
Boryl-substituted low-valent heavy group 14 compounds
Chenxi
Duan
and
Chunming
Cui
 *
*
State Key Laboratory of Elemento-Organic Chemistry and Frontiers Science Center of New Organic Matter, Nankai University, Tianjin 300071, China. E-mail: cmcui@nankai.edu.cn
First published on 12th December 2023
Abstract
Low valent group 14 compounds exhibit diverse structures and reactivities. The employment of diazaborolyl anions (NHB anions), isoelectronic analogues to N-heterocyclic carbenes (NHCs), in group 14 chemistry leads to the exceptional structures and reactivity. The unique combination of σ-electron donation and pronounced steric hindrance impart distinct structural characteristics to the NHB-substituted low valent group 14 compounds. Notably, the modulation of the HOMO–LUMO gap in these compounds with the diazaborolyl substituents results in novel reaction patterns in the activation of small molecules and inert chemical bonds. This review mainly summarizes the recent advances in NHB-substituted low-valent heavy Group 14 compounds, emphasizing their synthesis, structural characteristics and application to small molecule activation.
 Chenxi Duan | Chenxi Duan is currently a master's student under the supervision of Prof. Chunming Cui at the State Key Laboratory of Elemento-Organic Chemistry, Nankai University, China. She received her bachelor's degree in 2021 from the Department of Chemistry at Yanbian University, China. Her current research focussed on the synthesis, structures and reactivity of novel low-valent silicon compounds stabilized by boryl substituents. |
 Chunming Cui | Chunming Cui obtained his PhD degree in 2001 from Department of Chemistry of the University of Goettingen, Germany. Then, he conducted postdoctoral research at the University of California at Berkeley with Prof. John Arnold and Davis with Prof. Philip P. Power. Since 2004, he has been a professor at the State Key Laboratory of Elemento-Organic Chemistry, Nankai University, China. His primary research has focussed on synthesis and reactivity of novel low-valent main group and lanthanide element compounds and their catalytic applications. |
1. Introduction
Low valent main group compounds such as heavy carbenes and multiple bonding species constitute unique classes of highly reactive synthetic reagents with unusual structures and reaction patterns.1 They have also been employed as ligands for metal complexes and homogeneous catalysis as well as reagents for the activation of inert small molecules with unusual selectivity.2 In this context, the substituent effects on the structures and reactivity of these low valent species have been extensively examined with various carbon, nitrogen and oxygen-based organic substituents. In recent years, the employments of boryl substituents in the synthesis of these highly reactive species have drawn a great deal attention.Organoboranes with a three-coordinate boron atom are typical Lewis acids that have been widely studied as electrophilic reagents and Lewis acidic catalysts because of the existence of an empty p orbital on the boron atom. Transition metal boryl complexes (R2B-M) with one or more M–B σ bonds have also been investigated for many years as intermediates or catalysts for borylation reactions.3,4 These boryl complexes, in which the boryl ligands are still predominantly electrophilic, have been traditionally prepared by the reaction of anionic metal fragments with organoborane halides or the oxidative-addition of B–H and B–B bonds to low valent transition metal centers.3 Early transition and main-group metal boryl complexes could not be easily obtained until the synthesis of the first nucleophilic N-heterocyclic boryl reagent by Nozaki and co-workers (Scheme 1).5 The boryl fragment of the diazaborolyl C2N2B framework is electronically distinct from common boryl groups (R2B) because of the two nitrogen lone pairs and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π electrons are effectively delocalized with the boron empty orbital in the five-membered framework, leading to the stable six-electron aromatic system, which is isoelectronic to N-heterocyclic carbenes (NHCs). The electron delocalization over the five-membered skeleton manifests the primarily Lewis basic characteristics at the boron center.
C π electrons are effectively delocalized with the boron empty orbital in the five-membered framework, leading to the stable six-electron aromatic system, which is isoelectronic to N-heterocyclic carbenes (NHCs). The electron delocalization over the five-membered skeleton manifests the primarily Lewis basic characteristics at the boron center.
 | ||
| Scheme 1 Synthesis of N-heterocyclic boryllithium 1. | ||
The synthesis of the nucleophilic diazaborolyllithium [HCNDip]2BLi(thf)21 (represented as NHBLi(thf)2, Scheme 1) was accomplished by the reduction of the N-heterocyclic boron bromide with lithium in THF at low temperature.5 The strong σ-donating character of this family of NHB anions compared to NHCs and low electronegativity of the boron atom are attractive features, which could lead to unusual electronic structures and chemical and physical properties of the boryl-substituted compounds. The reactivity of NHB anion 1 towards a variety of electrophiles, such as water, hydrocarbons, acyl chlorides, aldehydes, ketones, and carbon dioxide, underlines the nucleophilic nature of the boron atom. In addition, boryl anion 1 could be employed for the synthesis of alkaline, rare earth and transition metal complexes with novel structures and reactivity.6
The inherent advantages of the stable ring system suggest that modifications to this skeleton could significantly enrich boryl anion chemistry. Following the Nozaki's N-heterocyclic boryllithium 1, Kinjo and co-workers unveiled the synthesis of triazaborolyl anion 3 by the reduction of 2-bromo-1,3,4,2-triazaborole 2 (Scheme 2). Boryllithium 3 has been successfully employed for the synthesis of novel NHB-substituted Al, Au, Zn, Mg, Sb, and Bi complexes.7 DFT calculations indicate that the B–Li bond in 3 exhibits enhanced polarization compared to NHB anion 1.
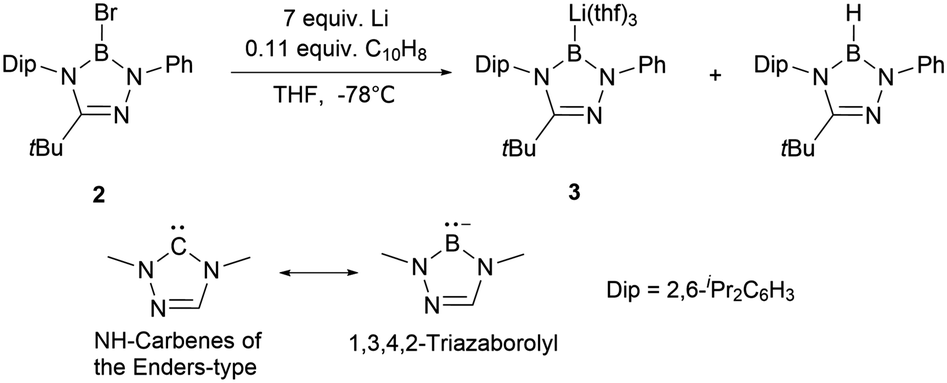 | ||
| Scheme 2 Synthesis of N-heterocyclic boryllithium 3. | ||
Because of the unique electronic effects and steric hindrance of anionic N-heterocyclic boryl reagents, they have also been studied as ligands for the stabilization of low-coordinate and low-valent transition metal and main group compounds.8 The chemical characteristics of these compounds are largely governed by the σ-electron donating properties of the N-heterocyclic boryl substituents, manifesting as the elevated energies of the highest occupied molecular orbitals (HOMO) and enhanced reactivity toward small molecules. Furthermore, because of the isoelectronic relationship of aminoborane R2BNR2 with alkene R2CCR2, the electron delocalization of the BN π systems in the ring with low valent group 14 fragments may exist, which could provide the additional electronic stabilization effects and thus exhibit distinct reactivity and electronic property.
In contrast, common three-coordinate organoboranes (R3B) exhibit remarkable Lewis acidity. The organoboryl R2B group-substituted metal and main group compounds (R2B-M and R2B-E) display quite different electronic structures from those substituted by NHBs because they could attract electrons from low valent species via the electron delocalization to the empty p orbital.
The activation of small molecules and inert bonds is a cornerstone for the understanding many catalytic processes, with such reactivity traditionally being a hallmark of transition metal complexes.9 Over the past two decades, the resurgence of low-valent main group chemistry has led to many new discoveries in the activation of small molecules and inert chemical bonds.10 It has been shown that their reactivity could be effectively controlled by tuning the nature of the substituents. One notable strategy involves the oxidative-addition of E–H σ-bonds mediated by carbenes and heavy analogues EX2 (E = Si, Ge, or Sn).11 Taking carbene (X2C:) as an example, the activation of the E–H bond is highly dependent on the nature of the X-substituent because the substituents with different electronic properties strongly affect the HOMO–LUMO gaps. The carbenes with a wide HOMO–LUMO gap could be obtained by the employments of electronegative nitrogen and oxygen-based groups in cyclic backbones. The substituents based on more electropositive elements such as carbon, boron and silicon tend to yield wider X–E–X angles and amplified HOMO energies.12 Thus, acyclic (amino)alkyl carbenes, possessing relatively small HOMO–LUMO separation compared to diaminocarbenes, excel in activating various chemical bonds, such as H–H and N–H.13
DFT calculations indicated that the boryl substituents in NHBs exhibit stronger electron-donating ability and π-accepting ability. The calculated Tolman and the percent buried volume demonstrate that substitution, backbone saturation, ring size, and heteroatoms all influence the electronic and spatial properties of NHBs.14,15 Therefore, the manipulation of the energies of the HOMO and LUMO orbitals in the compounds through the design of rational NHB structures is the key to achieving small molecule activity. This review primarily concentrated on the contemporary advancements in N-heterocyclic boryl (NHB)-stabilized low valent group 14 compounds, focusing on their synthesis, structural features, and their applications to the activation of small molecules and inert chemical bonds. In addition, the other types of boryl-substituted low valent group 14 compounds also included in the review for comparison.
2. Heavy group 14 carbene analogues
2.1 Silylenes
In 1981, West and co-workers reported the transient silylene, SiMes2 (Mes = 2,4,6-Me3C6H2), which undergoes dimerization at 77 K, leading to the formation of disilene Mes2SiSiMes2.16 Following this seminal work, a fair number of stable silylenes have been obtained with the investigations focussing on their novel structures and reactivity.17,18 In 2012, Aldridge's group reported the first synthesis of NHB-substituted silylene Si{B(NDipCH)2}{N(SiMe3)Dip} 4 by reaction of SiIV trihalide Si{N(SiMe3)Dip}Br3 with two equiv. of NHB-lithium reagent 1.19 In this reaction, 1 served as a reductant as well as a substituent (Scheme 3).
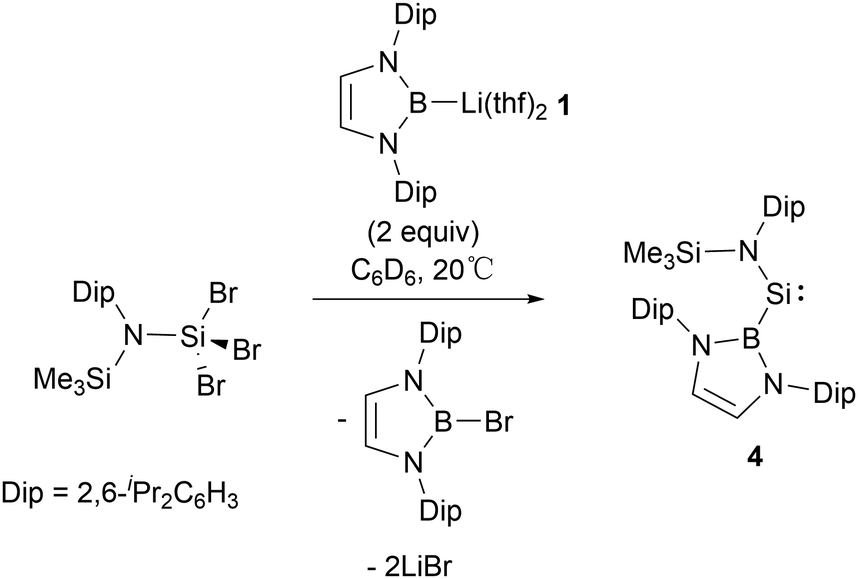 | ||
| Scheme 3 Synthesis of acyclic silylene 4. | ||
The 29Si NMR spectrum of 4 at an exceedingly low field (δSi = 439.7 ppm) is in line with a two-coordinate silylene, as seen in acyclic silylenes Si{C(SiMe3)2CH}2 and Si{N(SiMe3)2}2 with δSi values of 567 and 224 ppm, respectively.20,21 X-ray crystallographic analysis revealed that the Si–B and Si–N bond lengths are 2.066(1) Å and 1.731(1) Å, respectively. The B–Si–N bond angle of 109.7(1)° is close to that computed value for acyclic diaminosilylene Si{N(SiMe3)2}2 (110.2°).21 However, these Si(II) angles are much wide compared to N-heterocyclic silylene Si(NtBuCH)2 (90.5(10)°).22 Theoretical analyses suggested that silylene 4 features a narrow singlet–triplet gap of 103.9 kJ mol−1, which markedly enhances its reactivity. Silylene 4 possesses both a π-donor amido and a π-acceptor boryl substituent, with the amine substituent being nearly parallel to the B–Si–N plane (interplane torsion angle = 9.9°). DFT calculations indicated that the HOMO and LUMO are predominantly located on the divalent silicon atom.
In 2019, the group of Cui fused an NHB substituent with ethylenediamine, leading to the synthesis of a sterically demanding boryl-substituted ethylenediamine [CH2NH(NHB)]2 (NHB = (CHDipN)2B, Dip = 2,6-iPr2C6H3, (Scheme 4)).23 The reaction of the dilithium salt of the ethylene-linked diamido ligand with SiCl4 followed by reduction with two equiv. of lithium in the presence of 5% naphthalene in THF yielded the saturated N-heterocyclic silylene 5 in good yield. The structural validation of 5, as derived from single crystal X-ray diffraction studies, indicated a folded SiN2C2 ring with the N1–Si–N2 angle of 91.82(7)°. The Si–N bond lengths of 1.7313 and 1.7397(15) Å are marginally elongated compared to those (1.72 Å) in Si(NButCH2)2.24 The C–C bond length of 1.509(3) Å in the five SiN2C2 membered ring is somewhat shortened relative to that in Si(NButCH2)2 (1.520 Å). The NMR spectrum of 5 shows a 29Si shift at 137.8 ppm. Theoretical explorations indicated that the lone pair of silicon atom in 5 resides in the HOMO−2 orbital. In contrast, the LUMO mainly consist of the unoccupied silicon p-orbital with some contributions from the N–B π orbitals.
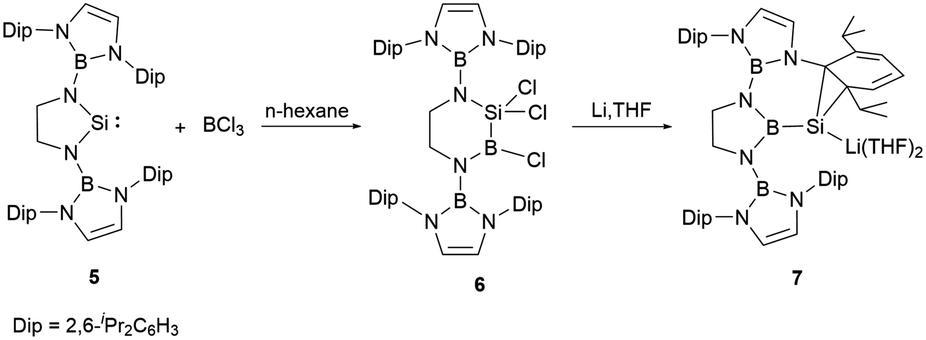 | ||
| Scheme 4 Reaction of silylene 5 with BCl3 and the subsequent reduction reaction. | ||
Compound 5, upon treatment with BCl3, underwent a ring-expansion transformation,25 leading to the formation of the six-membered silicon-boron heterocycle 6. Reduction of 6 with four equivalents of lithium in THF at −78 °C over 2 days yielded the intriguing N-heterocyclic boryl-substituted silanorcaradienyllithium 7 (Scheme 4). In this reaction, the boron-silicon exchange took place, leading to the N-heterocyclic boryl-substituted silicon compound. This B–Si exchange reaction opened a new entry to the development of N-heterocyclic boryl-substituted low valent silicon chemistry.
The 29Si NMR spectrum of 7 shows a pronounced broad signal at −161.5 ppm, which stands in proximity to the silacyclopropenyllithium (−153.9 ppm).26 The ring contraction of the six-membered C2N2SiB ring to the five-membered C2N2B boryl-substituted silicon compound with maintaining the B–Si bond is unprecedented. It is very likely that the reduction of 6 may generate a highly reactive SiB double bond intermediate, which then isomerize to the stable five-membered C2N2B ring. X-ray single-crystal diffraction studies confirmed its structure, in which the cycloaddition of the silicon atom with one CC bond of the pendant aromatic Dip ring forming a SiC2 ring was observed.
At ambient temperature, compound 7 reacted with the nucleophiles MeOTf and Et3NHCl, leading to a ring expansion at SiC2 ring with the formation of silacycloheptatrienes 8 and 9 in high yields (Scheme 5).23 Compound 9 reacted with NHCMe (1,3,4,5-tetramethylimidazol-2-ylidene) at room temperature, leading to the liberation of the silicon atom from the seven-membered ring with the formation of NHC-stabilized hydrido-silylene 10. The 29Si NMR spectrum of 10 features a signal at −122.1 ppm, which lies within the range for NHC-stabilized hydridosilylenes (−137.8 to −80.5 ppm).27
 | ||
| Scheme 5 Reactions of 7 with electrophiles and reaction of 9 with NHCMe. | ||
2.2 Germylenes
In 2012, a collaborative effort involving Aldridge, Jones and Mountford groups led to the successful synthesis of NHB-substituted (amino)(boryl)germylene 11.19 Following the work, Aldridge and co-workers enriched the chemical landscape with a myriad of bory-substituted germylenes. By 2015, utilizing Lappert's diamidogermylene [{(Me3Si)2N}2Ge] in tandem with the NHB lithium reagent, NHB-substituted aminogermylene [{(Me3Si)2N}Ge{B(NDipCH)2}] 12 was synthesized.28 In 2016, N-heterocyclic carbene stabilized (NHB)(chloro)germylene 13 and (aryl)(NHB)germylene 14 (Scheme 6) were reported.29,30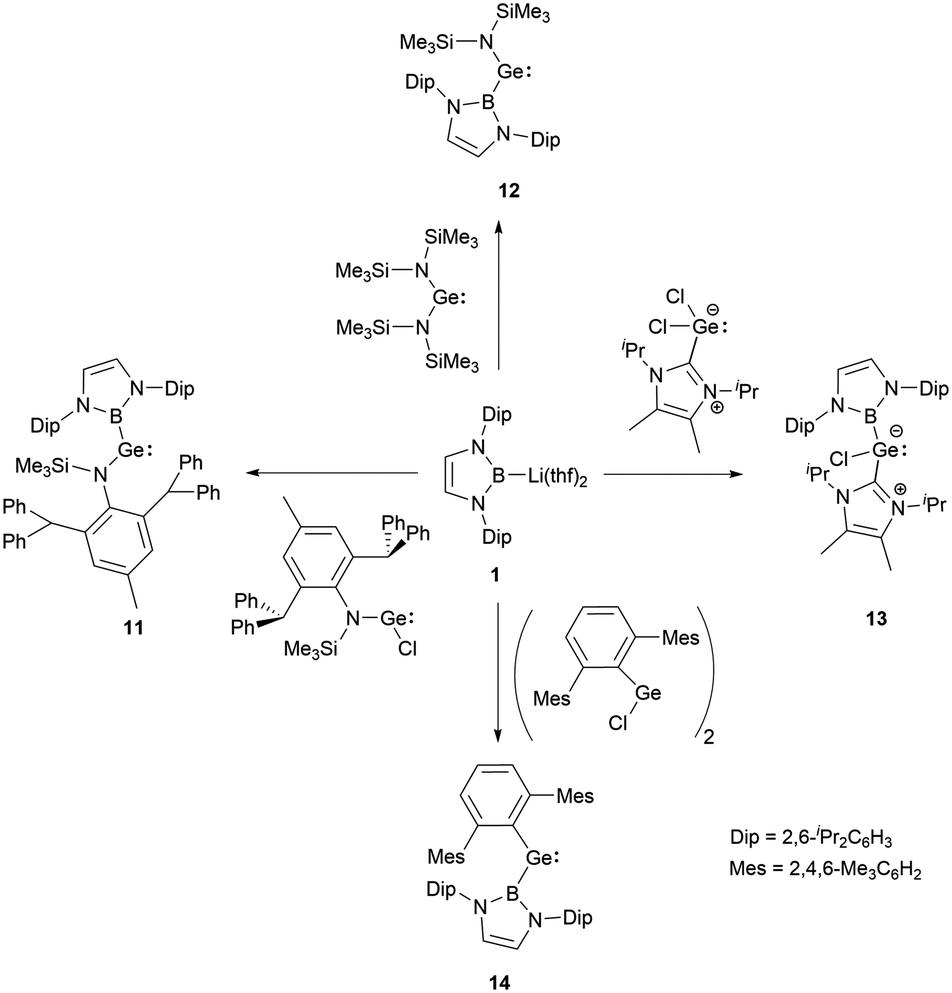 | ||
| Scheme 6 Synthesis of NHB-substituted germylenes. | ||
The Ge–B bond distances of 2.141(2), 2.129(6), 2.111(3), and 2.127(3) Å in germylenes 11–14 are very close. As strong σ-donors ascend the HOMO, compound 14 features the narrowest HOMO–LUMO gap. At ambient conditions, 14 readily participates in intramolecular C–H activation, leading to the formation of germanium hydride 15 (Scheme 7).30
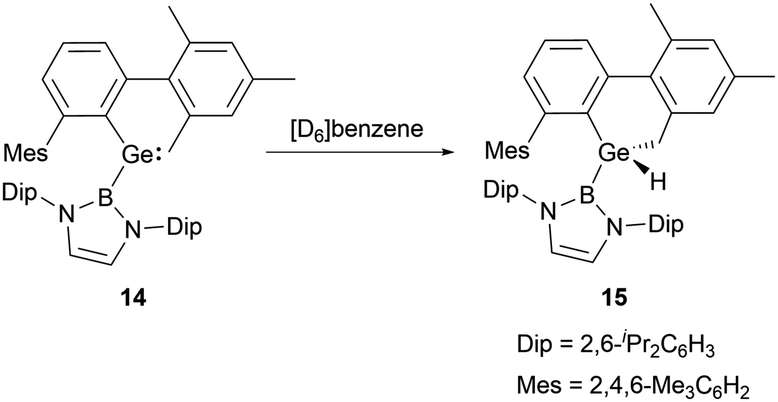 | ||
| Scheme 7 Intramolecular C–H activation mediated by germylene 14. | ||
The employments of N-heterocyclic boryl ligands in low valent germanium chemistry predominantly necessitate the boryllithium reagents, which reacted suitable divalent germanium precursors to yield NHB-substituted germylenes. The strong σ-electron donation capacities coupled with significant steric encumbrance of NHB substituents are pivotal in modulating the HOMO–LUMO gaps and stability.
On the other hand, Kinjo and his co-workers synthesized a six-membered C3NBGe cyclic system 16 by reaction of a cyclic (alkyl)(amino)germylene (cAAGe) with an arylboron dibromide under ambient conditions in 2019. The subsequent reduction with one equivalent of KC8 in the presence of an excess of PMe3 yielded phosphine-stabilized divalent germylene 17.31 Similarly, the reduction in the presence of 1,3,4,5-tetramethylimidazol-2-ylidene (MeNHC) yielded NHC-stabilized germylene 18 (Scheme 8).
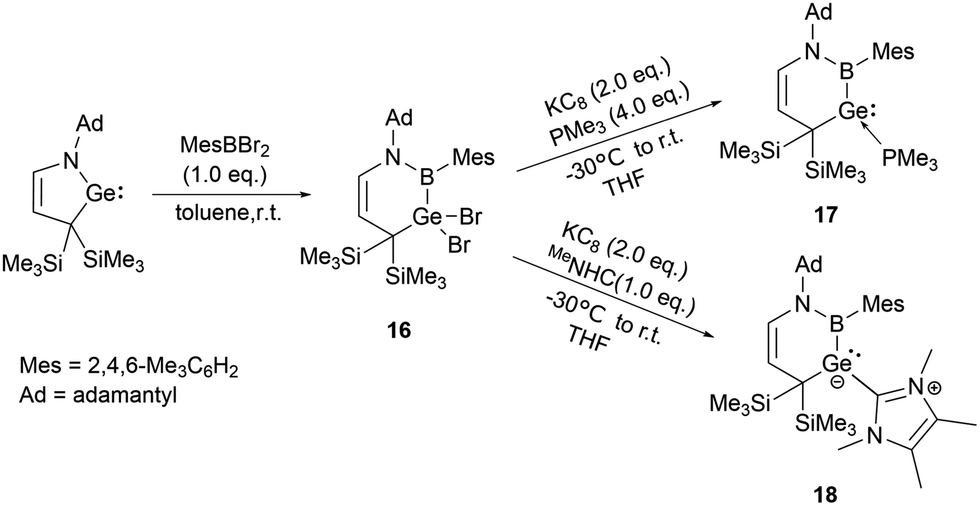 | ||
| Scheme 8 Preparation of 16 and reduction of 16 in the presence of a base. | ||
2.3 Stanylenes
In low-valent silicon chemistry, the synthesis of diboryl-substituted silylenes represents a challenge. However, tin chemistry presents a contrasting scenario, enabling easier realization of such compounds probably due to the large ionic radii of divalent tin atom. A noteworthy development in 2012 saw N-heterocyclic boryllithium reagents reacting with either SnCl2 or [Sn{N(SiMe3)Dip}(μ-Cl)]2, leading to the formation of NHB-substituted stannylenes, namely NHB-stabilized stannylene Sn{B(NDipCH)2}{N(SiMe3)Dip} 19 and di(NHB)-stabilized Sn{B(NDipCH)2}220 (Scheme 9).19 Insights from X-ray single crystal diffraction analysis and Density Functional Theory (DFT) calculations highlighted the following: the resultant systems manifest bent monomeric conformations [with angles: ∠N–Sn–B = 106.7(2)° and ∠B–Sn–B = 118.8(3)°]. The calculated singlet and triplet energy gaps (ΔEs→t) for 19 and 20 stand at 121.7 and 39.2 kJ mol−1, respectively. As seen in compound 19, the HOMO centered on tin distinctly showcases 5p orbital traits with a contribution of 38.8% from Sn 5p and 18.3% from Sn 5s, respectively.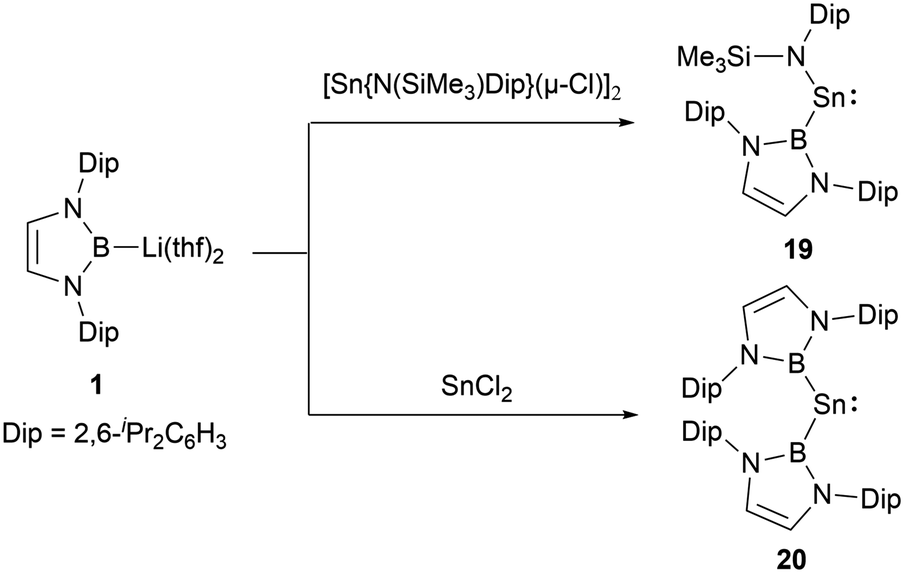 | ||
| Scheme 9 Synthesis of NHB-substituted stannylenes. | ||
3. Multiple bonding species
3.1 Silicon
Disilenes, compounds featuring a SiSi double bond, have been the subject of extensive research due to their distinct electron structures and geometries.32 Boryl-substituted disilenes could be obtained by the substitution reaction of disilene anions with boron halides, hydroboration of disilynes and reduction of boryl-substituted halosilanes. The substituents on the boron atom significantly affect the interaction of the silicon–silicon double bonds with the boron atom (s).
In 2008, Sekiguchi and co-workers reported boryl-substituted disilenes 22a and 22b (Scheme 10).33 The team achieved the synthesis via a salt elimination reaction between the sp2 silicon anion 21 and the corresponding halogenated boranes. X-ray single-crystal structural analysis of 22b revealed that, due to decreased steric hindrance, the SiSi bond length of 2.192(2) Å is marginally shorter than that of 21 [2.1983(18) Å]. The geometry of 22b is almost planar, with the sum bond angles around the silicon skeletal atoms being 359.98° and 359.99°. While the SiSi bond exhibits a slight twist with a torsion angle of 7.1°, the boron substituents stand perpendicular to the SiSi double bond. This orientation suggests the absence of π-conjugation between the SiSi double bond and the boryl group.
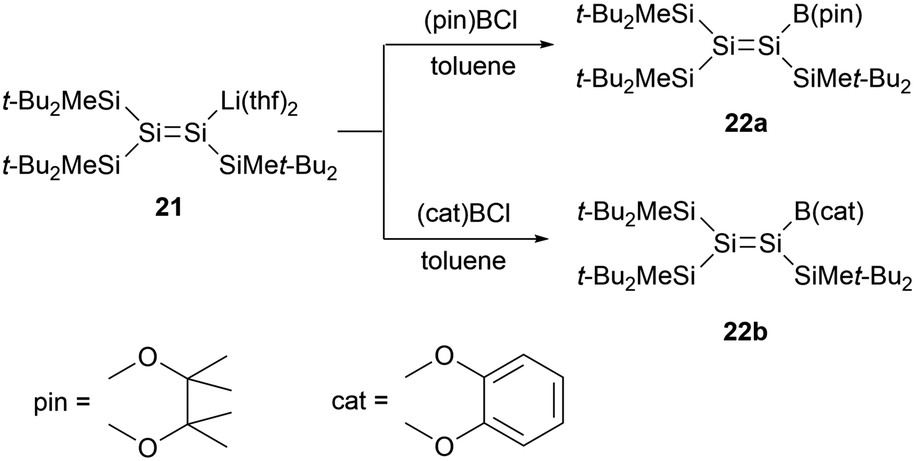 | ||
| Scheme 10 Synthesis of the first boryl-substituted disilenes. | ||
In 2010, the same group reported hydroboration of disilyne RSi![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiR (R = SiiPr[CH(SiMe3)2]2) 23 with borabicyclo[3,3,1]nonan-9-yl (9-BBN) for the synthesis of boryl-substituted disilene compound 24a (Scheme 11).34 The single-crystal structural analysis elucidates that the boryl group and the Si–Si double bond intersect to form a dihedral angle of approximately 18°. The B–Si bond length of 2.002(4) Å is notably shorter than that in 22b [2.022(8) Å], while the Si–Si double bond length of 2.1838(12) Å is within the range for Si–Si double bonds (2.138–2.280 Å).35 These structural features suggest the presence of π-conjugation between the π orbitals of the Si–Si double bond and the unoccupied p orbitals of the boron atom in compound 24a.
SiR (R = SiiPr[CH(SiMe3)2]2) 23 with borabicyclo[3,3,1]nonan-9-yl (9-BBN) for the synthesis of boryl-substituted disilene compound 24a (Scheme 11).34 The single-crystal structural analysis elucidates that the boryl group and the Si–Si double bond intersect to form a dihedral angle of approximately 18°. The B–Si bond length of 2.002(4) Å is notably shorter than that in 22b [2.022(8) Å], while the Si–Si double bond length of 2.1838(12) Å is within the range for Si–Si double bonds (2.138–2.280 Å).35 These structural features suggest the presence of π-conjugation between the π orbitals of the Si–Si double bond and the unoccupied p orbitals of the boron atom in compound 24a.
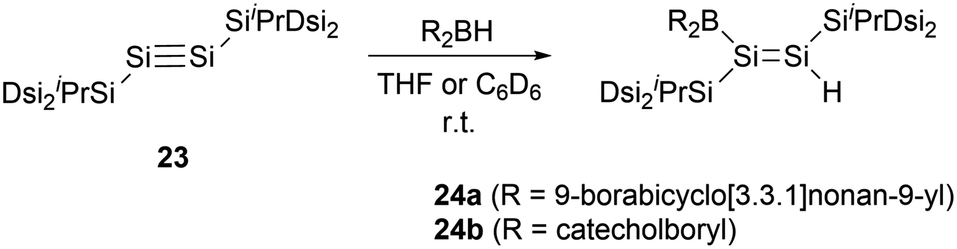 | ||
| Scheme 11 Synthesis of boryl-substituted disilenes 24a and 24b. | ||
In 2011, the team embarked on an investigation to ascertain the influence of varying boron substituents on the structures and properties of disilenes.36 They conducted a series of hydroboration reactions of disilyne 23 with diverse hydroboranes for the synthesis of boryl-substituted disilenes with different substituents on the boron atom. Notably, when compound 23 was reacted with catecholboryl hydride, compound 24b was successfully produced (Scheme 11).
By comparing single-crystal structures of compounds 24a and 24b, distinct structural differences could be seen. Compound 24b exhibited a dihedral angle of 52° between the boron substituent and the Si–Si double bond. This angle is markedly larger than the corresponding angle of 18° observed in 24a. This variation could be very likely attributed to the steric hindrance of the substituents on the boron atom and the interaction between the oxygen lone pair electrons and the boron atom. Such factors may hinder effective π-conjugation between the boron atom and the SiSi double bond. From the UV-vis spectra, 24a displayed a red-shifted maximum absorption wavelength (λmax: 469 nm) in comparison to that of 24b (λmax: 411 nm), suggesting that the π-conjugation effect in 24a is more pronounced than that in 24b. Theoretical calculations further supported the observation, indicating the influence of the dihedral angle on the π-conjugation between the π orbitals of the SiSi and the vacant p orbitals of the boron atom.
Drawing on these structural characteristics, the incorporation of both electron acceptors and donors in π systems emerges as a pivotal factor. It is instrumental in fine-tuning the photovoltaic attributes of the system. Such a phenomenon is colloquially termed the “push–pull effect”.37
In 2017, Iwamoto group reported a stable boryl-substituted disilene, demonstrating the “push–pull effect”.38 By reduction of the cyclic (alkyl)(amino)silylene (CAASi) 25 and TipSiCl3 with potassium graphite KC8, anionic disilene 26 was generated (Scheme 12). Subsequently treatment of 26 with the chloroborane in benzene yielded the BBN-substituted disilene 27 in 59% yield.
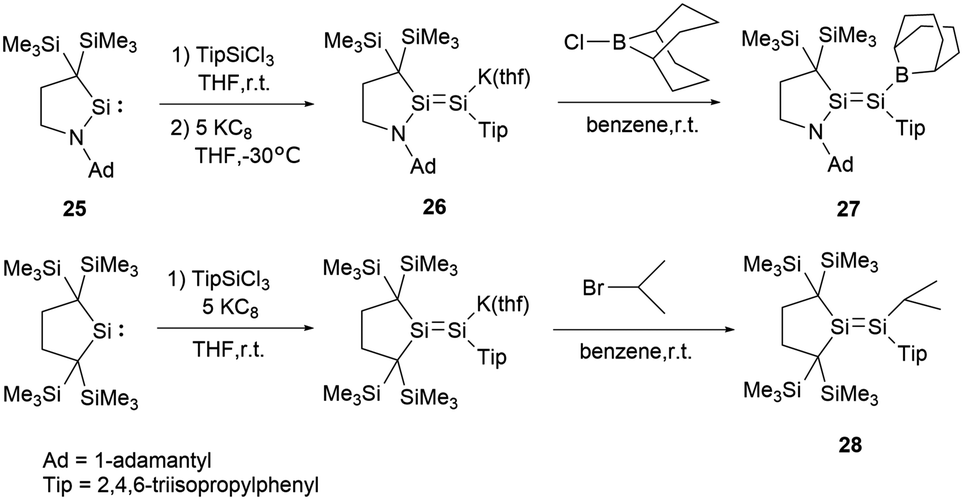 | ||
| Scheme 12 Synthesis of disilene 27 with “push–pull effects” and alkyl-substituted disilene 28. | ||
To elucidate the roles of the amino and borane groups in 27, the team synthesized the alkyl-substituted disilene 28 using a similar method for the comparison of its properties with those of 27. Notably, in 27, the boron and amine groups are positioned in trans to each other. The Si–Si bond length of 2.2146(6) Å in 27 is longer than that of 2.1828(6) Å in 28. The Si–N bond length of 1.7215(15) Å in 27 exceeds the typical Si–N double bond lengths in the range of 1.55–1.59 Å.39 However, the Si–N bond length is slightly shorter than found in amino-substituted disilenes (1.703–1.775 Å),40 which aligns closely with that observed in 25. When comparing the Si–B bond length of 1.945(2) Å in 27 to typical Si–B double bonds [1.8379(17) and 1.859(2) Å],41 it's longer but notably shorter than that found in boryldisilenes [1.996–2.022 Å].33,34,36 A pronounced difference in the 29Si NMR chemical shift between the two silicon atoms suggests notable polarization of the Si–Si double bond. The structural data supports the resonance structures shown in Scheme 13 due to the contributions from N+Si and SiB− double bonds. Theoretical analyses also highlighted that the 2p orbitals of the nitrogen and boron atoms in 27 play a crucial role in contributing to both the HOMO (π(SiSi)) and LUMO (π*(SiSi)).
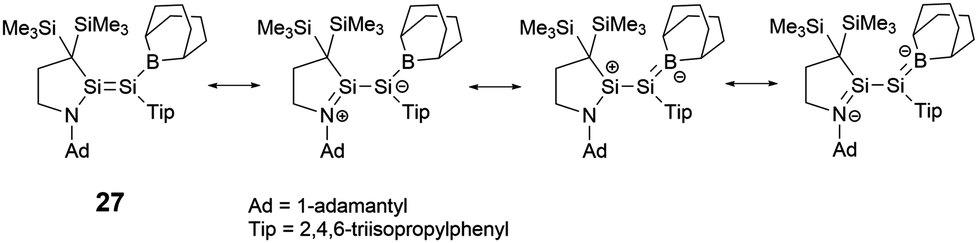 | ||
| Scheme 13 Resonance forms of boryl-substituted disilene 27. | ||
Recently, Iwamoto and co-workers reported that the monatomic silicon compound 29 undergoes a reaction with 9-BBN (BBN = borabicyclo[3.3.1]nonane) hydride and chloride, resulting in 1-amino-2-boryldisilenes 30a and 30b (a = H, b = Cl) (Scheme 14).42 The work demonstrated that “push–pull” effects serve as effective means to manipulate SiSi bond.
 | ||
| Scheme 14 Synthesis of 1-amino-2-boryldisilenes 30a and 30b. | ||
The single-crystal structure analysis of 30 revealed the twist angles of 48.11 and 60.91° for the Si–Si double bonds. The Si–B bond lengths measure 1.935(3) Å and 1.931(3) Å, which are marginally shorter than 27.38 Further, the Si–N bond lengths of 1.699 and 1.702(2) Å are longer than standard SiN double bonds. The Si–Si bond lengths of 2.2184(11) and 2.2541(10) Å are consistent with a SiSi double bond. Due to the influence of amino and boron substituents, the SiSi double bonds are highly polarized. As the twist angle increases, the polarization degree of the SiSi bond also escalates. A natural resonance theory (NRT) predicted that the contribution from III surpasses that from structure I in compounds 30a and 30b, while the contributions of the two structures in 27 are approximately equal (Scheme 15).
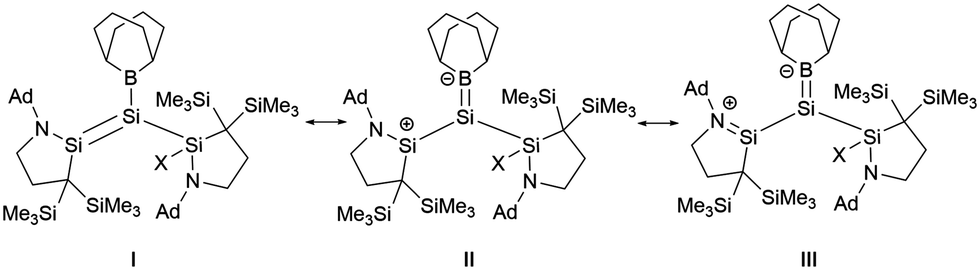 | ||
| Scheme 15 Resonance forms of boryl-substituted disilene 30. | ||
Compared to other organic group-substituted disilenes, those based on boryl substituents stand out. This distinction is attributed mainly to the conjugation between the SiSi π and boron 2p orbitals, leading to unique structures and reactivities. On the other hand, the boryl groups in the diazaborole frameworks feature strong σ-donating properties with relatively weak π-acceptor capacities, which result in unusual electronic structures and reactivity.
In 2020, Cui and co-workers synthesized a series of diazaborolyl (NHB) substituted silicon halides by taking advantage of exchange reactions between Si–OMe bonds with BX3 (X = Cl, Br).43 When NHB-substituted dibromosilane 31 was subjected to reduction with potassium graphite (KC8) in DME for 12 hours, the reaction generated bis(NHB)disilane 32, which was presumably formed by the abstraction of hydrogen from THF by a hydrosilylene intermediate. The NHB substituent in 32 is oriented perpendicularly to the B2Si2 plane. Compound 32 exhibits a notably shorter Si-Si bond length of 2.2796(15) Å, which is even shorter than several long SiSi double bond (2.289 Å).44 Attempts to generate divalent silicon species by dehydrohalogenation of 31 with N-heterocyclic carbene (NHC) led to the unexpected formation of the NHC-stabilized cationic species [(NHB)SiHBr(IiPr)]+Br−33 (Scheme 16).
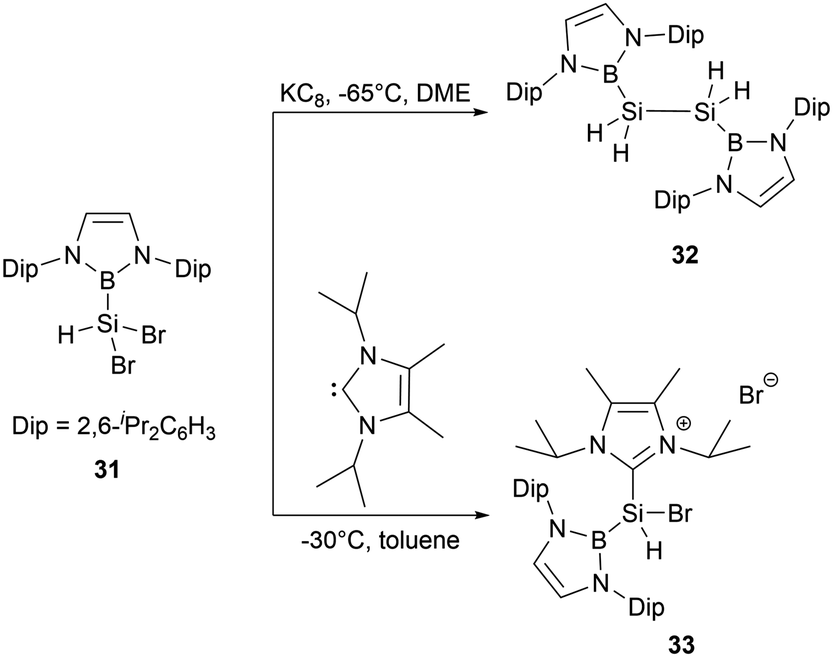 | ||
| Scheme 16 Synthesis of bis(NHB)disilane 32 and silyl cation 33. | ||
Reduction of dibromosilane NHB(Ph)SiBr234 bearing a NHB and a phenyl groups with KC8 in THF yielded NHB-substituted disilene (NHB)PhSi = SiPh(NHB) 35 (Scheme 17). The 29Si NMR spectrum of disilene 35 exhibits a singlet resonance at δ 200 ppm, which is downfield shifted compared to common disilenes (50–155 ppm) with organic substituents. Furthermore, its UV-vis absorption spectrum exhibits an absorption maximum of 447 nm, similar to those reported for tetraaryl-substituted disilenes. X-ray crystallographic analysis confirmed that disilene 35 adopts a trans-configuration for diboryl substituents. Upon reaction with MeOH, compound 35 was converted to methoxydisilane 36 without cleaving the B–Si bond. This reactivity is in contrast to that of compound 24a, which could undergo the cleavage of the B–Si bond under similar conditions.36
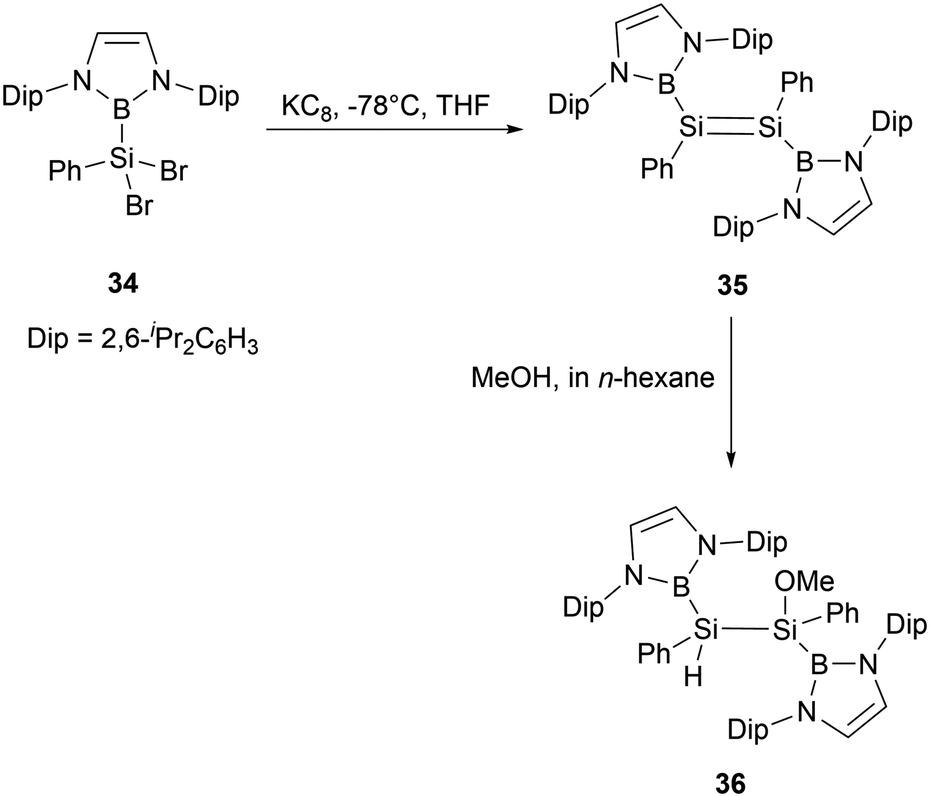 | ||
| Scheme 17 Synthesis of NHB-substituted disilene 35 and its reaction with MeOH. | ||
In the same year, Cui group successfully conducted the reduction reaction of (NHB)SiBr337 with magnesium. The reaction yielded a MgSi2 three-membered ring system 1-magnesium-2,3-disilacyclopropene [(NHB)Si]2Mg(THF)338 (Scheme 18).45 X-ray single crystal analysis of 38 elucidated that the molecule adopts a Mg–Si–Si tricyclic structure with the Si–Mg bond lengths of 2.574(18) and 2.600(2) Å. The Si–Si bond length of 2.223(17) Å is in the range for a typical Si–Si double bond. Notably, the two NHB groups adopt a cis configuration with the B, Si1, Si2, and Mg atoms residing on the same plane. This geometry is in contrast to a palladium disilyne complex previously described, in which trans-bend geometry in the disilyne segment and a pyramidal geometry of the silicon atom were observed.46 DFT calculations indicated that 38 has significant dianionic character. On the other hand, the Si–Mg bonds in the Si–Si–Mg tricyclic core structure was predicted to have some degree of covalent bond characters.
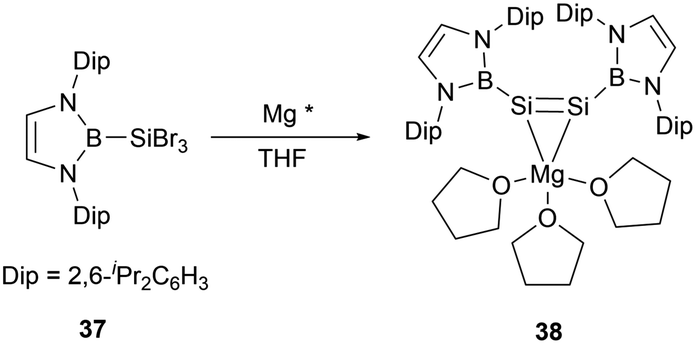 | ||
| Scheme 18 Preparation of magnesium disilene 38 by reduction reaction. | ||
Following these findings, the reduction of the diazaborolyl trichlorosilane (NHB)SiCl3 with three equivalents of KC8 led to the synthesis of the anionic bis(disilene) (THF)K(boryl)Si = Si(boryl)O(CH2)4(boryl)Si = Si(boryl)KTHF (39, Scheme 19) with two different disilene units.45 The formation of this compound is hypothesized to involve a disilene silicon radical anion intermediate, which could cleave the C–O bond of THF molecule.
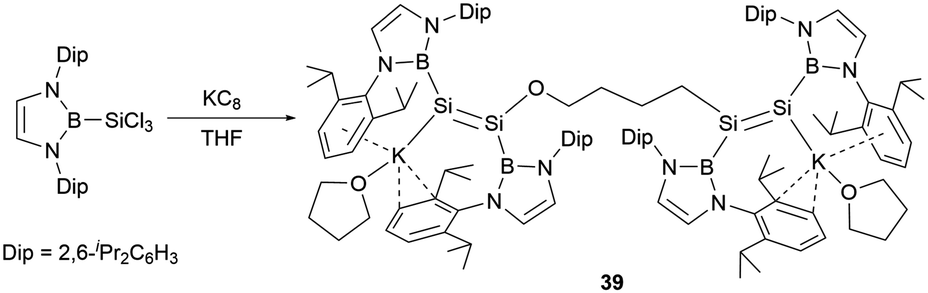 | ||
| Scheme 19 Synthesis of NHB-substituted bis-disilene dianion 39. | ||
Furthermore, Cui and co-workers also accomplished the reduction of (NHB)SiBr3 by utilizing four equivalents of lithium, leading to the synthesis of the first dilithiodisilene (NHB)LiSi = SiLi(NHB) 40 (Scheme 20).47 The 29Si NMR spectrum for 40 exhibited a resonance at 297.0 ppm, which is downfield shifted in comparison to that of 38 (204.1 ppm).
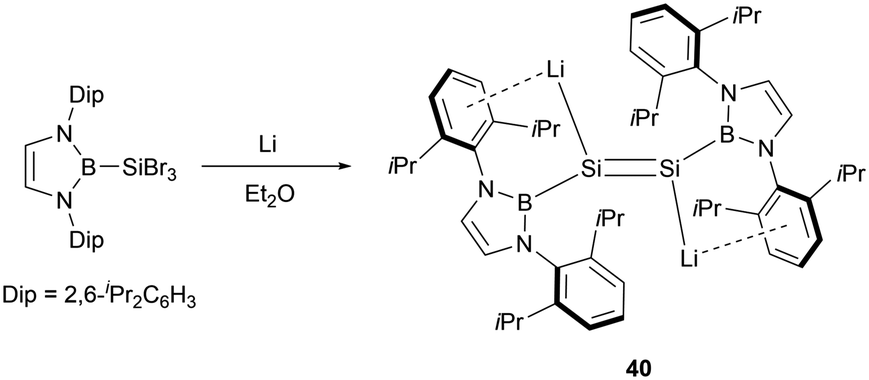 | ||
| Scheme 20 Synthesis of NHB-substituted dilithiodisilene 40. | ||
X-ray crystallographic analysis revealed a Si–Si bond length of 2.223(4) Å in 40. Additionally, the Si–Li bond length of 2.482(12) Å in 40 is slightly shorter than that found in the disilene lithium salt (2.85 Å).48 The near coplanar angle of 1.87° involving the N-heterocyclic boryl plane and the Si–Si double bond indicates the possible electron delocalization of the boryl plane with the SiSi π bond. The dihedral angle of 2.01° defined by B1–Sil–Sil′–Lil supports that the four atoms are almost in the same plane. Electronic effects from the positively charged boryl and lithium substituents in 40 may greatly contribute to the planar conformation.49 The HOMO of 40 predominantly comprises Si lone pair electrons, while the LUMO is primarily represented by the Si–Si π* bond.
The dianionic disilene 40 exhibited reactivity towards electrophiles through a salt elimination pathway. Reaction of 40 with dihaloboranes cleanly yielded BSi2 three-membered ring compounds 41a–c (Scheme 21).47 The unsaturated Si–Si–B tricyclic system is isoelectronic with both the aromatic cyclopropenyl cations ([C3R3]+) and borirenes (R2C2BR). X-ray crystallographic analysis disclosed that three-membered rings 41a–c feature tri-coordinated planar silicon and boron atoms. The 29Si NMR spectra for compounds 41a–c show a singlet at 112.2, 142.0, and 141.1 ppm, respectively. In the 11B NMR spectra, two broad peaks were observed for each of the rings (41a: δB = 24.2 and 50.1 ppm, 41b: δB = 23.5 and 72.9 ppm, 41c: δB = 23.4 and 73.3 ppm). Both single crystal analysis and theoretical calculations support that compounds 41a–c are strained 2π aromatic system.
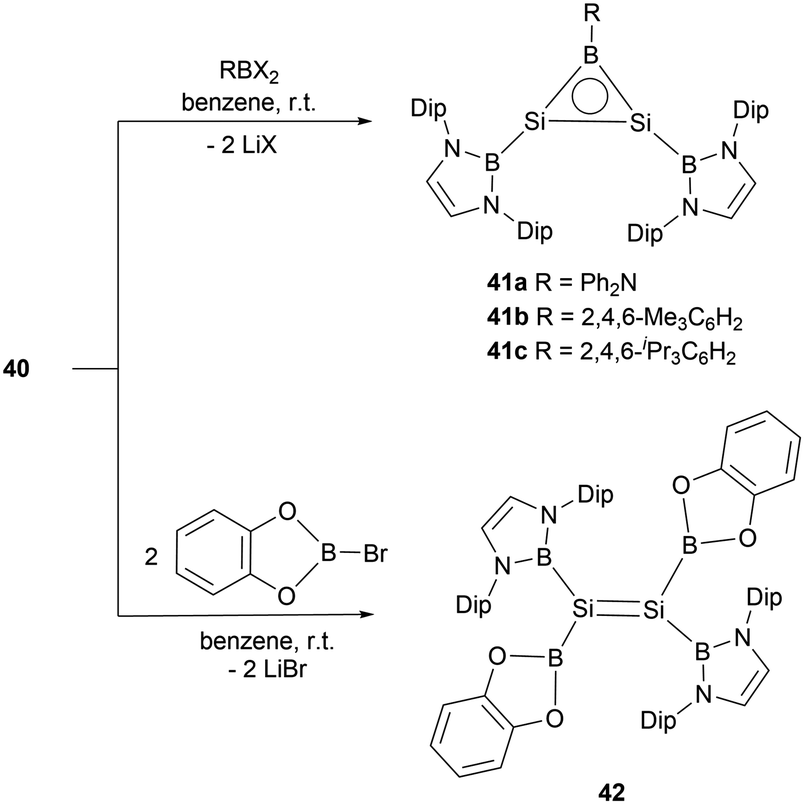 | ||
| Scheme 21 Reactions of 40 with haloboranes. | ||
Reaction of dilithiodisilene 40 with two equivalents of (cat)BBr yielded the tetraboryl disilene 42 (Scheme 21). X-ray single-crystal diffraction studies disclosed the Si–Si double bond length of 2.1654(12) Å. Further structural analysis indicated that disilene 42 features an essentially planar SiSi bond, defined by a torsion angle of 0° and a trans-bend angle of 1.86°, highlighting the effects of the boryl-substituent (σ-donor) on the geometry of the SiSi double bond. Disilene 42 represents a planar disilene with the smallest trans-bend angle and zero torsion angle.50
Remarkably, Cui and co-workers synthesized the NHB-substituted disilyne, the first boryl-substituted group 14 alkyne analogue.51 The synthesis involved the reaction of an N-heterocyclic silylene with BBr3 followed by reduction with lithium (Scheme 22). Compound 43 is stable in solid state and in n-hexane but slowly decomposed in benzene.
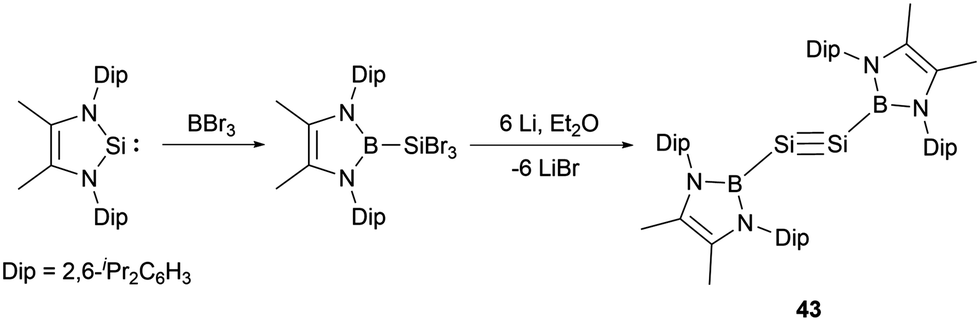 | ||
| Scheme 22 Synthesis of the first NHB-stabilized group 14 alkyne analogue 43. | ||
Disilyne 43 (NHB)SiSi(NHB) has been characterized by spectroscopic methods and X-ray single-crystal diffraction studies. The 29Si NMR spectrum of 43 delineates a singlet at δSi = 70.0 ppm, which is slightly upfield shifted compared with the known silyl-substituted disilynes, resonating at δSi 89.9 and 77.1 ppm respectively.52a,b The Si−Si bond length of 2.107(9) Å is consistent with those (2.0569(12) to 2.108(5) Å) reported for Si–Si triple bonds.52 The NHB rings align nearly coplanar with the B–Si–Si–B plane. This structural feature suggests an intriguing electronic delocalization spanning the π bond and the NHB ring. The HOMO and HOMO−2 are predominantly sculpted by the two Si–Si π bonds, also integrating contributions from the π bond of the NHB ring. The electronic features of the triple bond indicate that the electron delocalization between the Si–Si π bond and the NHB ring plays important roles in the stabilization and controlling geometry of the whole system.
In the same year, Wang and co-workers synthesized the neutral homoaromatic diboradisilacyclobutene 45 by reduction of borylaminobromosilane 44 with KC8 (Scheme 23).53 The 11B NMR spectrum exhibits two singlets at 24.4 ppm and 11.8 ppm. In the 29Si NMR spectrum of 45, signals are observed at −14.0 and −80.6 ppm, respectively.
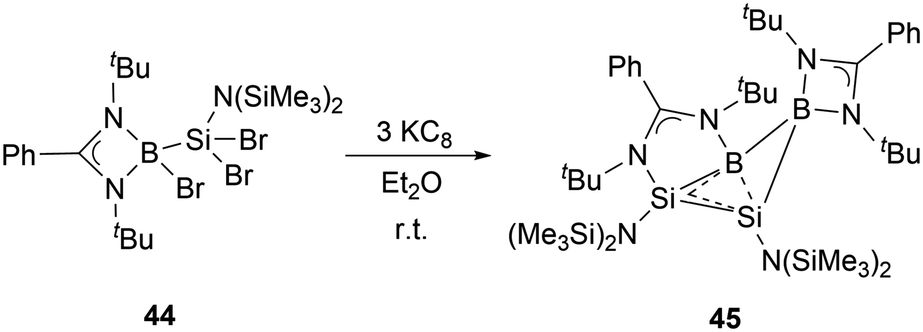 | ||
| Scheme 23 Synthesis of diboradisilacyclobutene 45. | ||
X-ray single-crystal analysis of 45 revealed a puckered four-membered B2Si2 ring with an angle of 26.2° as well as an unusually short transannular B–Si distance of 2.306(3) Å. Density functional theory (DFT) calculations, along with structural analyses, suggest that the relatively long Si–Si and short B–Si bonds are likely due to the delocalization of 2π electrons over the BSi2 moiety. These observations indicated that 45 possesses a significant 2π-homoaromatic character.
At ambient conditions, the reaction of compound 45 with two equivalents of CuCl in THF yielded diboradisilacyclobutane 46 (Scheme 24),53 the first instance of a cyclobutane analogue featuring both boron and silicon. The reaction is likely to go through a single electron transfer (SET) mechanism. The formation of 46 validates the existence of the internal Si–Si double bond in 45, which exhibits strong reducing capabilities. Treatment of compound 45 with an equimolar amount of HCl in THF at −78 °C resulted in the formation of 47via the 1,3-addition of HCl to the B1 and Si2 atoms in the B2Si2 ring. This reaction further demonstrates strong 1,3-interactions and polarization between B1 and Si2 atoms.
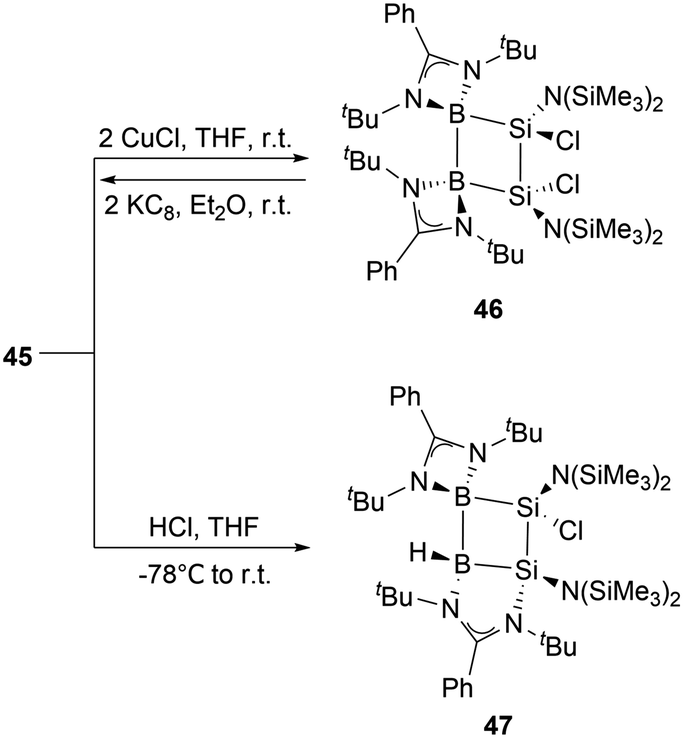 | ||
| Scheme 24 Reactivity of 45. | ||
3.2 Germanium
The valence electrons of Group 14 elements display an increasing lone-pair character as the atoms become heavier. This trend is evident from carbon to lead when these atoms form multiple bonds.In 2016, Aldridge and co-workers successfully synthesized the first NHB-stabilized digermavinylidene by reduction NHC-stabilized (NHC)(NHB)GeCl 13.29 The reduction products vary based on different reducing agents. Using Jones' reagent, the reaction led to the formation of an asymmetric NHC-stabilized digermene (NHC)(NHB)GeGe(NHB) 48. On the other hand, with highly reductive metal potassium or potassium graphite, the formally Ge0 species K2[(NHB)GeGe(NHB)] 49 was generated. An interesting transformation occurred to 49 in the presence of the oxidant [Cp2Fe][BArf4], where one of the two NHB groups migrates to give digermavinylidene (NHB)2GeGe 50, the first example of isolable group 14 vinylidene (Scheme 25).
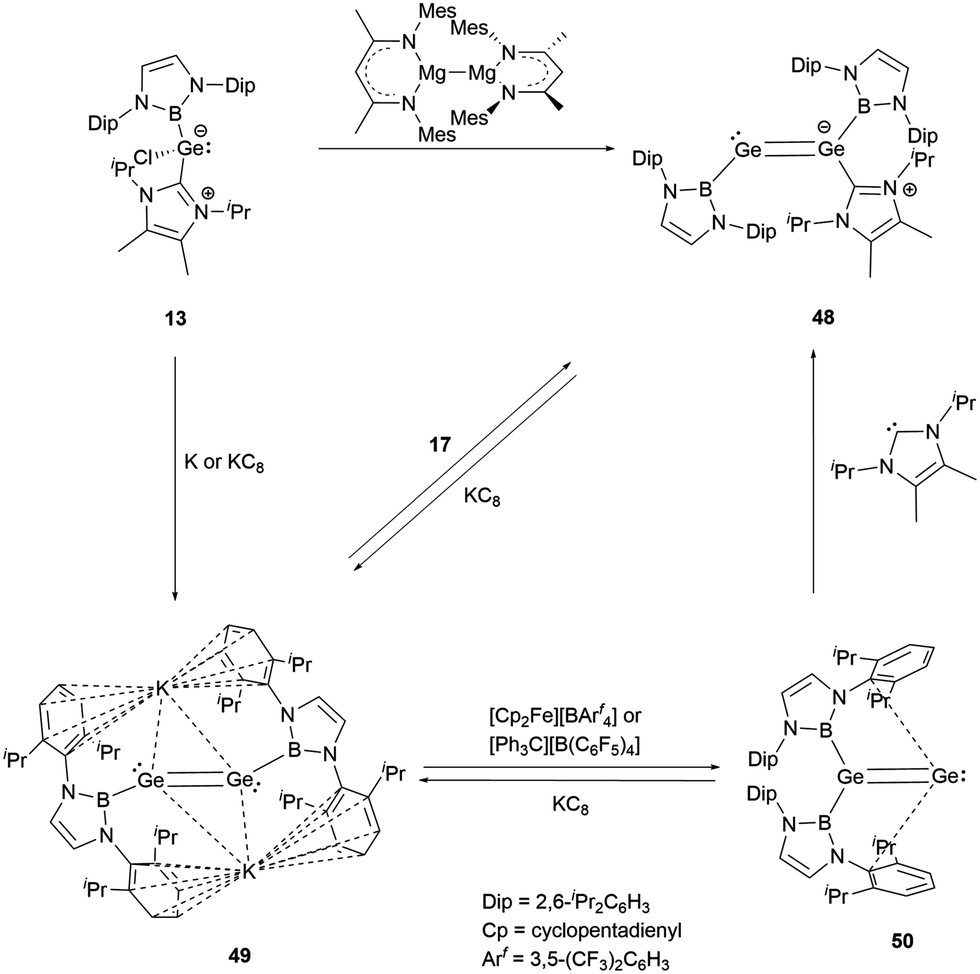 | ||
| Scheme 25 Synthesis of digermavinylidene 50 and the related digermanium systems 48 and 49. | ||
X-ray single crystal analysis revealed that 50 has a distinctive 1,1-disubstituted GeGe fragment. The Ge–B and Ge–Ge bond distances are 2.033(2) and 2.312(1) Å, respectively. The latter distance is typical for a GeGe double bond. DFT calculations disclosed the HOMO orbital of 50 corresponds to the Ge–Ge π bond, which is different from H2CC, where the HOMO is a lone pair on the carbon.54 This difference suggests that the Ge–Ge π bond molecular orbitals are inherently less stable than the C–C π bond. The LUMO orbital aligns with the B2GeGe plane and points to the side aromatic π system. The migration of an NHB substituent from one germanium atom to the other indicates the flexibility of a B–Ge bond. Alternatively, adding a suitable NHC molecule to 50 also led to the migration of the NHB group to yield mono-NHC adduct 48.
Dianionic digermene 49 reacted with NHC-stabilized dichlorogermane to give the cyclic compound Ge3(NHB)2(NHC) 51.55 Although the X-ray single-crystal structure has not been obtained for 51, the structure could be primarily drawn from the 1H and 11B NMR spectra. Notably, the structure of 51 has been characterized to have two equivalent NHB groups and one NHC group. Because of the predisposition of the tricyclic germanium system to dimerize, treatment of 51 with triphenylborane yielded germanium cluster Ge6(NHB)452 (Scheme 26).
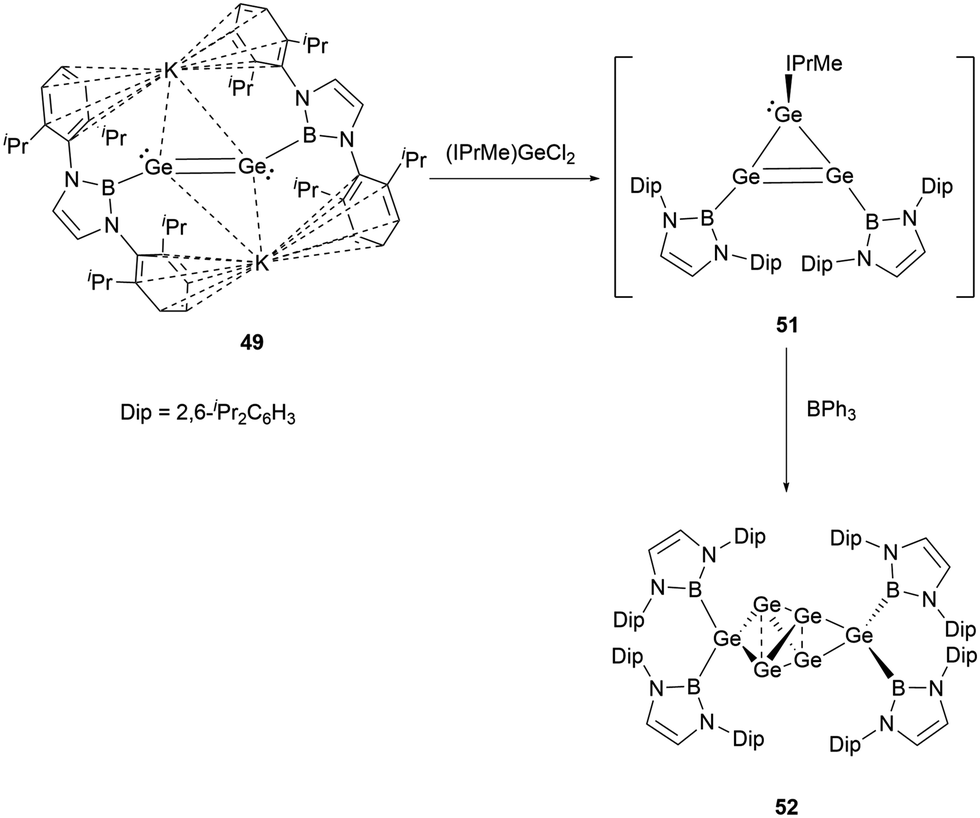 | ||
| Scheme 26 Synthesis of NHB-substituted Ge6 cluster Ge6{B(NDipCH)2}452. | ||
As shown in Scheme 27, treatment of 13 with either Na[BArf4] or Li[Al(OC(CF3)3)4] yielded the dimeric cationic compound [532]2+ (Scheme 27).56 The structure of [532]2+ includes NHB and NHC substituents with trans configuration around the two germanium centers. The Ge–Ge bond length of 2.300(2) Å is consistent with the established values for a GeGe double bond. However, this bond length is substantially shorter than that observed in the tungsten-substituted digermene complex {(NHC)[Cp*(CO)3W]GeGe[W(CO)3Cp*](NHC)}[BArf4]2 (2.429(1) Å).57
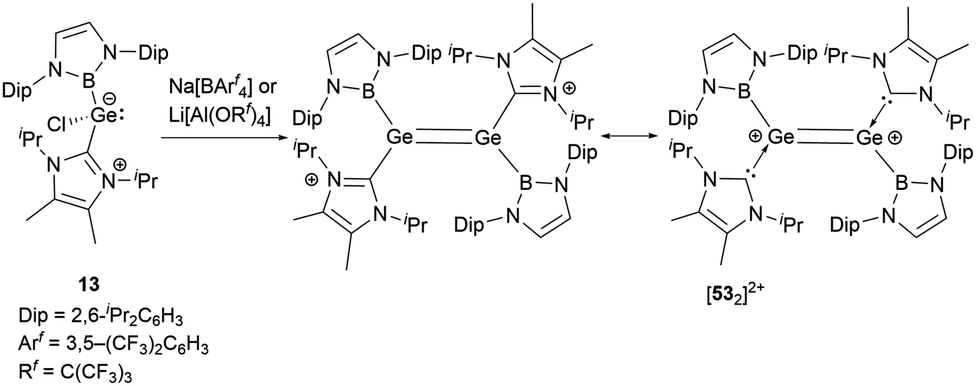 | ||
| Scheme 27 Synthesis of the dimeric cationic compound [532]2+. | ||
In solvents with an electron-donating atom, such as THF or pyridine, [532]2+ rapidly dissolves, resulting in the cleavage of the Ge–Ge bond with the generation of the cationic adducts [53·L]+ (Scheme 28). These compounds showcase a three-coordinated germanium with a pyramidal geometric configuration. In the mono cations, the angle formed between the neutral donors around germanium is proximate to 87.5(2)° and 90.2(1)°. On the other hand, the boryl substituent manifests considerably enlarged angles of 100.9(2) and 105.5(1)°. These observations might be indicative of the spatial demands of the NHB substituents and the implications of Bent's law, which emphasizes the concentration of p-orbital character in the bonds to the more electronegative atoms.58
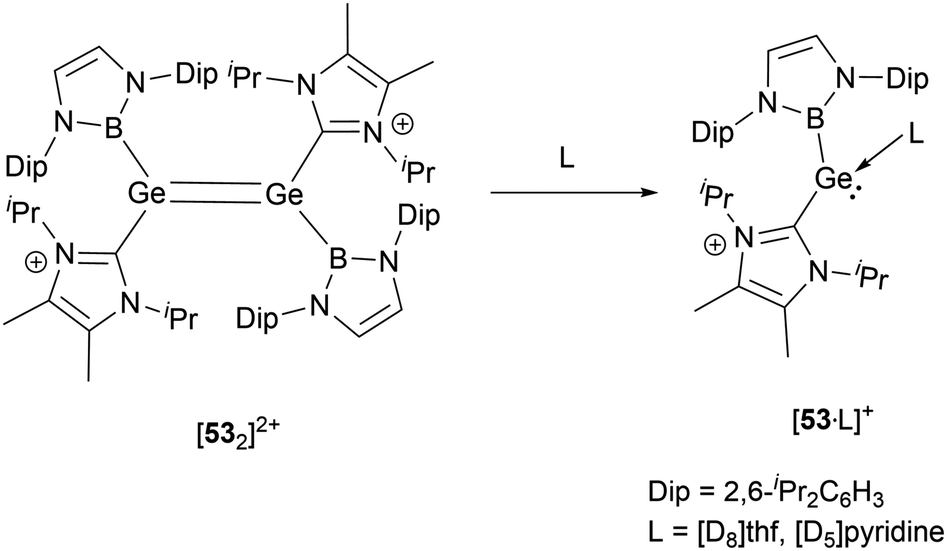 | ||
| Scheme 28 Cleavage of dimeric [532]2+ in the presence of Lewis bases. | ||
3.3 Tin
Recently, a novel NHC-stabilized boryl tin(II) bromide (NHC)(NHB)SnCl (54) was reported.59 Interestingly, it behaves chemically akin to similar germanium compounds, leading to several types of products under varying reduction conditions. When it was reduced with Jones's Mg(I) reagent, the unsymmetrical NHC-stabilized distance NHBSnSn(NHC)(NHB) 55 was formed. On the other hand, reduction with an excess of potassium graphite (KC8) or potassium naphthalenide (NpK) produced the formal Sn0 compound K2[Sn2(NHB)2] (56, Scheme 29).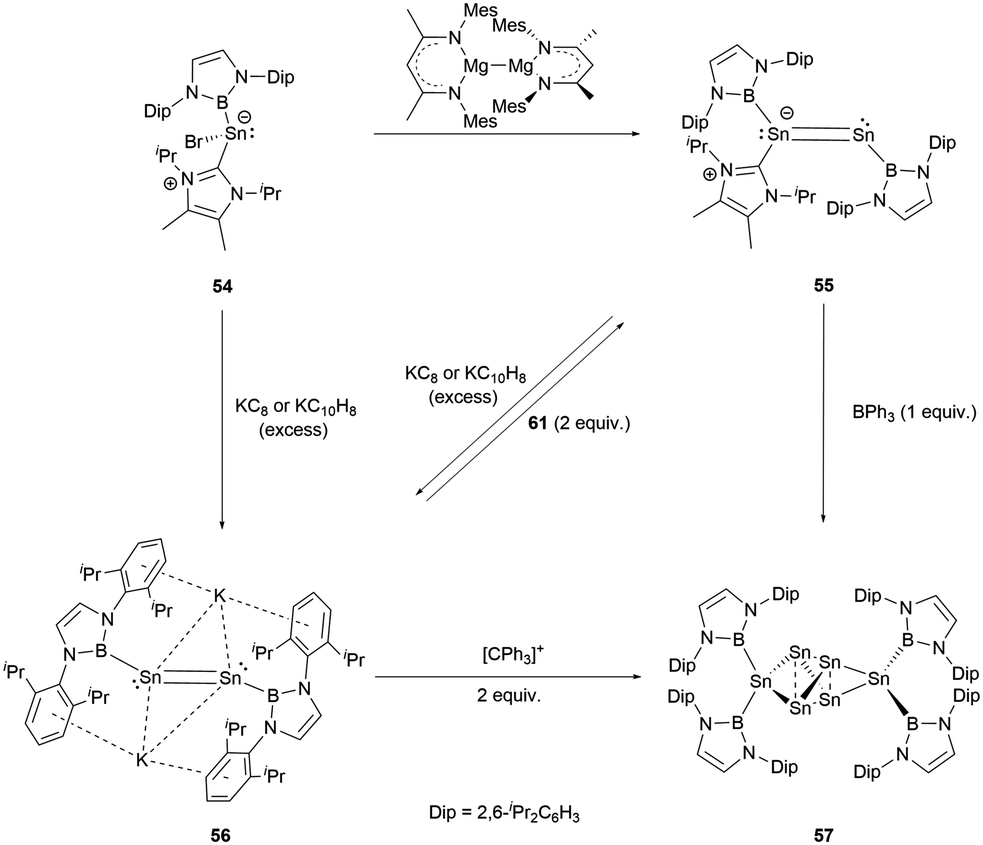 | ||
| Scheme 29 Reactions of 54 with different reducing agents. | ||
X-ray single-crystal analysis revealed the Sn–Sn distances in 55 and 56 to be 2.581 and 2.749(1) Å, respectively. Notably, 56 exhibited a trans configuration involving its Sn2 unit with the Sn–Sn–B angle of 96.5(1)°. The addition of BPh3 expunges the NHC ligand from 55. The product thus generated is indistinguishable from that formed by the reaction of 56 with an oxidizing agent. These reactions collectively yield a previously unidentified compound Sn6[NHB]457 (Scheme 29).
The structure of 57 was confirmed by X-ray single-crystal analysis. The central four tin atoms adopt a slightly distorted tetrahedral geometry. Intriguingly, the opposite edges of the tetrahedron are extended symmetrically and connected by [Sn(NHB)2] entities. Both the intra-tetrahedral Sn–Sn bond distances (2.848 and 2.831(1) Å) and those involving the [Sn(NHB)2] units (2.841(1), 2.834(1) Å) are in the expected values for a Sn–Sn single bond. Additionally, the Sn–B bond lengths (2.259 and 2.269(6) Å) closely match those found in Sn(NHB)2.
The mechanism for the formation of 57 is very likely to involve a vinylidene intermediate, which appears to easily undergo atom transfer, leading to the generation of Sn(NHB)2 and [Sn3(NHB)2]. Remarkably, the trinuclear compound exhibits the trend to dimerize, a process that is paired with the migration of the boryl ligand.
Furthermore, modulating the amount of the reducing agent KC8 could lead to some novel species. Compound 54 reacted with 1.5 equivalents of KC8 to yield a NHB-stabilized dianionic Sn4 cluster K2[Sn4(NHB)4] 58 (Scheme 30).59 X-ray single-crystal analysis of 58 revealed that the four symmetrical tin atoms are disposed in a quasi-square geometry with the Sn–Sn bond distances of 2.9018(9) and 2.9010(6) Å and Sn–B bond length of 2.306(5) Å. The Sn–Sn–Sn angle of 88.41(3)° is close to 90°. The two potassium ions, situated both above and beneath the Sn4 unit (K⋯Sn contacts of 3.915 and 3.510 Å), are encapsulated between the Dip aromatic rings from the NHB frameworks with the K⋯aromatic contacts of 3.088 and 3.413 Å. Consecutively alternating boron atoms reside either above or below the approximate Sn4 plane. As a result, each tin atom adopts a pyramidal geometry (the sum of angles around tin atom = 318.6°). Interestingly, this structure bears some resemblance to cyclobutadiene.
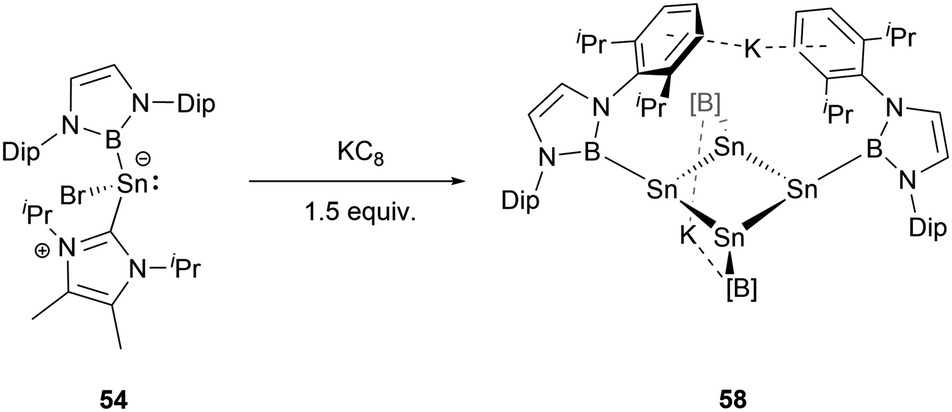 | ||
| Scheme 30 Synthesis of compound 58. | ||
From the structural analysis of NHB and other boryl-substituted group 14 multiple bonding species, it could be concluded that diazaborolyl substituents feature strong σ electron-donating properties, which could stabilize neutral, anionic and cationic group 14 multiple bonding species. In addition, because of the relatively weak B–E (E = Si, Ge, Sn) bond, boryl substituent may undergo intramolecular migration to yield novel multiple bonding species or molecular clusters.
4. Application to the activation of small molecules
4.1 Activation of dihydrogen
At ambient conditions, NHB-substituted silylene 4 exhibits a remarkable ability to directly cleave H2 σ-bond, resulting in the formation of the dihydrosilane H2Si(NHB){N(Dip)SiMe3} 59 (Scheme 31).19 The direct activation of dihydrogen by 4 presents the first experimental validation of H2 activation by a silylene. DFT calculations supports a cooperative bimolecular mechanism. The activation of HD by 4 exclusively yielded H(D)Si(NHB){N(Dip)SiMe3}, in line with the proposed bimolecular process.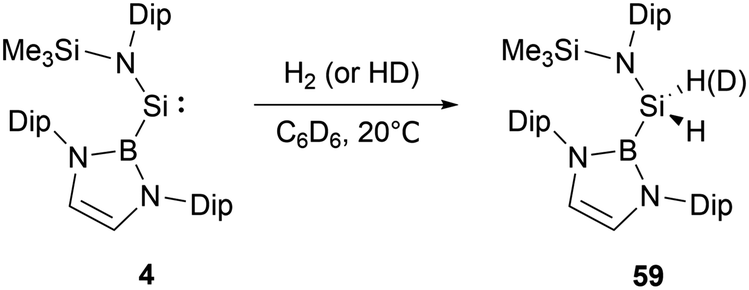 | ||
| Scheme 31 Activation of H2 by acyclic NHB-substituted silylene 4. | ||
In contrast to the straightforward oxidative addition of H2 to the silicon center in silylene 4, the reaction of the corresponding germylene 12 with H2 is more complex (Scheme 32).30 The 1H NMR spectrum of the reaction product exhibited three Ge–H signals at δH 1.73, 2.53 and 4.92 ppm and two different boryl and one N(SiMe3)2 signals. X-ray single-crystal diffraction studies confirmed an asymmetrical digermane product 60 with a Ge–Ge single bond distance of 2.431(1) Å. One germanium atom is attached to the NHB substituent, N(SiMe3)2 group and one hydrogen atom, whereas the other Ge atom is bonded with one NHB and two hydrogen substituents. The mechanistic analysis suggested the initial activation of H2 by 12, leading to the formation of the low valent germanium intermediate [(NHB)GeH]n and the by-product HN(SiMe3)2. The germanium intermediate subsequently reacted with H2 to yield (NHB)GeH3, which then reacted with the excess of 12 to give 60. Concurrently, the generated amine HN(SiMe3)2 from the initial step underwent a reaction with 12 in the latter phase, yielding by-products [Ge{N(SiMe3)2}2] and N-heterocyclic hydroborane.
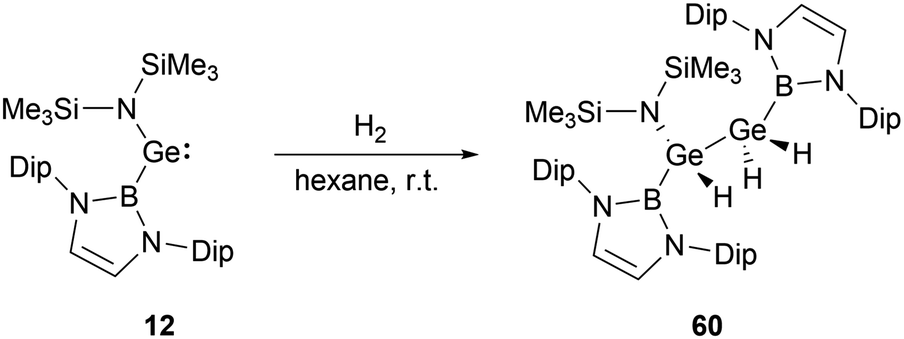 | ||
| Scheme 32 Activation of dihydrogen by (amido)borylgermylene 12. | ||
The NHC-stabilized cationic germylene [53·thf]+ could react with dihydrogen even under relatively mild conditions, resulting in the formation of the NHC-stabilized dihydrogermane cation 61 (Scheme 33).56 Single-crystal structural analysis disclosed significant variations in geometric parameters: compared with other Ge(IV) species, the Ge–B and Ge–C bond lengths of 2.048 and 1.991(2) Å are shortened, whereas the B–Ge–C angle of 114.4(1)° is expanded.
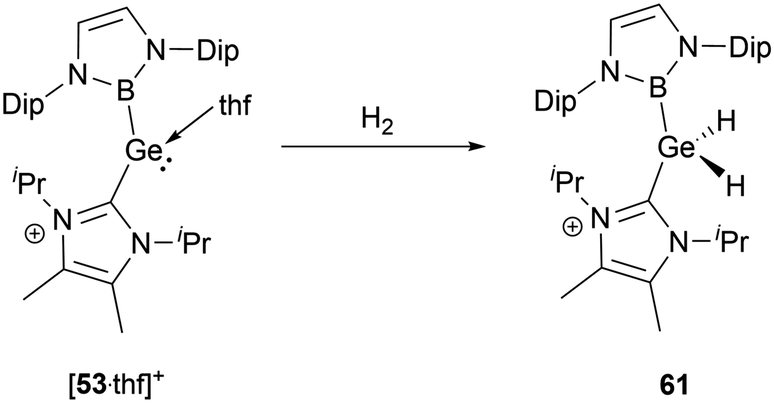 | ||
| Scheme 33 Activation of H2 by the cationic complex [53·thf]+. | ||
The cyclic boryl-stabilized germylene 17 also exhibits the capability to activate H2.31b Exposure of 17 to dihydrogen in C6D6 solution at 50 °C over 12 hours resulted in the formation of the boryl-substituted dihydrogermane 62 (Scheme 34).
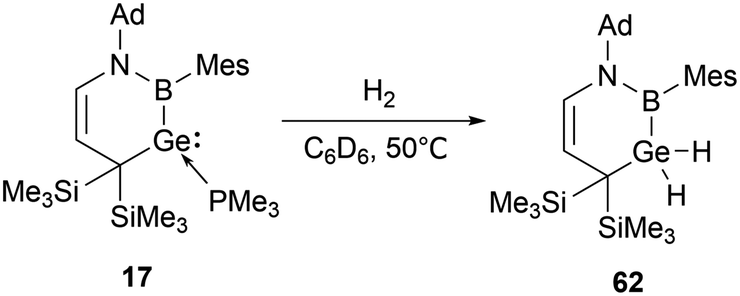 | ||
| Scheme 34 Activation of H2 by cyclic boryl-stabilized germylene 17. | ||
NHB and amino-substituted stannylene 19 (Scheme 9) does not react with H2 under normal conditions.60 However, at ambient conditions, H2 partakes in oxidative addition at the tin center of di(NHB)-substituted stannylene 20,61 leading to the formation of the diboryl dihydro tin(IV) compound 63 (Scheme 35). A distinct NMR signal attributable to the SnH2 unit appears at δH = 2.22 ppm. Notably, the B–Sn–B angle in 63 is appreciably expanded to 135.6(1)° from the 118.8(3)° observed in the 20. This may be attributed to the contraction of the Sn–B bond as the oxidation state varying from SnII to SnIV, leading to a greater B–Sn–B angle to release steric hindrance presented by the two NHB substituents.
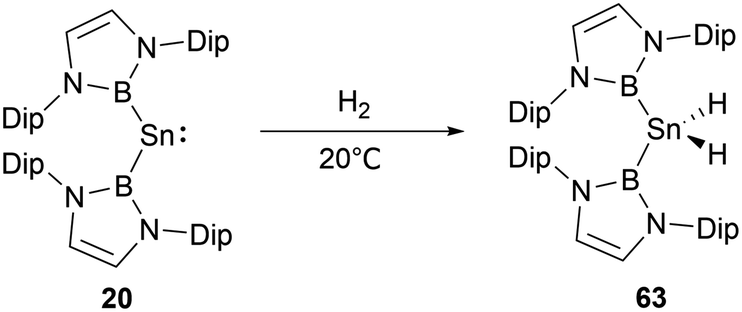 | ||
| Scheme 35 Reactivity of bis(NHB) stannylene 20 with H2. | ||
Group 14 EE double bonds with boryl substituents display enhanced reactivity toward dihydrogen activation, largely attributed to their elevated HOMO energy levels coupled with narrow HOMO–LUMO gaps. Upon exposure to H2, 27 underwent a reaction to form the trihydrodisilane 64 (Scheme 36).62 DFT calculations indicate that the initial cleavage of the Si–B bond leads to the formation of the termed INT1N. The intermediate, INT1N, subsequently experienced a 1,2-hydrogen shift and was converted to the acyclic silylene, INT2N. The final stage involves the oxidative addition of H2 to silylene intermediate INT2N to give the final product 64. For comparison, disilene 65 could not activate H2 under similar conditions. However, disilene 66 bearing a boryl substituent could slowly react with dihydrogen to give trihydrodisilane 67 in a comparable fashion with 27 (Scheme 36). The investigations on the mechanism and the comparisons indicated the pivotal roles of amino and boron constituents on the SiSi double bond in the course of dihydrogen activation.
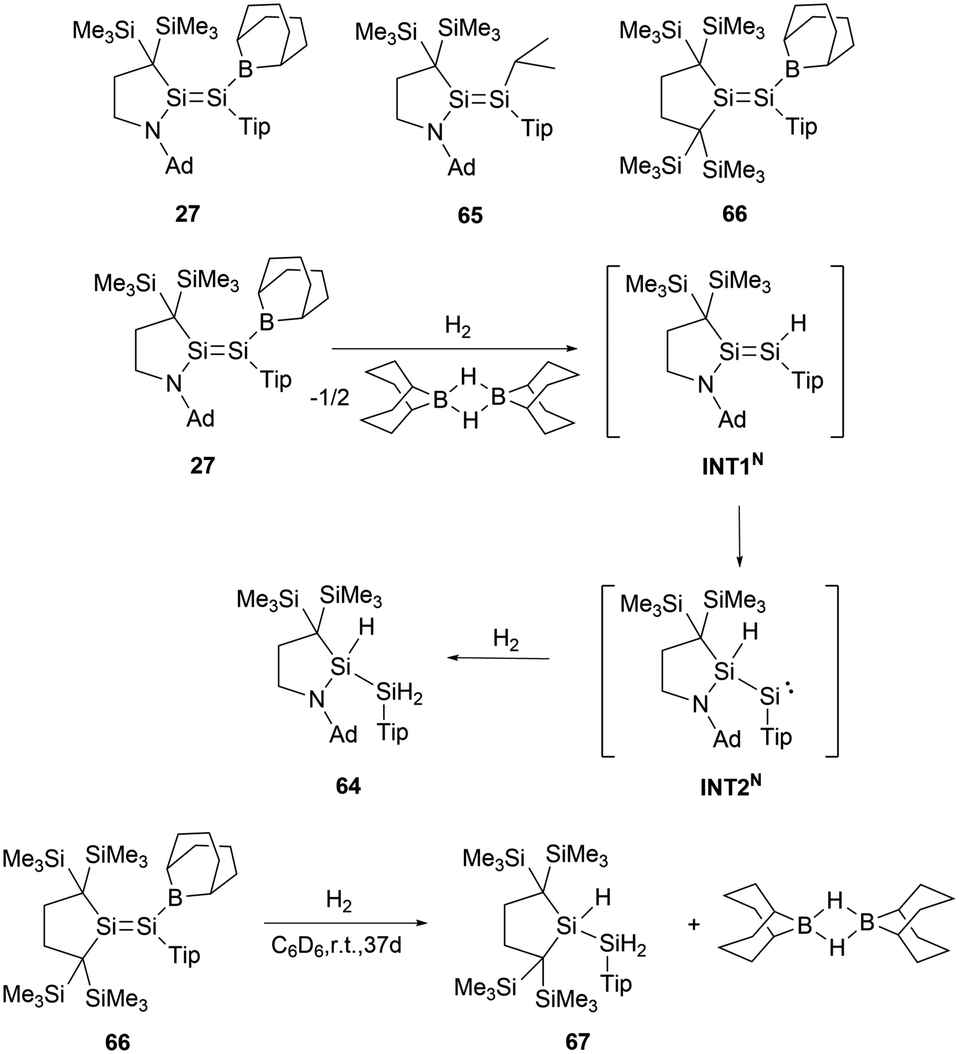 | ||
| Scheme 36 Activation of H2 by aminoboryldisilene 27 and monoboryldisilene 66. | ||
Disilyne 43 exhibits pronounced reactivity with H2, undergoing a reaction at ambient temperature to give NHB-stabilized 1,2-dihydrodisilene 68 (Scheme 37).51 In contrast to digermyne Ar′GeGeAr′ (Ar′ = 2,6-Dip2C6H3, Dip = 2,6-iPr2C6H3), which could react with dihydrogen to yield a mixture containing digermene and digermanes,63 neither prolonged reaction time nor the use of an excess of H2 induces further reactions with dihydrodisilene 68, suggesting the robust nature of the SSi double bond in 68. The Si–Si bond length of 2.1489(9) Å in 68 is lengthened relative to 43 but distinctly shorter than other reported 1,2-dihydrodisilenes [2.1708(6) Å].64 These structural data indicate that the NHB group has a significant electronic effect on the geometries of disilenes and disilynes. Mechanistically, the reaction might be driven by the LUMO of 43 interfacing with the H–H σ-bond. The reaction with H2 went through oxidative-addition at the silicon center through a SiH2 three-membered ring transitional state followed by intramolecular hydrogen shift to give the final dihydrodisilene product 68 (Scheme 37).
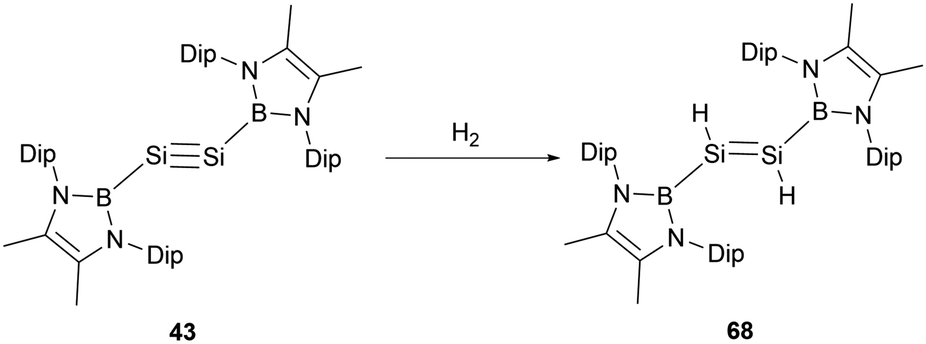 | ||
| Scheme 37 Activation of dihydrogen by disilyne 43. | ||
Digermavinylidene 50 stands out due to its highly unsaturated nature and a small HOMO–LUMO gap of 150 kJ mol−1.65 Such characteristics enable 50 to activate dihydrogen under milder conditions.29 The activation pathway involves a 1,2-diboryl migration, ultimately leading to the formation of the symmetrical diboryl tetrahydrogermane (NHB)H2GeGeH2(NHB) 69 (Scheme 38).
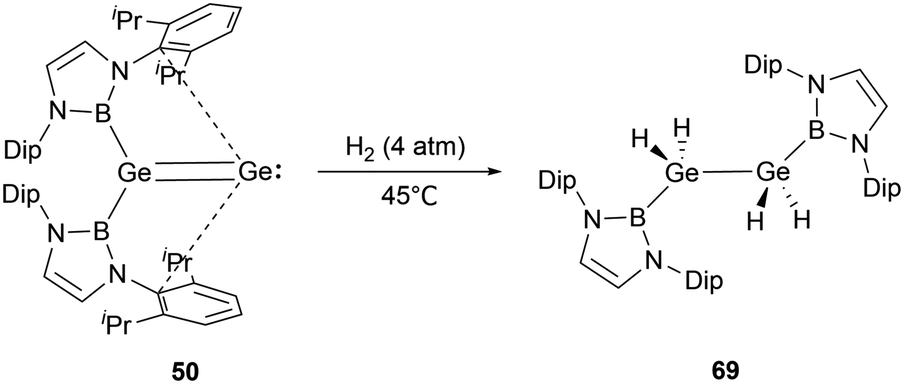 | ||
| Scheme 38 Activation of H2 by digermavinylidene 50. | ||
4.2 Ammonia
When [53·thf]+ is exposed to 1 atm. of NH3, a rapid reaction occurs, resulting in a complete conversion to a novel product in less than 5 minutes (Scheme 39).56 The NMR signals suggested the formation of the N–H activation product 70. Over 2 days, this initially formed N–H activation product underwent a full decomposition to give [(IPrMe)H]+ and H2N(NHB), likely owing to the reductive elimination of the B–N bond at the germanium center.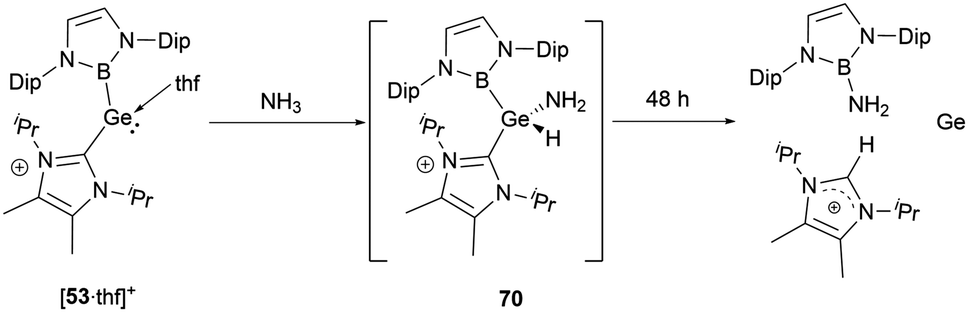 | ||
| Scheme 39 Activation of NH3 by the cationic complex [53·thf]+. | ||
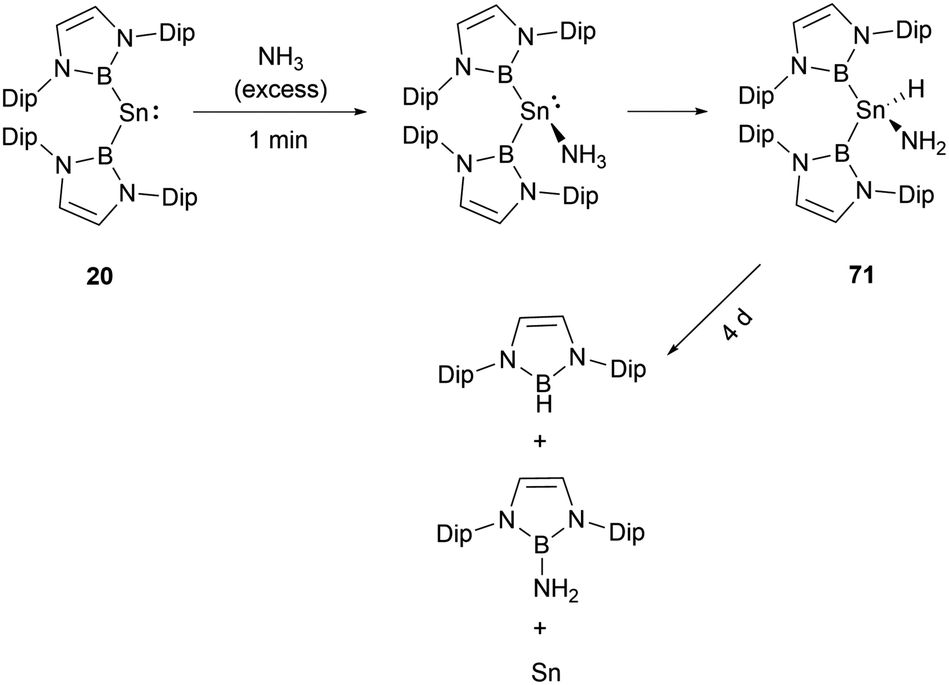
Stannylenes, upon treatment with NH3, showed distinctive and unique reaction pathways. When an excess of NH3 is introduced to a solution of 20 in benzene, stannylene 20 initially behaved as a Lewis acid to give the Lewis acid–base adduct 20·NH3 (Scheme 40).61 In the next stage, hydrogen shift to tin center led to the oxidative-addition process with the formation of the amido(IV) hydride (NHB)2SnH(NH2) 71. Over a duration of 4 days, it decomposed to a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (NHB)NH2 and (NHB)H. Similar to the reaction between [53·thf]+ and ammonia (Scheme 39), the π-donor amino substituent associates with NHB substituent to produce aminoborane (NHB)NH2. Similarly, the reductive elimination of BH at tin center led to the formation of hydroborane and metal tin (Scheme 40).
:1 mixture of (NHB)NH2 and (NHB)H. Similar to the reaction between [53·thf]+ and ammonia (Scheme 39), the π-donor amino substituent associates with NHB substituent to produce aminoborane (NHB)NH2. Similarly, the reductive elimination of BH at tin center led to the formation of hydroborane and metal tin (Scheme 40).
 | ||
| Scheme 40 Activation of NH3 by bis(NHB) stannylene 20. | ||
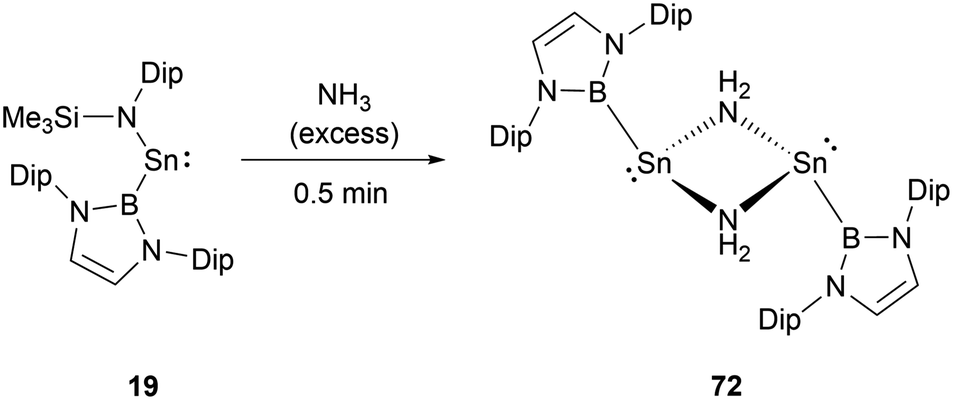
While stannylene 19 is inert to dihydrogen, its interaction with an excess of NH3 results in a different outcome.61 Initially, the reaction eliminates HN(SiMe3)2 with the formation of the NHB-substituted tin amide (NHB)SnNH2, which then underwent rapid dimerization to yield amino-bridged dimeric tin species [(NHB)Sn(μ-NH2)]272 (Scheme 41).
 | ||
| Scheme 41 Reactivity of amidostannylene 19 toward NH3. | ||
4.3 Carbon monoxide, carbon dioxide and nitrous oxide
The acyclic NHB-substituted amonosilylene 4, upon exposure to an excess of CO at room temperature for 48 hours, yielded a dimeric molecule 73 (Scheme 42).66 In this molecule, two tetravalent silicon centers form part of a nearly planar Si2C2O2 six-membered ring. Each silicon center is bonded to a carbon atom and two oxygen atoms derived from CO, as well as an NHB substituent. This structure can be envisioned as arising from the insertion of the C–C triple bond of the oxalate anion [OCCO]2− into the Si–B bond of 4. This action subsequently establishes molecular skeletons comprising both 4 and 6-membered rings via intra- and inter-molecular Si–O interactions. The 29Si NMR signals of 73, ranging from δSi 439.7 to −30.4 ppm, exhibit a marked high-field shift, consistent with the transition of oxidation states from SiII to SiIV. Such a reductive coupling of CO, yielding the oxalate dianion [OCCO]2−, is precedented in d- and f-block metal systems, including alkali metals.67 This reaction mode stands out as a novel approach in the reaction chemistry of silylenes.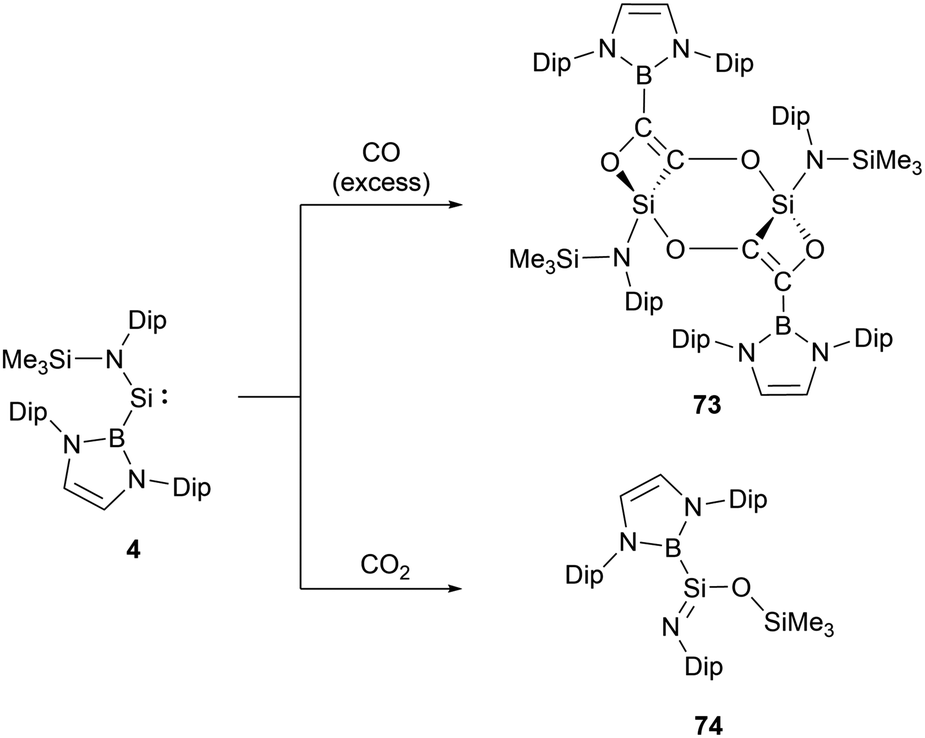 | ||
| Scheme 42 Activation of CO and CO2 by acyclic silylene 4. | ||
The analysis of low-valent group 14 species suggests that certain systems can extract one oxygen atom from CO2 to produce CO. If this system follows above mentioned mechanism, there might be potential pathways to transform CO2 into [C2O2]2−via CO production. When 4 reacted with an excess of CO2 at ambient temperature, the predominant product formed is iminosilane 74 (Scheme 42).66 The 29Si NMR shift of 74 at δSi −25 ppm suggests the oxidized silicon center. The X-ray single-crystal structural analysis of 74 disclosed the NHB-stabilized iminosilane structure (NHB)(Me3SiO)SiNDip with trimethylsiloxyl group. Iminosilane 74, possessing a planar triangular silicon center with the Si–B bond, was obviously formed by the abstraction of one oxygen atom from CO2 followed by the Me3Si group migration from nitrogen to oxygen atom. The iminosilane features a short Si–N bond length of 1.578(1) Å and a wide Si–N–C angle of 149.8(1)°, consistent with the previously reported structures with a three-coordinate silicon atom featuring a SiN double bond.68 Reaction of 4 with N2O yielded the same compound with the liberation of N2.66
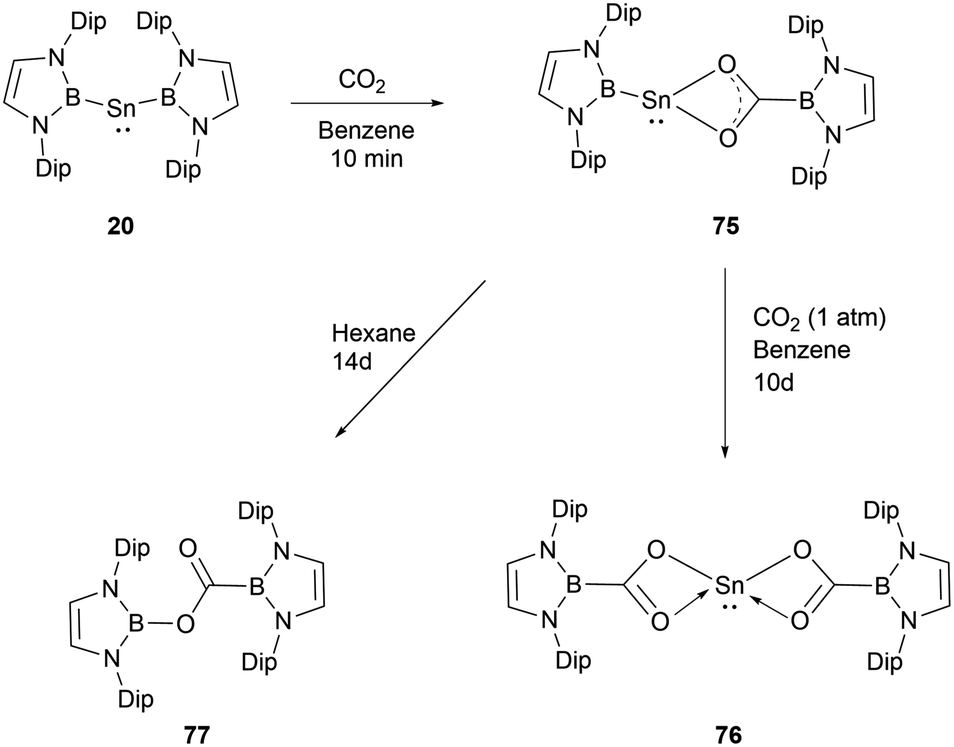
In contrast to the reaction mechanism proposed for the previously discussed silyene with CO2, the reaction of NHB-stabilized stannylene 20 with both CO2 and N2O go through the formal insertion of the oxygen atom into the B–Sn bond rather than the oxidative addition to Sn(II).69 Remarkably, the resulting products maintain the oxidation state of Sn(II). When stannylene 20 reacted with CO2, the mono-insertion product 75 formed rapidly, with the entire process taking roughly 10 minutes as monitored by 1H and 11B NMR spectroscopic methods. The asymmetric coordination in 75 could be seen from the two distinct NMR signals for the NHB segments. Compound 75 was not stable in n-hexane and was converted to borylcarboxylate ester 77, resulting from the reductive elimination of the B–O bond at the tin center in 75. Notably, compound 75 is stable in C6D6, and the similar reaction in n-hexane was not observed under similar conditions. Compound 75 remained stable for several days under CO2 atmosphere before transforming into the double insertion compound 76 (Scheme 43). The investigations involving quantum chemical studies of similar transformations, especially the CO2 activation by low-valent silicon species, indicate that the reaction can be modulated significantly by aromatic solvents through C–H/π interactions between the reactants and solvents.70
 | ||
| Scheme 43 Activation of CO2 by the bis(NHB) stannylene 20. | ||
At ambient conditions, bis(NHB) stannylene 20 exhibits a pronounced reactivity towards N2O.69 The initial asymmetric product formed under the conditions demonstrated its instability. When subjected 20 to an excess of N2O, compound 78 was obtained by the insertion of one oxygen atom into one of the B–Sn bonds (Scheme 44). 78 may slowly underwent further oxygen insertion to give Sn{OB(NDipCH)2}2.
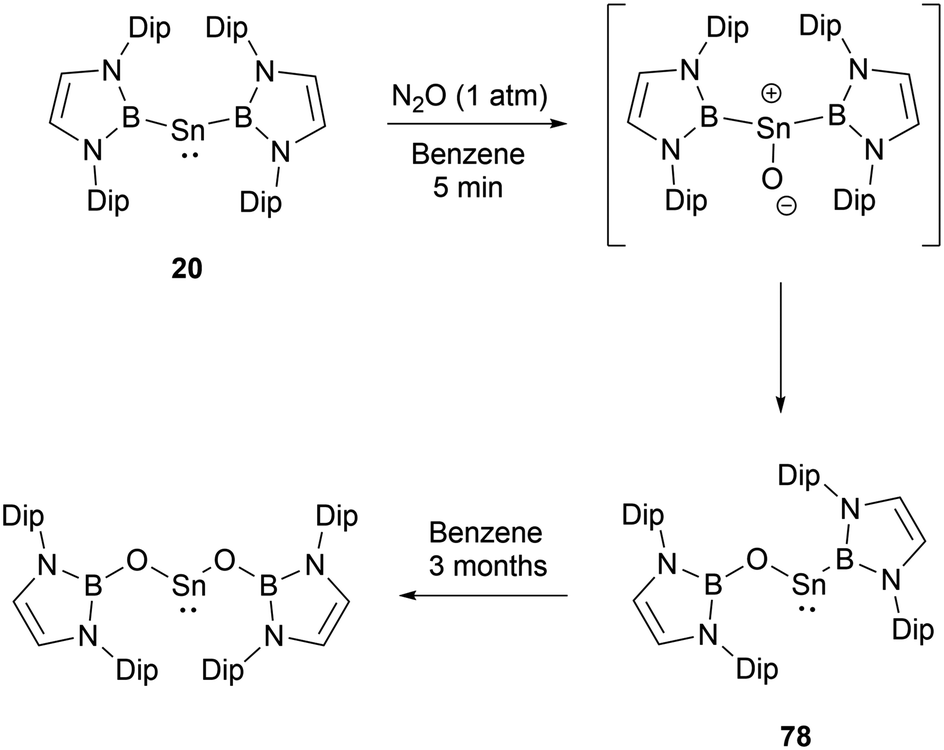 | ||
| Scheme 44 Activation of N2O by bis(NHB) stannylene 20. | ||
In 2022, Cui and co-workers reported on the reaction of an NHB-substituted disilyne with CO.71 Previous studies had suggested that the stability of transition metal carbonyl compounds is primarily attributed to the back-donation of d electrons into the π* orbitals of CO. Main group elements, due to the lack of d-valence electrons, are typically reluctant to generate stable carbonyl complexes under standard conditions. However, when a solid sample of disilyne 43 was exposed to CO at room temperature, it led to the formation of the unusual disilene dicarbonyl compound (NHB)(CO)Si–Si(CO)(NHB) 79 (Scheme 45). Compared to 43, the 29Si NMR spectrum of 79 exhibits a markedly shielded signal at δSi −200 ppm. The IR spectrum of 79 presents two stretch vibrations at 1895 and 1961 cm−1, which are significantly shifted compared to 2143 cm−1 for free CO.
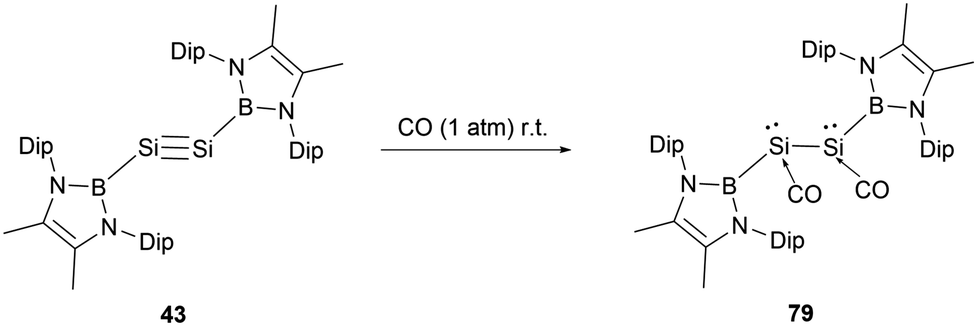 | ||
| Scheme 45 Synthesis of disilicon dicarbonyl compound 79. | ||
The X-ray single crystal analysis of 79 disclosed that the central B–Si–Si–B unit adopts a trans-bent configuration with a dihedral angle 169.2°. The Si–Si bond length of 2.3521(15) Å is consistent with a typical Si–Si single bond. The Si1–C1 and C1–O1 bond lengths of 1.799 and 1.134(4) Å indicate the relatively strong bonding between CO and Si atoms. The almost linear O–C–Si angle of 173.1(4)° combined with the pyramidal silicon atoms (total internal angles around Si summing up to 319.47°) indicated a carbonyl-stabilized bis(silylene) structure. The HOMO of 79 encompasses Si–C π orbitals, oxygen p-orbitals, and π-bonds of the NHB ring. In contrast, the LUMO is mainly anchored on the pair of SiCO groups. The pronounced σ-donor of the NHB group significantly alters the hybrid orbitals of the Si atom, reinforcing the back bonding from Si towards the CO π* orbital.
4.4 Activation of element-hydrogen bond
With a small singlet–triplet gap, 20 stands out in its reactivity towards E–H bonds, prominently B–H and Si–H bonds.61 Interacting with silanes and boranes, such as PhSiH3 and SiH4, or Me3N·BH3, it undergoes an E–H oxidative addition at the tin center, yielding the corresponding silylstannane 80 or borylstannane 81 (Scheme 46). This specific oxidative-addition reaction resulting in the formation of SnIV end products is novel. A noteworthy distinction exists between the final products 80 and 81 and the bis(NHB) stannylene 20. Silylstannane 80 possess an expanded B–Sn–B angle of 135.6 and 137.0(1)° compared to 118.8(3)° for 20. This expansion correlates with the Sn–B bond contraction during the tin oxidation state transition from SnII to SnIV, a necessary adjustment to diminish steric repulsion. In contrast, borylhydrostannane 81 features a smaller B–Sn–B central angle of 114.4(1)° coupled with an elongated Sn–B bond because of the steric demands of the H2B·NMe3 ligand.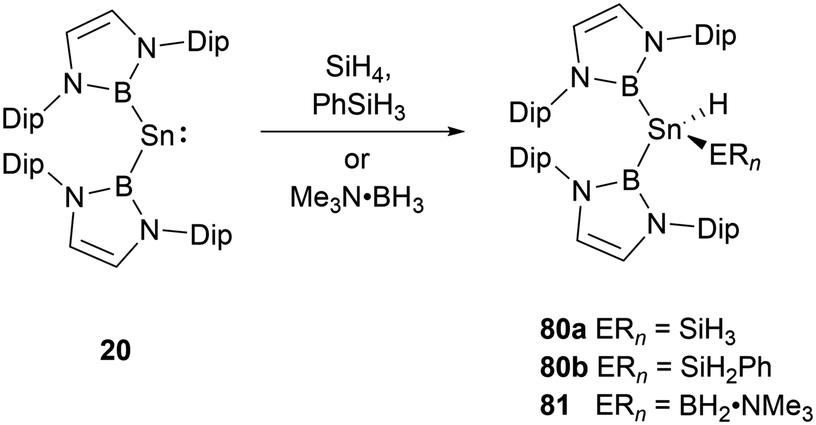 | ||
| Scheme 46 E–H bond activation by bis(NHB) stannylene 20. | ||
NHB-substituted germylene 12 reacted with SiH4 and Me3N·BH3 giving germanium hydrides 82 and 83 (Scheme 47) through oxidative-addition process.30
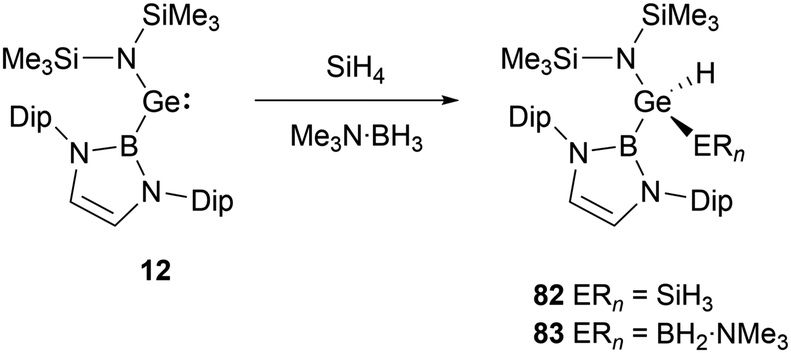 | ||
| Scheme 47 E–H bond activation by NHB amidogermylene 12. | ||
[53·thf]+ shows a similar propensity to react with silane derivatives.56 When Et3SiH or PhSiH3 was mixed with the same amounts of [53·thf]+, the resultant compounds have been identified as cationic silyl hydrides, namely 84a and 84b (Scheme 48). Further evidence for the formation of the silyl hydrides is provided by the X-ray single-crystal structural analysis. In an exemplary case with 84a, the shift in oxidation states from GeII to GeIV leads to a pronounced reduction in the Ge–B and Ge–C bond lengths of 2.042 and 2.000(3) Å compared to those (2.127 and 2.074(5) Å) observed for [53·thf]+. Simultaneously, the B–Ge–C angle undergoes an enlargement to 110.4(1)° compared to 100.9(2)° for [53·thf]+.
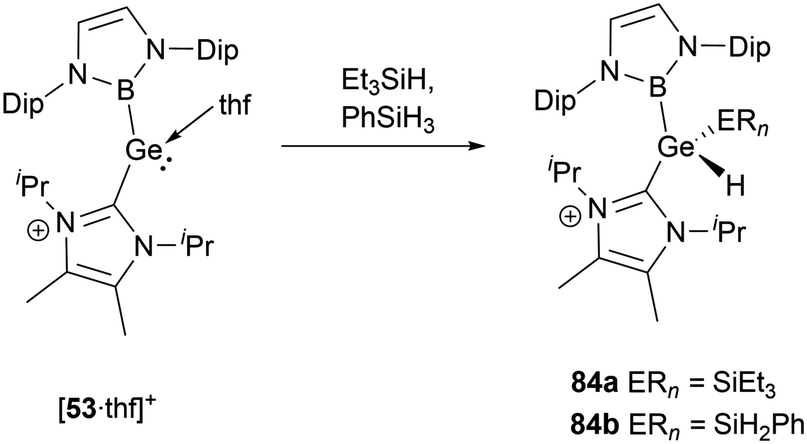 | ||
| Scheme 48 E–H bond activation by the cationic complex [53·thf]+. | ||
4.5 Unsaturated organic molecules
Group 14 carbene analogues and multiple bonding species could react with alkenes and alkynes to give cycloaddition products. NHB-substituted stannylenes 19 and 20, upon treatment with acetylenes, resulted in the formation of vinylstannylenes 85 and 86via the insertion of the acetylene into the B–Sn bonds.28 In contrast, stannylene 20 interacts with 2,3-dimethylbutadiene via a [4+1] cycloaddition, leading to the formation of stannacyclopentene 87 (Scheme 49).72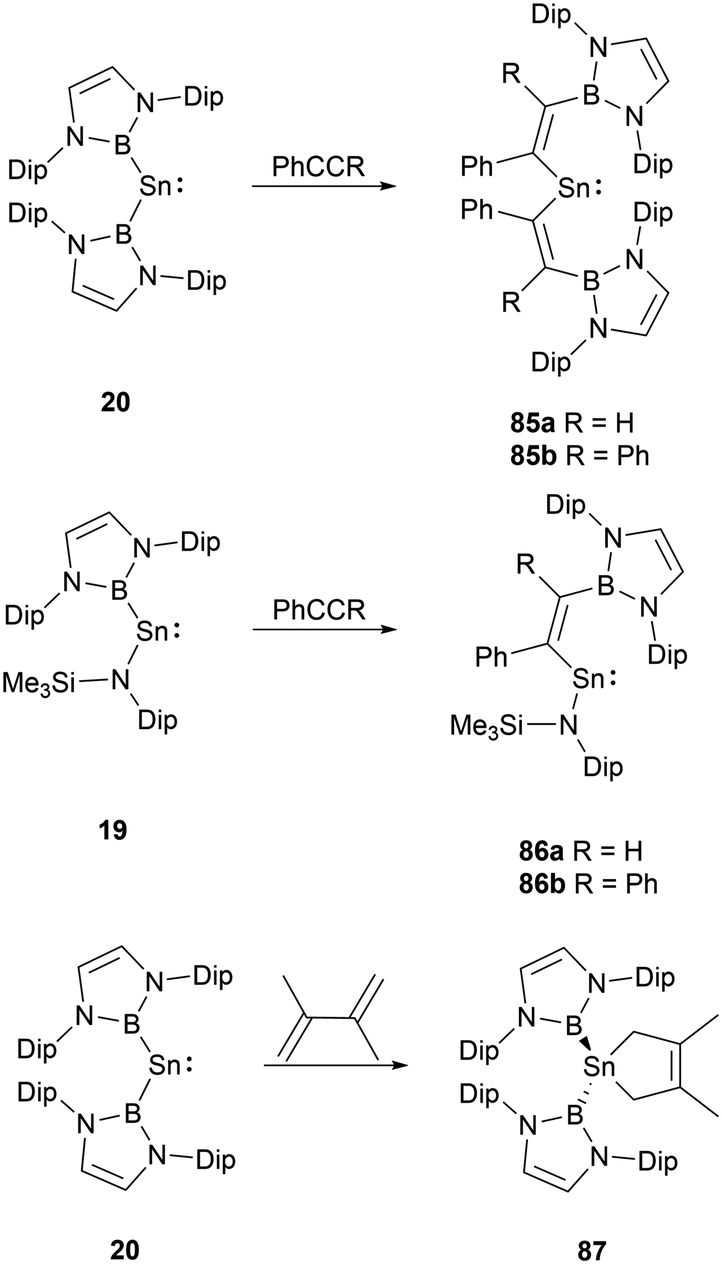 | ||
| Scheme 49 Reactivity of 19 and 20 toward carbon-based electrophiles. | ||
Insight into the structure of 86a, it's evident that the tin-acetylene segment aligns almost coplanar with the amino plane (C–Sn–N–Si) with a twist angle of 16.4(1)°. The twist angle corresponding to the ethylene substituent is markedly larger at 45.9(1)°, suggesting a minimal interaction between the CC double bond and the tin formal pz orbital. Conversely, there's a near coplanar alignment between the boryl and ethylene units, characterized by an N–B–C–C twist angle of 17.5(1)°. Observing the diboryl-substituted 85b, the boryl and ethylene units are less clearly aligned, but the ethylene substituent in 85b showcases improved π-conjugation with the C–C–Sn framework.
The NHB-stabilized silicon anion 7, owing to its distinctive electronic configuration, could be viewed as a masked lithium-substituted silylene. Upon reaction with AdNC and DipNC, silaketenimine anions 88a and 88b (Scheme 50) were obtained.73 These compounds feature a short SiC bond with a delocalized negative change over SiCN unit. The 29Si NMR spectra distinctly display singlets at δSi −144.0 and −163.4 ppm. X-ray single-crystal analysis validated their formation. Distinctly, 88a and 88b possess Si–C multiple bond lengths of 1.725(4) and 1.730(2) Å and C–N double distances of 1.285(4) and 1.279(3) Å. The SiCN angles of 160.6(3) and 157.4(18)°, respectively, deviate from the anticipated 180° for an allene unit. The shortened SiC and elongated CN double bond lengths underscore an allylic SiCN structure. Computational analyses suggested that the negative charge predominantly rest on the B–Si−CN allylic framework. When 88a was further treated with AdNC, it underwent a [2+2] cycloaddition reaction, leading to the formation of a highly delocalized system 89 with a central SiC2 three-membered ring.
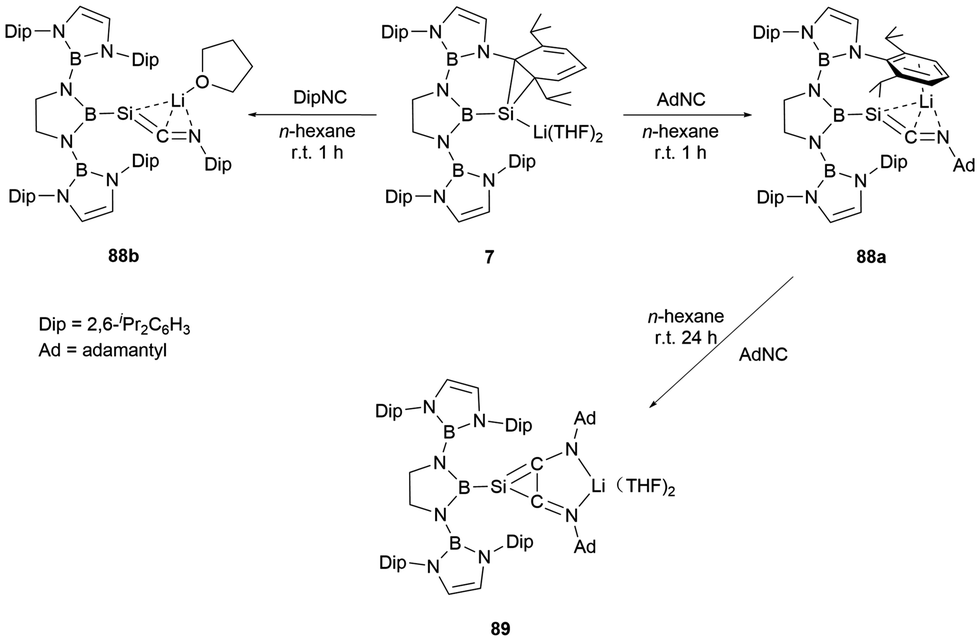 | ||
| Scheme 50 Reactions of Silanorcaradienyllithium 7 with isocyanides. | ||
5. Conclusions
In recent years, the reactivity patterns of low valent heavy main group 14 compounds towards activation of small molecules that may mimic those of low-coordinate transition metal complexes have attracted a great deal of attention for the development of sustainable chemical processes. From a kinetic standpoint, the activation process of these small molecules is intrinsically linked to the energy differential between the HOMO and LUMO orbitals. N-heterocyclic boryl substituents as strong σ-electron donating groups could effectively raise the HOMO energy level of the low valent compounds, which consequently narrows the energy disparity between the HOMO and LUMO, thus playing a pivotal role in tuning the energy gap. The distinctive electronic effects and bonding modes of low-valent heavy main group 14 compounds underscore their dependence on the substituents. Indeed, the impact of the substituents on the structural integrity and reactivity often exceeds our preliminary estimations and comprehension of the system as a whole. NHB ligands, characterized by their unique electronic attributes and considerable steric protections, are pivotal for the stabilization of highly reactive group 14 and, more broadly, main-group compounds. By comparison, the other boryl substituents presented in this review have quite different electronic effects on the resulting group 14 compounds due to the strong π-accepting properties of the boryl groups arising from the empty p orbitals on the boron atom. Presently, there are still a limited number of NHB and related strong σ-donating boron-based substituents. In addition, the synthetic pathways to the NHB substituted group 14 compounds still meet some challenges. It is anticipated that the emergence of a more diverse collection of boryl ligands with strong electron donating properties would significantly expand the chemical space of the low-valent group 14 chemistry.On the other hand, the applications of NHB-substituted group 14 anionic species, carbene analogues and multiple bonding species for the synthesis of novel group 14 compounds, especially functionalized molecular materials and selective catalysts, are still in their infancy. With the systematic investigations on the chemistry of NHB-substituted low-valent group 14 compounds, the innovations on group 14 bonding theories, reaction patterns and the related applications could be realized in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the National Key R&D Program of China (2021YFA1500200) and the National Natural Science Foundation of China (Grant no. 22131009 and 22221002) for the generous support.Notes and references
- (a) L. Dostál, Coord. Chem. Rev., 2017, 353, 142–158 CrossRef; (b) Z. Feng, S. Tang, Y. Su and X. Wang, Chem. Soc. Rev., 2022, 51, 5930–5973 RSC; (c) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (d) L. Wang, Y. Li, Z. Li and M. Kira, Coord. Chem. Rev., 2022, 457, 214413 CrossRef CAS; (e) Y. Wang and G. H. Robinson, J. Am. Chem. Soc., 2023, 145, 5592–5612 CrossRef CAS PubMed; (f) C. Weetman, Chem. – Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed; (g) C. Yan and R. Kinjo, J. Am. Chem. Soc., 2023, 145, 12967–12986 CrossRef CAS PubMed; (h) A. Agarwal and S. K. Bose, Chem. – Asian J., 2020, 15, 3784–3806 CrossRef CAS PubMed; (i) F. Dankert and C. Hering-Junghans, Chem. Commun., 2022, 58, 1242–1262 RSC; (j) M. Soleilhavoup and G. Bertrand, Chem, 2020, 6, 1275–1282 CrossRef CAS.
- (a) P. Chen, T. Xiong, Y. Pan and Y. Liang, Asian J. Org. Chem., 2022, 11, e202100738 CrossRef CAS; (b) P. Frisch and S. Inoue, Dalton Trans., 2020, 49, 6176–6182 RSC; (c) S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed; (d) C. Romain, S. Bellemin-Laponnaz and S. Dagorne, Coord. Chem. Rev., 2020, 422, 213411 CrossRef CAS; (e) M. M. D. Roy, A. A. Omana, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed; (f) X. Tan and H. Wang, Chem. Soc. Rev., 2022, 51, 2583–2600 RSC; (g) S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed; (h) R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 3488–3498 CrossRef CAS PubMed.
- For selected reviews on transition metal boryl complexes, see (a) B. Marciniec, C. Pietraszuk, P. Pawluc and H. Maciejewski, Chem. Rev., 2022, 122, 3996–40490 CrossRef CAS PubMed; (b) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed; (c) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS.
- For selected reviews on boryl complexes for catalysis, see (a) I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed; (b) I. A. I. Mkhalid, J. H. Narnard, T. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
- (a) M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377–1392 CrossRef CAS; (b) L. Weber, Eur. J. Inorg. Chem., 2017, 3461–3488 CrossRef CAS; (c) L. Weber, Coord. Chem. Rev., 2021, 431, 213667 CrossRef CAS; (d) L. Weber and L. Böhling, Coord. Chem. Rev., 2015, 284, 236–275 CrossRef CAS; (e) M. Yamashita and K. Nozaki, Pure Appl. Chem., 2008, 80, 1187–1194 CrossRef CAS.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (d) D. V. Partyka, Chem. Rev., 2011, 111, 1529–1595 CrossRef CAS PubMed; (e) H. G. Cheng, S. Chen, R. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2019, 58, 5832–5844 CrossRef CAS PubMed; (f) C. Liu and M. Szostak, Chem. – Eur. J., 2017, 23, 7157–7173 CrossRef CAS PubMed.
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed; (c) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC; (d) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (e) C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC; (f) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- (a) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (b) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- L. J. Romeo, A. Kaur, D. J. D. Wilson, C. D. Martin and J. L. Dutton, Inorg. Chem., 2019, 58, 16500–16509 CrossRef CAS PubMed.
- Y. Jing, J. Jiang, Y. Liu and Z. Ke, Organometallics, 2022, 41, 627–633 CrossRef CAS.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- (a) P. Zark, A. Schäfer, A. Mitra, D. Haase, W. Saak, R. West and T. Müller, J. Organomet. Chem., 2010, 695, 398–408 CrossRef CAS; (b) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (c) I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 16141–16144 CrossRef CAS PubMed; (d) T. Kosai, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2016, 55, 15554–15558 CrossRef CAS PubMed; (e) A. Rosas-Sanchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10549–10554 CrossRef CAS PubMed; (f) N. Weyer, M. Heinz, J. I. Schweizer, C. Bruhn, M. C. Holthausen and U. Siemeling, Angew. Chem., Int. Ed., 2021, 60, 2624–2628 CrossRef CAS PubMed.
- (a) S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC; (b) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed; (c) S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311–12316 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1999, 121, 9722–9723 CrossRef CAS.
- G. H. Lee, R. West and T. Muller, J. Am. Chem. Soc., 2003, 125, 8114–8115 CrossRef CAS PubMed.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 2002, 116, 2691–2692 CrossRef.
- L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed.
- R. West and M. Denk, Pure Appl. Chem., 1996, 68, 785–788 CrossRef CAS.
- (a) N. Metzler and M. Denk, Chem. Commun., 1996, 2657–2658 RSC; (b) Y. Suzuki, S. Ishida, S. Sato, H. Isobe and T. Iwamoto, Angew. Chem., Int. Ed., 2017, 56, 4593–4597 CrossRef CAS PubMed.
- T. Tanaka, M. Ichinohe and A. Sekiguchi, Chem. Lett., 2004, 33, 1420–1421 CrossRef CAS.
- (a) S. Inoue and C. Eisenhut, J. Am. Chem. Soc., 2013, 135, 18315–18318 CrossRef CAS PubMed; (b) D. Lutters, C. Severin, M. Schmidtmann and T. Muller, J. Am. Chem. Soc., 2016, 138, 6061–6067 CrossRef CAS PubMed.
- A. V. Protchenko, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, Organometallics, 2015, 34, 2126–2129 CrossRef CAS.
- A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed.
- (a) B. Rao, L. Wang and R. Kinjo, Angew. Chem., Int. Ed., 2019, 58, 231–235 CrossRef CAS PubMed; (b) B. Rao and R. Kinjo, Angew. Chem., Int. Ed., 2019, 58, 18150–18153 CrossRef CAS PubMed.
- (a) C. Prasang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900–921 RSC; (b) F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2021, 12, 2001–2015 RSC; (c) A. Rammo and D. Scheschkewitz, Chem. – Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed; (d) D. Scheschkewitz, Chem. Lett., 2011, 40, 2–11 CrossRef CAS.
- S. Inoue, M. Ichinohe and A. Sekiguchi, Chem. Lett., 2008, 37, 1044–1045 CrossRef CAS.
- K. Takeuchi, M. Ikoshi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2010, 132, 930–931 CrossRef CAS PubMed.
- M. Weidenbruch, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, UK, 2001, ch. 5, pp. 391–424 Search PubMed.
- K. Takeuchi, M. Ichinohe and A. Sekiguchi, Organometallics, 2011, 30, 2044–2050 CrossRef CAS.
- H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482–2506 CrossRef CAS PubMed.
- T. Kosai and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 18146–18149 CrossRef CAS PubMed.
- (a) J. Niesmann, U. Klingebiel, M. Schäfer and R. Boese, Organometallics, 1998, 17, 947–953 CrossRef CAS; (b) T. Iwamoto, N. Ohnishi, Z. Gui, S. Ishida, H. Isobe, S. Maeda, K. Ohno and M. Kira, New J. Chem., 2010, 34, 1637–1645 RSC.
- (a) T. A. Schmedake, M. Haaf, Y. Apeloig, T. Müller, S. Bukalov and R. West, J. Am. Chem. Soc., 1999, 121, 9479–9480 CrossRef CAS; (b) K. Takeuchi, M. Ikoshi, M. Ichinohe and A. Sekiguchi, J. Organomet. Chem., 2011, 696, 1156–1162 CrossRef CAS; (c) P. Willmes, L. Junk, V. Huch, C. B. Yildiz and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 10913–10917 CrossRef CAS PubMed.
- N. Nakata and A. Sekiguchi, J. Am. Chem. Soc., 2006, 128, 422–423 CrossRef CAS PubMed.
- T. Koike and T. Iwamoto, Chem. Commun., 2022, 58, 8061–8064 RSC.
- Z. Liu, J. Zhang, H. Yang and C. Cui, Organometallics, 2020, 39, 4164–4168 CrossRef CAS.
- T. A. Schmedake, M. Haaf, Y. Apeloig, T. Müller, S. Bukalov and R. West, J. Am. Chem. Soc., 1999, 121, 9479–9480 CrossRef CAS.
- M. Tian, J. Zhang, H. Yang and C. Cui, J. Am. Chem. Soc., 2020, 142, 4131–4135 CrossRef CAS PubMed.
- S. Ishida, R. Sugawara, Y. Misawa and T. Iwamoto, Angew. Chem., Int. Ed., 2013, 52, 12869–12873 CrossRef CAS PubMed.
- M. Tian, J. Zhang, L. Guo and C. Cui, Chem. Sci., 2021, 12, 14635–14640 RSC.
- Y. Heider, P. Willmes, V. Huch, M. Zimmer and D. Scheschkewitz, J. Am. Chem. Soc., 2019, 141, 19498–19504 CrossRef CAS PubMed.
- M. Karni and Y. Apeloig, J. Am. Chem. Soc., 1990, 112, 8589–8590 CrossRef CAS.
- (a) A. Fukazawa, Y. Li, S. Yamaguchi, H. Tsuji and K. Tamao, J. Am. Chem. Soc., 2007, 129, 14164–14165 CrossRef CAS PubMed; (b) M. Kobayashi, T. Matsuo, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2010, 132, 15162–15163 CrossRef CAS PubMed.
- Y. Ding, Y. Li, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2022, 61, e202205785 CrossRef CAS PubMed.
- (a) A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed; (b) Y. Murata, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2010, 132, 16768–16770 CrossRef CAS PubMed; (c) T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856–13857 CrossRef CAS PubMed; (d) S. Ishida, R. Sugawara, Y. Misawa and T. Iwamoto, Angew. Chem., Int. Ed., 2013, 52, 12869–12873 CrossRef CAS PubMed; (e) N. Wiberg, S. K. Vasisht, G. Fischer and P. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823–1828 CrossRef CAS.
- Y. Zhang, L. Wu and H. Wang, J. Am. Chem. Soc., 2022, 144, 22446–22450 CrossRef CAS PubMed.
- N. M. Kostic and R. F. Fenske, Organometallics, 1982, 1, 974–982 CrossRef CAS.
- X. Zheng, A. E. Crumpton, A. V. Protchenko, A. Heilmann, M. A. Ellwanger and S. Aldridge, Chem. – Eur. J., 2023, 29, e202203395 CrossRef CAS PubMed.
- R. J. Mangan, A. Rit, C. P. Sindlinger, R. Tirfoin, J. Campos, J. Hicks, K. E. Christensen, H. Niu and S. Aldridge, Chem. – Eur. J., 2020, 26, 306–315 CrossRef CAS PubMed.
- K. Inomata, T. Watanabe and H. Tobita, J. Am. Chem. Soc., 2014, 136, 14341–14344 CrossRef CAS PubMed.
- H. A. Bent, Chem. Rev., 1961, 61, 275–311 CrossRef CAS.
- X. Zheng, A. E. Crumpton, A. V. Protchenko, M. A. Ellwanger, A. Heilmann and S. Aldridge, Chem. – Eur. J., 2023, 29, e202300006 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- T. Kosai and T. Iwamoto, Chem. – Eur. J., 2018, 24, 7774–7780 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- T. Agou, Y. Sugiyama, T. Sasamori, H. Sakai, Y. Furukawa, N. Takagi, J. D. Guo, S. Nagase, D. Hashizume and N. Tokitoh, J. Am. Chem. Soc., 2012, 134, 4120–4123 CrossRef CAS PubMed.
- W. P. Leung, Y. C. Chan and C. W. So, Organometallics, 2015, 34, 2067–2085 CrossRef CAS.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. A. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
- (a) P. A. Bianconi, I. D. Williams, M. P. Engeler and S. J. Lippard, J. Am. Chem. Soc., 1986, 108, 311–313 CrossRef CAS; (b) A. S. Frey, F. G. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed; (c) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC; (d) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- (a) T. J. Hadlington, T. Szilvasi and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 14282–14286 CrossRef CAS PubMed; (b) R. S. Ghadwal, H. W. Roesky, K. Propper, B. Dittrich, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2011, 50, 5374–5378 CrossRef CAS PubMed; (c) L. Kong and C. Cui, Organometallics, 2010, 29, 5738–5740 CrossRef CAS; (d) P. P. Samuel, R. Azhakar, R. S. Ghadwal, S. S. Sen, H. W. Roesky, M. Granitzka, J. Matussek, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2012, 51, 11049–11054 CrossRef CAS PubMed.
- A. V. Protchenko, M. A. Fuentes, J. Hicks, C. McManus, R. Tirfoin and S. Aldridge, Dalton Trans., 2021, 50, 9059–9067 RSC.
- S. Jain and K. Vanka, Phys. Chem. Chem. Phys., 2019, 21, 14821–14831 RSC.
- Y. Ding, J. Zhang, Y. Li and C. Cui, J. Am. Chem. Soc., 2022, 144, 20566–20570 CrossRef CAS PubMed.
- A. V. Protchenko, D. Dange, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 10902–10905 CrossRef CAS PubMed.
- L. Zhu, J. Zhang, H. Yang and C. Cui, J. Am. Chem. Soc., 2019, 141, 19600–19604 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |