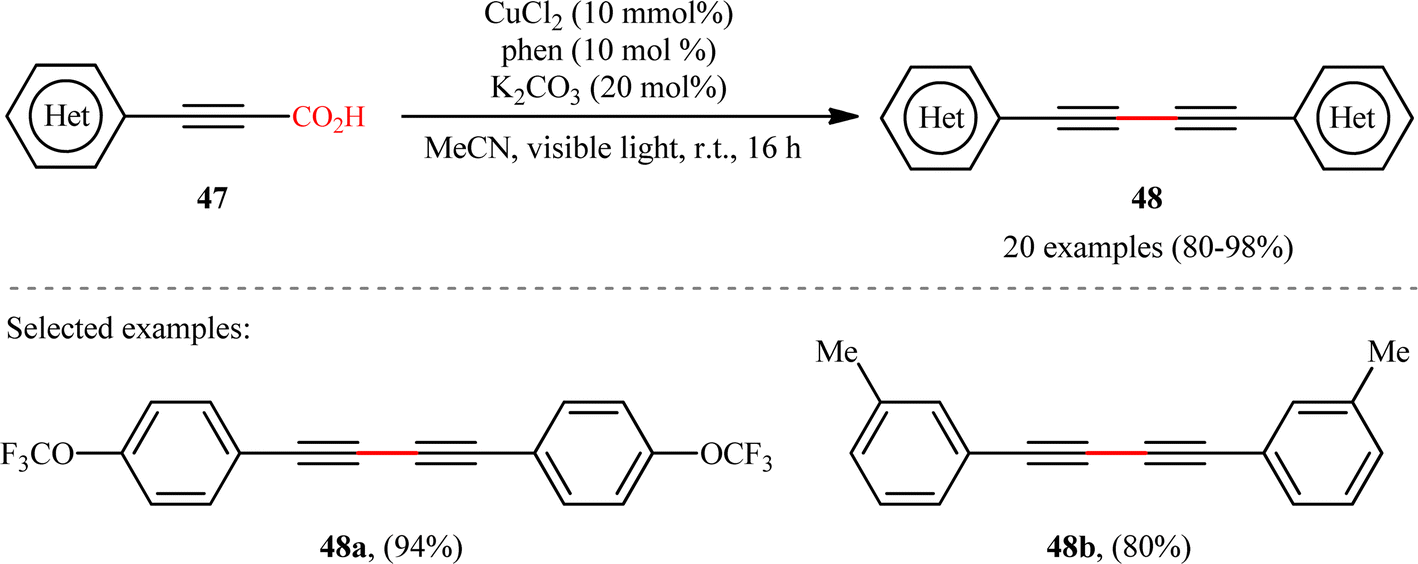
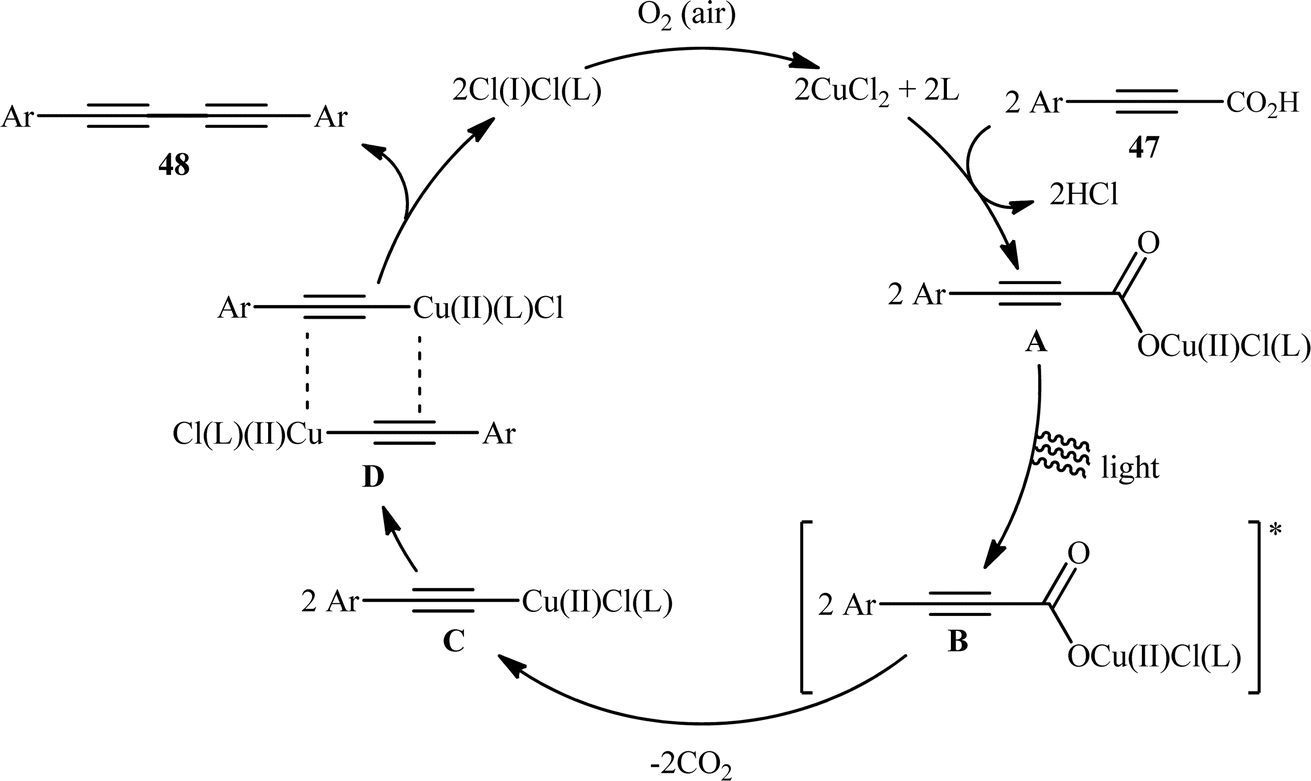
With the objective of designing a milder procedure to 1,3-diynes through the oxidative decarboxylative homocoupling of propiolic acids, very recently Pathania and Roy were able to demonstrate that a library of symmetrical 1,4-di(hetero)arylsubstituted 1,3-diynes 48 could be obtained in high to excellent yields (up to 98%) from the respective (hetero)aryl propiolic acids 47 using CuCl2/phen/K2CO3 combination as a catalytic system under the irradiation of visible light at room temperature (Scheme 24).26 Notably, the presence of catalyst, ligand or light was crucial for the success of this transformation. The reaction was completely shut down in the absence of any of them. Unfortunately, alkyl-substituted propiolic acids were incompatible in this protocol. It is noteworthy that when the cross-over experiment was performed using 3-phenylpropiolic acid and 3-(p-tolyl)propiolic acid, the mixture of three possible products were obtained in the ratio 1:1:1, indicating that cupric acetylides may have participated in an intermolecular fashion to produce the observed 1,3-diyne products. The plausible mechanism for this homocoupling reaction as proposed by the authors is depicted in Scheme 25.
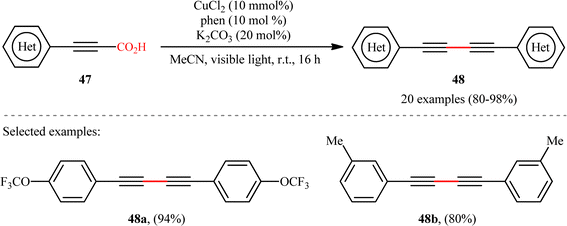 |
| | Scheme 24 Photoinduced Cu-catalyzed decarboxylative homocoupling of (hetero)aryl propiolic acids 47.26 | |
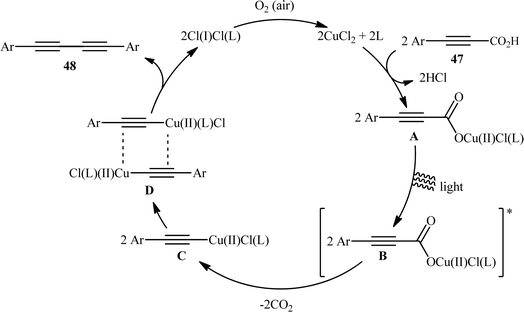 |
| | Scheme 25 Plausible mechanism for the formation of 1,4-di(hetero)arylsubstituted 1,3-diynes 48. | |
8. Conclusion
As shown in this review, over the last fourteen years (2010–2023), several interesting double decarboxylative homo- and cross-coupling reactions of carboxylic acids were developed that allow the direct and efficient creation of various C–C bonds [i.e., C(sp3)–C(sp3), C(sp3)–C(sp2), C(sp3)–C(sp), C(sp2)–C(sp2), C(sp2)–C(sp), and C(sp)–C(sp)], without the requirement for any pre-functionalization of the starting materials. The major advantage of this new page of C–C bond formation is the low-cost, non-toxic, shelf-stable, and readily available starting materials along with the generation of non-toxic and easily removable CO2 as the only waste by-product. Nevertheless, there are still many challenges to be addressed in this methodology. Some of these are listed below: (i) most of the reported examples on this chemistry were performed at elevated temperatures; thus, the exploration and discovery of novel catalytic systems, which can allow these reactions under milder conditions would be highly desirable; (ii) the majority of reactions covered in this review are limited to the use of silver catalysts. Therefore, the exploration of naturally abundant and inexpensive first row transition metal catalysts on this chemistry will be highly desirable from economy and sustainable chemistry point of view; (iii) as illustrated, almost all reported examples on the titled reactions most likely proceeds through a radical pathway. Considering the large number of advantages of electrochemically driven radical transformations, the investigation and development of efficient double decarboxylative couplings under electrochemical conditions is highly desirable; (iv) in general, cross-coupling reactions between two similar types of carboxylic acids (e.g., two benzoic acids) suffer from formation of unwanted homocoupling side products. Therefore, more efforts are expected to develop highly chemoselective protocols towards cross-coupling over homo-coupling reactions; and (v) the reported examples of the formation of some C–C bond types (e.g., sp3–sp3 and sp–sp) are generally limited to homocoupling reactions with no successful report on cross-coupling reactions.
Conflicts of interest
There are no conflicts to declare.
References
- Selected reviews:
(a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC;
(b) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed;
(c) T. Zhang, N. X. Wang and Y. Xing, J. Org. Chem., 2018, 83, 7559–7565 CrossRef CAS PubMed;
(d) G. J. Perry and I. Larrosa, Eur. J. Org. Chem., 2017, 3517–3527 CrossRef CAS PubMed;
(e) N. A. A. Salman, A. H. Adhab, H. Bahir, M. H. Sami and R. sadeghzadeh, Chem. Rev. Lett., 2023, 6, 139–149 CAS;
(f) H. Tariq, T. A. Qassem, H. Bashir, F. Behmagham and F. Shahimi, Chem. Rev. Lett., 2023, 6, 235–244 Search PubMed;
(g) N. A. A. Salman, A. H. Adhab, H. Bahir, M. H. Sami and R. sadeghzadeh, Chem. Rev. Lett., 2023, 6, 139–149 CAS;
(h) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8 CAS;
(i) S. Majedi, S. Majedi and F. Behmagham, Chem. Rev. Lett., 2019, 2, 187–192 Search PubMed.
- Selected reviews:
(a) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(b) R. Sharma and M. R. Yadav, Org. Biomol. Chem., 2021, 19, 5476–5500 RSC.
- Selected reviews:
(a) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS;
(b) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed;
(c) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC;
(d) C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed. Engl., 2012, 51, 5062–5085 CrossRef CAS PubMed;
(e) M. R. J. Sarvestani, N. Mert and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed.
- Selected reviews:
(a) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(b) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(c) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(d) J. Li, L. Li and E. Vessally, J. Chin. Chem. Soc., 2021, 68, 13–26 CrossRef CAS.
- Selected reviews:
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed;
(b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed;
(c) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef PubMed;
(d) I. Söğütlü, E. A. Mahmood, S. A. Shendy, S. Ebrahimiasl and E. Vessally, RSC Adv., 2021, 11, 2112–2125 RSC;
(e) S. K. Saraswat, R. Seemaladinne, H. Zaini, N. Ahmad, N. Ahmad and E. Vessally, RSC Adv., 2023, 13, 13642–13654 RSC.
-
(a) Y. Peng, R. Isshiki, K. Muto and J. Yamaguchi, Chem. Lett., 2022, 51, 749–753 CrossRef CAS;
(b) K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719–10722 RSC;
(c) J. K. Li, B. Zhou, Y. C. Tian, C. Jia, X. S. Xue, F. G. Zhang and J. A. Ma, Org. Lett., 2020, 22, 9585–9590 CrossRef CAS PubMed;
(d) P. Das, S. Gondo, P. Nagender, H. Uno, E. Tokunaga and N. Shibata, Chem. Sci., 2018, 9, 3276–3281 RSC;
(e) R. A. Daley and J. J. Topczewski, Org. Biomol. Chem., 2019, 17, 1709–1713 RSC;
(f) T. Maji and J. A. Tunge, Org. Lett., 2015, 17, 4766–4769 CrossRef CAS PubMed.
- J. Choi, J. Lim, F. M. Irudayanathan, H. S. Kim, J. Park, S. B. Yu, Y. Jang, G. C. E. Raja, K. C. Nam, J. Kim and S. Lee, Asian J. Org. Chem., 2016, 5, 770–777 CrossRef CAS.
- H. X. Zou, Y. Li, Y. Yang, J. H. Li and J. Xiang, Adv. Synth. Catal., 2018, 360, 1439–1443 CrossRef CAS.
- Y. Wang, H. Zhao, X. Xie, H. Jiang, H. Deng, J. Hao and W. Wan, Synth. Commun., 2019, 49, 2961–2970 CAS.
- W. P. Mai, G. Song, G. C. Sun, L. R. Yang, J. W. Yuan, Y. M. Xiao, P. Mao and L. B. Qu, RSC Adv., 2013, 3, 19264–19267 RSC.
- J. Cornella, H. Lahlali and I. Larrosa, Chem. Commun., 2010, 46, 8276–8278 RSC.
- Z. Fu, Z. Li, Q. Xiong and H. Cai, RSC Adv., 2015, 5, 52101–52104 RSC.
- K. Xie, S. Wang, Z. Yang, J. Liu, A. Wang, X. Li, Z. Tan, C. C. Guo and W. Deng, Eur. J. Org. Chem., 2011, 5787–5790 CrossRef CAS.
- Á. L. Mudarra, S. M. de Salinas and M. H. Pérez-Temprano, Org. Biomol. Chem., 2019, 17, 1655–1667 RSC.
- P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed. Engl., 2012, 51, 5945–5949 CrossRef CAS PubMed.
- N. Rameau, S. Cadot, A. Paquet, C. Pinel and L. Djakovitch, Top. Catal., 2014, 57, 1430–1437 CrossRef CAS.
- F. Pu, L. Y. Zhang, Z. W. Liu and Y. Y. Shi, Adv. Synth. Catal., 2018, 360, 2644–2649 CrossRef CAS.
- N. Zhang, D. Yang, W. Wei, L. Yuan, F. Nie, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 3258–3263 CrossRef CAS PubMed.
- Q. Jiang, J. Jia, B. Xu, A. Zhao and C. C. Guo, J. Org. Chem., 2015, 80, 3586–3596 CrossRef CAS PubMed.
- S. Mondal, S. P. Midya, S. Mondal, S. Das and P. Ghosh, Chem.–Eur. J., 2023, e202303337 Search PubMed.
- M. Meng, G. Wang, L. Yang, K. Cheng and C. Qi, Adv. Synth. Catal., 2018, 360, 1218–1231 CrossRef CAS.
- A. Hossian, K. Manna, P. Das and R. Jana, ChemistrySelect, 2018, 3, 4315–4318 CrossRef CAS.
- D. X. Liu, F. L. Li, H. X. Li, J. Gao and J. P. Lang, Tetrahedron, 2014, 70, 2416–2421 CrossRef CAS.
- D. X. Liu, F. L. Li, H. X. Li, W. J. Gong, J. Gao and J. P. Lang, Eur. J. Org. Chem., 2014, 4817–4822 CrossRef CAS.
- S. Ghosh and S. K. Chattopadhyay, Tetrahedron Lett., 2022, 102, 153908 CrossRef CAS.
- V. Pathania and S. R. Roy, Asian J. Org. Chem., 2023, 12, e202300300 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence i
i