
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H bond activation at antimony(III): synthesis and reactivity of Sb(III)–oxyaryl species†‡
Gabriel
Duneş
ab,
Marie
Cordier
a,
Samia
Kahlal
a,
Alpar
Pöllnitz
b,
Jean-Yves
Saillard
*a,
Cristian
Silvestru
 *b and
Yann
Sarazin
*a
*b and
Yann
Sarazin
*a
aUniversité de Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, Campus de Beaulieu, 35042 Rennes, Cedex, France. E-mail: yann.sarazin@univ-rennes.fr; jean-yves.saillard@univ-rennes.fr
bDepartment of Chemistry, Supramolecular Organic and Organometallic Chemistry Centre (SOOMCC), Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, 11 Arany Janos, 400028 Cluj-Napoca, Romania. E-mail: cristian.silvestru@ubbcluj.ro
First published on 5th June 2024
Abstract
We report on the synthesis, structure and reactivity of [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3), an organoantimony(III)-oxyaryl species obtained upon Csp2–H bond activation in a phenolate ligand and stabilised by the monoanionic pincer {NCNMe4}−. The mechanism leading to the formation of 3 is highly sensitive to steric considerations. It was probed experimentally and by DFT calculations, and a number of intermediates and related complexes were identified. All data agree with successive heterolytic bond cleaving and bond forming processes involving charged species, rather than a pathway involving free radicals as previously exemplified with congeneric bismuth species. The nucleophilic behaviour of the oxyaryl ligand in 3, a complex that features both zwitterionic and quinoidal attributes, was illustrated in derivatisation reactions. In particular, insertion of CS2 in the Sb–Coxyaryl bond generates [{NCNMe4}Sb(S2C-C6H2-tBu2-3,5-O-4)].
Introduction
The activation of C–H bonds is essential in organic and organometallic synthesis, catalysis, and industrial processes. While this arena has been dominated by late transition metals, main group compounds have also entered the game in recent years.1 Salient examples with metals from groups 2, 13 and 14 include C–H bond activation in aryl, allylic, and benzylic positions, in hydropyrimidine and in α-positions of cyclic thioethers.2–8 In group 15, bismuth compounds have also been investigated, as this metal is a component of the “nMnO3/Bi2O3” catalyst of the SOHIO process.9 BiCl3 activated the para-C–H position in the phenolate 2,6-tBu2-C6H3-O−, yielding C–C coupled quinoidal (A) and biphenolic (B) products via a free radical mechanism (eqn (1)).10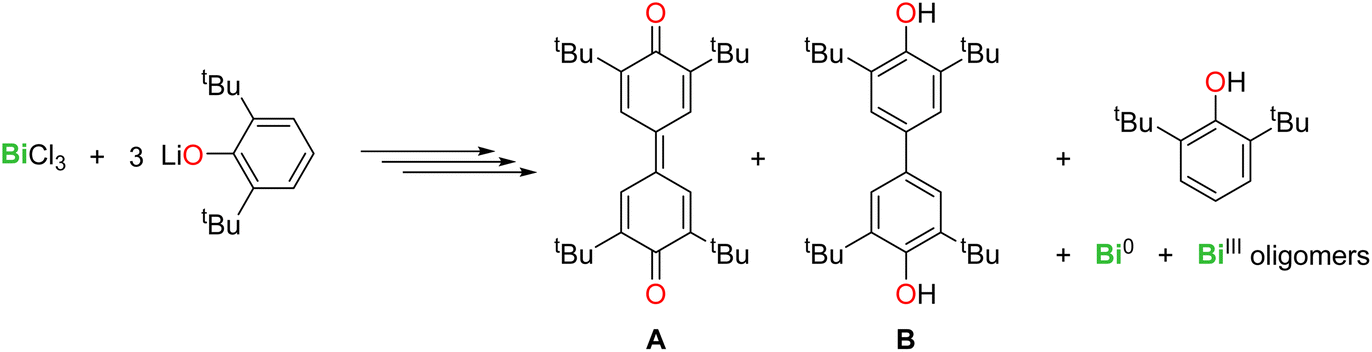 | (1) |
The organobismuth(III) [{NCNMe4}BiCl2], where NCNMe4 is the pincer 2,6-(Me2NCH2)2-C6H3−, generated equimolar amounts of the Bi-oxyaryl [{NCNMe4}Bi(C6H2-tBu2-3,5-O-4)] (Fig. 1, I) and of 2,6-tBu2-C6H3OH upon reaction with two equivalents of [{2,6-tBu2-C6H3O}K];11,12 based on Hanna's BiCl3 precedent,10 the mechanism was suggested to involve radical species. A case of Bi-mediated intramolecular C–H activation in benzylic position was recently reported in the planar Bi(III) compound [{NCNDiPP2}Bi] (II) supported by a trianionic pincer ligand, with concomitant extrusion of H2.13 Astounding work by Cornella has demonstrated the exquisite ability of Bi(III) to engage in controlled redox cycles with Bi(I) and Bi(V) and to catalyse a suite of organic reactions.14–18
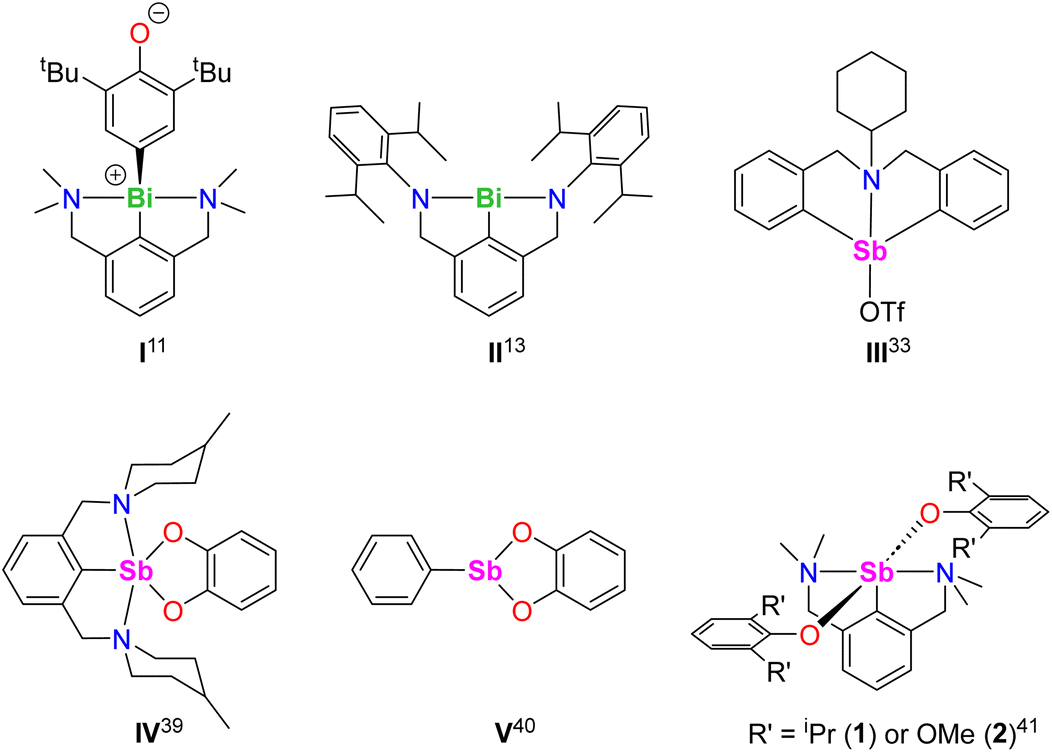 | ||
| Fig. 1 Selected examples of known Sb(III) and Bi(III) relevant to the present work. | ||
The chemistry of the lighter metalloid, antimony(III), is comparatively less developed.19 Recent breakthroughs in organoantimony(III) chemistry stem from the implementation of supporting ligands that stabilise highly reactive mononuclear species.20–24 Pincer ligands have been key to the developments of organoantimony compounds,19,20,25 and NCN− pincers have a leading role among these. Although SbF5 or SbF6− superacids are known to promote C–H bond activation and functionalisation processes,26–32 examples of metal-mediated reactions involving discrete organoantimony complexes for C–H bond activation are seldom. The azastibocine triflate [{C6H11N(CH2C6H4)2}SbOTf] (Fig. 1, III) is an efficient Lewis acid catalyst for Mannich reactions; [{C6H11N(CH2C6H4)2}SbF] triggers cyclisation-aromatisation from arylacetylenes, anilines and aldehydes.33,34 Antimony(V)-oxo porphyrins mediate C–H to C–C bond conversion in Michael acceptors under photoirradiation, without variation of the high-valent oxidation state.35
As part of our program targeting p-block complexes with alkoxides and other O-based derivatives,36–38 we have taken an interest in heteroleptic Sb(III)-phenolato complexes. Although their Sb(V) congeners are plethoric, few examples of heteroleptic arylantimony(III) phenolates are known. They include the catecholates [{2,6-(MeN[CH2CH2]2NCH2)2C6H3}Sb(O2-1,2-C6H4)] (IV) and [PhSb(O2-1,2-C6H4)] (V).39,40 Besides, we have recently disclosed the bis(phenolate)s [{NCNMe4}Sb(OC6H3-iPr2-2,6)2] (1) and [{NCNMe4}Sb(OC6H3-(OMe)2-2,6)2] (2).41 However, we are showing herein that under identical conditions, their derivative with tBu substituents, [{NCNMe4}Sb(OC6H3-tBu2-2,6)2], cannot be obtained, and instead the oxyaryl [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3) is isolated quantitatively. We are reporting here on the syntheses and structures of 3 and related Sb(III) complexes. Mechanisms, bonding features and reactivity patterns, including a case of insertion into a Sb(III)–C bond, are discussed. Investigations probing whether a radical-based mechanism could account for the formation of 3 provide a different perspective.
Results and discussion
General syntheses and characterisation
The reaction of [{NCNMe4}SbCl2]42 with two equivalents of [{2,6-tBu2-C6H3O}K] affords clean formation of the Sb(III)-oxyaryl complex [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3) upon release of 2,6-tBu2-C6H3OH and KCl (Scheme 1). This outcome contrasts with the analogous reactions involving the less bulky phenolates [{2,6-iPr2-C6H3O}K] and [{2,6-(OMe)2-C6H3O}K], which were shown to afford the anticipated [{NCNMe4}Sb(OC6H3-iPr2-2,6)2] (1) and [{NCNMe4}Sb(OC6H3-(OMe)2-2,6)2] (2).41 The overall behaviour was reminiscent of that observed with bismuth, where a radical mechanism was suggested for the formation of the congeneric [{NCNMe4}Bi(C6H2-tBu2-3,5-O-4)].11,12,43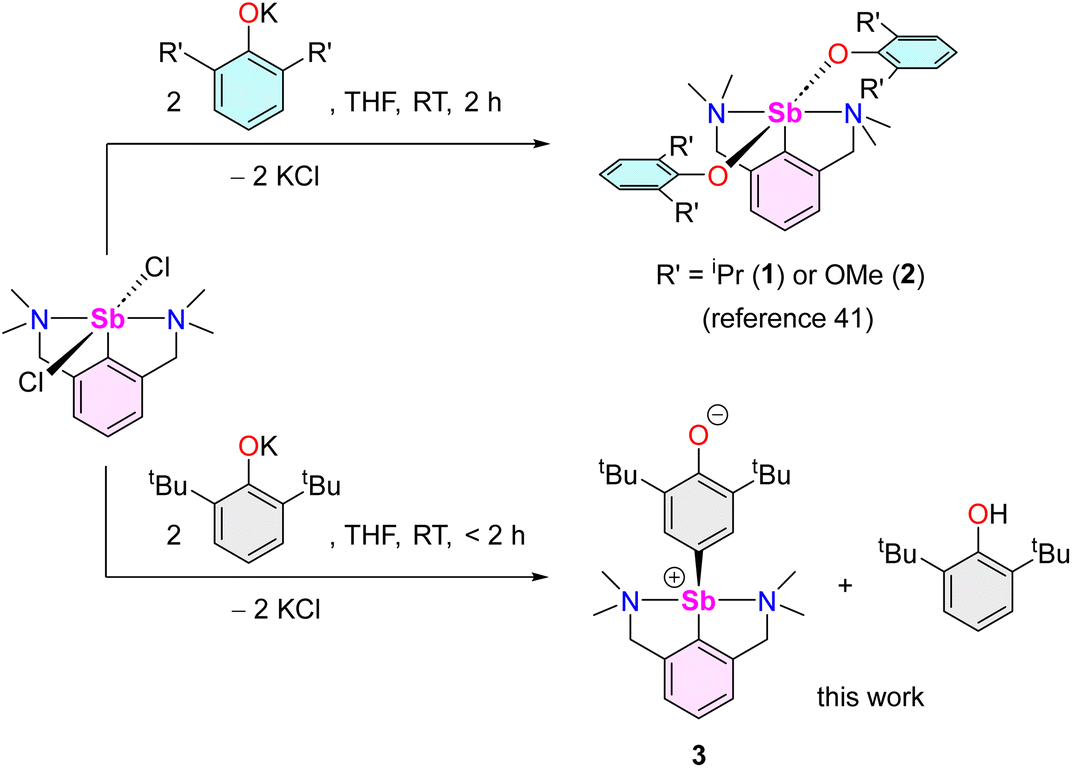 | ||
| Scheme 1 Synthesis of [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3). | ||
The air-sensitive 3 forms in yields over 85% within 2 h in THF at room temperature. The complex can be depicted as a zwitterion with a positively charged Sb(III) centre, or its quinoid-like, neutral-at-metal mesomer (Fig. 2). The presence of the oxidation products resulting from free radical couplings, i.e. 3,3′,5,5′-tetra-tert-butyldiphenoquinone (A) and 3,3′,5,5′-tetra-tert-butyl-[1,1′-biphenyl]-4,4′-diol (B) as reported in the reaction of BiCl3 with [{2,6-tBu2-C6H3O}Li] (see eqn (1)),10 was not detected in the synthesis of 3.
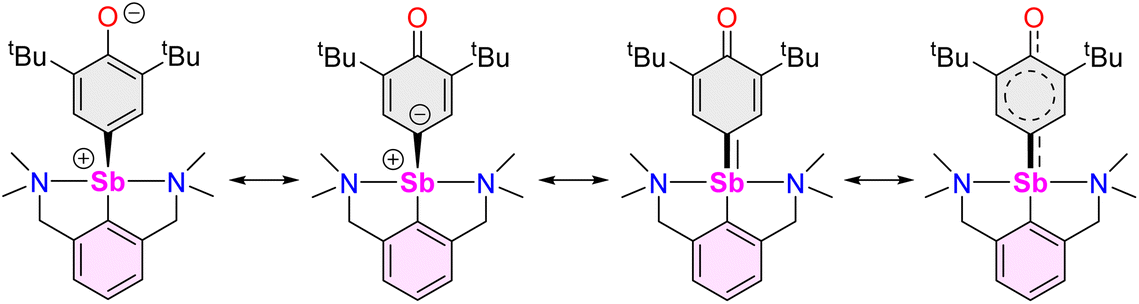 | ||
| Fig. 2 Mesomeric forms of [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3). | ||
Measurements showed that as expected for zwitterions,44 the conductivity of a THF solution of 3 (Λm = 27.2 mS cm2 mol−1) is relatively low. It is comparable to that for the neutral bis(phenolate) 1 (21.2 mS cm2 mol−1), and only marginally higher than that for [Sb(mesityl)3] and [{NCNMe4}SbCl2] (resp. 0.0 and 9.1 mS cm2 mol−1). By comparison, the dissociated ion pair [Na(OEt2)4]+·[H2N{B(C6F5)3}2]− (ref. 45) gives rise to much greater conductivity under otherwise identical conditions (22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mS cm2 mol−1). Complex 3 is diamagnetic. The NMR data of 3 recorded in CD3CN and CD2Cl2 were very different from those in THF-d8 although they corroborated in all cases the identity and the purity of the complex (see ESI, Fig. S1–S6‡). The VT 1H NMR spectra in THF-d8 were consistent with free rotation of the oxyaryl fragment around the Sb–Coxyaryl bond at high temperature (338 K),46 and with a frozen edge-inversion processes47 at 263 K (see ESI‡ for VT NMR). The FTIR spectrum of 3 (Fig. S7‡) features a strong band at 1559 cm−1 for the stretching of the C–O bond. It is below the values for carbonyl stretching in quinones, ca. 1660–1690 cm−1, and lower even than the remarkably low out-of-phase carbonyl stretching in substituted diphenoquinones, e.g. 1602 cm−1 in A.48 Such low νC
000 mS cm2 mol−1). Complex 3 is diamagnetic. The NMR data of 3 recorded in CD3CN and CD2Cl2 were very different from those in THF-d8 although they corroborated in all cases the identity and the purity of the complex (see ESI, Fig. S1–S6‡). The VT 1H NMR spectra in THF-d8 were consistent with free rotation of the oxyaryl fragment around the Sb–Coxyaryl bond at high temperature (338 K),46 and with a frozen edge-inversion processes47 at 263 K (see ESI‡ for VT NMR). The FTIR spectrum of 3 (Fig. S7‡) features a strong band at 1559 cm−1 for the stretching of the C–O bond. It is below the values for carbonyl stretching in quinones, ca. 1660–1690 cm−1, and lower even than the remarkably low out-of-phase carbonyl stretching in substituted diphenoquinones, e.g. 1602 cm−1 in A.48 Such low νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency is consistent with a partial CO double bond order, and agrees with the dual zwitterionic and quinoid-like nature of 3.
O stretching frequency is consistent with a partial CO double bond order, and agrees with the dual zwitterionic and quinoid-like nature of 3.
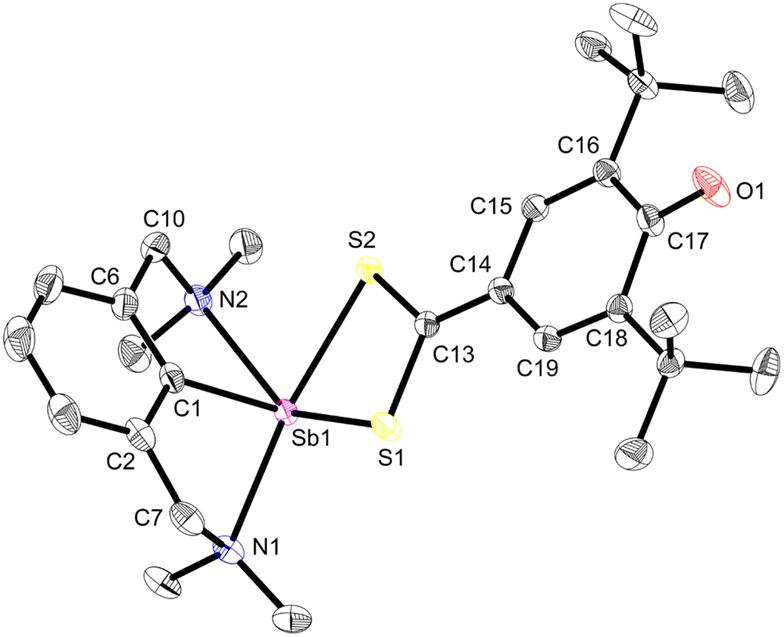
The molecular solid-state structure of 3 features a four-coordinate metalloid in a pseudotrigonal bipyramidal arrangement, with the (pRN1, pRN2) enantiomer49 depicted in Fig. 3. The short Sb1–N1 and Sb1–N2 interatomic distances (2.4458(11) and 2.4436(11) Å) testify to two strong N→Sb dative bonds,42 and the coordination geometry about Sb is best described as see-saw (τ4 = 0.66).50 The Sb–Coxyaryl bond length (Sb1–C13 = 2.0862(12) Å) is below the low end of Sb–Caryl distances in four-coordinate arylantimony(III) complexes.51–53 The Sb–Coxyaryl interatomic distance in 3 is comparable with the value found for the few reported SbC double bonds.54–56 The Sb–C13 bond in 3 is hence intermediary between single and double Sb(III)–carbon bonds. The C16–O1 distance in 3 (1.2708(16) Å) also ranges between the values of single and double carbon–oxygen bonds, e.g. as in B (1.385(4) and 1.391(3) Å)57vs. 2,6-di-tert-butyl-1,4-benzoquinone (1.2456(6) and 1.2570(6) Å)58 or 2,4,6-tri-tert-butylphenoxyl radical (1.246(2) Å).59 It hence feels legitimate to consider that this fragment exhibits partial quinoidal behaviour. A more detailed description of the structure of 3 is provided in ESI.‡
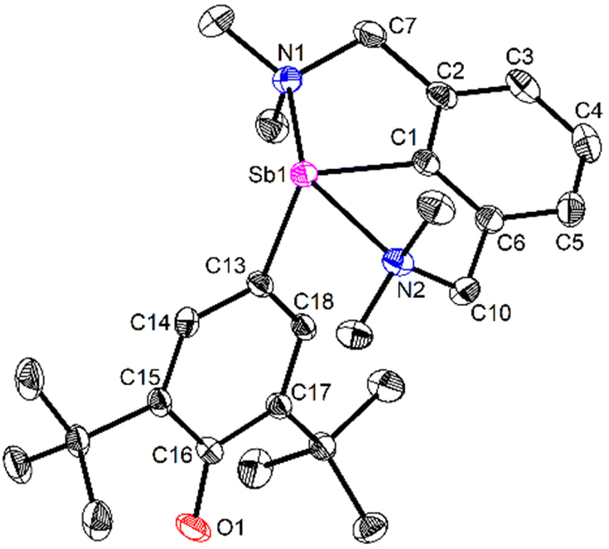 | ||
| Fig. 3 View of the molecular structure of [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3). H atoms omitted for clarity. Ellipsoids at the 50% probability level. Only one of the two enantiomers (pRN1, pRN2)49 in the asymmetric unit is shown. Selected distances (Å) and angles (°): Sb1–C1 = 2.1134(13), Sb1–C13 = 2.0862(12), Sb1–N1 = 2.4458(11), Sb1–N2 = 2.4436(11), C13–C14 = 1.4063(17), C13–C18 = 1.4047(17), C14–C15 = 1.3803(17), C15–C16 = 1.4572(18), C16–C17 = 1.4559(17), C17–C18 = 1.3777(17), C16–O1 = 1.2708(16); C1–Sb1–C13 = 101.02(5), C1–Sb1–N1 = 73.94(4), C1–Sb1–N2 = 74.16(4), C13–Sb1–N1 = 96.54(4), C13–Sb1–N2 = 89.09(4), N1–Sb1–N2 = 148.10(4). | ||
Synthetic and NMR insight
Beyond the fact that it requires tert-butyl groups in positions 2 and 6 of the phenolate instead of smaller substituents (see Scheme 1), we investigated the mechanism leading to 3. Monitoring of the reaction in THF-d8 by 1H NMR spectroscopy indicated clean and quantitative production of 3 and 2,6-tBu2-C6H3OH, without detectable presence of A or B (Fig. S37 and S42‡). Complex 3 is EPR silent (as a crystalline solid or in solution in THF); only minor signals corresponding to an organic fragment, most likely resulting from minute decomposition during sample preparation, were detected. The sequential NMR-EPR-NMR spectroscopic analysis of the same sample of crystalline 3 dissolved in THF-d8 confirmed the sample remained intact in solution, within the accuracy allowed by NMR, over the course of 2 h (Fig. S11‡). Addition of TEMPO or galvinoxyl to a mixture of [{NCNMe4}SbCl2] and [{2,6-tBu2-C6H3O}K] in THF did not prevent formation of 3, although TEMPO ultimately led to an oxo-bridged dimer (vide infra). Moreover, NMR monitoring showed that the rates of formation of 3 in the dark and in ambient light are near-identical, which further militates against the involvement of radicals. Based on these observations (also corroborated by DFT calculations, vide infra), we propose that unlike earlier suggestions for bismuth,113 does not result from homolytic bond cleavages and recombination of free radicals.The identity of the precursors required to achieve the C–H activation leading to 3 was probed (Scheme 2). The complex can be obtained from [{NCNMe4}Sbl2], although the reaction is slow in this case (12 h in THF-d8, Scheme 2a). The two –NMe2 side-arms in the pincer ligand can be replaced by bulkier and stronger –NiPr2 donors; the Sb(III)-oxyaryl resulting from C–H activation of the phenolate, 4 (Scheme 2b), is near-quantitatively isolated. However, with the dichloro precursor [{CNMe2}SbCl2],53 where the CNMe2 bidentate ligand bears a single –NMe2 tether, the reaction with two equivalents of [{2,6-tBu2-C6H3O}K] gives the bis(phenolate) [{CNMe2}Sb(OC6H3-tBu2-2,6)2] (5) akin to 1 and 2 (Scheme 2c). No C–H activation of the phenolate occurred, indicating that the presence of two Lewis bases is mandatory for the process to occur. Accordingly, addition of N,N-dimethyl-aminopyridine (DMAP) to a solution of pristine 5 affords the tetramer [{CNMe2}Sb(C6H2-tBu2-3,5-O-4)]4 (64) as insoluble crystals.601H NMR monitoring of this reaction in THF-d8 showed disappearance of all Sb-based species, with regeneration of DMAP and stoichiometric release of 2,6-tBu2-C6H3OH (Fig. S19‡). We assume the addition of DMAP to 5 (or, more generally, the presence of two stabilising Lewis bases) is required in order to stabilise the transient, positively charge antimony centre en route towards the formation of 64 (or, in the seminal case with the {NCNMe4} ligand, the production of 3), as pointed out by our DFT calculations (vide infra).
 | ||
| Scheme 2 Reactions of Sb(III) dihalides with potassium phenolates. | ||
Compound 64 can be seen as the tetrameric equivalent of zwitterionic 3 (Fig. 4). Each Sb centre is tetracoordinated. The O- and N-atoms are in trans position (N1–Sb1–O1 = 157.25(12)°), whereas the two carbon atoms are in cis (C4′–Sb1–C15 = 98.36(16)°). The Sb–Coxyaryl distance is noticeably larger than that in 3 (2.121(5) vs. 2.0862(12) Å). The Sb1–O1 bond in 64 (2.088(4) Å) is much shorter than in the four-coordinate Sb-bis(phenolate) 1 (2.187(1) Å).41 However, the amino side-arm in 64 is binding with antimony considerably more weakly (Sb1–N1 = 2.574(4) Å) than in both 3 and 1 (Sb–N = 2.4436(11)–2.4458(11) Å and 2.426(1) Å,41 respectively). Complex 64 is the utmost expression of the zwitterionic form of 6 (a complex that we could not isolate nor detect spectroscopically), yielding a charge-neutral tetramer through binding of the negatively-charged O atoms to Sb cations.
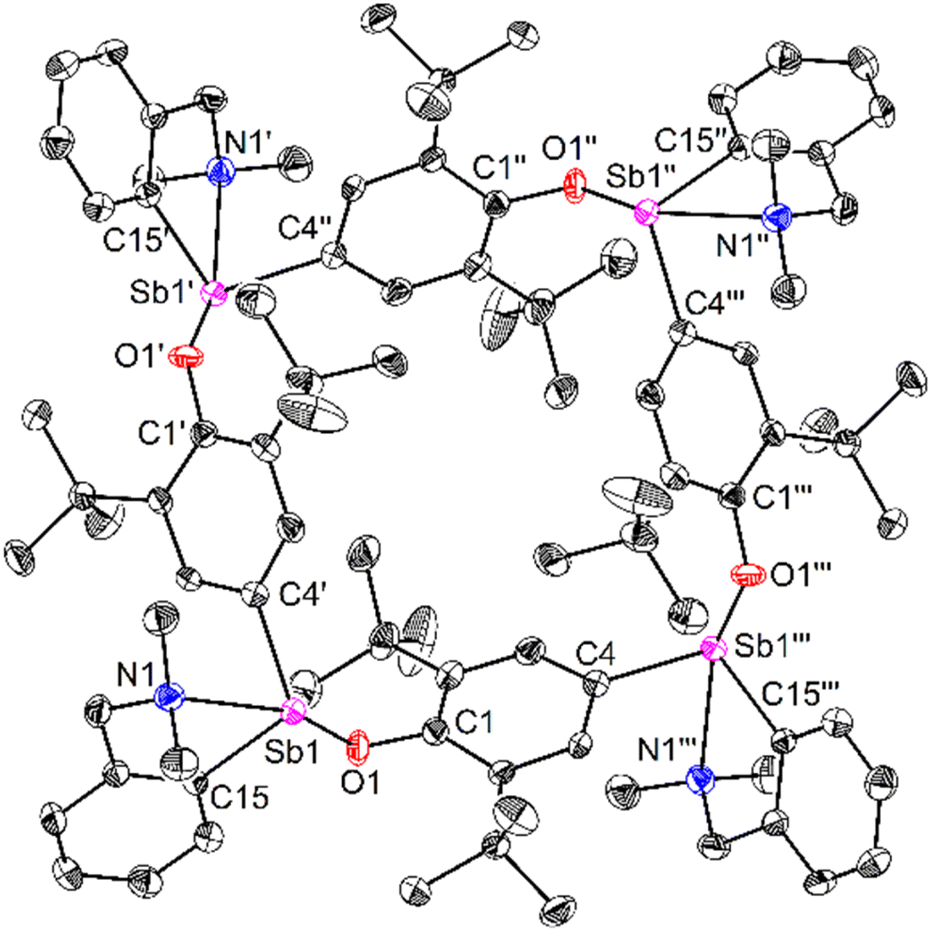 | ||
| Fig. 4 Representation of the molecular structure of [{CNMe2}Sb(C6H2-tBu2-3,5-O-4)]4 (64). H atoms omitted for clarity. Ellipsoids at the 50% probability level. Selected distances (Å) and angles (°): Sb1–C4′ = 2.121(5), Sb1–C15 = 2.157(4), Sb1–N1 = 2.574(4), Sb1–O1 = 2.088(4), C1–O1 = 1.346(5); C4′–Sb1–O1 = 99.36(16), C4′–Sb1–C15 = 98.36(16), C4′–Sb1–N1 = 82.47(15), C15–Sb1–N1 = 72.87(15), C15–Sb1–O1 = 84.46(15), N1–Sb1–O1 = 157.25(12). | ||
The reaction of [{NCNMe4}SbCl2] with two equivalents of [{2,4,6-tBu3-C6H2O}K] affords [{NCNMe4}Sb(OC6H2-tBu3-2,4,6)2] (7, Scheme 2d), demonstrating that the presence of tBu groups in positions 2 and 6 of the phenolate is compatible with the formation of regular organoantimony(III) bis(phenolate)s. Finally, the bis(phenolate) [{NCNMe4}Sb(OC6H2-tBu2-2,4)2] (8) was obtained with [{2,4-tBu2-C6H2O}K], even at 65 °C (Scheme 2d); hence, C–H activation only occurs in position 4 of di-tert-butylphenolate. This last finding also confirms that steric hindrance in the direct vicinity of the Ophenolate atom is not an impeding factor for the C–H activation step, as inferred from the opposite reactivity observed during the formations of 1 and 2vs. 3 from differently substituted 2,6-dialkylphenolates (Scheme 1).
Overall, it transpires from these experimental data that the behaviour of Sb(III) cannot be extrapolated from that of its heavier congener, Bi(III). Besides, in our hands, the reaction of SbCl3 with three equivalents of [{2,6-tBu2-C6H3O}K] in THF gave [{2,6-tBu2-C6H3O}3Sb] quantitatively; no traces of A or B were detected.10,61
Several model reactions were performed in THF-d8 and monitored by NMR to probe the behaviour of the system (Scheme 3). First, although potassium phenolate was fully consumed, the reaction of [{NCNMe4}SbCl2] with an equimolar amount of [{2,6-tBu2-C6H3O}K] did not produce [{NCNMe4}Sb(Cl)(OC6H3-tBu2-2,6)] (9). Instead, it yielded half equivalents of 3, 2,6-tBu2-C6H3OH and unreacted [{NCNMe4}SbCl2] within the first point of analysis (30 min; Scheme 3e and Fig. S38‡). This suggests that 9 is either never formed, or that it is an intermediate in the formation of 3. By contrast, in a two-step process (Scheme 3f), the reaction of [{NCNMe4}SbCl2] with equimolar [{2,4,6-tBu3-C6H2O}K] first generated a mixture of unreacted dichloride, bisphenolate 7 and a small amount of, at least, one other phenolate thought to be [{NCNMe4}Sb(Cl)(OC6H2-tBu3-2,4,6)] (10). This intractable mixture, assumed to be an equilibrium between 10vs. [{NCNMe4}SbCl2] and 7, was treated with one equivalent of [{2,6-tBu2-C6H3O}K], generating 3 and 2,4,6-tBu3-C6H2OH (Fig. S43‡). We hence reasoned that 3 may form through reaction of the monochloro, monophenolato intermediate 10, with one equivalent of [{2,6-tBu2-C6H3O}K]. Consumption of 10 would drive the reaction towards complete formation of 3 and release of 2,4,6-tBu3-C6H2OH, with concomitant disappearance of [{NCNMe4}SbCl2] and 7. In agreement with this hypothesis, in a separate experiment, the combination of [{NCNMe4}SbCl2] with equimolar 7 in THF-d8 at RT led to an equilibrated mixture with an NMR signature (Fig. S44‡) matching that of the first step of reaction (3.f); the same reaction at −35 °C gave a single, new species, assumed to correspond to pure 10. However, it is also possible that 3 results from direct action of [{2,6-tBu2-C6H3O}K] on 7; indeed, a native sample of 7 reacts with one equivalent of [{2,6-tBu2-C6H3O}K] to give 3, 2,4,6-tBu3-C6H2OH and [{2,4,6-tBu3-C6H2O}K] (Fig. S46‡). Both mechanisms may well be simultaneously at play; yet, regardless of the prevailing manifold, production of 3 and release of 2,4,6-tBu3-C6H2OH according to reaction (3.f) are clean and quantitative.
 | ||
| Scheme 3 Reactions monitored by 1H NMR spectroscopy in THF-d8. | ||
The synthesis of a Bi(III)-oxyaryl species by reaction of [{NCNMe4}BiCl2] with a 1:1 combination of [{2,6-tBu2-C6H3O}K] and [{2,6-iPr2-C6H3O}K] was reported to have failed.11 In this light, and in view of the outcome of reaction (3.f), it was surprising to observe that in a two-step process, [{NCNMe4}SbCl2] reacts with [{2,6-iPr2-C6H3O}K] and then [{2,6-tBu2-C6H3O}K] to afford half-equivalents of 3, [{NCNMe4}Sb(OC6H2-iPr2-2,6)2] (1) and 2,6-tBu2-C6H3OH (Scheme 3g and Fig. S47‡). Besides, the reaction of [{NCNMe4}SbCl2] with equimolar [{2,6-iPr2-C6H3O}K] was consistent with the formation of 1 along unreacted dichloride. The reason behind the different behaviour of [{2,4,6-tBu3-C6H2O}K] and [{2,6-iPr2-C6H3O}K] towards [{NCNMe4}SbCl2] originates from the fact that unlike 7 (vide supra), complex 1 is inert toward [{NCNMe4}SbCl2]; the 1:1 mixture of the two compounds in THF-d8 remains essentially unchanged after 24 h at 65 °C. Unexpectedly, treatment of 3 with one equivalent of 2,6-iPr2-C6H3OH generated the exact same mixture of 1, 3 and 2,6-tBu2-C6H3OH (half equivalents of each; Scheme 3h and Fig. S49‡) as that obtained in reaction (3.g), in a process postulated to evolve through the intermediate [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)(OC6H2-iPr2-2,6)] (11). Note also that 3 and 2,6-tBu2-C6H3OH engage in an equilibrium in solution; the 1H and 2H monitoring of a 1:1 mixture of 3 and 2,6-tBu2-C6H3OD returned a solution that contained 3 along 2,6-tBu2-4-D-C6H2OH, 2,6-tBu2-4-D-C6H2OD and 2,6-tBu2-C6H3OH (Fig. S50 and S51‡).
We then attempted to prepare a complex amenable to nucleophilic substitution. Crystals of [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)][Cl] (12) were obtained in 75% yield by reaction of [Me3NH]+Cl− with 3 (Scheme 4), a reaction that illustrates the zwitterionic character of 3. The Sb(III)-hydroxyaryl 12, formally the product of C–H activation on 2,6-di-tert-butylphenol, is, to our knowledge, unique; there is no related antimony complex in the CCDC database.62 Similarly, treatment of 3 with Bochmann's acid45 afforded the loose ion pair [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)]+[H2N{B(C6F5)3}2]− (12′).
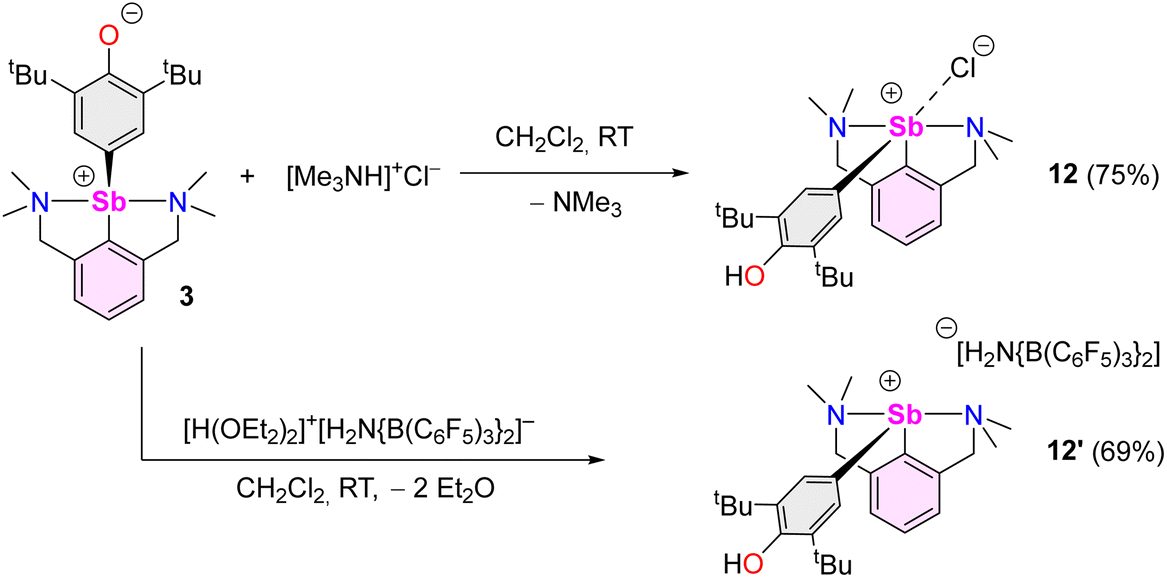 | ||
| Scheme 4 Synthesis of [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)][X] (X = Cl, 12; H2N{B(C6F5)3}2, 12′). | ||
The geometry about the Sb atom in 12 (Fig. 5) is pseudo-trigonal bipyramidal (see ESI‡ for details). Complex 12 forms a loose ion pair with a long Sb1–Cl1′ distance (3.6761(14) Å). The Cl− anion is engaged in H-bonding with the OH group (OH⋯Cl = 2.1538(341) Å).63 Complexes 12 and 3 present noticeable structural differences. In particular, the C–Ohydroxyl distance in 12 (C16–O1 = 1.373(4) Å) is longer than the corresponding C–Ooxy one in 3 (1.2708(16) Å). The discrepancies in the metric parameters from 3 to 12 indicate protonation of the O atom, re-aromatisation of the C13–C14–C15–C16–C17–C18 ring, and loss of quinoidal character. The molecular solid-state structure of 12′ was also elucidated (Fig. S62‡). Characteristically, there is no interaction between the cation and [H2N{B(C6F5)3}2]−.64,65
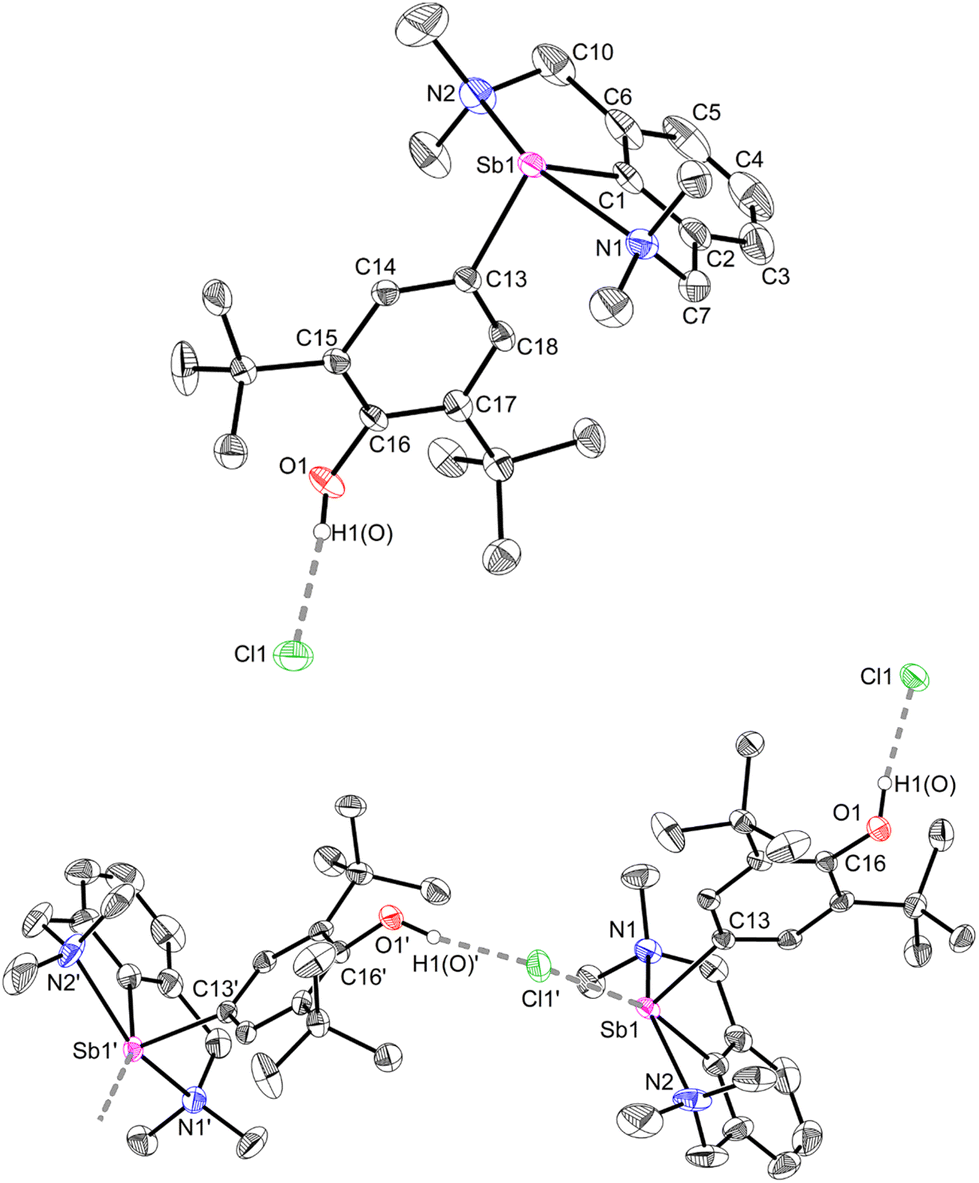 | ||
| Fig. 5 Top: representation of the molecular structure of [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)][Cl] (12). Bottom: arrangement of two adjacent units in the lattice. H atoms (except that on O atoms) and non-interacting CH2Cl2 molecules omitted for clarity. Ellipsoids at the 50% probability level. Selected distances (Å) and angles (°): Sb1–C1 = 2.106(4), Sb1–C13 = 2.141(4), Sb1–N1 = 2.398(4), Sb1–N2 = 2.453(4), Sb1–Cl1′ = 3.6761(14), C13–C14 = 1.387(5), C13–C18 = 1.389(5), C14–C15 = 1.404(5), C15–C16 = 1.410(5), C16–C17 = 1.422(5), C17–C18 = 1.387(5), O1–C16 = 1.373(4), O1–H1(O) = 0.909(6), Cl1–H1(O) = 2.154 (34); C1–Sb1–C13 = 95.52(15), C1–Sb1–N1 = 74.59(15), C1–Sb1–N2 = 74.41(18), C13–Sb1–N1 = 90.80(13), C13–Sb1–N2 = 90.55(14), N1–Sb1–N2 = 148.95(14). | ||
Complex 12 reacts with equimolar [{2,6-tBu2-C6H3O}K] in THF-d8 to afford a 1:1 mixture of 3 and 2,6-tBu2-C6H3OH (Fig. S52‡). Two pathways can be envisaged. The reaction may occur via salt metathesis that implicates an intermediate [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)(OC6H3-tBu2-2,6)] akin to putative 11. Alternatively, since this intermediate could not be isolated nor seen spectroscopically, deprotonation of 12 by [{2,6-tBu2-C6H3O}K] with subsequent extrusion of KCl could also take place. To distinguish between these two pathways, the reaction of 12 with one equivalent of [{2,6-iPr2-C6H3O}K] was carried out. It afforded half equivalents of 3, 1 and 2,6-tBu2-C6H3OH (Scheme 5; Fig. S53‡).
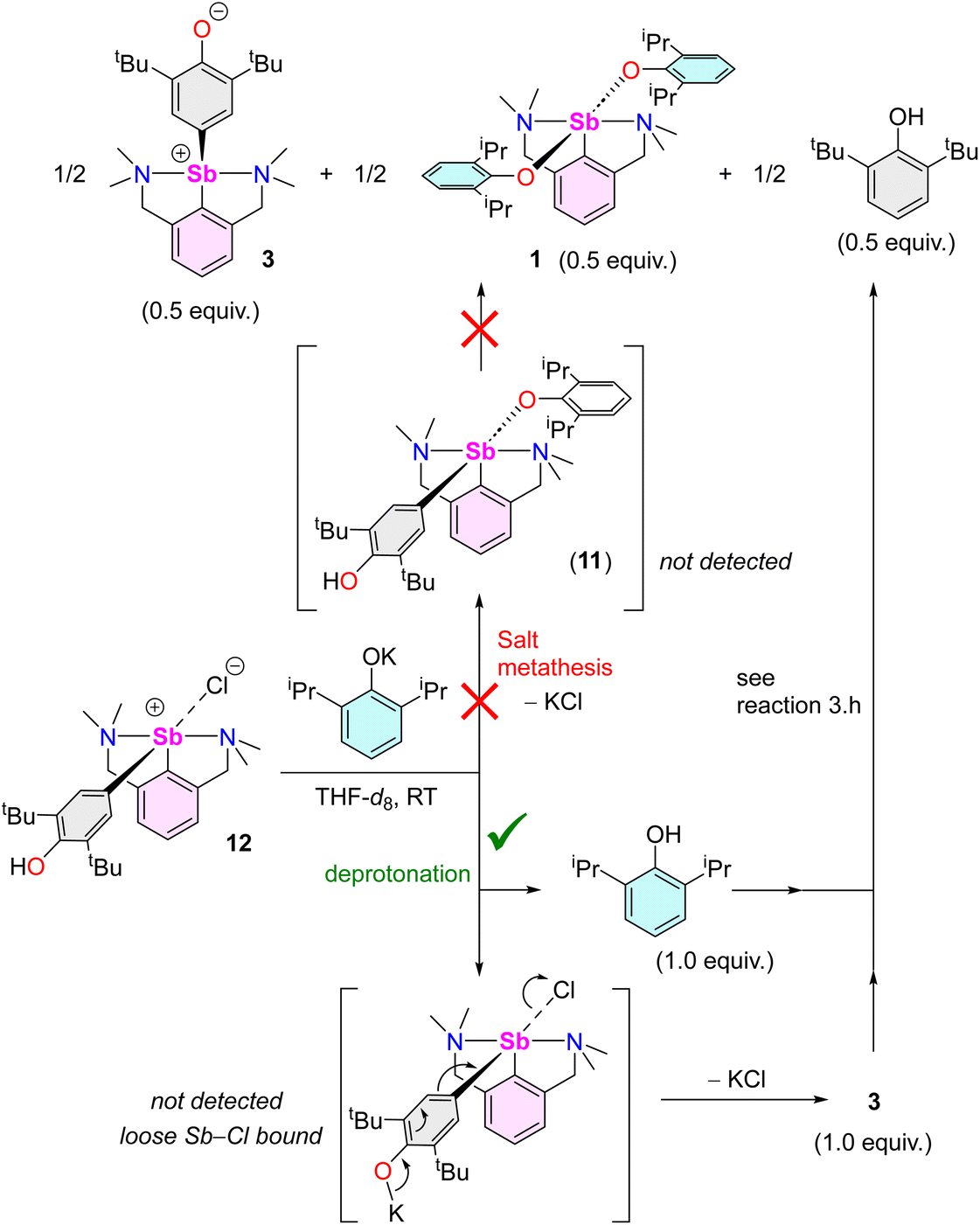 | ||
| Scheme 5 Reaction of [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)(Cl)] (12) with [{2,6-iPr2-C6H3O}K], highlighting the two potential mechanistic pathways. | ||
Again, initial steps consisting of salt metathesis progressing through 11 or deprotonation of 12 by [{2,6-iPr2-C6H3O}K] could both account for this outcome. If generated upon deprotonation of 12, complex 3 and 2,6-iPr2-C6H3OH66 would indeed further react to give half equivalents of 1, 3 and 2,6-tBu2-C6H3OH, as learnt from reaction (3.h). Low temperature NMR in THF-d8 provided conclusive evidence. The equimolar reaction of 12 with [{2,6-iPr2-C6H3O}K] at −40 °C led to a ca. 1:1 mixture of 3 and 2,6-iPr2-C6H3OH (Fig. S54‡). Upon warming to 20 °C, this mixture evolved toward the final production of 3, 1 and 2,6-tBu2-C6H3OH in equal concentrations. In addition, the DOSY spectrum recorded at −40 °C in THF-d8 indicates the presence of two major species, assigned as 3 and 2,6-iPr2-C6H3OH; it was independently confirmed by cross-referencing with the 1H NMR spectrum of a 1:1 mixture of these two compounds (THF-d8, −40 °C). At any rate, the fact that two species are detected in substantial concentrations rules out a mechanism involving the formation of a single intermediate 11. Hence, the reaction of 12 with [{2,6-iPr2-C6H3O}K] proceeds via initial deprotonation, followed by reaction of the resulting 2,6-iPr2-C6H3OH with 3 (Scheme 5).67
Mechanistic proposal
DFT calculations at the PBE0/Def2TZVP-D3(BJ) level were performed to provide insights into the stability of the complexes and intermediates discussed therein and to shed some light on the mechanism leading to 3. Calculations on the isolated bisphenolates [{NCNMe4}Sb(OC6H3-iPr2-2,6)2] (1), [{NCNMe4}Sb(OC6H2-tBu3-2,4,6)2] (7) and [{NCNMe4}Sb(OC6H2-tBu2-2,4)2] (8), as well as the non-observed [{NCNMe4}Sb(OC6H3-tBu2-2,6)2] analogue and the non-sterically hindered [{NCNMe4}Sb(OC6H3-Me2-2,6)2] species, indicate rather similar structural and electronic properties for the five complexes (Table S3‡). The Sb–O distance in [{NCNMe4}Sb(OC6H3-tBu2-2,6)2] is only 0.060 and 0.056 Å longer than in [{NCNMe4}Sb(OC6H3-Me2-2,6)2] and 1, respectively. However, the corresponding Wiberg bond indices suggest somehow weaker covalency in the case of the first, more encumbered complex. Complex 7 exhibits similar bonding features as [{NCNMe4}Sb(OC6H3-tBu2-2,6)2], whereas the less sterically hindered 8 presents stronger Sb-phenolate bonding. The atomic charges are indicative of a non-negligible ionic bonding component, especially for [{NCNMe4}Sb(OC6H3-tBu2-2,6)2] and 7. The related single –NMe2 tethered [{CNMe2}Sb(OC6H3-tBu2-2,6)2] (5) exhibits similar bonding features, but with a somewhat lower HOMO–LUMO gap, owing to its unsaturation. To summarise, all computed bisphenolato species have similar stability and bonding mode, the two more sterically hindered species [{NCNMe4}Sb(OC6H3-tBu2-2,6)2] and 7 having moderately weaker Sb–ligand bonds. Similarly, calculations on 3 and its hypothetical oxyaryl analogues [{NCNMe4}Sb(C6H2-R2-3,5-O-4)] (R = Me and iPr) did not reveal any noticeable stability or structural differences between them (Table S4‡). In particular, they all exhibit partial double bond character of the Sb–C(aryl) bond (compare the Sb–C(aryl) and Sb–C(NCNMe4) Wiberg bond indices in Table S3‡), together with non-negligible zwitterionic character (see the Sb and O NBO charges on Table S4‡). The single –NMe2 tethered elusive species 6 also presents similar bonding features. Its lower HOMO–LUMO gap is related to its LUMO lower energy, an accepting orbital whose main lobe points in a direction approximately trans to N (Fig. S64‡), thus explaining the oligomerisation of 6. Accordingly, formation of 64 from 6 is estimated to be exothermic by 27 kcal mol−1 in THF (total energy).On the basis of our computed results and all the above-reported experimental findings, we suggest that 3 is the outcome of a succession of heterolytic bond cleaving and forming processes (Scheme 6), in a mechanism that bears some resemblance with that described for the double C–H activation of bismuth-bound diphenyl amide.68 A process of formation of 3 by reaction of [{NCNMe4}SbCl2] with [{2,6-tBu2-C6H3O}K] involving free radicals, similar to what was seen with BiCl310 or proposed in the case of [{NCNMe4}BiCl2],11 should be excluded. Instead, our mechanistic proposal involves partially charged species. Complex 3 itself has a pronounced ionic character, as indicated by the conductivity measurements. In this context, it is worth noting that related bismuth(III) bis(phenolate)s bearing NCN tridentate ligands have been shown to present unusually long Bi–OAr interatomic distances in the solid state and, in fact, that they dissociate in THF solution to generate [{NCN}Bi(OAr)]+[OAr]− separated ion pairs;43,69 similar phenomena can be considered for the lighter Sb(III) derivatives. Since the Sb-bis(phenolate)s 5 and 7 can be synthesised and calculations indicate identical stability for 7 and [{NCNMe4}Sb(OC6H3-tBu2-2,6)2], we propose this later species can be obtained by salt metathesis and acts as a transient, non-observable intermediate (step 1). Our computational search for a subsequent intramolecular rearrangement leading to the formation of an intermediate in which one of the phenolato ligand has undergone dearomatisation and is now bonded through Coxyaryl was unsuccessful. Rather, our calculations indicate that decoordination of one phenolate should occur, followed by its head-to-tail reversal and the formation of a Sb–Coxyaryl(sp3) bond accompanied by the decoordination of the second phenolate ligand. Two ion pairs have been successively identified during this process, namely Int1 and Int2 in Scheme 6. Even if one cannot be certain that the last ion pair structure is its global energy minimum, it suggests that the leaving phenolate tears out the not-too-remote proton attached to Cpara, to produce 2,6-tBu2-C6H3OH and 3. Owing to the complexity of the potential energy surface associated with the ion pair ground state and that of the subsequent C–H activation reaction, it was not possible to evaluate the energy barrier of the latter. However, the almost thermoneutral computed thermodynamic balance of the whole process consisting in the formation of 3 agrees with its observation (Scheme 6). Since no important electronic differences were found in our various computed species between those bearing iPr and tBu substituents, the fact that this reaction is not observed in the case of iPr (and OMe) suggests that the formation of 3 is to some extent favoured by the weaker Sb–O bonds in the corresponding bisphenolate intermediate. Yet, we propose that it is chiefly dictated by the specific shape and volume of the tBu groups. They control the specific topology of the Int2 ion pair (Scheme 6; note that this intermediate is reminiscent of Sb-promoted Friedel–Crafts reactions70,71) in a way that favours, among several types of non-covalent interactions, the proper Coxyaryl–H⋯OC6H3-tBu2-2,6 one, thus preparing the C–H activation step. It should be noted that the reaction free energies given in Scheme 6 were computed with solvent (THF) corrections. The computed thermodynamic balance of the whole reaction in vacuum is much higher (23.7 kcal mol−1). This result stresses the importance of solvent effect, which could not be considered explicitly in the calculations; note also that KCl could only be treated, once formed, as a non-interacting spectator. Both factors may play a role in the critical discrimination between the reaction product formations in Scheme 1. Our mechanistic proposal involves ionic intermediates rather than neutral radicals. Consistent with this, the formation of 3 is much slower in toluene than in the more polar THF: where complete formation of the complex is observed within the first point of analysis in THF-d8 (ca. 10 min), the reaction is not yet over after 24 h in tol-d8.
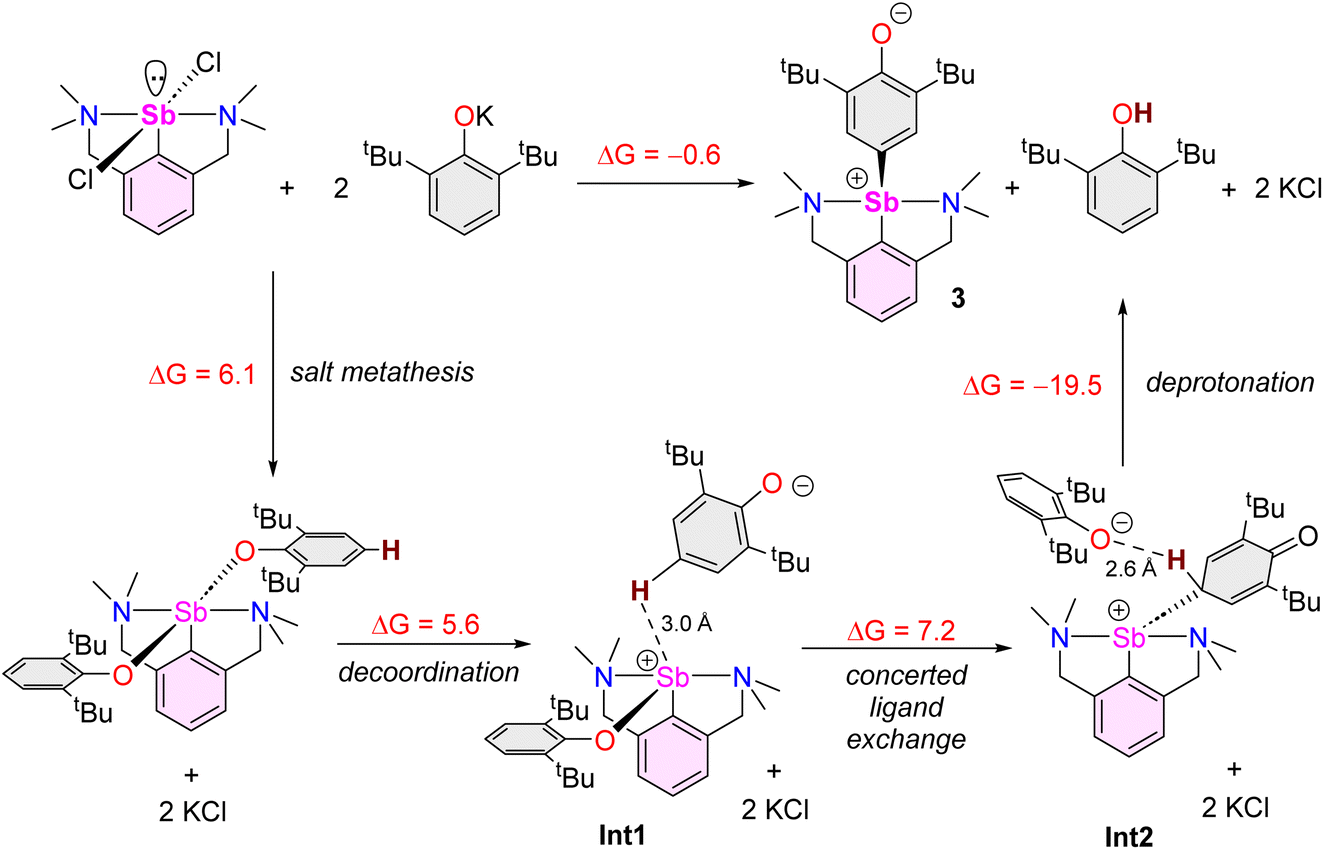 | ||
| Scheme 6 Proposed pathway for the formation of 3 based on both experimental and computational data. The free energies at 298 K are given in kcal mol−1. They were calculated assuming solvent (THF) effects (see Computational details). | ||
Further reactivity studies
Beyond the reactions leading to 12 and 12′, the reactivity of complex 3 towards various reagents was examined (Scheme 7, Fig. S58–S60‡). The nucleophilic nature of the Coxyaryl atom was evidenced by reaction of 3 with excess CS2. It proceeded to return [{NCNMe4}Sb(S2C-C6H2-tBu2-3,5-O-4)] (13), the product of insertion of CS2 in the Sb–Coxyaryl bond. By contrast, no reaction takes place between 12 and CS2, hence highlighting that the greater nucleophilic character of the Coxyaryl atom in zwitterion-like 3 is required for insertion to occur. The synthesis of 13 is a rare example of insertion chemistry into a Sb(III)–C bond.72 Our attempts to assess potential radical behaviour in 3 gave mitigated results. The reaction with S8 gave a mixture of the diphenoquinone A, [{NCNMe4}Sb(μ2-S)]2 and crystalline [({NCNMe4}Sb(μ-S5))2],73 but we were unable to delineate the details of this reaction. Treatment of 3 with excess TEMPO gave mixtures of unreacted 3, tetramethylpiperidine (TMP) and crystals of A and of [{NCNMe4}Sb(μ2-O)]2,74 the latter being the outcome of abstraction of the oxygen in TEMPO and formation of TMP. Upon reaction with N-iodosuccinimide, 3 generated a mixture of [{NCNMe4}Sb(I)(succinimide)] (ca. 80%), crystalline [{NCNMe4}Sbl2] (ca. 20%) and A.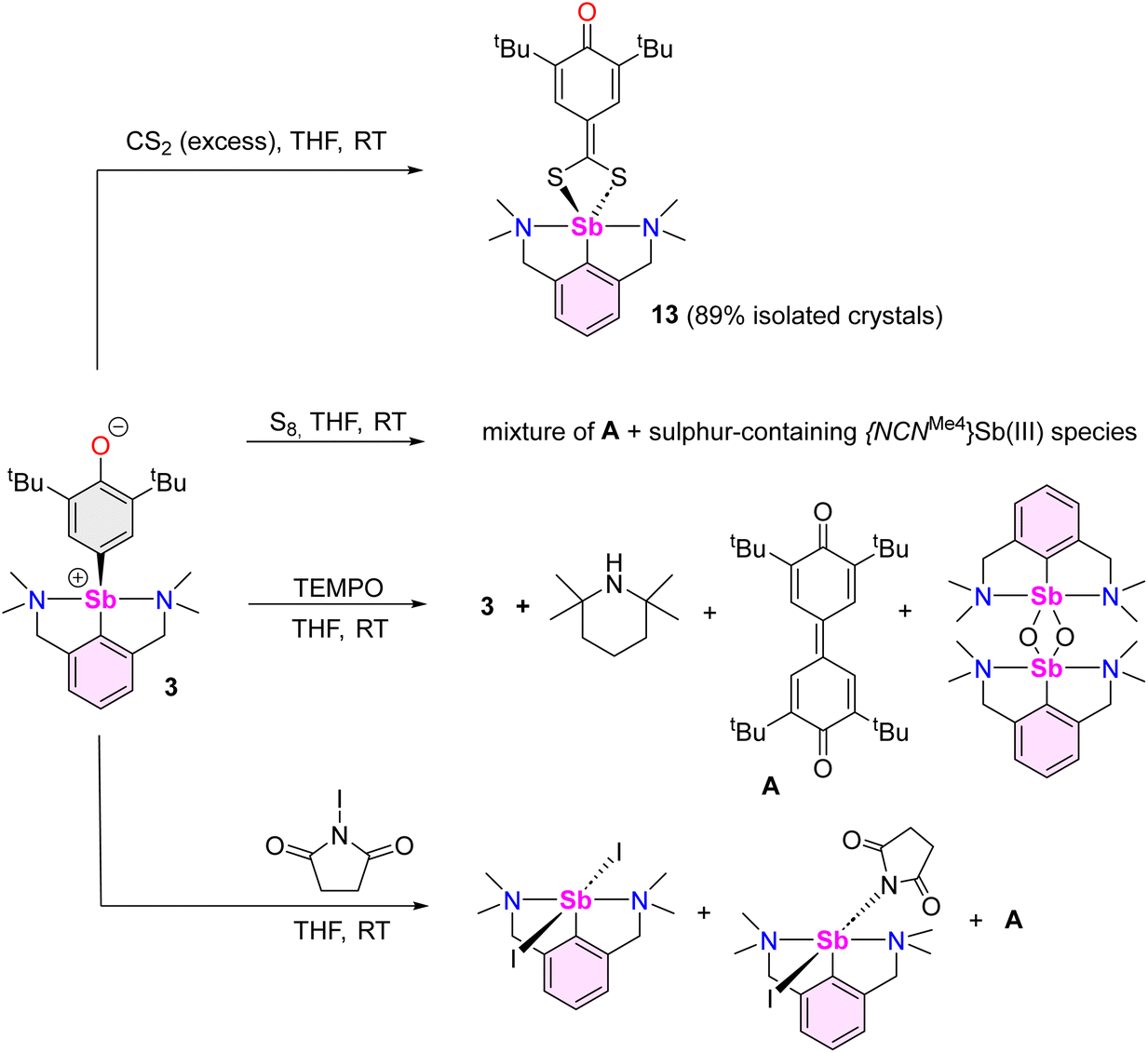 | ||
| Scheme 7 Probing the reactivity of 3. | ||
Compound 13 recrystallised as a five-coordinate hemidirected complex (Fig. 6). The metric parameters around Sb(III) match those in [{NCNMe4}Sb(S3C)].72 The C17–O1 bond length (1.248(2) Å) in 13 is marginally shorter than the corresponding one in 3 (1.2708(16) Å). The substantial variations of the Ci–Ci+1 bond lengths in the S2C-C6H2-tBu2-3,5-O-4 ligand (compare for instance C17–C18, 1.475(2) Å, and C18–C19, 1.355(2) Å) indicate dearomatisation of the cyclic core, although the atoms S1, S2, C13 to C19 and O1 remain coplanar. Compound 13 hence adopts a quinoid-like arrangement. By comparison, the Ci–Ci+1 interatomic distances in [(p-tol-CS2)3Sb], in the range 1.3594(91)–1.4233(88) Å,75 agree better with those expected in an aromatic ring. The C13–C14 interatomic distance matches that for a double bond, e.g. it is identical to that in a Pd-(methylene-1,1′-dithiolato)-cyclohexanedione complex (1.395(6) Å),76 and hence the bidentate ligand in 13 must be regarded as a dithiolate.
 | ||
| Fig. 6 Representation of the molecular structure of [{NCNMe4}Sb(S2C-C6H2-tBu2-3,5-O-4)] (13). H atoms and non-interacting pyridine molecules omitted for clarity. Ellipsoids at the 50% probability level. Selected distances (Å) and angles (°): Sb1–C1 = 2.1429(17), Sb1–N1 = 2.5492(15), Sb1–N2 = 2.6003(15), Sb1–S1 = 2.5505(5), Sb1–S2 = 2.5723(5), C17–O1 = 1.248(2), S1–C13 = 1.7369(17), S2–C13 = 1.7364(17), C13–C14 = 1.395(2), C14–C15 = 1.429(2), C14–C19 = 1.431(2), C15–C16 = 1.358(2), C16–C17 = 1.474(2), C17–C18 = 1.475(2), C18–C19 = 1.355(2); C1–Sb1–N1 = 72.36(6), C1–Sb1–N2 = 71.69(6), C1–Sb1–S1 = 96.31(5), C1–Sb1–S2 = 96.96(5), N1–Sb1–N2 = 126.95(5), N1–Sb1–S1 = 78.32(4), N1–Sb1–S2 = 144.59(4), S1–Sb1–N2 = 143.01(4), S1–Sb1–S2 = 69.240(15), S2–Sb1–N2 = 77.48(3). | ||
Conclusion
The mechanism of C–H bond activation leading to the selective formation of [{NCNMe4}Sb(C6H2-tBu2-3,5-O-4)] (3) starting from [{NCNMe4}SbCl2]42 and [{2,6-tBu2-C6H3O}K] has been probed. Our experimental and DFT data indicate it proceeds through heterolytic bond breaking and bond forming steps involving charged species; no evidence for a radical-based pathway was detected by EPR, and 3 is formed even in the presence of galvinoxyl or TEMPO. This contrasts with the pertaining chemistry of the larger congener, Bi(III).10,11 Complex 3 reacts with CS2 to afford [{NCNMe4}Sb(S2C-C6H2-tBu2-3,5-O-4)] (13), following a migratory insertion step that highlights the highly nucleophilic character of the Coxyaryl atom in 3. By contrast, [{NCNMe4}Sb(C6H2-tBu2-3,5-OH-4)]+[Cl]− (12) is inert towards CS2.Representative interatomic distances for 3, 12, 13 and 3,3′,5,5′-tetra-tert-butyldiphenoquinone (A) are summarised in Fig. 7.77 Key comparative structural features include the followings: (i) the {C6H2-tBu2-3,5-OH}− ligand in 12 features an aromatic ring, with C–O distances as expected for a phenol, (ii) the S2C-C6H2-tBu2-3,5-O-4 ligand in 13 adopts a quinoid-like pattern, with large variations between C–C bond lengths, and (iii) the main bond distances in 3 are intermediate between those in A or 13 and those in 12. The oxyaryl fragment in 3 is hence intermediate between quinoidal and aromatic, and the Sb–Coxyaryl bond features some double bond character. We note that the length of the SbVC(R)(R′) double bond reported for a triphenylstibonium(V) ylide, 2.049(4) Å,78 is only slightly shorter than that in 3; however, the different oxidation states between the compounds limits the usefulness of this comparison.
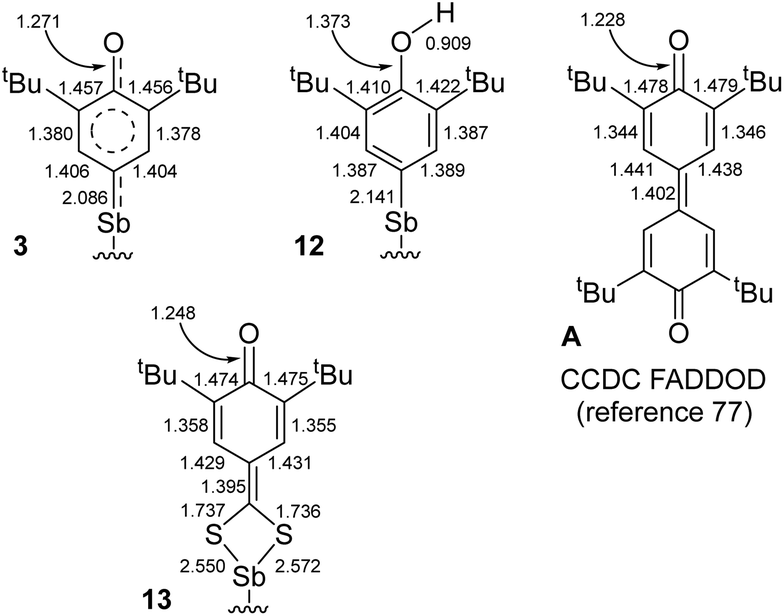 | ||
| Fig. 7 Selected interatomic distances in 3, 12, 13 and A. Supporting ligand framework {NCNMe4}− and ESD's omitted for clarity. | ||
Driven by the impressively successful implementation of bismuth in molecular catalysis in recent years,14–18 and by the intriguingly different behaviours of antimony(III) and bismuth(III) towards phenolates disclosed herein, we will next assess how 3 and related phenolato complexes can be exploited in efficient catalytic manifolds, and whether further specificities of antimony with respect to its heavier congener, bismuth, can be highlighted. The discovery and the understanding of elementary reactions such as those presented herein, associated to the mechanistic changeover observed between congeneric Sb and Bi complexes, constitute a necessary first step in developing applications in fields such as catalysis.
Data availability
The experimental and theoretical data that supports the findings and conclusions of this study are available in the ESI‡ of this article.Author contributions
G. D. carried out the synthetic and spectroscopic experimental work and participated to the data analysis and their processing, and to the preparation of the manuscript. M. C. solved the crystallographic structures. A. P. participated to the synthetic experimental work. S. Kahlal and J.-Y. Saillard performed the theoretical calculations and their analysis, and participated to the preparation of the manuscript. C. Silvestru participated to data analysis, planning of experimental work and manuscript preparation. Y. Sarazin was the lead scientist; he secured the funding, participated to experiment planning, to data analysis, and to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the French Agence Nationale de la Recherche for the provision of a research grant to G. D. (BiMeDep project, ANR-21-CE07-0045-02). The financial support through a grant of Romanian Ministry of Research and Innovation, CNCS – UEFISCDI, project number PN-III-P4-ID-PCE-2020-2651, is highly acknowledged. We thank Dr Albert Soran as well as the support provided by the National Centre for X-ray Diffraction (Babeş-Bolyai University, Cluj-Napoca, Romania) for the elucidation of the XRD structure of complex 4. The French Grand Equipment National de Calcul Intensif is acknowledged for HCP support (Project a0010807367).References
- C. I. Raţ, A. Soran, R. A. Varga and C. Silvestru, Adv. Organomet. Chem., 2018, 70, 233–311 CrossRef.
- H. Braunschweig, C. Drost, P. B. Hitchcock, M. F. Lappert and L. J.-M. Pierssens, Angew. Chem., Int. Ed. Engl., 1997, 36, 261–263 CrossRef CAS.
- G. Tan, T. Szilvási, S. Inoue, B. Blom and M. Driess, J. Am. Chem. Soc., 2014, 136, 9732–9742 CrossRef CAS PubMed.
- N. Maudoux, J. Fang, T. Roisnel, V. Dorcet, L. Maron, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2014, 20, 7706–7717 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed.
- J. C. Mullins, K. Yuvaraj, Y. Jiang, G. P. Van Trieste, III, A. Maity, D. C. Powers and C. Jones, Chem. – Eur. J., 2022, 28, e202202103 CrossRef CAS PubMed.
- J. Mai, M. Morasch, D. Jędrzkiewicz, J. Langer, B. Rösch and S. Harder, Angew. Chem., Int. Ed., 2023, 62, e202212463 CrossRef CAS PubMed.
- T. A. Hanna, Coord. Chem. Rev., 2004, 248, 429–440 CrossRef CAS.
- T. A. Hanna, A. L. Rieger, P. H. Rieger and X. Wang, Inorg. Chem., 2002, 41, 3590–3592 CrossRef CAS PubMed.
- I. J. Casely, J. W. Ziller, M. Fang, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 5244–5247 CrossRef CAS PubMed.
- D. R. Kindra, I. J. Casely, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 7777–7787 CrossRef CAS PubMed.
- T. Hynes, J. D. Masuda and S. S. Chitnis, ChemPlusChem, 2022, 87, e202200244 CrossRef CAS PubMed.
- F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235–4240 CrossRef CAS PubMed.
- O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- M. Mato, D. Spinnato, M. Leutzsch, H. Won Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CrossRef CAS PubMed.
- C. I. Raţ, C. Silvestru and H. J. Breunig, Coord. Chem. Rev., 2013, 257, 818–879 CrossRef.
- P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef PubMed.
- C. Ganesamoorthy, C. Wölper, A. S. Nizovtsev and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 4204–4209 CrossRef CAS PubMed.
- R. J. Schwamm and M. P. Coles, Chem. – Eur. J., 2019, 25, 14183–14191 CrossRef CAS PubMed.
- K. Dollberg, S. Schneider, R.-M. Richter, T. Dunaj and C. von Hänisch, Angew. Chem., Int. Ed., 2022, 61, e202213098 CrossRef CAS PubMed.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, Angew. Chem., Int. Ed., 2023, 62, e202302071 CrossRef CAS PubMed.
- L. Dostál, R. Jambor, M. Aman and M. Hejda, ChemPlusChem, 2020, 85, 2320–2340 CrossRef PubMed.
- G. A. Olah and R. H. Schlosberg, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS.
- G. A. Olah, G. Klopman and R. H. Schlosberg, J. Am. Chem. Soc., 1969, 91, 3261–3268 CrossRef CAS.
- G. A. Olah and Y. K. Mo, J. Am. Chem. Soc., 1972, 94, 6864–6865 CrossRef CAS.
- G. A. Olah, R. Renner, P. Schilling and Y. K. Mo, J. Am. Chem. Soc., 1973, 95, 7686–7692 CrossRef CAS.
- G. A. Olah, N. Yoneda and D. G. Parker, J. Am. Chem. Soc., 1976, 98, 5261–5268 CrossRef CAS.
- A. Tanoue, W.-J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 269–273 CrossRef CAS.
- P. J. F. de Rege, J. A. Gladysz and I. T. Horváth, Adv. Synth. Catal., 2002, 344, 1059–1062 CrossRef CAS.
- J. Xia, R. Qiu, S. Yin, X. Zhang, S. Luo, C.-T. Au, K. Xia and W.-Y. Wong, J. Organomet. Chem., 2010, 695, 1487–1492 CrossRef CAS.
- J. Lei, L. Peng, R. Qiu, Y. Liu, Yi Chen, C.-T. Au and S.-F. Yin, Dalton Trans., 2019, 48, 8478–8487 RSC.
- L. Capaldo, M. Ertl, M. Fagnoni, G. Knör and D. Ravelli, ACS Catal., 2020, 10, 9057–9064 CrossRef CAS PubMed.
- L. Wang, S. Fadlallah, C. Bellini, C. Orione, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Organometallics, 2015, 34, 1321–1327 CrossRef CAS.
- A.-A. Someşan, E. Le Coz, T. Roisnel, C. Silvestru and Y. Sarazin, Chem. Commun., 2018, 54, 5299–5302 RSC.
- A.-A. Someşan, E. Le Coz, C. I. Raţ, V. Dorcet, T. Roisnel, C. Silvestru and Y. Sarazin, Chem. – Eur. J., 2019, 25, 16236–16240 CrossRef PubMed.
- G. Strîmb, A. Pöllnitz, C. I. Raţ and C. Silvestru, Dalton Trans., 2015, 44, 9927–9942 RSC.
- M. Wieber and N. Baumann, Z. Anorg. Allg. Chem., 1973, 402, 43–46 CrossRef CAS.
- G. Duneş and C. Silvestru, New J. Chem., 2024, 48, 5523–5529 RSC.
- D. A. Atwood, A. H. Cowley and J. Ruiz, Inorg. Chim. Acta, 1992, 198–200, 271–274 CrossRef CAS.
- I. J. Casely, J. W. Ziller, B. J. Mincher and W. J. Evans, Inorg. Chem., 2011, 50, 1513–1520 CrossRef CAS PubMed.
- E. Stellwagen, J. D. Prantner and N. C. Stellwagen, Anal. Biochem., 2008, 373, 407–409 CrossRef CAS PubMed.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. L. Hughes and M. Bochmann, Organometallics, 2002, 21, 451–453 CrossRef CAS.
- A. Ahmed, R. A. Bragg, J. Clayden, L. W. Lai, C. McCarthy, J. H. Pink, N. Westlund and S. A. Yasin, Tetrahedron, 1998, 54, 13277–13294 CrossRef CAS.
- Y. Yamamoto, X. Chen, S. Kojima, K. Ohdoi, M. Kitano, Y. Doi and K.-Y. Akiba, J. Am. Chem. Soc., 1995, 117, 3922–3932 CrossRef CAS.
- R. A. Nyquist, Appl. Spectrosc., 1982, 36, 533–535 CrossRef CAS.
- J. Rigaudy and S. P. Klesney, Nomenclature of Organic Chemistry, Sections A, B, C, D, F and H, Pergamon Press, Oxford, 1979 Search PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- R. Kather, T. Svoboda, M. Wehrhahn, E. Rychagova, E. Lork, L. Dostál, S. Ketkov and J. Beckmann, Chem. Commun., 2015, 51, 5932–5935 RSC.
- G. Duneş, A. Soran and C. Silvestru, Dalton Trans., 2022, 51, 10406–10419 RSC.
- L. M. Opris, A. Silvestru, C. Silvestru, H. J. Breunig and E. Lork, Dalton Trans., 2003, 4367–4374 RSC.
- C. Jones, J. W. Steed and R. C. Thomas, J. Chem. Soc., Dalton Trans., 1999, 1541–1542 RSC.
- J. Krüger, C. Wölper and S. Schulz, Organometallics, 2022, 41, 3788–3793 CrossRef.
- J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 1669–1678 CrossRef.
- M. A. Jackisch, F. R. Fronczek, C. C. Geiger, P. S. Hale, W. H. Daly and L. G. Butler, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 919–922 CrossRef.
- G. G. Aleksandrov, Y. T. Struchkov, D. I. Kalinin and M. G. Neigauz, Zh. Strukt. Khim., 1973, 14, 797–803 Search PubMed.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- Complex 64 is insoluble in hydrocarbons, dichloromethane, THF, pyridine, acetonitrile, DMSO and water. We could not record its NMR data.
- X. Kou, X. Wang, D. Mendoza-Espinosa, L. N. Zakharov, A. L. Rheingold, W. H. Watson, K. A. Brien, L. K. Jayarathna and T. A. Hanna, Inorg. Chem., 2009, 48, 11002–11016 CrossRef CAS PubMed.
- CSD version 5.43, last update November 2023.
- Note that the position of H1(O) atoms in 12 was not idealised but localised by Fourier difference map analysis.
- Y. Sarazin, D. L. Hughes, N. Kaltsoyannis, J. A. Wright and M. Bochmann, J. Am. Chem. Soc., 2007, 129, 881–894 CrossRef CAS PubMed.
- The only significant difference between the metric parameters of the cations in 12 and 12′ is the length of the O–H bond, which is much larger in 12 (0.909(6) Å) because of OH⋯Cl hydrogen bonding than in 12′ (0.76(3) Å); the O1–H1(O) bond in 12′ is coplanar with the aromatic ring, whereas it was orthogonal in 12. See ESI‡ for details.
- We have checked by NMR monitoring in THF-d8 that complex 1 does not react with 2,6-tBu2-C6H3OH (3 equivalents), even after overnight reflux.
- Consistent with this conclusion, the reaction of 12 with KN(SiMe3)2 in THF-d8 gives 3, KCl and HN(SiMe3)2, without formation of [{NCNMe4}Sb{N(SiMe3)2}(C6H2-tBu2-3,5-O-4)K], see Fig. S56.‡.
- B. Ritschel, J. Poater, H. Dengel, F. M. Bickelhaupt and C. Lichtenberg, Angew. Chem., Int. Ed., 2018, 57, 3825–3829 CrossRef CAS PubMed.
- H. W. Moon, F. Wang, K. Bhattacharyya, O. Planas, M. Leutzsch, N. Nöthling, A. A. Auer and J. Cornella, Angew. Chem., Int. Ed., 2023, 62, e202313578 CrossRef CAS PubMed.
- G. A. Olah, P. Schilling and I. M. Gross, J. Am. Chem. Soc., 1974, 96, 884–892 CrossRef CAS.
- G. A. Olah and J. Nishimura, J. Am. Chem. Soc., 1974, 96, 2214–2220 CrossRef CAS.
- For the insertion of CS2 into Sb(III)–S bonds, see: L. Dostál, R. Jambor, A. Růžička, R. Jirásko, E. Černošková, L. Beneš and F. de Proft, Organometallics, 2010, 29, 4486–4490 CrossRef.
- L. Dostál, R. Jambor, A. Růžička, R. Jirásko, V. Lochař, L. Beneš and F. de Proft, Inorg. Chem., 2009, 48, 10495–10497 CrossRef PubMed.
- L. Dostál, R. Jambor, A. Růžička, M. Erben, R. Jirásko, E. Černošková and J. Holeček, Organometallics, 2009, 28, 2633–2636 CrossRef.
- M. Kimura, A. Iwata, M. Itoh, K. Yamada, T. Kimura, N. Sugiura, M. Ishida and S. Kato, Helv. Chim. Acta, 2006, 89, 747–783 CrossRef CAS.
- K. K. Manar, A. N. Gupta, A. K. Gupta, L. B. Prasad, P. Srivastava, M. G. B. Drew and N. Singh, ChemistrySelect, 2017, 2, 2655–2664 CrossRef CAS.
- M. A. Khan, A. Osman and D. G. Tuck, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1399–1402 CrossRef.
- G. Ferguson, C. Glidewell, I. Gosney, D. Lloyd, S. Metcalfe and H. Lumbroso, J. Chem. Soc., Perkin Trans. 2, 1988, 1829–1837 RSC.
Footnotes |
| † Dedicated to Prof. Hans J. Breunig, in recognition of his immense contribution to the chemistry of organopnictogens. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, spectroscopic characterisations, crystallographic and computational data. CCDC 2335992–2335998. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01400f |
| This journal is © The Royal Society of Chemistry 2024 |