
A ligand-modulated photostable Mn(I)–carbonyl complex for preferential conversion of CO2 to CO in water†
Chandan
Das
a,
Suchismita
Ghosh
a,
Rathindranath
Biswas
 a,
Goutam K.
Lahiri
a and
Arnab
Dutta
*abc
a,
Goutam K.
Lahiri
a and
Arnab
Dutta
*abc
aChemistry Department, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: arnab.dutta@iitb.ac.in
bInterdisciplinary Program for Climate Studies, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India
cNational Center of Excellence-Carbon capture and utilization, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India
First published on 23rd August 2024
Abstract
A strategically designed redox-active ligand ensures proper electronic balance in an Mn(I)–carbonyl template to induce photostability and water solubility. This newly designed Mn–carbonyl complex showcased rapid CO2 reduction with preferential production of CO (faradaic efficiency ∼88%) as the only C1 product in pure water, even from a flue gas resource.
Natural photosynthetic machinery remains the inspiration in this pursuit as it highlights the possibility of expertly coupling CO2 reduction with a solar-driven electrochemical pathway.1 Following this cue, a plethora of electrocatalytic CO2 reduction methods have been developed as an attractive and sustainable gateway for generating essential and market-ready C1-feedstocks such as CO, HCOOH, CH3OH, and CH4. The direct one-electron reduction of CO2 to reactive CO2˙− is highly energy-demanding (requires −1.48 V vs. RHE).2 However, the proton-coupled reduction of CO2 to relatively stable product CO can be realized at moderate potentials (CO2 + 2H+ + 2e− → CO + H2O, −0.11 V vs. RHE).3,4 During this CO2/CO conversion, the competitive hydrogen evolution emerges as a major side reaction due to its operation at a favourable potential window (2H+ + 2e− → H2, 0 V vs. RHE),4 especially in protic solvents. Hence, the search for an efficient and selectively CO2-reducing electrocatalyst in aqueous media with minimal interference from H2 evolution remains the holy grail in the pursuit of a viable CO2 utilization strategy. The emergence of electrocatalytic activity by the manganese5 analogues of the conventional Re(I) catalysts6–8 in CO2 reduction has spurred the advancement of non-noble metal-driven CO2 activation. One of the distinct differences between the Re and Mn-based electrocatalysts lies in the tendency for dimerization for the leading Mn–carbonyl complexes.5,9,10 Classically, [MnBr(bpy)(CO)3] complexes display two successive one-electron irreversible reduction steps (separated by ∼200–300 mV) during a cathodic scan to produce a penta-coordinated monoanionic [Mn(bpy)(CO)3]− species, which acts as the active catalyst for the CO2RR. [Mn(bpy)(CO)3]− can further react with the parent[MnBr(bpy)(CO)3] complex to form a [Mn–Mn]0 dimer.11 However, generating this highly electron-dense Mn-species under reducing conditions typically requires a substantially high overpotential, which lowers the overall efficiency of the catalyst. Hence, the subtle modifications of the ligand scaffold are probed to develop energy-efficient electrocatalysts.12–14 However, the majority of these CO2-reducing Mn-based electrocatalysts are reported to be operational in organic media, while they display acute light sensitivity in the presence of water.15–19 Here, we have portrayed a tactical ligand design strategy to modulate the electronic balance between the carbonyl and the auxiliary ligand in a Mn(I)–carbonyl template. Following this strategy, a photostable and water-soluble Mn(I)–carbonyl complex was crafted that preferentially electro-reduces variable CO2 resources (including flue gas) exclusively to CO in water.
The [2,6-diphenyl(di-azo)] pyridine (dipap) ligand (L1) was utilized for generating a new genre of Mn(I) complex via the initial reaction between L1 and Mn(CO)5Br (Fig. S1–S6, ESI†). The X-ray data of 1 reveals (Fig. 1a and Table S1a, ESI†) an octahedral Mn(I) centre ligated to L1 in a cis-bidentate geometry with Nazo and Npyridinyl sites, while one azo group remains uncoordinated. The rest of the four coordination spots are filled by three CO and one Br− ion. The CO groups occupy the trans-positions for both the N-ligations in L1. The electron-dense Mn(I) center is stabilized in 1 with a strong π-back donation from Mn(I) (dπ) to coordinated Nazo and Npyridinyl(π*), which was evident from the relatively shorter Mn–Nazo (1.975 Å) and Mn–Npyridinyl (2.042 Å) bonds (Fig. 1a and Table S1a, ESI†). Interestingly, the CO groups positioned trans to Mn–Nazo and Mn–Npyridinyl bonds displayed a relatively longer Mn–CO bond distance (1.842 Å and 1.808 Å, respectively). This data showcases a dominant influence of the ligand scaffold L1 over the CO groups in stabilizing the electron-rich Mn(I) motif.20 This data indicates that both ligand L1 and CO groups are in competition to draw the Mn(I) electron density via π-back bonding, where L1 prevails over CO in complex 1. Such a situation leads to vulnerable Mn–CO bonds in complex 1 in the presence of light and converts 1 to a decarbonylated complex 1a under ambient conditions. The crystal structure of 1a (Fig. S6 and Table S2a, ESI†) demonstrates the formation of trigonal bipyramidal [MnIIBr2(L1)], containing a Mn(II)-coordinated to a N,N,N-tridentate L1 along with two cis-oriented Br−. The central Mn(II) binds with two Nazo and one Npyridinyl atom of L1 in 1a, albeit with a limited π-back donation, which was corroborated by the longer Mn–Nazo (2.393 and 2.363 Å) and Mn–Npyridinyl (2.141 Å) bonds (Fig. S6 and Table S2a, ESI†). The subtle variations in the coordination environment in the ligands and complexes L1, 1, and 1a have also affected deep into the ligand framework, which was monitored by the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nazo stretching band (Fig. S5, ESI†).
Nazo stretching band (Fig. S5, ESI†).
 | ||
| Fig. 1 SCXRD structures of complexes (a) 1, and (b) 2. C, N, O, Br, and Mn atoms are displayed as grey, blue, red, brown, and purple spheres, respectively. The hydrogen atoms are omitted for clarity. CCDC: (1) 2063724, (2) 2236211. (c) IR-SEC studies of 1 and 2 under Ar in DMF. The resting species of 1 (black trace) forms 2e− reduced anionic species (red trace) at −0.85 V. The resting species 2 (blue trace) forms Mn0 species (green trace) upon initial reduction at −1.5 V and finally 2e− reduced anionic species (purple trace) at −1.9 V. | ||
These results indicated that the Mn–CO bonds are required to be strengthened to induce better photostability into the Mn(I) carbonyl motif. Such a scenario can be created by deploying a ligand with relatively poorer π-back donation ability compared to CO. Following this cue, a new ligand scaffold L2 was designed with a redox-active triazole ligand containing an electron-donating –NH2 group and flanked with two pyridines (Fig. S7–S9, ESI†). This ligand produces [Mn(L2)(CO)3Br] complex (2), one of the best photostable Mn(I)–tricarbonyl complexes reported to date (Fig. 1b and Fig. S10–S12, ESI†). The presence of the peripheral amine also induces water solubility for 2, allowing us to follow its electrochemical reactivity even in the aqueous solution beyond the conventional organic realm. Crystals of 2 grown from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3OH/CH2Cl2 mixture illustrated the presence of a Mn(I) core in an octahedral cavity. Here, L2 stabilizes the Mn(I) centre via cis-oriented Npyridinyl and Ntriazole ligations while the rest of the coordination spots are filled by three CO and one Br− ion (Fig. 1b and Table S3a, ESI†).
:1 CH3OH/CH2Cl2 mixture illustrated the presence of a Mn(I) core in an octahedral cavity. Here, L2 stabilizes the Mn(I) centre via cis-oriented Npyridinyl and Ntriazole ligations while the rest of the coordination spots are filled by three CO and one Br− ion (Fig. 1b and Table S3a, ESI†).
The crystal structures of 1 and 2 were compared to further probe the differences in the electronic distribution around the Mn core. Interestingly, it was observed that the Mn–CO bonds trans to Mn–NL1/L2 are 0.01–0.05 Å shorter in 2 (1.734 Å and 1.793 Å) compared to 1 (Fig. 1a, b and Table S4, ESI†). This data suggests a better π-back bonding for Mn(I) → CO compared to Mn(I) → L2 in 2, which was further corroborated by relatively longer Mn–Ntria (2.092 Å) and Mn–Npyridinyl (2.132 Å) bonds. The FTIR data also demonstrated red-shifted asymmetric νCO stretching bands at 1937 and 1914 cm−1 in 2 compared to 1, indicating an improved Mn(I) → CO π-back bonding in the presence of an L2 scaffold (Fig. 1c and Table S4, ESI†). The optical properties and physical appearance of 2 remain the same even after its continuous exposure to white light for 24 hours under ambient conditions and upon 440 nm LED light irradiation as well (Fig. S11, ESI†). This data suggests that 2 retains its structural integrity under light irradiation, possibly due to stronger Mn–CO bonds with subtle modulation of the auxiliary ligand.
Complexes 1 and 1a are only soluble in organic solvents such as DMF and DMSO, while 2 displays a wide solubility ranging from organics to water. Thus, the electrochemistry studies of 1 and 1a were performed in DMF, while the same for 2 has been executed in both DMF and water. Here, all the data in organic media were reported vs. the ferrocenium/ferrocene couple and the aqueous data are represented against the standard hydrogen electrode (SHE) unless otherwise mentioned. The cyclic voltammogram (CV) of 1 displays an irreversible cathodic peak at −0.73 V, followed by four subsequent reversible signals in the range of −1.2 to −2.5 V during a cathodic scan (Fig. 2a and Fig. S13, ESI†). The stoichiometric redox signals were further analyzed via a complementary differential pulse voltammetry (DPV) experiment (Fig. 2a). The formation of this reduced Mn-species was also monitored via the significant red-shift of the asymmetric νCO bands during a spectroelectrochemical-IR (SEC-IR) experiment executed at −0.85 V primarily due to an enhanced π-back donation from the highly reduced metal center to strongly π-accepting CO ligands (Fig. 1c). The SEC-IR data were collected for each of the four subsequent of the reversible redox signals where no significant change was observed for the νCO bands but a sharp drop in νNN stretching (Fig. S15, ESI†). This observation prompted the assignment of these signals as ligand-based redox events. The assignment of the Mn(I/−I) process followed by four consecutive ligand-based reductions was also supported by the spectroelectrochemical-optical (SEC-optical) spectral change under analogous conditions, with a decrease in ligand-centred π–π* (375 nm) and metal-based d–d transition (590 nm) and simultaneous increase in MLCT band (450 nm) (Fig. S15b, ESI†).
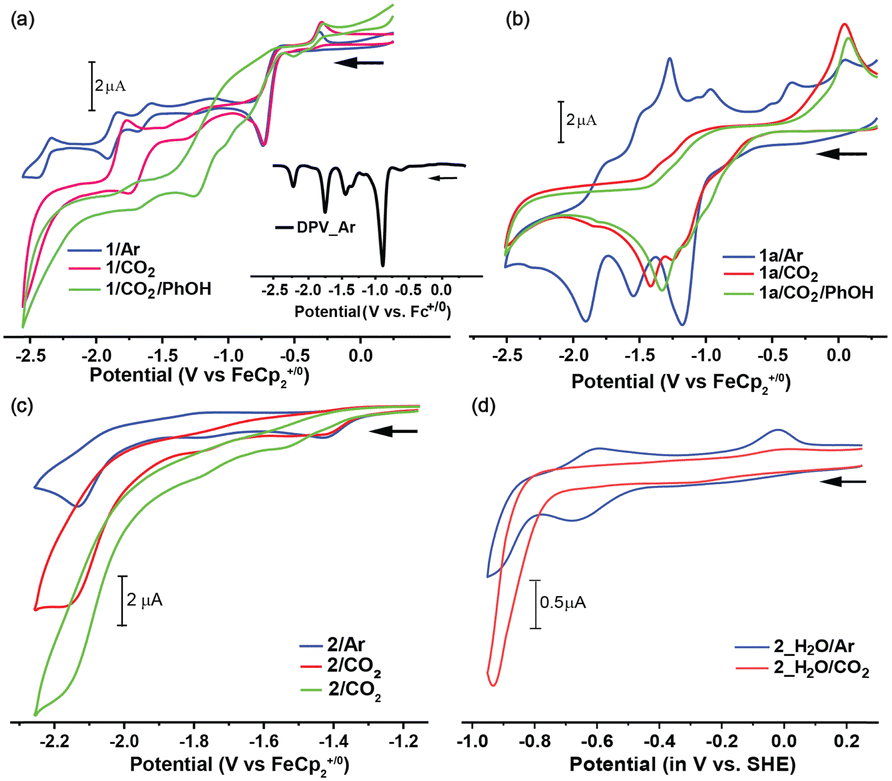 | ||
| Fig. 2 Cyclic voltammograms of complexes (a) 1 (in DMF), (b) 1a (in DMF), (c) 2 (in DMF), and (d) 2 in water, pH 7.0 recorded under Ar (blue trace), CO2 (red trace) and CO2 with added phenol (for 1 and 1a) or water (for 2) (green trace). A 1 mM catalyst in 0.1 M TBAF (in DMF) or 0.1 M KHCO3 (in water) was used with a glassy carbon working electrode, Pt counter electrode, and Ag/AgCl reference electrode. The inset in (a) displays the DPV spectra of 1 in DMF in the reduction direction (black trace). | ||
Under a CO2 atmosphere, the CV profile of 1 exhibited a significant enhancement in the cathodic current at −1.9 V, indicating a possible catalytic reduction of CO2. The molecular interaction of CO2 with 1 was followed with SEC-IR studies, where the IR spectra showed no discernible changes at constant potentials of −0.85 V and −1.5 V under CO2 compared to an Ar atmosphere in DMF (Fig. 1c and Fig. S16, S35, ESI†).
Additionally, no new bands appeared in the 2100–2200 cm−1 region, where a free CO stretching band is typically observed. However, the scenario changed when a potential of −1.95 V was applied, where the free CO band (νCO-asym) centred around ∼2140 cm−1 appeared along with a concomitant decrease in CO2 asymmetric stretching band (νCO2-asym) ∼2350 cm−1 (Fig. S35a, ESI†). This data indicates the consumption of CO2 followed by the production of CO during the catalysis. The catalytic nature of this signal was confirmed by the deviation from linearity of the current vs. √(scan rate) plot (Fig. S14, ESI†). The addition of the weak Brønsted acid phenol (PhOH) to a CO2-saturated DMF solution of 1 demonstrated a slight change in CO2 reduction signal along with the generation of a new catalytic signature beyond −1.0 V (Fig. 2a). This signal is primarily due to the electrocatalytic H2 production, which was confirmed by the complementary gas chromatography (GC) study executed with the headspace gas of an air-tight cell setup (Fig. S31, ESI†). Hence, 1 generates CO during CO2 reduction along with H2 in the presence of acid in organic media. The evolved CO was measured against time for 1 in DMF by GC analysis of the headspace of the air-tight electrolysis cell during controlled potential electrolysis (CPE) (Fig. S31, ESI†). The bulk electrolysis of 1 (Fig. S30 and S31, ESI†) at −1.95 V in DMF generated CO with a turnover number for CO (TONCO) ∼ 121 with a CO/H2 selectivity of 75:11 while displaying a faradaic efficiency (FE) of 86% (Table S5, ESI†).
On the other hand, complex 1a displayed a strong reduction peak at −1.2 V, probably due to the overlap of metal-centred and ligand-based redox events followed by two solely ligand-based reduction signals between −1.6 and −2.12 V (Fig. 2b). This allocation was further proven through SEC-optical investigations of 1a with appropriate change in metal and ligand-based transitions (Fig. S27, ESI†). The ligand-based reduction signals were further identified during a comparative CV of free ligand L1 sample (Fig. S26, ESI†). Under a CO2 atmosphere, the stoichiometric signals of 1a change, although no significant current enhancement leading to CO2 catalysis is observed even in the presence of external acid PhOH (Fig. 2b). The CPE experiments with 1a under CO2 at −1.5 V in PhOH-added DMF showcased majorly H2 production with no detectable CO (Table S5, ESI†). Thus, it was concluded that 1a remains dormant for electrocatalytic CO2RR.
The CV of 2 in DMF demonstrated that an irreversible reduction peak at −1.4 V is assigned due to the possible formation of Mn0 species (Fig. 2c and Fig. S18, ESI†). A reduction peak was noticed next at −1.7 V, presumably due to the formation of [MnL2(CO)3]−/Mn(−I) species along with an additional ligand-based reduction peak at −2.1 V (Fig. S17, ESI†). These assignments are supported by optical-SEC studies for 2 recorded under Ar conditions (Fig. S28, ESI†). The SEC-IR at −1.5 and −1.9 V showcased the generation of Mn(0) species and Mn(−I) species, respectively (Fig. 1c).21 Under a CO2 atmosphere, a significant current enhancement in the cathodic wave was observed for 2 in DMF along with the loss of reversibility of the peak at −1.7 V without the addition of any external acid. This current response was attributed to electrocatalytic CO2 reduction, ably supported by its dependence on scan rate and altering pattern of CO2 and Ar atmosphere (Fig. S19 and S20, ESI†). The SEC-IR data of 2 under CO2 atmosphere displayed a sharp decrease in νCO2-asym band along with the evolution of the free νCO-asym band at −2.2 V, indicating the consumption of CO2 during the catalysis along with the production of CO analogous to 1 (Fig. S35, ESI†). Finally, the rinse test experiments were conducted for 2 in DMF that verified the homogeneous nature of the electrocatalytic CO2 reduction (Fig. S25, ESI†). This CO2 reduction signal displayed a sharp increase when water was added externally as a proton source (Fig. 2c). The changes in CV data showcased the key role of the proton source (Fig. S21 and S22, ESI†) in electrocatalytic CO2 reduction. The GC-study of 2 in the DMF/H2O blend displayed majorly the production of CO under CO2 (Fig. S31, ESI†). This data provides conclusive evidence that 2 selectively reduces CO2 to CO even in the presence of water without any significant interference with H2 evolution. The CPE experiment performed with 2 in water-added DMF solution showcased significant CO production (TONCO ∼ 534) over H2 (CO:H2 ∼ 88:11) with 99% FE (Table S5, ESI†). During this CPE experiment 2 demonstrates no significant degradation, highlighting its remarkable stability under electrocatalytic conditions and a desirable feature for sustainable electrocatalysts (Fig. S32, ESI†). The inherent water solubility of 2 led us to its electrochemical studies in pure water. The CV of 2 in aqueous conditions (pH ∼ 6.8, 0.1 M carbonate) under Ar showed a reduction signal at −0.2 and −0.68 V due to the Mn(0) and penta-coordinated Mn(−I) formation, respectively, similar to DMF behaviour (Fig. 2d and Fig. S23, ESI†). A drop in ligand-centred π–π* (∼380 nm) and a slight increase in MLCT bands (∼570 and 600 nm) backed the redox signal assignments (Fig. S29, ESI†). In the presence of CO2, a strong cathodic response was observed beyond −0.85 V, indicating electrocatalytic CO2 reduction by 2 in water (Fig. 2d and Fig S24, ESI†). The CPE experiment for 2 in aqueous media (−0.85 V vs. SHE) displayed a continued preference for CO production over H2 (73:22; TONCO ∼ 113) while maintaining an appreciable 95% FE and low overpotential requirement (0.27 V) (Table S5, ESI†). Currently, the majority of CO2 is produced during energy conversion around the globe, where fossil fuel-derived CO2 is emitted with multiple other components (such as SOx, NOx, O2, and N2). Typically, this flue gas is treated first via a CO2 capture process to generate a pure stream of CO2 before it can be exposed to a CO2 utilization method. Hence, the source of CO2 was changed from a pure stream (100%) to a flue gas (15% CO2 along with 5% O2, 500 ppm SO2, and 100 ppm NO2 v/v), and the electrocatalytic CO2 reduction reactivity of complex 2 was investigated. The CV data of 2 under flue gas displayed the CO2 reduction signature at −0.85 V, while a new peak appeared at −0.40 V (Fig. S33, ESI†). Bulk electrolysis experiments were performed at both these signals to investigate their origin. The complementary GC followed by CPE showcased the formation of only H2 in the −0.4 V region, while both H2 and CO signals were noticed at −0.85 V (Fig. S34, ESI†). This data indicates that complex 2 maintains its remarkable CO production ability over H2 (CO:H2 ∼ 62:37, 99% FE) in aqueous solution with an appreciable rate (TONCO ∼ 32) even with a lean CO2-containing flue gas feed (Table S5, ESI†). This data indicates that the unique catalyst design strategy leads to an all-weather-ready electrocatalyst 2 that can handle untreated flue gas as it is, which will reduce the cost and overall process complexity of CO2 management in the absence of any CO2 capture step.
The CV and SEC-IR experiments indicated that 1 follows a 2 e−-concerted reduction step (path A) while 2 adheres to the formation of Mn(0) species (path B), a reaction to generate the catalytically active penta-coordinated Mn(−I) species S-1 (Scheme 1). The Mn(−I) species can generate a [Mn–Mn]0 dimer upon a comproportionation reaction with the parent complex in the case of less sterically hindered 1.11 The involvement of the redox-active ligand motifs alters electron density on the Mn center, which is underlined by the high overpotential requirement for electrocatalytic CO2 reduction by 2 compared to 1 under analogous conditions (Table S5, ESI†). This common intermediate S-1 can further bind to CO2, followed by protonation to produce S-2 species. The appearance of unique features ∼1800 cm−1 in the SEC-IR experiment under a CO2 atmosphere for both 1 and 2 supports the formation of this Mn–COOH intermediate (Fig. S35, ESI†). S-2 can proceed further via either an initial proton (PTET) or electron transfer (ETPT) step to form an S-3 intermediate. Both these pathways require the cleavage of a C–OH bond, which is expedited with an external proton source. Both PTET and ETPT pathways converge to generate a neutral tetracarbonyl species S-3. Finally, S-3 undergoes one e− reduction along with CO dissociation to complete the catalytic cycle, which was demonstrated by the evolution of the νCO-asym band in SEC-IR (Fig. S35, ESI†).
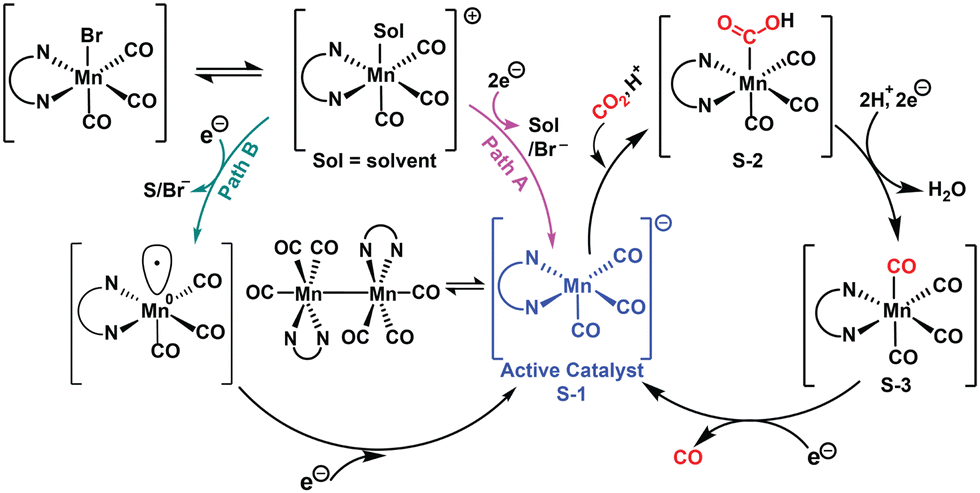 | ||
| Scheme 1 Proposed mechanism for electrocatalytic CO2RR for complexes 1 and 2. | ||
The intrinsic light sensitivity of the vulnerable Mn–CO bonds has impeded the practical applications of CO2-reducing Mn–carbonyl complexes. In this work, a tactically designed redox-active ligand was included in the catalyst framework that subtly tuned the electronic distribution, leading to an improved p-back bonded Mn–CO motif. This feature significantly improved the photostability of this new complex 2, as it rapidly converted CO2 to CO even from an untreated flue gas resource in pure water under electrocatalytic conditions with minimal H2 generation. Hence, this complex represents a robust and practically viable CO2 reduction catalyst that can be used for large-scale CO2 management.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
C. D. and A.D. have filed a patent application together with IIT Bombay (Indian Patent Application No. 202221049949) based on this work.Notes and references
- J. J. Concepcion, et al. , Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560–15564 CrossRef CAS PubMed.
- A. J. Morris, et al. , Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- E. E. Benson, et al. , Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- J. J. Walsh, et al. , Faraday Discuss., 2015, 183, 147–160 RSC.
- M. Bourrez, et al. , Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- J. Hawecker, et al. , J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- K. A. Grice and C. P. Kubiak, Advances in Inorganic Chemistry, Elsevier, 2014, vol. 66, pp. 163–188 Search PubMed.
- K.-Y. Wong, et al. , J. Electroanal. Chem., 1998, 453, 161–170 CrossRef CAS.
- J. M. Smieja, et al. , Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- C. Riplinger, et al. , J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- M. H. Rønne, et al. , ChemElectroChem, 2021, 8, 2108–2114 CrossRef.
- Q. Zeng, et al. , Organometallics, 2014, 33, 5002–5008 CrossRef CAS.
- C. Riplinger and E. A. Carter, ACS Catal., 2015, 5, 900–908 CrossRef CAS.
- D. C. Grills, et al. , J. Am. Chem. Soc., 2014, 136, 5563–5566 CrossRef CAS PubMed.
- C. Costentin, et al. , Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS PubMed.
- M. Beley, et al. , J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- G. Neri, et al. , Chem. Commun., 2016, 52, 14200–14203 RSC.
- A. Nakada and O. Ishitani, ACS Catal., 2018, 8, 354–363 CrossRef CAS.
- P. Kang, et al. , Chem. Sci., 2013, 4, 3497–3502 RSC.
- B. K. Ghosh and A. Chakravorty, Coord. Chem. Rev., 1989, 95, 239–294 CrossRef CAS.
- M. D. Sampson, et al. , J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC (1) 2063724, (2) 2236211. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03202k |
| This journal is © The Royal Society of Chemistry 2024 |