
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure of the caffeine–pyrogallol complex: revisiting a pioneering structural analysis of a model pharmaceutical cocrystal†‡
Okba
Al Rahal
 a,
Michael
Ferguson
a,
Cameron B.
Lennox
ab,
Louise
Male
a and
Tomislav
Friščić
*ab
a,
Michael
Ferguson
a,
Cameron B.
Lennox
ab,
Louise
Male
a and
Tomislav
Friščić
*ab
aSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: t.friscic@bham.ac.uk
bDepartment of Chemistry, McGill University, 801 Sherbrooke St. W., H3A 0B8 Montreal, Canada
First published on 4th June 2024
Abstract
The 1967 attempt of structural analysis of the solid-state complex of caffeine and pyrogallol was a pioneering structural investigation in the supramolecular chemistry of caffeine, of what today would easily be considered an archetype of a model pharmaceutical cocrystal. Re-investigating this historically important system demonstrates that this long overlooked complex is most likely a tetrahydrate with a different structure and composition than initially proposed, and provides the crystal structure of the anhydrous cocrystal.
Cocrystals have emerged as a means to develop new materials, by modifying the solid-state environment and properties of molecules without disrupting their covalent structure.1 Cocrystallisation has found particular value in controlling solid-state reactivity,2 and the creation of materials with novel optical,3 emissive,4 electrical,5 responsive and other behaviours.6 Arguably the most important application of cocrystals is in pharmaceutical materials science, allowing the optimisation of diverse solid-state properties of active pharmaceutical ingredients (APIs).7 The development of pharmaceutical cocrystals has heavily relied on model systems, such as commercial drugs, but also readily accessible molecules with functional groups typical of APIs. Among the latter, caffeine (caf, Fig. 1a) is among the most popular, providing a sandbox for exploring cocrystal design,8 polymorphism,9 moisture sensitivity,10 nucleation,11 screening methods,12 formulation stability, etc.13 The Cambridge structural database (CSD) contains >330 caf-based structures, a testament to the popularity and importance of caf in the structural science of organic solids.14 Complexation of caf is a persistent topic of supramolecular chemistry in solution and the solid state, and has been attracting the attention of chemists, and pharmaceutical and food scientists for over 120 years.15,16 Much groundbreaking work in caf complexation has focused on carboxylic acids,17 notably the work of Higuchi in the 1950s,18 and the earliest crystallographic report of a caf cocrystal in the CSD appears to be by Shefter, who in 1968 reported the structure of a complex with 5-chlorosalicylic acid: a pioneering entry of X-ray crystallography to understand caf complexation.19
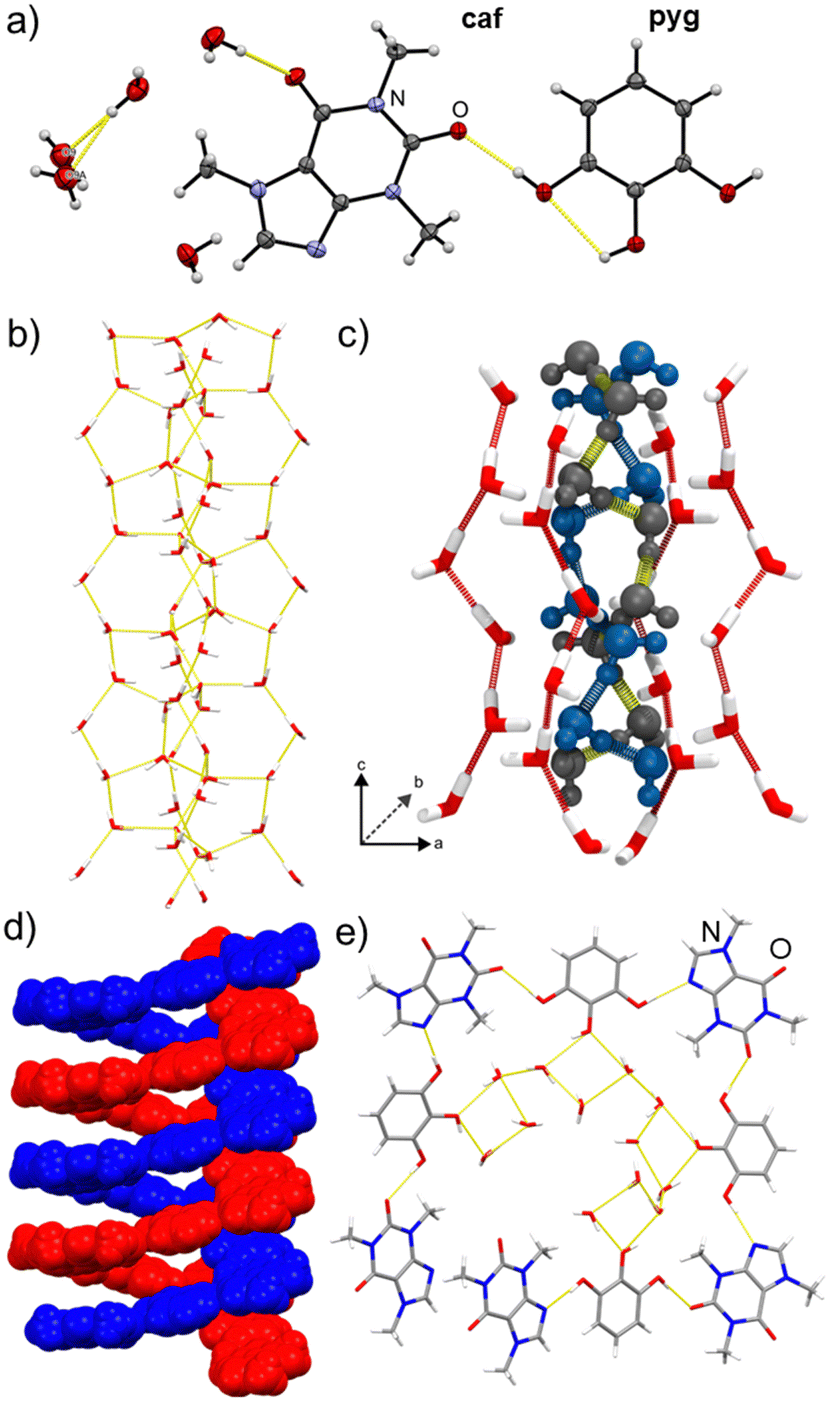 | ||
| Fig. 1 (a) ORTEP view of the asymmetric unit of caf·pyg·4H2O, with non-hydrogen atoms shown as ellipsoids of 50% probability. Views of the crystal structure of caf·pyg·4H2O: (b) a single hydrogen-bonded column; (c) two disordered core chains, shown in ball-and-stick style and colored blue and black, surrounded by an envelope of water molecules shown in stick style; (d) two interwoven hydrogen-bonded caf·pyg helices and (e) repeat unit of a caf·pyg helix viewed along the crystallographic c-axis. | ||
Here, we revisit an even earlier attempt of crystallographic analysis of a solid-state caf complex, which appears to have remained largely overlooked. In a thesis published in 1967, followed by a subsequent 1968 report, Arnone and Marchessault described a hydrated cocrystal with pyrogallol (pyg, Fig. 1a), of composition caf·pyg·5H2O.20 Whereas low data quality prevented reliable analysis (R-value > 0.5), the structure was proposed to consist of alternating caf and pyg layers, connected by hydrogen bonding with water molecules. Unexpectedly, a recent report outlined the structure of a hydrated cocrystal of composition 2caf·3pyg·2.5H2O, with crystallographic parameters very different from those of Arnone and Marchessault.21 While the original 1967 work appears to have remained largely unknown to the crystal engineering community, its historical significance in pharmaceutical and food science as one of the earliest, if not the earliest, crystallographic explorations of the supramolecular chemistry of caf, and also a prototype of a model pharmaceutical cocrystal, mandate re-visiting this system. We now show that the phase reported by Arnone and Marchessault is readily reproduced, but is in fact a tetrahydrate of a different structure than initially proposed, and report the previously not known anhydrate.
Cocrystallisation of caf and pyg was explored by ball-milling and by SpeedMixing,22i.e. rapid mixing in the presence of a liquid additive. Upon processing an equimolar mixture of caf and pyg in the presence of small amounts of nitromethane (MeNO2), both methods provided a material whose powder X-ray diffraction (PXRD) pattern was distinct from any of the starting materials or caf hydrate (see ESI‡). Varying the amount of liquid (expressed as the ratio of liquid additive volume to weight of solid material, η23) enabled this material to be obtained free of any starting materials, as evidenced by PXRD. Recrystallisation of the product by slow evaporation of a solution in ethyl acetate (EtOAc) produced rod-shaped crystals suitable for single crystal X-ray diffraction. Crystallographic analysis revealed a tetragonal unitcell in space group P42/n, with parameters a = 23.1123(5) Å, c = 6.8449(2) Å. These measured crystallographic parameters are consistent with those reported (a = 23.26 Å, c = 6.99 Å) for tetragonal caf·pyg·5H2O by Arnone and Marchessault. Analysis, however, reveals that the structure and composition are significantly different from those proposed for caf·pyg·5H2O.
Most significantly, the crystal structure (Fig. 1) was found to be a channel hydrate with a tetrahydrate (caf·pyg·4H2O), rather than pentahydrate, composition. The asymmetric unit of caf·pyg·4H2O contains a single molecule of caf and pyg, as well as four water molecules, with one being split equally over two mutually exclusive sites (oxygen atoms O9 and O9A, Fig. 1a). The water molecules arrange into columns extending along the crystallographic c-axis, exhibiting edge-fused hydrogen-bonded pentagons and heptagons (Fig. 1b, also ESI‡). Each water column comprises a core helical chain of alternating, hydrogen-bonded O9- and O9A-based water molecules, surrounded by an envelope of other water molecules. The 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 disorder of O9 and O9A atoms, validated by periodic density-functional theory (DFT), results in two types of such core chains, identical in structure but offset by a helix half-turn (Fig. 1c, also ESI‡). Each water column is further surrounded by helices of hydrogen-bonded alternating molecules of caf and pyg, again propagating in the c-direction (Fig. 1d). As a result of the centrosymmetric space group P42/n, the structure of caf·pyg·4H2O contains an equal number of left- and right-handed caf·pyg helices, as well as left- and right-handed O9⋯O9A hydrogen-bonded helices. The repeat unit of the caf·pyg helix is approximately rectangular in shape, and is based on O–H⋯N and O–H⋯O hydrogen bonds between 1,3-positioned –OH groups of each pyg with neighbouring caf molecules. The pyg molecules adopt a planar anti-conformation with only one intramolecular hydrogen bond.24 In this conformation, the –OH group in the 2-position of pyg attaches the caf·pyg helix to the self-assembled column of water molecules via O–H⋯O bonds (Fig. 1e).
:50 disorder of O9 and O9A atoms, validated by periodic density-functional theory (DFT), results in two types of such core chains, identical in structure but offset by a helix half-turn (Fig. 1c, also ESI‡). Each water column is further surrounded by helices of hydrogen-bonded alternating molecules of caf and pyg, again propagating in the c-direction (Fig. 1d). As a result of the centrosymmetric space group P42/n, the structure of caf·pyg·4H2O contains an equal number of left- and right-handed caf·pyg helices, as well as left- and right-handed O9⋯O9A hydrogen-bonded helices. The repeat unit of the caf·pyg helix is approximately rectangular in shape, and is based on O–H⋯N and O–H⋯O hydrogen bonds between 1,3-positioned –OH groups of each pyg with neighbouring caf molecules. The pyg molecules adopt a planar anti-conformation with only one intramolecular hydrogen bond.24 In this conformation, the –OH group in the 2-position of pyg attaches the caf·pyg helix to the self-assembled column of water molecules via O–H⋯O bonds (Fig. 1e).
The formation of a caf·pyg·4H2O single crystal from EtOAc was not expected, as no water was added either during the synthesis of the bulk material or recrystallisation. Moreover, the PXRD pattern simulated for the caf·pyg·4H2O structure differs from that of the bulk material before recrystallisation from EtOAc, indicating that the formation of a hydrate crystal is due to moisture absorption during crystal growth.
The caf·pyg·4H2O was also accessible in bulk form, by SpeedMixing or ball milling in the presence of water (see ESI‡). Specifically, milling at η = 0.25 μL mg−1 (corresponding to caf:pyg:H2O molar ratio of ca. 1:1:4.4) gave a material whose PXRD pattern, according to Rietveld refinement, was an excellent match to that simulated for caf·pyg·4H2O (see ESI‡). Attempted indexing of the PXRD pattern gave a tetragonal unitcell with a = 23.165(4) Å and c = 6.944(1) Å, in agreement with the parameters for the herein reported caf·pyg·4H2O structure, and those previously noted for caf·pyg·5H2O. To validate whether the bulk material could fit to the previously proposed caf·pyg·5H2O model, we attempted to find the structure solution from PXRD data using DASH (see ESI‡). Rietveld fitting of the three best simulated annealing solutions for the formula caf·pyg·5H2O gave unsatisfactory results, evidenced by significant residual features in the difference plot and poor Rwp (16–20%) and Rp (11–14%) indices (see ESI‡). In contrast, Rietveld refinement of the herein reported caf·pyg·4H2O structure against bulk PXRD data gave a much better fit, with Rwp = 7.7% and Rp = 5.3%.
Simultaneous thermogravimetric and differential scanning calorimetry analysis (TGA/DSC, see ESI‡) of the material revealed a weight loss of ca. 18% in the range 22–60 °C, consistent with the formula caf·pyg·4H2O. The weight loss is accompanied by a broad endothermic event with a maximum at ∼43 °C, followed by a sharper one at ∼127 °C. Hot-stage microscopy (see ESI‡) revealed that the weight loss step and the first endothermic signal are associated with sample liquefaction, which then solidifies and melts again. These observations suggest that heating of caf·pyg·4H2O leads to deliquescence and water loss, followed by crystallisation of an anhydrous phase. This agrees with TGA/DSC of the material prepared by milling or SpeedMixing with MeNO2, which revealed no weight loss before ca. 175 °C, indicating that it should be the anhydrous cocrystal caf·pyg. The DSC thermogram of this material exhibits a single, sharp endothermic event at ∼127 °C, confirming the melting point of the anhydrous cocrystal. Overall, these observations show that the herein obtained hydrate, as well as the material described by Arnone and Marchessault, is a tetrahydrate of composition caf·pyg·4H2O.
Besides the difference in water content, the herein determined structure of the caf·pyg hydrate differs significantly from the previously proposed pentahydrate model. In the latter, the molecules of caf and pyg are not directly hydrogen-bonded, but occupy separate layers connected through hydrogen bonds to chains of water molecules.25 One supramolecular aspect of the proposed caf·pyg·5H2O structure, however, matches the herein reported caf·pyg·4H2O structure: the orientation of π-stacked caf and pyg molecules in caf·pyg·4H2O corresponds well to that in the proposed pentahydrate model (see ESI‡). A deeper comparison of caf·pyg·4H2O and the pentahydrate model was, however, not possible, as the poor data quality from the original report meant that even the geometries of the component molecules were distorted (see ESI‡).
In order to directly compare the herein determined caf·pyg·4H2O and previously proposed caf·pyg·5H2O structures, we conducted a DFT optimisation using the PBE functional and D3 semi-empirical dispersion correction. Upon optimisation, the putative pentahydrate structure underwent significant (>15%) volume expansion. Detailed analysis of the optimised crystal structure also revealed a number of unusual characteristics, including the pyg molecule adopting an unusual conformation24 with only one intramolecular hydrogen bond, the central hydroxyl moiety twisted outside of the molecular plane, as well as several water molecules involved in very long (>3 Å) O–H⋯O hydrogen bonds or engaging in hydrogen bonding through just one –OH group. Attempted Rietveld fitting of the optimised pentahydrate structure to the PXRD pattern of bulk caf·pyg·4H2O was unsuccessful (see ESI,‡Rwp = 46.5% and Rp = 33.7%), reinforcing the view that the reported20 pentahydrate complex of caf and pyg was the herein described caf·pyg·4H2O structure.
To explore if previous work20 might have been dealing with another hydrated phase, the anhydrous caf·pyg was studied by dynamic vapour sorption (DVS, see ESI‡), which revealed a reversible weight change step of 18.8%, consistent with gain or loss of four equivalents of water from caf·pyg·4H2O (calculated: 18.4%). Analysis of the PXRD pattern of bulk caf·pyg·4H2O after the desorption step was consistent with complete transformation into anhydrous caf·pyg and, after exposure to 90% relative humidity (RH), it revealed complete conversion back to caf·pyg·4H2O. The PXRD pattern of an equimolar physical mixture of caf and pyg after exposure to 100% RH over 24 h revealed Bragg reflections consistent with caf·pyg·4H2O, caf·pyg, pyg·0.25H2O (CSD QQQBKD01) and caf hydrate (CSD CAFINE01), but no other solid forms of caf and/or pyg. Moreover, in situ PXRD monitoring of the thermal desolvation of caf·pyg·4H2O by heating in the range 20–100 °C revealed only the disappearance of Bragg reflections of caf·pyg·4H2O and the appearance of those of caf·pyg (see ESI‡).
It is surprising that TGA/DSC, VT-PXRD and DVS experiments so far did not provide any indication of the recently reported 2caf·3pyg·2.5H2O. Consequently, we attempted a targeted synthesis, by milling caf and pyg in the respective 2:3 stoichiometric ratio, with the addition of ∼2.5 equivalents of water. After 20 minutes, this experiment yielded caf·pyg·4H2O, as seen by PXRD. Reducing the amount of water to ca. 1.7 equivalents, however, gave a mixture of caf·pyg·4H2O and 2caf·3pyg·2.5H2O phases (see ESI‡). These experiments indicate a strong preference for the formation of the caf·pyg·4H2O phase, even when the amount of water is low.
We have also determined the structure of the previously not reported anhydrous cocrystal of caf and pyg (Fig. 2). Single crystals of this anhydrous phase were produced by slow evaporation of a solution of the product of SpeedMixing from a chloroform and acetone mixture (see ESI‡).
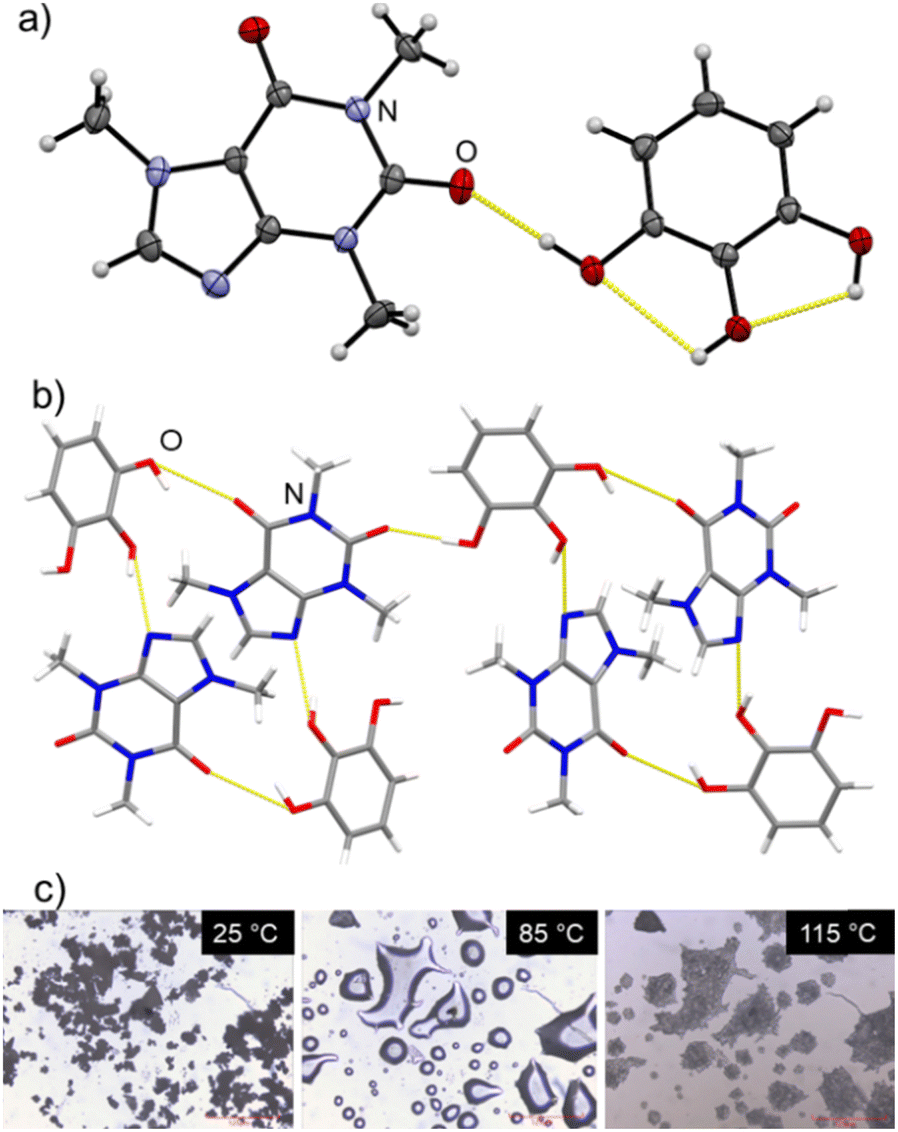 | ||
| Fig. 2 (a) ORTEP representation of the asymmetric unit of caf·pyg, with non-hydrogen atoms show as 50% probability ellipsoids; (b) view of the caf·pyg crystal structure, illustrating the hydrogen-bonded assembly of molecules; (c) hot-stage microscopy images of caf·pyg·4H2O, illustrating the liquefaction and recrystallisation (also see ESI‡). | ||
Structural analysis confirmed the composition caf·pyg, and the PXRD pattern simulated for the structure agreed with that of bulk caf·pyg (see ESI‡). The structure of caf·pyg is monoclinic (space group P21/n), and consists of tetrameric (caf)2(pyg)2 units reminiscent of the MacGillivray synthon,26 with the two ortho-neighbouring –OH groups of each pyg molecule forming O–H⋯N and O–H⋯O hydrogen bonds with imidazole and the C6-keto moieties of caf molecules, respectively, forming a R44(22) motif. The remaining –OH group on each pyg molecule forms a hydrogen bond to a keto group of a neighbouring (caf)2(pyg)2 unit, resulting in layers parallel to the crystallographic (101) set of planes. In contrast to caf·pyg·4H2O, the pyg molecules in anhydrous caf·pyg adopt the most stable syn1-conformation,24 with two intramolecular O–H⋯O hydrogen bonds.
In summary, we have re-investigated what is likely to be one of the earliest reported attempts of crystallographic analysis of a caffeine cocrystal. We show that the initially reported crystalline complex with pyrogallol and its associated crystallographic parameters are readily reproduced. Structure analysis, however, shows that the complex is actually a tetrahydrate of a caffeine cocrystal with pyrogallol, with the anhydrous form also reported here. The tetrahydrate crystal packing is different than initially proposed: whereas the original model anticipated that caffeine and pyrogallol would interact solely via π-stacking,25 the herein reported structures show direct hydrogen bonding between the components. Beyond the historical relevance in caffeine complexation, which is an area of significance in the development of supramolecular chemistry and has attracted interest for over a century, we note that this is also a contribution to the still poorly developed understanding of caffeine complexation with polyphenols.14,27
The data supporting this article have been included as part of the ESI.‡ Crystallographic data in CIF format has been deposited with the Cambridge Structural Database (CSD), deposition codes CCDC 2344173 and 2344174.‡
We are grateful to Prof. A. Arnone for valuable discussions and comments, and for the support of Leverhulme International Professorship (TF), NSERC Doctoral scholarship (CBL), University of Birmingham, and the BlueBEAR HPC service.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047 RSC; (b) G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed; F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638 Search PubMed.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030 CrossRef CAS PubMed.
- R. Kuroda, Y. Imai and N. Tajima, Chem. Commun., 2002, 2848 RSC.
- J. Vainauskas, T. H. Borchers, M. Arhangelskis, L. J. McCormick McPherson, T. S. Spilfogel, E. Hamzehpoor, F. Topić, S. J. Coles, D. F. Perepichka, C. J. Barrett and T. Friščić, Chem. Sci., 2023, 14, 13031 RSC.
- C.-H. Liu, M. R. Niazi and D. F. Perepichka, Angew. Chem., Int. Ed., 2019, 58, 17312 CrossRef CAS PubMed.
- T. H. Borchers, F. Topić, M. Arhangelskis, J. Vainauskas, H. M. Titi, O. S. Bushuyev, C. J. Barrett and T. Friščić, J. Am. Chem. Soc., 2023, 145, 24636 CAS.
- (a) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef CAS PubMed; (b) S. L. Childs and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 4208 CrossRef CAS.
- T. Leyssens, N. Tumanova, K. Robeyns, N. Candoni and S. Veesler, CrystEngComm, 2014, 16, 9603 RSC.
- S. J. Diez, M. D. Eddleston, M. Arhangelskis, M. Milbled, M. J. Müller, A. D. Bond, D.-K. Bučar and W. Jones, Cryst. Growth Des., 2018, 18, 3263 CrossRef CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013 CrossRef CAS.
- D.-K. Bučar, G. M. Day, I. Halasz, G. G. Z. Zhang, J. R. G. Sander, D. G. Reid, L. R. MacGilivray, M. J. Duer and W. Jones, Chem. Sci., 2013, 4, 4417 RSC.
- S. Aher, R. Dhumal, K. Mahadik, A. Paradkar and P. York, Eur. J. Pharm. Sci., 2010, 41, 597 CrossRef CAS PubMed.
- P. MacFhionnghaile, C. M. Crowley, P. McArdle and A. Erxleben, Cryst. Growth Des., 2020, 20, 736 CrossRef CAS.
- CSD Version 5.45 (March 2024 update) contains 338 structures with caf molecules, of which 247 do not have any protonation or metal bonding on the N-3 atom. Of the latter, 155 involve also a carboxylic acid, and 40 include a phenol group only.
- (a) A. Regenbogen and N. Schoorl, Pharm. Weekbl., 1924, 61, 34 CAS; (b) A. Kwisda, Z. Allg. Oesterr. Apoth.-Ver., 1897, 35, 90 Search PubMed; (c) I. Horman and R. Viani, J. Food Sci., 1972, 37, 925 CrossRef CAS.
- (a) Y. Shen, Y. Xiao, R. M. Edkins, T. G. A. Youngs, T.-L. Hughes, J. Tellam and K. Edkins, Int. J. Pharm., 2023, 647, 123520 CrossRef CAS PubMed; (b) N. J. Baxter, M. P. Williamson, T. H. Lilley and E. Haslam, Farad. Trans., 1996, 92, 231 RSC.
- (a) G. P. Stahly, Cryst. Growth Des., 2009, 9, 4212 CrossRef CAS; (b) M. Donbrow and Z. A. Jan, J. Pharm. Pharmacol., 1965, 17, 129S CrossRef CAS.
- T. Higuchi and D. A. Zuck, J. Am. Pharm. Assoc., 1952, 41, 10 CrossRef CAS PubMed.
- (a) E. Shefter, J. Pharm. Sci., 1968, 57, 350 CrossRef CAS PubMed; (b) E. Shefter, J. Pharm. Sci., 1968, 57, 1163 CrossRef CAS PubMed.
- (a) A. Arnone, The Crystal Structure of the Caffeine-Pyrogallol Complex, MSc thesis, State University College of Forestry at Syracuse University, 1967; (b) A. Arnone and R. H. Marchessault, Structure of the Caffeine—Pyrogallol Complex in Molecular Association in Biological and Related Systems, ed. E. D. Goddard, Adv. Chem. Ser., ACS, vol. 84, 1968, pp. 235–258 Search PubMed.
- F. Molajafari, T. Li, M. Abbasichaleshtori, M. H. Z. D., A. F. Cozzolino, D. R. Fandrick and J. D. Howe, CrystEngComm, 2024, 26, 1620 RSC.
- Y. Teoh, G. Ayoub, I. Huskić, H. M. Titi, C. W. Nickels, B. Herrmann and T. Friščić, Angew. Chem., Int. Ed., 2022, 61, e202206293 CrossRef CAS PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418 RSC.
- (a) R. Thakuria, S. Cherukuvada and A. Nangia, Cryst. Growth Des., 2012, 12, 3944 CrossRef CAS; (b) H. Gier, W. Roth, S. Schumm and M. J. Gerhards, Mol. Struct., 2002, 610, 1 CrossRef CAS; (c) I. Vedernikova, D. Salahub and E. Proynov, J. Mol. Struc. THEOCHEM, 2003, 663, 59 CrossRef CAS.
- A similar structure is, however, observed in a hydrate of caf cocrystal with gallic acid: L. Vella-Zarb and U. Baisch, Acta Cryst., 2018, E74, 559 CrossRef PubMed.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS PubMed.
- The herein reported structures differ notably from those of caf with polyphenols gallic acid and methyl gallate, that are layered or column-based:25 (a) R. Martin, T. H. Lilley, N. A. Bailey, C. P. Falshaw, E. Haslam, D. Magnolato and M. J. Begley, Chem. Commun., 1986, 105 RSC; (b) H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152 CrossRef CAS.
Footnotes |
| † This article is dedicated to the memory of Professor Robert H. Marchessault. |
| ‡ Electronic supplementary information (ESI) available: Details of experimental and computational procedures, and PXRD, crystallographic, thermal analysis and FTIR-ATR data. CCDC 2344173 and 2344174. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02289k |
| This journal is © The Royal Society of Chemistry 2024 |