
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bio-sourced liquid resins and their photopolymerization with poly(ethylene glycol) diacrylate in the roadmap to more sustainable digital light processing technologies†
Ľudmila
Hodásová‡
ab,
Isaac
Isarn‡
c,
Fernando
Bravo
*c,
Carlos
Alemán
 abd,
Núria
Borràs
ab,
Gemma
Fargas
e and
Elaine
Armelin
*ab
abd,
Núria
Borràs
ab,
Gemma
Fargas
e and
Elaine
Armelin
*ab
aIMEM-BRT Group, Departament d'Enginyeria Química, EEBE, Universitat Politècnica de Catalunya (UPC), C/d'Eduard Maristany, 10-14, Edifici I, 08019, Barcelona, Spain. E-mail: elaine.armelin@upc.edu
bBarcelona Research Center for Multiscale Science and Engineering, Universitat Politècnica de Catalunya (UPC), C/d'Eduard Maristany, 10-14, Edifici IS, 08019, Barcelona, Spain
cInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: fbravo@iciq.cat
dInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, 08028, Barcelona, Spain
eCIEFMA Group, Departament de Ciencia i Enginyeria dels Materials, EEBE, Universitat Politècnica de Catalunya (UPC), C/d'Eduard Maristany, 10-14, Edifici I, 08019, Barcelona, Spain
First published on 26th January 2024
Abstract
Non-fossil feedstocks for the production of photocurable resins have attracted growing interest from the scientific community and industry in order to achieve more sustainable 3D-printing technologies. Herein, we report the successful photopolymerization process of three diallyl ester monomers, derived from succinic acid, D,L-malic acid and L-(+)-tartaric acid (natural acids), with poly(ethylene glycol) diacrylate, a petroleum-based co-monomer well-known for its fast UV light reaction response. The existence of hydroxyl groups beside the ester units in the malic and tartaric compounds did not influence either the kinetics or the thermal stability of the thermoset polymers. Therefore, the most prominent composition was formed by 50 wt% of the bio-derived diallyl succinate, and 50 wt% of the synthetic, having excellent thermal stability and very good dimensional resolution and transparency in DLP printed samples after light curing, and most importantly, such samples promptly undergo hydrolytic degradation thanks to the presence of the ester linkages that are incorporated by the natural monomer.
1. Introduction
Plastics that are both bio-based and biodegradable, such as poly(lactides) (PLA, PLLA, PGLA)1 and poly(hydroxyalkanoates) (PHAs),2 or even bioplastic blends,3 as is the case of modified naturally occurring carbohydrate-based polymers4 (starch, cellulose, and others), have given way to an explosion of products (plastic cutlery, bottles, bags, plant pots, food packaging, etc.) made with these macromolecules.5–9 In this way, polymers from bio-based sources are in the crosshairs of companies to make their way in an increasingly competitive market of sustainable plastics.10–13There are many commercially available plastics synthesized from bio-based sources, nylon 11 being one example. This polymer can be synthesized from monomers produced from vegetable oils, particularly from castor oil, and fatty acids.14,15 In another example, the DuPont Company has developed a polyester (poly(trimethylene terephthalate)) from renewable resources.16 The polymer is particularly employed in the production of fibres and it is obtained from the reaction between terephthalic acid (synthetically-based) and 1,3-propanediol (PDO) (bio-sourced). There are certain microorganisms able to ferment the sugars produced from corn, to convert them into useful molecules for plastic manufacturing, as in the case of PDO, which can be isolated from bacteria fermentation reactions. However, in such reactions, one of the biggest disadvantages is the purification process, which limits the production, giving a low efficiency and resulting in high production costs.
Therefore, the need for innovation in sustainable polymers sets the focus in the chemical supply chain. The difficulty lies in finding appropriate raw materials necessary to achieve good polymerization reactions (fast kinetics, high molecular weight, good crosslinking, etc.), mechanically stable end-pieces (good processability) and recyclability. The abovementioned examples are bio-based thermoplastics useful for extrusion, injection, fused deposition modelling (FDM) and other processing technologies, but not for photopolymerization.
In digital light processing (DLP), photocurable liquid resins are required.17–19 Unlike stereolithography (SLA), which employs an optical scanning module to focalize a laser to light up a single image, DLP uses ultraviolet (UV) light (and visible light) to project a mask of a completely cross-sectional layer at a time to obtain the final prototype.20 In such technologies, at least five key factors should be taken into account: (i) the light wavelength; (ii) the power of the light; (iii) an open and transparent vat; (iv) the irradiation time per layer; and (v) the necessity of liquid resins that are typically designed for fast reaction at a specific wavelength. Additionally, in order to have a fast curing (or material solidification), a photoinitiator is generally required for the radical polymerization, as well as difunctional (or polyfunctional) oligomers and monomers with reactive double bond end groups. Ideally, at least one of these compounds should be derived from biomass to help to decrease the environmental footprint of fossil fuel raw materials.
There are several possible green resins that are of utmost relevance to achieve a positive impact in additive manufacturing.21–24 Acrylate- and epoxide-vegetable oil derivatives are among the most commonly reported resources for biobased resins. Cui et al.25 have described a series of photopolymer resins composed of urethane epoxidized soybean oil (SBO-URE) and poly(ethyleneglycol) diacrylate (PEGDA) for SLA printing. They obtained optimal strength and flexural mechanical properties for prototypes containing 20% SBO-URE, proving that it can be used as an environmental and renewable material for stereolithographic 3D printing.
Another important obstacle in plastic sustainability is the fact that bio-based polymers are not always biodegradable products.26–28 Thus, more investigations into degradability and composting properties would be profitable.29 A new class of poly(propylene fumarate) tri-block copolymer composites, developed by Becker and co-workers,30–32 is one good example of a bio-based and degradable 3D-prototyping system. In this case, the hydrolytic degradation is promoted by the presence of labile thiol crosslinker groups.
In our laboratories, we have also developed different classes of resins for DLP and SLA manufacturing with high degradability properties, promoted by the presence of ester-amide groups.33–35 An unsaturated polyesteramide (UPEA), derived from amino acid (L-phenylalanine), presented a degradation rate of 16.5% in 90 days, even though the polymerization kinetics with PEGDA were very slow due to the poor reactivity of UPEA.34 In a more recent study, an acrylamide resin, also derived from L-phenylalanine (codified as PADEG), combined with the commercially available PEGDA co-monomer, and a dimethyl acrylamide (DMAM) reactive diluent, gave rise to a photocured copolymer with an even higher hydrolytic degradation rate of 28.5% in 90 days, with non-toxic degraded compounds, as proved by cytotoxicity studies.35
Nevertheless, many of the examples described before have the disadvantages of long synthetic routes and cumbersome purification processes being required to obtain the photocurable blend. Circumventing these limitations, herein, we describe the direct synthesis (one-step) of three plant-based resins. Conveniently, these resins are obtained as liquids easily homogenized with the PEGDA co-monomer, thus avoiding solubility issues sometimes observed in similar blends. This facilitates their copolymerization by employing UV-light printing. Along with performance, the thermal, mechanical and spatial resolution of the 3D-printed dumbbell pieces were investigated. Moreover, following our previous approaches in the development of biodegradable acrylate copolymers, in the present study we compare the biodegradability of the most prominent poly(ester ether) material with others reported in the literature.
2. Experimental procedure
2.1. Materials
Succinic acid (ACS reagent, ≥99.0%), D,L-malic acid (ACS reagent, >99.0%), L-(+)-tartaric acid (ACS reagent, ≥99.5%), poly(ethylene glycol) diacrylate (molecular weight, 250 g mol−1), and allyl bromide (reagent grade, 97%) were purchased from Sigma Aldrich. The photoinitiator used was phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO), supplied by TCI chemicals. Ethyl acetate, dichloromethane, acetone and other solvents used in the synthesis were provided by Panreac (ITW Reagents). Sodium hydroxide pellets were also purchased from Panreac company.2.2. Synthesis of photocurable liquid resins: diallyl succinate (DAS) as a model
In a 500 mL round-bottom flask, an aqueous solution of NaOH (80 mL, 10 wt%) was added dropwise to a succinic acid (11.81 g, 0.10 mol) and allyl bromide (26.62 g, 0.22 mol, excess of 10%) mixture, in 100 mL acetone (pure grade). The mixture was then stirred at 65 °C for 8 h before diluting with water (500 mL) and sequentially washing three times with dichloromethane (DCM, 100 mL). Then the excess of solvent was evaporated under an air flow, using vigorous stirring to eliminate the excess of allyl bromide in the same step. The final product, diallyl succinate monomer (DAS), was isolated as a colourless liquid, with a yield of about 85%. FTIR-ATR (cm−1): 3081 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H, stretching); 2880–2995 (C–H, stretching); 1732 (CO ester, stretching); 1649 (CC stretching); 1377, 1415 (CH2, –CH2, in-plane bending); 1100–1250 (C–O–C ester, stretching); 834 and 801 (C–H, out-of-plane bending and wagging). 1H-NMR (400 MHz, CDCl3, δ ppm): 5.90 (m, 2H, CH), 5.31 (dd, 4H; CH2), 4.60 (d, 4H, –O–CH2), 2.63 (s, 4H, –CH2–CO).
C–H, stretching); 2880–2995 (C–H, stretching); 1732 (CO ester, stretching); 1649 (CC stretching); 1377, 1415 (CH2, –CH2, in-plane bending); 1100–1250 (C–O–C ester, stretching); 834 and 801 (C–H, out-of-plane bending and wagging). 1H-NMR (400 MHz, CDCl3, δ ppm): 5.90 (m, 2H, CH), 5.31 (dd, 4H; CH2), 4.60 (d, 4H, –O–CH2), 2.63 (s, 4H, –CH2–CO).
The syntheses of the other two liquid acrylate resins, diallyl malate (DAM) and diallyl tartrate (DAT), are described in the ESI.†36–38 The complete scheme of synthesis is depicted in Fig. 1A.
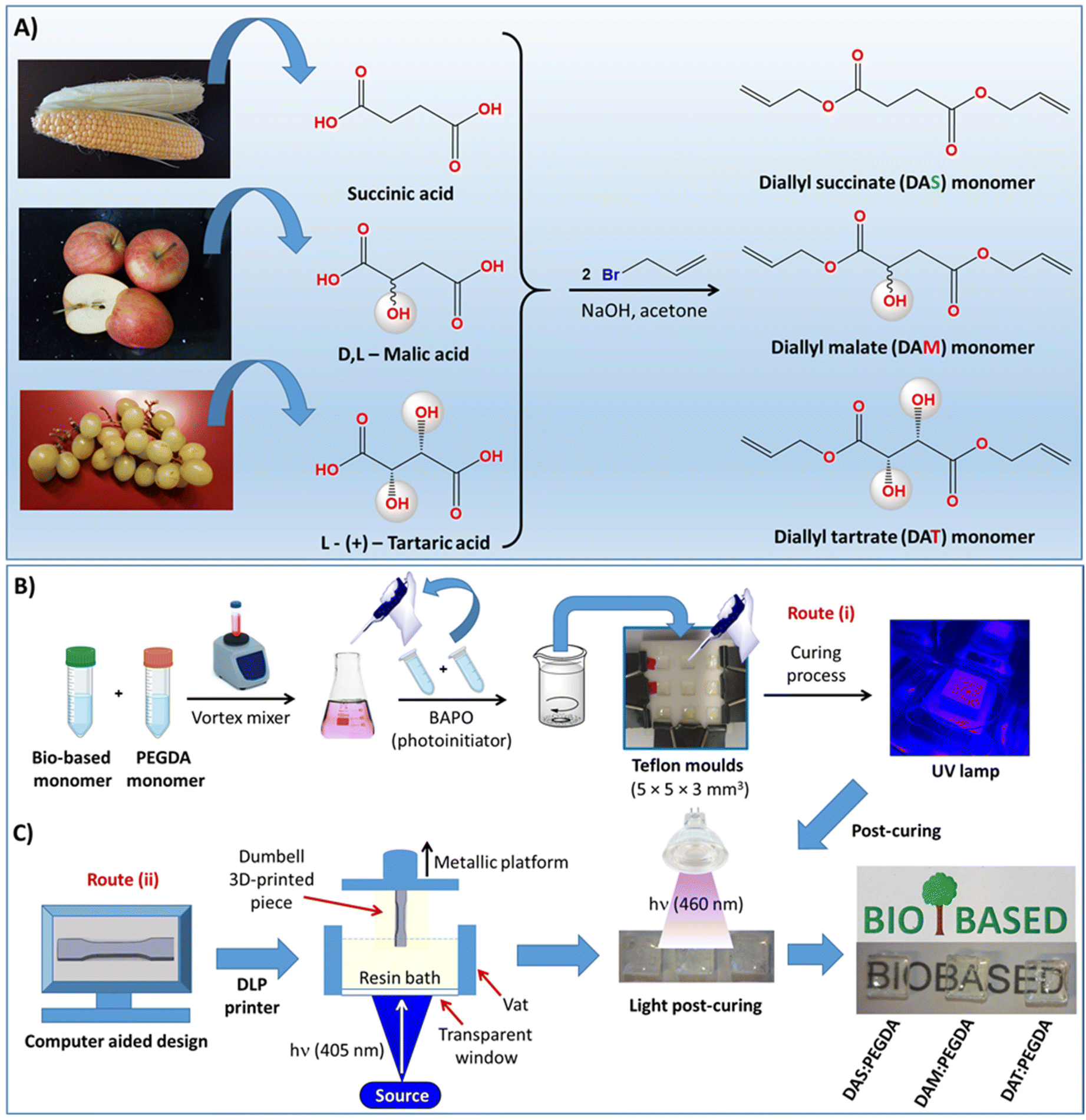 | ||
| Fig. 1 (A) Illustration of the synthetic route followed to prepare the bio-based monomers: DAS, DAM and DAT. (B) Schematic representation of the procedure carried out for the copolymerization of the bio-based monomers and PEGDA co-monomer and the subsequent deposition inside Teflon moulds and curing under light. (C) Representation of the scale-up production of dumbbell test specimens in a DLP printer and the post-curing process in controlled visible light. | ||
2.3. Copolymerization of bio-based resins with poly(ethylene glycol)diacrylate (PEGDA) co-monomer under UV light
Two procedures were used: (i) small-scale by using small Teflon moulds with the dimensions indicated in Fig. 1B, and (ii) laboratory-scale by using a 3D DLP printer, as indicated in Fig. 1C. In the first case (route i), 10 g of bio-based resin (DAS, DAM or DAT) (0.05 mol) was initially dispersed in 11.26 mL of PEGDA co-monomer (0.05 mol) in a small vial with a tap and mixed with the help of a magnetic stirrer for 1 h, while protected against light and heating. After dispersion, 2.0 wt% of BAPO was added into the glass tube and left to homogenize for 30 min under magnetic stirring. The samples were then poured into Teflon moulds, such as that depicted in Fig. 1B, and irradiated with 405 nm UV light (FungDo UV source) for 10 min. After this time, the solid samples (Fig. 1C) were moved to a higher wavelength source (460 nm, 35 μW cm−2) to ensure complete curing. Therefore, post-curing was carried out with the samples facing the lamp for 6 h each side (bottom and top). All samples were cleaned, washed with isopropanol, dried under vacuum and properly characterized. The samples are transparent and practically colourless.2.4. Scale-up production of mechanical test samples with a 3D DLP printer
In the laboratory scale-up production (route ii, Fig. 1C), a commercially available 3D ANYCUBIC Photon S UV LCD printer was used. The printer is composed of a resin vat at the bottom and a metallic platform at the upper part. The liquid resin, previously mixed in the same proportions as those described above, is poured into the vat and irradiated at 405 nm wavelength (0.5 mW cm−2), from the bottom of the bath. The photopolymerization occurs between the resin and the metallic platform, on which the samples are printed, thus avoiding oxygen inhibition. This machine allows precise control of the layer thickness. Therefore, a 50 μm layer thickness was fixed, with the first four layers being irradiated for 400 s and 60 s for the overall depositions to achieve 3 mm of total thickness. These parameters (time/layer) were determined to be optimal after many tests. For thicker pieces, a thermal post-curing was required. The samples were placed in a vacuum oven for 12 h at 170 °C.2.5. Reaction conversion approached by gel fraction studies
The gel fraction of square cured pieces (0.6 × 0.6 × 0.3 cm3) was determined by swelling and gravimetric evaluations, by adapting method C described in the ASTM D2765-16 standard.39 The method consists in measuring the weight increase in xylene solvent, at 110 °C for 24 h, and the weight losses of the specimens. The results are expressed as follow: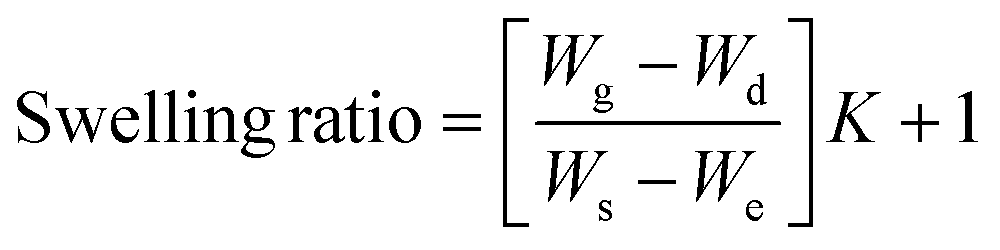 | (1) |
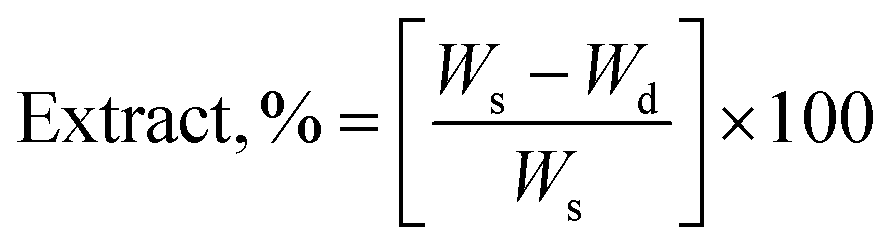 | (2) |
For the mechanical tests, the pieces underwent the same light post-curing treatment as that described in section 2.3 (Fig. 1C). The samples were washed with isopropanol in a sonication bath for 30 min to remove any unreacted monomer.
To compare the effect of the concentration of the photoinitiator (BAPO) during the curing time, two formulations were tested: one with 0.5 wt% and the other with 2.0 wt%. The polymerization reaction was followed with FTIR-ATR and the curing kinetics were measured by rheology, using the shrinkage of the pieces with time. Experimental details are reported in the ESI.†
2.6. Characterization techniques
The bio-based monomers were characterized with infrared (FTIR-ATR) spectroscopy and with proton nuclear magnetic resonance (1H-NMR). The chemical compositions of the copolymers, before and after the post-curing processes, were also evaluated with FTIR-ATR. The thermal stability and glass transition temperatures of the bio-based copolymers were also evaluated with thermogravimetry (TGA), differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA). Furthermore, the photopolymerization reaction of the 3D-printed pieces was followed with photorheology. The morphology of the samples was investigated with scanning electron microscopy (SEM). Experimental details of all the above mentioned procedures are supplied in the ESI, section 2.5.†2.7. Enzymatic degradation
Enzymatic degradation studies were carried out by placing the cured samples (25 mg, 5 × 5 × 3 mm3) in vials containing 0.1 mg mL−1 of lipase Rhizopus oryzae (30 U mg−1, Sigma Aldrich) in 5 mL of phosphate buffered saline (PBS) solution, supplemented with 0.1 mg mL−1 of NaN3 (ACROS Organics) to prevent contamination. The vials were closed with screw caps and sealed with parafilm to avoid loss of the solution by evaporation, and the samples were incubated at 37 °C in a shaking incubator set at 100 rpm for a total of 90 days. The PBS solution was replaced every 48 h. The variation of the weight loss against the exposure time was used to evaluate the enzymatic degradability from a quantitative point of view. For this purpose, samples (in triplicate) were removed every week from the solution and weighed. Degradation was quantitatively monitored as weight loss (in %), by applying the following formula (eqn (3)):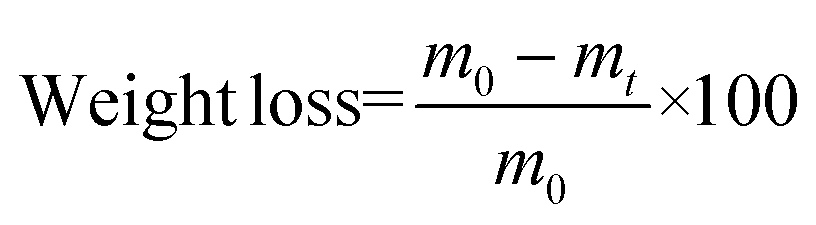 | (3) |
3. Results and discussion
3.1. Synthesis of bio-based monomers and characterization of the photopolymerized copolymers
The synthesis of the DAS, DAM and DAT diallyl monomers was planned for their further copolymerization with PEGDA co-monomer, a well-known photopolymerizable compound with fast reactivity. The presence of the hydroxyl groups in the bio-based raw materials DAM and DAT would affect the kinetics of polymerization. This is the reason why they were chosen to compare with DAS (bio-sourced derived molecule without OH groups). The kinetics of their reaction with PEGDA were followed by rheology studies and will be discussed later. Regarding the chemical structure, as can be seen in Fig. 2, the reduction ofC–H groups from the stretching (3080 cm−1) and out-of-plane bending (∼810 cm−1) absorption bands was used to ensure correct copolymerization, as well as the increase of the intensity of –C–H bonds (2940–2820 cm−1 and 1440 cm−1) belonging to CH2 groups.
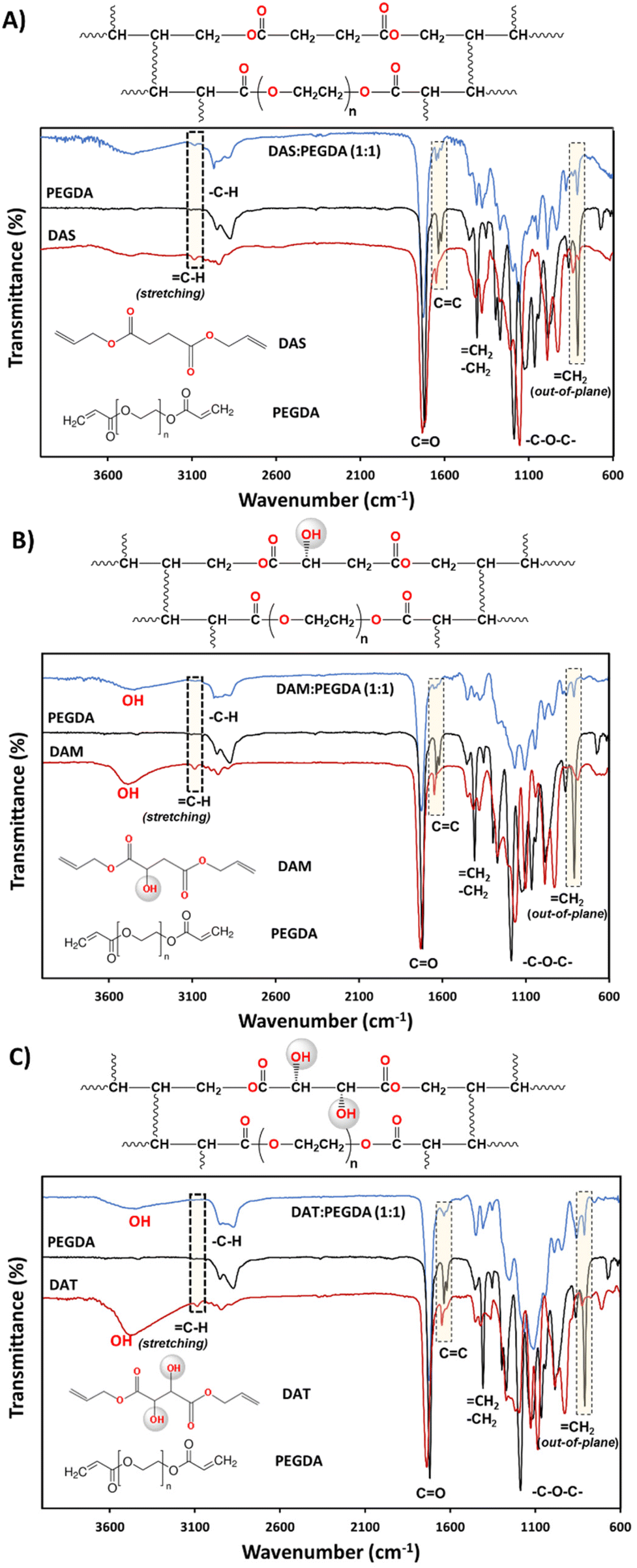 | ||
Fig. 2 Sequence of FTIR-ATR spectra comparing the monomers and the copolymer main functional groups: (A) DAS, PEGDA, DAS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEGDA (1:1); (B) DAM, PEGDA, DAM:PEGDA (1:1); and (C) DAT, PEGDA, DAT:PEGDA (1:1). The reduction of double bond end groups, after UV photopolymerization, and the chemical structures of the copolymers are highlighted. :PEGDA (1:1); (B) DAM, PEGDA, DAM:PEGDA (1:1); and (C) DAT, PEGDA, DAT:PEGDA (1:1). The reduction of double bond end groups, after UV photopolymerization, and the chemical structures of the copolymers are highlighted. | ||
The presence of hydroxyl groups in the DAM:PEGDA and DAT:PEGDA copolymers was evidenced by the broad and intense absorption bands at 3400 cm−1 (stretching) after the photopolymerization reaction. Such groups originate from the bio-based monomers used as raw materials (Fig. 2B and C).
The structures of the bio-based monomers were corroborated by comparison of the 1H-NMR spectra with previously published works.38,40 DAS is a symmetric molecule with only four signals in 1H-NMR (Fig. S1.A†): one singlet at 2.63 ppm, from 4Hs of methylenes beside the carbonyl groups; 4Hs at 4.60 ppm, from methylene linked to the CH and oxygen from the ester group; a multiplet at ∼5.90 ppm attributed to 2Hs from CH– bonds; and two doublets at 5.31 ppm, corresponding to the 4Hs of the terminal double bond CH2.
In the DAM monomer, the last three groups appear in the same region, 4.60 ppm (–CH2–O–), 5.90 ppm (CH–) and 5.30 ppm (CH2). The DAM bio-based monomer is not symmetrical; therefore, three distinctive peaks appear at 2.70 ppm, which integrates 2H corresponding to methylene beside one of the carbonyls, 3.40 ppm corresponding to the broad signal of the OH group, and 4.43 ppm (1H) corresponding to the doublet signal of the CH group besides the other carbonyl bond (Fig. S1B†).
As in the case of DAS, the DAT monomer is symmetrical and a reduced set of NMR peaks is evidenced. Some of them are similar to those of DAS. However, DAT presents a broad singlet of the two OH groups at 3.40 ppm, and 2Hs from the chiral C–H centres as a doublet at 4.48 ppm (Fig. S1C†). It was not possible to perform the chemical characterization of the copolymers with the PEGDA co-monomer by NMR due to their insolubility in many solvents.
In order to monitor the photopolymerization reaction and to determine a proper irradiation time per layer in the 3D printer, an indirect method was used. This method is considered a quantitative tool for monitoring photopolymerization reactions.41 Stress relaxation tests of uncured monomer mixtures were recorded with a rheometer. Shrinkage was determined as the volume reduction of a gap layer of 50 μm, from the beginning to the end of the experiment, following eqn (S1) (section 2.5, ESI†). For this assay, 60% of shrinkage was arbitrarily taken for comparison purposes of the different samples and photoinitiator concentrations. This percentage reveals the appropriate mechanical consistency to go on to print the next layer and for the piece not to collapse. The setup of the rheometer and the UV light used to irradiate the liquid samples, as well as the irradiation power versus wavelength, are depicted in Fig. S2 and S3,† respectively.
As introduced in the experimental procedure, two different concentrations of the photoinitiator were employed (0.5 wt% and 2.0 wt%) for DAS and PEGDA photopolymerizations. Comparing the two curves (Fig. 3A and B), 2 wt% of initiator seems to slightly accelerate the beginning of the polymerization. The mixture starts to solidify after only 4 s of the lamp being switched on, compared to 10 s needed with the lowest BAPO concentration. Therefore, 2 wt% was fixed as the photoinitiator content to reduce the curing time needed.
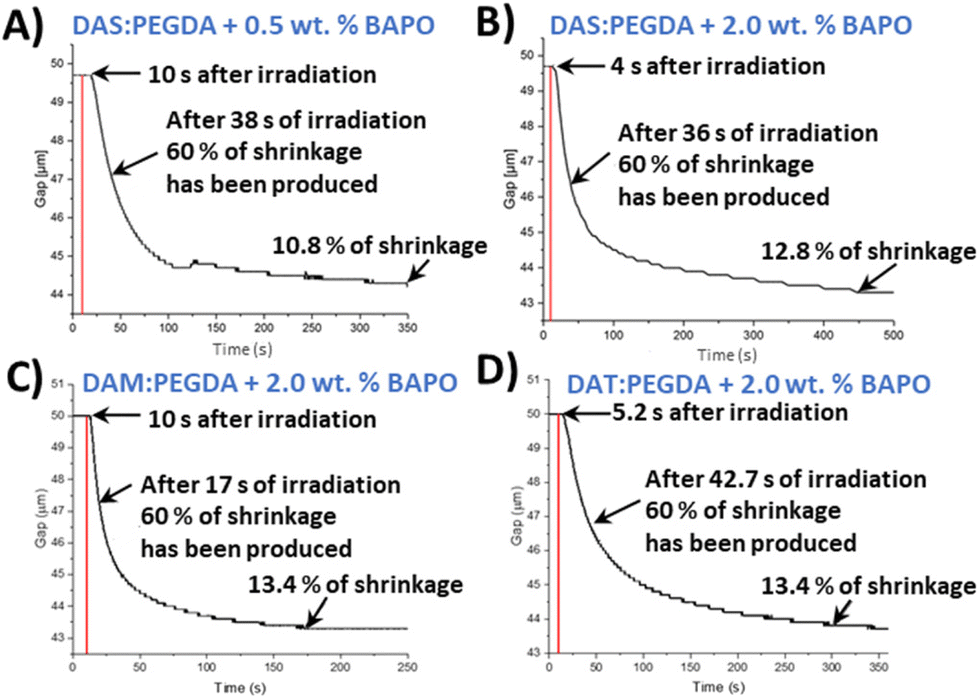 | ||
| Fig. 3 Rheometer-monitored graphs of the bio-based acrylate copolymers over time under LED light (405 nm): (A) DAS:PEGDA with 0.5 wt% of BAPO (photoinitiator); (B) DAS:PEGDA with 2 wt% of BAPO; (C) DAM:PEGDA with 2 wt% of BAPO; and (D) DAT:PEGDA with 2 wt% of BAPO. The red line indicates the time when the lamp was turned on. | ||
The reaction of the DAM bio-based monomer with PEGDA kicks off quickly, with fast decay of the gap variation change curve with time (Fig. 3C) and achieving 60% shrinkage in only 17 s. On the contrary, when the bio-sourced monomer has two hydroxyl groups in its chemical structure (DAT), the copolymerization starts very rapidly after light activation (4 s). However, the time necessary to achieve 60% of shrinkage was worse than for the other two monomers, with a more prolonged time (42.7 s) (Fig. 3D).
From those plots, it is deduced that the asymmetric bio-based monomer DAM, with only one hydroxyl lateral group (D,L-malic acid), offers the faster conversion with the lowest irradiation time (160 s). Our hypothesis is that the electron-donor group beside the ester unit benefits the nucleophilicity of the double-bond end group in the radical polymerization. Moreover, this monomer (DAM) does not have the same steric hindrance than dat monomer, with two hydroxyls, favouring the kinetics of polymerization. In all cases, the copolymers maintained a reduced shrinkage behaviour after the complete irradiation (∼10–13%), which is in agreement with the usual range for common photopolymerizable polymers.42,43 Furthermore, these results indicate that, in particular for the case of DAS, the irradiation time should not be lower than 400 s per 50 μm layer thickness to ensure the maximum compaction of the successive layers in the printer machine.
Having characterized the monomers and copolymers, the next section will discuss the efficacy of the post-curing process.
3.2. Curing process of the bio-based allyl copolymers with UV light: effects on the chemical composition and thermal properties
Acrylate polymers prepared by DLP and SLA prototyping usually require a post-curing process, i.e. further light irradiation or thermal treatments.18 This protocol can be applied to polyesters and poly(ester amides), as long as the optimal properties of the final product are maintained. However, often the use of high temperatures and prolonged curing times can cause sample degradation. In this work, before printing, some tests were carried out to ensure the correct copolymerization (curing process) between the two monomers. First, we tried to perform a post-curing treatment on the freshly polymerized samples (UV light at 405 nm, 0.5 mW cm−2) by placing them in an oven at high temperature (170 °C, 12 h). Unfortunately, the DAM and DAT-derived samples became dark and brittle (Fig. S4†). The DAS:PEGDA solid samples maintain a homogenous aspect, before and after the thermal-curing process. The second post-curing method is that depicted in Fig. 1C, which involved light irradiation with a 460 nm wavelength of visible light (35 μW cm−2), for 12 hours, under an LED lamp. This method was preferred. It is effective when having transparent and colourless samples like ours. Higher wavelengths manage to penetrate better throughout the thickness of the sample than lower wavelength visible light.
The efficacy of the dual-light curing process was followed by FTIR, using the absorption bands corresponding to CC at 1636 cm−1, and C–H at ∼3080 cm−1 and ∼809 cm−1 (Fig. 4, dashed boxes highlighted). These bands are clearly visible before the post-curing. Close inspection of the 1000–600 cm−1 wavenumber range (Fig. 4, on the right) proves that the double bonds from the out-of-plane vibration bands have been reduced considerably for all compositions after the visible light irradiation (460 nm).
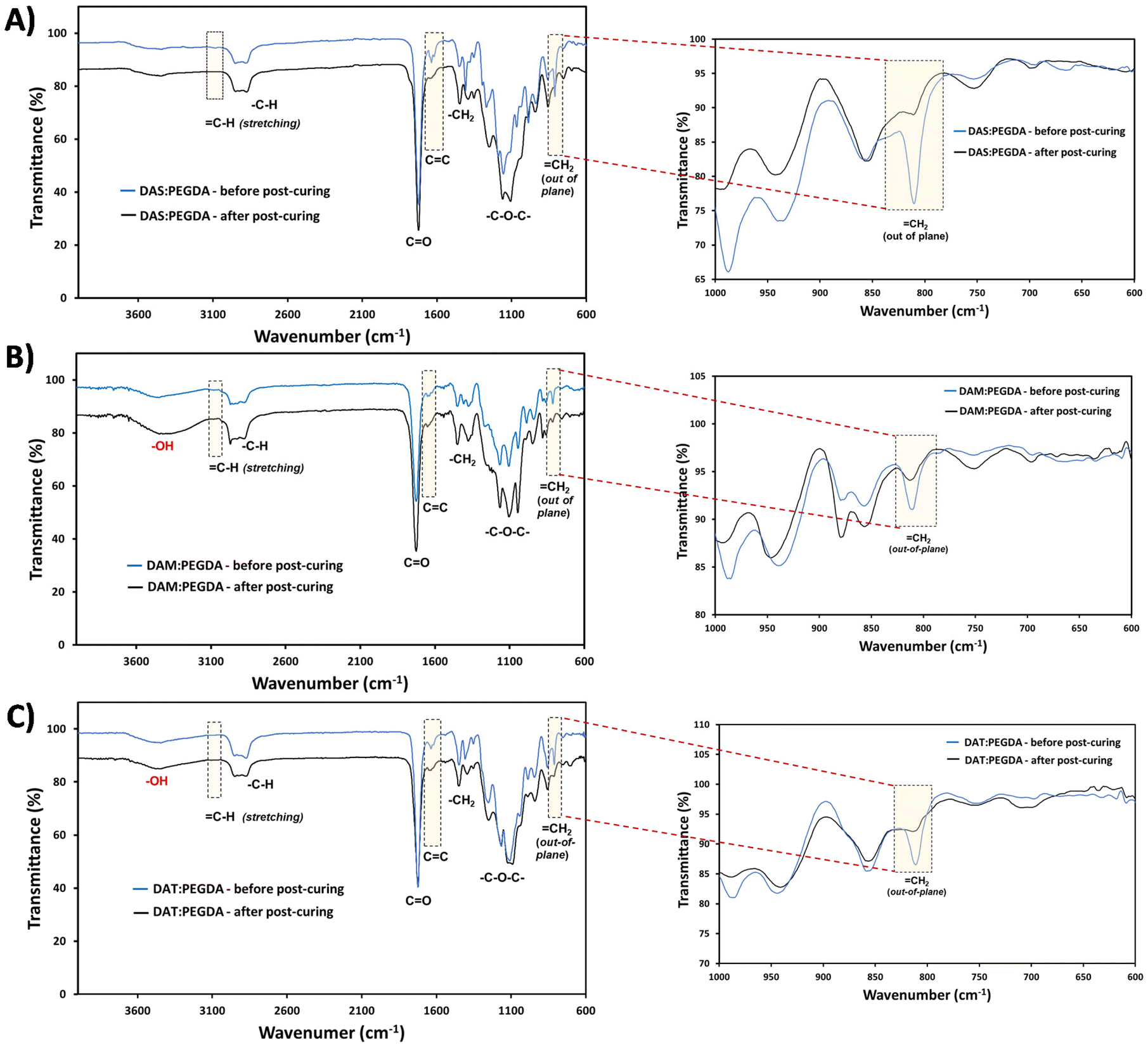 | ||
| Fig. 4 FTIR-ATR spectra comparing the samples before and after the post-curing process: (A) DAS:PEGDA; (B) DAM:PEGDA; and (C) DAT:PEGDA copolymers. On the right, the detail of the CH2 out-of-plane bending bands, which overlapped evidencing that the polymerization reaction was complete. | ||
In order to prove the good crosslinking between the monomers, the gel content was investigated by measuring the swelling properties of the acrylate copolymer in xylene and the amount of soluble polymer not crosslinked, i.e. the extract percent. The results are expressed in Table S1.† After the test, the samples were removed intact from the solvent bath. No loss of the smallest particles, yellowing of the product, or changes in the FTIR absorption bands were observed, proving that there was no chemical or physical degradation within 24 hours. The very low swelling ratios obtained (∼1.0–1.2) confirm the high degree of crosslinking and the stability of the samples from chemical attack. It is possible to certify that the stoichiometric mixtures are well-cured presenting values higher than 93% solid content (wt% of insoluble polymer from the extract percent data). In summary, the pre-curing by UV/visible lamps and the thermal post-curing processes were highly efficient.
Having established the optimal conditions for the bio-based monomer:PEGDA copolymer curing, the thermal properties were investigated. In all cases, the solids obtained correspond to vitreous materials with glass transition temperatures (Tg) slightly above the ambient temperature (Fig. 5A–C). The very low Tg and heat capacity per gram of sample (ΔCp) observed are probably due to the long length of the bi-functional monomer PEGDA, which leads to a larger free-volume between the covalent bonds with the bio-based monomer, thus offering a reduced steric hindrance to chain mobility. Examples of glass transition temperatures and heat capacities of different acrylate polymers have been reported.44 Regarding the thermal stability determined by TGA, the maximum decomposition temperature (Td,max) is higher than 430 °C in all cases (Fig. 5D–F), showing highly stable materials. However, slight differences can be observed before and after the visible light curing. Before post-curing, all samples lose 10% of their weight at temperatures around 200 °C. Furthermore, DAT:PEGDA uncured samples presented three decomposition decays before post-curing (Fig. 5F), which can be attributed to a more irregular microstructure and phase separation. Even after light irradiation at 460 nm for 12 h, DAM:PEGDA and DAT:PEGDA still lose weight at around 240 °C, whereas DAS:PEGDA remains stable until 330 °C with only one decay at a maximum T of 440 °C and a char yield of 3% at 600 °C. Cortés-Guzmán et al.45 recently described similar thermal properties for a monofunctional guaiacol glycidyl ether acrylate monomer (GuGEA) photopolymerized with diglycidyl ether of vanillyl alcohol (DGEVDA), a bi-functional crosslinker. In their example, the thermal properties were slightly improved with annealing treatment and they could also be modulated with a higher content of DGEVDA. The disadvantage of such compounds is the complexity of the preparation of the bio-based monomers, if compared to the one-step route described to obtain DAS, DAM and DAT.
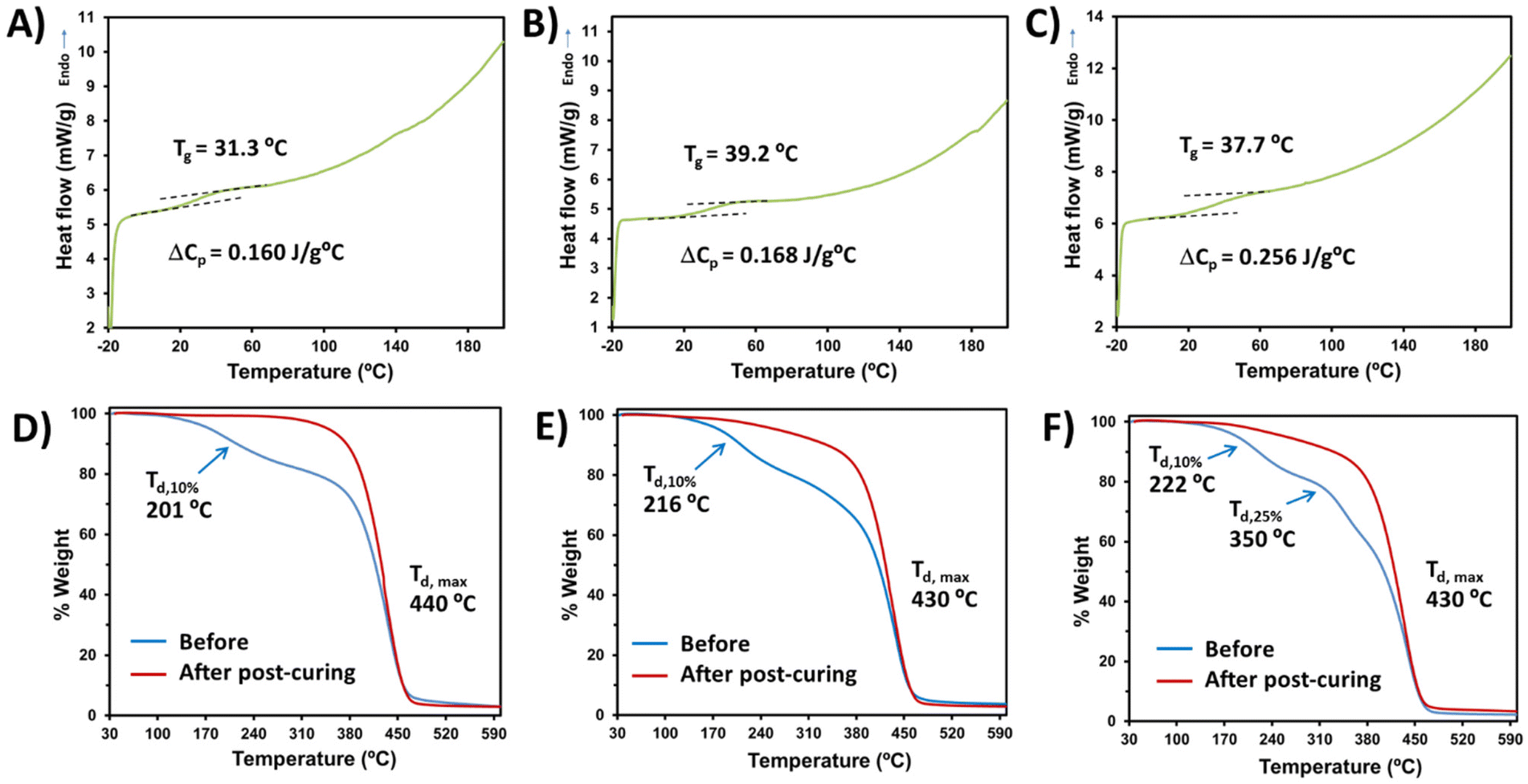 | ||
| Fig. 5 DSC analysis showing the second heating curves used to determine the glass-transition temperatures and heat capacities of: (A) the DAS:PEGDA sample; (B) the DAM:PEGDA sample; and (C) the DAT:PEGDA sample. TGA curves, illustrating the main degradation steps, before and after complete curing, for: (D) the DAS:PEGDA sample; (E) the DAM:PEGDA sample; and (F) the DAT:PEGDA sample. | ||
Overall, after considering the comparisons and justification of the not significant influence of the hydroxyl groups in the light-induced polymerization with PEGDA, as well as their thermal responses, we decided to exclude the DAM and DAT bio-based derived monomers in the DLP printing of mechanical- and degradability-test specimens. The second reason for this decision was also based on the synthesis yields of DAM and DAT diallyl molecules, which were much lower than that of DAS (50–60% for DAM and DAT, compared to 85% for DAS). From a commercial point of view, mass production of such raw materials for 3D-printing applications could be impractical and not-cost effective.
3.3. Dimensional stability of 3D-printed pieces, thermomechanical analysis and stress–strain behaviour of the bio-based DAS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :PEGDA copolymer
:PEGDA copolymer
For 3D printing of DAS:PEGDA mixtures, a commercial DLP 3D Anycubic Photon printer (405 nm, 0.5 mW cm−2) was employed, with an irradiation time of 60 s per layer (50 μm). This ensures structural integrity of the pieces, as previously seen in the rheology tests, whereas a light-irradiation post-curing at 460 nm (35 μW cm−2) for several hours ensures the maximum conversion of the double bonds. The following images (Fig. 6A and B) show the forms that were printed for the biodegradability (square format) and stress–strain experiments (dog-bone format), respectively. It should be noted that the printed samples had some burrs in the bottom corners of the pieces (Fig. 6A, left), which can be easily removed with a cutter tool (Fig. 6A, right). The DAS:PEGDA solids obtained are practically colourless and essentially maintain the transparent aspect before and after the dual-curing process with visible light. Most importantly, no cracks or fissures were observed when compared to DAM:PEGDA and DAT:PEGDA (Fig. S4†). Fig. 6C and D represent a resolution piece prototype usually used to observe the dimensional quality of printed samples, before (C) and after the post-curing treatment (D). As can be seen in the images (Fig. 6D, left and E), thin pillars and bigger shapes are correctly printed, if the separation among them is sufficient to avoid the merging of consecutive structures as visualized in the columns (Fig. 6D, right and F).
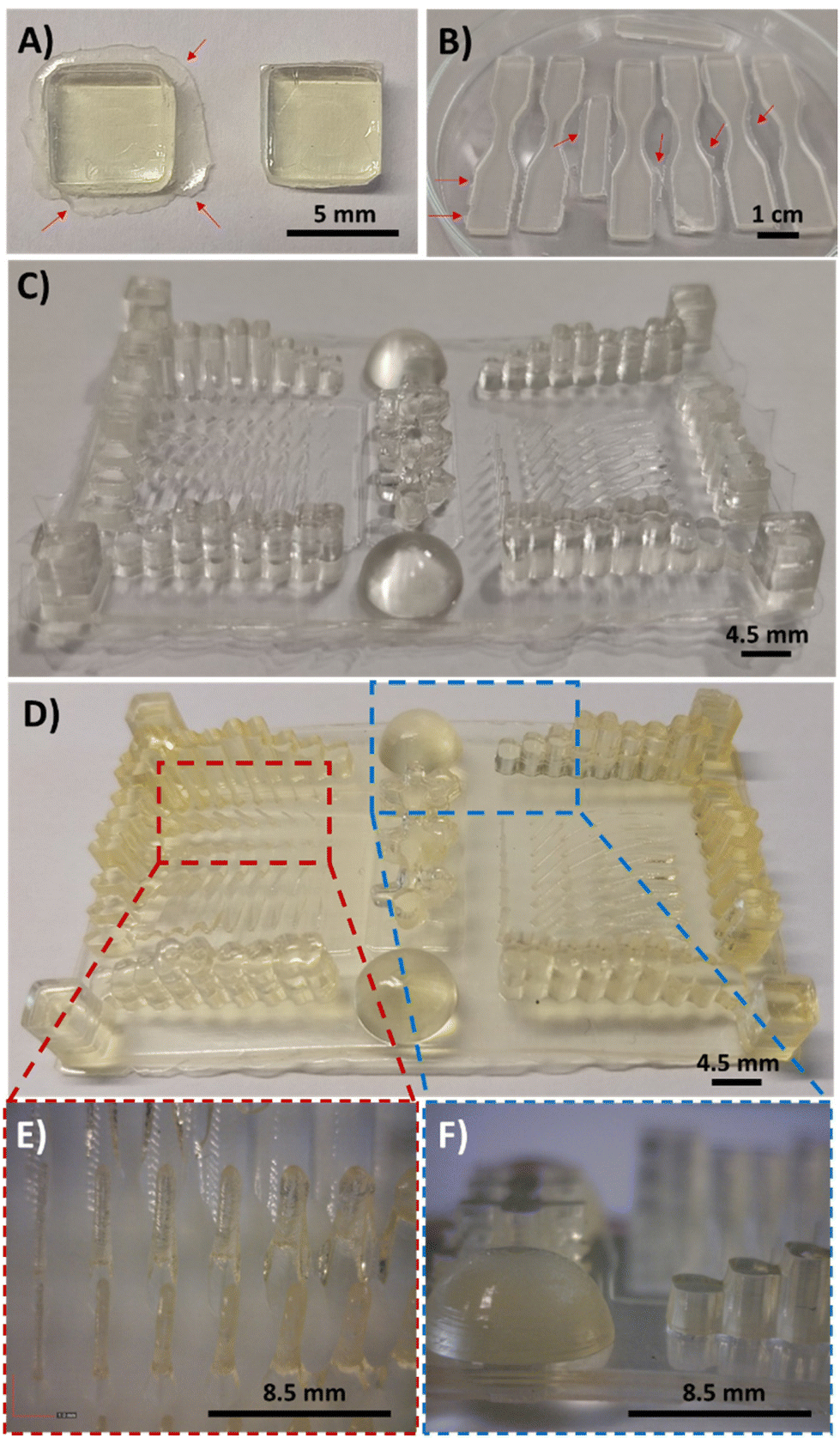 | ||
| Fig. 6 Examples of some pieces printed with the DLP 3D printer employing the bio-based resin DAS and PEGDA monomer: (A) square bar samples used for biodegradability assays. (B) Dumbbell-shaped probes for stress–strain experiments. (C) Details of the dimensional stability of the resolution piece. Arrows in (A and B) indicate the bottom membrane that can be easily removed. (D) Resolution piece after the post-curing processes. (E and F) Details from the pillars and shapes printed. | ||
The success of DAS:PEGDA photoprinting can be attributed to the high homogeneity of the mixtures with an appropriate amount of photoinitiator. Fig. 7A depicts the main infrared absorption bands for such samples, characterized by the absence of the terminal double bond groups belonging to the two monomers. Although the real proportion of each component in the cured pieces could not be determined by the H-NMR technique, the thermal properties and the mechanical stability were found to be good enough for the scale-up production of bigger prototypes, as will be discussed below.
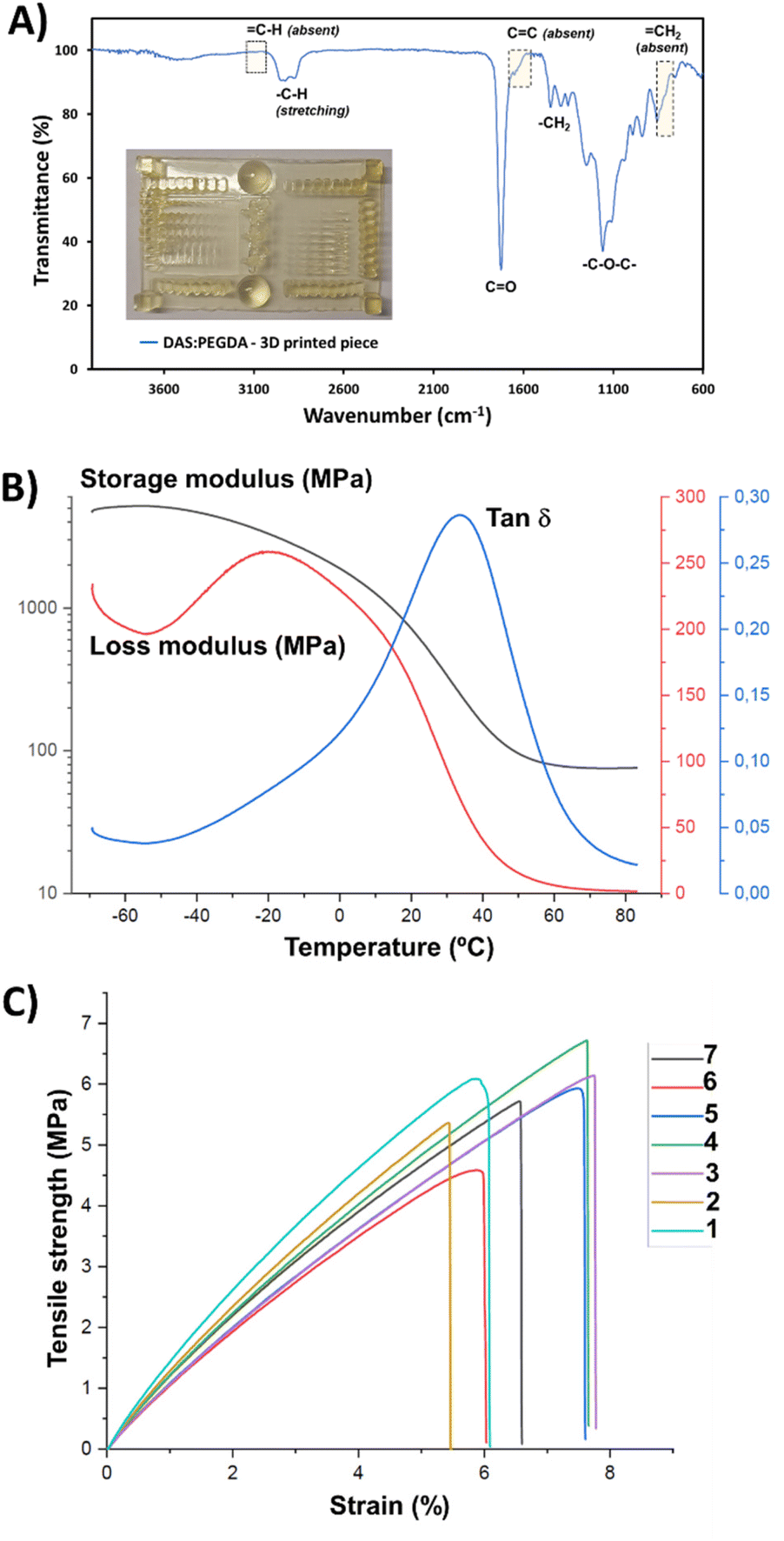 | ||
| Fig. 7 (A) FTIR-ATR spectrum of the 3D-printed piece shown in the inset image. (B) DMA analysis of the DAS:PEGDA copolymer. (C) Stress–strain curves of DAS:PEGDA copolymer dumbbell samples. Both assays were performed with samples DLP printed under UV light and after post-curing processes. | ||
The Tg value of the DAS:PEGDA copolymer was also measured for the cured samples that were assessed by three-point bending dynamic mechanical analysis (DMA) (Fig. 7B). A Tg of 34.0 °C was found by determining the maximum of the loss factor curve (tanδ).46 The elastic modulus was calculated to be 127.2 ± 5.8 MPa, according to eqn (S2).† This value is the average from 3 replicas and the ranges provided indicate a confidence level of 95%. From the storage modulus curve, a rubbery plateau of 65–80 °C can be appreciated, confirming the well-cured thermostable material.
Based on previous works and our experience,35,47 where the anisotropy feature of the poly(ester amide)s 3D-printed in-plane in the X and Z directions was proved with the tensile tests results, the building orientation of the 3D-printed specimens used in this work and employed in the stress–strain mechanical assays was uniquely in the X–Y in-plane direction (flat). The elastic modulus (E), determined from the linear curve of the strain–stress graphs (Fig. 7C), and the maximum tensile strength (σmax) were 105.6 ± 13.4 MPa and 5.8 ± 0.7 MPa, respectively, corresponding to rigid polymeric materials. The behaviour of the curves corroborates the absence of plasticity of the DAS:PEGDA probes, with a low elongation at break (εb) of 6.7 ± 0.9%. Considering that our material is not a highly crosslinked polymer, its reinforcement would be desirable. Some of the additives described in a recent review reported by Darji et al.,48 such as methacrylated wood flour powder,49 could help to improve the strength and toughness of our photo-polymerized bio-based copolymer. Undoubtedly, additives and plasticizers will affect the material properties and their employment in the DAS:PEGDA composition would be a task for future research.
3.4. Enzymatic biodegradability of the bio-based DAS:PEGDA polyester copolymer
Commercially available lipases, proteases and cutinase enzymes, acquired from fungal or bacterial sources, can promote a variety of chemical reactions, including esterification, transesterification, ester hydrolysis, and aminolysis, among others. They act as catalysts, accelerating the process. Among them, lipases are the preferred class for the hydrolytic degradation of polyesters, such as PLA, PHAs, polycaprolactone (PCL), and poly(ester amides) (PEAs). Moreover, Sander and co-workers50 revealed that Rhizopus oryzae lipase and Fusarium solani cutinase are better able to hydrolyse flexible and lineal aliphatic and aliphatic–aromatic polyesters, in which the ester groups are highly accessible sites. Greene et al.27 have also proven that, when the enzyme (0.1–5.0 wt%) is embedded in the polymer matrix forming a composite, it greatly accelerates the degradation rate, compared to the same material with the enzyme dispersed in the buffer media, thus confirming that the ester groups are even more accessible to be hydrolysed.
Nevertheless, not all enzymes resist the high temperatures of 3D-printer machines or the high-temperatures of post-curing treatments. The authors used Amano lipase (AL) and the fused deposition modelling of PCL/AL composites at 90 °C.
In this study, we employed Rhizopus oryzae lipase in PBS solution at 37 °C, in order to compare the biodegradation rate of DAS:PEGDA 3D-printed samples with other polymers synthesized with photocuring processes.
Fig. 8A exemplifies the weight loss percentage (eqn (3)) evolution with time. Although the biodegradation rate is slow, it follows an upward trend, reaching about 16% mass loss in 3 months. The enzyme/PBS solution was renewed every 48 h. The experiments were carried out in triplicate and a linear correlation factor (〈R2〉) of 0.9783 was obtained (Fig. S5A†). This means that an estimate of 5 years can be predicted for 100% total degradation of these bio-based DAS:PEGDA DLP prototypes, in this enzymatic medium. If we compare the weight loss to other similar poly(ether ester) polymers that include PEG in their chemical structure, the hydrolysis rate is very similar,34 whereas the PEGDA homopolymer does not degrade. However, this behaviour is affected by the presence of monomers more suitable to degrade, as is the case of PADEG:PEGDA:DMAM, which includes α-amino acid units.35 The degradation of the bio-based DAS:PEGDA copolymer is even better compared with well-known biodegradable polymers, including caprolactone units.27
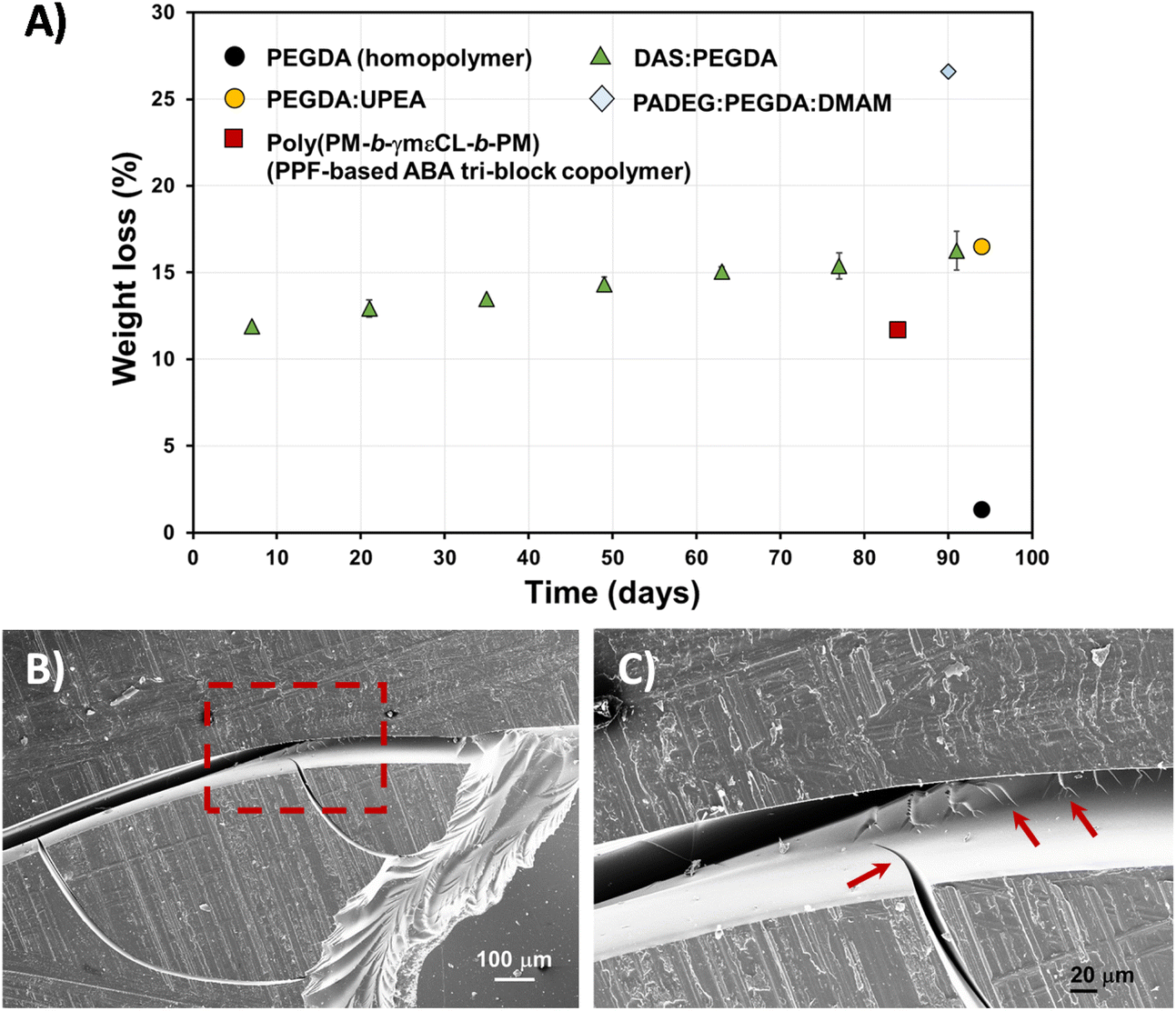 | ||
| Fig. 8 (A) Hydrolytic degradation of DAS:PEGDA 3D-printed pieces, in PBS buffer with Rhizopus oryzae lipase, with time (solid triangles), calculated from normalized weight loss (eqn (3)). The results were compared to other printed samples, for which the data were extracted from the following references: PEGDA homopolymer and PADEG:PEGDA:DMAM;35 PEGDA:UPEA;34 and poly(PM-b-γmεCL-b-PM).27 In the ultimate example, the authors did not employ enzymes. (B) SEM micrographs of the DAS:PEGDA degraded surface, at low magnification (75×). (C) SEM micrograph of the dashed square shown in (B), at high magnification (267×). Arrows indicate the crack propagation. | ||
The degradation rate has an enormous dependency on the polymer's chemical structure, buffer temperature and enzyme typology. Therefore, it can be modulated to accelerate or shorten the hydrolysis reaction as exemplified in an acrylate–methacrylate–carboxymethyl cellulose copolymer, printed by DLP (AMSx–MC examples, in Fig. S5B†).4 An increase in the natural polysaccharide content (lower “x” value) in the polymer network leads to an enhanced weight loss because Cellulase enzymes have high affinity to cellulose.
After the 3-month period of the assays, the DAS:PEGDA sample's morphology was investigated by SEM. The fracture behaviour is frequently found in fragile polymeric materials. The cracks start at the edges of the pieces and propagate over the surface (Fig. 8B) and interior of the sample (Fig. 8C), which also explains the increasing degradability of the copolymer with time.
4. Conclusions
The diverse and expanding 3D-prototyping industry increasingly demands resins free from petroleum-based sources. In this work, we have demonstrated the feasibility of obtaining well-cured bio-based poly(ether ester) solid pieces composed of 50 wt% of diallyl ester resins and 50 wt% of low molecular weight PEGDA. The bio-based monomers (DAS, DAM and DAT) were obtained in an easy single-step procedure, providing components that are liquid. These facts facilitate their use as blend components, and allows an increase in the biobased content to a remarkable 50 wt%.The presence of hydroxyl groups in the DAM and DAT resins did not show any substantial improvement in either the kinetics of polymerization or in the thermal properties of the final copolymers, with respect to the monomer without hydroxyl groups. Moreover, the detection of too brittle materials, after the thermal post-curing process with high temperatures, led us to discard such monomers for the 3D-printing and the biodegradation studies. Big and thick acrylate prototypes usually require thermal curing for complete crosslinking, as proved in the present work.
The fact that the thermal and mechanical properties of the copolymer containing DAS bio-based and PEGDA monomers are somewhat worse than those of other poly(ether ester)s reported in the literature does not hinder its potential applicability in additive manufacturing processes. The cured samples had very good dimensional stability and a promising biodegradation rate in a lipase medium.
While natural-sourced feedstock selection is very important when considering sustainability, evaluating the material's biodegradability is equally important for its end-of-life uses. Future work will be conducted to increase the bio-based content with respect to the synthetic one and explore how to improve the toughness of the DAS:PEGDA copolymer.
Looking into the future of introducing more sustainable plastics to the 3D-printing markets, the diallyl succinate monomer could help to decrease the dependency on fossil-based formulations in DLP and SLA technologies.
Author contributions
Conceptualization: F.B. and E.A.; investigations: L.H. and I.I.; writing—original draft preparation: E.A. and F.B.; writing, review and editing: E.A., F.B., and C.A.; supervision: N.B. and G.F.; funding acquisition: F.B. and E.A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project (BASE3D 001-P-001646), which is co-funded by the European Regional Development Fund (ERDF) within the framework of the ERDF Operational Program of Catalonia 2014–2020. The research leading to these results received funding from “La Caixa Foundation”, under the grant agreement LCF/PR/PR20/51150010. We thank Ph.D. Sofia Paulo and Ph.D. Ina Keridou, from UPC, for their help with SEM and NMR analyses, respectively; and S. de la Flor from Universitat Rovira i Virgili for facilitating the access to the DMTA equipment.References
- J. D. Badia and A. Ribes-Greus, Eur. Polym. J., 2016, 84, 22–39 CrossRef CAS.
- T. C. Guimarães, E. S. Araújo, M. L. Hernández-Macedo and J. A. López, J. Polym. Environ., 2022, 30, 2669–2684 CrossRef.
- N. T. Dintcheva, G. Infurna and F. D'Anna, Sci. Total Environ., 2021, 763, 143044 CrossRef CAS PubMed.
- J. Zhang, D. Huang, S. Liu, Z. Yang, X. Dong, H. Zhang, W. Huang, S. Zhou, Y. Wei, W. Hua, Y. Jin, W. Zhou and W. Zheng, J. Appl. Polym. Sci., 2022, 139, 52155 CrossRef CAS.
- K. Ghosh and B. H. Jones, ACS Sustainable Chem. Eng., 2021, 9, 6170–6187 CrossRef CAS.
- M. Zhang, G. M. Biesold, W. Choi, J. Yu, Y. Deng, C. Silvestre and Z. Lin, Mater. Today, 2022, 53, 134–161 CrossRef CAS.
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS.
- T. Tábi, T. Ageyeva and J. G. Kovács, Polym. Test., 2021, 101, 107282 CrossRef.
- X. Qi, Y. Ren and X. Wang, Int. Biodeterior. Biodegrad., 2017, 117, 215–223 CrossRef CAS.
- S. A. Backer and L. Leal, Acc. Chem. Res., 2022, 55, 2011–2018 CrossRef CAS PubMed.
- L. Jasinska-Walc, M. Bouyahyi and R. Duchateau, Acc. Chem. Res., 2022, 55, 1985–1996 CrossRef CAS PubMed.
- M. Zumstein, G. Battagliarin, A. Kuenkel and M. Sander, Acc. Chem. Res., 2022, 55, 2163–2167 CrossRef CAS PubMed.
- Y. Xie, S. Gao, D. Zhang, C. Wang and F. Chu, Resour. Chem. Mater., 2023, 2, 223–230 CAS.
- L. Martino, L. Basilissi, H. Farina, M. A. Ortenzi, E. Zini, G. Di Silvestro and M. Scandola, Eur. Polym. J., 2014, 59, 69–77 CrossRef CAS.
- T.-H. Kim, S.-H. Kang, J.-E. Han, E.-J. Seo, E.-Y. Jeon, G.-E. Choi, J.-B. Park and D.-K. Oh, ACS Catal., 2020, 10, 4871–4878 CrossRef CAS.
- Q. Xie, X. Hu, T. Hu, P. Xiao, Y. Xu and K. W. Leffew, Macromol. React. Eng., 2015, 9, 401–408 CrossRef CAS.
- A. Andreu, P.-C. Su, J.-H. Kim, C. S. Ng, S. Kim, I. Kim, J. Lee, J. Noh, A. S. Subramanian and Y.-J. Yoon, Addit. Manuf., 2021, 44, 102024 CAS.
- A. Al Rashid, W. Ahmed, M. Y. Khalid and M. Koç, Addit. Manuf., 2021, 47, 102279 CAS.
- A. Nazir, O. Gokcekaya, K. Md. Masum Billah, O. Ertugrul, J. Jiang, J. Sun and S. Hussain, Mater. Des., 2023, 226, 111661 CrossRef.
- E. Rossegger, K. Moazzen, M. Fleisch and S. Schlögl, Polym. Chem., 2021, 12, 3077–3083 RSC.
- F. S. Al-Jahwari and T. Pervez, in Encyclopedia of Renewable and Sustainable Materials, Elsevier, 2020, vol. 1–5, pp. 200–206 Search PubMed.
- E. Skliutas, M. Lebedevaite, S. Kasetaite, S. Rekštytė, S. Lileikis, J. Ostrauskaite and M. Malinauskas, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed.
- V. S. D. Voet, J. Guit and K. Loos, Macromol. Rapid Commun., 2021, 42, 1–11 CrossRef PubMed.
- J. Guit, M. B. L. Tavares, J. Hul, C. Ye, K. Loos, J. Jager, R. Folkersma and V. S. D. Voet, ACS Appl. Polym. Mater., 2020, 2, 949–957 CrossRef CAS.
- Y. Cui, J. Yang, D. Lei and J. Su, Ind. Eng. Chem. Res., 2020, 59, 11381–11388 CrossRef CAS.
- S. Choe, Y. Kim, G. Park, D. H. Lee, J. Park, A. T. Mossisa, S. Lee and J. Myung, ACS Appl. Polym. Mater., 2022, 4, 5077–5090 CrossRef CAS.
- A. F. Greene, A. Vaidya, C. Collet, K. R. Wade, M. Patel, M. Gaugler, M. West, M. Petcu and K. Parker, Biomacromolecules, 2021, 22, 1999–2009 CrossRef CAS PubMed.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897 RSC.
- S. Choe, Y. Kim, G. Park, D. H. Lee, J. Park, A. T. Mossisa, S. Lee and J. Myung, ACS Appl. Polym. Mater., 2022, 4, 5077–5090 CrossRef CAS.
- A. Kirillova, T. R. Yeazel, K. Gall and M. L. Becker, ACS Appl. Mater. Interfaces, 2022, 14, 38436–38447 CrossRef CAS PubMed.
- S. R. Petersen, J. Yu, T. R. Yeazel, G. Bass, A. Alamdari and M. L. Becker, Biomacromolecules, 2022, 23, 2388–2395 CrossRef CAS PubMed.
- T. R. Yeazel-Klein, A. G. Davis and M. L. Becker, Adv. Mater. Technol., 2023, 2201904 CrossRef CAS.
- G. Ruano, J. Tononi, D. Curcó, J. Puiggalí, J. Torras and C. Alemán, Soft Matter, 2020, 16, 8033–8046 RSC.
- S. I. Macías, G. Ruano, N. Borràs, C. Alemán and E. Armelin, J. Polym. Sci., 2022, 60, 688–700 CrossRef.
- I. Isarn, Ľ. Hodásová, M. M. Pérez-Madrigal, F. Estrany, E. Armelin and F. Bravo, Macromol. Rapid Commun., 2023, 2300132, 1–13 Search PubMed.
- H. Umakoshi, N. Iwasa and H. Yamamoto, European Patent, 3438135, 2019 Search PubMed.
- R. N. Guthikonda, L. D. Cama, M. Quesada, M. F. Woods, T. N. Salzmann and B. G. Christensen, J. Med. Chem., 1987, 30, 871–880 CrossRef CAS PubMed.
- C. Ye, V. S. D. Voet, R. Folkersma and K. Loos, Adv. Mater., 2021, 33, 2008460 CrossRef CAS PubMed.
- ASTM, Standard Test Methods for Determination of Gel Content and Swell Ratio of Crosslinked Ethylene Plastics, United States, 2016.
- K. I. K. Herath, L. P. Tan, C. L. L. Chai and M. J. M. Abadie, J. Biomater. Sci., Polym. Ed., 2010, 21, 1459–1481 CrossRef CAS PubMed.
- P. Szymaszek, W. Tomal, T. Świergosz, I. Kamińska-Borek, R. Popielarz and J. Ortyl, Polym. Chem., 2023, 14, 1690–1717 RSC.
- C. Schmidt and T. Scherzer, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 729–739 CrossRef CAS.
- J. Świderska, Z. Czech and A. Kowalczyk, Pol. J. Chem. Technol., 2013, 15, 81–85 Search PubMed.
- J. Wen, in Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer, New York, NY, 2nd edn, 2007, pp. 145–154 Search PubMed.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, R. M. Johnson, L. Adegoke, M. Ecker, W. E. Voit and R. A. Smaldone, Polym. Chem., 2023, 14, 2697–2707 RSC.
- I. Binyamin, E. Grossman, M. Gorodnitsky, D. Kam and S. Magdassi, Adv. Funct. Mater., 2023, 33, 2214368 CrossRef CAS.
- W. Tomal and J. Ortyl, Eur. Polym. J., 2022, 180, 111588 CrossRef CAS.
- V. Darji, S. Singh and H. S. Mali, Polym. Compos., 2023, 44, 4370–4419 CrossRef CAS.
- J. Yao and M. Hakkarainen, Compos. Commun., 2023, 38, 101506 CrossRef.
- M. T. Zumstein, D. Rechsteiner, N. Roduner, V. Perz, D. Ribitsch, G. M. Guebitz, H. P. E. Kohler, K. McNeill and M. Sander, Environ. Sci. Technol., 2017, 51, 7476–7485 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00207a |
| ‡ Both co-authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |