
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence-based pH-shift assay with wide application scope for high-throughput determination of enzymatic activity in enzyme mining and engineering†‡
Avinash Vellore
Sunder
,
Marie-Luise
Reif
and
Wolf-Dieter
Fessner
 *
*
Department of Organic Chemistry & Biochemistry, Technical University of Darmstadt, Peter-Grünberg-Straße 4, 64287 Darmstadt, Germany. E-mail: fessner@tu-darmstadt.de
First published on 2nd August 2024
Abstract
A number of enzymes important for biocatalyst development or as drug targets are associated with a pH shift during their catalytic reaction, owing to the concommitant release or uptake of protons. Here, we show that an enzyme assay developed using the fluorescent pH indicator HPTS can be adapted for reliable and continuous activity determination of representative enzymes from multiple EC classes that operate in the viable pH range 5.5–8.5, using ratiometric measurement (F485/F405). Kinetic measurements obtained with this method closely match literature values determined using other assay types. Further, the assay was employed to screen variants of transketolase from Geobacillus stearothermophilus (TKgst) aimed at engineering substrate promiscuity and remote enantioselectivity for 3-hydroxyaldehydes. The fluorescence-based assay displayed 70-fold improved sensitivity in comparison to an absorption-based assay for transketolase screening, with a limit of detection of 0.044 mM and Z-factor of 0.52. Double-site mutagenesis at the G264 and S385 positions yielded variants with 5–15-fold increased activity on the tested 3-hydroxyaldehydes compared to the TKgst (L382F) base variant. Although the directed evolution engineering strategy did not achieve significant remote enantioselectivity in this first round of mutagenesis, the simple fluorescence-based pH-shift assay was shown to be useful as a versatile primary high-throughput screen for in vitro enzyme engineering.
Introduction
Monitoring enzyme activity is of fundamental importance in enabling drug development and accelerating industrial biocatalysis.1 Activity assays play an important role in facilitating the discovery of new enzymes, profiling substrate or inhibitor scope, and rapid screening of directed evolution libraries to identify improved enzyme variants.2 It is therefore essential to develop simple and convenient methods that enable continuous monitoring of enzyme activity with high-throughput parallelization.3 In particular, a generic assay principle can facilitate application of the assay to a wide range of enzymes and alternative substrates. Several enzymatic reactions are accompanied by the release or uptake of protons, thereby leading to a change in pH of the reaction medium.4 The use of a pH indicator in a weakly buffered reaction medium therefore offers a rapid and convenient means of monitoring enzyme activity continuously by using a general-purpose pH-shift assay. Most importantly, employing such a generic chemical reaction principle for analysis uncouples the assay from structural requirements concerning the substrate/product or inhibitor, thereby allowing the direct use of unmodified native probes for the screening of enzyme variants.2 Indeed, several reports have detailed the development of pH-shift assays to monitor the activity of esterases,5 lipases,6 nitrilases,7 dehalogenases,8 decarboxylases,9 transketolases,10 kinases,11 nucleotidyl-,12 and glycosyltransferases13 in the presence of colorimetric pH indicators.However, fluorescence-based methods allow for lower background, higher sensitivity and greater dynamic range of an assay in comparison to conventional absorption measurements.14 Development of an intensive signal can be achieved despite a low concentration of the fluorescent dye, yet from very low substrate conversion under initial rate conditions where enzyme activity is usually measured.15 In a recent report, a derivative of the fluorescent pH indicator 8-hydroxypyrene-1,3,6-trisulfonate (HPTS, pyranine) has been employed in the determination of activity of haloalkane dehalogenases.16 HPTS has several favourable properties including a high fluorescence quantum yield, dual excitation nature facilitating ratiometric measurements, and pH sensitivity in the near neutral region,17 which makes it attractive for the development of a generic enzyme assay system (Scheme 1).
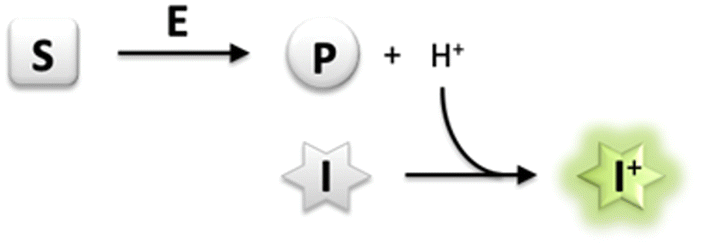 | ||
| Scheme 1 Generic principle of a pH-shift assay; exemplary illustration for dissociable product formation (S, substrate; E, enzyme; P, product; I, pH-responsive indicator dye). | ||
Therefore, in this study we have evaluated the suitability of HPTS as a general fluorescence indicator in pH-shift assays for the rapid quantitative activity determination of several representative enzymes from different EC classes, including esterase, decarboxylase, kinase, nucleotidyltransferase, and glycosyltransferase. In particular, we have focused on transketolase, a thiamine diphosphate (ThDP) dependent enzyme involved in carboligation of aldehyde substrates.18In vivo, transketolases catalyse the reversible transfer of a 2-carbon hydroxyacetyl unit from a donor ketose sugar phosphate to an acceptor aldose phosphate.19 However, they have garnered significant attention in synthetic biocatalysis in recent years with respect to their broad substrate scope. Transketolases from E. coli or Geobacillus stearothermophilus (TKgst) have been modified by semi-rational engineering to facilitate the binding of non-hydroxylated aliphatic20,21 and aromatic aldehydes,22,23 stereoinverted 2-hydroxyaldehydes,24 and diastereomeric aldoses25 for carboligation by using hydroxypyruvate as a carbonyl donor to drive an irreversible conversion. Transketolases have also been engineered for acceptance of alternative carbonyl donors such as pyruvate26 or non-native acceptors such as nitrosoarenes.27 In this context, we have herewith evaluated the alternative fluorescent pH-shift assay with HPTS in high-throughput screening format to screen a small TKgst mutagenesis library against chiral 3-hydroxyaldehydes in an attempt to further engineer substrate promiscuity and remote enantioselectivity.
Results and discussion
Use of a fluorescent pH indicator to measure enzyme activity
The estimation of enzyme activity by measuring a pH shift during the catalytic reaction requires the use of a sensitive and facile pH indicator. Most enzymes relevant for preparative applications are active under neutral to slightly alkaline physiological conditions (pH 7.0–8.5), and the quantification of reaction progress requires dyes with compatible pKα for optimal sensitivity. In addition, fluorescence measurements offer better dynamic range and sensitivity with low background as compared to reading absorbance. HPTS is a fluorescent pH indicator that has been extensively applied in the preparation of immobilized pH sensors.28 Besides being inexpensive, non-toxic, and water soluble, HPTS displays a very high quantum yield and photostability. The two excitation maxima of HPTS at λ = 405 and 450 nm facilitate ratiometric measurements, with emission in the fluorescein range allowing the use of a basic filter set for readout. More importantly, the pH sensitivity range of HPTS matches that of HEPES buffer (6.8–8.2), allowing the use of a single indicator/buffer system for application to multiple enzymes that operate within this pH range. Indeed, the use of a HPTS derivative has been recently demonstrated in the measurement of haloalkane dehalogenase activity.16 Although other fluorescent indicators are commercially available or have been synthesized,29 these are either expensive, limited to an acidic pH range or show poor water-solubility and quantum yield when compared to HPTS. Although we observed that fluorescent dyes like carboxyfluorescein or 4-methylumbelliferone were also suitable to monitor enzyme activity in the desired pH range, they did not lead to significant change in fluorescence intensity nor offered the advantage of ratiometric measurement (data not shown). We therefore attempted to extend the utility of HPTS to enable the development of a general-purpose assay suitable for activity measurement in multiple enzyme classes.The presence of three sulfonate groups makes the pKα of HPTS dependent on the chemical composition of the molecular environment and the ionic strength (IS).17 In our measurements in HEPES buffer (pH 5.5–9.5), the pKα of HPTS was determined to be 7.46 at 30 mM IS (Fig. 1), which matches that of HEPES buffer (pKα 7.48 at 25 °C). This can be easily maintained in the enzyme assay either by adjusting the concentration of required cofactors (e.g., MgCl2 or NTPs) or the addition of NaCl. At 100 mM IS the HPTS pKα decreased to 7.26 (at 25 °C), in which case MOPS buffer (pKα 7.2 at 25 °C) could be used to measure enzyme activity, extending the usable pH range for the assay.
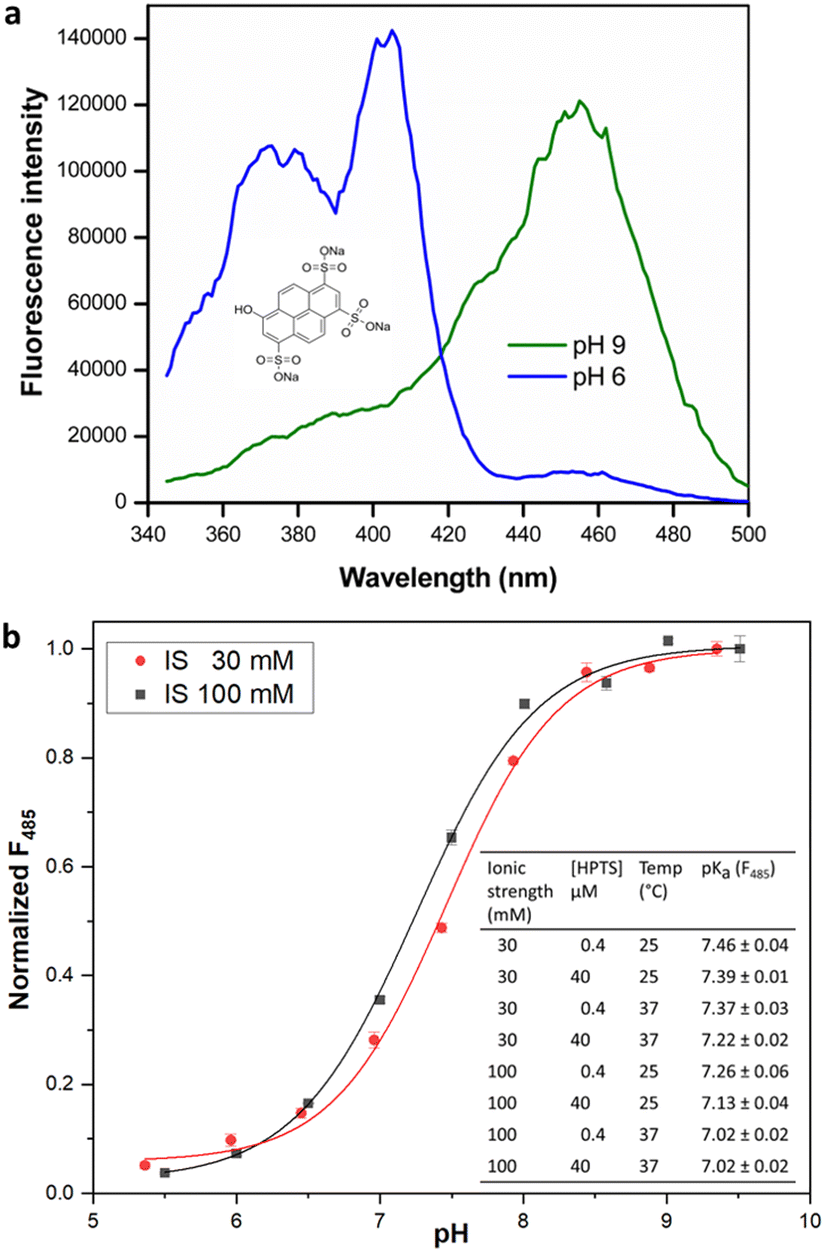 | ||
| Fig. 1 (a) HPTS structure (inset) and fluorescence excitation spectra, λem = 511 nm. (b) Dependence of fluorescence intensity (485 nm) of HPTS on pH at 25 °C. Inset: pKα estimates for HPTS under different reaction conditions. Fluorescence measurements were made as described in methods. pKα values are given as mean ± SD (n = 3). | ||
Further, the change in HPTS pKα upon temperature increase to 37 °C closely followed the change in buffer pKα, confirming that the assay can be utilized in the biologically relevant temperature range. Interestingly, we observed that increase in HPTS concentration from 0.4 to 40 μM in 20 mM HEPES buffer of uniform IS did not significantly alter its pKα. This is in contrast to a previous report,16 where the pKα of soluble HPTS was found to change with increased dye concentration (up to 380 μM in phosphate buffer; however, we did not check such rather high concentrations), while immobilized or ion-paired HPTS did not show altered pH sensitivity.16
Transketolase activity and substrate scope
We initially investigated the assay protocol to monitor the progress of the transketolase reaction in an attempt to improve the sensitivity of the established chromogenic assay, which is based on phenol red as an indicator dye. The use of hydroxypyruvate (HPA) as ketol donor with glycolaldehyde as the most reactive acceptor in the TK catalytic reaction releases CO2, in turn leading to an increase in pH that allows the continuous measurement of TK activity.10 Because CO2 is involved in an equilibrium with bicarbonate, and the corresponding partial consumption of hydroxide causes a damped response, this rendered a sensitive fluorescent signal more attractive. A buffer concentration of 2 mM HEPES was used as the reaction medium to provide a balance between sensitivity and accuracy. The starting pH was set at 7.0, as TKgst can maintain maximum activity over a broad pH range (7–8).30 The enzyme reaction with glycolaldehyde as the model acceptor showed a significant increase in fluorescence intensity ratio (λex 485 nm/λex 405 nm) over time, compared to blanks with substrate or enzyme omitted (Fig. 2). A slight increase (2%) in signal intensity was observed in the control measurements compared to those with only buffer, which could be attributed to slow thermal degradation of hydroxypyruvate in HEPES buffer.30 The assay calibration curve (Fig. 2) showed good linearity over 0–1 mM HCO3−, and was used to calculate the increase in HCO3− concentration over the course of the enzyme reaction. Extrapolation gave a specific activity of 6.11 U mg−1 with 20 mM glycolaldehyde (Table 1), which was in good agreement with values obtained from the earlier colorimetric pH assay utilizing phenol red in 2 mM TEA buffer.10,23 The limit of detection (LOD) and limit of quantification (LOQ) values derived from the calibration curve were 0.044 and 0.14 mM HCO3−, respectively. These are comparable to the corresponding values of 0.04 and 0.11 mM HCO3− obtained with 28 μM phenol red.10 The higher susceptibility, and lower interference from background effects, of the fluorimetric measurement allows reducing the dye concentration (0.4 μM) significantly, which provides adequate sensitivity with a 70-fold lower concentration of indicator compared to the colorimetric assay using phenol red (28 μM).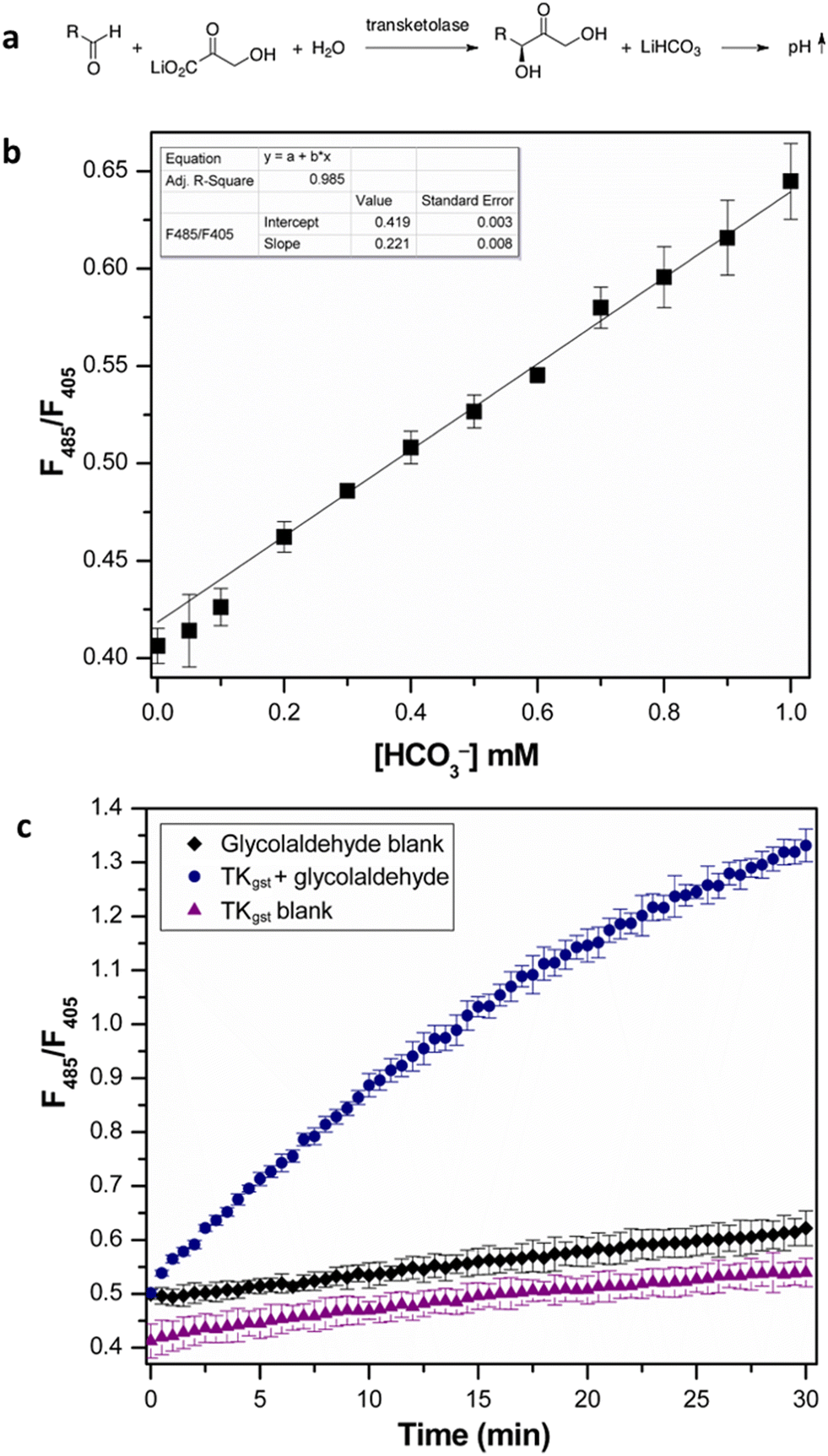 | ||
| Fig. 2 (a) Transketolase reaction with pH shift. (b) Calibration curve of fluorescence intensity ratio (λex 485 nm/(λex 405 nm) with NaHCO3 standard (0–1 mM). All measurements were performed in triplicate. (c) Progress of transketolase reaction with 20 mM glycolaldehyde and 50 mM LiHPA. Error bars represent SD from triplicate measurements. | ||
| Aldehyde acceptor | [S] (mM) | TKgst (μg) | Specific activity (U mg−1) |
|---|---|---|---|
| Glycolaldehyde | 20 | 4.8 | 6.11 ± 0.20 |
| D-Glyceraldehyde | 20 | 12 | 5.14 ± 0.06 |
| D-Lactaldehyde | 20 | 27 | 0.63 ± 0.02 |
| D-Erythrose | 50 | 27 | 0.76 ± 0.03 |
| D-Ribose | 200 | 54 | 0.37 ± 0.005 |
| Propanal | 200 | 54 | 0.63 ± 0.005 |
| 4-Hydroxybutanal | 200 | 81 | 0.12 ± 0.002 |
| rac-3-Hydroxybutanal | 20 | 54 | 0.18 ± 0.004 |
| L-Glyceraldehyde | 20 | 81 | 0.10 ± 0.003 |
Next, we used the assay to determine the substrate scope and specific activity of wt TKgst against a set of aldehyde acceptors with varying hydroxylation (Table 1). As expected from its natural specificity, TKgst showed a preference for (2R)-hydroxylated aldehydes. The highest activity was observed with glycolaldehyde and D-glyceraldehyde, and significantly lower activity with L-glyceraldehyde. The quantitative activity data obtained using the fluorescent pH assay were similar to those from earlier reports.23,30 TKgst showed lower activity (5–10% relative to glycolaldehyde) with poly-hydroxylated sugars such as D-ribose and D-erythrose and aliphatic aldehydes such as propanal, even at higher substrate concentrations (50–200 mM).
Kinetic constants were also determined for the acceptors D-glyceraldehyde, glycolaldehyde, and donor LiHPA (Table S2†). Respective KM values of 7.36 mM, 4.5 mM, and 1.8 mM were obtained, which were in conformity with reported values.25 Although the values for kcat were slightly lower than reported, this can be explained by a difference in purity of the enzyme sample or changes in the reaction environment that could have interfered with the pH measurement.
Versatility of the assay with enzymes from other EC classes
To explore the versatility of the HPTS-based pH assay, we further investigated a broad range of enzymes from different EC classes (Table 2 and Fig. S1†) that have been extensively developed for industrial biocatalysis or pharmaceutical drug development; most of these enzymes are also active in the HTPS relevant 5.5–8.5 pH range.§| Enzymea | EC class | Fluorimetric pH-shift assay conditionsb | Specific activity (U mg−1) | Absorption assay referencec | Specific activityd (U mg−1) | Ref. |
|---|---|---|---|---|---|---|
| a Enzyme was expressed in E. coli with C-terminal His tag as described in ESI† and desalted or diluted into the corresponding buffer before assay. b Details of substrate and cofactors in the assay mixture are included in text and in ESI.† c Other components and conditions were used as mentioned in literature. d Specific activity was measured using the reference assays for enzyme preparations that were produced and purified in our lab. e Specific activity as specified in product information of commercial lyophilized enzyme preparations obtained from Merck (Sigma-Aldrich; Germany). No reference assays were performed for hexokinase and AChE. f Activity was measured in the absence (CMP-Neu5Ac hydrolysis) or presence (sialyl transfer) of 5 mM lactose in the catalytic reaction. | ||||||
| Transketolase (Geobacillus stearothermophilus) | EC 2.2.1.1 | 2 mM HEPES | 6.1 ± 0.1 | pH assay (phenol red, 2 mM TEA, pH 7.5) | 4.4 ± 0.2 | 10 |
| pH 7.0 | ||||||
| 25 °C | ||||||
| pH assay (phenol red, 2 mM TEA, pH 7.0) | 5.8 ± 0.2 | |||||
| Hexokinase (Saccharomyces cerevisiae) | EC 2.7.1.1 | 2 mM HEPES | 28.7 ± 1.2 | G6PDH coupled assay (NADH oxidation) | 25e | 11 |
| pH 8.2 | ||||||
| 25 °C | ||||||
| Acetylcholinesterase (electric eel) | EC 3.1.1.7 | 2 mM HEPES | 1530 ± 110 | Ellman's assay | 1527e | — |
| 30 mM NaCl | ||||||
| pH 8.0 | ||||||
| 37 °C | ||||||
| Pyruvate decarboxylase (Zymobacter palmae) | EC 4.1.1.1 | 2 mM MOPS | 26.1 ± 0.4 | ADH coupled assay | 19.2 ± 0.2 | 31 |
| 100 mM NaCl | K M = 1.85 ± 0.02 mM | K M = 2.5 ± 0.2 mM | ||||
| pH 6.5 | ||||||
| 25 °C | ||||||
| N-Acylneuraminate cytidylyltransferase (Neisseria meningitidis) | EC 2.7.7.43 | 2 mM HEPES | 29.1 ± 2.5 | pH assay (cresol red, 2 mM Tris, pH 8.6) | 32.6 ± 2.7 | 12 |
| pH 8.2 | ||||||
| 37 °C | ||||||
| 2,3-Sialyl transferasef (Photobacterium phosphoreum) | EC 2.4.99.6 | 2 mM HEPES | 2.6 ± 0.1 (transferase) | pH assay (phenol red, 2 mM Tris, pH 8.0) | 1.9 ± 0.03 (5 mM lactose) | 13 |
| 30 mM NaCl | ||||||
| pH 8.0 | 1.4 ± 0.07 (hydrolysis) | 1.03 ± 0.02 (hydrolysis) | ||||
| 30 °C | ||||||
Similar to transketolase, the assay fared well in the detection of hexokinase activity on D-glucose, showing good correlation with the coupled assay employing glucose-6-phosphate dehydrogenase and a commercial hexokinase preparation. Hexokinase is a well-studied metabolic kinase that phosphorylates sugars, where the transfer of a phosphate group from ATP to the terminal hydroxyl group is accompanied by the release of a proton. Hexokinase finds application both in the screening of kinase inhibitors and in biocatalysis for the synthesis of sugar phosphates. The activity of yeast hexokinase on different sugar substrates was examined using the fluorimetric pH assay (Fig. S2†). In conformity with literature reports,11,32 the enzyme was most active on D-fructose, D-glucose (28.7 U mg−1), and 2-deoxy-D-glucose, with 50% activity on D-mannose (relative to D-glucose) and 15% activity on sucrose.11 The assay also showed adequate sensitivity to discern a low activity on D-arabinose (0.1 U mg−1).
Acetylcholinesterase is an important representative of hydrolase enzymes, with therapeutic applications in treatment of neurodegenerative disorders.33 Employing the fluorimetric pH assay, AChE specific activity could be quantified with a detection limit of 5 mU mL−1, using 1 mM acetylcholine chloride as the substrate and acetic acid as the calibration standard (Fig. S3†). The ionic strength of the assay mixture was adjusted by the addition of 30 mM NaCl. Further, using the assay an IC50 value of 1.9 μM could be determined for the known AChE inhibitor galantamine hydrobromide,33 which was similar to reported values.34 Although the standard Ellman assay uses acetylthiocholine as substrate, we assume comparable AChE specific activity using the fluorimetric assay with acetylcholine (Table 2), because both substrates have been shown to have similar KM and kcat using microcalorimetric assays.35 It is therefore evident that the fluorescence assay has good potential for utilization in the screening and identification of kinase and AChE inhibitors.
Similar to kinases, the assay could also be used for other enzymes that catalyse phosphoryl transfer reactions that are important for preparative biocatalysis, such as in the synthesis of oligosaccharides. For instance, CMP-sialate synthetase (CSS) and 2,3-sialyl transferase (SiaT) are employed in the synthesis of sialylated glycoconjugates that are associated with the regulation of several developmental and immunological processes, as well as microbial pathogenesis.36 In both cases, laborious or discontinuous methods are required to follow the enzyme reaction. However, alternative colorimetric pH-shift assays have been developed recently (using cresol red or phenol red with Tris buffer) that are simple and advantageous for continuous activity measurement of these enzymes.12,13 Activity measurements using the fluorescent pH assay gave quantitatively comparable values for CSS, 2,3-SiaT from Photobacterium phosphoreum (with lactose as acceptor) and mechanistically related sialidase activities (Table 2 and Fig. S4 and S5†).
Yet another enzyme studied was pyruvate decarboxylase (PDC), an enzyme that has been extensively employed in biofuel production as well as carboligation in biocatalytic synthesis.31 As the majority of PDCs are most active at acidic pH with a significant decrease in activity beyond pH 7.5, MOPS buffer was employed for the assay at a starting pH of 6.5 (Fig. S6†). The ionic strength was adjusted by the inclusion of 100 mM NaCl to match the pKα of HPTS and buffer. Specific activity and KM values obtained with pyruvate (Table 2) for the PDC from Zymobacter palmae were comparable to measurements made using the alcohol dehydrogenase-coupled assay,31 attesting to the versatility of the fluorimetric pH assay.
Application of the assay for engineering remote enantioselectivity in TKgst
We further applied the fluorescent assay in the screening of a TKgst variant library in an attempt to engineer selectivity for enantiomers of 3-hydroxyaldehyde acceptors. The carboligation products are deoxysugars having interesting bioactivity,37 and the resultant structural 1,3-diol motifs are frequently found in various natural products including polyketides.38 As noted above, transketolases have been engineered to accept aldehyde acceptors structurally lacking or containing a stereoinverted 2-hydroxyl group, but modifying the TK specificity and selectivity on the more remote 3-hydroxyl group has not been studied so far. In the TKgst active site, several amino acid residues that form a channel for substrate entrance to the catalytic core at the ThDP cofactor are highly conserved. These residues (H462, R521, D470, H28, G264, H263, L191, L382, and S385) bind the aldehyde acceptor and position the electrophilic carbonyl group toward a nucleophilic attack. In order to generate a TK variant library for 3-hydroxyaldehydes, we used the L382F variant as the starting template, because it is known to accept aliphatic aldehydes missing a 2-hydroxyl group.21 The S385 residue particularly interacts with and stabilizes a (3R)-configurated hydroxyl moiety in aldose phosphate acceptors via hydrogen bonding (Fig. 3). Therefore, we reasoned that a modification at S385 should modify (improve or even cancel) the preference for the (3R)-hydroxyl substrate. The G264 residue is situated opposite to S385, and therefore could be modified to serve as a potential complementary hydrogen-bonding donor when attempting to invert the stereoselectivity toward the (3S)-hydroxyl configuration. Accordingly, the G264 residue was first mutated to the amino acids Ala, Val or Ser, so as to induce a change in substrate orientation without causing steric hindrance for substrate binding. The final TKgst library was generated from the L382F/G264(A,S,V) variants, by mutating S385 using NNK codon degeneracy.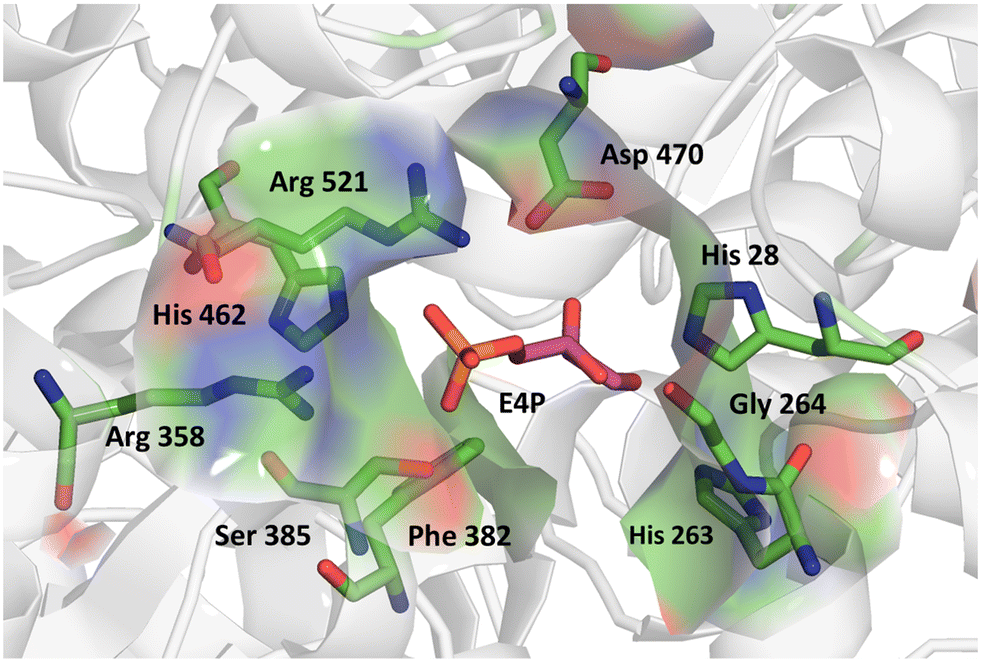 | ||
| Fig. 3 Acceptor binding site of TKgst L382F with bound erythrose-4-phosphate (purple) illustrating the residues involved in substrate interaction. Structure model was constructed based on the X-ray structure of TK from Bacillus anthracis (PDB 3M49). Graphic was produced with PyMOL.39 | ||
Assays employed to screen enzyme variants generated by directed evolution require adequate sensitivity, reproducibility, robustness, and accuracy, in order to enable fast high-throughput identification of improved hits. The nature of the fluorescent pH assay makes it advantageous as a primary screening technique to discern active variants over a broad substrate range, while also allowing the relatively accurate determination of specific activity, selectivity, or kinetic parameters. To screen the new TKgst variant library, we adapted an optimized method for cell growth in microplates as reported earlier10 and lysis using only lysozyme and nuclease in 2 mM HEPES buffer. First, the screening protocol was validated using statistic parameters like coefficient of variance (CV) and Z-factor.40 A CV of 14.6% was achieved for the whole process involving cell growth and lysis, protein estimation, and fluorescent assay, with 10.5% CV for the assay alone. A CV value <15% enhances the sensitivity of hit detection by the screening system, allowing identification of variants with at least 2-fold improved activity.41 We also determined the Z-factor by analysing control samples of wild-type TKgst and E. coli host with the empty vector, assaying them using 5 mM glycolaldehyde and LiHPA (Fig. S7†). Negligible activity was observed with the E. coli background from low expression of its native transketolase. A Z-factor of 0.64 was observed for the assay alone and 0.52 for the complete process, which is in the range for an “excellent” assay even with the rate-limiting concentration of glycolaldehyde; implying that the assay fulfils requirements for use in HTS.
Following assay validation, the L382F/G264X/S385X variant library was first subject to preliminary screening to detect variants displaying enhanced activity with 10 mM rac-3-hydroxybutanal. While several variants exhibited higher specific activity of up to 5-fold compared to the L382F base variant, a considerable fraction of such variants belonged to the G264V sub-library (Fig. 4). On the other hand, many inactive variants were observed when the G264 residue was either left unchanged or modified to Ala or Ser. Regardless, the top 10% active mutants from each sub-library were chosen and subjected to a second round of screening using 5 mM enantiomerically pure (3R)- or (3S)-hydroxybutanal (Fig. S8†),42 with the specific activity ratio used to evaluate potential improvement in enantioselectivity. Unfortunately, significant enhancement in selectivity was not observed, with only 15% of the mutants showing slight improvement in selectivity for (3S)-hydroxybutanal (specific activity ratio (S/R) > 2; Fig. 5a).
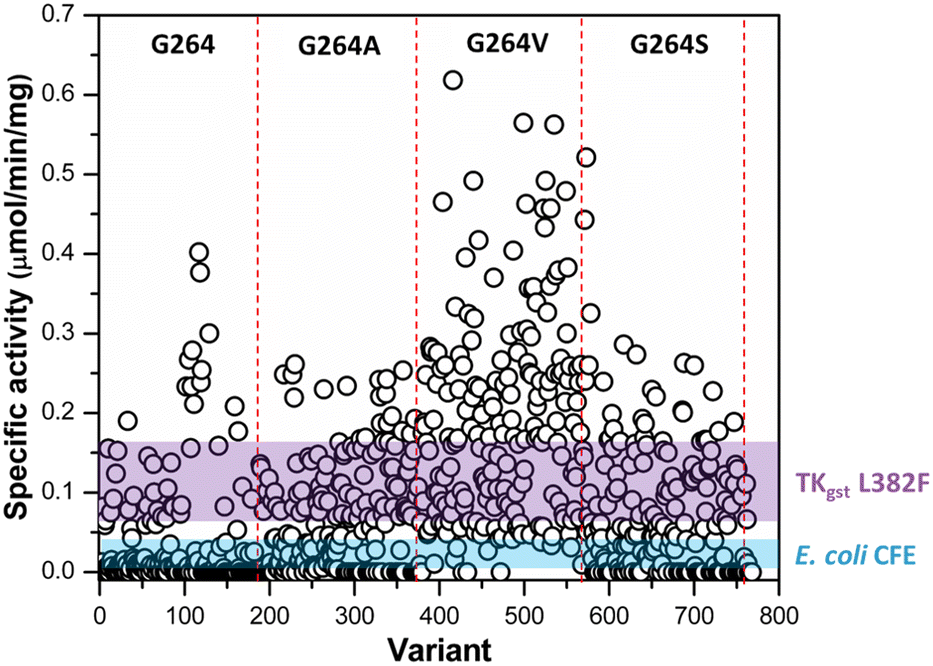 | ||
| Fig. 4 Preliminary activity screening of TKgst variant library (L382F/G264X/S385X) with rac-3-hydroxybutanal. The coloured bands represent mean ± 3SD of the specific activity of TKgst (L382F) mutant and E. coli cell-free extract control. | ||
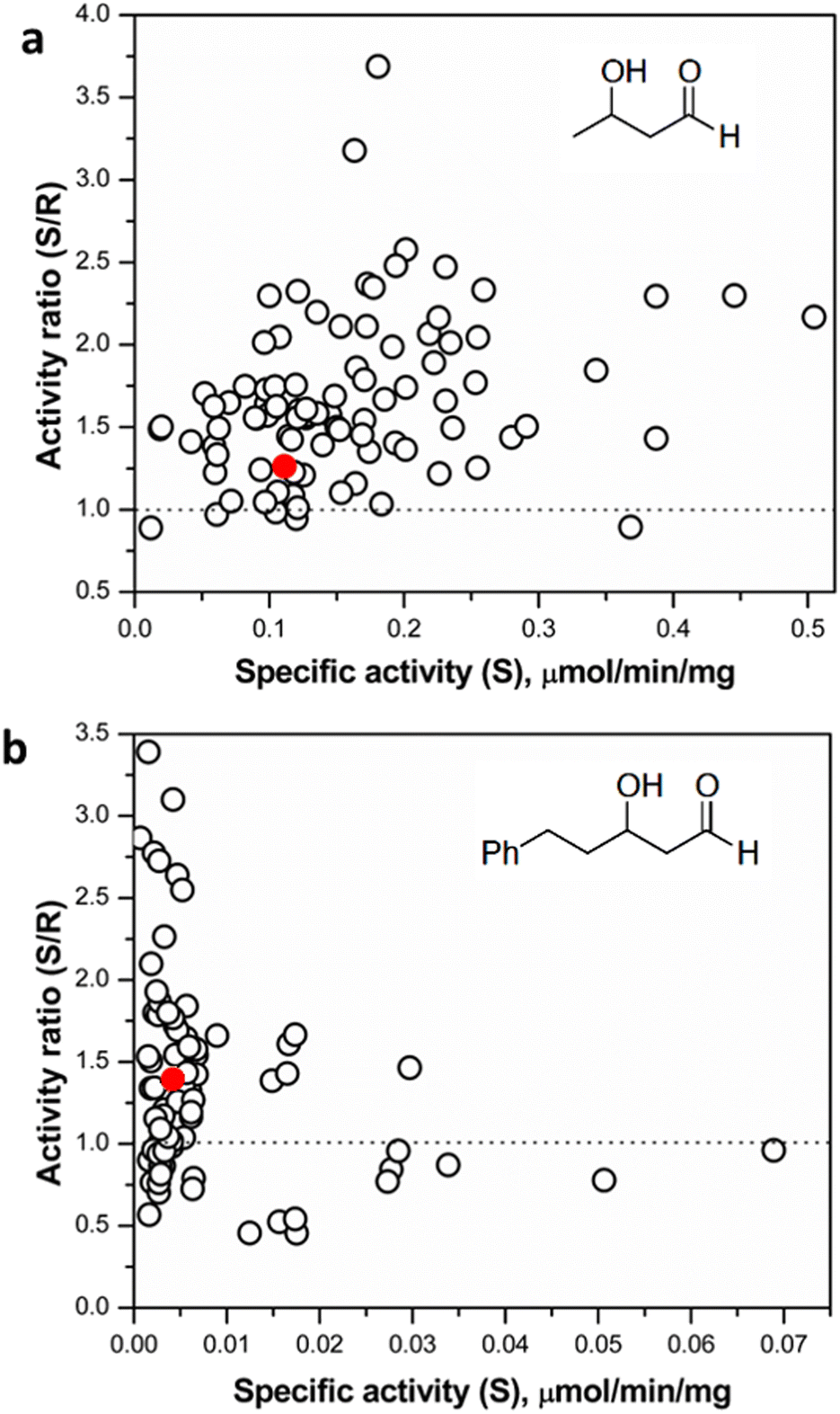 | ||
| Fig. 5 (a) Screening of selected hits (top 10% active variants from preliminary screening round) with (3R)- and (3S)-hydroxybutanal. (b) Screening of selected hits with (3R)- and (3S)-hydroxyphenylpentanal. The red dots denote the reference starting mutant TKgst (L382F). | ||
It is plausible that 3-hydroxybutanal, being a relatively small and flexible substrate, has a greater freedom of movement in the TK active site, consequently reducing the potential for selective binding of the enantiomers. Similar observations have been made in studies directed at an enantioselectivity switch for 2-hydroxylated aldehydes upon screening libraries with D- or L-glyceraldehyde, which both may productively bind in more than one alternative orientations.24 The innate low stability of small chain aliphatic aldehydes might also interfere with the measurements. To address this potential lacuna, we further tested the TKgst hit plate against enantiomers of an arylated 3-hydroxyaldehyde (3-hydroxy-5-phenylpentanal, HPP; Fig. 5 and S8†).43 We hypothesized that the addition of the bulky phenyl moiety should enforce an extended-chain binding orientation of the isomers, thereby improving distinctive substrate–enzyme interactions and thus better discrimination of the 3-hydroxyl configuration. Rescreening of the 3-hydroxybutanal hit plate using the fluorescent assay revealed that HPP indeed was accommodated by 16 variants. The most active variants exhibited up to 15-fold improved activity over TKgst (L382F), indicating that the beneficial effects of G264X/S385X mutations are amplified by the use of a bulky substrate. However, only minor improvement in enantioselectivity was observed even with this substrate, where all the active variants exhibited a specific activity ratio in the range 0.5–3.2 only. Sequencing revealed that the favoured mutations were changes to Ala or Val at both the 264 and 385 positions. The S385V mutation resulted in a slightly higher selectivity for (3R)-HPP. An analytical scale reaction using 15 mM rac-HPP and the active variant L382F/G264A/S385A (heat-purified enzyme) verified formation of the carboligated adduct according to TLC control (see ESI†).
Although the introduction of remote enantioselectivity could not be successfully completed in this initial study, the mutations at G264 and S385 were able to improve enantiocomplementary binding of 3-hydroxyaldehydes in the active site and their acceptance by transketolase, leading to higher activity. We expect that construction and screening of larger libraries by including further residues with substrate contact should provide enhanced stereoselectivity.
Experimental
Materials and methods
HPTS and aldehyde acceptors for TK were purchased from Sigma-Aldrich, and used freshly as supplied. Hydroxypyruvate (LiHPA) was prepared according to the literature procedure.44 Enantiomers of 3-hydroxybutanal were synthesized from 4,4-dimethoxybutanone,42 and 3-hydroxy-5-phenylpentanal from hydrocinnamaldehyde.43 Transketolase from Geobacillus thermophilus (TKgst) was expressed and purified as reported earlier21 and stored as lyophilized powder until use. Expression and purification protocols for other enzymes used in the study are provided in the ESI.† Fluorescence measurements were performed using a BMG Labtech FLUOstar instrument equipped with 485 nm and 405 nm excitation and 520 nm emission filters. All fluorescence measurements and enzyme assays were performed in 200 μL final volume using flat bottom Nunc Immuno 96-well black microplates.Characterization of the fluorescent indicator
HPTS fluorescence excitation spectra were measured in 20 mM HEPES buffers of varying pH (5.0–9.5), with emission at 511 nm, on a microtiter plate reader (Infinite F200; Tecan, Crailsheim, Germany). The pH sensitivity of HPTS was determined by measuring fluorescence in 20 mM HEPES buffers of varying pH (5.0–9.5) and constant ionic strength (30 mM or 100 mM). A Knick 761 Calimatic pH meter with a double pore pH electrode calibrated using reference buffers (pH 4.01 and pH 7.00; Carl Roth, Germany) was used for pH measurements. The pKα values were determined from the inflection point of a sigmoidal fit to fluorescence intensity (485 nm) versus pH using Origin 8.5 (OriginLab, USA).Determination of enzyme activity
For activity determination with pure enzymes, the respective enzyme was desalted into 2 mM buffer of corresponding pH and ionic strength (Table 2, ESI†) before the assay.Assay mixtures for other enzymes were set up as given in the ESI† and Table 2.
TKgst variant library construction and screening
Conclusions
The fluorimetric pH-shift assay using HPTS is a practical, efficient and reliable method that can be applied to measure activities of a wide range of enzymes that are important for advanced applications in biocatalysis. Its generic chemical reaction principle renders the assay particularly suitable to evaluate an enzyme's substrate (or inhibitor) scope under consistent reaction conditions. The method is also useful for high-throughput screening of enzyme libraries from enzyme engineering by directed evolution in vitro (as demonstrated here for transketolase) or from metagenomic prospecting.While our results demonstrate its versatility, limitations of the assay include (i) a workable pH range of 5.5–8.5 for direct continuous activity monitoring due to the nature of the pH-shift principle; (ii) a need for measurements at two distinct excitation wavelengths to render a single ratiometric data point, thereby not allowing a direct readout; and (iii) a sensitivity limit when using filter-based plate readers if the available filters do not match the HTPS maxima. However, we expect that these limitations can be tackled with advanced instrumentation and the identification of modified fluorescent dyes having lower or higher pKα values for use in more acidic or more alkaline ranges, respectively.17,29 Additionally, the presence of buffering substrates or co-factors, and multiple products that may cause pH changes, may limit the applicability of the assay. Therefore, it is important to maintain uniform starting conditions including pH, temperature, and ionic strength to obtain reliable data from kinetic parameter measurements. Although the assay can be efficiently performed using a standard microtitre plate protocol, we suggest that the high responsiveness and excellent water solubility of HPTS should qualify even as a potential detection system in microfluidic systems for monitoring pH-sensitive reactions under uHTS conditions.
Data availability
The data supporting this article are available within the article and its ESI.†Author contributions
Avinash Vellore Sunder: investigation, formal analysis, and writing – original draft. Marie-Luise Reif: investigation and formal analysis. Wolf-Dieter Fessner: conceptualization, funding acquisition, formal analysis, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Dedication
It is with great pleasure that we dedicate this paper to George M. Whitesides on the occurrence of his 85th anniversary. George was my (WDF) postdoctoral advisor from 1986 to 1987 for working on enzymatic asymmetric C–C bond formation by using aldolase catalysis, a topic of major promise for synthetic organic chemistry. After completing my PhD in hardcore hydrocarbon chemistry on realizing the first non-Paquette dodecahedrane synthesis via the C20H20 isomer “pagodane”, the transition to enzyme catalysis in aqueous solution, carbohydrate stereochemistry and sugar phosphate analysis posed quite some challenge. For some years the Whitesides group pioneered the field of enzymatic carboligation with a number of key developments in asymmetric aldol and acyloin synthesis (e.g., ref. 18), but George's interest soon became captured by other important problems that awaited his fundamental insights and visionary impact. I myself returned to Germany to start my independent research program on enzyme catalysis (guess the topic? carboligation!).Thus, we needed some special reason to get in touch again. At the Technical University of Darmstadt I headed the academic selection committee for the biennial Emanuel-Merck Lectureship Award (https://en.wikipedia.org/wiki/Emanuel_Merck_Lectureship) from 2000 to 2022, and George was selected as the recipient of the 2005 award. In my laudatio speech, I addressed many facets of George's impressive achievements, but since the president of our university was attending as well, I then posed the challenge that we at a Technical University should have the courage to do even better engineering work than the ingenious micro-contact printing technology developed by George's group in an academic setting at Harvard. Why not scale down by another order of magnitude: nano-contact printing? In my office I had carved a mirror-image stamp out of a big potato and was therefore able to stamp the letters “NANO” on some index cards using a large ink bath! The problem was solved (Fig. 6), and George played along nicely by kindly hand-signing a few of these cards for us.
 | ||
| Fig. 6 Illustration of “nano-contact printing” technology on the occasion of George M. Whitesides receiving the Emanuel-Merck Lecureship award 2005 at the Technical University of Darmstadt. | ||
Needless to mention that George then presented three excellent award lectures on highly stimulating topics, as diverse as on microfluidic technology, soft lithography, and protein ligand design. Happy Anniversary, George!
Acknowledgements
This project has received funding through The German Federal Ministry of Education and Research (BMBF, grant no. 031B0595) and the initiative ERA CoBioTech (European Union Horizon 2020 Research and Innovation Programme, grant no. 722361). We gratefully acknowledge Alexander Mertsch for help with the synthesis of HPP.Notes and references
- S. Slagman and W.-D. Fessner, Chem. Soc. Rev., 2020, 49, 1968 Search PubMed.
- Y. V. Sheludko and W.-D. Fessner, Curr. Opin. Struct. Biol., 2020, 63, 123 CrossRef CAS PubMed.
- M. G. Acker and D. S. Auld, Perspect. Sci., 2014, 1, 56 CrossRef CAS; W.-D. Fessner, Nat. Catal., 2019, 2, 738 CrossRef; U. Markel, K. D. Essani, V. Besirlioglu, J. Schiffels, W. R. Streit and U. Schwaneberg, Chem. Soc. Rev., 2020, 49, 233 RSC.
- R. J. Kazlauskas, in: Enzyme Assays, ed. J.-L. Reymond, Wiley-VCH, Heidelberg, 2006, pp. 15–39 Search PubMed.
- L. E. Janes, A. C. Löwendahl and R. J. Kazlauskas, Chem. – Eur. J., 1998, 4, 2324 CrossRef CAS; M. F. Paye, H. B. Rose, J. M. Robbins, D. A. Yunda, S. Cho and A. S. Bommarius, Anal. Biochem., 2018, 549, 80 CrossRef PubMed.
- F. Morís-Varas, A. Shah, J. Aikens, N. P. Nadkarni, J. Rozzell and D. C. Demirjian, Bioorg. Med. Chem., 1999, 7, 2183–2188 CrossRef CAS PubMed; M. de los Angeles Camacho-Ruiz, J. C. Mateos-Díaz, F. Carrière and J. A. Rodriguez, J. Lipid Res., 2015, 56, 1057 CrossRef PubMed.
- A. Banerjee, P. Kaul, R. Sharma and U. Banerjee, J. Biomol. Screening, 2003, 8, 559 CrossRef CAS PubMed.
- P. Holloway, J. T. Trevors and H. Lee, J. Microbiol. Methods, 1998, 32, 31 CrossRef CAS; S. Marvanová, Y. Nagata, M. Wimmerová, J. Sýkorová, K. Hynková and J. Damborský, J. Microbiol. Methods, 2001, 44, 149 CrossRef PubMed; T. M. Phillips, A. G. Seech, H. Lee and J. T. Trevors, J. Microbiol. Methods, 2001, 47, 181 CrossRef PubMed; L. Tang, Y. Li and X. Wang, J. Biotechnol., 2010, 147, 164 CrossRef PubMed.
- K. Yu, S. Hu, J. Huang and L. H. Mei, Enzyme Microb. Technol., 2011, 49, 272 CrossRef CAS PubMed.
- D. Yi, T. Devamani, J. Abdoul-Zabar, F. Charmantray, V. Helaine, L. Hecquet and W.-D. Fessner, ChemBioChem, 2012, 13, 2290 CrossRef CAS PubMed.
- E. Chapman and C.-H. Wong, Bioorg. Med. Chem., 2002, 10, 551 CrossRef CAS PubMed.
- N. He, D. Yi and W.-D. Fessner, Adv. Synth. Catal., 2011, 353, 2384 CrossRef CAS; D. Yi, N. He, M. Kickstein, J. Metzner, M. Weiß, A. Berry and W.-D. Fessner, Adv. Synth. Catal., 2013, 355, 3597 CrossRef; A. Mertsch, S. Poschenrieder and W.-D. Fessner, Adv. Synth. Catal., 2020, 362, 5485 CrossRef.
- C. Deng and R. R. Chen, Anal. Biochem., 2004, 330, 219 CrossRef CAS PubMed; A. Mertsch, N. He, D. Yi, M. Kickstein and W.-D. Fessner, Chem. – Eur. J., 2020, 26, 11614 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- T. Scheidt, H. Land, M. Anderson, P. Berglund, Y. Chen, D. Yi and W.-D. Fessner, Adv. Synth. Catal., 2015, 357, 1721 CrossRef CAS; R. Roldán, I. Sanchez-Moreno, T. Scheidt, V. Hélaine, M. Lemaire, T. Parella, P. Clapés, W.-D. Fessner and C. Guérard-Hélaine, Chem. – Eur. J., 2017, 23, 5005 CrossRef PubMed; Y.-C. Thai, A. Szekrenyi, Y. Qi, G. W. Black, S. J. Charnock and W.-D. Fessner, Bioorg. Med. Chem., 2018, 26, 1320 CrossRef PubMed; H. Land, F. Ruggieri, A. Szekrenyi, W.-D. Fessner and P. Berglund, Adv. Synth. Catal., 2020, 362, 812 CrossRef.
- S. Nevolova, E. Manaskova, S. Mazurenko, J. Damborsky and Z. Prokop, Biotechnol. J., 2019, 14, 1800144 CrossRef PubMed.
- O. S. Wolfbeis, E. Furlinger, H. Kroneis and H. Marsoner, Fresenius' Z. Anal. Chem., 1983, 314, 119 CrossRef CAS; Y. Avnir and Y. Barenholz, Anal. Biochem., 2005, 347, 34–41 CrossRef PubMed; R. Nandi and N. Amdursky, Acc. Chem. Res., 2022, 55, 2728 CrossRef PubMed.
- D. C. Myles, P. J. Andrulis III and G. M. Whitesides, Tetrahedron Lett., 1991, 32, 4835 CrossRef CAS; Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899 CrossRef.
- G. Schenk, R. G. Duggleby and P. F. Nixon, Int. J. Biochem. Cell Biol., 1998, 30, 1297 CrossRef CAS PubMed.
- E. G. Hibbert, T. Senussi, M. E. B. Smith, S. J. Costelloe, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2008, 134, 240 CrossRef CAS PubMed; J. L. Galman, D. Steadman, S. Bacon, P. Morris, M. E. B. Smith, J. M. Ward, P. A. Dalby and H. C. Hailes, Chem. Commun., 2010, 46, 7608 RSC.
- D. Yi, T. Saravanan, T. Devamani, F. Charmantray, L. Hecquet and W.-D. Fessner, Chem. Commun., 2015, 51, 480 RSC; C. Zhou, T. Saravanan, M. Lorilliere, D. Wei, F. Charmantray, L. Hecquet, W.-D. Fessner and D. Yi, ChemBioChem, 2017, 18, 455 CrossRef CAS PubMed.
- P. Payongsri, D. Steadman, H. C. Hailes and P. A. Dalby, Enzyme Microb. Technol., 2015, 71, 45 CrossRef CAS PubMed.
- T. Saravanan, M. L. Reif, D. Yi, M. Lorillière, F. Charmantray, L. Hecquet and W.-D. Fessner, Green Chem., 2017, 19, 481 RSC.
- J. Abdoul Zabar, M. Lorillière, D. Yi, S. Thangavelu, T. Devamani, L. Nauton, F. Charmantray, V. Hélaine, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2015, 357, 1715 CrossRef CAS; M. Lorilliere, M. De Sousa, F. Bruna, E. Heuson, T. Gefflaut, V. de Bernardinis, S. Thangavelu, D. Yi, W.-D. Fessner, F. Charmantray and L. Hecquet, Green Chem., 2017, 19, 425 RSC; H. Casajus, A. Lagarde, M. Leremboure, T. D. D. Miguel, L. Nauton, V. Thery, W.-D. Fessner, N. Duguet, F. Charmantray and L. Hecquet, ChemCatChem, 2020, 12, 5772 CrossRef.
- M. Lorillière, R. Dumoulin, M. L'Enfant, A. Rambourdin, V. Thery, L. Nauton, W.-D. Fessner, F. Charmantray and L. Hecquet, ACS Catal., 2019, 9, 4754 CrossRef.
- T. Saravanan, S. Junker, M. Kickstein, S. Hein, M. K. Link, J. Ranglack, S. Witt, M. Lorillière, L. Hecquet and W.-D. Fessner, Angew. Chem., Int. Ed., 2017, 56, 5358 CrossRef CAS PubMed; H. Yu, R. I. Hernández López, D. Steadman, D. Méndez-Sánchez, S. Higson, A. Cázares-Körner, T. D. Sheppard, J. M. Ward, H. C. Hailes and P. A. Dalby, FEBS J., 2020, 287, 1758 CrossRef PubMed; N. Ocal, G. Arbia, A. Lagarde, M. Joly, S. Gittings, K. M. Graham, F. Charmantray and L. Hecquet, Adv. Synth. Catal., 2023, 365, 78 CrossRef.
- I. Fúster Fernández, L. Hecquet and W.-D. Fessner, Adv. Synth. Catal., 2022, 364, 612 CrossRef; I. Fúster Fernández, M. Kickstein and W.-D. Fessner, Adv. Synth. Catal., 2023, 365, 3861 CrossRef.
- J. Santner, M. Larsen, A. Kreuzeder and R. N. Glud, Anal. Chim. Acta, 2015, 878, 9 CrossRef CAS PubMed.
- P. Horváth, P. Šebej, D. Kovář, J. Damborský, Z. Prokop and P. Klán, ACS Omega, 2019, 4, 5479 CrossRef.
- J. Abdoul-Zabar, I. Sorel, V. Hélaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marlière, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2013, 355, 116 CrossRef CAS.
- D. Gocke, T. Graf, H. Brosi, I. Frindi-Wosch, L. Walter, M. Müller and M. Pohl, J. Mol. Catal. B: Enzym., 2009, 61, 30 CrossRef CAS.
- A. Sols, G. de la Fuente, C. Villar-Palasi and C. Asensio, Biochim. Biophys. Acta, 1958, 30, 92 Search PubMed; H. K. Chenault, R. F. Mandes and K. R. Hornberger, J. Org. Chem., 1997, 62, 331 CrossRef CAS PubMed.
- J. I. da Silva, M. C. de Moraes, L. C. C. Vieira, A. G. Corrêa, Q. B. Cass and C. L. Cardoso, J. Pharm. Biomed. Anal., 2013, 73, 44 CrossRef PubMed.
- S. H. Lee, B. C. Kim, J. K. Kim, H. S. Lee, M. Y. Shon and J. H. Park, Bull. Korean Chem. Soc., 2014, 35, 1681 CrossRef CAS.
- P. A. A. de Almeida Neves, E. Novato Silva and P. S. L. Beirão, Adv. Enzyme Res., 2017, 5, 1 CrossRef.
- S. Dedola, S. Ahmadipour, P. de Andrade, A. N. Baker, A. N. Boshra, S. Chessa, M. I. Gibson, P. J. Hernando, I. M. Ivanova, J. E. Lloyd, M. J. Marın, A. J. Munro-Clark, G. Pergolizzi, S.-J. Richards, I. Ttofi, B. A. Wagstaff and R. A. Field, RSC Chem. Biol., 2024, 5, 167 RSC.
- C. J. Thibodeaux, C. E. Melançon and H.-W. Liu, Nature, 2007, 446, 1008 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, Version 1.3, Schrodinger LLC, 2002, https://www.pymol.org Search PubMed.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67 CrossRef PubMed.
- O. Salazar and L. Sun, Methods Mol. Biol., 2003, 230, 85 CAS.
- R. Roldán, K. Hernandez, J. Joglar, J. Bujons, T. Parella, I. Sánchez-Moreno, V. Hélaine, M. Lemaire, C. Guérard-Hélaine, W.-D. Fessner and P. Clapés, ACS Catal., 2018, 8, 8804 CrossRef PubMed.
- M. Narasimhulu, T. Srikanth Reddy, K. Chinni Mahesh, A. Sai Krishna, J. Venkateswara Rao and Y. Venkateswarlu, Bioorg. Med. Chem. Lett., 2009, 19, 3125 CrossRef CAS PubMed.
- F. Dickens and D. H. Williamson, Biochem. J., 1958, 68, 74 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00566j |
| ‡ Dedicated to George M. Whitesides on the occurrence of his 85th anniversary. |
| § Data reported in BRENDA database (https://www.brenda-enzymes.org). |
| This journal is © The Royal Society of Chemistry 2024 |