
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Productive chemistry induced by mechanochemically generated macroradicals
Chenxu
Wang
 *ab,
Cai-Li
Sun
a and
Roman
Boulatov
*b
*ab,
Cai-Li
Sun
a and
Roman
Boulatov
*b
aCollege of Chemistry and Chemical Engineering, Xi’an University of Science and Technology, Xi’an 710054, China. E-mail: chenxuwang@xust.edu.cn
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: boulatov@liverpool.ac.uk
First published on 16th August 2024
Abstract
Large or repeated mechanical loads degrade polymeric materials by accelerating chain fragmentation. This mechanochemical backbone fracture usually occurs by homolysis of otherwise inert C–C, C–O and C–S bonds, generating highly reactive macroradicals. Because backbone fracture is detrimental on its own and the resulting macroradicals can initiate damaging reaction cascades, a major thrust in contemporary polymer mechanochemistry is to suppress it, usually by mechanochemical release of “hidden length” that dissipates local molecular strain. Here we summarize an emerging complementary strategy of channelling mechanochemically generated macroradicals in reaction cascades to form new load-bearing chemical bonds, which enables local self-healing or self-strengthening, and/or to generate mechanofluorescence, which could yield detailed quantitative molecular understanding of how material-failure-inducing macroscopic mechanical loads distribute across the network. We aim to identify generalizable lessons derivable from the reported implementations of this strategy and outline the key challenges in adapting it to diverse polymeric materials and loading scenarios.
 Chenxu Wang | Chenxu Wang received his BEng degree from Sichuan University and PhD degree from University of Liverpool focusing on polymer mechanochemistry under supervision of Prof. Roman Boulatov. He is currently a lecturer in Xi’an University of Science and Technology with interests including understanding mechanochemical phenomena through experiments and quantum mechanics computation and developing stress-responsive polymer materials. |
 Cai-Li Sun | Cai-Li Sun received her PhD from the Technical Institute of Physics and Chemistry, Chinese Academy of Sciences in 2017 under the supervision of Prof. Qing-Zheng Yang and Prof. Chen-Ho Tung. After a postdoc at University of Liverpool with Prof. Roman Boulatov, she joined the College of Chemistry and Chemical Engineering at Xi’an University of Science and Technology in 2021. Her current research interest focuses on photochemistry, mechanochemistry and energy conversion using molecular photoswitches. |
 Roman Boulatov | Roman Boulatov earned a PhD at Stanford for work on metalloporphyrins, particularly for catalytic low-temperature oxygen reduction under the supervision of Prof. James Collman. After a postdoc at Harvard with Prof. George Whitesides, where he explored unconventional means of energy conversion, he started his independent research program in polymer mechanochemistry at the UIUC before moving to Liverpool in 2012. |
1. Introduction
Mechanochemical reactions are induced by mechanical loading, such as stretching, shearing and compression. Polymeric materials experience diverse mechanical loads throughout their lifetime: during synthesis, processing, service and recycling, which makes their mechanochemical reactions ubiquitous. Hence, polymer mechanochemistry is central to understanding how polymers respond to mechanical loads in multiple areas of polymer science, including melt processing, jet injection, ageing, material failure, and tribology.1–4 It also has the potential to revolutionize the design and synthesis of polymers, leading to more sustainable processes, novel materials, and improved control over polymer properties.5–8The underlying basis of polymer mechanochemistry is the dramatically increased reactivities of macromolecular chains confined to highly non-equilibrium stretched geometries (Fig. 1a), including opening of bond angles and elongations of backbone bonds.4,9 These geometries result from interactions between a chain and its surroundings under stretching, compression, or shear and change the thermodynamic and kinetic stabilities of individual bonds and monomers, usually (but not necessarily10) accelerating reactions that are unobservably-slow in strain-free chains.11,12 Mechanical loads have been demonstrated to accelerate (usually homolytic) backbone bond scissions,13,14 which fracture overstretched polymer chains, isomerisation of backbone cis-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds to trans configuration, formally electrocyclic reactions, and dissociation of non-covalent interactions. All these reactions have been exploited to inhibit macroscopic failure of bulk polymers (for example, by releasing “hidden length”15–17) and/or to develop stress-reporting, self-healing and/or self-strengthening and other mechanoresponsive materials.8,18
C bonds to trans configuration, formally electrocyclic reactions, and dissociation of non-covalent interactions. All these reactions have been exploited to inhibit macroscopic failure of bulk polymers (for example, by releasing “hidden length”15–17) and/or to develop stress-reporting, self-healing and/or self-strengthening and other mechanoresponsive materials.8,18
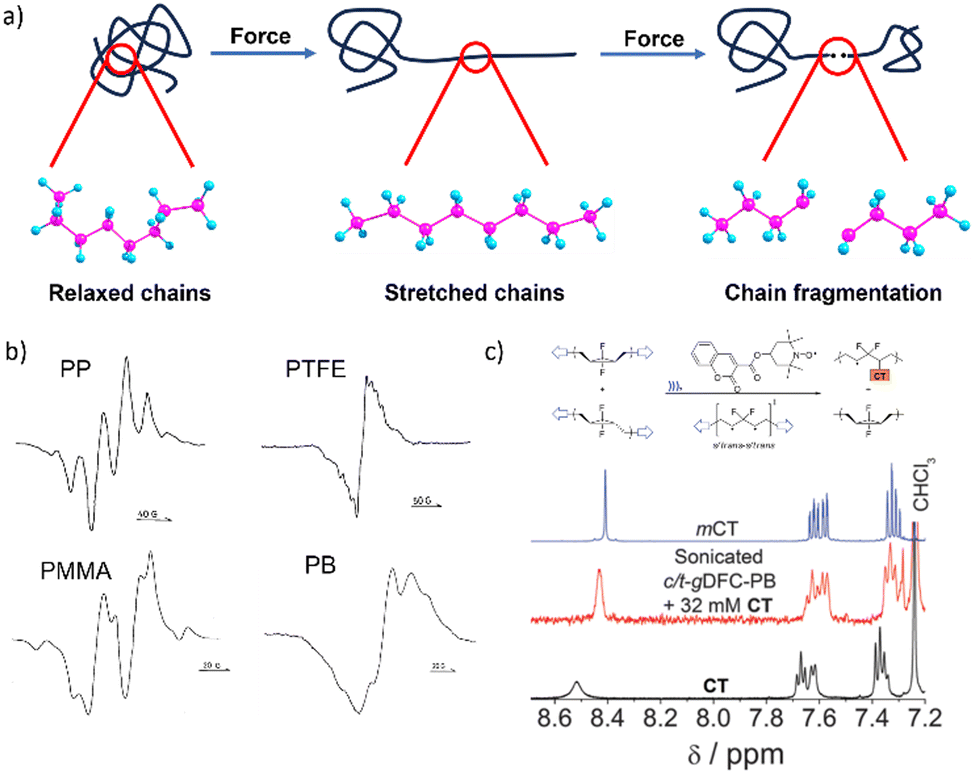 | ||
| Fig. 1 Mechanochemical generation of macroradicals. (a) A cartoon representation of the events underlying mechanochemical generation of macroradicals by stretching a polymer chain. (b) ESR spectra of several polymers milled at 77 K in vacuum confirming the generation of radicals. Adapted from ref. 19 with permission. (c) 1H-NMR spectrum of the polymer-radical scavenger (coumarin-2,2,6,6-tetramethylpiperidine1-oxyl, CT) adduct, showing shifts in the CT resonances that are consistent with carbon addition to the oxygen-centred radical (mCT: the O-methylated CT model compound). Bimolecular trapping of macroradicals in sonicated solutions is very rare and in this case is probably enabled by the relative inertness of the putative internal macrodiradical, which resides in a deep energy well over a broad range of forces. Adapted from ref. 20 with permission. | ||
Mechanochemical reactions are governed by the coupling of dynamic processes across multiple length and timescale.21,22 This makes the relationship between the macroscopic control parameters, such as stress tensors, and microscopic reaction probabilities intractable. Consequently, both mechanistic and microkinetic descriptions of mechanochemical reactions of polymers are far more challenging than that of purely thermal reactions.23 Despite important recent advances, conceptually robust and generalizable approaches to mechanistic and quantitative descriptions of polymer mechanochemistry, particularly at the molecular scale, remain to be developed.24
Load-induced backbone fragmentation is the most common and best-understood mechanochemical polymer reaction (Fig. 1a). Suggested by Staudinger in the 1930s to rationalize the reduction of the molecular weights of masticated polymers,25 it was subsequently studied by Kauzmann and Eyring, who proposed that the kinetics of mechanochemical chain fracture in solution increases exponentially with the force stretching the chain.26 Subsequently, this postulate was generalized to bulk polymers (with stress acting as the macroscopic equivalent of the single-chain force) and to single-molecule force spectroscopy, and rationalized in terms of the Kramers interpretation of the transition state theory.
Mechanochemical chain fracture, particularly by homolysis of C–C backbone bonds of commodity polymers such as polystyrene (PS)27 and polyethylene (PE),28 has been studied extensively. The expected generation of macroradicals has been demonstrated repeatedly in diverse loading scenarios by electron spin resonance (ESR)19 or inferred from the incorporation of radical scavengers in polymer chains (Fig. 1b and c).20,29 The high bond-dissociation energy of the C–C bond (75–90 kcal mol−1) makes its spontaneous homolysis under ambient conditions extremely unlikely. Quantum-chemical calculations suggest that the same bond in a polymer chain stretched to 5 nN has the half-life in the microsecond range at 300 K (vs. 1038 years for strain-free chain).4 Homolysis of a backbone bond is now accepted as being the primary mechanism of mechanochemical chain fracture in bulk elastomers, glasses and melts under stretching,2,6 grinding30 and shearing;29 in rapid flows of polymer solutions (including those generated by sonication);31 and during freeze–thaw cycles.32 Macroscopic manifestations of mechanochemical chain fracture include reduction of tensile strength and elastic modulus of solids, and viscosity of melts and solutions. Reduction of polymer molar masses accompanying these changes has been documented by size-exclusion chromatography.
The primary focus of contemporary polymer mechanochemistry is to divert the reactivity of an overstretched chain away from macroradical-generating backbone bond homolysis (so-called non-selective mechanochemistry) towards more complex reactions of specifically designed monomers. Yet, such mechanochemical macroradical generation is almost certainly unavoidable because the capacity of polymer backbones to support large changes in their end-to-end and even contour lengths underlies the technological utility of polymers. In theory, this makes the idea of extracting additional functionality from these inevitable macroradical byproducts resemble free lunch. In practice, however, the compositions, topologies and microstructures of commercial polymers represent a complex compromise among many often-contradictory requirements (e.g., cost, processibility, mechanical properties). How to engineer these productive reaction channels into a polymer while preserving or enhancing its technological or economic value remains an exceptionally challenging and largely unsolved problem.
Generating macroradicals by intentional application of mechanical load offers at least two advantages over irradiation or heating. The most obvious is universality: existing empirical and computational evidence suggest that a macromolecule of any composition fractures by backbone-bond homolysis with detectable probability if stretched hard or fast enough. This eliminates the complications (added cost, reduced shelf-life, handling restrictions) inherent in the requirement for specific (photo)reactive moieties either at a chain terminus or within the backbone to generate macroradicals by irradiation or heating. More subtle and still-underappreciated is the role of molecular recoil in suppressing macroradical disproportionation and recombination in high-viscosity environments, such as concentrated solutions, melts and glasses.2,33 Such disproportionation and recombination can lower the efficiency of non-mechanochemical generations by 50% of more.34,35
Only overstretched backbones fracture mechanochemically on practical timescales. Most of the molecular strain energy of such backbones is stored in non-reactive molecular degrees of freedom,36 such as larger-than-equilibrium bond angles (Fig. 1a). Once the chain traverses the scissile transition state, this energy dissipates by retraction of the relatively mobile radical-bearing termini away from each other to restore the strain-free backbone conformers. An example of the importance of chain recoil in mechanochemical chain fracture comes from mechanochromism of polymers containing dimers of stabilized radicals derived from (multi)aryl- or multi(carbonitrile)-substituted benzofuranones, fluorenes, imidazoles, or succinonitriles (Fig. 2). Under ambient conditions the radicals exist as colourless dimers, thanks to the highly favourable and nearly barrierless dimerization (ΔGo < −30 kcal mol−1, ΔG‡ ∼10 kcal mol−1).37 When stretched to >2 nN, the dimers dissociate at sub-ms timescales, producing persistent colour (for reference, single-chain force of ∼5 nN is required to reduce the half-life of an aliphatic C–C bond to ∼1 ms). Once the macroscopic load is released, the colour fades at a rate consistent with slow diffusion-limited recombination. Scrambling of dimers in loaded mixtures of chains derivatized with distinct homodimers suggests that recombination of a pair of radicals arising from fracture of two separate chains is kinetically competitive with recombination of the same fractured chain. These observations strongly suggest chain recoil following mechanochemical backbone fracture.
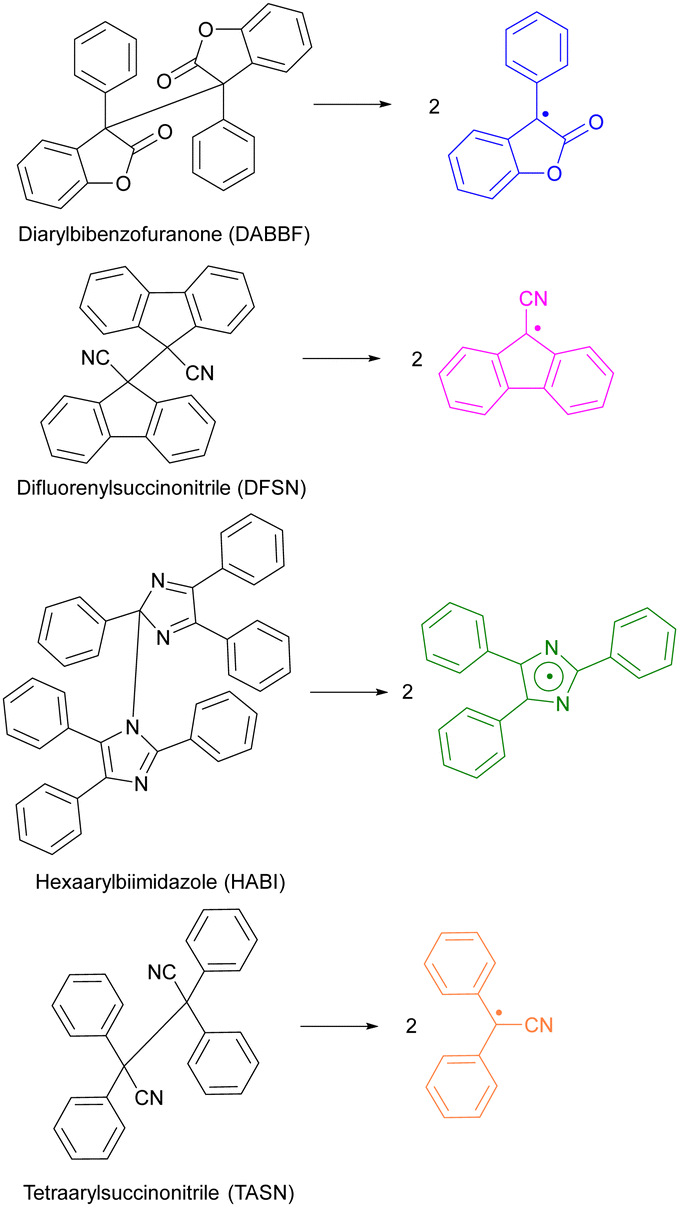 | ||
| Fig. 2 Reported scissile mechanochromic reactions: the dimers are colourless, the radical products span the colour from yellow to green. Adapted from ref. 32 and 38–40. | ||
In this review we aim to analyse the reported approaches to converting the usually destructive process of non-selective mechanochemical homolysis of a polymer backbone to productive uses. In the next section we summarize how these are applied to synthetic problems. Subsequently, we discuss how they have been adapted to scenarios where mechanochemical radical generation would normally be incidental to the intended material use (although the reported implementations still rely on mechanical loading designed to maximize the production of macroradicals). Our review excludes the few recent papers41 suggesting biological role of mechanochemical chain fracture of structural proteins, and of macroradical derived from such fracture, because the available data are too limited to support generalizable inferences. We conclude the review by analysing the challenges of harnessing mechanical loads to initiate productive chemistry and point out the possible further developments in such macroradical-driven productive mechanochemistry.
2. Synthetic application of mechanochemically generated macroradicals
Mechanochemistry is becoming an increasingly widespread tool of synthetic chemistry because it often allows elimination of reaction solvents, reduction in reaction temperatures and attendant lowering of energy consumption.42–46 Mechanochemical synthesis is usually carried out in ball mills or similar mechanical devices, whose internal moving parts grind, shear, and compress powders; or in sonicated solutions. These processes often generate reactive intermediates (e.g., radicals and ions) either small molecules, such as hydroxyl radicals from water sonolysis,47 or macroradicals from homolysis of polymer chains, which drive useful chemistry. Reported mechanochemical syntheses, including of polymers, have been extensively reviewed.18,48–52 This section aims at inverting the perspective of these reviews from the product or loading mechanism to the fate of the mechanochemically-generated macroradical.The reported examples of synthetic exploitations of mechanochemically-generated macroradicals include (Fig. 3): (a) initiation of polymerisation of free monomers; (b) generation of block copolymers by recombination of macroradicals of different chain composition; (c) synthesis of semi-telechelic polymers; (d) grafting of chains on surfaces and (e) generation of reactive oxygen species. Compared to other polymerization approaches, mechanochemically generated macroradicals have the advantage of simplicity and convenience (e.g., no need for end-derivatization), but often require more optimization of reaction conditions to maximize reaction selectivity.
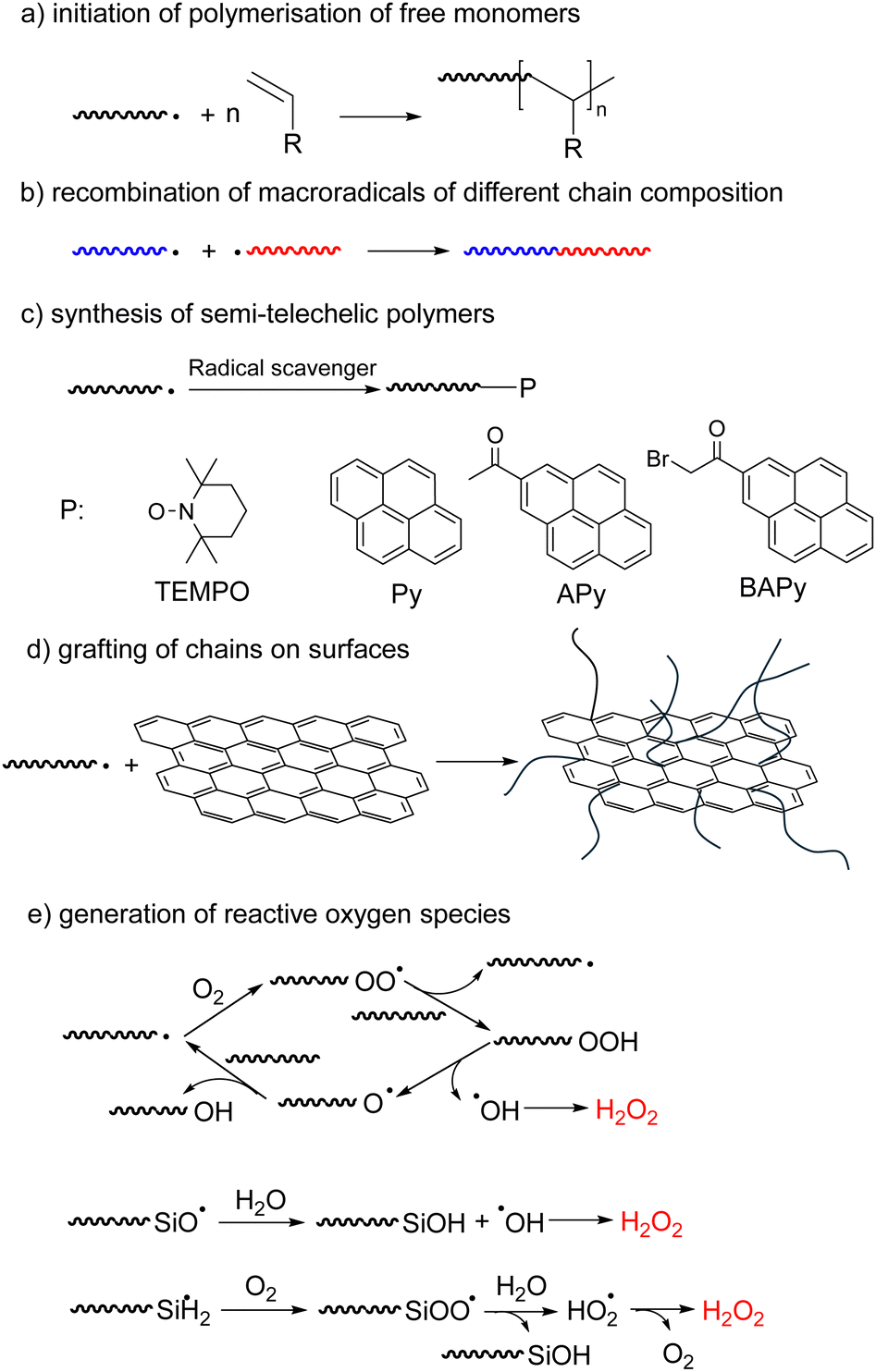 | ||
| Fig. 3 Five types of synthetic applications of mechanochemically-generated macroradicals: (a–c) homogeneous formation of new backbone bonds; (d) interphasial bond formation; (e) chemistries mediated by small molecules. Wavy lines represent polymer chains. | ||
The earliest demonstrations of the synthetic utility of mechanochemically generated macroradicals were to initiate polymerisation of monomers (Fig. 3a), starting with the 1954 discovery that sonicating solutions of polymethyl methacrylate (PMMA) and styrene produced PMMA-polystyrene (PS) block copolymers (Fig. 4a).53 The work was rapidly extended to sonicating mixed solutions of polyacryl-amide and acrylonitrile to yield polyacrylamide-polyacrylonitrile block copolymers (Fig. 4b);54 and of aqueous solutions of hydroxyethyl cellulose (HEC) and methyl methacrylate or acrylic acid (AA) to generate HEC/PMMA and HEC/PAA copolymers, respectively.55 Vibratory ball-milling of powdered mixtures of HEC and AA yielded HEC–PAA graft copolymers (Fig. 4c).56 Many studies in this area aim to demonstrate the utility of mechanochemically-generated radicals for the preparation of materials with otherwise difficult-to-access properties, as illustrated by HEC copolymers mentioned above, or a polybutadiene-PS block copolymer, generated by sonicating a styrene solution of polybutadiene (Fig. 4d), for manufacturing high impact PSs.57
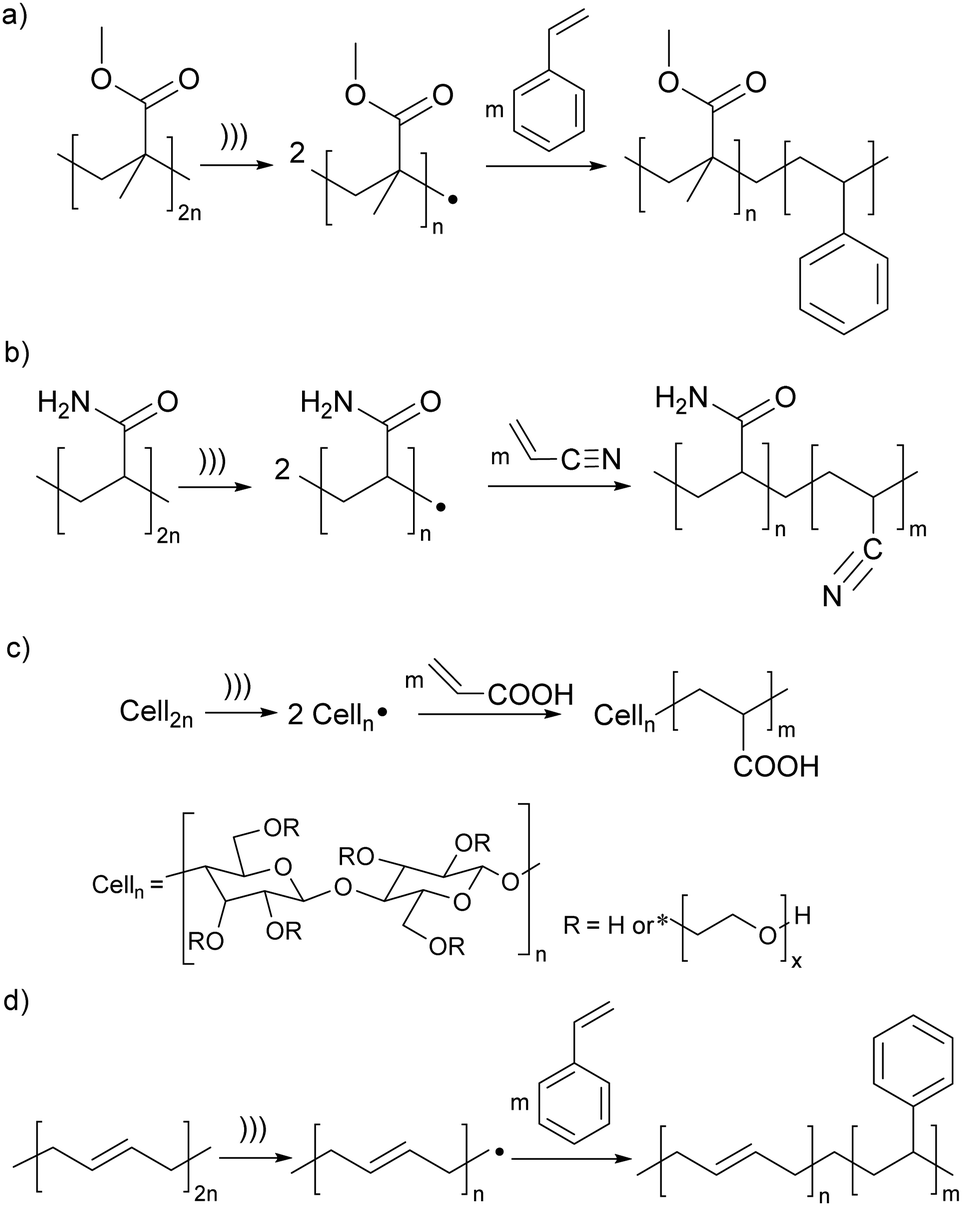 | ||
| Fig. 4 Examples of polymerisation of monomers initiated by mechanochemically-generated macroradicals from: (a) acrylates; (b) polyacrylamide; (c) cellulose; (d) butadiene. ))) signifies ultrasound; radical chain terminations are not shown. Adapted from ref. 54, 55, 57 and 58. | ||
Synthetic applications of sonication can suffer side-reactions from small-molecule radicals generated sonolitically and from uncontrollable flow fields from cavitation shock waves, making high-speed agitation an attractive alternative for synthetic applications. Early detailed kinetic simulations of polyvinyl acetate-vinyl acetate solutions59 were critical in enabling systematic identification of optimal reaction conditions and improved control of kinetics and yield.60 As expected, initiation of polymerization requires more viscous solutions than in sonication4 and as a result is unobservable below threshold polymer concentrations.61,62
Mechanochemically-generated macroradicals appear to offer particular advantages for the synthesis of block copolymers of immiscible homopolymers, by recombination of macroradicals derived from each component of the polymer mixtures (Fig. 3b). Such block copolymers are commercially valuable as additives that improve the mechanical properties of polymer blends. Illustrative examples include sonicating solutions of PS with poly(n-vinyl carbazole),63 poly(alkyl methacrylates),58 poly(cis-butadiene) or poly(methyl phenyl silane)64 in THF or toluene (Fig. 5a), or of mixed aqueous solutions of poly(vinyl chloride) and poly(vinyl alcohol).65 As is the case for high-speed agitation, mechanistic and kinetic analyses are usually required to optimize copolymer yields. Interpolymer radical recombination was confirmed either by IR65 or by fluorescence-detection SEC using a homopolymer component labelled with chromophores such as pyrene (Fig. 5b).66
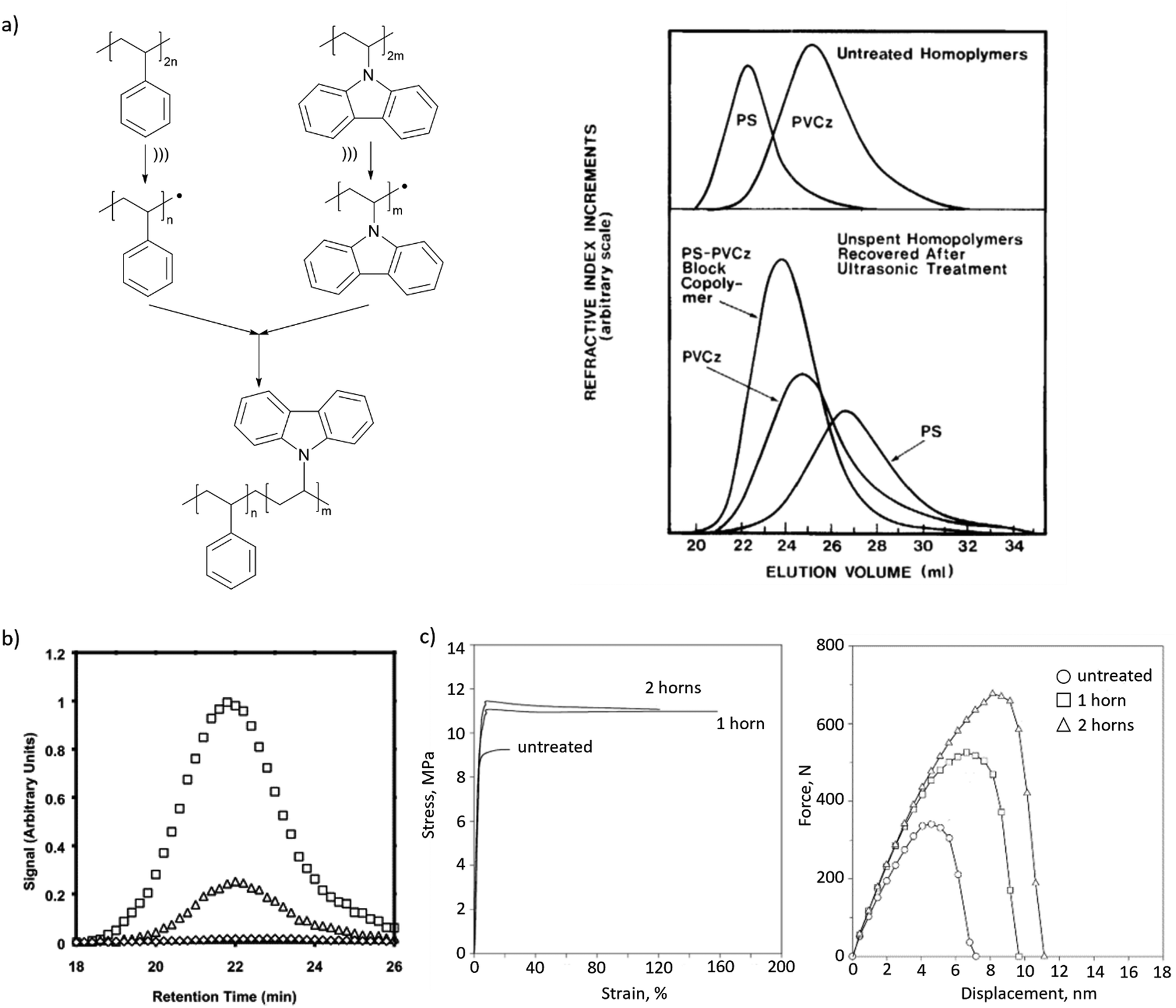 | ||
| Fig. 5 Block copolymers produced by recombination of mechanochemically generated macroradicals. (a) Sonication of solutions of PS and PVCz produced their copolymer, as confirmed by GPC. Adapted from ref. 63 with permission. (b) Fluorescence-detector gel-permeation chromatograms revealing pyrene emission of PnBMA/PS copolymer obtained in sonication with pyrene-labelled PS. Adapted from ref. 66 with permission. (c) Tensile and impact behaviour of PP/NR blends with or without (‘untreated’) ultrasonication during blending. Adapted from ref. 67 with permission. | ||
Recombination of chemically-distinct macroradicals was observed during shearing pulverization of PMMA/pyrene-labelled PS solid mixtures68 and by applying ultrasound during twin-screw extrusion of immiscible melts of polypropylene (PP) and natural rubber (NR). The latter yielded material with improved mechanical properties compared to that generated without ultrasonication, which was attributed, based on viscosity tests, SEM and AFM imaging, tensile and impact test (Fig. 5c), to the formation of copolymers.67 A remaining road-block for widespread uses of these approaches is the continued lack of means to control the microstructure (e.g., the number of blocks and their lengths) of the resulting products.
Macroradicals generated in milled solids can be trapped by diverse small molecules to produce well defined semi-telechelic polymers (Fig. 3c).69 This process has been used most often to install end-groups suitable for reversible-deactivation radical polymerisation. Most extensively studied are stable-free-radical mediated polymerization starting with TEMPO-terminated chains,70–72 but examples of atom-transfer radical polymerizations, using Cl-terminated chains, were also reported.73 Chain extensions and synthesis of block-copolymers have been demonstrated and the nature of both the initial polymer and the radical scavenger used to trap its mechanochemically-generated radicals on subsequent polymerization kinetics have been studied.
Mechanochemically generated macroradicals offer a practical means of derivatizing diverse surfaces with covalently-anchored macromolecules (Fig. 3d).74,75 For example, sonicating a PVA solution containing suspended graphene grafted PVA onto the graphene, which improved its mechanical properties.75 Superhydrophobic (likely covalently-bound) PDMS coatings of SiO2, TiO2, ZnO and Al2O3 nanoparticles were obtained by ball-milling a mixture of the two components for <1 h at ambient temperature,74 albeit at lower surface chain density than is achievable by alternative methods. The complexity of the processes during ball-milling makes the relative contributions of radical vs. nucleophilic anchoring chemistries uncertain.
A clever attempt to expand the range of reactions inducible by mechanically loading an elastomer exploited the long-known capacity of mechanochemically-generated macroradicals in contact with H2O and air to produce reactive oxygen species (ROS, Fig. 3e).76–78 Squeezing a polymer hose made of PDMS, Tygon or PVC and filled with an aqueous solution of an AuIII salt, Neutral Red dye or latent fluorophore (boronic ester-protected umbelliferone) produced Au nanoparticles, bleached the dye and turned on fluorescence, respectively. The speculated mechanism included homolysis of an overstretched polymer backbone to primary macroradicals, which bound O2 to yield superoxyl macroradicals ROO˙, followed by H abstraction from neighbouring macrochains, thermal decomposition of the resulting alkyl hydroperoxides ROOH to RO˙ and HO˙, or hydrolysis to HO2˙, and recombination and disproportionation of these ROS to H2O2. A Joule of input mechanical energy was claimed to yield on the order of tens of mg of H2O2 per m2 of the polymer/water interface, with the yield scaling linearly with the surface area, suggesting ROS generation at the water–polymer interface.
Depolymerization of certain self-immolative polymers79 was triggered by mechanochemical backbone bond homolysis, but extending this observation to support recycling of commercial polymers has proven difficult.80 Technological utility of mastication rests in its capacity to facture backbone mechanochemically, but the quadratic or steeper dependence of the fragmentation probability on chain contour length4,33 makes ball-milling impractical (or even untenable) for reducing the chain sizes to below hundreds of monomers.81 Hydrocarbon macroradicals can undergo β scission, which shortens the backbone by 2 C atoms and generates an olefin monomer, but the high activation free energies make this reaction kinetically non-competitive with recombination except at conditions of anaerobic pyrolysis.
In summary, mechanochemically generated macroradicals initiate polymerization of activated olefins, such as acrylates or styrenes; recombine with other (macro)radicals and/or add to nearby surfaces. These simple reactions produced diverse block copolymers and graft polymers, allowed installation of complex end groups and effected diverse redox transformations. Synthetic applications of mechanochemically generated macroradicals remain fewer and less diverse than those of small-molecule reactants. For certain targets, for example in the preparation of topologically complex and compositionally heterogeneous chains by grafting, the combination of operational simplicity, low cost and useful product distributions make mechanochemical macroradical generation attractive, if underexploited, alternatives to non-mechanochemical syntheses.
3. Applications of mechanochemically generated radicals for stress-responsive polymers
Polymeric materials are routinely subject to mechanical loads throughout their lifecycles, making exploitation of mechanical force an attractive approach to endowing them with productive autonomic responses. Demonstrated examples include self-healing, self-strengthening and stress-reporting materials (Table 1).| Productive response | Reaction of mechanochemically generated macroradicals | Polymers | Mechanophores or dopants | Ref. |
|---|---|---|---|---|
| Autonomic growth, Self-strengthening | Initiation of polymerization (Fig. 3a) | DN hydrogel | AMPS, NIPAAm | 82 and 83 |
| Surface patterning | Initiation of polymerization (Fig. 3a) | PNaAMPS + PAAm DN hydrogel | Various monomers | 84 |
| Self-strengthening | Chain-to-chain addition (Fig. 7) | Styrene-butadiene copolymers | None | 33 |
| Self-strengthening | Chain-to-chain addition | Polyisoprene | None | 85 |
| Mechanochromism, self-strengthening | Initiation of polymerization (Fig. 3a) | Polyurethanes | DFSN/TASN/HABI/TASN | 39, 40, 86 and 87 |
| Mechanochromism | None | Polycaprolactone | TASN | 88 |
| Mechanochromism | None | Multinetwork elastomer | DFSN | 38 |
| Mechanochromism | H abstraction from a stabilizer (Fig. 9) | Polystyrene | H-DAAN, derivatives | 89 and 90 |
| Mechanochromism | O2 binding | DN hydrogels | ANS | 91 |
| Mechanochromism | Initiation of polymerization (Fig. 3a) | DN hydrogels | NIPAAm and ANS | 92 |
Polymers capable of constructive mechanochemical remodelling
Mechanical stress degrades polymers by promoting scission of polymer chains, which decreases the average molar mass and hence viscosities of solutions and melts, and the density of backbone bonds across which macroscopic load distributes in elastomers. Preventing this degradation and even turning it into a constructive response (i.e., autonomic molecular reinforcement or self-strengthening by forming new load-bearing bonds) are long-lasting challenges of polymer science, where mechanochemically generated macroradical offer a practical solution. So far, these macroradicals were shown to initiate polymerisation of monomeric dopants or crosslinking of adjacent chains.A double-network (DN) hydrogel comprising interpenetrating stretchable and brittle networks (red and blue lines, respectively, Fig. 6a) and swollen with a mixed solution of mono- and bis acrylate monomers, grows when stretched82 under N2. Homolysis of the backbones of the brittle network generated macroradicals, which initiated polymerisation of the monoacrylate monomers and the bisacrylate cross-linkers, forming a new network (red lines). At sufficiently high ratios of the mono and bis acrylates the new network had a higher cross-linking density, and was more brittle, than the original primary network. In subsequent stretching this new network fractured preferentially, which under constant external supply of the monomers caused the gel to grow in volume and increased its elastic modulus (Fig. 6b) until the whole sample fractured.
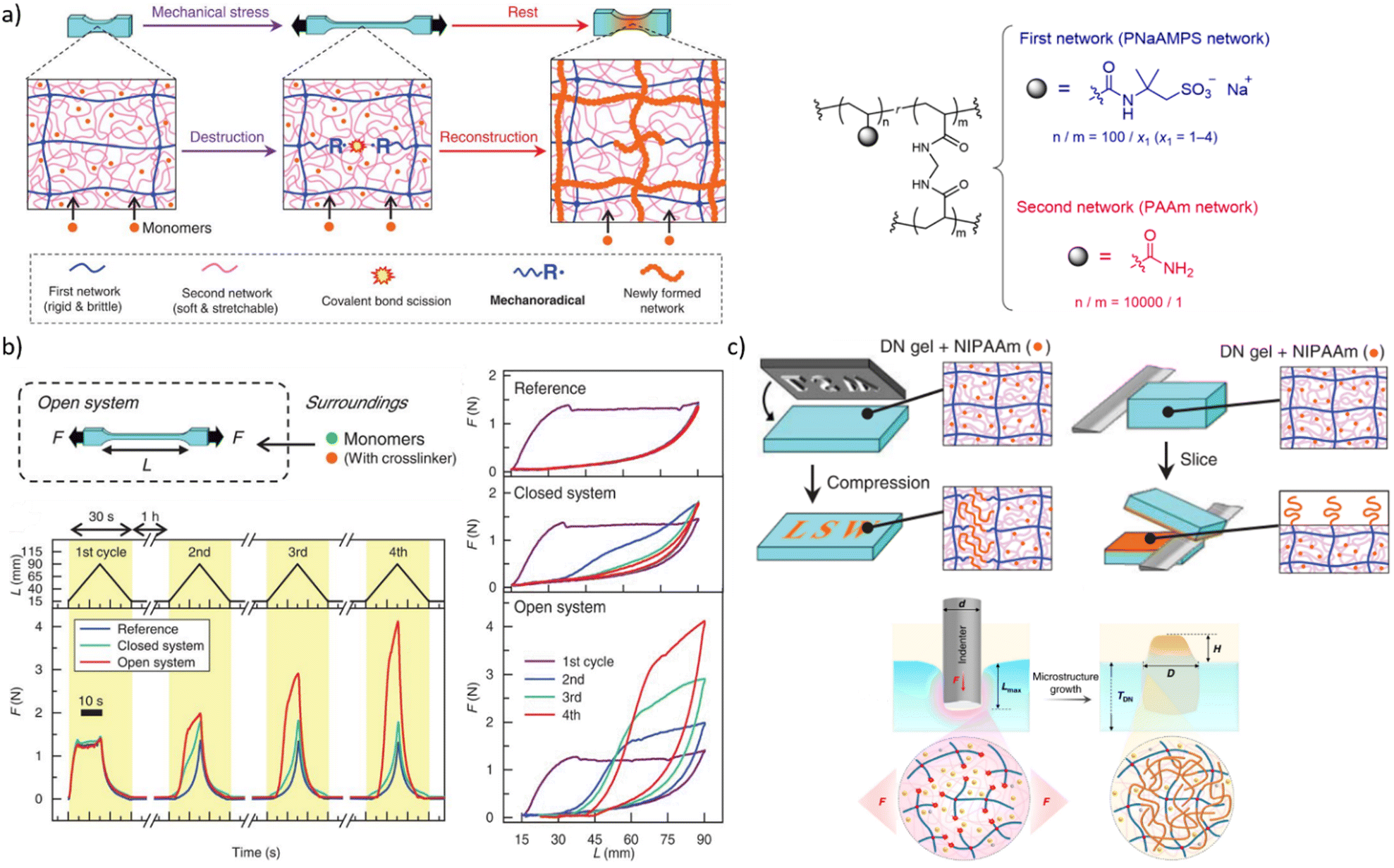 | ||
| Fig. 6 Double-network (DN) hydrogels that initiate polymerization during or after mechanical loading. (a) Left: the schematics of the processes enabling load-induced growth. Right: Chemical composition of the DN gels. (b) Cycling a gel between stretching/relaxation/rest caused the gel to expand, but only when continuously supplied with acrylate monomers under N2 (“open system”). (c) Localized functionalization of the DN hydrogel by compression-triggered polymerization. Adopted from ref. 82 and 84 with permission. | ||
A dissociatively labile azoalkane crosslinker in the brittle network increased the concentration of macroradicals produced by stretching the gel 5-fold compared to the original bisamido linked material (Fig. 6a), which accelerated the polymerization and increased the yield of newly formed polymer per stretching cycle in the plastically-deformed region83 of the gel. A lumped kinetic model93 of these gels related the concentration of the primary macroradicals and the rates of incorporation of the monomers and crosslinkers into growing chains to the growth kinetics of the network under repetitive load–release-rest cycles.
The non-diffusive nature of the macroradicals formed by network fracture allows localized grafting. Its exploitable potential was demonstrated84 with complex microstructuring of a hydrogel surface by applying localized axial compression to the gel, and feeding different functional monomers to each compressed area (Fig. 6c). The resulting surfaces manifested spatially varying chemical compositions in diverse sizes and shapes, which supported oriented growth of cells, directional transportation of surface water droplets and localized thermally-induced phase separation. A major unaddressed question is how to extend this approach beyond multinetwork gels, given its reliance on maintaining high concentrations of macroradicals to achieve useful kinetic chain lengths in monomer polymerizations.
A practical self-strengthening polymer would likely require neither externally-supplied activated monomers, nor high flux of mechanochemically generated radicals but instead rely on macroradical-initiated sequential addition of intact polymer chains to compensate for mechanochemical loss of backbone bonds. Certain unsaturated polyolefins, e.g., polyisoprene (PE), bear sufficient concentration of suitably reactive macroradical-accepting moieties (CC) to make them plausible starting points for creating self-strengthening elastomers of broad engineering and commercial interest.
Initial support for this conclusion came from observations that ball milling of unsaturated polyolefins under certain conditions yielded insoluble material, in contrast to milling of polymers lacking CC bonds that remained soluble. In these experiments, however, the average molar mass of the soluble fraction of the milled polymer decreased even when polymer grafting, signifying chain-to-chain additions, was achieved, suggesting net loss of backbone bond density.85 Subsequent measurements of the viscosity of sheared melts of styrene–butadiene copolymers (SBC, Fig. 7a) proposed macroradical-mediated crosslinking as the origin of the insoluble material.29 Quantitative mechanistic interpretation of these measurements, is precluded by the estimated rate of macroradical production being much lower than the rate of chain fracture. The authors attributed the discrepancy to chain fractures producing “caged” macroradicals, which preferentially disproportionate into a vinyl-terminated and sp3-C terminated daughter chains before they react with any other component of the melt (e.g., an adjacent chain, a radical trap or O2), or are detected spectroscopically. This explanation is however implausible, as explained in the introduction.
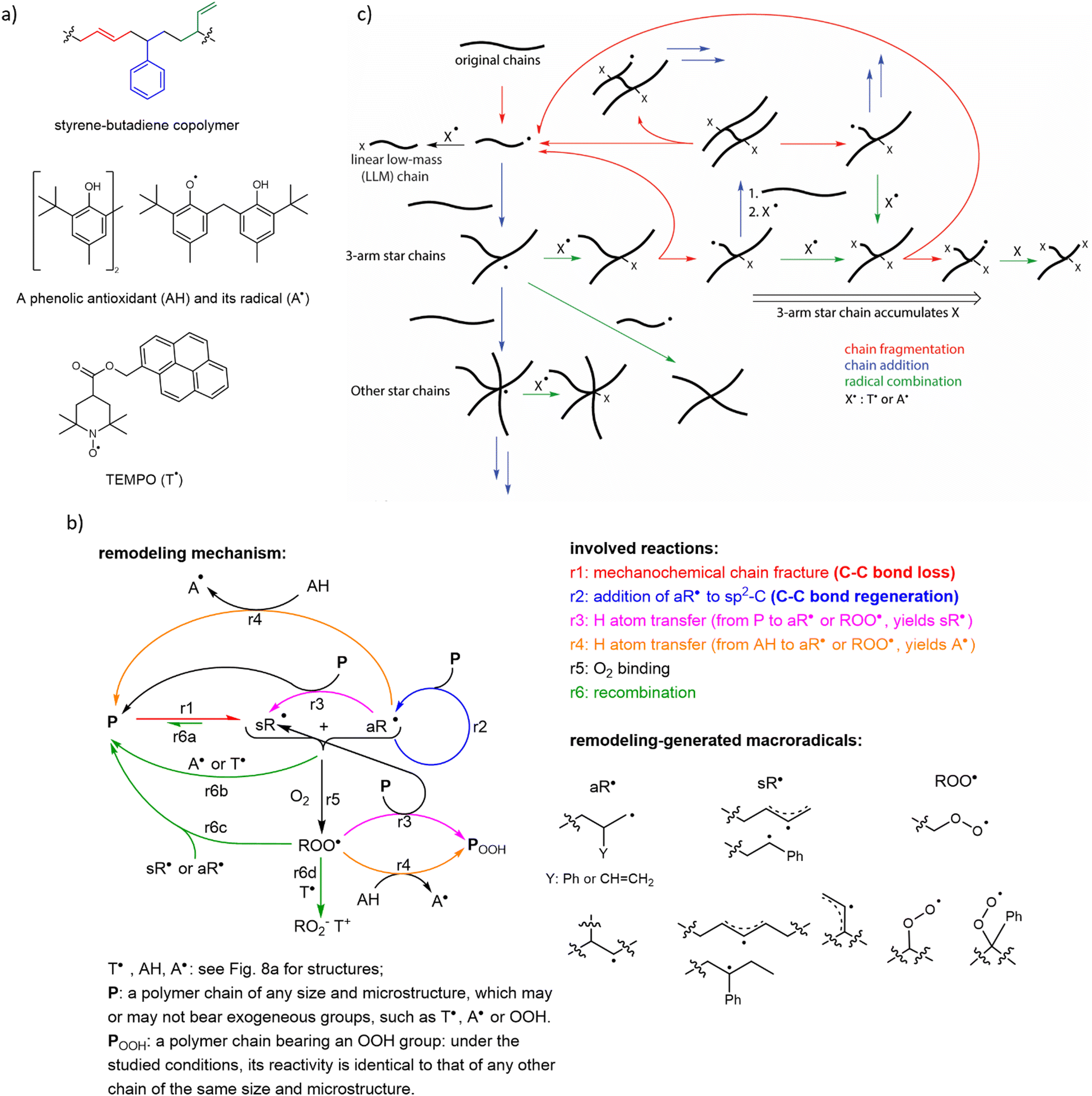 | ||
| Fig. 7 The complex reaction networks enable constructive remodeling of unsaturated polyolefins (exemplified here by styrene–butadiene copolymer, SBC) driven by mechanochemically-generated macroradicals. (a) Chemical structures of SBC indicating its 3 repeat units (derived from styrene, 1,4- and 1,2-enchained butadiene), a representative commercial phenolic antioxidant (and its radical) and pyrene modified TEMPO typically used in mechanistic studies of mechanochemical remodelling. (b) The simplest mechanism of low-temperature mechanochemical remodelling of an unsaturated polyolefin, P, under various practically-relevant loading conditions. Note the distinct roles played by alkyl- (aR˙) and stabilized (allyl and benzylic, in case of styrene copolymers, sR˙) macroradicals. The fraction of aR˙ macroradicals in the products chain fracture depends on the polymer composition, the fracture force, and the small-molecule dopants, with the maximum fraction of aR˙ varying from ∼0 to ∼0.5 for commercial unsaturated polyolefins under practically-relevant conditions. The lower yield of aR˙ per chain fracture favours material degradation. (c) The primary reactions responsible for increasing topological complexity of chains in a mechanically loaded remodeling unsaturated polyolefin, due to the repeated formation and fracture of branched chains, which also leads to accumulation of small-molecule radical solutes (e.g., O2 or radical traps) in remodelled chains. Adapted from ref. 33 with permission. | ||
Recent work33 identified a complex reaction network and kinetic feedback loops that determine if a polymer remodels constructively or degrades under repeated mechanical loading and argued that similar reaction networks govern mechanochemical remodelling of any polymer mixture whose composition supports radical reaction cascades (Fig. 7b). The resulting quantitative molecular mechanistic framework allows the seemingly contradictory bulk mechanochemistry of unsaturated polyolefins to be rationalized and suggests approaches to developing practical self-strengthening elastomers based on unsaturated polyolefins (Fig. 7c). The following conclusions can be drawn from the reported data:
(1) Self-strengthening by chain-to-chain addition is self-limiting thanks to a steep increase in the probability of mechanochemical chain fracture with the degree of chain branching and its total contour length. In other words, the more chains a mechanochemically-generated radicals adds before inactivating (either by recombination or radical chain transfer), the more likely the resulting macromolecule mechanochemically fragments (Fig. 7c). This observation suggests that uncontrolled embrittlement of the previously reported self-strengthening elastomers94,95 is not intrinsic to constructive mechanochemical remodelling.
(2) A single parameter, the number of new backbone bonds generated per chain fracture, is sufficient both to predict the evolution of the composition and of the cross-linking density of a loaded unsaturated polyolefin with useful accuracy, and to quantify the effect of dopants (e.g., AH or T˙ in Fig. 7a), and the relative fractions of different C-atom types on this evolution. The ratio <1 corresponds to material degradation, whereas >1 means self-strengthening. This simplicity facilitates identification of candidate compositions, microstructures, and chain topologies of prospective self-repairing materials for further experimental evaluation and optimization.
(3) Constructive remodelling does not need small-molecule dopants (e.g., monomers or cross-linkers) to regenerate its mechanically-fractured load-bearing network and therefore is not limited by the amount of such dopants that the material can contain or by their externally-maintained flux.
(4) Even short kinetic chain lengths (the number of backbone bonds formed per initiator) allow macroradical-to-chain addition to maintain constant density of load-bearing bonds in the stressed material. This obviates the need for activated CC bonds of acrylates or styrene, which were thought necessary for self-strengthening in the absence of small-molecule crosslinkers86 but are difficult to incorporate into chains. It also suggests that practically-achievable concentrations of CC bonds suffice to counteract the chain-growth-terminating effects of O2 (Fig. 7b). Autonomic response that is insensitive to O2 is more practical than the one limited to anaerobic conditions.
The above conclusions form the foundation for a mechanistically-based approach to designing polymers capable of productive autonomic molecular-level self-repair. Practical applications of this approach likely require extending these insights to accommodate mechanochemistry of common commercial crosslinks (i.e., polysulfides) and to higher temperatures.
Stress-reporting polymers
Catastrophic failure of an elastomer originates in a small volume of material where highly localized mechanical stress causes sufficient number of backbones to fracture for the network to depercolate.2 Mechanochromism is thought to offer a practical means of visualizing these overstressed volumes of a loaded material prior to its failure.6 A mechanochromic material changes its optical properties (e.g., colour or fluorescence efficiency) when loaded above a threshold. Load-to-colour transduction relies on load either disrupting weakly bound aggregates, or accelerating rearrangement of covalent bonds. The former is reversible on a practical timescale, whereas the latter is usually not (with some notable exceptions). Most reported mechanochromic reactions don’t involve macroradicals or chain fracture but the minority that exploits mechanochemical bond homolysis offer important advantages. The latter mechanochromism results from either (1) chain fracture yielding macroradicals that absorb visible light (with or without subsequent emission) or (2) the macroradicals initiating a reaction cascade that yields a chromophore or fluorophore.The reported latent mechanochromes activated by C–C bond homolysis are dimers of (multi)aryl- or multi(carbonitrile)-substituted benzofuranones, fluorenes, imidazoles, or succino-nitriles (Fig. 2). Their scissile bonds are much more mechanochemically labile than C–C bonds of typical polymers, producing mechanochromism under diverse loading scenarios, including melt crystallization,88 gel freezing,32,38–40 swelling of a multi-network gel (when the dimer is incorporated in the brittle network),38 grinding a powder,30 and stretching an elastomer37 (Fig. 8a). The formation of the radicals accompanying the appearance of the colour was confirmed and quantified by ESR in several cases. The colour, which spans from yellow to green, depending on the mechanochrome, persists in the glassy state and slowly fades in melt or when the load is removed, due to diffusion-limited recombination of the macroradicals (including recombination of dangling chains in a molecular network of an elastomer37,96). Conversely, rapid recombination precludes detectable mechanochromism in fast solvent flows, e.g., in sonicated solutions, of these polymers.
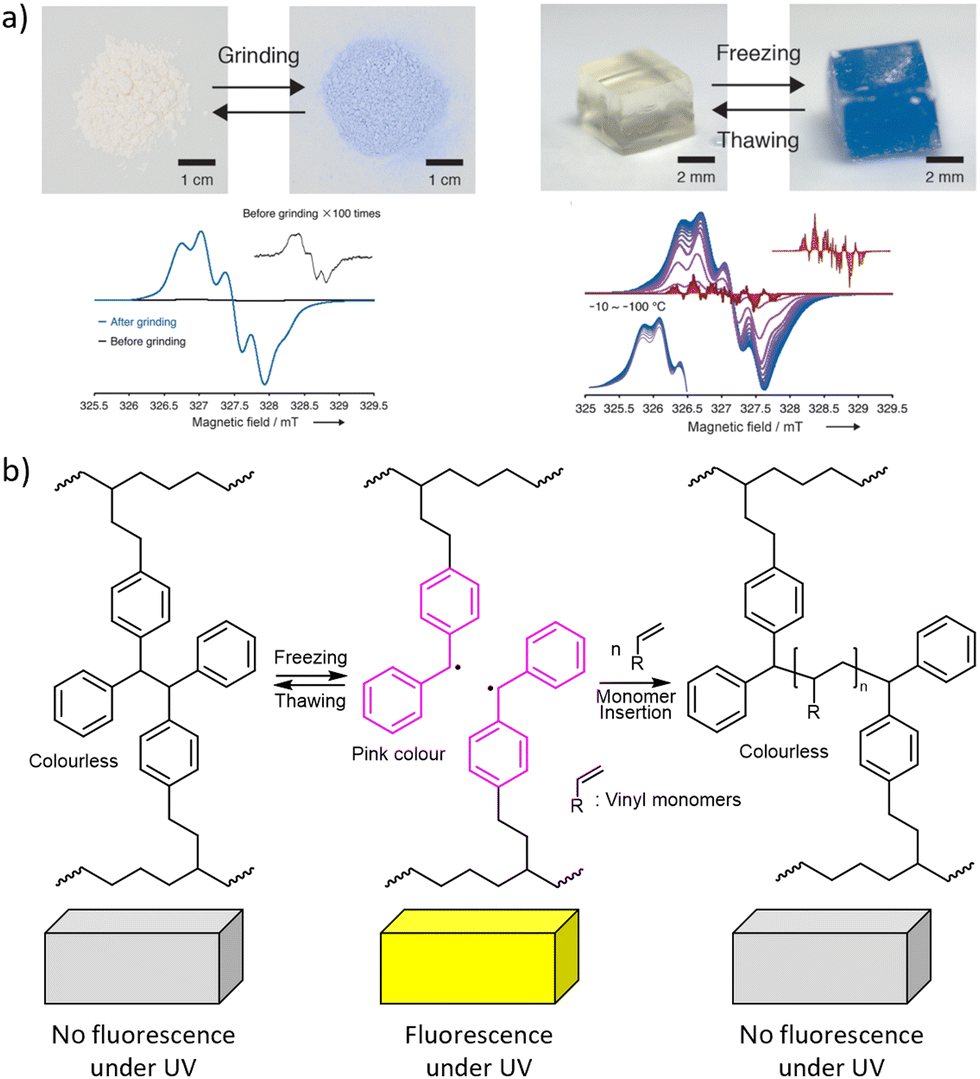 | ||
| Fig. 8 Mechanochromism from chain fracture to macroradicals that absorb visible light. (a) Top: DABBF-linked polymers change colour after grinding their powders or freezing their hydrogels. Bottom: The ESR spectra confirming the formation of radicals. Adapted from ref. 32 with permission. (b) Polymerisation of acrylate solutes initiated by TASN radicals during freeze–thaw cycles. See Fig. 2 for the chemical structures. | ||
Both hexaarylbisimidazole (HABI) and tetraarylsuccinonitrile (TASN) radicals87 can initiate radical polymerization of an acrylate, making them rare examples of so-called multi-modal mechanophores,15,22,97i.e., moieties manifesting multiple productive responses to mechanical load (Fig. 8b).
The reversible coloration of these mechanochromes that fades with diffusion-limited kinetics is unique and offers considerable potential to quantify at the molecular level non-equilibrium chain dynamics,24 as well as dynamics of distribution and dissipation of macroscopic load in viscous media.2 This potential, however, remains to be exploited. Simultaneously, the high mechanochemical lability of the scissile C–C bonds in these dimers means that their mechanochromism occurs at load that are too low to inform on the molecular mechanisms responsible for bulk fracture of polymeric materials, which require homolysis of much more inert C–C bonds.
A secondary mechanochromism, whereby a macroradical generated by homolysis of the backbone of an arbitrary composition reacts with a latent fluorophore to make it emissive, increases the likelihood that the temporospatial distribution of mechanofluorescence in a loaded material corresponds to the distribution of single-chain forces of the magnitude that cause material failure. The first demonstration of such secondary mechanochromism was the yellow fluorescence of a ball-milled mixture of polystyrene and diarylacetonitrile (H-DAAN), which was attributed to DAAN˙, generated by H-atom transfer from H-DAAN to polystyrene-derived macroradical90 (Fig. 9a). Bulky substituents at the aromatic rings of H-DAAN enhanced fluorescent intensity of the milled powders, presumably by inhibiting DAAN˙ recombination.89
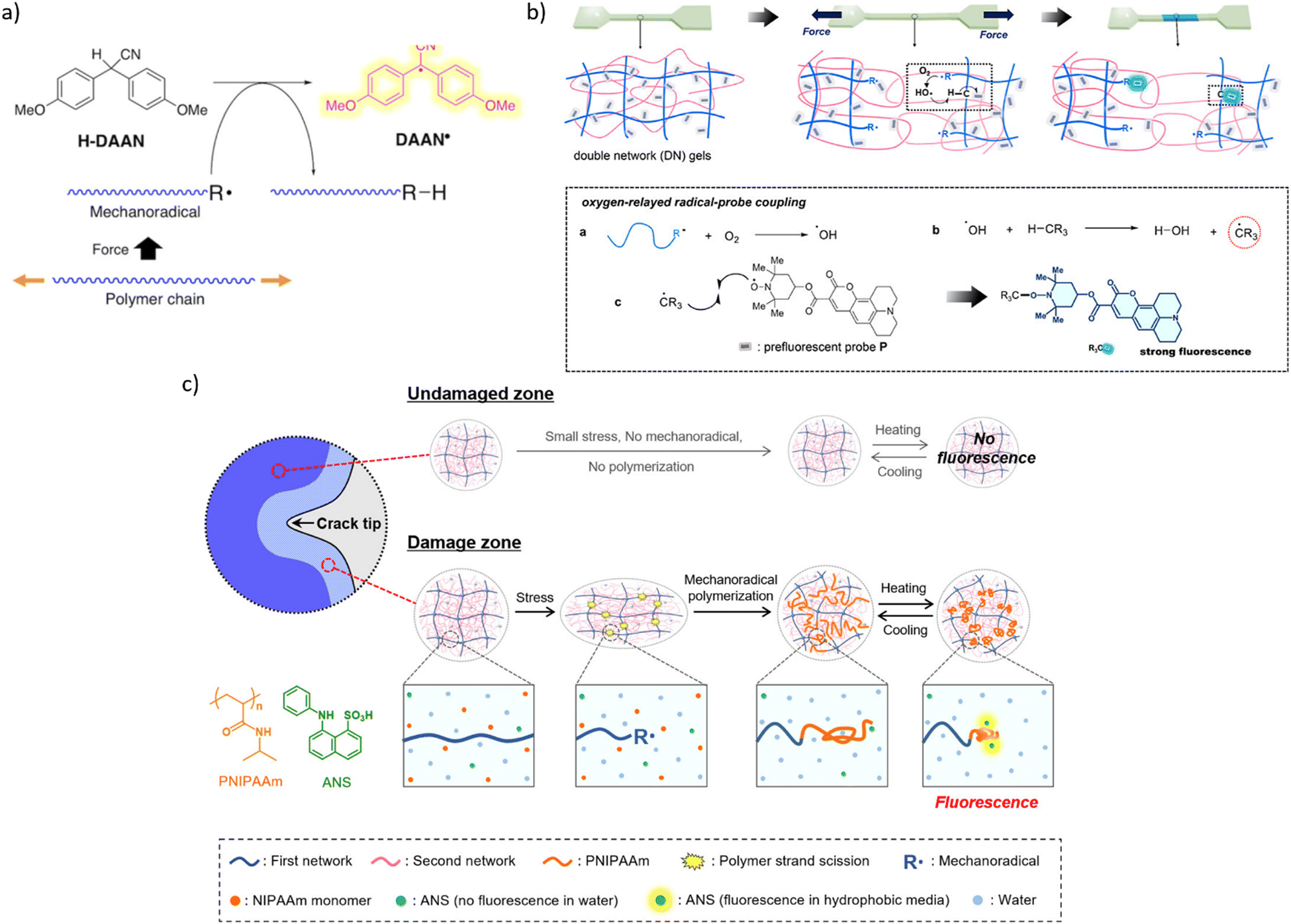 | ||
| Fig. 9 Mechanochromism from chromophores generated in secondary reactions of macroradicals. (a) Generation of fluorescent diarylacetonitrile radical (DAAN˙) by a chain-transfer reaction of diarylacetonitrile (H-DAAN) with a mechanochemical produced macroradical. Adapted from ref. 90 with permission. (b) Schematics of secondary mechanofluorescence based on ROS-mediated recombination of a latent fluorophore and macroradical. Adapted from ref. 91 with permission. (c) A cartoon representation of mechanofluorescent visualization of chain fracture around a propagating crack by enhanced fluorescence of an anilinonaphthalenesulfonate sequestered in the newly formed low-polarity polymer from macroradical-initiated polymerization of NIPAAm (orange structure). Adapted from ref. 92 with permission. | ||
The only other reported manifestations of secondary mechanochromism we are aware of take advantage of the high concentration of mechanochemically-generated macroradicals achievable upon plastic deformation a double-network hydrogel. In one example,91 deformation of a gel containing a latent hydrophobic fluorophore (a coumarin derivative bound to a TEMPO moiety, Fig. 9b) in air produced higher-intensity emission than the similar distortion under N2 did. The authors attributed this observation to a combination of the low concentration and the low mobility of the latent fluorophore in the hydrogel.
The latent fluorophore becomes emissive upon recombining with a C-based radical, e.g., a macroradical. The emission intensity depends on the fraction of macroradicals that bind the latent fluorophore vs. that are consumed in non-fluorogenic reactions (such as recombination of a pair of radical-terminated dangling chains), which is governed in part by the relative mobilities of the recombining radicals. The authors attributed the enhanced fluorescence in air to the aerobically promoted migration of the backbone C-centred radical along the molecular network by a combination of 1,n-H shifts and diffusion of ˙OH. Briefly, a primary macroradical binds O2, followed by an 1,n-H shift to yield ROOH (and moving the radical to another backbone C atom), and the subsequent decomposition of the latter by several mechanisms to yield ˙OH. The high solution mobility of ˙OH combined with its capacity to abstract any H atom of the polymer is equivalent to a rapid diffusion of a macroradical across the network. The cycle of O2 binding, 1,n-H shift, ˙OH generation, and backbone-H abstraction, continues until thus-wandering macroradical recombines with the dye, or another C-based (macro)radical (including any adventitious open-shell solutes).
Because a continuous flux of mechanochemically-generated macroradicals can create locally hypoxic or anoxic environments,33 molecular interpretation of the temporospatial distribution of the mechanofluorescent intensity that depends on local O2 concentration is likely to be ambiguous. A water soluble latent fluorophore would eliminate such difficulties by obviating the need for a ROS-reliant radical relay mechanism of recombination of non-diffusive radicals.
The other reported example of secondary mechanochromism exploited the strong increase of the fluorescent quantum yield of anilinonaphthalenesulfonates on reduction of the dielectric constant of its surroundings. Under N2 the damage zone around a propagating crack of a hydrogel saturated with NIPAAm and 8-anilino-1-naphthalenesulfonic acid (ANS, green structure in Fig. 9c) fluoresced because mechanochemically generated macroradicals initiated polymerization of NIPAAm, which created a separate microphase with lower polarity than water, thereby increasing the fluorescence of the acid sequestered in it.92
In summary, autonomic self-healing and self-strengthening that exploit the reactivity of mechanochemically generated macroradicals to maintain or increase the cross-linking density of an elastomer under load offer considerably advantages over other backbone bond-forming chemistries, including compositional simplicity, O2 tolerance, and self-regulation which eliminates uncontrolled embrittlement and the need for small-molecule additives.33 The molecular origin of these advantages is sufficiently well understood to facilitate the expansion of this approach beyond unsaturated polyolefins. Macroradical-initiated mechanofluorescence, in theory, eliminates the problem inherent to molecular interpretation of optomechanochemical response of conventional mechanofluorophores and mechanochromes,2 and could enable faithful mapping of the distribution of chain-level forces in bulk polymers under mechanical loads up to global-fracture-inducing thresholds. The small number of reported examples of such secondary mechanofluorescence, however, mean that this potential remains speculative.
4. Conclusions and perspective
Under sufficient mechanical loads polymer chains fracture by (usually) homolysis of the covalent bonds comprising the backbone, forming macroradicals. This degrades the mechanical (and often chemical, optical and thermal) properties of known polymers. Yet, the high reactivity of such macroradicals can also be exploited to initiate or drive useful chemistries. Earliest, and still most numerous, examples are synthetic applications, including post-polymerization modifications and chain-extending homopolymerizations, to yield compositions and microstructures that are sometimes difficult to obtain by other methods. These rely on the intentional generation of macroradicals by griding or milling of polymer powders or sonicating their solutions, or occasionally, melts. Several approaches have been developed to control the balance of broken and formed backbone bonds, which determines the molar mass distribution of the products. Synthetic applications of mechanochemically generated macroradicals can now be viewed as a well-established part of “green chemistry”.Recently, several examples of materials, of varying degrees of compositional and microstructural complexity, have been described in which mechanochemically generated macroradicals initiate productive chemistries, including autonomic localized self-healing, self-strengthening and internal-stress visualization. They illustrate how macroradicals generated incidentally to the primary use of the material can be channelled to endow the material with additional exploitable characteristics, without dedicated energy inputs or limiting current uses. Full realization of the plausible potential of these emerging applications of mechanochemically generated macroradicals is predicated on resolving numerous outstanding challenges of both fundamental and applied nature. The former include methodological, conceptual and empirical advances to handle the mechanistic and kinetic complexity that appears to enable the macroscopic manifestations of constructive molecular remodeling of a mechanically loaded polymers. Simultaneously, the compositions, topologies and microstructures of commercial polymers represent a complex compromise among many often-contradictory requirements (e.g., cost, processibility, mechanical properties). It remains to be determined how or even if new reaction channels can be engineered into an existing engineering or specialty polymer while preserving or enhancing its technological or economic value.
The search for strategies that capture the reactivity of macroradicals to drive useful chemistry highlighted in this review complements the primary focus of contemporary polymer mechanochemistry on directing the chemistry of overstretched polymer backbones away from homolytic bond scission and towards more complex chemistry at specially designed sites.
Data availability
All data discussed in this Review is taken from referenced literature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (22303065) and the Shaanxi Province Postdoctoral Science Foundation (2023BSHEDZZ303).Notes and references
- Q. Mu and J. Hu, Phys. Chem. Chem. Phys., 2024, 26, 679–694 RSC.
- R. T. O'Neill and R. Boulatov, Angew. Chem., Int. Ed., 2024, 63, e202402442 CrossRef.
- R. T. O’Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef PubMed.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS PubMed.
- X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 2108327 CrossRef CAS.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- A. Krusenbaum, S. Gratz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2020, 6, 84–98 CrossRef.
- L. Anderson and R. Boulatov, Adv. Phys. Org. Chem., 2018, 52, 87–143 CrossRef CAS.
- S. Akbulatov, Y. Tian, Z. Huang, T. J. Kucharski, Q.-Z. Yang and R. Boulatov, Science, 2017, 357, 299–303 CrossRef CAS PubMed.
- P. S. Weiss, Science, 2023, 380, 1013 CrossRef CAS PubMed.
- Y. Tian, T. J. Kucharski, Q.-Z. Yang and R. Boulatov, Nat. Commun., 2013, 4, 2538 CrossRef PubMed.
- S. Garcia-Manyes and A. E. Beedle, Nat. Rev. Chem., 2017, 1, 0083 CrossRef CAS.
- P. Glynn, B. Van der Hoff and P. Reilly, J. Macromol. Sci., Chem., 1972, 6, 1653–1664 CAS.
- H. Zhang, X. Li, Y. Lin, F. Gao, Z. Tang, P. Su, W. Zhang, Y. Xu, W. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef.
- Y. Tian, X. Cao, X. Li, H. Zhang, C.-L. Sun, Y. Xu, W. Weng, W. Zhang and R. Boulatov, J. Am. Chem. Soc., 2020, 142, 18687–18697 CrossRef CAS PubMed.
- Z. Wang, X. Zheng, T. Ouchi, T. B. Kouznetsova, H. K. Beech, S. Av-Ron, T. Matsuda, B. H. Bowser, S. Wang, J. A. Johnson, J. A. Kalow, B. D. Olsen, J. P. Gong, M. Rubinstein and S. L. Craig, Science, 2021, 374, 193–196 CrossRef CAS PubMed.
- H. Hu, X. Cheng, Z. Ma, R. P. Sijbesma and Z. Ma, J. Am. Chem. Soc., 2022, 144, 9971–9979 CrossRef CAS PubMed.
- M. Sakaguchi and J. Sohma, J. Polym. Sci., Part B: Polym. Phys., 1975, 13, 1233–1245 CAS.
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Science, 2010, 329, 1057–1060 CrossRef CAS PubMed.
- Z. Huang and R. Boulatov, Chem. Soc. Rev., 2011, 40, 2359–2384 RSC.
- S. Akbulatov, Y. Tian and R. Boulatov, J. Am. Chem. Soc., 2012, 134, 7620–7623 CrossRef CAS PubMed.
- R. T. O’Neill and R. Boulatov, Synlett, 2022, 851–862 CrossRef.
- R. T. O’Neill and R. Boulatov, Nat. Chem., 2023, 15, 1214–1223 CrossRef.
- H. Staudinger and H. F. Bondy, Ber. Dtsch. Chem. Ges., 1930, 63, 734–736 CrossRef.
- W. Kauzmann and H. Eyring, J. Am. Chem. Soc., 1940, 62, 3113–3125 CrossRef CAS.
- J. Li, S. Guo and X. Li, Polym. Degrad. Stab., 2005, 89, 6–14 CrossRef CAS.
- Y. Li, J. Li, S. Guo and H. Li, Ultrason. Sonochem., 2005, 12, 183–189 CrossRef CAS.
- D. Dondi, A. Zeffiro, A. Buttafava, C. Marciano, M. Bianchi and A. Faucitano, Polym. Degrad. Stab., 2013, 98, 392–407 CrossRef CAS.
- K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 556–560 CrossRef CAS.
- P. S. Sathiskumar and G. Madras, Ultrason. Sonochem., 2012, 19, 503–508 CrossRef CAS.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- C. Wang, S. Akbulatov, Q. Chen, Y. Tian, C.-L. Sun, M. Couty and R. Boulatov, Nat. Commun., 2022, 13, 3154 CrossRef CAS PubMed.
- Y. Nakamura and S. Yamago, Macromolecules, 2015, 48, 6450–6456 CrossRef CAS.
- R. Vinu and L. J. Broadbelt, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 29–54 CrossRef CAS.
- T. J. Kucharski and R. Boulatov, J. Mater. Chem., 2011, 21, 8237–8255 RSC.
- H. Yokochi, R. T. O'Neill, T. Abe, D. Aoki, R. Boulatov and H. Otsuka, J. Am. Chem. Soc., 2023, 145, 23794–23801 CrossRef CAS PubMed.
- T. Watabe and H. Otsuka, Angew. Chem., Int. Ed., 2023, 62, e202216469 CrossRef CAS PubMed.
- F. Verstraeten, R. Gostl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- S. Kato, D. Aoki and H. Otsuka, ACS Appl. Polym. Mater., 2020, 3, 594–598 CrossRef.
- B. Rennekamp, C. Karfusehr, M. Kurth, A. Ünal, D. Monego, K. Riedmiller, G. Gryn’ova, D. M. Hudson and F. Gräter, Nat. Commun., 2023, 14, 2075 CrossRef CAS.
- Joseph L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS.
- L. E. Wenger and T. P. Hanusa, Chem. Commun., 2023, 59, 14210–14222 RSC.
- S. M. Zeitler and M. R. Golder, Chem. Commun., 2024, 60, 26–35 RSC.
- A. K. Padmakumar, N. K. Singha, M. Ashokkumar, F. A. Leibfarth and G. G. Qiao, Macromolecules, 2023, 56, 6920–6927 CrossRef CAS.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2016, 9, 135–139 CrossRef.
- J. Collins, T. G. McKenzie, M. D. Nothling, S. Allison-Logan, M. Ashokkumar and G. G. Qiao, Macromolecules, 2018, 52, 185–195 CrossRef.
- Z. Wang, J. Ayarza and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2019, 58, 12023–12026 CrossRef CAS PubMed.
- P. Yan, W. Zhao, F. McBride, D. Cai, J. Dale, V. Hanna and T. Hasell, Nat. Commun., 2022, 13, 4824 CrossRef CAS PubMed.
- S. Chen, C. Fan, Z. Xu, M. Pei, J. Wang, J. Zhang, Y. Zhang, J. Li, J. Lu, C. Peng and X. Wei, Nat. Commun., 2024, 15, 769 CrossRef CAS PubMed.
- P. Alexander and M. Fox, J. Polym. Sci., 1954, 12, 533–541 CrossRef CAS.
- A. Henglein, Makromol. Chemie-Macromol. Chem. Phys., 1954, 14, 128–145 CrossRef CAS.
- N. Xiuyuan, H. Yuefang, L. Bailin and X. Xi, Eur. Polym. J., 2001, 37, 201–206 CrossRef CAS.
- R. Chen, C. Yi, H. Wu and S. Guo, J. Appl. Polym., 2009, 112, 3537–3542 CrossRef CAS.
- G. Bouquet, R. Veraart and J. Peltier, J. Appl. Polym., 2009, 112, 1546–1551 CrossRef CAS.
- S. L. Malhotra and J. M. Gauthier, J. Macromol. Sci., Chem., 1982, 18, 783–816 Search PubMed.
- K. Goto and H. Fujiwara, J. Polym. Sci., Part B: Polym. Lett., 1963, 1, 505–508 CrossRef CAS.
- R. B. Seymour and G. A. Stahl, J. Polym. Sci., Part A: Polym. Chem., 1976, 14, 2545–2552 CAS.
- Y. Minoura, T. Kasuya, S. Kawamura and A. Nakano, J. Polym. Sci., Part A: Polym. Chem., 1967, 5, 43–56 CrossRef CAS.
- R. B. Seymour and G. A. Stahl, J. Macromol. Sci., Chem., 1977, 11, 53–64 Search PubMed.
- S. L. Malhotra, J. Macromol. Sci., Chem., 1982, 18, 1055–1085 Search PubMed.
- G. J. Price and P. J. West, Polymer, 1996, 37, 3975–3978 CrossRef CAS.
- H. Fujiwara, T. Ishida, N. Taniguchi and S. Wada, Polym. Bull., 1999, 42, 197–204 CrossRef CAS.
- A. H. Lebovitz, M. K. Gray, A. C. Chen and J. M. Torkelson, Polymer, 2003, 44, 2823–2828 CrossRef CAS.
- J. S. Oh, A. I. Isayev and M. A. Rogunova, Polymer, 2003, 44, 2337–2349 CrossRef CAS.
- A. H. Lebovitz, K. Khait and J. M. Torkelson, Macromolecules, 2002, 35, 9716–9722 CrossRef CAS.
- R. Schwarz and C. E. Diesendruck, Adv. Sci., 2023, 10, 2304571 CrossRef CAS PubMed.
- Ç.-G. Huceste and H. Yesim, J. Appl. Polym., 2000, 77, 1950–1953 CrossRef.
- M. Bartsch and G. Schmidt-Naake, Macromol. Chem. Phys., 2006, 207, 209–215 CrossRef CAS.
- G. Schmidt-Naake, M. Drache and M. Weber, Macromol. Chem. Phys., 2002, 203, 2232–2238 CrossRef CAS.
- M. Degirmenci, H. Catalgil-Giz and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2003, 42, 534–540 CrossRef.
- N. Celik, S. Akay, F. Sahin, G. Sezer, E. Dagasan Bulucu, M. Ruzi, H.-J. Butt and M. S. Onses, Adv. Mater. Interfaces, 2023, 10, 2300069 CrossRef CAS.
- B. Shen, W. Zhai, D. Lu, J. Wang and W. Zheng, RSC Adv., 2012, 2, 4713–4719 RSC.
- H. T. Baytekin, B. Baytekin and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 3596–3600 CrossRef CAS PubMed.
- A. V. Parameswar, K. R. Fitch, D. S. Bull, V. R. Duke and A. P. Goodwin, Biomacromolecules, 2018, 19, 3421–3426 CrossRef CAS PubMed.
- C. Zapp, A. Obarska-Kosinska, B. Rennekamp, M. Kurth, D. M. Hudson, D. Mercadante, U. Barayeu, T. P. Dick, V. Denysenkov, T. Prisner, M. Bennati, C. Daday, R. Kappl and F. Gräter, Nat. Commun., 2020, 11, 2315 CrossRef CAS PubMed.
- O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- J. Zhou, T.-G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300768 CrossRef CAS PubMed.
- S. Aydonat, A. H. Hergesell, C. L. Seitzinger, R. Lennarz, G. Chang, C. Sievers, J. Meisner, I. Vollmer and R. Göstl, Polym. J., 2024, 56, 249–268 CrossRef CAS.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- Z. J. Wang, J. Jiang, Q. Mu, S. Maeda, T. Nakajima and J. P. Gong, J. Am. Chem. Soc., 2022, 144, 3154–3161 CrossRef CAS PubMed.
- Q. Mu, K. Cui, Z. J. Wang, T. Matsuda, W. Cui, H. Kato, S. Namiki, T. Yamazaki, M. Frauenlob, T. Nonoyama, M. Tsuda, S. Tanaka, T. Nakajima and J. P. Gong, Nat. Commun., 2022, 13, 6213 CrossRef CAS PubMed.
- A. Smith, J. Shay, R. Spontak, C. Balik, H. Ade, S. Smith and C. Koch, Polymer, 2000, 41, 6271–6283 CrossRef CAS.
- K. Seshimo, H. Sakai, T. Watabe, D. Aoki, H. Sugita, K. Mikami, Y. Mao, A. Ishigami, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Angew. Chem., Int. Ed., 2021, 60, 8406–8409 CrossRef PubMed.
- S. Kato, D. Aoki and H. Otsuka, Chem. Lett., 2021, 50, 1223–1225 CrossRef CAS.
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed.
- T. Yamamoto and H. Otsuka, Polym. Chem., 2023, 14, 2464–2468 RSC.
- T. Yamamoto, S. Kato, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2020, 60, 2680–2683 CrossRef PubMed.
- Y. Zheng, J. Jiang, M. Jin, D. Miura, F. X. Lu, K. Kubota, T. Nakajima, S. Maeda, H. Ito and J. P. Gong, J. Am. Chem. Soc., 2023, 145, 7376–7389 CrossRef CAS PubMed.
- T. Matsuda, R. Kawakami, T. Nakajima and J. P. Gong, Macromolecules, 2020, 53, 8787–8795 CrossRef CAS.
- Z. Sheng, J. Ma, Z. Shen, S. Qu and Z. Jia, Macromolecules, 2023, 56, 9398–9409 CrossRef CAS.
- Y. Pan, H. Zhang, P. Xu, Y. Tian, C. Wang, S. Xiang, R. Boulatov and W. Weng, Angew. Chem., Int. Ed., 2020, 59, 21980–21985 CrossRef CAS PubMed.
- A. L. B. Ramirez, Z. S. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Nat. Chem., 2013, 5, 757–761 CrossRef CAS PubMed.
- H. Zhang, D. Zeng, Y. Pan, Y. Chen, Y. Ruan, Y. Xu, R. Boulatov, C. Creton and W. Weng, Chem. Sci., 2019, 10, 8367–8373 RSC.
- X. He, Y. Tian, R. T. O’Neill, Y. Xu, Y. Lin, W. Weng and R. Boulatov, J. Am. Chem. Soc., 2023, 145, 23214–23226 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |