
Nucleic acid degradation as barrier to gene delivery: a guide to understand and overcome nuclease activity
Heyang
Zhang
ab,
Jo
Vandesompele
cd,
Kevin
Braeckmans
 ae,
Stefaan C.
De Smedt
ade and
Katrien
Remaut
*ad
ae,
Stefaan C.
De Smedt
ade and
Katrien
Remaut
*ad
aLaboratory for General Biochemistry and Physical Pharmacy, Department of Pharmaceutical Sciences, Ghent University, 9000 Ghent, Belgium. E-mail: Katrien.remaut@ugent.be
bLeiden Academic Centre for Drug Research, Leiden University, 2333 CC Leiden, The Netherlands
cDepartment of Biomolecular Medicine, Ghent University, 9000 Ghent, Belgium
dCancer Research Institute Ghent (CRIG), Ghent, Belgium
eCentre for Nano- and Biophotonics, Ghent University, 9000 Ghent, Belgium
First published on 11th December 2023
Abstract
Gene therapy is on its way to revolutionize the treatment of both inherited and acquired diseases, by transferring nucleic acids to correct a disease-causing gene in the target cells of patients. In the fight against infectious diseases, mRNA-based therapeutics have proven to be a viable strategy in the recent Covid-19 pandemic. Although a growing number of gene therapies have been approved, the success rate is limited when compared to the large number of preclinical and clinical trials that have been/are being performed. In this review, we highlight some of the hurdles which gene therapies encounter after administration into the human body, with a focus on nucleic acid degradation by nucleases that are extremely abundant in mammalian organs, biological fluids as well as in subcellular compartments. We overview the available strategies to reduce the biodegradation of gene therapeutics after administration, including chemical modifications of the nucleic acids, encapsulation into vectors and co-administration with nuclease inhibitors and discuss which strategies are applied for clinically approved nucleic acid therapeutics. In the final part, we discuss the currently available methods and techniques to qualify and quantify the integrity of nucleic acids, with their own strengths and limitations.
 Heyang Zhang | Heyang Zhang obtained her Doctoral degree in Pharmaceutical Sciences in 2020 at Ghent University, Belgium. Since then she has been a postdoctoral fellow at Leiden University, The Netherlands. In 2022, she became senior researcher at Leiden Academic Center for Drug Research (LACDR) of Leiden University. Her research focuses on nanomaterials for biotherapeutics delivery and RNA-based (e.g., mRNA, siRNA) treatment for autoimmune diseases. |
 Jo Vandesompele | Jo Vandesompele obtained a Master of Science in Bioscience Engineering (1997) and a PhD in Medical Genetics (2002) at Ghent University. Since 2007, he has been professor in Functional Cancer Genomics and Applied Bioinformatics at Ghent University. He is co-founder of the Cancer Research Institute Ghent (CRIG). His main research focus is RNA quantification and non-coding RNA and to exploit RNA for diagnostic and therapeutic purposes. |
 Kevin Braeckmans | Kevin Braeckmans first studied physics before doing his doctoral studies in pharmaceutical sciences at Ghent University in Belgium. From early on he became passionate about developing biophotonics technologies for drug delivery and diagnostics. In 2008 he was appointed professor at Ghent University as the group leader of the Bio-Photonics Research Group. In 2015 he received a prestigious ERC Consolidator Grant and became full professor in 2018. His research presently focuses on studying biological barriers to nanomedicines by advanced microscopy techniques, and combining light with nanoparticles to enable light-triggered drug delivery and related therapeutic applications. |
 Stefaan C. De Smedt | Stefaan C. De Smedt obtained a PhD in Pharmacy from Ghent University in 1995. In 1999, he became Professor in Physical Pharmacy and Biopharmacy at Ghent University where he founded the Ghent Research Group on Nanomedicines. He served as Dean of his faculty from 2010–2014. Currently he is a Specially Appointed Professor of Nanjing Forestry University. In 2015, he became Editor of JCR for the region Europe-Middle East & Africa and Editor in chief of JCR in 2023. His research is situated at the interface between drug delivery, materials sciences, physical chemistry and biophysics. |
 Katrien Remaut | Katrien Remaut graduated as a Pharmacist at Ghent University in 2001 and obtained a PhD in Pharmaceutical Sciences in 2007. In 2013, Katrien was elected as a member of the Young Academy in Flanders. In 2014, she was appointed tenure track professor at the Lab General Biochemistry and Physical Pharmacy of Ghent University. Since 2020, she has been Editor at the European Journal of Pharmaceutics and Biopharmaceutics. Her research focuses on nucleic acid delivery, with a special interest in ocular mRNA delivery and the use of advanced microscopy methods to follow the degradation of nucleic acids. |
1. Introduction
Many diseases find their origin in the absence of correct proteins, or the expression of malfunctioning proteins. The human genome encodes about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins, yet only a minor fraction of disease-related proteins can be pharmaceutically targeted with currently approved small molecule drugs.1 Protein-replacement therapy is often challenging due to high manufacturing costs, the instability and short half-life of administered proteins and the difficulties of reaching intracellular targets. As a result, only 2% of the protein-coding human genome is considered druggable with conventional strategies. In contrast, nucleic acid drugs can target virtually any disease-related gene when the genetic defect underlying such a disease has been found.
000 proteins, yet only a minor fraction of disease-related proteins can be pharmaceutically targeted with currently approved small molecule drugs.1 Protein-replacement therapy is often challenging due to high manufacturing costs, the instability and short half-life of administered proteins and the difficulties of reaching intracellular targets. As a result, only 2% of the protein-coding human genome is considered druggable with conventional strategies. In contrast, nucleic acid drugs can target virtually any disease-related gene when the genetic defect underlying such a disease has been found.
Nucleic acid drugs can be classified as DNA or RNA molecules that can regulate protein expression in many ways, such as replacing the disease-causing gene with a healthy copy (e.g., plasmid DNA, messenger RNA), inactivating/silencing the disease-causing gene (e.g., small interfering RNA, microRNA, antisense oligonucleotides) or knock-out/correct the disease-causing gene (e.g., genome editing).2 Natural occurring DNA is mostly double-stranded and is relatively stable as it is compacted in the nucleus of cells with positively charged histone proteins. Therapeutic DNA consists of long, double-stranded (ds) plasmid DNA (pDNA), where the 5′ and 3′ ends of linear DNA are joined together to form a circular molecule that mostly exists in the more compact supercoiled form (Fig. 1A).
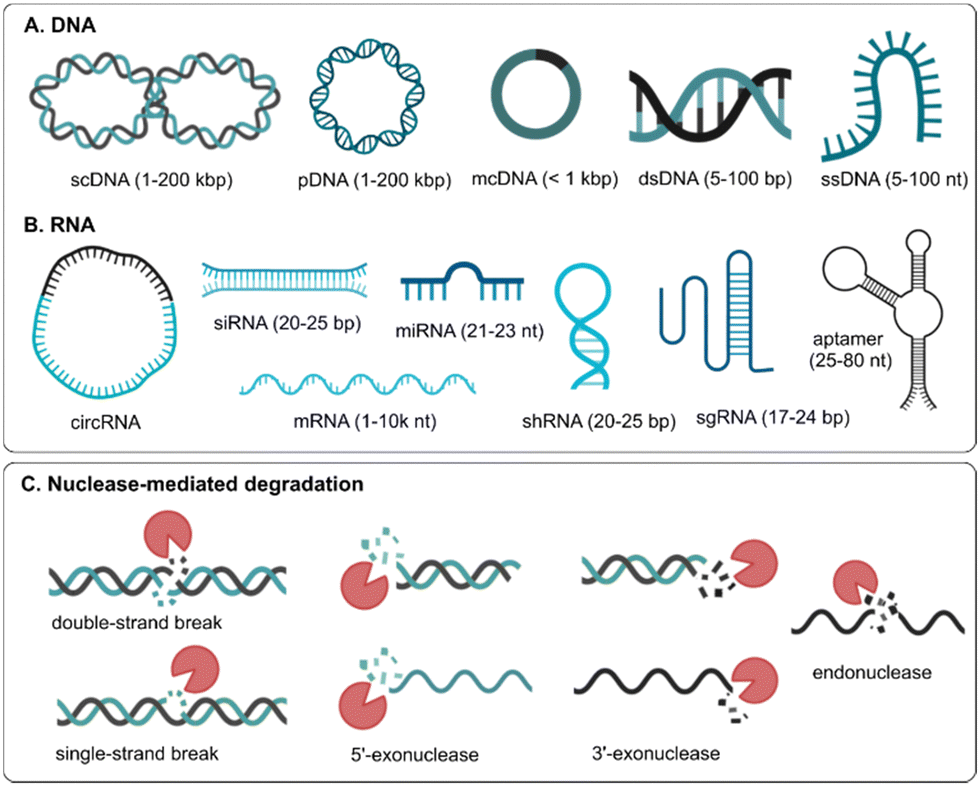 | ||
| Fig. 1 Schematic overview of different types of (A) DNA and (B) RNA based therapeutics. scDNA: supercoiled DNA; pDNA: plasmid DNA; mcDNA: mini circle DNA; dsDNA: double-stranded DNA; ssDNA: single-stranded DNA; circRNA: circular RNA; siRNA: small interfering RNA; miRNA: microRNA; mRNA: messenger RNA; shRNA: short hairpin RNA; sgRNA: small guide RNA. (C) Different types of nuclease-mediated degradation mechanisms of nucleic acids. | ||
Minicircle DNA (mcDNA) contains almost exclusively the gene of interest and its regulating sequence motifs, making it shorter than regular pDNA. pDNA and mcDNA need to reach the nucleus of the cells and employ the DNA transcription and translation apparatus to result in protein production. On the other hand, short, single-stranded (ss) DNA oligonucleotides or aptamers are used that should reach the cytosol or nucleus of cells to regulate disease-related protein expression. Unlike DNA, naturally occurring RNA is mostly single-stranded, of which the most known form is long, coding messenger RNA (mRNA) that is translated into protein or short, non-coding microRNAs (miRNAs) that can regulate protein expression by binding to complementary mRNA sequences. Short, double-stranded small interfering RNAs (siRNA) are extensively investigated to silence protein expression. When RNA is single-stranded and unstructured, it is more vulnerable to degradation, while the stability of dsRNA is increased. In some cases, RNA can also fold back on itself to form short stretches of double helix in regions of self-complementarity (i.e., stem-loop structure), such as short hairpin RNA (shRNA). Also, when the 3′ and 5′ ends of single-stranded linear RNA are joined together by covalent bonds, circular RNA (circRNA) is formed that is more resistant to degradation by RNA exonucleases3 (Fig. 1B).
As gene therapy is starting to take off for an increasing number of diseases with promising outcomes, nucleic acids are becoming the drug of the future.4 Of all extracellular and intracellular barriers that genetic drugs encounter (e.g., cellular uptake, endosomal escape, nuclear entry, degradation, off-target effect, immune stimulation and toxicity), the stability of genetic drugs is the least frequently studied and reported. Yet, just like any conventional drug, the half-life of genetic drugs after administration to the human body is of extreme importance. A variety of nucleases can result in single-stranded or double-stranded breaks in the nucleic acid sequence, which will abolish its activity (Fig. 1C). Where and when nucleases are encountered depends on the administration route, the identity of genetic drugs (DNA vs. RNA, single-stranded vs. double-stranded, short vs. long, unmodified vs. modified) and the formulation strategy (non-formulated vs. encapsulated in viral vs. non-viral vectors). In this review, we will overview the variety of nucleases in both the extracellular and intracellular environments that genetic drugs can encounter. Then, the most common strategies to prevent nucleic acid degradation are presented, together with the current state of clinically approved gene therapeutics and how nucleic acid identity and structure determines which options are most used to prevent degradation. To conclude, a number of techniques to quantify nucleic acid degradation will be discussed, with a special focus on their applicability to measure in situ degradation.
2. Nucleases as chemical barriers to gene delivery
With any route of administration, nucleic acids are susceptible to enzymatic degradation due to the presence of ubiquitous nucleases in the biological environment. Therefore, nuclease-mediated degradation is one of the major hurdles for efficient gene delivery, which is, however, still poorly understood. Generally, nucleases are enzymes capable of cleaving the phosphodiester bonds between the nucleotides of DNA and/or RNA. Exonucleases only work from the 5′ and/or 3′ end of the nucleic acids, while endonucleases can cut at different places in the polynucleotide strand, either through a single-strand or a double-strand break (Fig. 1C). In nature, nucleases play crucial roles in genetic quality control and host defense, but their action is (mostly) unwanted in gene therapy. Nucleases are diverse in their functions and selective for certain types of nucleic acid substrates. Numerous cellular endo- and exo-nucleases have been described, but often their function and subcellular localization are poorly documented.5 In the next paragraphs, nucleases (DNases, RNases) that are capable of cleaving nucleic acids will be reviewed.2.1 DNases
Deoxyribonucleases (DNases) are enzymes that hydrolyze the phosphodiester bonds in the ssDNA or dsDNA backbone. DNases are encoded by several genes and expressed in many tissues. They play a pivotal role in maintaining homeostasis and limiting inflammation by degrading both exogenous and endogenous DNA. DNase I and DNase II are the two main families of endonucleases, while three-prime repair exonucleases (TREX) 1 and TREX2 are sometimes referred as DNase III.6 A summary of the DNases with their most important characteristics can be found in Table 1.| Family | DNase I family | DNase II family | DNase III family | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | DNase I | DNase X | DNase1L2 | DNase γ | DNase IIα | DNase IIβ | L-DNase II | TREX1 | TREX2 |
| The preference of ssDNA and dsDNA was demonstrated as: +++, strong; ++, intermediate; +, weak; /, not detected or reported. |
|||||||||
| Coding gene | DNASE1 | DNASEIL1 | DNASEIL2 | DNASEIL3 | DNASE2 | DNASE2B | SERPINB1 | TREX1 | TREX2 |
| M w (kDa) | 44–60 | 60 | 37 | 32 | 32–40 | 40 | 27 | 33 | 26 |
| Optimal pH | 6.5–8.0 | 6.5–8.0 | 5.6 | 6.5–8.0 | 4.5–5.5 | 4.5–5.5 | 4.5–5.5 | 7.5–8.0 | 7.5–8.0 |
| Activators | Ca2+, Mg2+, Mn2+ | Ca2+, Mg2+, Mn2+ | Ca2+, Mg2+, Mn2+, CO2+, | Ca2+, Mg2+ | / | / | / | / | / |
| Inhibitors | Zn2+, Ni2+, EDTA, EGTA, G-actin | Zn2+, Ni2+, EDTA, EGTA | Zn2+, NaCl, EDTA, EGTA, ATA | Zn2+, Ni2+, EGTA, ATA, poly(ADP-ribosyl)ation | Zn2+, Na+, Cu2+ | Zn2+, Ni2+, ATA, Mg2+, Co2+ | / | / | / |
| Sequence-specific | ++ | / | / | + | Purine | / | / | 3′-overhang | 3′-overhang |
| ssDNA | + | / | / | +++ | ++ | ++ | ++ | +++ | / |
| dsDNA | +++ | / | / | +++ | +++ | +++ | +++ | / | +++ |
| Products | 5′-P/3′-OH | 5′-P/3′-OH | 5′-P/3′-OH | 5′-P/3′-OH | 5′-OH/3′-P | 5′-OH/3′-P | 5′-OH/3′-P | / | / |
| Extracellular site | GI tract, kidney, pancreas, salivary glands, stomach, thymus, bone marrow, small intestine, colon, duodenum | Lung, nasopharynx, bronchus, skeletal muscle, cardiomyocytes | Skin, salivary gland, epidermis | Lung, GI tract, liver, kidney, muscle, skin, bone marrow, spleen | All tissues, bone marrow, skin | Lung, lens, salivary gland | Bone marrow | Testis, lung, stomach, colon, placenta, skin, lymph node | Adrenal gland, skin, bone marrow, testis, lymph node |
| Subcellular site | Vesicle | Cytosol, cell surface, lysosome | ER, Golgi, mitochondria | Nucleus | Lysosomes | Lysosomes | Cytosol, nucleus | Cytosol, ER, nucleus | Nucleus, cytosol |
| Main organ and cell type | Pancreas | Heart/muscles | Skin, brain, lung, placenta | Spleen, liver | All tissues, skin | Eye lens, salivary glands | Spleen | All tissues | All tissues |
| Exocrine cells, Paneth cells | Myocytes | Keratinocyte | Macrophages, DCs | Macrophages | Fiber cell | Neutrophils | Melanocyte | Suprabasal keratinocytes | |
| Biological fluid | Blood, urine | / | / | Blood | / | / | / | / | / |
| Nuclease | Endo | Endo | Endo | Endo | Endo | Endo | Endo | Exo | Exo |
DNase I, an endonuclease encoded by the human gene DNASE1, is a secreted glycoprotein that is mainly produced by organs of the digestive system (e.g., pancreas, salivary glands) and accounts for 40–99% of the total endonuclease activity in most organs (e.g., kidney, parotid and submaxillary glands, stomach, pituitary gland, intestinal mucosa and pancreas) and biological fluids (e.g., blood, semen, saliva, breast milk, sweat and urine).9,10 DNase I represents the major nucleolytic activity in serum (Fig. 2B) and is responsible for the degradation of circulating DNA derived from apoptotic and necrotic cell death, thereby preventing immune stimulation. It plays a key role, together with DNase 1L3, in degrading neutrophil extracellular traps (NETs) to prevent the formation of blood clots during inflammation.11 DNase I preferentially introduces single-stranded breaks in protein-free dsDNA at phosphodiester linkages adjacent to a pyrimidine nucleotide (C,T). It can also degrade ssDNA and the DNA in RNA/DNA hybrids, but with a significantly reduced activity (500 to 50 times less, respectively) when compared to dsDNA.12 DNase I has some sequence preference as it is most effective on the B-form of DNA of mixed nucleotide composition, but less efficient at sites containing C or G at their 3′ ends, or extended A-T or G-C sites.13 Intracellularly, DNase I is inactivated by binding to the actin cytoskeleton (G-actin), which is thought to provide self-protection against degradation of genetic information.14
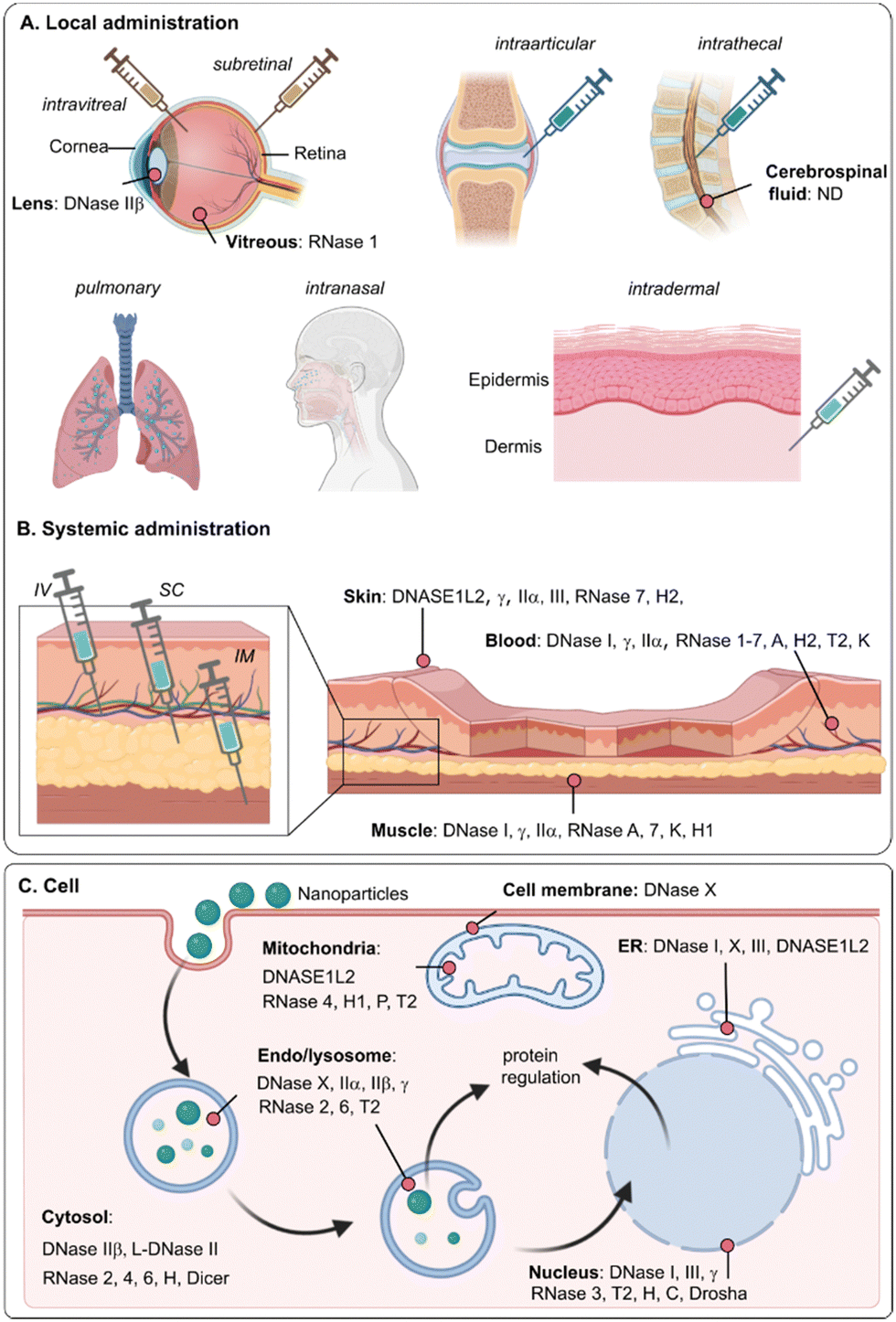 | ||
| Fig. 2 (A) Common administration routes for gene therapy and reported extracellular nucleases (B) and intracellular nucleases (C) that can be encountered in the human body. IV: intravenous injection; IM: intramuscular injection; SC: subcutaneous injection. | ||
DNASE1L1/DNase X is similar to DNase I, but has an extra conserved hydrophobic domain at its C-terminus, which is unique in the DNase I family. Together with an glycosylphosphatidylinositol (GPI) anchor, it plays a key role in locating DNase X to the cell surface, with its catalytic domain protruding in the extracellular space. DNase X has been found to greatly inhibit endocytosis-mediated gene transfer, serving as a major barrier to naked DNA transfer.15,16 As an ecto-enzyme, DNase1L1 is mainly expressed in muscular cells (e.g., heart, skeletal) and other human tissues (e.g., lung, placenta), and involved in the digestion of extracellular DNA in skeletal, muscle and different types of migrating cells. It was also detected in some tumor cell lines and shown to translocate to the nucleus during apoptosis. The intracellular distribution is associated with the endoplasmic reticulum (ER), sarcoplasmic reticulum, early endocytic vesicles, and Golgi apparatus (Fig. 2C).
DNASE1L2 is unique among the DNase family members as it exhibits maximal activity at acidic pH5.6 instead of in the neutral pH range. As such, it is the only acidic nuclease that is dependent on divalent cations (e.g., Ca2+, Mg2+) for its activity.8 The expression levels of DNAS1L2 are relatively low, but detectable in many human tissues (e.g., lung, placenta) and most strongly expressed in skin-derived cells (differentiated keratinocytes of the stratum corneum)17 (Fig. 2B). While Ca2+ and Mg2+ ions should both be present to activate DNASE1L2, Co2+ and Mn2+ ions can activate the enzyme even when present alone. The activity can be inhibited when increasing the concentration of NaCl but in contrast to DNase I, DNASE1L1 and DNASE1L3, no inhibitory effect of Ni2+ was found on DNASE1L2 activity.7,8 In the epidermis and skin surface, DNASE1L2 is thought to be involved in DNA degradation during keratinocyte differentiation and the prevention of the formation of bacterial biofilms, respectively. Intracellularly, DNASE1L2 is predominantly found in the ER and Golgi complex, although it is mainly considered as a secreted nuclease (Fig. 2C).
DNASE1L3/DNase γ is a Mg2+/Ca2+-dependent endonuclease that works both extracellularly and intracellularly. Just like DNase I, it is capable of cleaving ssDNA, dsDNA and DNA in chromatin, but in contrast to DNase I, DNASE1L3 has the ability to degrade protein-bound or liposome-coated DNA. As mentioned above, together with DNase I, DNASE1L3 is involved in degrading DNA from apoptotic and necrotic cells and NETs.18,19 Unlike DNase I, DNASE1L3 is resistant to G-actin inhibition. On the other hand, aurintricarboxylic acid (ATA), a general DNase inhibitor, strongly inhibits DNASE1L3 and DNASE1L2, but not DNase I and DNASE1L1. DNASE1L3 was also inhibited by poly(ADP-ribosyl)ation in vitro.20 DNASE1L3 is primarily expressed in lymphoid organs, such as lymph node, pituitary gland, adrenal gland, liver, spleen, and bone marrow, especially in myeloid cells such as macrophages of liver and spleen. The intracellular localization of DNASE1L3 is controversial, although it has been reported to be stored in the nuclear envelope in living cells and then translocated into the nucleus in dying cells to induce DNA fragmentation (Fig. 2C). Mutations in DNASE1L3 result in systemic lupus erythematosus, an autoimmune disease where mistakenly healthy tissue in the body is attacked by the immune system.
DNase IIα or shortly DNase II, is an intracellular and heavily glycosylated protein that is preferentially localized in lysosomes (Fig. 2C). Human DNase IIα consists of a single contiguous polypeptide with at least one intramolecular disulfide bridge maintaining protein structure. It contains six conserved cysteine residues and mutations of any of these cysteines completely ablates its enzymatic activity. N-glycosylation is critical for formation of the active enzyme and DNase IIα produced in the absence of glycosylation exhibits markedly reduced activity.26 DNase IIα is ubiquitously expressed in most human tissues (e.g., macrophages of most tissues and bone marrow) and biological fluids (e.g., blood, saliva, urine and testicular liquid). Due to its preferred location in lysosomes, DNase IIα plays a critical role in phagocyte-mediated apoptotic DNA degradation and the lysosomal clearance of DNA entering the cell via endocytosis.27–29 Together with DNASE1L2, DNase IIα was found as the major DNA-degrading enzyme in the stratum corneum and maintenance of homeostasis of skin cells30 (Fig. 2A).
DNase IIβ, also known as DNase II-like acid DNase (DLAD), is highly expressed in the eye lens and salivary glands, while lower expression levels are found in lung, trachea, prostate, and lymph nodes31 (Fig. 2A). Humans do not express DNase IIβ in the liver.27 DNase IIβ plays an important role in the clearance of DNA from the eye lens, in the course of terminal differentiation of lens fiber cells. Deficiency of this enzyme or the intravitreal injection of therapeutic antisense oligonucleotides might lead to DNA accumulation and cataract.32 DNase IIβ can mainly be found intracellularly, inside lysosomes.33 However, its function is still unclearly defined.
L-DNase II is strictly not part of the DNase II family, as it is encoded by the SERPINB1 gene, but like the other enzymes of the DNase II family, L-DNase II is active at acidic pH and can function in absence of cations. L-DNase II is derived from the leukocyte elastase inhibitor (LEI), which is present in the cytosol and has an anti-protease activity. When the molecular weight from LEI decreases, it loses its anti-protease activity, translocates to the nucleus and acquires an endonuclease activity. In this form, it is called L-DNase II, with a main role in the fragmentation of DNA of apoptotic cells. It is hypothesized that the transition from LEI to L-DNase II can also occur when the intracellular pH decreases.34,35 It is expressed in many cell types including keratinocytes. In contrast to DNase IIα, L-DNase II is dispensable for the acid DNases in murine stratum corneum.
TREX2 is an autonomous DNA 3′-5′ exonuclease that is responsible for removing the 3′-mismatched sequence of duplex DNA and functions in cell proliferation, genome integrity and skin homeostasis maintenance. In comparison with TREX1, TREX2 does not have the carboxyl-terminal hydrophobic domain that is responsible for intracellular localization, but contains a conserved DNA binding loop of three conserved arginine residues while the corresponding loop in TREX1 has a single conserved arginine residue with additional residues outside the loop region for binding.39,40 Although TREX1 and TREX2 share the similar substrate preference and optimal pH of 7.5–8.0, their binding affinity is distinct and cellular functions are not overlapped. TREX2 prefers to trim the single-stranded 3′-overhang of duplex DNA and specifically interacts with the non-scissile strand of dsDNA and chromosomal DNA.41 The endogenous TREX2 was reported to be widely expressed in human cell lines and mouse tissues including lung, liver, kidney, spleen, brain, testis and heart. Interestingly, TREX2 is one of the genes that is most highly enriched in psoriatic lesions compared with normal skin. Furthermore, TREX2 expression is restricted into the cytosol of keratinocytes, where it contributes to approximately half of the 3′-exonuclease activity of the epidermis, but also accumulates in their nucleus.42 For the subcellular localization, TREX2 was found in both the nucleus and cytosol of human cells.43,44
2.2 RNases
Ribonucleases (RNases) are very important enzymes for RNA metabolism in almost all organisms. They can hydrolyze ssRNA, dsRNA, and RNA/DNA duplexes. In physiological conditions, most extracellular RNases are protective against infections by degrading extracellular RNAs, while intracellular RNases have as main function to be cytotoxic against malignant cells, by degrading intracellular RNA.45 RNases are extremely common and degrade any unprotected RNA. A summary of mammalian RNases relevant towards gene therapy, and their most important characteristics can be found in Table 2.| Name | M w (kDa) | Activator | Inhibitor | pH | Sequence-specific | ssRNA | dsRNA | Extracellular sites | Subcellular sites | Biological fluid | Main cell type | Nuclease | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The preference of ssRNA and dsRNA was demonstrated as: +++, strong; ++, intermediate; +, weak; /, not detected or reported. |
|||||||||||||
| RNase A (Bovine pancreatic RNase 1) | 14 | Na+, sulfate | Uridine vanadate complexes, heavy metal ions, RI | 6–10 | Poly(C) > poly(U) > poly(A) | +++ | / | Lung, spleen, muscle, gut, pancreas, heart | / | Extracellular space, blood | / | Endo | |
| RNase A super family | RNase 1 (Pancreatic RNase) | 18 | / | RI | 7.3–8.0 | Poly(C) > poly(U) > poly(A) | ++ | +++ | Pancreas, testis, placenta, lung, adipose tissue, stomach, eye | Exosomes | Amniotic fluid, blood, serum, synovial fluid, CSF, urine, vitreous, retina, extracellular space | Hofbauer cells, alveolar cells type 1/2, macrophage, adipocytes | Endo |
| RNase 2 (EDN) | 29 | / | / | 6.5–7.0 | Poly(U) > poly(C) > poly(A) | +++ | / | Spleen, bone marrow, lung | Lysosome, cytosolic granules | Amniotic fluid, blood, CSF, urine, gut lavage fluid, extracellular space | Eosinophils, Granulocytes, Kupffer cells, DCs | Endo | |
| RNase 3 (ECP) | 16–22 | / | / | 7.0 | Poly(U) > poly(C) > poly(A) | +++ | / | Bone marrow | Exosomes, Azurophil granule lumen | Anniotic fluid, blood, CSF, sputum, tear, plasma, gut lavage fluid, serum, nasal fluid, bronchoalveolar lavage fluid, extracellular space | Neutrophils, eosinophils, monocytes, T cell | Endo | |
| RNase 4 | 18 | / | / | 7.0–8.0 | Poly(U) > poly(C) | +++ | / | Liver, spleen, pancreas, endocrine tissue, GI tract, kidney, lung, salivary gland, colon, bone marrow | Cytosolic granules, mitochondria, exosomes | Blood, CSF, extracellular space | Monocytes, B cell, T cell | Endo | |
| RNase 5 (ANG) | 14 | / | / | 7.0 | Low activity tRNA specific? | / | / | Liver, spinal cord neuros | Exosomes, extracellular space, nucleus, cytosolic vesicle, RNA stress granules | Amniotic fluid, blood, plasma, bronchoalveolar lavage fluid, serum, CSF | Hepatocytes, monocyte, myeloid DC, endothelial cells, T cells, mast cells | Endo | |
| RNase 6 (RNase K6) | 16 | / | / | 6.0–8.0 | Poly(U) > poly(C) | / | / | Lung, heart, placenta, kidney, spleen, lymph node, bone marrow, tonsil | Extracellular space, exosomes, cytosolic vesicle | Amniotic fluid, blood | Kuffer cells, macrophages, Hofbauer cells, B cells, DCs | Endo | |
| RNase 7 | 14 | / | NaCl, CaCl2 | 7.4 | / | +++ | / | Skin, liver, kidney, skeletal muscle | Exosomes, extracellular space, cytosol | Amniotic fluid, blood, skin washing fluid | Basal cells, keratinocytes, urothelial cells, cholangiocytes | Endo | |
| RNase 8 | 18–19 | / | / | / | / | +++ | / | Placenta | Extracellular space | Digestive fluid | / | Endo | |
| RNase C | Dicer | 82 | Mg2+, Ni2+, CO2+, Mn2+ | RI | / | / | / | +++ | Brain, lung, GI tract, kidney, bone marrow, epididymis, | Nucleus, P-bodies, cytosol | / | Kupffer cell, endothelial cells | Endo |
| Drosha | 160 | All tissues, lung, eye | Nucleoplasm, cytosol | / | Cardiomyocytes, ciliated cells | Endo | |||||||
| RNase H | H1 | 32 | Mg2+, Mn2+ | Sulfhydryl reagents | 7.0–8.0 | Non-labeled site | RNA or RNA/DNA hybrid | Lymph node, brain, kidney, muscle, testis | Mitochondria, nucleus | / | Monocytes, exocrine glandular cells | Exo- and endo | |
| H2 | 33 | Mg2+ | Mn2+, sulfhydryl reagents | 7.0–8.0 | junction at the 3′ end of the four ribonucleotides | RNA or RNA/DNA hybrid | Lung, stomach, skin, lymph node, GI tract, liver, brain, bone marrow, pancreas, kidney | Nucleus | Blood | / | |||
| RNase T2 | 30 | EDTA | Mg2+, Ca2+, Hg2+, Cu2+, Zn2+, heparin, EDTA, mononucleotides | 4.0–5.0 | Poly(A) | / | ++ | Temporal lobe, fetal brain, endometrium, pancreas, bone marrow | Lysosomes, mitochondria, vacuoles, ER | Blood | Eosinophil, neutrophil | Endo | |
| RNase K | 15 | / | / | / | ApU>ApG>UpU | / | / | Brain, endocrine tissue, GI tract, pancreas, kidney, muscle, stomach, small intestine | Cytosol, membrane | Blood | / | Endo | |
| RNase L | 83 | 2-5A | VAL, EA | / | UU > UA ≫ UG > UC | +++ | / | All cells | Cytosol | Blood | All cells | Endo | |
The family members that are structurally characterized (e.g. RNase 1–5; 7) show similarity to the bovine pancreatic RNase A, one of the most studied proteins in literature.51 RNase A is an endonuclease that cleaves specifically on the 3′-side of pyrimidine (C, U) bases, with poly(C) cleaved approximately 20-fold faster than poly(U) and 2000-fold faster than poly(A).52,53 RNase A is one of the most stable proteins in our body, that is difficult to inactivate. It still works well at 90 °C, for example, while other enzymes are already denatured.54 Some reducing agents (like dithiothreitol, dithioerythritol, glutathione and the amino acid cysteine) can be used in combination with denaturants to reduce the disulfide bonds in RNase A and inhibit the protein. Evolutionary, RNase 1, 4 and 5 and RNase 2/3 and 7/8 can be grouped together, with RNase I having the highest catalytic activity of the RNase A superfamily.
RNase 1, or human pancreatic RNase, has been isolated from the pancreas with highest expression, but is present in many tissues (e.g., kidney, brain, testis, ovary, mammary gland) and fluids (e.g., serum, urine, saliva, milk, and seminal plasma). It allows degradation of both ssRNA and dsRNA as well as DNA/RNA hybrids. Like RNase A, it has a substrate preference towards poly(C) over poly(U), while no activity towards poly(A), but differs from the bovine pancreatic RNase A by its high activity towards dsRNA, and significant lower activity towards ssRNA. RNase 1 is hypothesized to contribute to the normalization of serum viscosity and non-specific response to pathogenic RNA molecules as human defense.55,56 Intracellularly, RNase 1 can be efficiently bound by the human RI via formation of a tight RNase-inhibitor complex, thereby blocking the catalytic activity.57,58
RNase 2 (eosinophil-derived neurotoxin (EDN)) and RNase 3 (eosinophil cationic protein (ECP)) have 67% amino acid identity similarity. Both are present in cytosolic granules in eosinophils, specialized cells of the circulating innate immune system, from which they are secreted upon stimulation or activation. RNase 2 and 3 have preference of poly(U) over poly(C) as substrate (in contrast to RNase 1) and are completely inactive on poly(A) and dsRNA.59 RNase 2 is also expressed in other blood cell types, such as monocytes and dendritic cells. Its expression can be found in spleen, liver, lung, kidney, placenta leukocytes and body fluids. RNase 2 has antiviral properties against ssRNA viruses like HIV and RSV and functions as an endogenous ligand for the pathogen recognition receptor, TLR2. Also RNase 3 has antiviral properties and acts against bacteria, independent from its ribonucleolytic activity.60,61 The concentration of RNase 3 in normal blood was reported to be 3 ng mL−1, and 7 ng mL−1 in serum.62
RNase 4 has the shortest amino acids sequence (119 residues) and shares 43% and 31% structural identity with RNase 1 and RNase 2 respectively. It strongly prefers poly(U) over poly(C), with maximum RNase activity (more than 95%) between pH 7.0 and 8.0, and minimum RNase activity at pH 5.0.63 Human RNase 4 was found in some somatic tissues, such as liver (highly expressed), pancreas, kidney, lung, heart, and placenta but not brain.64,65 As reported, cytosolic granules of monocytes also express this RNase, although the cell lines that selectively express RNase 4 are still unclear.
RNase 5 or angiogenin (AGN) is similar to RNase 1 but with much lower catalytic activity. It is the only canonical RNase that has six cysteines (forming three disulfide bonds) instead of eight (forming four disulfide bonds) in the catalytic active site. As the name suggests, it has angiogenic properties and requires it endonuclease activity to induce blood vessel growth, but has hardly any RNase activity against standard RNA substrates.66 RNase 5 was found to have a wide tissue and organ distribution and was identified in normal human plasma with concentrations of 0.11–0.38 mg L−1.67 Some studies demonstrated an elevated RNase 5 expression in tumor cell lines, involving in cell proliferation and malignant cancer development.
RNase 6 exhibits an overall moderate relative catalytic efficiency, higher than that of RNase 3 but significantly lower than that of RNase 7.68 Unfortunately, the information concerning substrate preference, pH optimum and other catalytic properties is rather limited. The expression of RNase 6 was found in many tissues, including high expression in lung (with highest expression level), spleen, thymus as well as low expression in kidney, liver, brain, heart, pancreas, skeletal muscles, monocytes and neutrophils.69 As well, it was found in lysosomes, cytosolic granule and extracellular regions.
RNase 7 or skin-derived RNase, is secreted by keratinocytes and contributes to wound healing and tissue repair and is the most abundant RNase in human skin.70 It is also present in epithelial tissues and organs such as kidney, urinary tract, respiratory tract, genitourinary tract, liver, skeletal muscle and heart, contributing to urinary tract sterility and epidermis protection. Purified RNase 7 was found to have 50 times higher catalytic activity than RNase 3, while recombinant RNase 7 exhibited 8-fold lower ribonucleolytic activity than RNase 1 but twice that of RNase 8.71 It shares 78% amino acid similarity with RNase 8 which is expressed uniquely in the placenta, but has a very low RNase activity.72,73 The function of RNase8 is poorly understood, but it is hypothesized to play a role in placental host defense.
Dicer and Drosha are two specific members of the RNase III family that are worth to mention due to their involvement in the RNA interference pathway. Dicer is a human endonuclease that cleaves dsRNA or hairpin dsRNA regions of ssRNA (e.g., pre-miRNA) into short ds or ss 20–25 bp fragments (e.g., siRNA or miRNA respectively). Dicer facilitates the loading of the guide strand in RNA-induced silencing complexes (RISC) that together with the Argonaute nuclease are capable of degrading complementary mRNA.79 Dicer-derived siRNAs contain both a passenger and guide strand, where the passenger strand is degraded, while the guide RNA-loaded RISC can degrade many copies of the target mRNA. Exogenous administered siRNA executes its silencing effect through RISC, without the need to be pre-processed by Dicer. Naturally, Dicer plays a role in processing several types of non-coding RNAs. For example, small nucleolar RNAs can serve as a source of short regulatory RNA species generated by Dicer, which may be involved in the control of processing and translation of various mRNAs. In addition to processing endogenous RNAs, Dicer has been reported to process exogenous RNAs, such as viral-associated RNAs (e.g., adenovirus RNA and HIV trans-activation response element RNA).80 Dicer localizes to the endoplasmic reticulum.81 Drosha is a dsRNA-specific endoribonuclease that plays a key role in the initial miRNA maturation in the nucleus.82 Drosha is localized in the cell nucleus and cytosol and cleaves long RNA primary transcripts (pri-miRNAs) into 70 bp long hairpin-shaped pre-miRNAs that are subsequently cut by the cytosolic Dicer to generate the short 20–25 ss mature miRNAs that are further processed by the RNA interference pathway as mentioned above.83
| Name | RNA/DNA | Chemical modifications | Vector | Target (indication) mechanism | Administration | First approval | Ref. |
|---|---|---|---|---|---|---|---|
| DNA therapeutics | |||||||
| Recombinant human p53 adenovirus (Gendicine) | rAd-p53 (2.85 kb) | / | ADV5 (viral) | P53 expression (Head and neck cancer) | Intratumor injection | CFDA, 2003 | 454 and 455 |
| Cambiogenplasmid (Neovasculgen) | scDNA encoding VEGF165 with CMV promotor (4.86 kb) | / | / | VEGF165 expression Peripheral arterial disease | I.M. | Russia, 2011 | 456 |
| Alipogene tiparvovec (Glybera) | Human LPL gene variant LPLS447X (3.6 kb) | / | AAV1 (viral) | LPL expression Familial LPLD | I.M. | EMA, 2012 (withdrawn, 2017) | 457 |
| Voretigene neparvovec (Luxturna) | AAV2-hRPE65v2 (3.9 kb) | • A modified Kozak sequence | rAAV2 (viral) | RPE65 expression (LCA) | Subretinal injection | US FDA, 2017 | 458 |
| Onasemnogene abeparvovec (Zolgensma) | SMN1 gene (n.d.) | / | rAAV9 (viral) | SMN1 expression (SMA) | I.V. | US FDA, 2019 | 459 |
| ZyCoV-D | pDNA encoding spike protein of SARS-COV-2 (3.8 kb) | • Unmethylated CpG | / | Spike protein expression (SARS-COV-2 virus infection) | I.M. (Jet injector) | India, 2021 | 285 |
| DNA ASO therapeutics | |||||||
| Fomivirsen (Vitravene) | 21 mer ASO (ssDNA) | • PS backbone | / | CMV IE-2 mRNA (CMV retinitis) | Intravitreal injection | US FDA, 1998 (withdrawn, 2002) | 294 and 460 |
| • 5′ CpG motif | |||||||
| Defibrotide (Defitelio) | ssDNA/dsDNA (9–80 mer, av. 50 mer) | • PO backbone | / | Non-specific (sVOD) | I.V. | EMA, 2013 | 461 |
| Mipomersen (Kynamro) | 20 mer gapmer (DNA/RNA) | • PS backbone | / | ApoB mRNA (HoFH) | S.C. | US FDA, 2013 (withdawn, 2022) | 297 |
| • 2′-MOE 5-mer regions | |||||||
| Eteplirsen (Exondys 51) | 30 mer SSO (ssDNA) | • PMO | / | Dystrophin premRNA (DMD) | I.V. | US FDA, 2016 | 307 |
| Inotersen (Tegsedi) | 20 mer gapmer (ssDNA) | • Five 2′-MOE nucleotides at 5′ and 3′-ends | / | TTR mRNA (hATTR) | S.C. | EMA, US FDA, 2018 | 462 |
| • m5C | |||||||
| Golodirsen (Vyondys 53) | 25 mer SSO (ssDNA) | • PMO | / | Dystrophin premRNA (DMD) | I.V. | US FDA, 2019 | 308 |
| Casimersen (Amondys 45) | 22 mer SSO (ssDNA) | • PMO | / | Dystrophin premRNA (DMD) | I.V. | US FDA, 2021 | 309 |
| RNA ASO therapeutics | |||||||
| Pegaptanib (Macugen) | 27 mer aptamer (dsRNA) | • PS 3′-3′ deoxythymidine cap | / | VEGF-165 (retinal AMD) | Intravitreal injection | US FDA, 2004 (withdrawn, 2019) | 463 |
| • 2′-OMe purine ribose sugars | |||||||
| • 2′-F pyrimidine ribose sugars | |||||||
| • Two 20 kDa PEG conjugation | |||||||
| Nusinersen (Spinraza) | 18 mer SSO (ssRNA) | • PS-backbone | / | SMN2 pre-mRNA intron7 (SMA) | Intrathecal injection | US FDA, 2016 | 301 |
| • 2′-MOE | |||||||
| • m5C | |||||||
| Volanesorsen (Waylivra) | 20 mer ASO (ssRNA) | • PS backbone | / | Apo C-III mRNA (FCS) | S.C. | EMA, 2019 | 302 |
| • 2′-MOE | |||||||
| • m5U, m5C | |||||||
| Viltolarsen (Viltepso) | 21 mer SSO (RNA) | • PMO | / | Dystrophin premRNA (DMD) | I.V. | US FDA, 2020 | 306 |
| siRNA therapeutics | |||||||
| Patisiran (Onpattro) | 19+2 mer siRNA | • 2′-OMe uridines | LNP (DLin-MC3-DMA, DSPC, cholesterol, PEG2000-DMG, molar ratio: 50:10:38.5:1.5; molar N/P 3) |
TTR mRNA (hATTR) | I.V. | US FDA, EMA, 2018 | 317 and 464 |
| • 3′-3′ deoxythymidine cap | |||||||
| Givosiran (Givlaari) | 21/23 mer siRNA | • Partial PS backbone | / | ALAS1 mRNA (AHP) | S.C. | US FDA, 2019 | 465 |
| • Partial 2′-F | |||||||
| • Partial 2′-OMe | |||||||
| • GalNAc conjugation | |||||||
| Inclisiran (Leqvio) | 21/23 mer siRNA | • Six PS linkages | PCSK9 mRNA (ASCVD) | S.C. | EMA, 2020 | ||
| • 2′-F or 2′-Ome | |||||||
| • GalNAc conjugation | |||||||
| Vutrisiran (Amvuttra) | 21/23 mer siRNA | • 2′-F | / | TTR mRNA (hATTR) | S.C. | FDA, 2022 | 466 |
| • 2′-OMe | |||||||
| • PS linkages | |||||||
| • GalNAc conjugation | |||||||
| Lumasiran (Oxlumo) | 21/23 mer siRNA | • Partial PS backbones | / | HAO1 mRNA (PH1) | S.C. | EMA, 2020 | 318 |
| • Partial 2′-OMe | |||||||
| • Partial 2′-F | |||||||
| • GalNAc conjugation | |||||||
| mRNA therapeutics | |||||||
| BNT162b2 (Comirnaty) | mRNA encoding spike protein of SARS-COV-2 (4284 nt) | • m1ψ | LNP (ALC-0315, DSPC, cholesterol, ALC-0159, molar ratio: 46.3:9.4:42.7:1.6; molar N/P 6) |
Spike protein (SARS-COV-2 virus infection) | I.M. | US FDA, 2021 | 317 and 467 |
| • 5′-cap: Cap1 analog (m27,3′-O)Gppp(m2′-O)ApG | EMA (conditional marketing authorization), 2021 | ||||||
| • 5′-UTR: incorporation of HBA1 with Kozak consensus (GCCACCAUG), where 37-nt is shared by HBA1 and HBA2 mRNAs | |||||||
| • 3′-UTR: incorporation of segments derived from human mitochondrial 12S rRNA (139 nt), human AES/TLE5 gene (136 nt) and inserted 6-nt downstream the second stop codon | |||||||
| • Codon: ↑ CGG codons, replace GAA by GAG (14 GAA unchanged, in total 48 GAG) | |||||||
| • Two consecutive UGA stop codon of ΨGAΨGA | |||||||
| • 3′-end: 2 segmented poly(A) tracts (A30(GCATATGACT)A70) | |||||||
| mRNA 1273 (Spikevax) | mRNA encoding spike protein of SARS-COV-2 (4004 nt) | • m1ψ | LNP (SM102, DSPC, cholesterol, PEG2000-DMG, molar ratio: 50:10:38.5:1.5; molar N/P 6) |
Spike protein (SARS-COV-2 virus infection) | I.M. | US FDA, 2021 | 317 and 337 |
| • 5′-cap: Cap 1 | EMA (conditional marketing authorization), 2021 | ||||||
| • 5′-UTR: a GC-rich element (CCCCGGCGCC), Kozak sequence (GCCACCAUG) | |||||||
| • 3′-UTR: 110 nt, incorporation of HBA1 between the last stop codon and a poly(A) tail | |||||||
| • Codon: ↑ CGG codons, replace 20 GAA by GAG (in total 14 GAA, 34 GAG); | |||||||
| • Secondary structure flanking the start codon | |||||||
| • Stop codon of ΨGAΨAAΨAG | |||||||
2.3 Nucleic acid degradation as a barrier to gene delivery
Several factors determine to which extent the different nucleases in the human body will form an obstacle to gene delivery, including the type of nucleic acid (DNA or RNA), the administration route, the chemical modification of nucleic acids, the delivery form (naked or complexed) and the delivery system (viral or non-viral vector). Here, we overview the known extracellular and intracellular nucleases that can be encountered during gene delivery.One way to circumvent lysosomal degradation, is the direct delivery of nucleic acids into the cytosol of the cells via fusion with the cell membrane, or by using physical methods, such as microinjection, electroporation or photoporation.118–120 Nevertheless, PO-ONs and siRNA that were directly microinjected into the cytosol of cells were degraded, even when the endosomal compartment was avoided.117,121 For larger DNA fragments, Bamford et al. reported that degradation of DNA in CHO cells does not appear to be a major issue when microinjection or electroporation is used as the transfection technique.122 Escande and co-workers, however, reported that only 50% of naked pDNA remained intact within 2 h in the cytosol of COS-7 cells due to the presence of Ca2+-dependent and physiologically pH optimal endo-exonucleases. Complexation of pDNA with polyethyleneimine (PEI) prevented the in vitro pDNA degradation, indicating the cytosolic degradation as a critical barrier for transgene transport into the nucleus.123 Also Lechardeur et al. showed the metabolic instability of microinjected pDNA in the cytosol of cells, with a half-life of 90 min that was attributed to cytosolic DNases.124 The pDNA degradation caused by cytosolic nucleases has been further confirmed by Rattan et al., where the supercoiled pDNA was degraded into two fragments, a linear plasmid of 6.4 kb and nicked plasmids of 10 kb, after incubation with isolated cytosol.125 Raes et al. studied the integrity of mRNA delivered to cells through photoporation, and found that mainly extracellular degradation in the transfection medium was responsible for low transfection efficiencies, most likely mediated by excreted RNases like RNase I.126 Besides, we found a notable intracellular degradation of both pDNA and mRNA after administration into HeLa cells and SKOV-3 cells by nucleofection.107 Ligon et al. developed a mathematical model to simulate the kinetics of mRNA degradation after transfection in single cells, and found an average degradation rate of mRNA of 0.062 h−1 in the intracellular environment.127 In addition to cytosolic mRNA degradation, the nuclear mRNA degradation is extensive and ubiquitous in mammalian cells although its mechanism is less understood.128 Other subcellular compartments, such as mitochondria (e.g., human mitochondrial nuclease MGME1), exosomes and ribosomes, are involved into nucleic acids (e.g., mRNA) degradation.129–131 To sum up, nuclease-mediated cleavage is a major impediment to gene therapy that is currently still not fully understood. Therefore, more studies on nuclease activity, identity and localization are needed so that this hurdle can be better addressed to achieve more efficient gene delivery in the future.
2.4 Beneficial effects of nucleases
Although nuclease-mediated degradation is indeed one of the obstacles to efficient gene delivery, some nucleases can be used as therapeutics for genome-editing, digesting endogenous self-nucleic acids and so on. For example, endonuclease Cas9 from Streptococcus pyogenes (SpCas9) has been widely used as a powerful tool to modify genomes. Cas9 leverages the sequence specificity of the guide RNA with which it interacts to bind and cleave complementary DNA sequences. Due to the precise genome modification at a specific DNA locus, CRISPR/Cas9 has been extensively studied in a variety of diseases, such as cancer and hereditary hematological disorders.136,137 Currently, other CRISPR-Cas systems targeting RNA, such as the Cas13 family, have also demonstrated some exciting outcomes, such as mRNA knockdown, mRNA live imaging, RNA base editing, cleavage of viral RNAs within mammalian cells, reduction of influenza viruses and SARS-CoV-2.138,139 For a more elaborated overview on genome-editing nucleases, the reader is referred to recent publications.140–142Moreover, as mentioned above, nucleases play a key role in protecting human individuals from auto-immunogenicity and participate in the host defense. An excess of endogenous nucleic acids (NA) can be associated with inflammatory and autoimmune syndromes by activating NA sensing pathways. For example, cell free extracellular DNA, mainly originating from dying hematopoietic cells (e.g., granulocytes, lymphocytes), is abundantly present in the circulation of human plasma (5–10 ng mL−1) and can activate innate immune responses. Intracellular DNAs (e.g., mitochondrial DNA) and RNAs (e.g., mitochondrial dsRNA) also exhibit immunostimulatory properties, like activating innate immune receptors and stimulating the secretion of inflammatory cytokines. Therefore, nucleases together with hepatorenal clearance mechanisms are of importance in eliminating cell-free NAs and thus prevent self-NA-mediated autoimmunity. For instance, DNase I in plasma digests naked DNA and nucleosomal DNA in the presence of heparin and/or plasmin, and the highly immunogenic DNA originating from NETs.143 Intracellular DNase IIα is also critical for eliminating self-DNA and limiting its capacity to induce harmful inflammatory and autoimmune responses in both mice and humans. Likewise, RNase A and RNase T2 regulate the endogenous RNA abundance, immunostimulatory potential and their involvement in autoimmune and inflammatory disorders.144 Due to the antitumorigenic properties, RNase T2 has been studied as an antiangiogenic and anti-vascular drug, by blocking the blood supply in tumor-associated vessels via stopping angiogenesis.145 As TREX1 is supposed to degrade tumor-derived DNA that would activate cGAS-STING, TREX1 inhibition has been reported as a novel immunotherapeutic strategy.146 Targeting TREX2 pathways may become useful in treating psoriasis. As mentioned above RNases like Dicer, Drosha and RNase H are required for the therapeutic action of some gene therapeutics like siRNA, miRNA and ASOs. Also, DNase I is used as a therapeutic itself, as a mucolytic agent after inhalation for cystic fibrosis therapy, with DNASE1L2 as a promising alternative as well.147,148
3. Available strategies to improve nuclease resistance of exogenous nucleic acids
Several strategies have been explored to prevent degradation of nucleic acids, which can be widely categorized into modification of the nucleic acid cargo itself, or additional protection of nucleic acids by encapsulation into viral-or non-viral vectors (Fig. 3).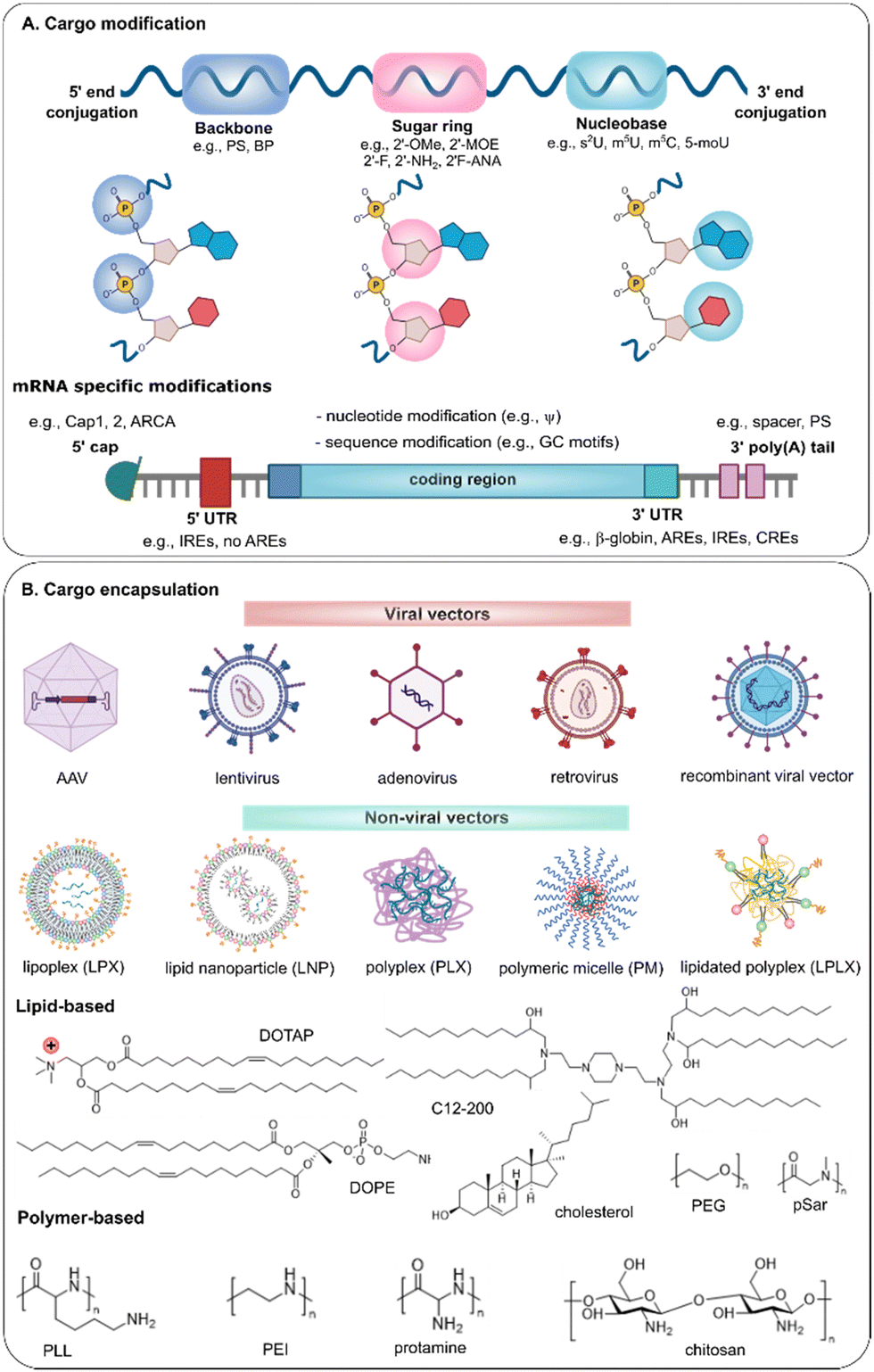 | ||
| Fig. 3 Schematic illustration of (A) structure modifications of nucleic acids and (B) the formulation toolbox for encapsulation of nucleic acids, with representation of commonly used lipids and polymers for non-viral gene delivery. | ||
3.1 Nucleic acid modification
To date, chemical modification represents one of the most effective approaches to enhance nucleic acid stability. A number of different strategies, as shown in Fig. 3A, are available to enhance the nuclease resistance of nucleic acids, which can be categorized into backbone, sugar ring or inter-nucleoside, nucleobase and 5′ or 3′ end modification.149–152 Both the type of modification, the location and the degree of substitution will determine the effect on nuclease stability and the remaining biological activity. Also, different modifications can be combined in one molecule.PS-modified siRNA was found more resistant to nucleases, but less effective than boranophosphate (BP) modification, where 25–75% BP modification can increase both nuclease resistance and RNAi efficiency.156 Other substitutions like phosphoramidates (replacing 3′-O atom with 3′-amino) and thio-phosphoramidates can directly bind to the active site of the enzyme and thus inhibit its activity, rendering high nuclease resistance of oligonucleotides. Imetelstat undergoing phase II/III clinical trial in patients with myelodysplastic syndromes, is a thio-phosphoramidates modified 13-mer ON.157,158 Further, the chirality of PS linkages, i.e., Sp and Rp, has shown influence on the nuclease stability, where Rp-gap ASOs and PS-siRNA with a higher fraction of Rp centers were more resistant to nuclease in FBS and cells than the Sp counterparts.159 In contrast, Sp-PS-ONs had an increased resistance to 3′-exonuclease in human plasma when compared with Rp-PS-ONs.160 A stereo-random mixture of Sp and Rp is suggested to achieve the balance between nuclease stability and silencing activity.161
Complete change of the backbone structure has been explored as well. Peptide nucleic acids (PNAs), for example, have a neutral charge backbone of repeating N-(2-aminoethyl)glycine units, instead of pentose sugar moieties or phosphate groups. They exhibit a notable duplex stability and nuclease resistance, especially in antisense constructs.162 Gaglione et al. reported that siRNA with one PNA-modified strand remained intact up to 6 h while siRNA with two PNA-modified strands was still visible on the gel after 24 hours’ incubation in full FBS.163
Extensive or complete 2′-OMe modification is mostly chosen when a high level of nuclease resistance is required. Choung et al., for example, found that siRNA with 2′-OMe or PS into the three terminal 3′ nucleotides remained intact up to 3 h in 10% human serum while unmodified siRNA was mostly degraded within 1 h. Nuclease resistance was further increased (up to 24 h) when a stretch of three 2′-OMe modified nucleotides was alternated with two or three unmodified nucleotides.164 Both the number and location of the introduced modifications contribute to an improved biological activity and nuclease resistance of siRNA. Compared with terminal modification, internally 2′-OMe modified siRNA showed extreme nuclease-resistance in serum.165 Of note, over 50% replacement of nucleotides with a 2′-OMe in siRNA affected its biological activity, unless alternated with non-modified nucleotides, while a fully modified 2′-OMe guide strand was completely inactive.166 Volkov et al. found that the main nuclease cleavage sites in serum were CpA, UpA and UpG and introducing 2′-OMe at these sites greatly increased stability.167 Partial introduction of 2′-OMe modified nucleotides has also been used into ASOs to increase the nuclease resistance and biological activity.168 As full modification does no longer support RNase H cleavage of the target RNA, hybrid ASOs are constructed in the form of a gapmer. A standard gapmer has a central region of PS-modified DNA bases to induce RNase H cleavage, flanked on both sides by RNA blocks with 2′-modifications to allow sufficient binding affinity and prevent unwanted exonucleases degradation.
2-O-Allyl and 2′-MOE are more voluminous substituents that can improve the stability of RNAs, but are also associated with more notable RNAi inhibition.169 Therefore, it is mostly applied for ASOs rather than siRNA. When combined with nucleobase modification, it was shown that the nuclease resistance of 2′-MOE-2-thiothymidine modified ONs was largely attributed to 2′-MOE.170 Further exploration on 2′-MOE resulted in 2′-O-[2-(methylamino)-2-oxoethyl] (2′-O-NMA), which, when incorporated at the 3′ end, resulted in enhanced exonuclease stability and binding affinity compared to 2′-MOE modified ASOs.171,172 The success of 2′-O-MOE has been well exemplified by its use in three FDA-approved ASOs (see Table 3 in Section 4).
2′-F substitution is commonly used to produce siRNA with increased serum stability, with only a slight effect on RNAi efficiency even after full replacement. In general, substitution is more frequently performed into the guide strand.173 While unmodified siRNA was fully degraded in 4 h (of which 70% already in the first minute), more than 50% and 25% of 2′-F-modified siRNA remained intact after 24 h and 48 h respectively, when exposed to human plasma.108 Chiu et al. found that 2′-fluoro-uridine (2′-FU) and 2′-fluoro-cytidine (2′-FC) modifications in either the antisense strand or both strands, over 70% of siRNA remained intact in cell extracts after 1 h, probably due to prevention of RNA digestion by RNase A.174 So far, four FDA-approved siRNA drugs incorporate extensive 2′-F substitution, in addition to 2′-OMe, with an improved nuclease stability and binding affinity to complementary RNA (see Table 3 in Section 4). It should be noted that the introduction of 2′-F modifications in siRNA is associated with toxicity. As also the contribution of 2′-OMe modifications to the nuclease resistance of siRNA is greater than that of 2′-F modifications, this leads to design of siRNA with alternating 2′-OMe and 2′-F modifications with efficient gene suppression.175 By changing the stereochemistry of the fluorine of 2′F-RNA, 2′F-ANA modifications are formed. Dowler et al. reported a fully modified 2′F-ANA strand into a siRNA duplex with a half-life of 6 h in 10% FBS when compared to an unmodified siRNA (half-life < 15 min).176
Ribose modifications are not limited to 2′ substitution in the sugar ring. Modifications on other positions in ribose, such as 4′-S,4′-C-aminomethyl-2′-O-methy and 4′-C-O-methyl-2′-O-methyl, efficiently protected siRNA from nuclease cleavage in vitro, but significantly inhibited RNAi.177–179 Also analogs like locked nucleic acid (LNA), unlocked nucleic acid (UNA) and ethylene-bridge nucleic acid (ENA), have demonstrated a robust nuclease resistance. As an example, LNAs are characterized by a methylene-bridge between 2′-OH and 4′-C of the sugar ring, thus structurally constraining the sugar moiety into the A-form helix and affecting the duplex folding. Typically, LNAs are often placed in the context of a DNA or a 2′-OMe backbone at every third position. The half-life of LNA-modified aptamers in human plasma was dramatically increased to 50 h, and LNA/DNA chimera remained intact within 20 h in bovine serum. As well, intact LNA-modified siRNA was still detectable after 48 hours’ incubation in undiluted human serum and mouse serum, but LNA modification strongly affected its interfering activity.180–182 UNA-modified siRNA demonstrated a prolonged circulation time (>30 min) when compared to the unmodified siRNA (1 min).183,184
In phosphorodiamidate morpholino oligonucleotide (PMO) modifications, the ribose sugar is even completely replaced by a six-membered morpholine ring, together with modification of the phosphatediester linkage to yield neutral instead of negatively charged nucleic acids. Two PMO drugs, Etepilrsen and Golodirsen, have been approved by the FDA. Such arsenal of chemical modifications, with a neutral charge, allows adapting a particular ON to a required mode of action and administration route.
The use of modified nucleotides in mRNA is widely employed to improve the nuclease resistance and limit the recognition by the innate immune system. For example, Kormann et al. reported that the replacement of 25% of U and C with 2-thiouridine (s2U) and 5-methylcytosine (m5C) dramatically increased the stability of the mRNA and decreased activation of the innate immune system both in vitro and in vivo.190 Li et al. showed that about 50% unmodified, N1-methylpseudouridine (m1ψ)- or ψ-modified mRNA was degraded, while approximately 75% 5-methoxyuridine (mo5U)-modified mRNA remained intact within 4.5 h in the transfected cells.191 It was also shown that introducing m1ψ modified nucleotides stabilized the mRNA secondary structure by stimulating the formation of double-stranded regions, resulting in an improved resistance against endonucleases when compared to non-modified uridine.192,193
Direct conjugation of ligands to the 5′ end and/or 3′ end of the sense strand without affecting RISC loading has been extensively used for siRNA drugs. Conjugation of palmitic acid at the 5′ end of the sense strand enhanced the nuclease stability of siRNA, with 10-fold higher half-life compared to naïve siRNA (16.8 h vs. 1.5 h) in 10% FBS. In a harsher environment (90% FBS), the unmodified siRNA was fully degraded within 1 h, while the intact palmitic acid-conjugated siRNA remained detectable after 3 hours’ incubation.199 Other aromatic compounds, such as phenyl, hydroxyphenyl and naphthyl, have also shown protection from nuclease degradation by conjugating them to siRNA, demonstrating comparable nuclease resistance as LNA-siRNA.200 Phosphorylation of the 5′ end of the guide strand has been successfully employed as a rational strategy to facilitate its loading into RISC, and stabilization of this moiety using phosphate analogs improves metabolic resistance and substantially enhances the duration of effect. N-acetylgalactosamine (GalNAc) conjugation is one of the leading strategies for the current ASO and siRNA therapeutics, where the GalNAc moiety is subsequently subject to enzymatic degradation and thus liberates the ONs.201 Conjugation of GalNAc to the passenger strand is typically preferred to not impair the on-target silencing activity and to reduce off-target silencing activity of the guide strand. Other bioconjugation strategies such as dynamic polyconjugates, or a new targeted RNAi molecule (TRiM) platform, composed of a highly potent siRNA and high affinity targeting ligands (e.g., GalNAc, RGD motifs, αvβ6 ligand), have been explored.202–205 In spite of significant advances in siRNA conjugates, the associated issues, such as low bioavailability, unfavorable pharmacokinetics and high cost, impede their translation in the clinic.194
When the 5′ and 3′ ends of nucleic acids are conjugated to each other, cyclic nucleic acids are formed that are resistant to exonuclease activity. Zhang et al. cyclized the 5′ and 3′ ends of the sense or antisense strand of siRNA into a 21-mer RNA ring, which was subsequently hybridized with a linear complementary RNA strand to form a circular siRNA duplex. When exposed to RNase A, only trace degraded fragments of circular siRNA was observed when compared with linear siRNA.206 Similarly, circular mRNA is more resistant against RNase and RNA exonuclease than linear mRNA, due to the covalently closed loop structures.207
Apart from 3′ and 5′ modifications, also the coupling of biomolecules internally has been explored. Astakhova et al. internally incorporated methionine- and leucine-enkephalin peptides into 21 mer LNA/DNA mixmer strands. The modified LNA/DNA was degraded within 1 h in 90% human serum, whereas the unmodified DNA was degraded within 10 min even in 10% human serum.208 Also attaching for example cationic (poly)amine groups to the nucleobase, sugar, backbone, 3′ or 5′ end or in the center of ASO to reduce the net negative charge of ASO could improve its nuclease resistance.209
Codon optimization. Besides the use of modified nucleotides as mentioned above, codon optimization is of interest to improve the nuclease resistance of mRNA, as the codon composition and identity are important to mRNA stability. For example, introducing rare codons into a small subset of a gene dramatically decreased mRNA stability. mRNA containing a HIS3 stop codon was stable while mRNA lacking an in-frame stop codon was unstable (half-life of 79 min vs. 9 min).211 mRNAs enriched in optimal codons tend to be more stable, have a higher translation efficiency and a longer poly(A) tail while mRNA enriched in non-optimal codons tends to be unstable, shows poor translation efficiency and a shorter poly(A) tail, which underlines the important role of codons in mRNA stability.212,213 Although a high G-C content may cause problems for mRNA secondary structure, a higher GC-rich element within the open reading frame (ORF) resulted in 100-fold translation efficiency, and an improved mRNA stability.214,215
5′ cap modification. Chemical modifications at the 5′ end of mRNA focus on the cap structure, as the cap is of great significance in maintaining mRNA stability and it also binds to the eukaryotic initiation factor eIF4F, thus influencing translation efficiency. The natural cap consists of a methylated G (m7G) that is linked to the first transcribed nucleotide via a 5′-5′ triphosphate bond to form the minimal Cap0 structure (m7GpppRNA). An additional methyl group on the ribose 2′-O position of the first transcribed nucleotide forms the Cap1 structure that helps recruit the ribosome and protect the mRNA from degradation. When the second nucleotide is also methylated, this yields the Cap2 structure. Further, nucleotides adjacent to the m7G Cap can be methylated to enhance mRNA stability, such as m6A or a 2′-O-dimethyladenosine (m6Am). Incorporating m6Am into the first nucleotide conferred a half-life of mRNA approximately 8.5 h in HEK293 cells, whereas the analogues with Am, 2′-O-methylcytidine, 2′-O-methylguanosine or 2′-O-methyluridine showed an average half-life around 6 h.216
It is of interest to develop alternative cap structures at the 5′ termini of mRNAs, in terms of mRNA stability and protein production. Current cap analogs include O-Me-m7GpppG, anti-reverse cap analogs (ARCA, m27,3-OGpppG), and modified ARCA. As an example, ARCA is a 2′-OMe modified m7G cap and has been incorporated into in vitro transcribed (IVT) mRNA to improve the stability. As reported, ARCA-capped mRNA was found to have a prolonged half-life, higher transfection efficiency in rabbit reticulocyte lysate and extended protein expression in cells.217–219 Recently, several cap analogues (e.g., PS, imidiphosphate, LNA and BP), also provide the mRNA with a longer half-life. For example, a higher stability of mRNA can be achieved by elongating the 5′-5′ bridge to a tetraphosphate and incorporating a single phosphorothioate.220–222
3′ end poly(A) tail modification. The poly(A) tail at the 3′ end is of importance in regulating mRNA quality and degradation, as mRNA degradation typically starts from deadenylation at the 3′ end. Typically, mRNAs in mammalian cells have a 150–250 nt long poly(A) tail, where stability of mRNA increases with increasing poly(A) tail length.223,224 Shortening a poly(A) tail in the cytosol to fewer than 15–20 nt leads to mRNA susceptible to degradation either through the decapping and 5′-3′ or 3′-5′ decay pathway.225 For instance, ARCA-capped luciferase mRNA with a poly(A60) and poly(A31) tail displayed a half-life around 282 min and 90 min in rabbit, respectively.217 Mockey et al. reported that co-delivery of free poly(A) chains could protect mRNA from degradation by balancing the deadenylation of the 3′ poly(A) tail and then the cleavage of the 5′ cap structure or the 5′-3′ exonucleotidic digestion.226 Further, a poly(thio-A) tail was studied to increase stability. It should be noted, however, that the optimal poly(A) tail length for efficient mRNA translation is still unclear, although around 100 nt poly(A) tail was suggested as optimal for synthesizing mRNA therapies.
Further, Trepotec et al. developed a segmented poly(A) approach, by splitting the most widely used but relatively unstable 120 nt poly(A) tail into two segments (each containing 60 adenosines) with a single or 6 nt (G/T) spacer, to support mRNA stability and protein expression.227 In addition to the poly(A) tail, a poly(G) tail was found to positively correlate between the G frequency and mRNA half-life in NIH3T3 cell and HeLa cell, as the 3′-terminal poly(G) tract may inhibit 3′-5′ degradation. However, such poly(G) modification did not result in any enhancement in mRNA level or translation rate.228
UTRs modification. 5′-UTRs and 3′-UTRs, consisting of regulatory sequence elements regulating the mRNA stability, are also important regions that influence the exogenous degradation of mRNA. Modification of UTRs represents an approach to enhance both mRNA stability and translation efficiency. The 5′-UTRs mainly affect the translation efficiency, although it also can inhibit both cap removal and 3′-5′ exonuclease degradation. Several strategies have been explored in 5′-UTR sequence to improve mRNA stability and translation accuracy, including (i) avoiding the start codons and non-canonical start codons in the 5′-UTR which disrupts translation, (ii) avoiding the highly stable secondary structures to prevent ribosome recruitment and codon recognition and (iii) use of a shorter 5′-UTRs. The 3′-UTRs assembles a group of unstable elements and thus play a key role in mRNA stabilization.229–231
A critical balance should be maintained between translation efficiency and mRNA stability, which respectively increase and decrease with increasing 3′-UTRs length.232 Currently, most mRNA therapeutics in clinical trials utilize 3′-UTRs derived from α- and/or β-globin mRNAs that harbor several sequence elements that increase the stability and translation of mRNA.233,234 Zarghampoor et al. demonstrated a half-life of mRNA with UTRs of human β-globin up to 48 h in HEK293T cells.235 The stabilizing effect of human β-globin 3′-UTRs sequences was further augmented by using two human β-globin 3′-UTRs arranged in a head-to-tail orientation. In addition, multiple sequence elements in the 3′-UTR have been reported to affect the stability of mRNA, such as AU-rich elements (AREs), GU-rich elements (GREs), iron response elements (IREs), C-rich elements (CREs) and AUUUA repeats. AREs, for example, result in a rapid mRNA degradation, and replacing them with β-globin 3′UTRs results in a significant increase in mRNA half-life.236–238 Unlike AREs, the effect of IREs depends on their precise location, as they regulate mRNA half-life when present at the 3′-UTRs and affect translation when located at the 5′-UTRs.
It should be noted that in addition to the above-mentioned modifications, the removal of double-stranded RNA contaminants during the production process of mRNA is of importance, as these very immunogenic by-products otherwise trigger innate immunity and enhance the mRNA degradation.229 This is especially true for self-amplifying mRNA, which in general do not contain modified nucleotides as they might interfere with the self-amplifying properties of the mRNA and are lost after one round of amplification.
3.2 Nucleic acid encapsulation to protect against enzymatic degradation
To further protect nucleic acids against enzymatic degradation, they are often delivered using viral or non-viral vehicles. Moreover, nucleic acid encapsulation can help to overcome additional extracellular and intracellular barriers such as cell targeting, cellular uptake and endosomal escape. Although many research papers focus on the biological effect of the delivered nucleic acids, we here overview the few of them that actually investigated to which extent the formulation protects the encapsulated nucleic acids against degradation.Lipids were introduced as carriers for therapeutic nucleic acid delivery over 30 years ago and still widely used for the encapsulation of small molecules and macromolecules244,245 (Fig. 3B). Liposomes are composed of a phospholipid bilayer with an aqueous core, where one or more rings of lipid bilayer surround an aqueous pocket. When employed as vehicles for nucleic acid delivery, the positively charged liposomes and negatively charged nucleic acids spontaneously form complexes referred as lipoplexes, where nucleic acids are assumed to be present between a lipid bilayer. This is consistent with the views from Gregoriadis that liposome-entrapped mRNA was fully shielded and protected from nuclease attack in the blood circulation.246 As shown in our previous work, however, only around 50% of mRNA was encapsulated in the core of lipoplexes and remained intact when incubated in human serum, while 50% mRNA was attached at the surface of the lipoplexes and thus rapidly degraded.107 Remaut et al. found that DOTAP/DOPE liposomes offered a good protection of phosphodiester ONs against degradation within the first 5 h, which was however followed by a spontaneous release and degradation of ONs on later time point.247 They also found that PEGylation of the liposomes’ surface lowered the protection against degradation when compared to non-pegylated liposomes, as complexed ONs were present on the outside of the lipoplexes, still accessible to DNase I.248 Lipid nanoparticles (LNPs) are typically formulated using ionizable lipids (e.g., C12-200 (Fig. 3), MC3 (Fig. 5 in Section 4), SM-102, ALC-0315 (Fig. 6 in Section 4), helper lipids (e.g., DOPE, DSPC), PEG-lipids and/or sterol lipids (e.g., cholesterol) and differ from liposomes as they do not have an aqueous core. At acidic pH, the protonated ionizable lipids can encapsulate negatively charged nucleic acids, while the resulting LNPs have an overall neutral surface at physiological pH.249 LNPs are formed by rapid (microfluidic) mixing of lipids in ethanol and nucleic acids (e.g., mRNA) in buffer, resulting in a lipid bilayer and some micelle-like structures in which nucleic acids are entrapped inside the LNPs, and protected from nuclease degradation. However, it has been shown that self-amplifying RNA (saRNA) complexed to the surface of LNPs was also protected from RNase degradation, depending on the lipid composition. The ionizable lipid, C12-200, protected around 100% of encapsulated saRNA (present inside the LNPs) and 10% of saRNA absorbed to the outside from RNase degradation. For DOTAP-composed LNPs, almost 100% of the encapsulated saRNA and 45% of the exteriorly complexed saRNA remained intact after RNase treatment, whereas 60% of the encapsulated saRNA and 95% of exterior saRNA of dimethyldioctadecylammonium (DDA)-LNPs were protected.250 Lou et al. found a comparable protection for saRNA of DDA-LNPs and DOTAP-LNPs when compared with DLin-DMA-LNPs, as visualized by agarose gel.251 Also a nanostructured lipid carrier (NLC), consisting of glyceryl trimyristate-dynasan 114, liquid oil (squalene), and nonionic surfactants including Span 60, PEGylated Tween 80 and DOTAP, was found to effectively protect against RNase degradation. Interestingly, the fraction of PEGylated Tween 80 played a key role in RNA protection, where NLCs with 35% Tween 80 did protect against RNase degradation while those with 70% Tween 80 did not, which was attributed to the PEG chains in Tween 80.252
 | ||
| Fig. 4 Scheme of AAV-bearing DNA delivery into cells. | ||
 | ||
| Fig. 5 Scheme of intracellular delivery of clinically approved therapeutics based on (A) ASO and (B) siRNA to modify protein expression, with (C) a representation of the clinically applied chemical modifications and LNP formulation. | ||
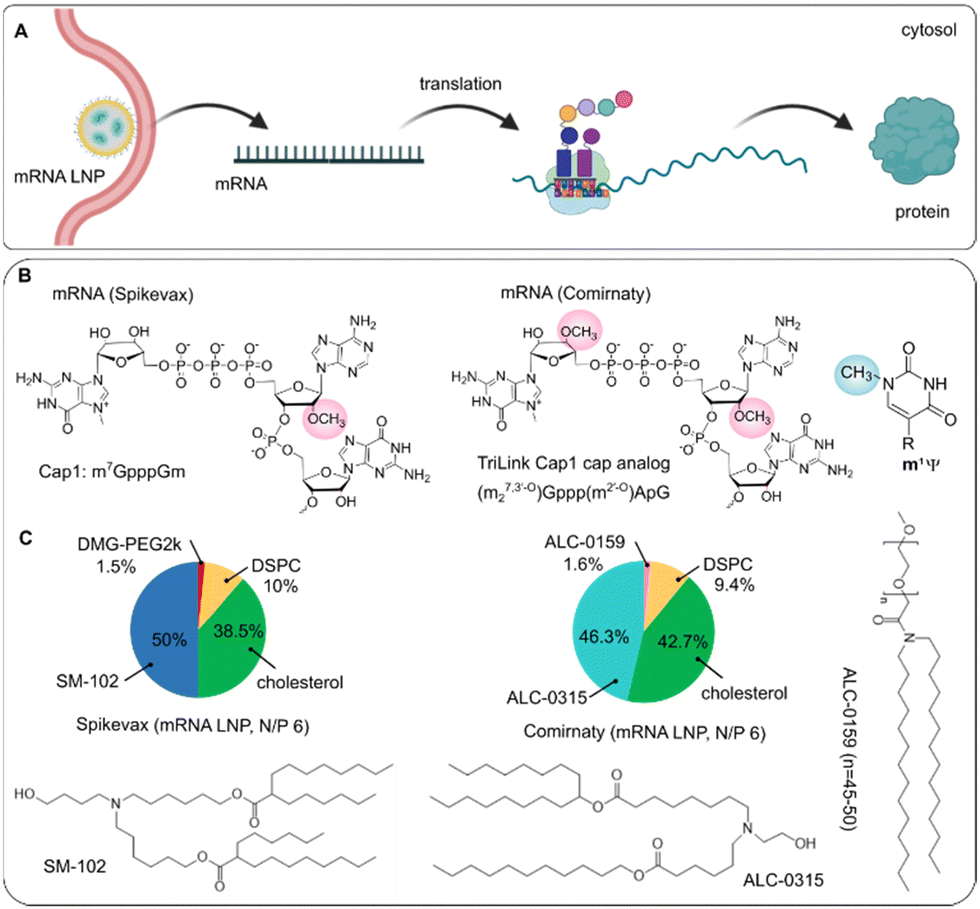 | ||
| Fig. 6 Scheme of (A) intracellular delivery of mRNA COVID-19 vaccines to produce protein of interest as well as (B) 5′-cap chemical modification of mRNA cargo, and (C) LNPs compositions from Spikevax and Comirnaty. | ||
While lipid-based nanoparticles are generally at least 70 nm in size, polymer-based particles have the advantage of being able to generate smaller sizes. Cationic polymers condense and pack negatively charged nucleic acids, which offers shielding from the environment (Fig. 3B). Poly-L-lysine (PLL) was the first cationic polymer investigated for DNA transfection, although poly-ethylenimine (PEI) is currently most widely used.253,254 Branched PEI (2 kDa) was proved to protect PS-ONs from DNase I cleavage, which was more effective than PEG–PEI, due to the stronger condensation.255 Zhang et al. demonstrated enhanced nuclease resistance of mRNA condensed with linear PEI (22 kDa), but found that the strong complexation prevented their cytosolic release and translation.256 Therefore, a critical balance should be maintained between encapsulation and protection of nucleic acids, while still allowing efficient cargo release. In this regard, stimuli-responsive delivery systems have been studied. For example, reduction-responsive polymers containing disulfide linkages that promote nucleic acid cargo release in the cytosol, as well-reviewed.257 Also pH-responsive polymers have attracted significant attention, to protect the nucleic acids in the endosomal compartment, while still allowing endosomal escape based on acidification of the endosomal compartment.258,259 Notwithstanding the great progress in both polymer synthesis tools and endosomal escape mechanism investigation, the integrity of nucleic acid cargo that escaped from endosomal compartments is rarely covered in the publications.
Yin et al. reported a hybrid system with PEI, namely a hydrogel composed of PEI (1.8 kDa) and graphene oxide (GO) that protected mRNA in serum for 48 h, whereas free mRNA was fully degraded within 6 h.260 As well, hybrid nanoparticles composed of a core of PEI-complexed mRNA and a DOPA (1,2-dioleoyl-sn-glycero-3-phosphate) shell were capable of providing an efficient protection of mRNA from RNase A degradation.261
Other systems that have been reported to protect nucleic acids include biodegradable poly(β-amino esters). The DNase I resistance of complexed PS-ONs was improved, while mRNA that was absorbed on the surface of a poly(β-amino ester) polymeric core had an increased resistance to nucleases degradation in comparison with naked mRNA in mice.262 Protamine-complexed shRNA remained intact after being exposed to DNase I for 1 h or FBS for 8 h, whereas naked shRNA was degraded.263 Brito et al. showed that a cationic nano-emulsion, composed of DOTAP and emulsion adjuvant MF59, had protective effects on saRNA and pDNA stability when incubated with RNase A and DNase respectively.264 He et al. developed amino-modified silica nanoparticles to protect DNA from DNase I cleavage, without compromising the biological properties of pDNA.265 Proteins that wrap up DNA resulted in an enhanced half-life around 158 min in human serum, which was 5-fold higher than that of the naked DNA.105
Recently, Yoshinaga developed a novel technology to provide protection of mRNA against RNases under physiological conditions, without using cationic materials. A complementary RNA oligonucleotide (OligoRNA, 17 nt) possessing a PEG strand was hybridized with mRNA, resulting in PEG strands surrounding the mRNA that inhibited recognition by RNases, without compromising its translational activity. Of interest, this strategy is only composed of PEG and mRNA that both have been approved for clinical usage.266 However, the nuclease resistance of PEG-mRNA was evaluated only in 1% FBS, which may overestimate the protection in representative environment.
3.3 Co-delivery with nuclease inhibitors
Although not widely used, it is useful to mention that pre-clinically, the use of pharmaceutical agents to inhibit nuclease activity has been explored to some extent. For example, EDTA and citrate are commonly used chelators that capture divalent cation cofactors that are required by some nucleases, such as the DNase I family. However, such chelators are typically quite toxic at the doses required to reduce extracellular nuclease activity, as divalent cations (e.g., Ca2+) are also required for many other biologic functions like cardiac contraction. Alternatively, agents like nucleotide analogs ATP and ATA, can directly inhibit the nucleases activity.267 ATA exhibits a broad spectrum of nucleases inhibition as it acts on DNase I, RNase A, restriction exonuclease and endonucleases III, and has been explored to prevent degradation of nucleic acids during gene delivery.268 By combining ATA with naked pDNA, an enhanced transgene expression was found in pigs, macaques and mice skin after intradermal administration.269,270 Likewise, intratracheal administration of ATA with naked pDNA resulted in a 86-fold and 54-fold increase of transfection efficiency in macaque lung and mouse lung, respectively.271 Walther et al. demonstrated the prevention of DNA degradation after jet-injecting ATA with naked pDNA in human mammary and colon carcinoma models, by increased gene expression and improved reduction in tumor growth.272 As a polyketide metabolite, DMI-2 was also found to inhibit the activity of porcine DNase II. An improved transgene expression in human pulmonary adenocarcinoma cells was observed when DMI-2 was included in the transfection medium containing lipofectamine–DNA complexes.273 Interestingly, anti-nuclease antibodies, by injecting the purified antigens against RNase A, RNase T1, RNase 1 and DNase 1, into rabbits together with complete or incomplete Freund's adjuvant, were found as powerful inhibitors of nucleases, without any biological application yet.274 It is also relevant to note that the addition of RNase inhibitors to the injection cocktails did not yield any notable enhancement of naïve siRNA stability in mice.275 Remarkably, research on the strategy of co-delivering nuclease inhibitors to reduce the degradation/clearance of nucleic acids is very limited when compared with chemical modifications and the encapsulation in vectors, as discussed above. A potential reason is the broad, non-specific action of nuclease inhibitors after administration. Targeted and in vivo stable nano-delivery systems containing both the genetic material and nuclease inhibitor could be an interesting approach to achieve safe and efficient co-delivery of nucleic acids and nuclease inhibitors.4. Most used strategies to prevent degradation of clinically approved gene therapeutics
While the previous section overviewed the possibilities to enhance stability of nucleic acids against degradation in general, this section will focus on the gene therapeutics that eventually reached the clinic, together with their applied strategies to prevent degradation, which are clearly largely depending on the type of nucleic acid to be delivered (Table 3).4.1 Viral delivery of large, double stranded DNA fragments
Back in 1971, DNA molecules firstly demonstrated their potential as a novel class of therapeutics after injection into human fibroblast cells to restore the missing enzyme activity.276 As naked DNA does not easily enter cells, mostly viral vectors are used for its delivery (Fig. 4). In the 1990s, the first long-term clinical trial was performed using retroviral-mediated transfer of the adenosine deaminase gene into T cells of two children with severe combined immunodeficiency.277 In recent years, DNA-based therapeutics have yielded promising outcomes in the treatment of a wide variety of disorders, such as cancer, cardiovascular diseases, neurodegenerative diseases, retinal diseases, etc.278Table 3 shows an overview of DNA-based therapeutics that received clinical approval. Notably, none of the approved formulations uses chemically modified sequences. Instead, all, except Neovasculgen and ZyCoV-D, use viral vectors to deliver DNA. Gendicine is a recombinant human p53 adenovirus and has been approved by the China Food and Drug Administration (CFDA) for the treatment of head and neck cancer in 2003. Adenoviruses (∼100 nm) contain a linear double-stranded DNA genome up to 35 kDa, which can be transcribed and replicated in the nucleus of the host cell. Its over 15 years of commercial use has shown an exemplary safety record, a wide indication (different cancer types and stages) and a significantly higher response rate in combination with chemotherapy and radiotherapy than standard therapies alone.279 Other widely used viral vectors are AAVs, small (∼25 nm) replication-deficient viruses with a linear dsDNA genome up to 4.7 kDa, that can persist in an extrachromosomal state without integrating into the genome of the host cell. Glybera, containing the human lipoprotein lipase (LPL) gene variant LPLS447X in a vector that comprised a protein shell derived from AAV1, CMV promoter, a woodchuck hepatitis virus post-transcriptional regulatory element and AAV2 derived inverted terminal repeats, has received marketing authorization for patients with LPL deficiency from the European Commission in 2012. As the most expensive drug (around $1 million) worldwide, it has been withdrawn in 2017 due to the limited demand and the concerned clinical effectiveness.280 Luxturna has been approved by the U.S. Food and Drug Administration (FDA) in 2017 to restore the vision of patients with Leber's congenital amaurosis, by subretinal injection of an AAV2 vector to deliver a normal human retinal pigment epithelium 65 (RPE65) gene to retinal cells.281 Recently, Zolgensma has received its approval from FDA and a conditional marketing authorization valid through the Europe for the treatment of children less than 2 years of age with spinal muscular atrophy caused by the mutation of survival motor neuron 1 (SMN1), by delivering the SMN1 gene with AAV9 via intravenous infusion.282 To fight against COVID-19, DNA vaccines have also been explored. JNJ-78436735 (AstraZeneca) contains a recombinant, replication-incompetent adenovirus serotype 26 (Ad26) vector encoding spike protein of SARS-CoV-2. It was safe and immunogenic in adults according to the interim study and has received conditional marketing authorization in Europe.283Some naked DNA drugs have received market approval. Neovasculgen, a supercoiled pDNA encoding a 165 amino acid form of vascular endothelial growth factor (VEGF) with CMV promotor, was authorized for atherosclerotic peripheral arterial disease in 2011 in Russia.284 As the first needle-free COVID-19 DNA vaccine, ZyCoV-D comprising DNA encoding spike protein and IgE signal peptide has received emergency use authorization in India.285 Other DNA vaccines, such as INO-4800 and COVID-eVax that are based on DNA encoding spike protein, as well as GX-19N containing two DNAs encoding spike protein and nucleoprotein, are being evaluated in clinical trials.286–288 Additionally, non-viral vectors have gained attention for DNA vaccines. For example, Covigenix VAX-001, where a spike protein encoded DNA and two genetic adjuvants are encapsulated into proteo-lipid formulations, is undergoing phase II trial.289 It remains however an open question whether DNA vaccines will provide more durable protection than their mRNA counterparts or any other type of vaccine.290
4.2 Naked delivery of modified ASO and siRNA therapeutics
First generation ASOs are synthesized by replacing one of the non-bridging oxygen atoms in the phosphate group (PO) with either a sulfur group (PS), methyl group (methyl phosphonates) or amines (phosphoramidates), thus resulting in enhanced nuclease resistance and improved plasma half-life. The first approved ASO drug, Fomivirsen is a PS-modified ODN (21 nt) complementary to mRNA transcripts of the major immediate-early region (IE2) proteins of human cytomegalovirus (CMV), and is a potent and selective anti-viral agent for CMV retinitis.293 Nevertheless, notable nuclease digestion was observed in the vitreous by 2 days following intravitreal injection in human.294,295 To further improve the nuclease resistance and affinity of target RNA, second generation ASOs, comprising of 2′-MOE modified nucleotides on both terminal ends, a gap of deoxynucleotides to support RNase H1 activity and PS at each inter-nucleotide linkage, have been widely studied. 2′-MOE is extensively used to enhance the nuclease resistance and reduce toxicity by increasing the binding affinity of ASOs to the target RNAs, particularly in combination with other modifications. Mipomersen with five 2′-MOE-modified nucleotides and PS modifications, has been developed as an ASO inhibitor of apolipoprotein B (ApoB)-100 synthesis. Its terminal elimination t1/2 in plasma increases as dose increases, ranging from 20 to 30 days after single SC injection.296,297 Inotersen, a 20 mer ASO inhibitor of human transthyretin (TTR) production and approved for the treatment of polyneuropathy in adults with TTR amyloidosis, contains five 2′-MOE-modified nucleotides at both 5′ and 3′ ends, ten DNA nucleotides and PS modifications throughout the whole sequence, and exhibits an elimination t1/2 around 32 days in human plasma following SC injection.298,299 Nusinersen, approved by FDA in 2016, is a PS and 2′-MOE-modified ON with an estimated half-life of 135–177 days in cerebrospinal fluid and 63–87 days in blood plasma.300 It is composed of an 18-nucleotide 2′-MOE SSO with a fully modified PS backbone and has been approved for the treatment of spinal muscular atrophy (SMA). Its mean terminal elimination half-life is estimated to be 135–177 days in cerebrospinal fluid (CSF), and 63–87 days in plasma following intrathecal injection.300,301 Volanesorsen, approved for treatment with familial chylomicronemia syndrome, is also modified with 2′-MOE and PS, with a human plasma t1/2 over 2 weeks after single SC administration.302–304 Third generation ASOs contain different modifications, such as PMOs, PNAs and LNAs. One example are SSOs that bind specific RNA-target sequences and modulate pre-mRNA splicing by sterically blocking the binding of splicing factors to the pre-mRNA.305 SSOs modified with PS backbone, 2′-OMe, 2′-MOE or PMO, have been widely used for two inherited diseases. For duchenne muscular dystrophy (DMD), PMO-modified SSOs include Golodirsen (plasma t1/2 of 3.4 h), Casimersen (plasma t1/2 of 3.5 h), Viltolarsen (t1/2 of 2.5 h) and Eteplirsen (plasma t1/2 of 3–4 h).306–309 As PMO-modified SSOs are associated with rapid kidney clearance and lower accumulation in tissues when compared to a negatively charged PS backbone, some conjugates have also been explored to reduce the rapid clearance. For instance, Macugen (27 mer anti-VEGF-RNA aptamer) was approved by the FDA in 2004 for the treatment of all types of neovascular age-related macular degeneration (AMD).310,311 It employs various modifications, where (i) almost all of the nucleobases are modified with 2′-fluoropyrimidines and 2′-O-methylpurines, (ii) the 3′-thymidine contains a 3′-3-linkage to the penultimate 2′-methoxyguanosine, and (iii) the 5′-end is conjugated to two 20 kDa PEG units. Due to a reduction in renal clearance, t1/2 in blood and uptake in organs were greatly improved.311 It should be noted that for all FDA approved ASO therapeutics some mild adverse effects, like inflammation at the injection site, can occur. Also more severe toxicity can occur, through mechanisms that are only partially understood, but involve either hybridization dependent (on-target and off-target), or independent (protein binding) events.312 Mipomersen, for example, has been withdrawn from the market as hepatotoxicity occurred in 10–20% of patients. Both preventing off-target effects and gathering more insights into mechanism of toxicity will remain of importance for developing chemically modified ASOs.304,313–316
Patisiran (Onpattro) is the first commercially available siRNA drug, where 2′-OMe modified siRNA targeting TTR is encapsulated into LNPs composed of DLin-MC3-DMA, DSPC, cholesterol, DMG-PEG2k at molar ratio of 50/10/38.5/1.5, and a LNP/siRNA N/P ratio of 3, and has been approved for the treatment of hereditary TTR induced amyloidosis by intravenous infusion in USA and Europe.317 These LNPs mainly target the liver due to apolipoprotein E (ApoE) mediated binding to the low-density lipoprotein receptor (LDLR) on the surface of hepatocytes. A notable reduction in the symptoms of the disease was detected after administration every 3 weeks for 18 months, when compared with patients taking placebo. However, the induced pro-inflammatory effect by the lipids as well as other side effects, such as redness, nausea, headache and breathing difficulties, supported the subsequent development of siRNA drugs conjugated with GalNAc instead of using LNPs. Tris-GalNAc mainly targets the liver as well, due to binding with the asialoglycoprotein receptor that is highly expressed on hepatocytes. Givosiran, a hepatic δ-aminolevulinic acid synthase 1 (ALAS1)-directed siRNA conjugated with GalNAc, has been approved for the treatment of acute hepatic porphyria (AHP). Inclisiran (siRNA targeting proprotein convertase subtilsin/kexin type 9 (PCSK9) mRNA) and Lumasiran (siRNA targeting hydroxyacid oxidase 1 gene (HAO1) mRNA) are both conjugated to tris-GalNAc.318 A robust pipeline of GalNAc-conjugated therapeutics is progressing into clinic. For example, 6 siRNA candidates from Alnylam that have the same structure and chemical modifications (e.g., 2′-OMe, 2′-F, PS) but different sequence and chemical modification patterns, are undergoing clinical trials. The success of GalNAc-conjugates is highly likely attributed to several beneficial properties including, but not limited to, its ability to actively target the liver via the asiaglycoprotein receptor, its modular nature which enables its direct ligation to desired siRNAs and in vivo tolerability, while for LNP encapsulation, also the potential toxicity of the excipients should be taken into account. However, also for GalNAc-conjugates, the chemical structure and safety profile have yet to reach maturity.319
Alternatively, other delivery vehicles have been explored for siRNA delivery by for example, making use of histidine-lysine co-polymer peptide nanoparticles, biodegradable poly(lactic-co-glycolic) acid (PLGA) particles, exosomes, gold NPs, cyclodextrin NPs, pseudoviral (SV40) particles, and so on.320–324 Especially strategies to target siRNA beyond the liver are needed to expand the therapeutic horizon.
4.3 Lipid nanoparticle mediated delivery of (modified) mRNA therapeutics
mRNA can express virtually any protein of interest, as soon as the cytosol of cells is reached (Fig. 6A). Due to its single-stranded structure, mRNA is however more prone to nuclease-mediated degradation than siRNA or pDNA. Also the immune-stimulatory properties of mRNA can result in its rapid degradation, even though several modifications can limit this immune response.135 Nevertheless, the year 2020 has witnessed a landmark in mRNA-based therapeutics as a highly appealing new drug class. As can be seen in Table 3, current approved mRNA vaccines (Comirnaty and Spikevax) combine both extensive mRNA modification and additional encapsulation mRNA into LNPs, as the current state of the art non-viral delivery system to protect mRNA and help in cytosolic delivery. With over 140 mRNA candidates currently in clinical trials, it is evident that the mRNA-based nano-delivery platform is a promising tool for the development of novel therapeutics and vaccines against a wide range of diseases. It is believed that mRNA stability is the current bottleneck that determines the storage conditions and shelf-life of the mRNA vaccines.325 Therefore, especially for RNA-based therapeutics, limiting degradation will be key.Both mRNA vaccines employ N1-methylpseudouridine (m1ψ) as modified nucleotide (Fig. 6B). On the contrary, the clinical phase I study of mRNA vaccines from CureVac has reported some disappointing results that might be related to the lack of modification of mRNA and enhanced degradation.326 Both mRNA vaccines use a Cap1 or Cap1 analog inserted at the 5′ end (Fig. 6B). The BNT162b2 mRNA vaccine contains a 5′-UTR derived from the human β-globin mRNA with an optimized Kozak sequence that helps drive high levels of translation from the correct start codon, as well as a 3′-UTR consisting of two sequences derived from the amino-terminal enhancer of split mRNA (i.e., ARES and mtRNR1 3′-UTR motives) and the mitochondrial encoded 12S rRNA, which aids high levels of protein expression by stabilizing the RNA.327–329 Also the 110 poly(A) tail is split in two segmented poly(A) stretches with a nucleotide-linker (GCAUAUGACU), to improve mRNA stability and reduce unwanted recombination during plasmid production.227,329
Apart from the chemical modifications of mRNA, efforts have been made to optimize LNPs for mRNA delivery. All ionizable lipids have a pH sensitive hydrophilic headgroup, a linker region and hydrophobic tails. While the Dlin-MC3-DMA used for siRNA delivery has unsaturated and non-branched tails, the new generation of ionizable lipids used in mRNA vaccines (SM-102 and ALC-0315) have saturated, branched tails that enhance endosomal escape and contain cleavable ester linkers to increase biodegradability.330,331 Although LNPs can shield mRNA from chemical degradation by nucleases, it has been suggested that water present in the core of mRNA LNPs results in non-enzymatic mRNA hydrolysis, which is now considered as the main mechanism for mRNA degradation and the limited storage stability of mRNA LNPs.317,332
To prevent hydrolysis, the currently approved Comirnaty should be kept at −80 °C, having a large impact on the distribution and storage. Freeze drying of mRNA LNPs has been explored to enable storage at higher temperature, where both the type of cryoprotectants and the buffers used greatly impact the stability of mRNA LNPs.333 Spikevax can be stored for 9 months at −50 °C to −15 °C and should be used within 14 days once thawed and stored at 2 °C to 8 °C. Zhang et al. reported a liquid LNP-mRNA formulation that can be kept at 25 °C for at least 1 week.334 Recently, mRNA LNPs from NeoVac have demonstrated stability at 4 °C for over 1 year and at room temperature for over 1 month. Unfortunately, the authors did not mention any key factors for achieving such thermostable formulations. Given that naked mRNA can be stable at 4 °C for only a few days, the improved thermostability is likely attributed to the optimal excipients (e.g., pH, buffer, antioxidants) and lyophilization.317 Also the administration route is of significance to prevent mRNA degradation. As zinc-finger antiviral proteins (ZAP), that recognize RNA and result in their rapid degradation, are rarely present in skeletal muscle, intramuscular injection of mRNA vaccines is preferred.335–337 ln addition, the use of analytical methods to experimentally determine stability of mRNA (briefly overviewed by Poveda et al. and Schoenmaker et al.) and theoretical predictions on mRNA stability as well as newly developed algorithms for mRNA codon and sequence optimization are expected to give further insights into the key determinants of mRNA LNP stability, resulting in an increased efficacy and an enhanced shelf life.193,317,338,339
4.4 Long-term safety aspects of viral and non-viral mediated gene delivery
Given that the remarkable advances in gene therapy have been achieved within the last 20 years, the full extent of long-term clinical effects of both viral and non-viral vectors is not yet fully elucidated. Viral vectors, such as AAV, adenovirus and lentivirus, may lead to reduced efficacy or immune responses in individuals upon repeated administration, or because of preexisting antibodies to the viral vector. Gendicine, for example, as the first gene therapy approved in 2003, is found therapeutic effective, well tolerated and safe, apart from a few serious adverse effects, such as platelet crisis likely due to the immune dysfunction of gene therapy vectors, and self-limited fever due to the strong immune responses induced by the adenoviral vector.340,341Also the repeated administration of non-viral vectors can induce the generation of neutralizing antibodies and thus alter the biodistribution and prevent the therapeutic efficacy. For example, immunologic concerns related to PEG, such as severe allergic reactions and the accelerated blood clearance (ABC) phenomenon, impairs the bioavailability, passive targeting and biodistribution of injected PEGylated particles, especially when repeated administration is required.342–345 As the approved gene therapies based on non-viral vectors have only been on the market for a couple of years, time will tell the long-term effect in terms of safety and efficacy. Onpattro - siRNA LNPs, for example, were approved in 2018, and its long-term effect is under assessment. Also for the recently approved mRNA-LNPs, the long-term safety remains to be seen, even though they have been widely used and are generally considered as safe, apart from mild side effects such as headache, fatigue, and soreness at the injection site and some rare severe adverse effects like myocarditis, pericarditis and anaphylaxis, a severe type of allergic reaction, that can occur after any kind of vaccination.346,347
5. Methods and techniques to quantify nuclease-mediated degradation
As the sequence/integrity of nucleic acids is a prerequisite for their biological effects, nucleic acids should remain intact during any step of the delivery pathway. To understand where and when nucleic acids are degraded, the ability to detect nucleic acids degradation and quantify the half-life is crucial, but extremely challenging, especially in living cells. This part overviews the different methods and techniques that are available to monitor the integrity of nucleic acids, with a special focus on the possibilities and limitations of each technique as summarized in Table 4.| Method | UV-vis (NanoDrop 3300) | Gel electrophoresis | CE (Agilent 2100 Bioanalyzer) | HPLC | PCR | Fluorescence-based assay | AFM | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AGE | PAGE | FISH | FCS | FRET | MB | ||||||
| Analysis | Absorbance | Size/length | Size/length | Size/hydrophobicity | Sequence | Sequence | Size | Sequence | Imaging | ||
| DNA/RNA sequence specificity | N | N | N | Partially | Partially | Y | Y | N | Y | Y | N |
| DNA/RNA length | / | 100–25 kb | 10–3000 nt | 25–12 kb (dsDNA) | <2 kb | 60 nt-40 kb | / | <4 kb | <10 nm | / | >20 nm |
| 6–6000 nt (RNA) | |||||||||||
| Sample amount | 0.5–2 μL | 100 ng | 1–5 μL | 10 μL | 10 μL | / | 1–50 μL | 1–50 μL | 10 μL | 30 μL | |
| Detection limit | 0.05 pg μL−1 | EB: >34 ng | nl | 0.02 ng μL−1 | <10 copies | 0.2 μg | μg | / | 0.2 nmol | 100 ng | |
| SYBR Green II: >14 ng | |||||||||||
| GelRed: >0.1 ng DNA | |||||||||||
| Reagent toxicity | N | Y | N | N | N | N | Y | ||||
| Equipment cost | Low | Low | High | High | Intermediate | High | High | ||||
| Cost per assay | Low | Low | High | High | Intermediate | Low | High | ||||
| User bench skill | Low | Low | Low | High | High | High | High | ||||
| Assay time | <1 min | 10–120 min | 1 h | 1 h | 1–2 h | <5 min | >5 min | ||||
| Hands-on time | <1 min | 5–15 min | 30 min | 1 h | 15–30 min | 1 h | >1 h | ||||
| Ability to automate | Y | N | N | Y | Y | Y | N | ||||
| Living cells | N | N | N | N | N | N | Y | Y | Y | N | |
| Advantage | Ease-of-use low-cost | Ease-of-use low-cost | No cross-contamination, no extra waste removal needed, | High sensitivity, tiny amount of sample | Tiny amount of sample | High sensitivity, high reproducibility, insensitive to pH, no contamination, no sample pre-treatment | Visualization | ||||
| Disadvantage | Susceptibility to pH and impurities, large variability below 5 μg mL−1 | Unfeasible to protein-rich medium, time-consuming, possible degradation during electrophoresis, large volume/sample | Susceptibility to impurities (e.g., phenol, DNA), new chips for each run, time-consuming | Contamination of column, unsuitable to large nucleic acids | Sample pre-treatment, unsuitable for long RNA | Susceptibility to impurities (e.g., phenol, DNA), large volume/sample | Clean sample, size limit (over 20 nm), toxicity | ||||
5.1 Nucleic acid degradation in buffer, extracellular fluids or cell extracts
 | ||
| Fig. 7 Techniques and methods currently available to investigate the integrity of nucleic acids. Scheme of (A) gel electrophoresis (GE), (B) capillary gel electrophoresis (CE), (C) high-performance liquid chromatography (HPLC) and (D) (q)PCR to detect fragments of nucleic acids. (E) Scheme of atomic force microscopy (AFM) assay and representative AFM images of naked DNA exposed to DNase I for 0 min and 9 min respectively. Reproduced with permission.416 Copyright © 2003, Oxford University Press. (F) Schematic principle of fluorescence based assays, including (F1) fluorescence in situ hybridization (FISH) and representative image of fixed cells (nucleus in blue), with mRNA in transcription or 3′-5′ degradation in red, 5′-3′ degradation in green and intact mRNA in yellow. Reproduced with permission.423 Copyright © 2016, © The Author(s) 2016. Published by Oxford University Press on behalf of nucleic acids research. (F2) Molecular beacons (MB) and dual-FRET MB (F: reporter fluorophore, Q: quencher molecule, D: donor, A: acceptor). (F3) Fluorescence resonance energy transfer (FRET) in nucleic acids dual-labeled with a donor (D) and acceptor (A) fluorophore. Representative confocal image of PO-ON double labeled by rhodamine green and Cy5 delivered by liposomes, showing intact (red, arrows) or degraded (green) ONs, and corresponding intracellular FRET-FCS measurements. (G) Diffusion-based assays, including single-color fluorescence correlation spectroscopy (FCS) for single fluorophore-labeled molecules, dual-color fluorescence correlation spectroscopy (FCCS) or FRET-FCS for dual fluorophores-labeled molecules, single particle tracking (SPT) and fluorescence recovery after photobleaching (FRAP) for single fluorophore-labeled molecules. (H) Representative decision flow chart to determine which measurement techniques are compatible with a specific research question to measure nucleic acid degradation. | ||
Not only the fragment lengths, but also the agarose concentration determines the electrophoretic mobility, where higher concentrations are more suitable to separate smaller molecules, and vice versa. Different conformations of pDNA move through the gel at different rates, where the compact, supercoiled form of pDNA moves faster, followed by the linear and open circular pDNA conformation. As demonstrated by Remaut et al., DNase I treatment of supercoiled pDNA resulted in single-stranded breaks and the formation of relaxed open circular pDNA, while DNase II resulted in a double-strand break and the direct conversion of supercoiled pDNA into linear pDNA.354 In general, 0.8–1% (w/v) agarose gels are used for DNA and mRNA, while up to 4% agarose is used to separate short nucleic acids like siRNA and ONs.255,355,356 Traditional agarose gel electrophoresis (AGE) is most effective to separate DNA fragments ranging from 100 bp to 25 kb. For the separation of DNA fragments >25 kb, pulse field gel electrophoresis is an alternative, where the larger DNA fragments are separated by the speed at which they reorient themselves with the changes in current direction.357
When short nucleic acids fragments need to be visualized with higher accuracy, polyacrylamide gel electrophoresis (PAGE) is often used. The pore size of a PAGE gel is controlled by modulating the concentrations of acrylamide, and is suitable to separate nucleic acids in the range of 10–3000 nt, or <500 nt ssDNA or ssRNA for denaturing PAGE.358 Depending on the gel (between 8–12%), even DNA molecules differing in size by only a single base pair can be resolved. It should be noted however, that the moderately hazardous polyacrylamide powders and gels require protection during handling.
Although gel electrophoresis is easy to perform, there are also some limitations to the technique. (i) This method is impractical for routine or high throughput DNA or RNA quantitation as it takes about 30 min to 1 h and is cumbersome for measuring degradation in function of time, as each time point has to be added as a separate lane on the gel. (ii) Degradation may proceed during sample preparation thus leading to overestimation of the enzymatic cleavage. (iii) Gel electrophoresis will detect all nucleic acids in a sample, including contaminating sequences in extracellular environment or cell extracts and is therefore mostly used to study nucleic acids in buffer and reduced protein-containing medium (e.g., Opti-MEM).107,126,359,360 As the relative quantification, by comparing band intensity values of the intact nucleic acids with those obtained in the degradation smear, is unreliable in more complex samples, such as human serum or cell extracts. (iv) The linear range of detection is rather limited (EtBr (>34 ng), SYBR Green II (>14 ng) and GelRed (>0.1 ng)), and thus the initial degradation might be underestimated.361 This especially holds true when exonuclease activity results in hydrolysis of small fragments from a large RNA or DNA sequence, resulting in degradation products that are below the size detection limit one the one hand, and still very close to that of the parent RNA or DNA on the other hand. Also single-stranded breaks that do not lead to a change in fragment size or conformation will remain unnoticed.
000 nt in polyacrylamide (PA) solutions with varying molecular weights and concentrations and found that increasing the PA concentration resulted in a better resolution of small fragments (<1000 nt) whereas large fragments (>3000 nt) could no longer be resolved.364 CSE with low-viscous sieving media of hydrophilic polymers polyvinylpyrrolidone (PVP, 1.3 MDa) has been shown to detect the purity and stability of mRNA (100–1000 nt).365 However, the application of CE to measure mRNA integrity is limited, as mRNAs are large in size/length and they natively form various secondary structures causing size heterogeneity and peak broadening. To improve the resolution of large RNAs, CSE has been developed to be ran under denaturing conditions to disrupt all secondary structures of RNA, and thus yield size-based separation. For example, Lu et al. separated large RNA molecules (2000 nt) by size and length using CSE under strongly denaturing and non-aqueous conditions.366 Using CSE with a sieving matrix containing acetic acid as denaturant, Sumitomo et al. reported that total RNA (10–10000 nt) extracted from NIH 3T3 cells was simultaneously denatured and separated in a small sample volume (<10 nL) without in vitro sample preparation.367 To further automate the process and increase throughput, several highly efficient and automated instruments for CE are available, such as Fragment Analyzer (Advanced Analytical Technologies), QIAxcel Advanced System (Qiagen) and Agilent 4200 TapeStation system (Agilent Technologies). Integration of CE into a microchip remarkably advances the development of miniaturized, rapid, automated, and integrated analytical systems with potential to be more cost-effective, faster and more sensitive. Examples are the Agilent 2100 Bioanalyzer (Agilent Technologies) and Experion (Bio-Rad Laboratories). For most of these instruments, commercial kits are available for optimized determination of DNA and RNA quality and quantity. Recently, Raffaele et al. developed an assay to evaluate the purity and integrity of mRNA encapsulated in LNPs using a commercial microchip (LabChip GXII Touch) with a short run time (∼70 s) that supports high-throughput formulation development.368
In addition to the capillary, the detector plays a key role in the sample size determination. Compared with UV, a laser induced fluorescence (LIF) detector confers high sensitivity and more selectivity but requires fluorescent nucleic acid molecules, which can be obtained by labeling them with dyes (e.g., EtBr, SYBR Green) before or during the electrophoresis (by incorporating the dyes in the matrix).369 Yang and Chang investigated the RNA integrity by CE equipped with cyan light-emitted diode-induced fluorescence. The capillary was coated with 0.1% poly(vinylpyrrolidone) (Mw of 1300 kDa) and filled with 0.4% poly(ethylene) oxide (Mw of 4000 kDa) and SYTO9 (fluorescent dye), and provided a cost-effective, rapid and sensitive method to detect RNA degradation induced by RNases or temperature.370 When integrated with MS/MS, CE was used to separate the degraded nucleosides of mRNA extracted from HeLa cells.371 A rapid, high-throughput fluorescence capillary gel electrophoresis method was reported to study DNA degradation mediated by varying metabolic enzymes.372 However, ssDNA has found to be degraded in Tris buffer during the CE measurements, indicating the importance of the buffer for CE.373 To improve the sensitivity of the detection mode, Qin et al. developed a microchip method with laser induced fluorescence detection (MCE-LIF) to study the nuclease activity. FAM-labeled ssDNA was degraded into 5-FAM-nucleoside monophosphates with greatly increased fluorescence intensity, and short non-labeled oligonucleotide fragments. After separation of the different fragments with CE, the fluorescence intensity of 5-FAM-nucleoside monophosphates was used to quantify the nuclease activity and the effect of nuclease inhibitors.374
CE is highly efficient, versatile, reproducible, ease-to-use and requires low sample and buffer volume to quantify and analyze the length distribution of nucleic acids. The combination of CE with advanced detection techniques like mass spectroscopy, is becoming an indispensable analytical tool to separate and quantify nucleic acids. Microchip CE has several advantages, including rapid analysis (approximately 10 times faster), less reagents and sample consumption, higher throughputs, faster and cost-effective. However, there are some drawbacks of microchip CE compared to conventional CE, including a lower peak capacity because of the shorter separation channels and a lack of compatibility with versatile UV detectors because microchip fabrication materials are typically not transparent to UV light.375–377
In reverse-phase high-performance liquid chromatography (RP-HPLC), molecules in the mobile phase are separated based on their polarity. As a hydrophobic stationary phase is used, polar (hydrophilic) molecules elute first while non-polar (hydrophobic) molecules are being retained in the column for longer times. As the degree of hydrophobicity of the bases is A > G > C ≈ T/U, nucleic acids of equal length but with a higher percentage of A and G will elute later.381 Ullio-Gamboa et al. reported isocratic RP-HPLC as a sensitive and precise tool to quantify PS-ONs containing unmethylated CpG motifs and found that the degradation by 3′-exonuclease I followed a pseudo first order kinetic with a half-life of 1.27 h, while upon encapsulation in ascorbyl palmitate coagel the half-life doubled.382 However, the resolution of RP-HPLC decreases with increasing length of ONs. To this end, denaturing RP-HPLC has been developed by heating the column, using high pH buffer or introducing organic additives. Urea-induced denaturing RP-HPLC resolved the two strands of 20 mer dsON.383
Alternatively, ion-pair reverse-phase-high-performance liquid chromatography (IP-RP-HPLC) has been reported to increase the resolution of separation, by adding ion-pair agents in the mobile phase (e.g., triethylammonium acetate (TEAA), triethylamine/hexafluoro-isopropanol (TEA/HFIP)). Nucleic acid fragments associate with the pairing ion with an opposite charge, which reduces charge-interactions with the column and increases differences in hydrophobicity.384,385 IP-RP-HPLC can separate nucleic acids ranging from a few nucleotides to several thousands of base pairs. Longer nucleic acids, like RNA, are typically analyzed under denaturing conditions, where shorter (degraded) fragments were eluted prior to the full-length RNA.386,387 Nwokeoji et al. provided a high resolution RNase mapping and fingerprint of ssRNA and dsRNA using IP-RP-HPLC.388 The resolution of IP-RP-HPLC can be fine-tuned by the pore sizes of the silica particles in the stationary phase. With a C18 column containing 2.6 μm solid-core particles with 80 Å pore sizes, a good separation of ssRNA digests by RNase A was achieved.389 IP-RP-HPLC was employed to separate dsDNA (17–51 bp) with a single base resolution, demonstrating its potential to study DNA degradation products.390 As well, IP-RP-HPLC has been reported to accurately and rapidly separate dsDNA fragments ranging from 50–2000 bp with highly reproducible retention time, where the separation of DNA fragments was length-dependent and sequence-independent.391
HPLC can be combined with numerous detection techniques, such as wavelength ratio analysis and IR-RP-HPLC-electrospray ionization mass spectrometry (ESI/MS). A general LC-MS-based RNA sequencing method has been developed to directly readout the complete sequence at single-base resolution for short synthetic RNA (<35 nt), indicating its potential to study RNA degradation.392 Also, HPLC-MS/MS was employed to separate and detect RNase U2-digested RNA products.393 Notably, HPLC is able to follow the secondary structure changes of nucleic acids, which, in turn is predictable to the nucleic acid integrity. Qiao et al. showed that the retention behavior of ONs and dsDNA in IR-RP-HPLC depends on their sequence and size. ONs that are more prone to self-dimerization have weaker retention, and the retention of dsDNA was influenced by the interactions of exposed bases in major or minor grooves with the hydrophobic alkyl chains of the stationary phase.394 Likewise, Betker et al. determined pDNA in each isoform (supercoiled, open-circular and linear) by using ultra-performance liquid chromatography (UPLC).395 LeRouge et al. developed an IR-RP-UPLC assay and investigated the structure difference of DNA- and RNA-ONs of equal length and sequence, which either formed a 4 base-pair or 2 base-pair tetraloop secondary structure.381 In addition, utilization of UPLC has decreased analysis time and thus increased the throughput for analysis.
Other techniques, such as ion-exchange chromatography (IEC) and size-exclusion chromatography (SEC), are excellent approaches for separations of nucleic acids. By using porous stationary phases that sieve the nucleic acids based on their size or hydrodynamic radius, SEC has been explored to separate nucleic acids based on their molecular weight and secondary structure.396 By using a Picogreen-based fluorescence-detection SEC tandem with AGE, Tan et al. followed DNA degradation in cell culture supernatant, although it was not an absolutely quantitative assay.397
When compared to gel electrophoresis, HPLC enables to automatically load and separate all samples without operator intervention, and precisely control the separation conditions, like flow rate, buffer concentration and temperature. Moreover, a pre-packed cartridge is suitable for more than 4000 samples while gel electrophoresis is only used for one run. Furthermore, the samples separated and detected by HPLC can be retrieved for subsequent applications whereas the samples retrieved from gel are contaminated and require additional purification with risk of limited recovery. Also the ability to provide the structural conformation of nucleic acids, is a significant advantage over the other above mentioned techniques. However, non-purified nucleic acids in biological samples may interfere with the analysis and contaminate the column. Furthermore, HPLC is mostly used in the analysis of small nucleic acids, and it is still challenging to follow the decay kinetics of large nucleic acid molecules (e.g., mRNA), owing to the mixture of degradation fragments with comparable length that can be formed. In contrast, HPLC-based techniques (e.g., RP-HPLC, IR-RP-HPLC, SEC) are efficient and commonly used approaches to purify mRNA by separating it from contaminating short dsRNA fragments, thus reducing the innate immune stimulating of mRNA.135
To detect a target RNA, RNA molecules should first be converted into their cDNA by reverse transcriptase, followed by cDNA amplification by standard PCR procedures. The cDNA synthesis primers, including either an oligo(dT) primer, a random primer, a combination of both, or a sequence specific primer, are of importance in priming cDNA synthesis.401 In particular, oligo(dT) primers anneal to the poly(A) tail of most mRNAs and could generate full length cDNA. Random primers (6–10 nt) anneal to all RNA (e.g., rRNA, tRNA, mRNA, IncRNA) and provide high cDNA yield even for little starting material, while sequence specific primers are designed to target specific transcript, yielding high sensitivity and specificity.402 Again, if the target is degraded, this will affect the cDNA that is being transcribed from it, as only intact RNA sequences will lead to the full complementary target cDNA sequence. To assess the fragmentation status of a polyadenylated transcript, a 3′:5′ integrity assay has been developed. Here, reverse transcription of RNA to cDNA is started from the 3′ poly(A) tail of the transcript using an oligo(dT) primer. In case of degradation, this results in shorter cDNA fragments that contain the complementary sequence to the 3′ end, while only cDNA coming from full-length RNA contains the complementary sequence to the 5′ end. By quantifying the 5′ end and 3′ end by RT-qPCR, the 3′:5′ ratio can be used to estimate mRNA integrity.403 Vermeulen et al. demonstrated the ability of this assay in combination with microfluidic electrophoresis to assess the integrity of RNA in tumor tissues.404 As well, Nolan et al. utilized the 3′:5′ assay to estimate mRNA integrity, with three target amplicons that were spatially separated with one towards the 5′ end, the second towards the center and the third towards the 3′ end of the mRNA sequence.405 Hence, the ratio of amplicons reflects the relative success of the oligo-dT primed RT to proceed along the entire length of the transcript. Cheyne et al. further optimized the 3′:5′ integrity assay to qualify mRNA with digital PCR.406 Interestingly, Björkman et al. provided a novel approach to quantify the RNA integrity by measuring the differential amplification (ΔAmp) of an RNase resistant endogenous marker relative to a reference gene (i.e., 18S RNA). This assay was able to detect 99.7% degradation and to precisely follow the decay kinetics of mRNA by RNase A, heating or UV irritation.407 In addition, standards of known concentration can be assayed to quantify mRNA in each sample. If suitable primers can be designed, then the target RNA can be directly quantified. Primers that generate amplicons of various lengths can be used to determine RNA integrity since larger amplicons become consistently more difficult to amplify as RNA degrades.338
Quantitative PCR (qPCR) or reverse transcriptase qPCR (RT-qPCR) have been considered as the ‘gold’ standard due to its high analytical sensitivity, high precision, and the fact that it allows a fast, high-throughput readout.408 However, there are also some limitations associated with qPCR for nucleic acid integrity measurements. It not only requires training, capital equipment and reagents and consumables, but also requires running multiple assays to evaluate different (c)DNA lengths within a sample, which leads to an iterative process or a need for multiple calibration curves. Hence the assays are often designed in a multiplexed format, but multiplex qPCR can be limited by poor compatibility of the various assays in the same reaction. Lastly, this approach can’t distinguish the different types of degradation. Therefore, the feasibility of qPCR to quantify the absolute concentration of amplifiable (c)DNA and the relative distribution of fragment lengths is limited by its imprecision. In contrast to qPCR, digital PCR enables absolute quantification with high precision of low amounts of amplifiable DNA, whereby amplicon lengths can be measured simultaneously without requiring calibration. Didelot et al., for example, utilized digital droplet PCR to detect the quality of DNA in clinical samples, including the length distribution of DNA fragments and the amount of PCR amplifiable DNA. Control samples containing artificially fragmented human genomic DNA (78 bp, 159 bp, 197 bp, 550 bp) were prepared to simulate a wide range of possible fragmentation sizes. As demonstrated, the quantification of formalin-fixed, paraffin-embedded samples of lung adenocarcinoma tumor tissues by this assay correlated well with the sequencing results for a subset of samples for DNA-quality assessment. This assay is simple, and no calibration is required.409
Obviously, PCR is highly sensitive to assess the integrity of nucleic acids. However, it is challenging to quantify the integrity of short nucleic acids (e.g., siRNA, aptamer, ASOs) as the primer has a comparable length to the template (e.g., 20 nt), thus leaving no remaining sequence to amplify. Special tricks are needed to quantify such small oligonucleotides, e.g. stem-loop RT primer for siRNA or splinted ligation qPCR for ASO.410,411 Of note, it is challenging or impossible to quantify or discriminate the shorter metabolized/degraded oligonucleotides. For the large nucleic acids (e.g., pDNA, mRNA), on the other hand, the distance between both primers can be very long if the full-length nucleic acid must be amplified. With mRNA, this is even more complicated as the whole sequence should first be converted to cDNA. Hence, multiple primer pairs can be designed to amplify shorter DNA or mRNA regions, for which only the equal amplification of all target regions reflects the presence of intact nucleic acids. Degradation outside the chosen target regions will however remain unnoticed. Moreover, quantification of RNA in biological samples can be complicated since even a trace amount of contaminating DNA can also be amplified and quantified. Hence, the choice of specific primer pairs is crucial to only amplify the target region of interest (e.g., by spanning a huge intron for a gene that has no retropseudogenes), or to verify that DNase treatment has effectively removed the DNA. Also, PCR is applicable to detect the degradation of nucleic acid in situ but not in living cells, as the target sequences first need to be isolated from living cells by the preparation of cell extracts. Up to 90% of transcripts can be lost during RNA purification, cDNA synthesis and other steps required for PCR, which limits its ability to provide important spatial-temporal information.412,413
:1) to 142 ± 37 min (1:1), depending on the G4/DNA ratio.415 Ten years later, the same group investigated the attack of RNase V1 on naked versus complexed siRNA. The RNase V1 enzyme was visualized after 1.5 min of adding the enzyme solution to free siRNA, which caused complete siRNA degradation within 3 min. With a flexible dendrimer (G4) at ratio of 2:1, the siRNA cargo remained intact for 28 min. The RNase resistance was further improved by prolonging the complex incubation time, most likely due to the formation of tightly packed and more stable complexes.416 While AFM has shown possibilities to visualize the decay of DNA/RNA or their complexes, there are some considerations including (i) the standard AFM tips with a radius of ∼10 nm set the detection limit of DNA fragment sizes. With an ultra-sharp tip, the grooves (2–3 nm) of DNA molecules can be observed in the high resolution images, although the detectable DNA fragment length remains limited to about 20 nm or even 50 nm (∼150 bp); (ii) clean samples with a minimum amount of interfering biological components (e.g., cellular proteins) are required to reliably identify short DNA fragments and obtain sufficient quality images, thus limiting its application in biological context; (iii) a high radiation dose (around kGy) is preferred to generate enough short DNA fragments to permit sampling in a reasonable number of AFM images, raising the obvious question of biological relevance, where typical therapeutic radiation doses are only a few Gy, and iv) AFM is used to visualize the radiation-induced DNA fragments, and rarely applicable to detect enzymatic DNA degradation in biological context, like in living cells. Therefore, AFM is presently useful mostly for the measurements of individual short DNA fragments ranging from tens to hundred nm, without any interference of biological components.417–420
5.2 In situ nucleic acid degradation in fixed and living cells
In this section, we overview the different fluorescence microscopy-based techniques that allow to track exogenous nucleic acids in fixed or living cells, based on fluorescent dyes that preferentially bind to the target nucleic acid, or by using fluorescently labeled nucleic acids.Fluorescent in situ hybridization (FISH). Fluorescent in situ hybridization (FISH) has been used to detect and localize specific DNA and RNA sequences in fixed cells and tissues, by employing fluorescent probes that bind to their target nucleic acids based on complementarity (Fig. 7F1).421 A large number of FISH probes are commercially available. Lechardeur et al. used FISH to assess the decay kinetics of pDNA in the cytosol of cells, with a half-life of around 90 min.124 Using the same approach, Pollard et al. studied the stability of naked pDNA in the cytosol of COS-7 cells and found that pDNA was degraded rapidly in the cytosol and only 50% of the microinjected pDNA remained intact within 2 h. When incubated with cell extracts, pDNA was almost completely degraded in 30 min.123 Seferos et al. prepared molecular probes consisting of a DNA sequence with a 3′ dabcyl quencher, which allowed to hybridize with fluorescein-labeled DNA complements. By plotting the fluorescence of the enzyme-catalyzed reaction as a function of time, the half-life of naked DNA and conjugated DNA in presence of DNase I was estimated as 23 ± 4 min and 100 ± 16 min respectively.422 Kramer exploited the metabolism of double-labeled mRNA (5′-green, 3′-red) in trypanosomes by using Affymetrix® single mRNA FISH probes, where yellow, red and green spots represent intact, 3′ to 5′direction decay and 5′ to 3′ direction decay, respectively.423 In order to obtain the detailed spatial and temporal information on RNA dynamics in single living cells, several fluorescent probes have been developed that recognize RNA targets with high specificity, sensitivity and signal to background ratio, as reviewed.412,424
FISH is mainly useful for the detection of nucleic acids in fixed samples and consequently provides static rather than dynamic views on nucleic acid quality and localization. Moreover, it requires denaturation, hybridization with a probe and washing before quantification by fluorescence microscopy. Thus the possible effects of the fixation agents and other supporting chemicals that are used for specimen preparation on the nucleic acid distribution or denaturation can’t be ruled out.425 As FISH is probe- and sample-specific for each type of nucleic acid, it requires optimizations on a case-by-case basis and thus is time-consuming and labor-intensive. Also, some of the relatively short FISH probes may non-specifically bind to endogenous nucleic acids and give false positive results. Of note, as most FISH probes are shorter than the whole length of the target nucleic acids, larger degradation fragments that still contain the target sequence cannot be distinguished from fully intact sequences. Therefore, in analogy with PCR, multiple FISH probes should be designed to identify consequent regions in the intact target DNA or RNA. In addition, detecting specific nucleic acids in low levels, without any interfering fluorescent background is challenging.426
Fluorescence resonance energy transfer (FRET). Fluorescence resonance energy transfer (FRET) is one of the most commonly used techniques to study molecular interactions in living cells on the nanoscale. In FRET, a donor molecule that is excited by a laser, transfers its absorbed energy to a nearby acceptor molecule, leading to a decrease in donor fluorescence and an increase in acceptor fluorescence (Fig. 7F3). As the energy transfer requires close proximity between donor and acceptor, any process that increases this distance will result in a decreased FRET efficiency. FRET has been mainly applied to study the degradation of short nucleic acids, such as siRNA and ONs (∼10 nm length).427 For example, in an intact ssON (10 mer) labeled by fluorescein (as donor, on the 3′ end) and rhodamine X (as acceptor, on the 5′ end) FRET occurs, giving rise to a high acceptor and low donor fluorescent signal. When ONs degraded, FRET was no longer possible and the fluorescence intensity of the donor increased whereas the fluorescence acceptor signal completely disappeared.428 Likewise, the degradation of ss- and ds-siRNA was followed in RNase A, cell extracts and living cells using FRET.121 Using acceptor photobleaching, Remaut et al. demonstrated that intact PO-ONs could be delivered by poly-β-amino esters.262 Also fluorescence correlation spectroscopy (FCS) can be used to follow FRET.
Although it has been demonstrated that FRET can be used to track nucleic acids in living cells, several studies have shown that measuring FRET can be quite problematic and requires specific expertise. In addition, various factors may impair the accuracy of FRET measurements, like cell movement.
Molecular beacons (MBs). A special form of energy transfer occurs in molecular beacons (MBs). MBs are ODNs with a stem-loop hairpin structure, where the donor molecule (usually at the 5′ end) internally quenches the fluorescence from the acceptor at the opposite end. When hybridized to its target site, the MB is stretched and quenching no longer occurs, resulting in detectable acceptor signal (Fig. 7F2).429 Combined with advanced imaging techniques, MBs are promising probes to track RNA in living cells, as well as RNA degradation. It should be noted in living cells, MBs, such as naturally occurring DNA (DNA MBs) and 2′-O-methyl RNA (2-OMe MBs), are susceptible to nuclease degradation themselves, what can lead to false negatives, or prone to nonspecific protein binding inside cells, leading to the generation of false positives. To improve the nuclease stability of MBs in the cytosol, several modifications have been studied, like 2-OMe, PS, PNA and LNA.429–431
To ensure that the MB hybridizes with the expected target, shared stem MBs (one 3′ shared stem and the other 5′ shared stem) are designed to eliminate false positives.431 As well, dual-fluorescence resonance energy transfer MB (dual-FRET MB) were designed, where two distinct MBs, one labeled with a donor fluorophore at the 3′ end and the other labeled with an acceptor fluorophore at the 5′ end, are used to target adjacent regions on the same target RNA. As the FRET signal can only be generated when the two MBs are bound to the same target, this strategy can avoid nonspecific signals emitted by single MBs. Other strategies, such as MBs linked to nanoparticles to minimizing their entry into the nucleus, have been used to reduce the false-positive signals of MBs.432 Despite the great progress made in the MB technology, several challenges still remain to be overcome, such as background fluorescence, off-target binding and rapid accumulation of the MBs in the nucleus of cells.433 Moreover, the hybridization-based methods are incompatible with the detection and tracking of DNA sequences in living cells, since denaturation of the dsDNA molecule is required as MBs only bind to single-stranded target sequences.425
Other strategies. Several strategies based on fluorescence difference have been developed to track the intracellular fate of exogenous nucleic acids. For example, a dual fluorescent labeled DNA (rhodamine and fluorescein) was used as a probe to follow intracellular DNA degradation. A single color (yellow) was found on the intact DNA and DNA with a single cut due to the close proximity of the fluorophores. When the DNA is cut into two fragments, the red and green colors are separately visible. Using this system, they tracked intracellular DNA degradation in CHO-K1 cells and found degradation in 41% of cells following 3 hours’ incubation. Longer degradation studies were limited by the photostability of the organic fluorophores.434 As another example, Reif et al. reported a method to monitor RNA degradation in real time in living cells using fluorogenic RNA in combination with RNA nanotechnology. Malachite green (MG), which is not fluorescent by itself, is capable of binding to its aptamer and emitting fluorescent light only if the RNA remains folded in the correct conformation. By fusing RNA aptamer that binds MG into RNA nanoparticles, the degradation and structure changes of RNA nanoparticles were assessed based on the MG aptamer fluorescence (in the presence of MG dye) in living cell. The half-life of electroporated RNA nanoparticle was 4.3 h in human nasopharyngeal carcinoma KB cells.435
Gong et al. presented a DNase-activatable fluorescence imaging probe (DFProbe) consisting of a dsDNA, stained with SYBR green that is also labeled with a fluorophore (ROX or Cy3) on one end, and a quencher on the other end. In intact DNA, only the green fluorescence from SYBR Green was observed. Upon degradation, SYBR Green was removed from the DFProbes resulting in decrease of green fluorescent signal, while the fluorophore was separated from the quencher, giving rise to a red fluorescence signal. This was proved to be an elegant technique to visualize the degradation of exogenous DNA in cells in real time, where enzymatic degradation of the DFProbe began 3 h post-transfection and increased over time.115
Fluorescence correlation spectroscopy (FCS). Fluorescence correlation spectroscopy (FCS) is a microscopy-based method that allows to determine the concentration and diffusion coefficient of fluorescently labeled molecules, by correlating fluorescent signals that arise due to movement of these molecules in and out of the detection volume of the FCS instrument (Fig. 7G). As previously reported, FCS is a non-invasive technique that is applicable to study DNA and/or RNA behavior in both biological contexts and living cells.436–438 By using single-color FCS, degradation of nucleic acids can be followed by an increase in diffusion coefficient. We studied the decay kinetics of pDNA and mRNA, and confirmed that lipid-based vectors protect pDNA and mRNA from nuclease-mediated degradation in undiluted biological fluids.107 However, it can only distinguish degradation products with a substantial difference (preferably factor 8) in molecular weight. Using fluorescence cross-correlation spectroscopy (FCCS), dual fluorescently labeled molecules can be studied. Kettling et al. studied the enzyme kinetics of the restriction endonuclease EcoRI in solution by using FCCS, and dual-labeled dsDNA (66 bp) with rhodamine green on one end, and Cy5 on the other end (Fig. 7G).439 Using a similar dual-labeled DNA, Sasaki and Kinjo followed enzymatic activity in both nuclease solution and living cells by FCS and FCCS.440
By combining FCS with FRET (FRET-FCS), our group studied the integrity of dual color-labeled ONs (40 mer, 3′-rodamine green, 5′-Cy5) in solution and living cells. In intact ONs, a high red to green fluorescence ratio (i.e., acceptor/donor ratio or R/G ratio) was observed upon donor excitation, whereas a gradual decrease in R/G ratio was observed upon degradation of the ONs. Using FRET-FCS the R/G ratio could be easily followed in buffer and living cells (Fig. 7F3). Also the protection of ONs by encapsulation into non-viral vectors, as well as the intracellular degradation of PO- and PS-ONs were followed and confirmed that PO-ONs were mainly degraded inside the lysosomal compartment.117,441 Interestingly, Heissig et al. studied the stability of dual-labeled RNA ONs (3′-Atto488, 5′-tetramethylrhodamine) in cellular extracts and in cultured cells by FCS, FCCS and FRET. As they found, FCCS was able to measure the integrity of RNA with high sensitivity, while FCS provided additional information on the interactions of ONs with cellular components but with decreased sensitivity. Further, they demonstrated the feasibility to monitor the cleavage of RNAs based on donor's lifetime (lifetime increased when RNA was cleaved) by fluorescence lifetime imaging microscopy, which provided not only the integrity but also the intracellular behavior of RNAs after transfection (e.g. endosomal release, liberation from complexes and localization).442
One limitation of FCS lies in the specialized instrumentation and the need for fluorescent labeling of the nucleic acids under investigation. Furthermore, FCS is generally less suitable for large molecules, due to the small size of the detection volume (about 1.9 μm in height and a half width of 0.2 μm), although it has been used to study large DNA molecules (up to 4 kbp) with dual-end labeling.443 Also, FCS and FCCS are limited to one-point measurements and different regions of interest in cells cannot be simultaneously compared. Since nucleic acid degradation progresses on the minute time scale, quantitative methods to visualize transportation and nuclease degradation of nucleic acids in living cells are required. Thus, raster image correlation spectroscopy (RICS) and raster image cross-correlation spectroscopy (ccRICS), which are image-based correlation methods, have been developed to monitor the process of nucleic acid degradation. With dual color ccRICS, Sasaki et al. studied the exogenous DNA degradation after microinjection into the cytosol of living cells. As they found, the injected DNAs were degraded within 10 min in the cytosol of MEF cells, and within 1 h in HEK293 cells, demonstrating dramatically different nuclease activity in those two cell lines.444
Single particle tracking (SPT) and fluorescence recovery after photobleaching (FRAP). Other fluorescence-based techniques, like single particle tracking (SPT) and fluorescence recovery after photobleaching (FRAP), may also be an alternative to assess the integrity of nucleic acids by diffusion analysis. SPT is a fast and sensitive technique to measure the mobility of individual fluorescent molecules.445 Briefly, the motion trajectories of fluorescently labeled molecules can be determined, from which the diffusion coefficient and concentration can be calculated. Due to the limitations in acquisition frame rates and detector sensitivity, we demonstrated that SPT is most easily applied to larger nucleic acids (e.g., pDNA, mRNA) which move more slowly and which can be labeled with multiple fluorophores, while detection of smaller degradation fragments is less reliable. Nevertheless, by using SPT, we found that Cy5 labeled pDNA (5.7 kb) degrades in human ascites, with a half-life of 0.9 h (unpublished data). FRAP is a microscopy-based technique to measure diffusion by photobleaching labeled molecules in a localized spot of a sample, after which the fluorescence recovers due to the diffusion of surrounding unbleached fluorescent molecules to the photobleached area. The diffusion coefficient is calculated based on the recovery rate and can be used to measure changes in the size of nucleic acids. Using FRAP, Lukacs et al. measured the translational diffusion of FITC-labeled dsDNA ranging from 21 bp to 6000 bp in the cytosol and nucleus of HeLa cells.446 Politz et al. showed that the diffusion coefficient of FITC-labeled ONs measured by FCS and FRAP was comparable, which was in agreement with what we found.447 Unlike FCS, FRAP is more suitable to detect highly concentrated solutions (>100 nM) and molecules over a wide size range.448,449 Compared to SPT, FRAP is better suited to detect intact DNA (5.7 kb) and mRNA (996 nt) in the presence of their degradation fragments.
Overall, fluorescent nucleic acids along with the detection techniques have been proved as a powerful tool to spatially and temporally follow the nucleic acids integrity. These fluorescence-based techniques have the great and unique benefit of real-time analysis in various media, including buffer solution, (undiluted) biological fluids and living cells. It is worth noting, however, that introducing (bulky) fluorophores comes with the risk of altering the distribution, interaction and/or dynamics of exogenous nucleic acids in cells and tissues. Therefore, the effect of the labeling strategy (e.g., type of dye, labeling density) on the investigated biological process should always be verified.
5.3 Theoretical prediction of nucleic acid degradation
Apart from experimentally measuring nucleic acid degradation, advances in deep learning allows to develop mathematical models by which the degradation of nucleic acids could be predicted. To date, a few models have been reported to predict the (non-enzymatic) degradation of mRNA, which benefited from the rapid development of mRNA vaccines. For example, Muneer et al. reported an artificial intelligence (AI)-based algorithm to predict degradation at each base of an RNA molecule, with a high speed and efficiency.450 Kaggle models were able to predict mRNA degradation with an excellent accuracy.451 Yaish and Orenstein used deep neural networks to predict mRNA degradation dynamics and identified known and novel cis-regulatory sequence elements of mRNA degradation.452 As well, DNA degradation in water was simulated by quantile models.453 The performance of such models might be restricted in predicting RNA/DNA degradation patterns due to the amount of data available for training and the limited accuracy of structure prediction. A better understanding of nucleic acids degradation and more training of the theoretical models based on experimental data would thus benefit the model development. It is up to date unclear how modeling could predict the protection of nucleic acids when encapsulated in viral or non-viral vectors, and how the interaction with the different extracellular and intracellular nucleases could be incorporated in the models.5.4 Practical guidelines for choosing a method to follow nucleic acid degradation
While the paragraphs above discuss in detail the pros and cons of each measurement technique, here we provide a guideline to estimate which methods are best compatible with a specific research question (Fig. 7H). First, buffer measurements allow to follow the degradation of nucleic acids without interference of contaminating nucleic acids sequences, which will be present in biological fluids, cell extracts or living cells. The available techniques then depend on whether sequence specific information on degradation is needed or not, and whether the nucleic acids themselves are fluorescently labeled (either directly, or after hybridization of fluorescent probes) or not. In biofluids and cell extracts, the possibilities to measure degradation of non-fluorescent nucleic acids are limited, due to the presence of contaminating nucleic acids. Therefore, the nucleic acid of interest is often fluorescently labeled, or specifically detected using PCR. In living cells, on the other hand, most techniques are based on detecting fluorescently labeled nucleic acids, where FCS, SPT and FRAP have the benefit to allow to follow degradation in real-time, while FISH often requires adding hybridization probes to fixed and permeabilized cells. Also the size of the nucleic acids to be detected, and the availability and user experience can steer the choice for one or another measurement technique. It should be noted that all techniques mentioned are mainly applicable to free nucleic acids. To predict the integrity of nucleic acids that are encapsulated into viral or non-viral vectors, a prior dissociation step is thus needed. The upcoming field of theoretical modelling could overcome these experimental limitations, although its suitability remains to be seen.6. General conclusions and future perspectives
Gene therapies for both inherent and acquired diseases are on the horizon and given the significant growing number of gene therapy clinical trials as well as the accelerated acceptance of mRNA vaccines, many more cell and gene therapy approvals are expected in the near future. However, the journey from concept to clinical application of gene therapeutics has been long and tortuous, involving low transgene efficiency, safety risks, manufacturing challenges (e.g., safety, quality, potency of final products, manufacturing process), market access and so on. In recent years, considerable achievements in chemical modifications of nucleic acids, development of novel delivery systems and searching new genetic candidates have been made. The fundamental research of nucleic acids degradation in extracellular and intracellular context, however, is lagging behind. The difficulties in mRNA vaccines’ stability and storage have confronted us with the need to better understand where, when and how nucleic acids are degraded. In the near future, advances in artificial intelligence and deep learning are expected to result in powerful mathematical models that can predict stability of nucleic acids in function of their sequence, used modifications, delivery methods and nucleases they are expected to encounter. Also, experimental research into nucleases that may attribute to exogenous nucleic acids degradation, as well as techniques to quantify the integrity of nucleic acids after administration, will continue to generate new fundamental insights. In conclusions, nucleic acid degradation has long been the elephant in the room for gene therapeutics, but we are entering an exciting new era where continued research efforts on nucleic acid stability will further improve the delivery efficiency in vivo.Author contributions
H. Z.: writing original draft, review & editing. J. V., K. B., S. D. S: writing, review & editing. K. R.: supervision, writing, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. Z. greatly acknowledges Sizhe Li for technical assistance. K. R. acknowledges financial support of the Research Foundation – Flanders (G0H7520N) and the China Scholarship Council (CSC) (201507060022).References
- K. T. G. Samarasinghe and C. M. Crews, Cell Chem. Biol., 2021, 28, 934–951 CrossRef PubMed.
- L. Naldini, Nature, 2015, 526, 351–360 CrossRef PubMed.
- R. Chen and S. K. Wang, Nat. Biotechnol., 2023, 41, 262–272, DOI:10.1038/s41587-022-01393-0.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef PubMed.
- D. Lechardeur, A. S. Verkman and G. L. Lukacs, Adv. Drug Delivery Rev., 2005, 57, 755–767 CrossRef PubMed.
- P. A. Keyel, Dev. Biol., 2017, 429, 1–11 CrossRef PubMed.
- D. Shiokawa and S. Tanuma, Biochemistry, 2001, 40, 143–152 CrossRef PubMed.
- D. Shiokawa, T. Matsushita, T. Kobayashi, Y. Matsumoto and S. Tanuma, Genomics, 2004, 84, 95–105 CrossRef PubMed.
- D. S. Jang, N. R. Penthala, E. O. Apostolov, X. Wang, T. Fahmi, P. A. Crooks and A. G. Basnakian, J. Biomol. Screening, 2015, 20, 202–211 CrossRef PubMed.
- M. Napirei, A. Ricken, D. Eulitz, H. Knoop and H. G. Mannherz, Biochem. J., 2004, 380, 929–937 CrossRef CAS PubMed.
- K. Samejima and W. C. Earnshaw, Nat. Rev. Mol. Cell Biol., 2005, 6, 677–688 CrossRef CAS.
- D. H. Sutton, G. L. Conn, T. Brown and A. N. Lane, Biochem. J., 1997, 321, 481–486 CrossRef CAS.
- H. Koohy, T. A. Down and T. J. Hubbard, PLoS One, 2013, 8, e69853 CrossRef CAS PubMed.
- M. C. Peitschl, B. Polzar2, H. Stephan2, T. Crompton3, H. Robson Macdonald3, H. G. Mannherz2 and J. Tschopp1′, EMBO J., 1993, 1, 371–377 CrossRef PubMed.
- D. Shiokawa, T. Matsushita, Y. Shika, M. Shimizu, M. Maeda and S. I. Tanuma, J. Biol. Chem., 2007, 282, 17132–17140 CrossRef CAS PubMed.
- M. Los, D. Neubüser, J. F. Coy, M. Mozoluk, A. Poustka and K. Schulze-Osthoff, Biochemistry, 2000, 39, 7365–7373 CrossRef CAS PubMed.
- L. Eckhart, H. Fischer and E. Tschachler, Front. Biosci., 2012, 17, 2461–2475 CrossRef PubMed.
- A. Wilber, M. Lu and M. C. Schneider, Mol. Ther., 2002, 6, 35–42 CrossRef CAS PubMed.
- V. Sisirak, B. Sally, V. D. Agati, W. Martinez-ortiz, Z. Birsin, J. David, A. Rashidfarrokhi, A. Yeste, C. Panea, A. Seema, M. Bogunovic, I. I. Ivanov, F. J. Quintana, I. Sanz, B. Keith, M. Tekin, F. Yalçınkaya, T. J. Cardozo, R. M. Clancy and P. Jill, Cell, 2017, 166, 88–101 CrossRef.
- A. Hamid Boulares, A. G. Yakovlev and M. E. Smulson, Madame Curie Bioscience Database, Landes Bioscience, 2013 Search PubMed.
- D. Pinto-González Howell, R. J. Krieser, A. Eastman and M. A. Barry, Mol. Ther., 2003, 8, 957–963 CrossRef.
- K. S. Maclea, R. J. Krieser and A. Eastman, Biochem. Biophys. Res. Commun., 2002, 292, 415–421 CrossRef CAS PubMed.
- A. Varela-Ramirez, J. Abendroth, A. A. Mejia, I. Q. Phan, D. D. Lorimer, T. E. Edwards and R. J. Aguilera, Nucleic Acids Res., 2017, 45, 6217–6227 CrossRef CAS PubMed.
- G. Bernard and G. Bernard, in The Enzymes, ed. P. D. Boyer, 1971, pp. 271–287 Search PubMed.
- M.-F. Counis and A. Torriglia, Biochimie, 2006, 88, 1851–1858 CrossRef CAS PubMed.
- K. S. Maclea, R. J. Krieser and A. Eastman, Biochem. J., 2003, 371, 867–876 CrossRef CAS PubMed.
- R. J. Krieser, K. S. Maclea, D. S. Longnecker, J. L. Fields, S. Fiering, A. Eastman and A. Eastman, Cell Death Differ., 2002, 9, 956–962 CrossRef CAS PubMed.
- P. Schafer, I. A. Cymerman, J. M. Bujnicki and G. Meiss, Protein Sci., 2007, 16, 82–91 CrossRef PubMed.
- K. S. MacLea, R. J. Krieser and A. Eastman, Biochem. Biophys. Res. Commun., 2002, 292, 415–421 CrossRef CAS PubMed.
- H. Fischer, J. Scherz, S. Szabo, M. Mildner and C. Benarafa, PLoS One, 2011, 6, 17581 CrossRef.
- R. J. Krieser, K. S. MacLea, J. P. Park and A. Eastman, Gene, 2001, 269, 205–216 CrossRef CAS PubMed.
- N. Sogo, K. Kohki, W.-F. Rie, F. Hidehiro, O. Yoshiyuki, U. Yasuo, H. Noriyasu, O. Nobuyuki, T. Yasuo, M. Takeshi, F. Yutaka and N. Shigekazu, Nature, 2003, 424, 1071–1074 CrossRef PubMed.
- M. Nakahara, A. Nagasaka, M. Koike, K. Uchida, K. Kawane, Y. Uchiyama and S. Nagata, FEBS J., 2007, 274, 3055–3064 CrossRef PubMed.
- A. Torriglia, P. Perani, J. Y. Brossas, E. Chaudun, J. Treton, Y. Courtois and M.-F. Counis, Mol. Cell. Biol., 1998, 18, 3612–3619 CrossRef PubMed.
- S. Altairac, S. C. Wright, Y. Courtois and A. Torriglia, Cell Death Differ., 2003, 10, 1109–1111 CrossRef PubMed.
- F. Yuan, T. Dutta, L. Wang, L. Song, L. Gu, L. Qian, A. Benitez, S. Ning, A. Malhotra, M. P. Deutscher and Y. Zhang, J. Biol. Chem., 2015, 290, 13344–13353 CrossRef PubMed.
- D. Chowdhury, P. J. Beresford, P. Zhu, D. Zhang, J. S. Sung, B. Demple, F. W. Perrino and J. Lieberman, Mol. Cell, 2006, 23, 133–142 CrossRef PubMed.
- C. J. Wang, W. Lam, S. Bussom, H. M. Chang and Y. C. Cheng, DNA Repair, 2009, 8, 1179–1189 CrossRef PubMed.
- D. Kavanagh, D. Spitzer, P. Kothari, A. Shaikh, M. K. Liszewski, A. Richards and J. P. Atkinson, Cell Cycle, 2008, 7, 12 CrossRef PubMed.
- W. O. Hemphill and F. W. Perrino, Methods in Enzymology, Academic Press Inc., 2019, vol. 625, pp. 109–133 Search PubMed.
- H.-L. Cheng, C.-T. Lin, K.-W. Huang, S. Wang, Y.-T. Lin, S.-I. Toh and Y.-Y. Hsiao, Nucleic Acids Res., 2018, 46, 12166–12176 CrossRef PubMed.
- A. R. Mathers, J. Invest. Dermatol., 2016, 136, 2337–2339 CrossRef PubMed.
- J. Manils, E. Casas, A. Viña-Vilaseca, M. López-Cano, A. Díez-Villanueva, D. Gómez, L. Marruecos, M. Ferran, C. Benito, F. W. Perrino, T. Vavouri, J. M. de Anta, F. Ciruela and C. Soler, J. Invest. Dermatol., 2016, 136, 2345–2355 CrossRef PubMed.
- M.-J. Chen, S.-M. Ma, L. C. Dumitrache and P. Hasty, Nucleic Acids Res., 2007, 35, 2682–2694 CrossRef PubMed.
- S. K. Gupta, B. J. Haigh, F. J. Griffin and T. T. Wheeler, Innate Immun., 2013, 19, 86–97 CrossRef PubMed.
- P. Koczera, L. Martin, G. Marx and T. Schuerholz, Int. J. Mol. Sci., 2016, 17 Search PubMed.
- G. Gotte and M. Menegazzi, Front. Immunol., 2019, 10, 1–26 CrossRef PubMed.
- A. M. Vicentini, B. Kieffer, R. Matthies, B. A. Hemmings, S. R. Stone, J. Hofsteenge, B. Kieffer and B. Meyhack, Biochemistry, 1990, 29, 8827–8834 CrossRef PubMed.
- K. A. Dickson, M. C. Haigis and R. T. Raines, Prog. Nucleic Acid Res. Mol. Biol., 2005, 80, 349–374 Search PubMed.
- D. M. Monti, N. Montesano Gesualdi, J. Matoušek, F. Esposito and G. D’Alessio, FEBS Lett., 2007, 581, 930–934 CrossRef PubMed.
- F. M. Richards and H. W. Wyckoff, in The Enzymes, ed. P. D. Boyer, 1st edn, 1971, pp. 647–806 Search PubMed.
- S. B. delCardayré and R. T. Raines, Biochemistry, 1994, 33, 6031–6037 CrossRef PubMed.
- S. Sorrentino and M. Libonati, Arch. Biochem. Biophys., 1994, 312, 340–348 CrossRef PubMed.
- T. Miyamoto, S. Okano and N. Kasai, Biotechnol. Prog., 2009, 25, 1678–1685 CrossRef PubMed.
- S. Sorrentino, M. Naddeo, A. Russo and G. D’Alessio, Biochemistry, 2003, 42, 10182–10190 CrossRef PubMed.
- J. B. P. Landré, P. W. Hewett, J.-M. Olivot, P. Friedl, Y. Ko, A. Sachinidis and M. Moenner, J. Cell. Biochem., 2002, 86, 540–552 CrossRef PubMed.
- S. P. Thomas, E. Kim, J.-S. Kim and R. T. Raines, Biochemistry, 2016, 55, 6359–6362 CrossRef PubMed.
- K. Bedenbender and B. T. Schmeck, Front. Cell Dev. Biol., 2020, 8, 576491 CrossRef PubMed.
- K. Mizuta, T. Yasuda, Y. Ikehara, W. Sato and K. Kishi, Z. Rechtsmed., 1990, 103, 315–322 Search PubMed.
- M. C. Munthe-Kaas, J. Gerritsen, K. H. Carlsen, D. Undlien, T. Egeland, B. Skinningsrud, T. Tørres and K. L. Carlsen, Allergy, 2007, 62, 429–436 CrossRef PubMed.
- D. Giusti, G. Gatouillat, S. Le Jan, J. Plée, P. Bernard, F. Antonicelli and B. N. Pham, Sci. Rep., 2017, 7, 4833 CrossRef PubMed.
- J. Bystrom, K. Amin and D. Bishop-Bailey, Respir. Res., 2011, 12, 10 CrossRef PubMed.
- A. Gupta and P. C. Rath, J. Biosci., 2012, 37, 103–113 CrossRef PubMed.
- S. Liang and K. R. Acharya, FEBS J., 2016, 283, 912–928 CrossRef PubMed.
- H. F. Rosenberg and K. D. Dyer, Nucleic Acids Res., 1995, 23, 4290–4295 CrossRef PubMed.
- S. K. Saxena, S. M. Rybak, R. T. Davey, R. J. Youle and E. J. Ackerman, J. Biol. Chem., 1992, 267, 21982–21986 CrossRef PubMed.
- J. Bläser, S. Triebel, C. Kopp and H. Tschesche, Eur. J. Clin. Chem. Clin. Biochem., 1993, 31, 513–516 Search PubMed.
- G. Prats-Ejarque, J. Arranz-Trullén, J. A. Blanco, D. Pulido, M. V. Nogués, M. Moussaoui and E. Boix, Biochem. J., 2016, 473, 1523–1536 CrossRef PubMed.
- H. F. Rosenberg and K. D. Dyer, Nucleic Acids Res., 1996, 24, 3507–3513 CrossRef PubMed.
- J. Rgen Harder and J.-M. Schrö, J. Biol. Chem., 2002, 277, 46779–46784 CrossRef PubMed.
- S. Sorrentino, FEBS Lett., 2010, 584, 2194–2200 CrossRef PubMed.
- J. Zhang, K. D. Dyer and H. F. Rosenberg, Nucleic Acids Res., 2002, 30, 1169–1175 CrossRef PubMed.
- K. D. Dyer and H. F. Rosenberg, Mol. Diversity, 2006, 10, 585–597 CrossRef PubMed.
- H. Wu, H. Xu, L. J. Miraglia and S. T. Crooke, J. Biol. Chem., 2000, 275, 36957–36965 CrossRef PubMed.
- D. Drider and C. Condon, J. Mol. Microbiol. Biotechnol., 2004, 8, 195–200 Search PubMed.
- A. W. Nicholson, Wiley Interdiscip. Rev.: RNA, 2014, 5, 31–48 CrossRef PubMed.
- D. L. Court, J. Gan, Y.-H. Liang, G. X. Shaw, J. E. Tropea, N. Costantino, D. S. Waugh and X. Ji, Annu. Rev. Genet., 2013, 47, 405–431 CrossRef PubMed.
- B. Lamontagne, S. Larose, J. Boulanger and S. A. Elela, Curr. Issues Mol. Biol., 2001, 3, 71–78 Search PubMed.
- S. M. Hammond, FEBS Lett., 2005, 579, 5822–5829 CrossRef PubMed.
- M.-S. Song and J. J. Rossi, Biochem. J., 2017, 474, 1603–1618 CrossRef PubMed.
- P. Provost, D. Dishart, J. Doucet, D. Frendewey, B. Samuelsson and O. Ra, EMBO J., 2002, 21, 5864–5874 CrossRef PubMed.
- S. Li and D. J. Patel, Cell Res., 2016, 26, 511–512 CrossRef PubMed.
- A.-J. Cheng, Cancer Sci. Res., 2014, 1, 1–5 CrossRef.
- M. Nowotny, S. A. Gaidamakov, R. J. Crouch and W. Yang, Cell, 2005, 121, 1005–1016 CrossRef PubMed.
- H. Nakamura, Y. Oda, S. Iwait, H. Inouet, E. Ohtsukat, S. Kanaya, S. Kimura, C. Katsuda, K. Katayanagi, K. Morikawa, H. Miyashiro and M. Ikehara, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 11535–11539 CrossRef PubMed.
- W. F. Lima, H. Wu and S. T. Crooke, Methods in Enzymology, 2001 Search PubMed.
- S. M. Cerritelli and R. J. Crouch, FEBS J., 2009, 276, 1494–1505 CrossRef PubMed.
- K. A. Majorek, S. Dunin-Horkawicz, K. Steczkiewicz, A. Muszewska, M. Nowotny, K. Ginalski and J. M. Bujnicki, Nucleic Acids Res., 2014, 42, 4160–4179 CrossRef PubMed.
- N. Luhtala and R. Parker, Trends Biochem. Sci., 2010, 35, 253–259 CrossRef CAS PubMed.
- A. Thorn, R. Steinfeld, M. Ziegenbein, M. Grapp, H.-H. Hsiao, H. Urlaub, G. M. Sheldrick, J. Gärtner and R. Krätzner, Nucleic Acids Res., 2012, 40, 8733–8742 CrossRef PubMed.
- Rajashree A. Deshpande and Vepatu Shankar, Crit. Rev. Microbiol., 2002, 28, 79–122 CrossRef PubMed.
- M. Irie, Pharmacol. Ther., 1999, 81, 77–89 CrossRef PubMed.
- L. Wu, Y. Xu, H. Zhao and Y. Li, Front. Immunol., 2020, 11, 1554 CrossRef PubMed.
- I. Øynebråten, K. Jung, F. Acquati, L. Mortara, A. De Vito, D. Baci, A. Albini, M. Cippitelli, R. Taramelli and D. M. Noonan, Front. Immunol., 2019, 10, 2587 CrossRef PubMed.
- E. Gusho, D. Baskar and S. Banerjee, Cytokine, 2020, 133, 153847 CrossRef CAS PubMed.
- C. S. Thakur, B. Kant Jha, B. Dong, J. Das Gupta, K. M. Silverman, H. Mao, H. Sawai, A. O. Nakamura, A. K. Banerjee, A. Gudkov and R. H. Silverman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9858–9890 CrossRef PubMed.
- F. S. D. Designed Research S, 2020, 117, 24802–24812.
- Y.-T. Wang, P.-H. Tseng, C.-L. Chen, D.-S. Han, Y.-C. Chi, F.-Y. Tseng and W.-S. Yang, Cardiovasc. Diabetol., 2017, 16, 46 CrossRef PubMed.
- H. J. Ezelle, K. Malathi and B. A. Hassel, Int. J. Mol. Sci., 2016, 17, 74 CrossRef PubMed.
- S. L. Liang, D. Quirk and A. Zhou, IUBMB Life, 2006, 58, 508–514 CrossRef CAS PubMed.
- A. Kladi-Skandali, K. Mavridis, A. Scorilas and D. C. Sideris, J. Cancer Res. Clin. Oncol., 2018, 144, 2049–2057 CrossRef CAS PubMed.
- B. A. Hackett, A. Yasunaga, D. Panda, M. A. Tartell, K. C. Hopkins, S. E. Hensley and S. Cherry, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7797–7802 CrossRef CAS PubMed.
- C. Gosse, J. B. Le Pecq, P. Defrance and C. Paoletti, Cancer Res., 1965, 25, 877–883 CAS.
- H. Takeshita, T. Naka-Jima, K. Mogi, Y. Kaneko, T. Yasuda, R. Iida and K. Kishi, Clin. Chem., 2004, 50, 446–448 CrossRef CAS PubMed.
- W. Yao, C. Mei, X. Nan and L. Hui, Gene, 2016, 590, 142–148 CrossRef CAS PubMed.
- J. H. Connolly, R. M. Herriott and S. Gupta, Br. J. Exp. Pathol., 1962, 43, 402–408 CAS.
- H. Zhang, K. Rombouts, L. Raes, R. Xiong, S. C. De Smedt, K. Braeckmans and K. Remaut, Adv. Biosyst., 2020, 4, 2000057 CrossRef CAS PubMed.
- J. M. Layzer, M. Rey, A. P. Mccaffrey, A. K. Tanner, Z. Huang, M. A. Kay and B. A. Sullenger, RNA, 2004, 10, 766–771 CrossRef CAS PubMed.
- L. Pitkä, M. Ruponen, J. Nieminen and A. Urtti, Pharm. Res., 2003, 20, 576–583 CrossRef PubMed.
- M. Zakrewsky, S. Kumar and S. Mitragotri, J. Controlled Release, 2015, 219, 245–256 CrossRef PubMed.
- E. Kovacs, J. Pediatr., 1955, 47, 340–346 CrossRef CAS PubMed.
- M. Buckell, Proc. Assoc. Din. Biochem., 1968, 5, 33 Search PubMed.
- E. Blanco, H. Shen, M. Ferrari and N. B. Author, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- Y. Fujiwara, K. Wada and T. Kabuta, J. Biochem., 2017, 161, 145–154 CAS.
- P. Gong, B. Shi, P. Zhang, D. Hu, M. Zheng, C. Zheng, D. Gao and L. Cai, Nanoscale, 2012, 4, 2454–2462 RSC.
- H. Pollard, J. S. Remy, G. Loussouarn, S. Demolombe, J. P. Behr and D. Escande, J. Biol. Chem., 1998, 273, 7507–7511 CrossRef CAS PubMed.
- K. Remaut, B. Lucas, K. Braeckmans, N. N. Sanders, S. C. De Smedt and J. Demeester, J. Controlled Release, 2005, 103, 259–271 CrossRef CAS PubMed.
- G. Takahashi, C. B. Gurumurthy, K. Wada, H. Miura, M. Sato and M. Ohtsuka, Sci. Rep., 2015, 5, 11406 CrossRef PubMed.
- L. Wayteck, R. Xiong, K. Braeckmans, S. C. De Smedt and K. Raemdonck, J. Controlled Release, 2017, 267, 154–162 CrossRef CAS PubMed.
- E. B. Ghartey-Tagoe, B. A. Babbin, A. Nusrat, A. S. Neish and M. R. Prausnitz, Int. J. Pharm., 2006, 315, 122–133 CrossRef CAS PubMed.
- K. Raemdonck, K. Remaut, B. Lucas, N. N. Sanders, J. Demeester and S. C. De Smedt, Biochemistry, 2006, 45, 10614–10623 CrossRef CAS PubMed.
- R. A. Bamford, Z. Y. Zhao, N. A. Hotchin, I. B. Styles, G. B. Nash, J. H. R. Tucker and R. Bicknell, PLoS One, 2014, 9, e95097 CrossRef PubMed.
- H. Pollard, G. Toumaniantz, J. L. Amos, H. Avet-Loiseau, G. Guihard, J. P. Behr and D. Escande, J. Gene Med., 2001, 3, 153–164 CrossRef CAS PubMed.
- D. Lechardeur, K.-J. Sohn, M. Haardt, P. B. Joshi, M. Monck, R. W. Graham, B. Beatty, J. Squire, H. O’brodovich and G. L. Lukacs, Gene Ther., 1999, 6, 482–497 CrossRef CAS PubMed.
- R. Rattan, A. U. Bielinska and M. M. Banaszak Holl, J. Gene Med., 2014, 16, 75–83 CrossRef CAS PubMed.
- L. Raes, S. Stremersch, J. C. Fraire, T. Brans, G. Goetgeluk, S. De Munter, L. Van Hoecke, R. Verbeke, J. Van Hoeck, R. Xiong, X. Saelens, B. Vandekerckhove, S. De Smedt, K. Raemdonck and K. Braeckmans, Nano-Micro Lett., 2020, 12, 185 CrossRef CAS PubMed.
- T. S. Ligon, C. Leonhardt and J. O. Rädler, PLoS One, 2014, 9, e107148 CrossRef PubMed.
- H. Liu, M. Luo and J. K. Wen, J. Zhejiang Univ., Sci., B, 2014, 15, 444–454 CrossRef CAS PubMed.
- C. Yang, R. Wu, H. Liu, Y. Chen, Y. Gao, X. Chen, Y. Li, J. Ma, J. Li and J. Gan, Nucleic Acids Res., 2018, 46, 11075–11088 CAS.
- J. LaCava, H. Jonathan, S. Cosmin, P. Elisabeth, T. Elizabeth, J. Alain and T. David, Cell, 2005, 121, 713–724 CrossRef CAS PubMed.
- A. L. Karamyshev and Z. N. Karamysheva, Front. Genet., 2018, 9, 431 CrossRef CAS PubMed.
- J. Connors, D. Joyner, N. J. Mege, G. M. Cusimano, M. R. Bell, J. Marcy, B. Taramangalam, K. M. Kim, P. J. C. Lin, Y. K. Tam, D. Weissman, M. A. Kutzler, M. G. Alameh and E. K. Haddad, Commun. Biol., 2023, 6, 188, DOI:10.1038/s42003-023-04555-1.
- A. M. Weiss, S. Hossainy, S. J. Rowan, J. A. Hubbell and A. P. Esser-Kahn, Macromolecules, 2022, 55, 6913–6937 CrossRef CAS PubMed.
- B. Briard, D. E. Place and T. D. Kanneganti, Physiology, 2020, 35, 112–124 CrossRef CAS PubMed.
- A.-K. Minnaert, H. Vanluchene, R. Verbeke, I. Lentacker, S. C. De Smedt, K. Raemdonck, N. N. Sanders and K. Remaut, Adv. Drug Delivery Rev., 2021, 176, 113900 CrossRef CAS PubMed.
- S. Rafii, E. Tashkandi, N. Bukhari and H. O. Al-Shamsi, Cancers, 2022, 14, 947, DOI:10.3390/cancers14040947.
- Y. Chen, R. Wen, Z. Yang and Z. Chen, Gene Ther., 2022, 29, 207–216 CrossRef CAS PubMed.
- T. R. Abbott, G. Dhamdhere, Y. Liu, X. Lin, L. Goudy, L. Zeng, A. Chemparathy, S. Chmura, N. S. Heaton, R. Debs, T. Pande, D. Endy, M. F. La Russa, D. B. Lewis and L. S. Qi, Cell, 2020, 181, 865–876 CrossRef CAS PubMed.
- E. L. Blanchard, D. Vanover, S. S. Bawage, P. M. Tiwari, L. Rotolo, J. Beyersdorf, H. E. Peck, N. C. Bruno, R. Hincapie, F. Michel, J. Murray, H. Sadhwani, B. Vanderheyden, M. G. Finn, M. A. Brinton, E. R. Lafontaine, R. J. Hogan, C. Zurla and P. J. Santangelo, Nat. Biotechnol., 2021, 39, 717–726 CrossRef CAS PubMed.
- H. Li, Y. Yang, W. Hong, M. Huang, M. Wu and X. Zhao, Signal Transduction Targeted Ther., 2020, 5, 1 CrossRef PubMed.
- F. D. Urnov, E. J. Rebar, M. C. Holmes, H. S. Zhang and P. D. Gregory, Nat. Rev. Genet., 2010, 11, 636–646 CrossRef CAS PubMed.
- A. V. Anzalone, L. W. Koblan and D. R. Liu, Nat. Biotechnol., 2020, 38, 824–844 CrossRef CAS PubMed.
- S. Wang, T. Xie, S. Sun, K. Wang, B. Liu, X. Wu and W. Ding, Sci. Rep., 2018, 8, 17788, DOI:10.1038/s41598-018-36198-2.
- P. Santa, A. Garreau, L. Serpas, A. Ferriere, P. Blanco, C. Soni and V. Sisirak, Front. Immunol., 2021, 12, 629922 CrossRef CAS PubMed.
- B. Schwartz, O. Shoseyov, V. O. Melnikova, M. McCarty, M. Leslie, L. Roiz, P. Smirnoff, G. F. Hu, D. Lev and M. Bar-Eli, Cancer Res., 2007, 67, 5258–5266 CrossRef CAS PubMed.
- W. O. Hemphill, S. R. Simpson, M. Liu, F. R. Salsbury, T. Hollis, J. M. Grayson and F. W. Perrino, Front. Immunol., 2021, 12, 660184 CrossRef CAS PubMed.
- R. L. Gibson, J. L. Burns and B. W. Ramsey, Am. J. Respir. Crit. Care Med., 2003, 168, 918–951 CrossRef PubMed.
- D. Delfino, G. Mori, C. Rivetti, A. Grigoletto, G. Bizzotto, C. Cavozzi, M. Malatesta, D. Cavazzini, G. Pasut and R. Percudani, Biomolecules, 2021, 11, 1–20 CrossRef PubMed.
- C. Chen, Z. Yang and X. Tang, Med. Res. Rev., 2018, 38, 829–869 CrossRef PubMed.
- G. Tavernier, O. Andries, J. Demeester, N. N. Sanders, S. C. De Smedt and J. Rejman, J. Controlled Release, 2011, 150, 238–247 CrossRef CAS PubMed.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1683 CrossRef PubMed.
- M. Gaglione and A. Messere, Mini-Rev. Med. Chem., 2010, 10, 578–595 CrossRef CAS PubMed.
- K. Hirano, H. Uchiyama and K. Taira, Tanpakushitsu Kakusan Koso, 1996, 41, 2139–2143 CAS.
- B. H. Yoo, E. Bochkareva, A. Bochakarev, T. C. Mou and D. M. Gray, Nucleic Acids Res., 2004, 32, 2008–2016 CrossRef CAS PubMed.
- J. Temsamani, J. Y. Tang, A. Padmapriya, M. Kubert and S. Agrawal, Antisense Res. Dev., 1993, 3, 277–284 CrossRef CAS PubMed.
- A. H. S. Hall, J. Wan, E. E. Shaughnessy, B. R. Shaw and K. A. Alexander, Nucleic Acids Res., 2004, 32, 5991–6000 CrossRef CAS PubMed.
- S. M. Gryaznov, Chem. Biodiversity, 2010, 7, 477–493 CrossRef CAS PubMed.
- Study to evaluate imetelstat (GRN163L) in subjects with international prognostic scoring system (IPSS) low or intermediate-1 risk myelodysplastic syndrome (MDS), https://clinicaltrials.gov/ct2/show/NCT02598661.
- H. Jahns, M. Roos, J. Imig, F. Baumann, Y. Wang, R. Gilmour and J. Hall, Nat. Commun., 2015, 6, 6317 CrossRef CAS PubMed.
- M. Koziołkiewicz, M. Wójcik, A. Kobylańska, B. Karwowski, B. Rȩbowska, P. Guga and W. J. Stec, Antisense Nucleic Acid Drug Dev., 1997, 7, 43–48 CrossRef PubMed.
- W. B. Wan, M. T. Migawa, G. Vasquez, H. M. Murray, J. G. Nichols, H. Gaus, A. Berdeja, S. Lee, C. E. Hart, W. F. Lima, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 13456–13468 CrossRef PubMed.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskii, M. Egholm, O. Buchard, S. H. Sonnichsen and P. E. Nielsen, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef PubMed.
- M. Gaglione, M. E. Mercurio, N. Potenza, N. Mosca, A. Russo, E. Novellino, S. Cosconati and A. Messere, BioMed Res. Int., 2014, 2014, 901617, DOI:10.1155/2014/901617.
- S. Choung, Y. J. Kim, S. Kim, H.-O. Park and Y.-C. Choi, Biochem. Biophys. Res. Commun., 2006, 342, 919–927 CrossRef PubMed.
- F. Czauderna, M. Fechtner, S. Dames, H. Aygün, A. Klippel, G. J. Pronk, K. Giese and J. Kaufmann, Nucleic Acids Res., 2003, 31, 2705–2716 CrossRef PubMed.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- A. A. Volkov, N. S. Kruglova, M. I. Meschaninova, A. G. Venyaminova, M. A. Zenkova, V. V. Vlassov and E. L. Chernolovskaya, Oligonucleotides, 2009, 19, 191–202 CrossRef CAS PubMed.
- S. K. Miroshnichenko, B. Amirloo, E. V. Bichenkova, V. V. Vlassov, M. A. Zenkova and O. A. Patutina, Russ. J. Bioorg. Chem., 2019, 45, 803–812 CrossRef CAS.
- H. Adachi, M. Hengesbach, Y. T. Yu and P. Morais, Biomedicines, 2021, 9, 1–26 CrossRef PubMed.
- K. G. Rajeev, T. P. Prakash and M. Manoharan, Org. Lett., 2003, 5, 3005–3008 CrossRef CAS PubMed.
- R. Pattanayek, L. Sethaphong, C. Pan, M. Prhavc, T. P. Prakash, M. Manoharan and M. Egli, J. Am. Chem. Soc., 2004, 126, 15006–15007 CrossRef CAS PubMed.
- T. P. Prakash, A. M. Kawasaki, E. V. Wancewicz, L. Shen, B. P. Monia, B. S. Ross, B. Bhat and M. Manoharan, J. Med. Chem., 2008, 51, 2766–2776 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- Y.-L. Chiu, RNA, 2003, 9, 1034–1048 CrossRef CAS PubMed.
- C. R. Allerson, N. Sioufi, R. Jarres, T. P. Prakash, N. Naik, A. Berdeja, L. Wanders, R. H. Griffey, E. E. Swayze and B. Bhat, J. Med. Chem., 2005, 48, 901–904 CrossRef CAS PubMed.
- T. Dowler, D. Bergeron, A.-L. Tedeschi, L. Paquet, N. Ferrari and M. J. Damha, Nucleic Acids Res., 2006, 34, 1669–1675 CrossRef CAS PubMed.
- S. Hoshika, N. Minakawa, A. Shionoya, K. Imada, N. Ogawa and A. Matsuda, ChemBioChem, 2007, 8, 2133–2138 CrossRef CAS PubMed.
- P. Dande, T. P. Prakash, N. Sioufi, H. Gaus, R. Jarres, A. Berdeja, E. E. Swayze, R. H. Griffey and B. Bhat, J. Med. Chem., 2006, 49, 1624–1634 CrossRef CAS PubMed.
- K. R. Gore, G. N. Nawale, S. Harikrishna, V. G. Chittoor, S. K. Pandey, C. Höbartner, S. Patankar and P. I. Pradeepkumar, J. Org. Chem., 2012, 77, 3233–3245 CrossRef CAS PubMed.
- K. S. Schmidt, S. Borkowski, J. Kurreck, A. W. Stephens, R. Bald, M. Hecht, M. Friebe, L. Dinkelborg and V. A. Erdmann, Nucleic Acids Res., 2004, 32, 5757–5765 CrossRef CAS PubMed.
- F. Darfeuille, J. B. Hansen, H. Orum, C. Di Primo and J.-J. Toulmé, Nucl. Med. Biol., 2004, 32, 3101–3107 CAS.
- J. Elmén, H. Thonberg, K. Ljungberg, M. Frieden, M. Westergaard, Y. Xu, B. Wahren, Z. Liang, H. Ørum, T. Koch and C. Wahlestedt, Nucleic Acids Res., 2005, 33, 439–447 CrossRef PubMed.
- M. B. Laursen, M. Malgorzata Pakula, A. Shan Gao, A. Kees Fluiter, O. R. Mook, F. Baas, N. Langklaer, S. L. Wengel, J. Wengel, J. Kjemsab and J. B. Bramsen, Mol. BioSyst., 2010, 6, 862–870 RSC.
- S. Gao, F. Dagnaes-Hansen, E. J. B. Nielsen, J. Wengel, F. Besenbacher, K. A. Howard and J. Kjems, Mol. Ther., 2009, 17, 1225–1233 CrossRef CAS PubMed.
- Y. Dong, D. J. Siegwart and D. G. Anderson, Adv. Drug Delivery Rev., 2019, 144, 133–147 CrossRef CAS PubMed.
- T. Aboul-Fadl, Curr. Med. Chem., 2005, 12, 2193–2214 CrossRef CAS.
- W. Brad Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef.
- M. Terrazas and E. T. Kool, Nucleic Acids Res., 2009, 37, 346–353 CrossRef CAS.
- N. Haginoya, A. Ono, Y. Nomura, Y. Ueno and A. Matsuda, Bioconjugate Chem., 1997, 8, 271–280 CrossRef CAS PubMed.
- M. S. D. Kormann, G. Hasenpusch, M. K. Aneja, G. Nica, A. W. Flemmer, S. Herber-jonat, M. Huppmann, L. E. Mays, M. Illenyi, A. Schams, M. Griese, I. Bittmann, R. Handgretinger, D. Hartl, J. Rosenecker and C. Rudolph, Nat. Biotechnol., 2011, 29, 154–157 CrossRef CAS PubMed.
- B. Li, X. Luo and Y. Dong, Bioconjugate Chem., 2016, 27, 853 Search PubMed.
- M. C. Kramer, K. A. Janssen, K. Palos, A. D. L. Nelson, L. E. Vandivier, B. A. Garcia, E. Lyons, M. A. Beilstein and B. D. Gregory, Plant Direct, 2020, 4, 1–22 CrossRef PubMed.
- H. K. Wayment-Steele, D. S. Kim, C. A. Choe, J. J. Nicol, R. Wellington-Oguri, A. M. Watkins, R. A. P. Sperberg, P. S. Huang, E. Participants and R. Das, Nucleic Acids Res., 2021, 49, 11405 CrossRef PubMed.
- I. V. Chernikov, V. V. Vlassov and E. L. Chernolovskaya, Front. Pharmacol., 2019, 10, 444 CrossRef PubMed.
- B. Godinho and A. Khvorova, Saúde Tecnologia, 2019, 5–17 Search PubMed.
- M. Hawner and C. Ducho, Molecules, 2020, 25, 5963 CrossRef PubMed.
- K. Klabenkova, A. Fokina and D. Stetsenko, Molecules, 2021, 26, 1–36 CrossRef PubMed.
- K. T. Shum and J. A. Tanner, ChemBioChem, 2008, 9, 3037–3045 CrossRef PubMed.
- T. Kubo, K. Yanagihara, Y. Takei, K. Mihara, Y. Morita and T. Seyama, Mol. Pharmaceutics, 2011, 8, 2193–2203 CrossRef PubMed.
- T. Kubo, K. Yanagihara, Y. Takei, K. Mihara, Y. Sato and T. Seyama, Biochem. Biophys. Res. Commun., 2012, 426, 571–577 CrossRef PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef PubMed.
- M. F. Yuen, I. Schiefke, J. H. Yoon, S. H. Ahn, J. Heo, J. H. Kim, H. Lik Yuen Chan, K. T. Yoon, H. Klinker, M. Manns, J. Petersen, T. Schluep, J. Hamilton, B. D. Given, C. Ferrari, C. L. Lai, S. A. Locarnini and R. G. Gish, Hepatology, 2020, 72, 19–31 CrossRef PubMed.
- D. B. Rozema, D. L. Lewis, D. H. Wakefield, S. C. Wong, J. J. Klein, P. L. Roesch, S. L. Bertin, T. W. Reppen, Q. Chu, A. V. Blokhin, J. E. Hagstrom and J. A. Wolff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12982–12987 CrossRef CAS PubMed.
- V. Soriano, P. Barreiro, E. Cachay, S. Kottilil, J. V. Fernandez-Montero and C. de Mendoza, Ther. Adv. Infect. Dis., 2020, 7, 1–11 Search PubMed.
- C. I. Wooddell, K. Blomenkamp, R. M. Peterson, V. M. Subbotin, C. Schwabe, J. Hamilton, Q. Chu, D. R. Christianson, J. O. Hegge, J. Kolbe, H. L. Hamilton, M. F. Branca-Afrazi, B. D. Given, D. L. Lewis, E. Gane, S. B. Kanner and J. H. Teckman, JCI Insight, 2020, 5, 1–19 CrossRef PubMed.
- L. Zhang, D. Liang, C. Chen, Y. Wang, G. Amu, J. Yang, L. Yu, I. J. Dmochowski and X. Tang, Mol. Ther.–Nucleic Acids, 2018, 10, 237–244 CrossRef CAS PubMed.
- X. Chen, T. Yang, W. Wang, W. Xi, T. Zhang, Q. Li, A. Yang and T. Wang, Theranostics, 2019, 9, 588–607 CrossRef CAS PubMed.
- I. Kira Astakhova, A. Lykke, H. Hansen, B. Vester and J. Wengel, Org. Biomol. Chem., 2013, 11, 4240 RSC.
- M. B. Danielsen and J. Wengel, Beilstein J. Org. Chem., 2021, 17, 1828–1848 CrossRef PubMed.
- P. J. Fairchild, RNA modifications, Human Press, 2021 Search PubMed.
- A. A. Klauer and A. van Hoof, Wiley Interdiscip. Rev.: RNA, 2012, 3, 649–660 CrossRef PubMed.
- Q. Wu, S. G. Medina, G. Kushawah, M. L. Devore, L. A. Castellano, J. M. Hand, M. Wright and A. A. Bazzini, eLife, 2019, 8, 1–23 Search PubMed.
- V. Presnyak, N. Alhusaini, Y. H. Chen, S. Martin, N. Morris, N. Kline, S. Olson, D. Weinberg, K. E. Baker, B. R. Graveley and J. Coller, Cell, 2015, 160, 1111–1124 CrossRef PubMed.
- M. Courel, Y. Clément, C. Bossevain, D. Foretek, O. V. Cruchez, Z. Yi, M. Bénard, M. N. Benassy, M. Kress, C. Vindry, M. Ernoult-Lange, C. Antoniewski, A. Morillon, P. Brest, A. Hubstenberger, H. R. Crollius, N. Standart and D. Weil, eLife, 2019, 8, 1–32 CrossRef.
- A. Thess, S. Grund, B. L. Mui, M. J. Hope, P. Baumhof, M. Fotin-Mleczek and T. Schlake, Mol. Ther., 2015, 23, 1456–1464 CrossRef CAS PubMed.
- J. Mauer, X. Luo, A. Blanjoie, X. Jiao, A. V. Grozhik, D. P. Patil, B. Linder, B. F. Pickering, J. J. Vasseur, Q. Chen, S. S. Gross, O. Elemento, F. Debart, M. Kiledjian and S. R. Jaffrey, Nature, 2017, 541, 371–375 CrossRef CAS PubMed.
- E. Grudzien, M. Kalek, J. Jemielity, E. Darzynkiewicz and R. E. Rhoads, J. Biol. Chem., 2006, 281, 1857–1867 CrossRef CAS.
- F. T. Zohra, E. H. Chowdhury, S. Tada, T. Hoshiba and T. Akaike, Biochem. Biophys. Res. Commun., 2007, 358, 373–378 CrossRef CAS PubMed.
- J. Stepinski, C. Waddell, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2001, 7, 1486–1495 CAS.
- M. Strenkowska, J. Kowalska, M. Lukaszewicz, J. Zuberek, W. Su, R. E. Rhoads, E. Darzynkiewicz and J. Jemielity, New J. Chem., 2010, 34, 993–1007 RSC.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. W. A. Grudzien-nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119–1131 CrossRef CAS PubMed.
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged and A. Thakur, Pharmaceutics, 2020, 12, 102 CrossRef CAS PubMed.
- D. Gagliardi and A. Dziembowski, Philos. Trans. R. Soc., B, 2018, 373, 20180160 CrossRef.
- C. R. Eckmann, C. Rammelt and E. Wahle, Wiley Interdiscip. Rev.: RNA, 2011, 2, 348–361 CrossRef CAS PubMed.
- B. Roy and A. Jacobson, Trends Genet., 2013, 29, 691–699 CrossRef CAS PubMed.
- M. Mockey, C. Gonçalves, F. P. Dupuy, F. M. Lemoine, C. Pichon and P. Midoux, Biochem. Biophys. Res. Commun., 2006, 340, 1062–1068 CrossRef CAS PubMed.
- Z. Trepotec, J. Geiger, C. Plank, M. K. Aneja and C. Rudolph, RNA, 2019, 25, 507–518 CrossRef CAS PubMed.
- H. Chang, J. Lim, M. Ha and V. N. Kim, Mol. Cell, 2014, 53, 1044–1052 CrossRef CAS.
- S. C. Kim, S. S. Sekhon, W. R. Shin, G. Ahn, B. K. Cho, J. Y. Ahn and Y. H. Kim, Mol. Cell. Toxicol., 2022, 18, 1–8, DOI:10.1007/s13273-021-00171-4.
- K. Leppek, R. Das and M. Barna, Nat. Rev. Mol. Cell Biol., 2018, 19, 158–174 CrossRef CAS PubMed.
- Y. Zhao, R. Shu and J. Liu, Mol. Ther.–Nucleic Acids, 2021, 26, 997–1013 CrossRef CAS PubMed.
- X. Wu and G. Brewer, Gene, 2012, 500, 10–21 CrossRef CAS PubMed.
- I. Peixeiro, A. L. Silva and L. Romão, Haematologica, 2011, 96, 905–913 CrossRef CAS PubMed.
- S. A. Waggoner and S. A. Liebhaber, Exp. Biol. Med., 2003, 228, 387–395 CrossRef CAS PubMed.
- F. Zarghampoor, N. Azarpira, S. R. Khatami, A. Behzad-Behbahani and A. M. Foroughmand, Gene, 2019, 707, 231–238 CrossRef CAS PubMed.
- C. Barreau, L. Paillard and H. B. Osborne, Nucleic Acids Res., 2005, 33, 7138–7150 CrossRef CAS.
- W. S. Koh, J. R. Porter and E. Batchelor, Sci. Rep., 2019, 9, 1–8 CrossRef CAS.
- E. L. Murray and D. R. Schoenberg, Mol. Cell. Biol., 2007, 27, 2791–2799 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS.
- P. Colella, G. Ronzitti and F. Mingozzi, Mol. Ther.–Methods Clin. Dev., 2018, 8, 87–104 CrossRef CAS.
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor, K. Chawla, S. Chattopadhyay, D. Chandra, N. Chilukuri and V. Betapudi, Front. Oncol., 2019, 9, 1–25 CrossRef PubMed.
- A. C. Nathwani, U. M. Reiss, E. G. D. Tuddenham, C. Rosales, P. Chowdary, J. McIntosh, M. Della Peruta, E. Lheriteau, N. Patel, D. Raj, A. Riddell, J. Pie, S. Rangarajan, D. Bevan, M. Recht, Y. M. Shen, K. G. Halka, E. Basner-Tschakarjan, F. Mingozzi, K. A. High, J. Allay, M. A. Kay, C. Y. C. Ng, J. Zhou, M. Cancio, C. L. Morton, J. T. Gray, D. Srivastava, A. W. Nienhuis and A. M. Davidoff, N. Engl. J. Med., 2014, 371, 1994–2004 CrossRef.
- L. M. Kattenhorn, C. H. Tipper, L. Stoica, D. S. Geraghty, T. L. Wright, K. R. Clark and S. C. Wadsworth, Hum. Gene Ther., 2016, 27, 947–961 CrossRef.
- G. J. Dimitriadis, Entrapment of ribonucleic acids in liposomes, 1978 Search PubMed.
- A. Gupta, J. L. Andresen, R. S. Manan and R. Langer, Adv. Drug Delivery Rev., 2021, 178 Search PubMed.
- G. Gregoriadis, Med. Drug Discovery, 2021, 12, 100104 CrossRef.
- K. Remaut, B. Lucas, K. Braeckmans, N. N. Sanders, J. Demeester and S. C. De Smedt, Biochemistry, 2006, 45, 1755–1764 CrossRef PubMed.
- K. Remaut, B. Lucas, K. Braeckmans, J. Demeester and S. C. De Smedt, J. Controlled Release, 2006, 117, 256–266 CrossRef PubMed.
- M. Cornebise, E. Narayanan, Y. Xia, E. Acosta, L. Ci, H. Koch, J. Milton, S. Sabnis, T. Salerno and K. E. Benenato, Adv. Funct. Mater., 2022, 32, 2106727 CrossRef.
- A. K. Blakney, P. F. McKay, B. I. Yus, Y. Aldon and R. J. Shattock, Gene Ther., 2019, 26, 363–372 CrossRef PubMed.
- G. Lou, G. Anderluzzi, S. T. Schmidt, S. Woods, S. Gallorini, M. Brazzoli, F. Giusti, I. Ferlenghi, R. N. Johnson, C. W. Roberts, D. T. O’Hagan, B. C. Baudner and Y. Perrie, J. Controlled Release, 2020, 325, 370–379 CrossRef PubMed.
- J. H. Erasmus, A. P. Khandhar, J. Guderian, B. Granger, J. Archer, M. Archer, E. Gage, J. Fuerte-Stone, E. Larson, S. Lin, R. Kramer, R. N. Coler, C. B. Fox, D. T. Stinchcomb, S. G. Reed and N. Van Hoeven, Mol. Ther., 2018, 26, 2507–2522 CrossRef PubMed.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1987, 262, 4429–4432 CrossRef CAS.
- A. P. Pandey and K. K. Sawant, Mater. Sci. Eng., C, 2016, 68, 904–918 CrossRef CAS.
- K. Remaut, B. Lucas, K. Raemdonck, K. Braeckmans, J. Demeester and S. C. De Smedt, Biomacromolecules, 2007, 8, 1333–1340 CrossRef CAS PubMed.
- H. Zhang, S. C. De Smedt and K. Remaut, Acta Biomater., 2018, 75, 358–370 CrossRef CAS.
- P. F. Monteiro, A. Travanut, C. Conte and C. Alexander, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1678 CAS.
- T. K. Georgiou, M. Ahmed and S. Wang, Front. Chem., 2021, 9, 645297 CrossRef PubMed.
- M. E. Jacobson, K. W. Becker, C. R. Palmer, L. E. Pastora, R. B. Fletcher, K. A. Collins, O. Fedorova, C. L. Duvall, A. M. Pyle and J. T. Wilson, ACS Cent. Sci., 2020, 6, 2008–2022 CrossRef CAS PubMed.
- Y. Yin, X. Li, H. Ma, J. Zhang, D. Yu, R. Zhao, S. Yu, G. Nie and H. Wang, Nano Lett., 2021, 21, 2224–2231 CrossRef CAS PubMed.
- F. Perche, R. Clemençon, K. Schulze, T. Ebensen, C. A. Guzmán and C. Pichon, Mol. Ther.–Nucleic Acids, 2019, 17, 767–775 CrossRef CAS PubMed.
- K. Remaut, N. Symens, B. Lucas, J. Demeester and S. C. De Smedt, J. Controlled Release, 2010, 144, 65–74 CrossRef CAS.
- M. Liu, B. Feng, Y. Shi, C. Su, H. Song, W. Cheng and L. Zhao, Nanoscale Res. Lett., 2015, 10, 134 CrossRef PubMed.
- L. A. Brito, M. Chan, C. A. Shaw, A. Hekele, T. Carsillo, M. Schaefer, J. Archer, A. Seubert, G. R. Otten, C. W. Beard, A. K. Dey, A. Lilja, N. M. Valiante, P. W. Mason, C. W. Mandl, S. W. Barnett, P. R. Dormitzer, J. B. Ulmer, M. Singh, D. T. O’Hagan and A. J. Geall, Mol. Ther., 2014, 22, 2118–2129 CrossRef CAS PubMed.
- X. Xiao He, K. Wang, W. Tan, B. Liu, X. Lin, C. He, D. Li, S. Huang and J. Li, J. Am. Chem. Soc., 2003, 125, 7168–7169 CrossRef PubMed.
- N. Yoshinaga, M. Naito, Y. Tachihara, E. Boonstra, K. Osada, H. Cabral and S. Uchida, Pharmaceutics, 2021, 13, 800 CrossRef CAS PubMed.
- N. L. Rosenberg, Mol. Cell. Biochem., 1987, 76, 113–121 CrossRef CAS PubMed.
- R. B. Hallick, B. K. Chelm and E. M. Orozco, Nucleic Acids Res., 1977, 4, 194 CrossRef PubMed.
- N. Inhibitors and P. Electrical, Methods Enzymol., 2002, 346, 72–91 Search PubMed.
- J. Glasspool-Malone, S. Somiari, J. J. Drabick and R. W. Malone, Mol. Ther., 2000, 2, 140–146 CrossRef CAS PubMed.
- J. Glasspool-Malone, P. R. Steenland, R. J. McDonald, R. A. Sanchez, T. L. Watts, J. Zabner and R. W. Malone, J. Gene Med., 2002, 4, 323–332 CrossRef PubMed.
- W. Walther, U. Stein, R. Siegel, I. Fichtner and P. M. Schlag, J. Gene Med., 2005, 7, 477–485 CrossRef CAS PubMed.
- G. F. Ross, M. D. Bruno, M. Uyeda, K. Suzuki, K. Nagao, J. A. Whitsett and T. R. Korfhagen, Gene Ther., 1998, 5, 1244–1250 CrossRef CAS PubMed.
- W. A. Kudlicki, M. M. Winkler and B. L. Pasloske, US20070148174A1, 2007.
- J. M. Layzer, A. P. McCaffrey, A. K. Tanner, Z. Huang, M. A. Kay and B. A. Sullenger, RNA, 2004, 10, 766–771 CrossRef CAS PubMed.
- C. R. Merril, M. R. Geier and J. C. Petricciani, Nature, 1971, 233, 398–399 CrossRef CAS PubMed.
- R. M. Blaese, K. W. Culver, A. D. Miller, C. S. Carter, T. Fleisher, M. Clerici, G. Shearer, L. Chang, Y. Chiang, P. Tolstoshev, J. J. Greenblatt, S. A. Rosenberg, H. Klein, M. Berger, C. A. Mullen, W. J. Ramsey, L. Muul, R. A. Morgan and W. F. Anderson, Science, 1995, 270, 475–480 CrossRef CAS PubMed.
- S. D. Patil, D. G. Rhodes and D. J. Burgess, AAPS J., 2005, 7, E61–E77 CrossRef CAS PubMed.
- W.-W. Zhang, L. Li, D. Li, J. Liu, X. Li, W. Li, X. Xu, M. J. Zhang, L. A. Chandler, H. Lin, A. Hu, W. Xu and D. Man-Kit Lam, Hum. Gene Ther., 2018, 29, 160–179 CrossRef CAS PubMed.
- L. M. Bryant, D. M. Christopher, A. R. Giles, C. Hinderer, J. L. Rodriguez, J. B. Smith, E. A. Traxler, J. Tycko, A. P. Wojno and J. M. Wilson, Hum. Gene Ther.: Clin. Dev., 2013, 24, 55–64 CAS.
- U.S.FDA, LUXTURNA, https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/luxturna.
- L. Urquhart, Nat. Rev. Drug Discovery, 2019, 18, 575 CrossRef CAS.
- J. Sadoff, M. Le Gars, G. Shukarev, D. Heerwegh, C. Truyers, A. M. de Groot, J. Stoop, S. Tete, W. Van Damme, I. Leroux-Roels, P.-J. Berghmans, M. Kimmel, P. Van Damme, J. de Hoon, W. Smith, K. E. Stephenson, S. C. De Rosa, K. W. Cohen, M. J. McElrath, E. Cormier, G. Scheper, D. H. Barouch, J. Hendriks, F. Struyf, M. Douoguih, J. Van Hoof and H. Schuitemaker, N. Engl. J. Med., 2021, 384, 1824–1835 CrossRef CAS.
- R. Deev, I. Plaksa, I. Bozo and A. Isaev, Am. J. Cardiovasc. Drugs, 2017, 17, 235–242 CrossRef CAS.
- T. Momin, K. Kansagra, H. Patel, S. Sharma, B. Sharma, J. Patel, R. Mittal, J. Sanmukhani, K. Maithal, A. Dey, H. Chandra, C. T. Rajanathan, H. P. Pericherla, P. Kumar, A. Narkhede and D. Parmar, eClinicalMedicine, 2021, 38, 101020 CrossRef.
- V. M. Andrade, A. Christensen-Quick, J. Agnes, J. Tur, C. Reed, R. Kalia, I. Marrero, D. Elwood, K. Schultheis, M. Purwar, E. Reuschel, T. McMullan, P. Pezzoli, K. Kraynyak, A. Sylvester, M. P. Mammen, P. Tebas, J. Joseph Kim, D. B. Weiner, T. R. F. Smith, S. J. Ramos, L. M. Humeau, J. D. Boyer and K. E. Broderick, npj Vaccines, 2021, 6, 1–4 CrossRef PubMed.
- A. Conforti, E. Marra, F. Palombo, G. Roscilli, M. Ravà, V. Fumagalli, A. Muzi, M. Maffei, L. Luberto, L. Lione, E. Salvatori, M. Compagnone, E. Pinto, E. Pavoni, F. Bucci, G. Vitagliano, D. Stoppoloni, M. L. Pacello, M. Cappelletti, F. F. Ferrara, E. D’Acunto, V. Chiarini, R. Arriga, A. Nyska, P. Di Lucia, D. Marotta, E. Bono, L. Giustini, E. Sala, C. Perucchini, J. Paterson, K. A. Ryan, A. R. Challis, G. Matusali, F. Colavita, G. Caselli, E. Criscuolo, N. Clementi, N. Mancini, R. Groß, A. Seidel, L. Wettstein, J. Münch, L. Donnici, M. Conti, R. De Francesco, M. Kuka, G. Ciliberto, C. Castilletti, M. R. Capobianchi, G. Ippolito, L. G. Guidotti, L. Rovati, M. Iannacone and L. Aurisicchio, Mol. Ther., 2021, 29, 1–16 Search PubMed.
- J. Y. Ahn, J. Lee, Y. S. Suh, Y. G. Song, Y.-J. Choi, K. H. Lee, S. H. Seo, M. Song, J.-W. Oh, M. Kim, H.-Y. Seo, J.-E. Kwak, J. W. Youn, J. W. Woo, E.-C. Shin, S.-H. Park, Y. C. Sung and J. Y. Choi, medRxiv, 2021, preprint, 2021.05.26.21257700 DOI:10.1016/S2666-5247(21)00358-X.
- Y. Li, R. Tenchov, J. Smoot, C. Liu, S. Watkins and Q. Zhou, ACS Cent. Sci., 2021, 7, 512–533 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2021, 39, 1479–1482 CrossRef PubMed.
- S. M. Gheibi-Hayat and K. Jamialahmadi, Biotechnol. Appl. Biochem., 2020, 1–9 Search PubMed.
- M. Mansoor and A. J. Melendez, Glasgow Biomedical Research Centre, Level, 2008, vol. 2, pp. 275–295 Search PubMed.
- C. A. Bradley, Nat. Milestones, 2019, 83, 59 Search PubMed.
- R. S. Geary, S. P. Henry and L. R. Grillone, Clin. Pharmacokinet., 2002, 41, 255–260 CrossRef PubMed.
- S. L. Hutcherson and R. Lanz, Am. J. Ophthalmol., 2002, 133, 467–474 CrossRef PubMed.
- S. T. Crooke and R. S. Geary, Br. J. Clin. Pharmacol., 2013, 76, 269–276 CrossRef CAS PubMed.
- R. S. Geary, B. F. Baker and S. T. Crooke, Clin. Pharmacokinet., 2015, 54, 133–146 CrossRef CAS PubMed.
- L. Gales, Pharmaceuticals, 2019, 12, 10–15 CrossRef PubMed.
- M. A. Gertz, M. Scheinberg, M. Waddington-Cruz, S. B. Heitner, C. Karam, B. Drachman, S. Khella, C. Whelan and L. Obici, Expert Rev. Clin. Pharmacol., 2019, 12, 701–711 CrossRef CAS PubMed.
- E. E. Neil and E. K. Bisaccia, J. Pediatr. Pharmacol. Ther., 2019, 24, 194–203 Search PubMed.
- I. Chen, Nat. Milestones, 2019, S17–S18 Search PubMed.
- J. Paik and S. Duggan, Drugs, 2019, 79, 1349–1354 CrossRef CAS PubMed.
- O. Esan and A. S. Wierzbicki, Drug Des., Dev. Ther., 2020, 14, 2623–2636 CrossRef CAS PubMed.
- Y. Masaki, Y. Iriyama, H. Nakajima, Y. Kuroda, T. Kanaki, S. Furukawa, M. Sekine and K. Seio, Nucleic Acid Ther., 2018, 28, 307–311 CrossRef CAS PubMed.
- J. Scharner, W. K. Ma, Q. Zhang, K. T. Lin, F. Rigo, C. Frank Bennett and A. R. Krainer, Nucleic Acids Res., 2020, 48, 802–816 CrossRef CAS PubMed.
- S. Dhillon, Drugs, 2020, 80, 1027–1031 CrossRef CAS PubMed.
- A. Aartsma-Rus and A. M. Krieg, Nucleic Acid Ther., 2017, 27, 1–3 CrossRef CAS PubMed.
- A. Aartsma-Rus and D. R. Corey, Nucleic Acid Ther., 2020, 30, 67–70 CrossRef CAS PubMed.
- M. Shirley, Drugs, 2021, 81, 875–879 CrossRef CAS PubMed.
- M. A. Havens and M. L. Hastings, Nucleic Acids Res., 2016, 44, 6549–6563 CrossRef PubMed.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS PubMed.
- F. Alhamadani, K. Zhang, R. Parikh, H. Wu, T. P. Rasmussen, R. Bahal, X. B. Zhong and J. E. Manautou, Drug Metab. Dispos., 2022, 50, 879–887 CrossRef CAS PubMed.
- T. Kasuya and A. Kugimiya, Nucleic Acid Ther., 2018, 28, 312–317 CrossRef CAS PubMed.
- P. J. Kamola, K. Maratou, P. A. Wilson, K. Rush, T. Mullaney, T. McKevitt, P. Evans, J. Ridings, P. Chowdhury, A. Roulois, A. Fairchild, S. McCawley, K. Cartwright, N. J. Gooderham, T. W. Gant, K. Moores, S. A. Hughes, M. R. Edbrooke, K. Clark and J. D. Parry, Mol. Ther.–Nucleic Acids, 2017, 8, 383–394 CrossRef CAS PubMed.
- A. Dieckmann, P. H. Hagedorn, Y. Burki, C. Brügmann, M. Berrera, M. Ebeling, T. Singer and F. Schuler, Mol. Ther.–Nucleic Acids, 2018, 10, 45–54 CrossRef CAS PubMed.
- Y. Mie, Y. Hirano, K. Kowata, A. Nakamura, M. Yasunaga, Y. Nakajima and Y. Komatsu, Mol. Ther.–Nucleic Acids, 2018, 10, 64–74 CrossRef CAS PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- M. M. Zhang, R. Bahal, T. P. Rasmussen, J. E. Manautou and X. Bo Zhong, Biochem. Pharmacol., 2021, 189, 114432, DOI:10.1016/j.bcp.2021.114432.
- H. Cui, X. Zhu, S. Li, P. Wang and J. Fang, ACS Omega, 2021, 6, 16259–16265 CrossRef CAS.
- M. Michael, J. Xu, D. M. Evans and P. Lu, J. Clin. Oncol., 2019, e14652–e14652 Search PubMed.
- T. Golan, E. Z. Khvalevsky, A. Hubert, R. M. Gabai, N. Hen, A. Segal, A. Domb, G. Harari, E. Ben David, S. Raskin, Y. Goldes, E. Goldin, R. Eliakim, M. Lahav, Y. Kopleman, A. Dancour, A. Shemi and E. Galun, Oncotarget, 2015, 6, 24560–24570 CrossRef PubMed.
- Y. Ramot, S. Rotkopf, R. M. Gabai, E. Zorde Khvalevsky, S. Muravnik, G. A. Marzoli, A. J. Domb, A. Shemi and A. Nyska, Toxicol. Pathol., 2016, 44, 856–865 CrossRef CAS PubMed.
- E. Z. Khvalevsky, R. Gabai, I. H. Rachmut, E. Horwitz, Z. Brunschwig, A. Orbach, A. Shemi, T. Golan, A. J. Domb, E. Yavin, H. Giladi, L. Rivkin, A. Simerzin, R. Eliakim, A. Khalaileh, A. Hubert, M. Lahav, Y. Kopelman, E. Goldin, A. Dancour, Y. Hants, S. Arbel-Alon, R. Abramovitch, A. Shemi and E. Galun, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20723–20728 CrossRef CAS PubMed.
- X. Liang, D. Li, S. Leng and X. Zhu, Biomed. Pharmacother., 2020, 125, 109997 CrossRef CAS PubMed.
- Y. Suzuki, K. Hyodo, Y. Tanaka and H. Ishihara, J. Controlled Release, 2015, 220, 44–50 CrossRef CAS PubMed.
- J. Cohen, Science, 2021, 372, 1381 CrossRef CAS.
- J. R. Babendure, J. L. Babendure, J. H. Ding and R. Y. Tsien, RNA, 2006, 12, 851–861 CrossRef PubMed.
- A. G. Orlandini von Niessen, M. A. Poleganov, C. Rechner, A. Plaschke, L. M. Kranz, S. Fesser, M. Diken, M. Löwer, B. Vallazza, T. Beissert, V. Bukur, A. N. Kuhn, Ö. Türeci and U. Sahin, Mol. Ther., 2019, 27, 824–836 CrossRef PubMed.
- K. D. Nance and J. L. Meier, ACS Cent. Sci., 2021, 7, 748–756 CrossRef PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef PubMed.
- E. Kon, U. Elia and D. Peer, Curr. Opin. Biotechnol, 2021, 2022, 329–336 Search PubMed.
- M. I. Henderson, Y. Eygeris, A. Jozic, M. Herrera and G. Sahay, Mol. Pharmaceutics, 2022, 19, 4275–4285 CrossRef CAS PubMed.
- S. Meulewaeter, G. Nuytten, M. H. Y. Cheng, S. C. De Smedt, P. R. Cullis, T. De Beer, I. Lentacker and R. Verbeke, J. Controlled Release, 2023, 357, 149–160 CrossRef CAS PubMed.
- N. N. Zhang, X. F. Li, Y. Q. Deng, H. Zhao, Y. J. Huang, G. Yang, W. J. Huang, P. Gao, C. Zhou, R. R. Zhang, Y. Guo, S. H. Sun, H. Fan, S. L. Zu, Q. Chen, Q. He, T. S. Cao, X. Y. Huang, H. Y. Qiu, J. H. Nie, Y. Jiang, H. Y. Yan, Q. Ye, X. Zhong, X. L. Xue, Z. Y. Zha, D. Zhou, X. Yang, Y. C. Wang, B. Ying and C. F. Qin, Cell, 2020, 182, 1271–1283 CrossRef CAS.
- Y. Zhu and G. Gao, RNA Biol., 2008, 5, 65–67 CrossRef CAS PubMed.
- X. Luo, X. Wang, Y. Gao, J. Zhu, S. Liu, G. Gao and P. Gao, Cell Rep., 2020, 30, 46–52 CrossRef CAS PubMed.
- X. Xia, Vaccines, 2021, 9, 1–19 Search PubMed.
- C. Poveda, A. B. Biter, M. E. Bottazzi and U. Strych, Vaccines, 2019, 7, 1–14 Search PubMed.
- H. Zhang, L. Zhang, A. Lin, C. Xu, Z. Li, K. Liu, B. Liu, X. Ma, F. Zhao, H. Jiang, C. Chen, H. Shen, H. Li, D. H. Mathews, Y. Zhang and L. Huang, Nature, 2023, 621, 396–403 CrossRef CAS PubMed.
- G. X. Chen, S. Zhang, X. Hua He, S. Y. Liu, C. Ma and X. P. Zou, OncoTargets Ther., 2014, 7, 1901–1909 CrossRef PubMed.
- Y. Li, W. Guo, X. Li, J. Zhang, M. Sun, Z. Tang, W. Ran, K. Yang, G. Huang and L. Li, Int. J. Oral Sci., 2021, 13, 38, DOI:10.1038/s41368-021-00145-1.
- E. T. M. Dams, P. Laverman, W. J. G. Oyen, G. Storm, G. L. Scherphof, J. W. M. Van Der Meer, F. H. M. Corstens and O. C. Boerman, J. Pharmacol. Exp. Ther., 2000, 292, 1071–1079 CAS.
- M. Estapé Senti, C. A. de Jongh, K. Dijkxhoorn, J. J. F. Verhoef, J. Szebeni, G. Storm, C. E. Hack, R. M. Schiffelers, M. H. Fens and P. Boross, J. Controlled Release, 2022, 341, 475–486 CrossRef PubMed.
- D. Shi, D. Beasock, A. Fessler, J. Szebeni, J. Y. Ljubimova, K. A. Afonin and M. A. Dobrovolskaia, Adv. Drug Delivery Rev., 2022, 180, 114079 CrossRef CAS PubMed.
- Y. Ju, W. S. Lee, E. H. Pilkington, H. G. Kelly, S. Li, K. J. Selva, K. M. Wragg, K. Subbarao, T. H. O. Nguyen, L. C. Rowntree, L. F. Allen, K. Bond, D. A. Williamson, N. P. Truong, M. Plebanski, K. Kedzierska, S. Mahanty, A. W. Chung, F. Caruso, A. K. Wheatley, J. A. Juno and S. J. Kent, ACS Nano, 2022, 16, 11769–11780 CrossRef CAS PubMed.
- M. Lamprinou, A. Sachinidis, E. Stamoula, T. Vavilis and G. Papazisis, Immunol. Res., 2023, 71, 356–372 CrossRef PubMed.
- S. M. Moghimi, Mol. Ther., 2021, 29, 898–900 CrossRef CAS PubMed.
- B. Yang, C. Qin, X. Hu, K. Xia, C. Lu, F. O. Gudda, Z. Ma and Y. Gao, Environ. Int., 2019, 132, 105087 CrossRef CAS PubMed.
- R. Aranda IV, S. M. Dineen, R. L. Craig, R. A. Guerrieri and J. M. Robertson, Anal. Biochem., 2009, 387, 122–127 CrossRef PubMed.
- S. J. Ahn, J. Costa and J. R. Emanuel, Nucleic Acids Res., 1996, 24, 2623–2625 CrossRef CAS PubMed.
- M. S. Levy, P. Lotfian, R. O’Kennedy, M. Y. Lo-Yim and P. A. Shamlou, Nucleic Acids Res., 2000, 28, e57, DOI:10.1093/nar/28.12.e57.
- S. N. Barnaby, A. Lee and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9739–9744 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, A. E. Prigodich, P. C. Patel and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 2072–2073 CrossRef CAS PubMed.
- K. Remaut, N. N. Sanders, F. Fayazpour, J. Demeester and S. C. De Smedt, J. Controlled Release, 2006, 115, 335–343 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, P. Dong, R. An, C. Xue, Y. Ge, L. Wei and X. Liang, Sci. Rep., 2015, 5, 11936 CrossRef CAS PubMed.
- A. H. S. Hall, J. Wan, A. Spesock, Z. Sergueeva, B. R. Shaw and K. A. Alexander, Nucleic Acids Res., 2006, 34, 2773–2781 CrossRef CAS PubMed.
- P. R. Walker, J. Leblanc, B. Smith, S. Pandey and M. Sikorska, Methods, 1999, 17, 329–338 CrossRef PubMed.
- L. M. Albright and B. E. Slatko, Curr. Protoc. Nucleic Acid Chem., 2000, A.3B.1–A.3B.5 Search PubMed.
- M. E. Hayes, D. C. Drummond, D. B. Kirpotin, W. W. Zheng, C. O. Noble IV, J. W. Park, J. D. Marks, C. C. Benz and K. Hong, Gene Ther., 2006, 13, 646–651 CrossRef PubMed.
- M. Oliveri, A. Daga, C. Cantoni, C. Lunardi, R. Millo and A. Puccetti, Eur. J. Immunol., 2001, 31, 743–751 CrossRef PubMed.
- R. S. Tuma, M. P. Beaudet, X. Jin, L. J. Jones, C. Y. Cheung, S. Yue and V. L. Singer, Anal. Biochem., 1999, 268, 278–288 CrossRef PubMed.
- E. Szántai and A. Guttman, Encyclopedia of Analytical Chemistry, 2015, pp. 1–26 Search PubMed.
- T. I. Todorov, O. De Carmejane, N. G. Walter and M. D. Morris, Electrophoresis, 2001, 22, 2442–2447 CrossRef PubMed.
- C. Liu, Y. Yamaguchi and X. Dou, Int. J. Mod. Phys. B, 2017, 31, 1–6 Search PubMed.
- L. De Scheerder, A. Sparén, G. A. Nilsson, P. O. Norrby and E. Örnskov, J. Chromatogr. A, 2018, 1562, 108–114 CrossRef PubMed.
- T. Lu, L. J. Klein, S. Ha and R. R. Rustandi, J. Chromatogr. A, 2020, 1618, 460875, DOI:10.1016/j.chroma.2020.460875.
- K. Sumitomo, M. Sasaki and Y. Yamaguchi, Electrophoresis, 2009, 30, 1538–1543 CrossRef PubMed.
- J. Raffaele, J. W. Loughney and R. R. Rustandi, Electrophoresis, 2022, 43, 1101–1106 CrossRef PubMed.
- É. Szöko and T. Tábi, J. Pharm. Biomed. Anal., 2010, 53, 1180–1192 CrossRef PubMed.
- T. H. Yang and P. L. Chang, J. Chromatogr. A, 2012, 1239, 78–84 CrossRef CAS PubMed.
- Y. Yu, S. H. Zhu, F. Yuan, X. H. Zhang, Y. Y. Lu, Y. L. Zhou and X. X. Zhang, Chem. Commun., 2019, 55, 7595–7598 RSC.
- L. Greenough, K. M. Schermerhorn, L. Mazzola, J. Bybee, D. Rivizzigno, E. Cantin, B. E. Slatko and A. F. Gardner, Nucleic Acids Res., 2015, 44, 15 CrossRef PubMed.
- A. Ric, V. Ong-Meang, V. Poinsot, N. Martins-Froment, F. Chauvet, A. Boutonnet, F. Ginot, V. Ecochard, L. Paquereau and F. Couderc, Electrophoresis, 2017, 38, 1624–1631 CrossRef CAS PubMed.
- Y. Qin, J. Zhao, Y. Huang, S. Li and S. Zhao, Anal. Methods, 2016, 8, 1852–1857 RSC.
- S. Hassan, Encyclopedia, 2020, 1, 30–41 CrossRef.
- S. F. Y. Li and L. J. Kricka, Clin. Chem., 2006, 52, 37–45 CrossRef CAS PubMed.
- M. A. A. Ragab and E. I. El-Kimary, Crit. Rev. Anal. Chem., 2021, 51, 709–741 CAS.
- Q. Huang, K. Kaiser and R. Benner, Limnol. Oceanogr.: Methods, 2012, 10, 608–616 CrossRef CAS.
- N. D. Sinha and K. E. Jung, Curr. Protoc. Nucleic Acid Chem., 2015, 2015, 10.5.1–10.5.39 Search PubMed.
- S. Shibayama, S. Ichiro Fujii and A. Takatsu, Chromatographia, 2014, 77, 1333–1338 CrossRef CAS.
- E. Lerouge, M. Basanta-Sanchez and S. V. Ranganathan, Honors Thesis, The Investigation of DNA and RNA Structural Differences Using Ultra High Performance Liquid Chromatography, University at Albany, 2016.
- G. V. Ullio-Gamboa, J. M. Llabot, M. F. Sanchez-Vallecillo, B. A. Maletto, S. D. Palma and D. A. Allemandi, Anal. Chem. Res., 2015, 4, 20–24 CrossRef CAS.
- S. Fueangfung, Y. Yuan and S. Fang, Nucleosides, Nucleotides Nucleic Acids, 2014, 33, 481–488 CrossRef CAS PubMed.
- M. J. Rocheleau, R. J. Grey, D. Y. Chen, H. R. Harke and N. J. Dovichi, Electrophoresis, 1992, 13, 484–486 CrossRef CAS PubMed.
- Z. Li, C. Liu, D. Zhang, S. Luo and Y. Yamaguchi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1011, 114–120 CrossRef CAS PubMed.
- A. Azarani and K. H. Hecker, Nucleic Acids Res., 2001, 29, E7 CrossRef CAS PubMed.
- M. J. Dickman, Chromatography Today, 2011, 4, 22–26 Search PubMed.
- A. O. Nwokeoji, M. E. Earll, P. M. Kilby, D. E. Portwood and M. J. Dickman, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1104, 212–219 CrossRef CAS PubMed.
- E. D. Close, A. O. Nwokeoji, D. Milton, K. Cook, D. M. Hindocha, E. C. Hook, H. Wood and M. J. Dickman, J. Chromatogr. A, 2016, 1440, 135–144 CrossRef CAS PubMed.
- C. L. Wysoczynski, S. C. Roemer, V. Dostal, R. M. Barkley, M. E. A. Churchill and C. S. Malarkey, Nucleic Acids Res., 2013, 41, e194 CrossRef CAS PubMed.
- K. H. Hecker, S. M. Green and K. Kobayashi, J. Biochem. Biophys. Methods, 2000, 46, 83–93 CrossRef CAS PubMed.
- N. Zhang, S. Shi, T. Z. Jia, A. Ziegler, B. Yoo, X. Yuan, W. Li and S. Zhang, Nucleic Acids Res., 2019, 47, e125 CrossRef CAS PubMed.
- W. M. Houser, A. Butterer, B. Addepalli and P. A. Limbach, Anal. Biochem., 2015, 478, 52–58 CrossRef CAS PubMed.
- J. Qiao, C. Liang, L. Wei, Z. Cao and H. Lian, J. Sep. Sci., 2016, 39, 4502–4511 CrossRef CAS PubMed.
- J. Betker, T. Smyth, W. Wang and T. J. Anchordoquy, J. Pharm. Sci., 2011, 101, 987–997 CrossRef PubMed.
- I. C. Santos and J. S. Brodbelt, J. Sep. Sci., 2021, 44, 340–372 CrossRef CAS PubMed.
- L. Tan, V. Yeo, Y. Yang and P. Gagnon, Anal. Bioanal. Chem., 2015, 407, 4173–4181 CrossRef CAS PubMed.
- N. Boulter, F. Garces Suarez, S. Schibeci, T. Sunderland, O. Tolhurst, T. Hunter, G. Hodge, D. Handelsman, U. Simanainen, E. Hendriks and K. Duggan, BMC Biotechnol., 2016, 16, 27 CrossRef PubMed.
- M. J. Brisco, S. Latham, P. A. Bartley and A. A. Morley, Biotechniques, 2010, 49, 893–897 CrossRef CAS PubMed.
- T. D. Schmittgen and K. J. Livak, Nat. Protoc., 2008, 3, 1101–1108 CrossRef CAS PubMed.
- M. N. Nikiforova and Y. E. Nikiforov, General Principles of Molecular Biology, 2010, pp. 42–57 Search PubMed.
- R. Vaidyanathan, Epicentre Forum, 2007, 12, 22 Search PubMed.
- D. V. Jose and B. Román, J. Exp. Bot., 2012, 63, 695–709 CrossRef PubMed.
- J. Lle Vermeulen, K. De Preter, S. Lefever, J. Nuytens, F. De Vloed, S. Derveaux, J. Hellemans, F. Speleman and J. Vandesompele, Nucleic Acids Res., 2011, 39, e63 CrossRef PubMed.
- T. Nolan, R. E. Hands and S. A. Bustin, Nat. Protoc., 2006, 1, 1559–1582 CrossRef CAS PubMed.
- C. Du Cheyne, Y. Chen, J. De Craene, O. Thas and W. De Spiegelaere, Anal. Biochem., 2021, 626, 114217 CrossRef PubMed.
- J. Björkman, D. Švec, E. Lott, M. Kubista and R. Sjöback, Biomol. Detect. Quantif., 2016, 6, 4–12 CrossRef PubMed.
- D. Klein, Trends Mol. Med., 2002, 8, 257–260 CrossRef PubMed.
- A. Didelot, S. K. Kotsopoulos, A. Lupo, D. Pekin, X. Li, I. Atochin, P. Srinivasan, Q. Zhong, J. Olson, D. R. Link, P. Laurent-Puig, H. Lè Ne Blons, J. B. Hutchison and V. Taly, Clin. Chem., 2013, 59, 815–823 CrossRef PubMed.
- S. Colombo, H. M. Nielsen and C. Foged, J. Controlled Release, 2013, 166, 220–226 CrossRef PubMed.
- M. Shin, P. Meda Krishnamurthy, G. Devi and J. K. Watts, Nucleic Acid Ther., 2022, 32, 66–73 CrossRef CAS PubMed.
- B. Gang, W. J. Rhee and A. Tsourkas, Annu. Rev. Biomed. Eng., 2009, 11, 25–47 CrossRef PubMed.
- M. Mitsuhashi, S. Tomozawa, K. Endo and A. Shinagawa, Clin. Chem., 2006, 52, 634–642 CrossRef CAS PubMed.
- M. Bezanilla, B. Drake, E. Nudler, M. Kashlev, P. K. Hansma and H. G. Hansma, Biophys. J., 1994, 67, 2454–2459 CrossRef CAS PubMed.
- H. G. Addelhady, S. Allen, M. C. Davies, C. J. Roberts, S. J. B. Tendler and P. M. Williams, Nucleic Acids Res., 2003, 31, 4001–4005 CrossRef PubMed.
- H. G. Abdelhady, Y. L. Lin, H. Sun and M. E. H. ElSayed, PLoS One, 2013, 8, e61710, DOI:10.1371/journal.pone.0061710.
- S. Santos, V. Barcons, H. K. Christenson, D. J. Billingsley, W. A. Bonass, J. Font and N. H. Thomson, Appl. Phys. Lett., 2013, 103, 063702 CrossRef.
- A. K. Mazur and M. Maaloum, Nucleic Acids Res., 2014, 42, 14006–14012 CrossRef CAS PubMed.
- T. Elsässer, S. Brons, K. Psonka, M. Scholz, E. Gudowska-Nowakc and G. Taucher-Scholz, Radiat. Res., 2008, 169, 649–659 CrossRef PubMed.
- D. Pang, A. R. Thierry and A. Dritschilo, Front. Mol. Biosci., 2015, 2, 1–7 CAS.
- Z. A. Ratan, S. Bin Zaman, V. Mehta, M. F. Haidere, N. J. Runa and N. Akter, Cureus, 2017, 9, e1325 Search PubMed.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308–311 CrossRef CAS PubMed.
- S. Kramer, Nucleic Acids Res., 2017, 45, e49 CrossRef PubMed.
- B. S. Morgan, J. E. Forte and A. E. Hargrove, Nucleic Acids Res., 2018, 46, 8025–8037 CrossRef CAS PubMed.
- R. W. Dirks and H. J. Tanke, Biotechniques, 2006, 40, 489–496 CrossRef CAS.
- R. W. Dirks, C. Molenaar and H. J. Tanke, Histochem. Cell Biol., 2001, 115, 3–11 CrossRef CAS PubMed.
- R. M. Clegg, Förster resonance energy transfer-FRET what is it, why do it, and how it's done, Elesvier B.V., 2009, vol. 33 Search PubMed.
- H. Uchiyama, K. ’ichi Hirano, M. Kashiwasake-Jibu and K. Taira, J. Biol. Chem., 1996, 271, 380–384 CrossRef CAS.
- K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856–870 CrossRef CAS PubMed.
- D. Zhao, Y. Yang, N. Qu, M. Chen, Z. Ma, C. J. Krueger, M. A. Behlke and A. K. Chen, Biomaterials, 2016, 100, 172–183 CrossRef CAS.
- R. Monroy-Contreras and L. Vaca, J. Nucleic Acids, 2011, 2011, 15 Search PubMed.
- S. Mao, Y. Ying, R. Wu and A. K. Chen, iScience, 2020, 23, 101801 CrossRef CAS PubMed.
- B. Charroux, F. Daian and J. Royet, iScience, 2020, 23, 101152 CrossRef CAS PubMed.
- C. Srinivasan, S. Siddiqui, L. K. Silbart, F. Papadimitrakopoulos and D. J. Burgess, Bioconjugate Chem., 2009, 20, 163–169 CrossRef CAS PubMed.
- R. Reif, F. Haque and P. Guo, Nucleic Acid Ther., 2012, 22, 428–437 CrossRef CAS.
- C. Hodges, R. P. Kafle and J.-C. Meiners, Single Molecule Spectroscopy and Superresolution Imaging X, SPIE, 2017, vol. 10071, p. 1007102 Search PubMed.
- R. Macháň and T. Wohland, FEBS Lett., 2014, 588, 3571–3584 CrossRef PubMed.
- S. A. Kim, K. G. Heinze and P. Schwille, Nat. Methods, 2007, 4, 963–973 CrossRef PubMed.
- U. Kettling, A. Koltermann, P. Schwille and M. Eigen, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1416–1420 CrossRef.
- A. Sasaki and M. Kinjo, J. Controlled Release, 2010, 143, 104–111 CrossRef PubMed.
- K. Remaut, B. Lucas, K. Braeckmans, N. Sanders, S. De Smedt and J. Demeester, J. Controlled Release, 2008, 132, e1–e18 CrossRef.
- P. Heissig, W. Schrimpf, P. Hadwiger, E. Wagner and D. C. Lamb, PLoS One, 2017, 12, 1–19 CrossRef PubMed.
- T. Takagi, H. Kii and M. Kinjo, Curr. Pharm. Biotechnol., 2005, 5, 199–204 Search PubMed.
- A. Sasaki, J. Yamamoto, T. Jin and M. Kinjo, Sci. Rep., 2015, 5, 14428 CrossRef PubMed.
- K. Braeckmans, D. Vercauteren, J. Demeester and S. C. De Smedt, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. S. Verkman, J. Biol. Chem., 2000, 275, 1625–1629 CrossRef PubMed.
- J. C. Politz, E. S. Browne, D. E. Wolf and T. Pederson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6043–6048 CrossRef.
- K. Remaut, N. N. Sanders, B. G. De Geest, K. Braeckmans, J. Demeester and S. C. De Smedt, Mater. Sci. Eng., R, 2007, 58, 117–161 CrossRef.
- S. C. De Smedt, K. Remaut, B. Lucas, K. Braeckmans, N. N. Sanders and J. Demeester, Adv. Drug Delivery Rev., 2005, 57, 191–210 CrossRef CAS.
- A. Muneer, S. M. Fati, N. Arifin Akbar, D. Agustriawan and S. Tri Wahyudi, J. King Saud Univ., 2022, 34, 7419–7432, DOI:10.1016/j.jksuci.2021.10.001.
- H. K. Wayment-Steele, W. Kladwang, A. M. Watkins, D. S. Kim, B. Tunguz, W. Reade, M. Demkin, J. Romano, R. Wellington-Oguri, J. J. Nicol, J. Gao, K. Onodera, K. Fujikawa, H. Mao, G. Vandewiele, M. Tinti, B. Steenwinckel, T. Ito, T. Noumi, S. He, K. Ishi, Y. Lee, F. Öztürk, A. Chiu, E. Öztürk, K. Amer, M. Fares, Eterna Participants and R. Das, Nat. Mach. Intell., 2022, 4, 1174–1184 CrossRef PubMed.
- O. Yaish and Y. Orenstein, Bioinformatics, 2022, 38, 1087–1101 CrossRef CAS PubMed.
- T. Saito and H. Doi, Front. Ecol. Evol., 2021, 9, 623831 CrossRef.
- Y. Li, B. Li, C. J. Li and L. J. Li, Expert Opin. Biol. Ther., 2015, 15, 437–454 CrossRef CAS.
- Z. Peng and X. Zhang, EP1642981A1, 2004.
- R. Deev, I. Plaksa, I. Bozo, N. Mzhavanadze, I. Suchkov, Y. Chervyakov, I. Staroverov, R. Kalinin and A. Isaev, Ther. Adv. Cardiovasc. Dis., 2018, 12, 237–246 CrossRef CAS.
- N. Miller, Nat. Rev. Drug Discovery, 2012, 11, 419 CrossRef CAS.
- A. M. Keeler and T. R. Flotte, Annu. Rev. Virol., 2019, 6, 601–621 CrossRef CAS PubMed.
- E. Dolgin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23866–23870 CrossRef CAS.
- S. L. Hutcherson and R. Lanz, Am. J. Ophthalmol., 2002, 133, 467–474 CrossRef.
- R. Chopra, J. D. Eaton, A. Grassi, M. Potter, B. Shaw, C. Salat, P. Neumeister, G. Finazzi, M. Iacobelli, K. Bowyer, H. G. Prentice and T. Barbui, Br. J. Haematol., 2000, 111, 1122–1129 Search PubMed.
- L. Gales, Pharmaceuticals, 2019, 12, 78 CrossRef PubMed.
- EMA, Pegaptanib, 2006.
- X. Zhang, V. Goel, H. Attarwala, M. T. Sweetser, V. A. Clausen and G. J. Robbie, J. Clin. Pharmacol., 2020, 60, 37–49 CrossRef PubMed.
- A. Kilanowska and S. Studzińska, RSC Adv., 2020, 10, 34501–34516 RSC.
- M. Friedrich and A. Aigner, BioDrugs, 2022, 36, 549–571 CrossRef.
- K. D. Nance and J. L. Meier, Cite This: ACS Cent. Sci., 2021, 7, 748–756 CAS.
| This journal is © The Royal Society of Chemistry 2024 |