
Effect of mussel-inspired polydopamine on the reinforced properties of 3D printed β-tricalcium phosphate/polycaprolactone scaffolds for bone regeneration
Chia-Che
Ho
ab,
Yi-Wen
Chen
cd,
Kan
Wang
e,
Yen-Hong
Lin
d,
Ta-Cheng
Chen
bf and
Ming-You
Shie
 *adg
*adg
aDepartment of Bioinformatics and Medical Engineering, Asia University, Taichung, Taiwan. E-mail: sethho@asia.edu.tw
bHigh Performance Materials Institute for x-Dimensional Printing, Asia University, Taichung City, Taiwan
cGraduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan
dx-Dimension Center for Medical Research and Translation, China Medical University Hospital, Taichung, Taiwan
eGeorgia Tech Manufacturing Institute, Georgia Institute of Technology, Atlanta, GA, USA
fDepartment of Information Management, National Formosa University, Yunlin, Taiwan
gSchool of Dentistry, China Medical University, Taichung, Taiwan
First published on 1st November 2022
Abstract
Bioceramic/polymer scaffolds have been considered as potential grafts used for facilitating bone healing. Unfortunately, the poor interfacial interaction between polymer matrices and bioceramic fillers limited their use in practical medicine. Thus, a facile strategy for reinforcing the three-dimensional printed β-tricalcium phosphate/polycaprolactone scaffolds through employing polydopamine modified-ceramics as fillers. The effects of the dopamine precursor on the compressive strength, degradability, cell proliferation, osteogenic differentiation, and in vivo osteogenicity were measured. The results indicated that the concentration of dopamine could remarkably affect the thickness and density of the polydopamine layer on fillers, further varying the compressive strength (1.23-fold to 1.64-fold), degradability, and osteogenicity of the scaffolds. More importantly, the presence of polydopamine in the three-dimensional printed composite scaffolds not only facilitated the proliferation, alkaline phosphatase activity and mineralization of mesenchymal stem cells, but also stimulated the formation of neo-bone tissue in femur defects. Taking together, the proposed scaffolds might serve as a candidate for bone regeneration.
1. Introduction
Synthetic polymers, such as polycaprolactone (PCL), polylactic acid (PLA) and polylactic-co-glycol acid (PLGA), have been considered as candidates for bone tissue engineering due to their combined advantages of biodegradability, biocompatibility, and workability.1,2 In particular, PCL has shown its applicability to the manufacturing of customized porous scaffolds with controlled porosities and pore structures by means of an extrusion-based three-dimensional (3D) printing strategy because of its low melting and crystallization temperature.3 Although the inert surface of PCL can hinder the adhesion and growth of cells, it can be remarkably enhanced through the incorporation of osteoconductive bioceramics, such as hydroxyapatite (HA), calcium phosphate-based and calcium silicate-based materials.4 Additionally, the inorganic particles can function as reinforcing agents to raise both stiffness and toughness of PCL through providing mechanical interlocking supports to the polymer matrix.5 However, the reinforcing effect of the bioceramic would gradually become irrelevant when the concentration of fillers exceeds the critical content in the composite owing to the absence of chemical bonds between hydrophilic ceramic fillers and the inert polymer matrix with hydrophobicity.6To overcome this issue, numerous attempts have been made, such as enhancing the exposed surface area of the bioceramic phase to leverage the osteoconductivity of composite scaffolds through deposition of calcium phosphate on the surface of polymeric scaffolds, as well as introducing additional fillers (e.g., graphene) in the composite system to promote the mechanical interlock bonding.7–9 Nevertheless, the effectiveness of the abovementioned routes on the simultaneous enhancement of both osteogenic activity and mechanical strength still remains challenging. Hence, performing suitable surface modification on the bioceramic fillers to enhance the interfacial strength between polymer matrices and fillers has been considered as a potential strategy to improve the mechanical properties of the composite with high filler contents.10 Wang et al. suggested that grafting of γ-methacryloxypropyl-trimethoxysilane on the surface of HA could be an effective strategy for enhancing the reinforcement effect of bioceramic dopants due to the improved interfacial interactions and better dispersity of fillers in polymer matrices.11 However, silane molecules leached from fillers due to unstable coating and/or degradation of substrates may lead to adverse effects on tissues surrounding the scaffolds.12
Since the last decade, the mussel-inspired polydopamine (PDA) surface modification has gained much attention due to its astounding material-independent adhesive properties and the ability to serve as a multifunctional platform for enabling the surface of PDA-modified substrates to be decorated effortlessly with a large pool of materials.13 Since its first emergence, the PDA modification strategy has contributed to many breakthroughs in the development of bone grafts and implants through immobilizing osteoconductive and osteoinductive matters on conventional biomaterials.14 However, limited research studies have focused on elucidating the effects of PDA modification on the reinforcing efficiency of bioceramics in the organic/inorganic composites despite the fact that PDA has been considered as an efficient interfacial reinforcement agent in different composite systems.15
Thus, the aim of this study was to assess the reinforcing efficiency of PDA the PCL/bioceramic composite. β-Tricalcium phosphate (β-TCP) was employed as the bioceramic filler due to its excellent biodegradability, biocompatibility, and osteoconductivity.16,17 The PDA-β-TCP/PCL composite was fabricated following a two-step process, including deposition of PDA on β-TCP powder and blending the modified powder into the PCL matrix. The morphologies and chemical structures of different PDA-β-TCPs obtained by performing the self-polymerization of the dopamine (DA) precursor at different DA concentrations. Furthermore, the porous scaffolds were fabricated through an extrusion-based robotic dispensing system, and the morphology, compressive strength, degradability, cytocompatibility, and osteogenicity of the scaffolds were analyzed. Finally, the in vivo femur bone defect regeneration experiments were presented to examine the osteogenic capability of PDA-β-TCP/PCL scaffolds, and we showed that the scaffolds can significantly enhance bone repair at the defect site.
2. Materials and methods
2.1. Preparation and characterization of PDA-β-TCP powder
Briefly, 10 g of β-TCP powder (Sigma-Aldrich, St. Louis, MO, USA) was immersed in 100 mL of trishydroxymethyl aminomethane buffer (tris-buffer; 50 mM, pH 8.5) containing various concentrations (2, 4, and 8 mg mL−1) of DA (Sigma-Aldrich) and vigorously stirred for 24 h at room temperature. After modification, the PDA-β-TCP was washed and dried at 100 °C for 1 day. The specimen codes “T”, “2PT”, “4PT” and “8PT” represent the pristine β-TCP and PDA-β-TCP powders obtained by performing the PDA modification at DA precursor concentrations of 2, 4, and 8 mg mL−1, respectively. The morphologies of different powders were examined using both a field emission scanning microscope (SEM; JSM-6700F, JEOL, Tokyo, Japan) and a high-resolution transmission electron microscope (HRTEM; JEM-1400, JEOL). The chemical structure of PDA-β-TCP was examined by 13C solid-state nuclear magnetic resonance (13C ssNMR; DSX400 WB, Bruker, Karlsruhe, Germany). The weight ratio of the PDA coated on β-TCP was determined by using a thermogravimetric analyzer (TGA; Q500, TA Instruments, New Castle, DE) over a temperature range from room temperature to 800 °C at a heating rate of 10 °C min−1 under atmosphere.2.2. Preparation of PDA-β-TCP/PCL composite scaffolds
To prepare β-TCP/PCL and PDA-β-TCP/PCL composites, 5 g of β-TCP or PDA- β-TCP powder was suspended in ethanol and the mixture was added dropwise to 5 g of molten PCL (Mw = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000, Polysciences, Warrington, PA) at 120 °C. After being vigorously stirred at 120 °C, the composite was dried at 100 °C for 24 h. The molten paste was transferred into a syringe barrel equipped with a metallic nozzle with an inner diameter of 0.4 mm. The specimen codes of “C”, “TC”, “2PTC”, “4PTC” and “8PTC” represent the pristine PCL, T/PCL, 2PT/PCL, 4PT/PCL, and 8PT/PCL, respectively. The scaffolds were fabricated by using a bioplotting system (Bioscaffolder 3.1, GeSim, Grosserkmannsdorf, Germany) at processing temperatures of 70 and 90 °C for neat PCL and composite scaffolds, respectively. The pneumatic pressures/nozzle moving rates used for the fabrication of C, TC, 2PTC, 4PTC, and 8PTC scaffolds were 1.5 mm s−1/230 kPa, 0.7 mm s−1/350 kPa, 0.6 mm s−1/400 kPa, 0.45 mm s−1/400 kPa, and 0.45 mm s−1/400 kPa, respectively. The layers with a thickness of 0.35 mm were stacked on each other at a 0°–90° orientation. The first two layers of the scaffold were both filled with seven parallel struts with a diameter of 500 μm and separated by 500 μm. Subsequent third and fourth layers were filled by six parallel struts which were located above the gaps of the first and second layers, respectively.
000–50000, Polysciences, Warrington, PA) at 120 °C. After being vigorously stirred at 120 °C, the composite was dried at 100 °C for 24 h. The molten paste was transferred into a syringe barrel equipped with a metallic nozzle with an inner diameter of 0.4 mm. The specimen codes of “C”, “TC”, “2PTC”, “4PTC” and “8PTC” represent the pristine PCL, T/PCL, 2PT/PCL, 4PT/PCL, and 8PT/PCL, respectively. The scaffolds were fabricated by using a bioplotting system (Bioscaffolder 3.1, GeSim, Grosserkmannsdorf, Germany) at processing temperatures of 70 and 90 °C for neat PCL and composite scaffolds, respectively. The pneumatic pressures/nozzle moving rates used for the fabrication of C, TC, 2PTC, 4PTC, and 8PTC scaffolds were 1.5 mm s−1/230 kPa, 0.7 mm s−1/350 kPa, 0.6 mm s−1/400 kPa, 0.45 mm s−1/400 kPa, and 0.45 mm s−1/400 kPa, respectively. The layers with a thickness of 0.35 mm were stacked on each other at a 0°–90° orientation. The first two layers of the scaffold were both filled with seven parallel struts with a diameter of 500 μm and separated by 500 μm. Subsequent third and fourth layers were filled by six parallel struts which were located above the gaps of the first and second layers, respectively.
2.3. Characterization of PDA-β-TCP/PCL composites
The rheological properties of composites were analyzed by using a rheometer (MCR302, Anton Paar, Graz, Austria) operated on a parallel plate geometry with a diameter of 25 mm and a gap of 0.5 mm. The oscillatory frequency sweep test was conducted to measure the changes in complex viscosity within an angular frequency range between 0.1 and 500 rad s−1 with a strain of 0.1% at different temperatures (60, 90, and 120 °C). The morphologies of different composite scaffolds were examined by SEM. The weight ratio of β-TCP or PDA-β-TCP in the composite was determined by using a thermogravimetric analyzer (TGA; Q400, TA Instrument, New Castle, DE) under a N2 atmosphere at a heating rate of 10 °C min−1. A differential scanning calorimeter (DSC; Q100, TA Instrument) was used to record the thermal history of the specimen that underwent two heat cycles from 10–85 °C under a N2 atmosphere at a temperature ramping rate of 5 °C min−1. The peak temperature recorded in the second heating scan and the integral area of the DSC curves were defined as the melting temperature and enthalpy change (ΔHm), respectively. The relative crystallinity (Xc) of the composite was calculated according to the following equation:where ΔHPCL and CPCL are melting enthalpy of fully crystalized PCL (139 J g−1) and the weight fraction of PCL, respectively.18
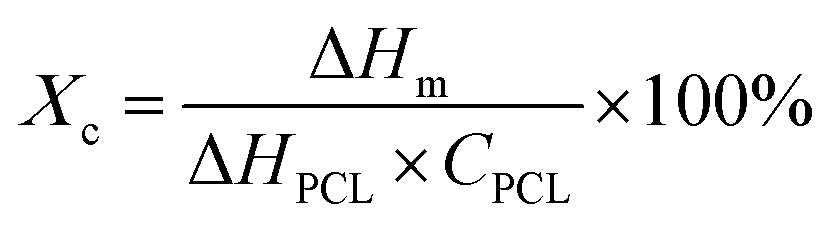
To measure the compressive strength and modulus of the composite scaffolds, the scaffold with a dimension of 6.5 × 6.5 × 10 mm3 was placed on a universal testing machine (MTS Exceed® E42.503 with 500 N load cell; MTS system, Eden Prairie, MN) at a loading rate of 1 mm min−1.
2.4. In vitro immersion experiments
To assess the effect of PDA-β-TCP on the degradation kinetics of PCL, the dry weight of the as-prepared scaffold with dimensions of 6.5 × 6.5 × 10 mm3 was recorded by using a digital balance. Subsequently, the scaffold was immersed in 40 mL of sterilized simulated body fluid (SBF) that was prepared according to the previous study.2 After being soaked in SBF at 37 °C for 1, 2, 3, and 4 weeks, the scaffolds were washed with distilled water three times, dried by freeze-drying, and weighed to calculate the weight loss of the individual sample. The compressive strengths and modulus of the scaffolds soaked in SBF for different periods were also tested.2.5. Cell adhesion and proliferation
Human Wharton's jelly mesenchymal stem cells (hWJMSCs; RM60596, BCRC, Hsinchu, Taiwan) were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Waltham, MA) supplemented with 10% fetal bovine serum (Gibco), 100 U mL−1 penicillin/100 μg mL−1 streptomycin (Gibco), 10−8 M dexamethasone (Sigma-Aldrich), 2.16 g L−1 glycerol 2-phosphate disodium salt hydrate (Sigma-Aldrich), and 0.05 g L−1L-ascorbic acid (Sigma-Aldrich) in an incubator with a humidified atmosphere and 5% CO2 at 37 °C, and the culture medium was changed every 2 days. Cells at passage 3–8 were used for further experiments. All scaffolds with dimensions of 6.5 × 6.5 × 1.4 mm3 were sterilized with 75% ethanol and ultraviolet light for 30 min. Afterwards, cells (10000 cells per well) were seeded on the scaffolds. After culturing for 6 and 24 h, the scaffolds were washed with phosphate buffer solution (PBS) three times and fixed in 4% paraformaldehyde at 4 °C overnight. Afterward, the cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 5 min. The cytoskeleton and nuclei of cells adhered on the scaffolds were stained with Alexa Fluor™ 488 phalloidin (Invitrogen, Carlsbad, CA) and 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen), respectively, according to the manufacturer's instruction. The fluorescent images of cells were captured by using an Olympus BX53 microscope equipped with a DP-50 digital camera (Olympus, Tokyo, Japan) at ×200 magnification. For cell proliferation analysis, the medium was replaced with fresh medium containing 0.5 mg mL−1 of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) at each culture period (1, 3, and 7 d), and then incubated with cells for 2 h. The precipitated formazan crystals were dissolved by DMSO and the absorbances at 570 nm with a reference wavelength of 650 nm were measured using a Tecan Infinite 200® PRO microplate reader (Tecan, Männedorf, Switzerland).
2.6. Osteogenic activity assay
The osteogenic differentiation of hWJMSCs cultured on different scaffolds was evaluated by measuring the alkaline phosphatase (ALP) activity and the level of matrix mineralization. For ALP activity assay, the cells cultured on scaffolds for 7 and 14 days were washed with 0.9% NaCl and lysed in 0.2% NP-40 (Sigma-Aldrich) solution. The cell lysate was incubated with p-nitrophenyl phosphate (pNPP; Sigma-Aldrich) for 1 h, after which the reaction was terminated by adding 5 M NaOH and the absorbance at 405 nm was measured using a microplate reader. Furthermore, the absorbance was normalized to the reaction time and the total protein of each sample which was determined using the BCA protein assay reagent (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. For matrix mineralization, the cells cultured on scaffolds for 7 and 14 days were fixed in 4% paraformaldehyde (Sigma-Aldrich) at room temperature for 2 h. After being washed with PBS, the scaffold was then stained by 0.5% Alizarin-Red S solution for 15 min at room temperature. After being extensively washed, the scaffold was then soaked in 250 μL of PBS containing 0.1 M cetylpyridinium chloride (Sigma-Aldrich) and the absorbance at 562 nm was measured. The absorbance of the dye-stained scaffold without culturing cells was used as the reference absorbance.2.7. In vivo bone healing
The in vivo bone healing experimental procedures employed in the present study were approved by the Animal Experimental Ethics Committee of China Medical University (CMUIACUC-2019-099-1). Male New Zealand white rabbits aged three months and weighing 1.5–2 kg were used and randomly divided into three groups, namely TC, 2PTC, and 8PTC. General anesthetization was induced by artificial respiration of oxygen containing 5% isoflurance. Disinfection was conducted by applying alcohol and iodine to the skin of the hind legs after being shaved. The skin and muscle fascia were carefully dissected to expose the distal femoral condyle without causing adverse nerve and blood vessel injuries. A bone defect with a 6 mm diameter and a 6 mm depth were made using a low-speed dental drill on the medial femoral condyle. Scaffolds sterilized with 75% ethanol disinfection were implanted in the condyle defects. Eventually, the wound was suture closed and an anti-inflammatory ointment was applied. The rabbits were sacrificed by carbon dioxide asphyxiation at 4 and 8 weeks post-surgery. The scaffold-implanted bone tissue was harvested and fixed in 10% normal formalin for 2 days. After being rinsed, gradient dehydrated, and embedded in an optimum cutting temperature compound (OCT®, KMA-0100-00A, CellPath Ltd, Newtown, Wales, UK), the specimens were sliced into 6 μm-thick sections and stained either with hematoxylin/eosin, Masson's Trichrome staining kit (MT; ScyTek Lab, West Logan, UT) or Von Kossa kit (VK, ScyTek Lab) following the manufacturer's instructions. Stained sections were visualized under a BX53 Olympus microscope equipped with a DP-50 digital camera. To better evaluate the ability of guiding bone regeneration for different scaffolds, the microcomputed tomographic images of the scaffold-implanted femurs were captured using a microcomputed tomography system (μCT, SkyScan 1076, SkyScan Inc., Kontich, Belgium) equipped with a 1.4 M X-ray CCD camera at the maximum tube current and voltage of 0.2 mA and 160 kV, respectively. The 3D models of the femur scans constructed from a 400 slice images with a slice increment of 30 μm were used for assessment and quantitative histomorphometric analysis. A SkyScan software was used to calculate the newly formed bone tissues in terms of the bone volume per tissue volume (BV/TV) and trabecular thickness (Tb.Th). The results were obtained in triplicate from three independent measurements.2.8. Statistical analysis
A one-way analysis of variance (ANOVA) was used to assess the significance of the differences between the mean values in the measured data obtained from triplicate experiments. A Scheffe's multiple comparison test was used to determine the significance of the deviations in the data for each specimen. In all cases, the results were considered statistically significant if the p-value <0.05.3. Results and discussion
3.1. Characterization of PDA-β-TCP powders
In the present study, PDA was modified on the surface of β-TCP based on the self-oxidation and polymerization of DA in a mild basic solution. During this process, the β-TCP surface could provide binding sites for the adhesion of monomer, oligomer, and polymer of DA through interactions between amine/catechol moieties of PDA and Ca2+/PO43− ions.19 To assess the effect of DA precursor concentration on the deposition of PDA, the microstructures of PDA-deposited powders were examined by SEM and TEM. As shown in Fig. 1, the SEM images illustrated that a smooth surface was observed on β-TCP particles. In contrast, the surface became rougher after PDA deposition due to the formation of a layer composed of granular aggregates. It seems that the size of aggregates deposited on the surface of particles gradually increased with increasing precursor concentration. It has been well-established that PDA deposition could be remarkably affected by a series of parameters, such as DA and dissolved oxygen concentration, solution pH, ionic strength, and temperature.20 Among these, it was suggested that the thickness of the deposited PDA layer could be increased by increasing the precursor concentration.21 Interestingly, the TEM micrographs indicated that a firm layer assembled by nanoparticles with a size of about 50 nm was observed on the surface of 2PT, yet the increase in the thickness of the deposited PDA layer was not observed while a higher concentration of the DA precursor was used. In contrast to 2PT, a loosened layer composed of nanoparticles with a diameter of ca. 110 nm was obtained in 4PT. However, the thickness of the PDA layer would remarkably decrease while the PDA deposition was performed at the DA concentration of 8 mg mL−1. To better elucidate the controversial results, the chemical structure and the content of PDA deposited on the β-TCP surface were examined by 13C ssNMR and TGA, respectively. | ||
| Fig. 1 SEM and TEM micrographs of pristine β-TCP and different PDA-β-TCP particles. | ||
The 13C ssNMR spectra of PDA-β-TCP powders (Fig. 2A) revealed good agreements with the typical spectra of PDA demonstrated in previous studies.22 where the characteristic peaks located at chemical shifts between 30 and 40 ppm could be assigned to carbon atoms present in the five-membered ring in the pyrrole moiety or the aminoethyl residue in catecholamine and catecholamine ortho-quinone; the board band in between 110 and 130 ppm was attributed to the existence of aromatic carbon atoms in catechol or quinone moieties; the characteristic peaks of bridgehead carbon atoms were located at 135 and 144 ppm, whereas the peaks of quinoid carbonyl carbon atoms were positioned between 170 and 180 ppm. These results illustrated that the major organic component in the powder was PDA and the chemical structure remained similar, regardless of whether the initial DA concentration was 2, 4, or 8 mg mL−1. The weight ratio of PDA in the β-TCP powder was determined by TGA. As seen in the TGA curves (Fig. 2B), there are two distinct regions of the temperature-dependent weight loss in the ranges of 50–200 °C and 200–650 °C, which could be attributed to the evaporation of adsorbed water and decomposition of PDA, respectively. By calculating the weight changes of the powders between 200 and 650 °C, the results indicated that the PDA content in the powder was increased from 0.53% (2PT) to 0.74% (4PT) and to 0.91% (8PT). This result, combined with the TEM observation, indicated that the amount of PDA formation indeed was determined by the initial concentration of DA as suggested by Ball et al., but there must be nucleation and growth sites other than the surface β-TCP for the formation of PDA.21 A previous study had demonstrated that DA and its oxidized derivatives unbound to the substrate could serve as nucleation and growth sites to facilitate the formation of nano-sized PDA aggregate dispersions in the solution.23 Della Vecchia et al. suggested that the formation kinetic of PDA aggregates could be encouraged while the oxidation and polymerization process was performed in the presence of phosphate ions due to the coordination between the protonated amino groups of DA and the dianion form of phosphate ions.24 Since phosphate ions could be dissipate from β-TCP powders during the PDA deposition process, it is postulated that the dissolved ionic products in a solution with a high content of DA may endorse the formation of PDA aggregates instead of the deposited PDA layer, resulting in the harvest of the PDA aggregate-rich composite powder.
 | ||
| Fig. 2 (A) 13C ssNMR spectra and (B) TGA curve of PDA-β-TCP synthesized at different concentrations of the DA precursor. | ||
3.2. Characterization of PDA-β-TCP/PCL composites
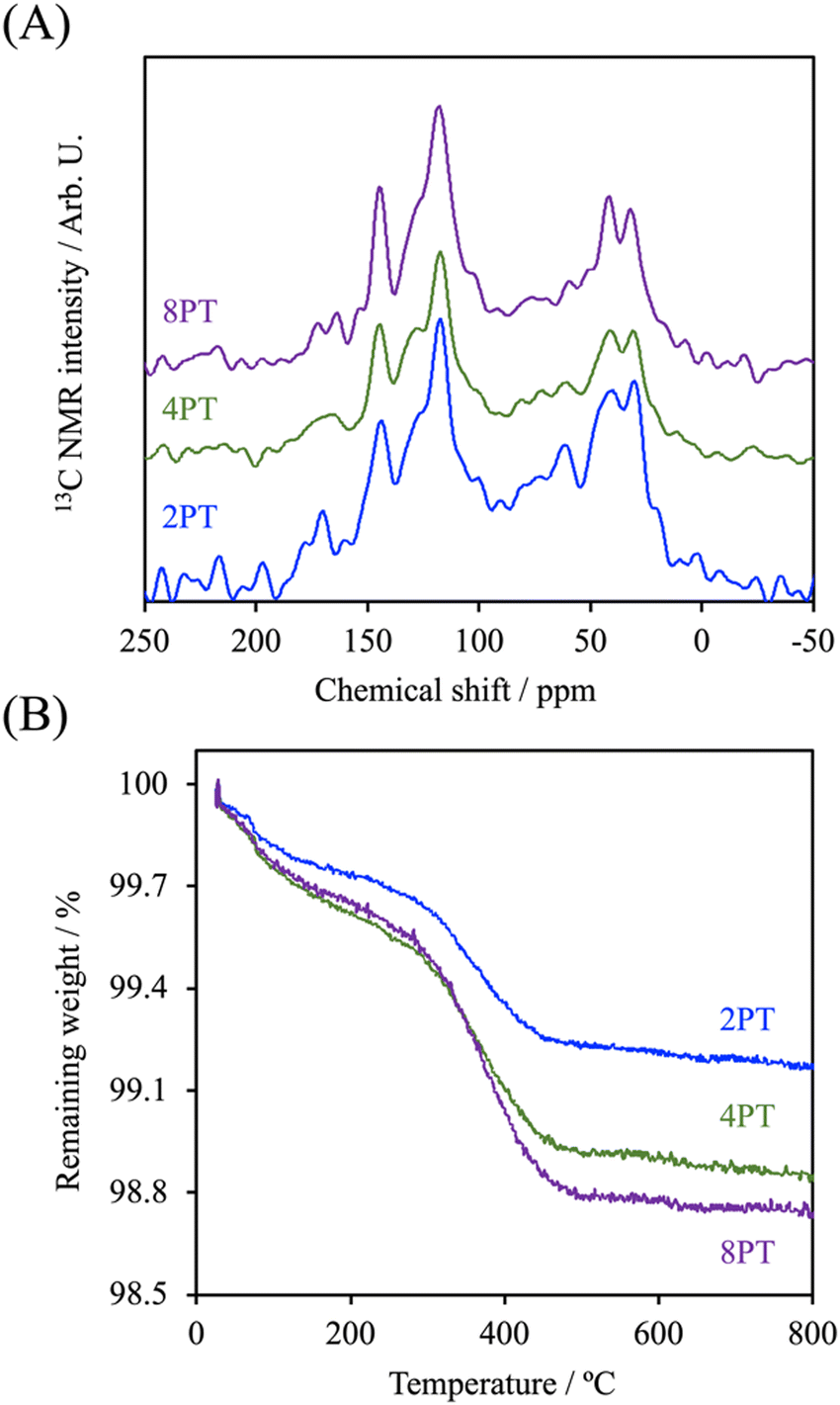
It is well documented that the rheological properties were the determinants that would remarkably influence the extrudability and printability of ceramic/polymer composite melts. To elucidate the effects of different fillers on the rheological properties of the composite melt, the evolution of complex viscosities of different groups as a function of angular shear rate at different temperatures was determined. As it is shown in Fig. 3, the complex viscosity of every group was increased with increasing processing temperature. The C group showed the lowest complex viscosity among all groups in an angular frequency sweep range between 0.5 and 500 rad s−1 at every temperature point of interest. Additionally, it possessed Newtonian plateau on approximately 620, 210, and 95 Pa s at 60, 90, and 120 °C in the experimental angular frequency range, respectively, except for that a shear-thinning region initiated at the angular frequency above 99.8 rad s−1 at 60 °C could be observed. In contrast, the composite melts at 60 °C revealed similar trends with significantly higher Newtonian plateau values increasing from 1700 to 2000 Pa s along with an order of the DA precursor concentration, which could be attributed to the existence of interactions between fillers and PCL molecules.25 At higher processing temperatures, it is obvious that the Newtonian plateau for complex viscosity vanished progressively in the presence of PDA, indicating a transition from an elastoviscous melt to a pseudoplastic one by the restrained relaxation of the PCL molecule. At 120 °C, 2PTC, 4PTC, and 8PTC groups revealed a clear shear-thinning behavior in particular in the low-frequency region, resulting in 1.6-, 2.7-, and 2.3-fold increament in zero-shear viscosities in contrast to the value of the TC group, respectively. This implies that the presence of PDA could be responsible for enhancing the affinity between fillers and matrix. Yang et al. indicated that the catechol moiety of PDA coated on reinforcing fillers could effectively increase dispersity of the fillers and interfacial strengths in the components by the formation of hydrogen bonds.25 It is noticed that 8PTC displayed a lower slope of complex viscosity as a function of angular frequency than that of 4PTC. It could be postulated that the coverage level and thickness of PDA on the fillers might be another prominent attribute that might alter the rheological behavior of the melts when associating this finding to the TEM micrographs.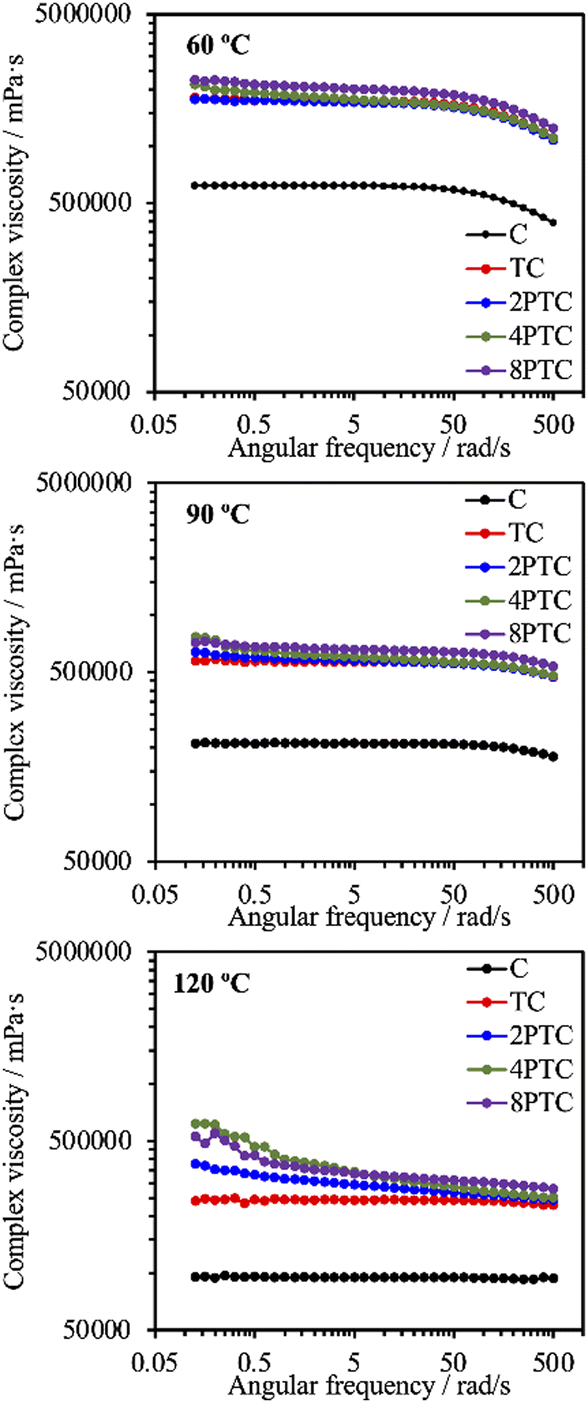 | ||
| Fig. 3 Complex viscosity evolutions of PCL and different composites as a function of angular frequency at 60, 90, and 120 °C. | ||
Despite the reduced flowability of PDA-β-TCP/PCL composites by the enhancement of interfacial strength of the components, the effect of the fillers on the rheological property was relatively weak. Proper printing windows could be addressed by tuning the nozzle temperature, extruding pneumatic pressure, and nozzle moving rate. Furthermore, molten composites possessed better shear-thinning performances with higher zero-shear viscosities are applicable for alleviating thermal instability concerns under appropriate dissipation conditions and consequently achieve immediate solidification to endorse fidelity of the printed layer.26 As seen in Fig. 4, different scaffolds sharing similar diameters of struts and pores were successfully obtained. It could be observed from SEM micrographs that some small pits with a size of several micrometers, which was a consequence of the cold contraction of the polymer matrix, could be observed on the surface of all scaffolds. For β-TCP/PCL and PDA-β-TCP/PCL groups, the fillers appeared near the outer surface of the scaffolds were covered with PCL due to the combination of extrusion procedures and the rheological characteristics of molten PCL. To better assess the distribution of the fillers in the composites, the scaffolds were broken in a liquid nitrogen bath and the cross-sections were visualized by SEM. A dense and homogeneous morphology was observed in PCL; whereas partially exposed β-TCP particles could be found in the PCL matrix for other groups. In contrast to the TC group, numerous small particles can be found in the matrix, which could be related to the existence of PDA/ionic product aggregates. Moreover, the dense structure of the matrix was retained while TC or 2PTC fillers were incorporated in PCL, but several porous regions were found in 4PTC and 8PTC, probably due to the presence of PDA aggregates in the composite, which could not be homogeneously disperse in the PCL matrix.
 | ||
| Fig. 4 Digital photographs and SEM micrographs of PCL and different β-TCP/PCL, composite scaffolds. | ||
Fig. 5A demonstrated the TGA results of different composites. It could be clearly observed that thermal decomposition of the pure PCL was initiated at about 190 °C, followed by a rapid decomposition kinetic starting at 300 °C until the complete decomposition of PCL was achieved at temperatures up to 500 °C.27 Similar thermal decomposition curves were obtained for all composite groups regardless of the precursor concentration. However, residual weights of 49.2%, 49.3%, 49.4%, and 49.0% for TC, 2PTC, 4PTC, and 8PTC, respectively, at 600 °C were obtained, which closely matched the designed filler contents in the present research (50%). To assess the interactions between the fillers and the PCL matrix, DSC analysis was employed to verify the effect of different fillers on the formation of PCL crystalline structures. The results of DSC demonstrated that the incorporation of TCP of PDA-TCP fillers did not remarkably alter the melting point of the PCL composites (Fig. 5B). The endothermic melting peaks of neat PCL and composites remained at 54.4 °C but increased to 56.1 °C while 8PTC was measured. However, it could be observed that the melting peaks gradually broadened with an increase in the initial DA concentration, which could have resulted from a wider range of crystallite sizes.28 The relative crystallinity (Xc) calculated from the DSC curves indicated that the Xc of PCL was dramatically increased from 47.8% to 60.8% while 50 wt% of TCP was blended in the matrix, indicating that TCP favoured the crystallization of PCL. However, a diminished crystallinity was obtained in the 2PTC group. This could have resulted from the elevated interaction between PCL molecules and the PDA deposited on the surface of the filler and consequently restrained the nucleation effect of TCP. Of note, the Xc was further increased to 47.6% and 50.0% for 4PTC and 8PTC, respectively. This might have originated from the presence of PDA aggregates in the PCL matrix that could act as heterogeneous fillers to encourage the nucleation effect.29
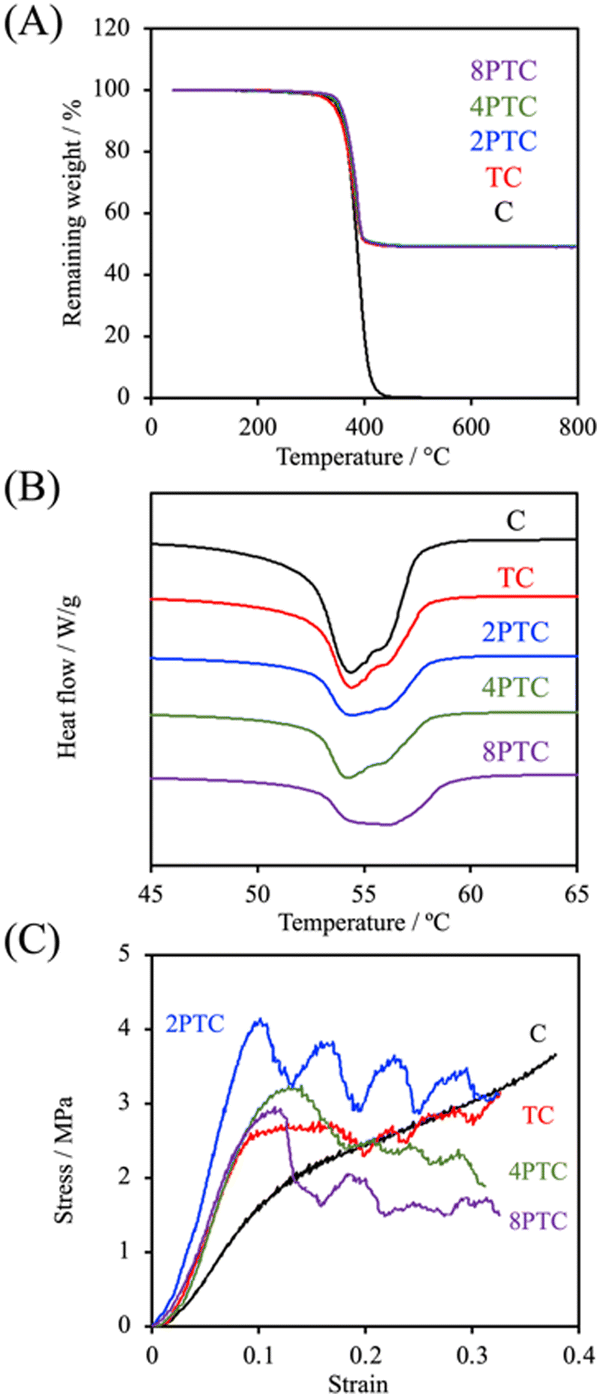 | ||
| Fig. 5 (A) TGA, (B) DSC, and (C) compressive stress–strain curves of different scaffolds. | ||
Furthermore, the results of compressive strength and modulus of the scaffolds fabricated by using an extrusion-based robotic dispensing system (as shown in Fig. 5C) indicated that compressive modulus of PCL was significantly improved while the fillers with or without PDA deposition were incorporated in the polymer matrix (p < 0.05). The compressive moduli of C, TC, 2PTC, 4PTC, and 8PTC were 20.4, 36.6, 59.7, 44.0, and 43.8 MPa, respectively; whereas the compressive strengths of TC, 2PTC, 4PTC, and 8PTC were 2.54, 4.16, 3.11, and 3.14 Mpa, respectively. The decreased strength of 4PTC and 8PTC in comparison to that of 2PTC could be attributed to the presence of loose structures in composites as observed from the SEM micrographs (Fig. 4). Nevertheless, it is worth noting that approximately 1.23 to 1.64-fold enhancement in compressive modulus and strength of TC scaffolds were achieved owing to the presence of PDA in the PCL matrix. Additionally, significant differences were observed while comparing the modulus and compressive strength of TC with each PDA-β-TCP/PCL group (p < 0.05), except for the strength between TC and 8PTC (p > 0.05). Although the mechanism of the PDA-reinforced β-TCP/PCL scaffolds was not clear, it is documented that the reinforcing ability of the PDA-deposited fillers would result from the enhancement of filler dispersion and formation of hydrogen bonds between fillers and the matrix.30 The compressive strengths of all scaffolds were in the range of the strength of cancellous bone (2–12 Mpa), but only the compressive moduli of PDA-β-TCP/PCL reached the lower-end of the cancellous bone (50–500 Mpa).31 Existing research suggested several opportunities that could be considered useful for optimizing the mechanical strength of the composite scaffolds in the future works. For instance, Soufivand et al. demonstrated that the 3D printed porous scaffolds with a hexagonal architecture exhibited significantly higher compressive strength than the rectangular-patterned scaffolds with similar porosity.32
3.3. In vitro immersion experiment
Despite the relatively lower melting point and higher toughness of PCL than other polyesters (e.g., PLA and PLGA) endorse it as a prominent printable biomaterial used for application of bone tissue engineering, the slow in vitro and in vivo degradation rate of PCL could result in an impaired efficiency of bone regeneration. This obstacle can be overcome by incorporating hydrophilic fillers into PCL to diminish the hydrophobicity of the matrix.33 To assess the effect of the fillers on the degradation rate of the PCL-based scaffolds, the evolution of weight change of different scaffolds after soaking in SBF over 4 weeks was recorded. As shown in Fig. 6A, all groups revealed slow degradation kinetics, and less than 2% of the PCL scaffolds were degraded over an immersion period of 2 weeks. However, a significantly faster degradation rate in the immersion period between 2 to 4 weeks could be observed in 4PTC and 8PTC while comparing with other groups. The remaining weights of C, TC, 2PTC, 4PTC, and 8PTC at an immersion period of 4 weeks were 98.5%, 98.4%, 98.6%, 95.5%, and 93.7%, respectively. The significantly lower remaining weights (p < 0.05) of 4PTC and 8PTC could be associated with the relatively higher contents of hydrophilic PDA aggregates buried in the composites, which could be exposed and contacted to water upon the occurrence of surface erosion on the scaffolds and subsequently altered the surface chemistry to promote the hydrolysis process.34 Additionally, the compressive strength results shown in Fig. 6B demonstrated that TC and 2PTC displayed the lowest and highest compressive strength values among all composite groups at each immersion period, respectively (p < 0.05). Although there were no significant differences between the strengths of each group before and after being soaking in SBF for 4 weeks (p > 0.05), a declining trend in the strength could be observed in both 4PTC and 8PTC during 2–4 weeks of immersion. This result was in agreement with the degradation result, implying that the presence of PDA aggregates and the porous matrix in 4TPC and 8TPC would be responsible for accelerating the degradation of the scaffolds.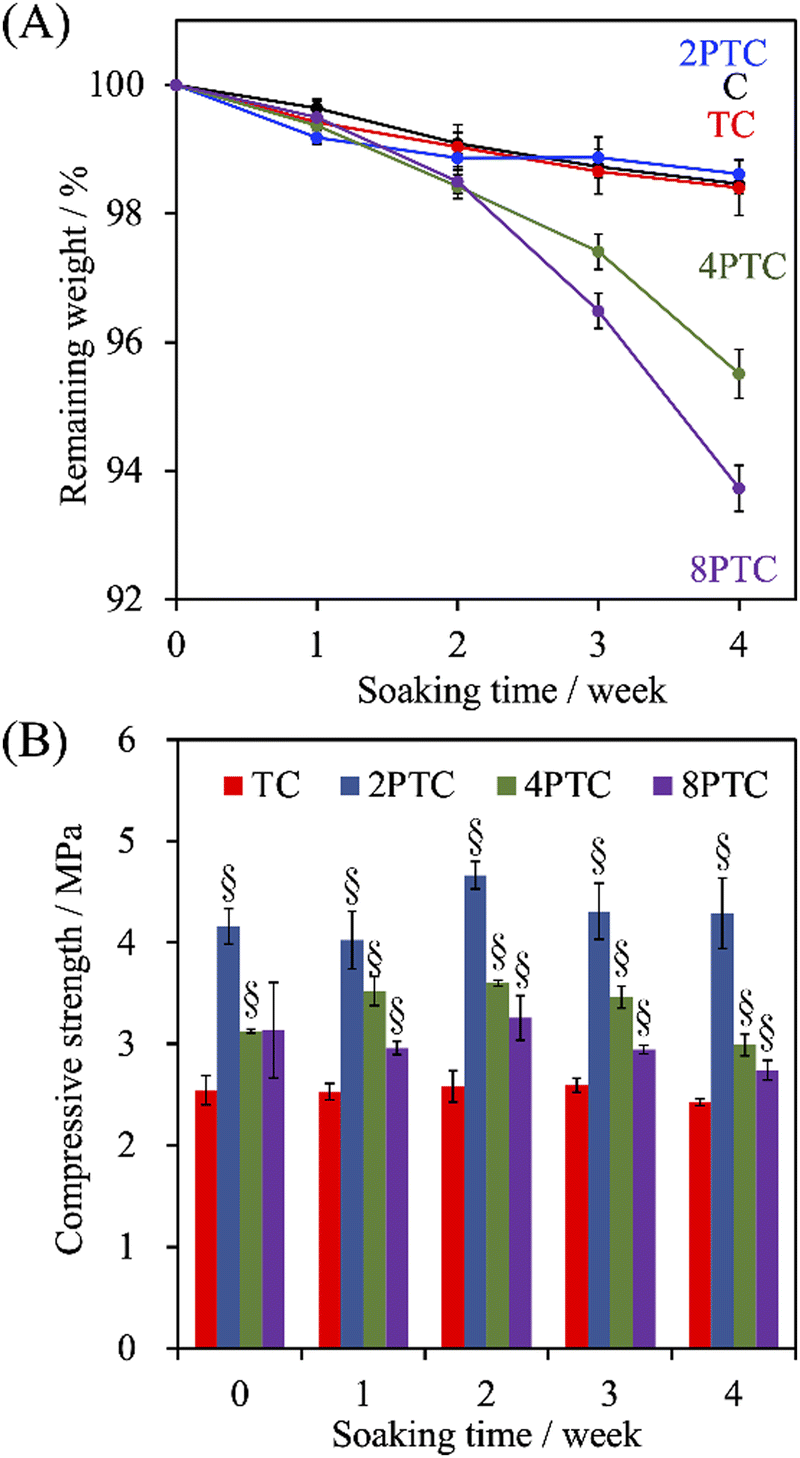 | ||
| Fig. 6 The evolution of (A) the weight loss and (B) compressive strength of PCL, β-TCP/PCL, and PDA-β-TCP/PCL scaffolds after being immersed in SBF for different periods. | ||
3.4. In vitro experiments
As evidenced by the previous reports, the deposition of the PDA layer on hydrophobic and inert surfaces indeed can facilitate cell adhesion and proliferation due to the suitable surface energy of the PDA layer.20 However, the presence of the PDA layer can barely contribute to the differentiation and mineralization of osteogenic cells if osteoconductive matters were lacking.35 In the present study, PDA was modified on an osteoconductive bioceramic, β-TCP. Differing from the PDA modification on inert polymers, the eluted ionic products of the ceramic powder can directly interact with DA and is oxidized through electrostatic interactions to form Ca/P-PDA layers or aggregate dispersions.19 To better investigate the effect of the PDA in the scaffolds on the fate of osteogenic cells, the adhesion, proliferation, osteogenic differentiation, and mineralization of hWJMSCs were conducted. As seen in Fig. 7A, the cells with a rounded shape with filopodia can be observed while culturing on the C group for 1 day, whereas cells cultured on the composite scaffolds revealed a larger spreading area with elongated stress fibers. Additionally, higher levels of the cell spreading area on PTC groups than that on TC one was observed, implying that the PDA-β-TCP/PCL composites could provide suitable surfaces for supporting the adhesion and cytoskeleton development of hWJMSCs. The result of quantitative measurement illustrated that the amounts of cells cultured on the scaffolds were gradually increased with increasing culture periods (Fig. 7B). Significantly higher amounts of cells cultured on the composite scaffolds could be measured while comparing with that on C group at all culture periods (p < 0.05). However, the amounts of cells cultured on TC, 2PTC, and 4PTC shared a similar level at each cultured period (p > 0.05). Of note, cells cultured on 8PTC scaffolds displayed the highest absorbance values than the polymer and composite scaffolds at each time point (p < 0.05). For example, the absorbances of cells cultured on 8PTC were 1.59- and 1.26-fold greater than those on C and TC scaffolds at the culture period of 1 day, and were 1.33- and 1.18-fold greater than those on C and TC at day 7.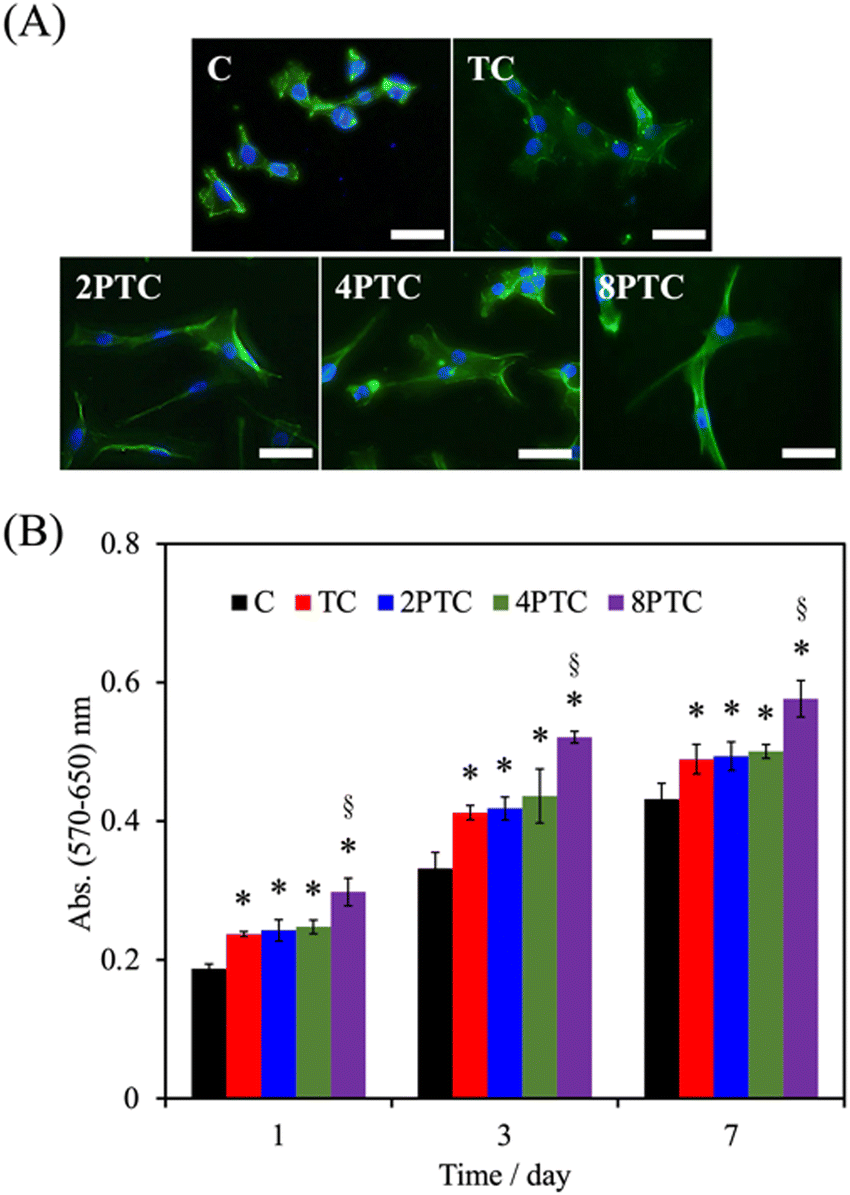 | ||
| Fig. 7 (A) Fluorescent cytoskeleton staining of hWJMSCs cultured on different scaffolds for 1 day. (B) The MTT assay of hWJMSCs cultured on different scaffolds for 1, 3, and 7 days. * and § indicate significant differences (p < 0.05) compared to C and TC, respectively. | ||
To further assess the presence of PDA in the composite scaffolds on the osteogenic differentiation of hWJMSCs, the ALP activity and mineralization of cells cultured on each scaffold were analyzed. ALP is a well-documented biomarker highly expressed during the early phase of bone formation.36 As shown in Fig. 8A, it could be observed that the ALP activities of cells in all groups were significantly upregulated during the cultured period from 7 days to 14 days (p < 0.05), except for that on C scaffolds (p > 0.05). Additionally, the ALP activity of cells cultured on the composite scaffolds was significantly greater than that on the C group (p < 0.05) during culture periods of 7 and 14 days. However, only 8PTC exhibited statistically higher ALP activities than TC scaffolds among all PDA-β-TCP/PCL scaffolds (p < 0.05), regardless of the culture periods. The quantitative results of alizarin red-S staining (Fig. 8B) demonstrated that the calcium nodules could barely be measured in cells cultured on the C group during culture periods of 7 and 14 days. In contrast, significantly higher levels of mineralization than the value derived from the C group were obtained on all composite scaffolds (p < 0.05). Although the values between the composite scaffolds were similar (p > 0.05) during the culture period of 7 days, significant differences appeared when the values of TC were compared with 4PTC and 8PTC (p < 0.05), implying better in vitro cell mineralization capacity of the PDA-β-TCP/PCL scaffolds.
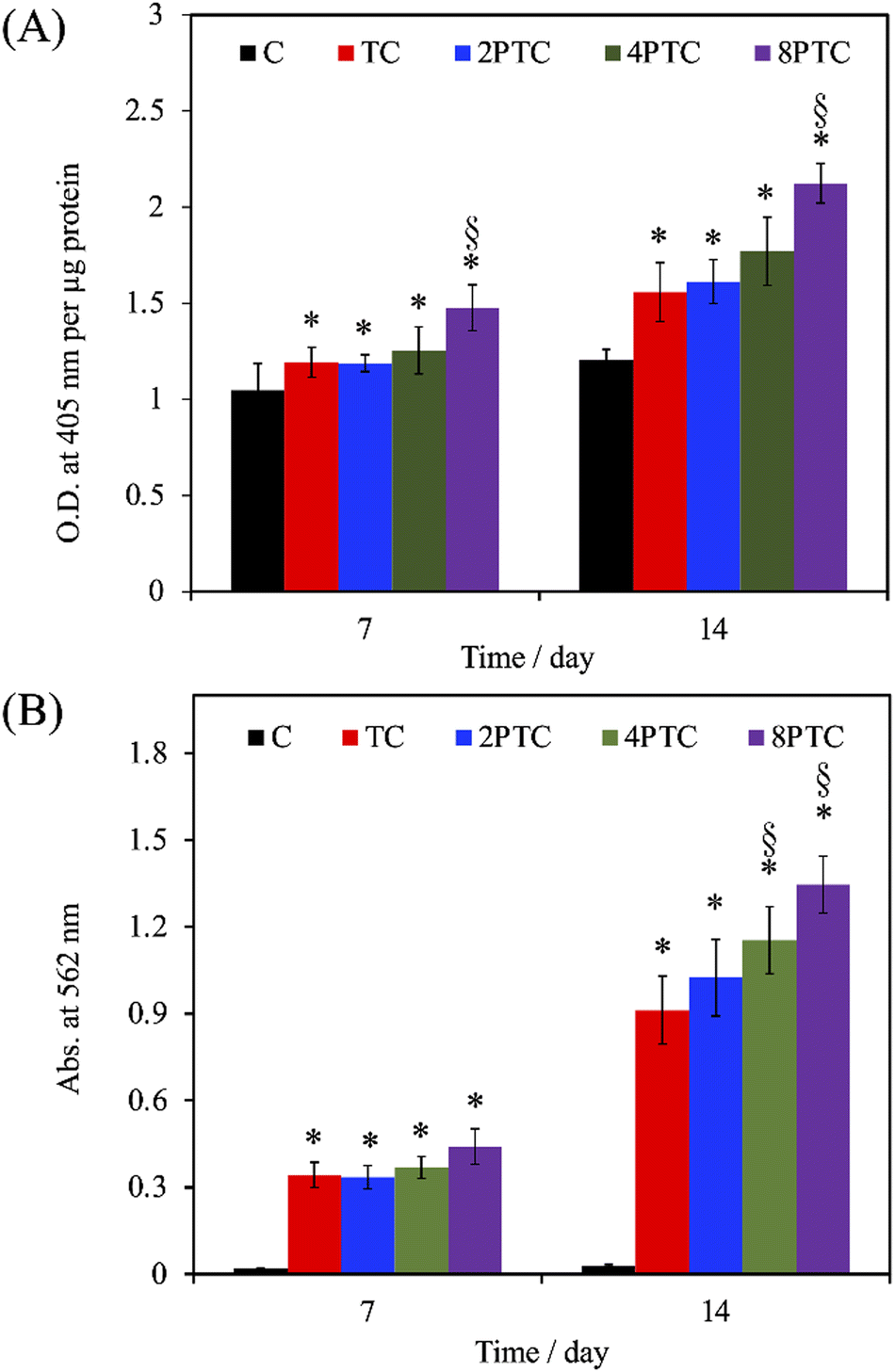 | ||
| Fig. 8 (A) ALP activity and (B) calcium deposition of hWJMSCs cultured on different scaffolds for 7 and 14 days. * and § indicate significant differences (p < 0.05) compared to C and TC, respectively. | ||
3.5. In vivo bone regeneration
The in vivo capabilities of our PDA-β-TCP/PCL were evaluated by implantation into the femoral condyle of rabbits. As seen from the CT images in Fig. 9A, after 4 weeks of implantations, there were increased bone tissue invasion into the center of the scaffolds for both 8PTC and 2PTC as compared to TC. In addition, the struts of 8PTC were discontinued and fractured due to the invasion of bone tissues and degradation of the scaffolds. It is important to note that an ideal scaffold should have equivalent mechanical strength and degradation rate so as to support bone regeneration.37 For TC wise, it could be seen that after 4 weeks of implantations, there was only bone growth at the top right corner of the scaffolds with no bone growth or tissue invasion at the rest of the scaffolds. Even after 8 weeks of implantations, there were still areas within the scaffolds that were not covered by new bone growth and invasion.38 Bone volume ratio (BV/TV) and trabecular thickness (Tb.Th) were further evaluated and shown in Fig. 9B and C respectively. As seen from the results, 8PTC was noted to have a significantly higher BV/TV ratio and Tb.Th as compared to both 2PTC and TC from 4 weeks of implantation. 2PTC was also noted to have a significantly higher BV/TV ratio and Tb.Th as compared to TC after 4 weeks of implantations. At 8 weeks of implantations, 8PTC was noted to have 40% BV/TV ratio and 0.36 mm Tb.Th as compared to 24% and 0.24 mm for 2PTC and 19% and 0.17 mm for TC.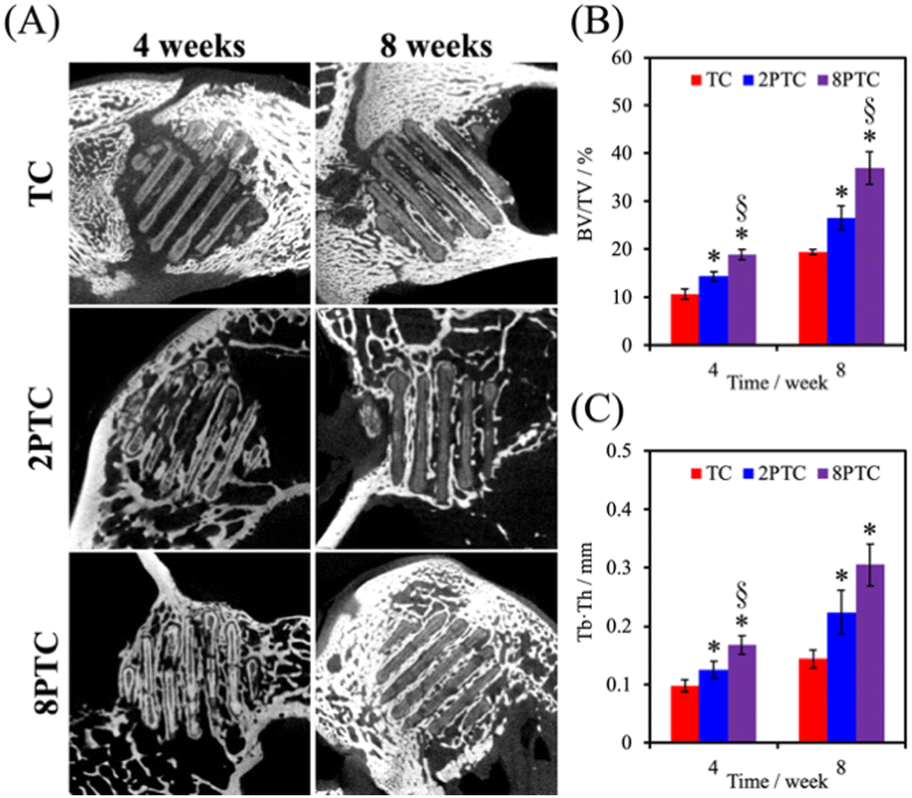 | ||
| Fig. 9 (A) μ-CT images of bone defects implanted with different scaffolds for 4 and 8 weeks. (B) Bone volume fraction (BV/TV) and (C) trabecular thickness (Tb.th) in different scaffolds. * and § indicate significant differences (p < 0.05) compared to TC and 2PTC, respectively. | ||
The harvested scaffolds were further stained with HE, MT, and VK stains to confirm bone regeneration using histological methods. The histological results (Fig. 10) correspond positively to the CT results above. Firstly, the photomicrographs of the VK stained sections of the 8PTC scaffold implanted bone show a larger area of newly generated bone than in 2PTC and TC after implantation for 4 and 8 weeks. In addition, the MT stained bone section of the 8PTC scaffolds indicates the formation of the maximum amount of new bone with observable bone cells compared to the 2PTC and TC groups.39 Secondly, it could be observed that the edges of the struts in 8PTC were irregular, rough, and discontinuous, thus further confirming the invasion of bone tissues into the center of the scaffolds.40 On the other hand, after 8 weeks of implantations, the majority of the microscopic visual field was covered with images of the struts with minimal staining surrounding the perimeter of the struts. Furthermore, the integrity of the struts was still present with smooth edges in the TC group.41 Finally, it could be seen that 8PTC had more intense VK stains as compared to the other two groups, thus strongly indicating that 8PTC was not only able to induce bone regeneration, it also enhances bone mineralization to form mature bones.42 Together with the above results, it could be confirmed that the presence of PDA enhanced in vivo bone regenerative capabilities of β-TCP/PCL scaffolds in a dose-dependent manner that confirmed that 8PTC could favor bone tissue growth from the periphery to the interior of scaffolds.43 Furthermore, the newly regenerate bone tissues could penetrate the interconnected porous structure of scaffolds as early as 4 weeks, which is important for improving the early osteointegration stage of the bone graft.44
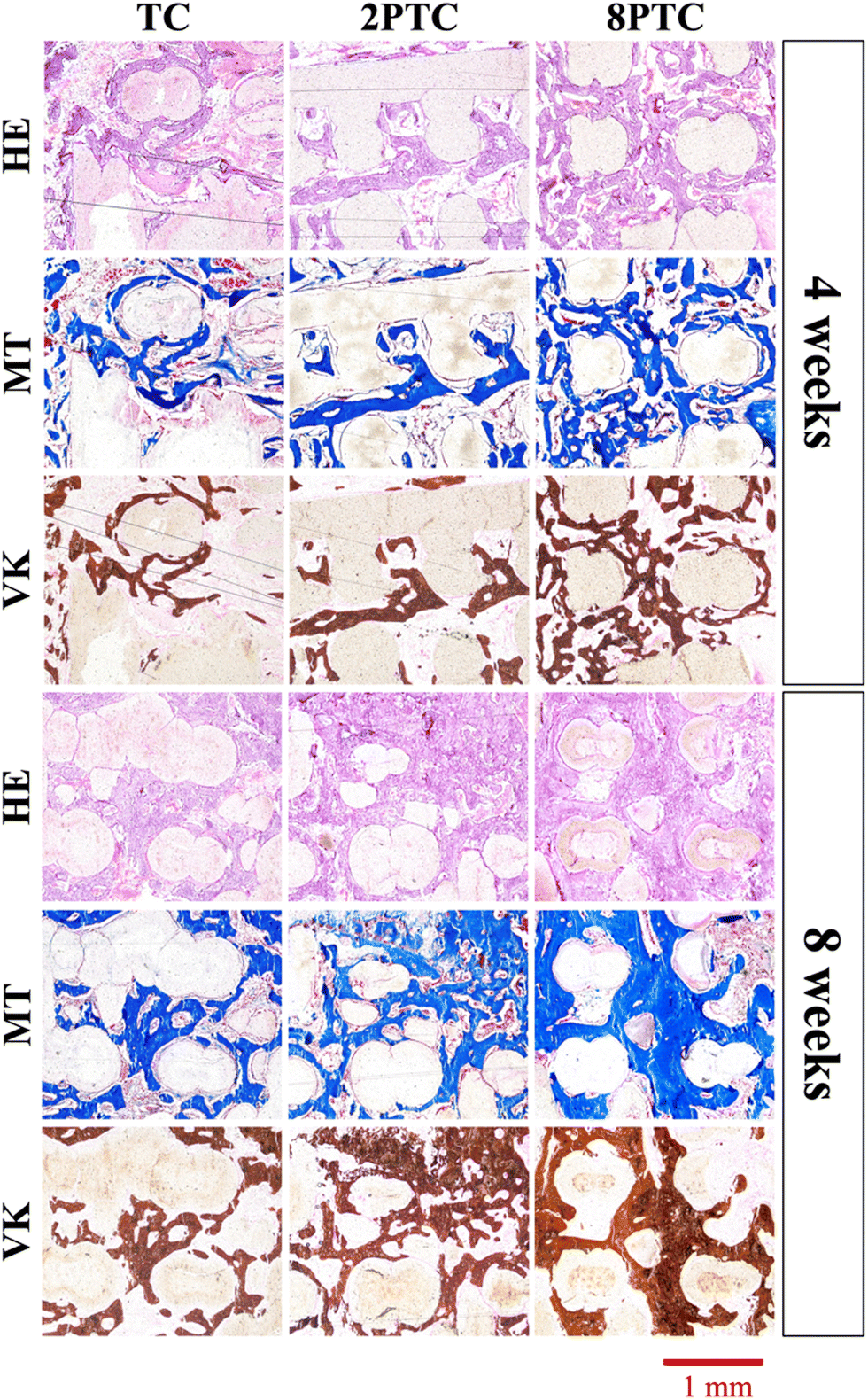 | ||
| Fig. 10 HE, MT, and VK staining of the distal femoral condyle defects after implantation with different composite scaffolds for 4 and 8 weeks. | ||
Although our in vivo results had confirmed that the incorporation of PDA-modified TCP exhibited a better capacity in accelerating the formation of neo-bone tissues in the rabbit femur defect model, detailed quantitative and qualitative analyses for investigating the expression levels of osteogenic differentiation markers, including Runx2, osteopontin, and osteocalcin (OC), to better validate the osteogenic effect and of the PDA-modified composite scaffolds, are performed. Although additional analysis should be conducted to assess the underlying mechanism of the elevated bone formation ability assisted by the proposed composite scaffolds, several pieces of existing evidence from the literature might be reasonable for elucidating the role of PDA. Wu et al. demonstrated that the eluted calcium and phosphate ions could associate with the as-formed PDA layer to alter the composition of the deposited layer, resulting in the upregulation of the expression of osteogenic differentiation genes.19 Kao et al. suggested that the release rate of Ca2+ from calcium silicate powders was decreased while decorated with PDA, and the PDA content displayed positive correlations with the adsorption of extracellular matrices (collagen I and fibronectin) that could further stimulate the expression of collagen I, Runx2, and OC.45 Zhou et al. demonstrated that the expression of α10, α11, and β1 integrin could be upregulated by the Ca2+-chelated PDA-PLGA/PCL composite scaffold, consequently encouraging the stem cell recruitment in a subcutaneous implantation model.46 Nevertheless, the ALP activity and cell mineralization results from in vitro experiments, as well as neo-bone formation results from the animal model reported here provide an initial insight into the versatile benefits of PDA on enhancing both physicochemical and biological properties of the 3D-printed β-TCP/PCL scaffolds.
4. Conclusion
In summary, we reported a facile route to reinforce the biodegradable PCL/β-TCP composite scaffolds fabricated by extrusion-based 3D printing through deposition of a PDA interfacial layer between organic and inorganic phases in the composite. A dense and continuous PDA layer could be formed on the surface of β-TCP and it could remarkably enhance the compressive strength and modulus without interfering with the degradability, cytocompatibility, and osteogenicity of the composite scaffold. The thickness and density of the layer would decrease with increasing concentration of the DA precursor and favoured the formation of nano-sized particles and aggregates, resulting in a diminished but comparable reinforcing efficiency of the composite scaffolds than pure PCL. However, this alternation in the structure of the PDA layer could lead to accelerated degradation which could be beneficial for the proliferation and production of early osteogenic differentiation indicator, ALP, of hWJMSCs. The histological findings revealed that there were more new bone tissues formed as well as some vessel tube formation of PCL/β-TCP occurred. Therefore, our results demonstrate that the facile PDA-reinforced β-TCP/PCL scaffolds could be considered as a promising bone graft for bone tissue engineering applications. We believe that 3D printing of porous scaffolds can be used to promote the application potential of such materials in bone tissue engineering applications.Author contributions
C. C. H., Y. W. C. and K. W. methodology, validation, investigation, formal analysis, writing – original draft, preparation, and writing – review & editing; Y. H. L. and T. C. C. helped with data analysis and interpretation; M. Y. S. conceptualization, funding acquisition, project administration, and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge receipt grants from the Ministry of Science and Technology (MOST 107-2314-B-039-002 and 107-2314-B-039-022) and the China Medical University (CMU111-ASIA-15). Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities, Office of Research & Development at China Medical University, Taichung, Taiwan.Notes and references
- Y. Lai, Y. Li, H. Cao, J. Long, X. Wang, L. Li, C. Li, Q. Jia, B. Teng, T. Tang, J. Peng, D. Eglin, M. Alini, D. W. Grijpma, G. Richards and L. Qin, Biomaterials, 2019, 197, 207–219 CrossRef CAS PubMed.
- C. T. Kao, C. C. Lin, Y. W. Chen, C. H. Yeh, H. Y. Fang and M. Y. Shie, Mater. Sci. Eng., C, 2015, 56, 165–173 CrossRef CAS PubMed.
- A. Abdal-hay, N. T. Raveendran, B. Fournier and S. Ivanovski, Composites, Part B, 2020, 197, 108158 CrossRef CAS.
- C. C. Ho, H. Y. Fang, B. Wang, T. H. Huang and M. Y. Shie, Int. Endod. J., 2018, 51, e291–e300 CrossRef PubMed.
- K. Liu, J. Wang, S. Fang, H. Wang, Y. Bai, Z. Zhao, Q. Zhu, C. Wang, G. Chen, H. Jiang, J. Sun and P. Zhang, Mater. Des., 2022, 220, 110856 CrossRef CAS.
- D. Liu, W. Nie, D. Li, W. Wang, L. Zheng, J. Zhang, J. Zhang, C. Peng, X. Mo and C. He, Chem. Eng. J., 2019, 362, 269–279 CrossRef CAS.
- Y. H. Lin, T. Y. Chuang, W. H. Chiang, I. W. P. Chen, K. Wang, M. Y. Shie and Y. W. Chen, Mater. Sci. Eng., C, 2019, 104, 109887 CrossRef CAS.
- P. S. P. Poh, D. W. Hutmacher, B. M. Holzapfel, A. K. Solanki, M. M. Stevens and M. A. Woodruff, Acta Biomater., 2016, 30, 319–333 CrossRef CAS.
- K. Y. Chen and C. H. Yao, Biomedicine, 2011, 1, 29–32 CrossRef CAS.
- J. J. Lee, H. Y. Ng, Y. H. Lin, E. W. Liu, T. J. Lin, H. T. Chiu, X. R. Ho, H. A. Yang and M. Y. Shie, Biomater. Adv., 2022, 142, 213132 CrossRef CAS PubMed.
- X. Wang, G. Song and T. Lou, Med. Eng. Phys., 2010, 32, 391–397 CrossRef.
- Y. Gu, L. Wei, Z. Zhang, J. V. Dessel, R. B. Driesen, I. Lambrichts, R. Jacobs, L. Tian, Y. Sun, Y. Liu and C. Politis, Mater. Des., 2022, 215, 110443 CrossRef CAS.
- T. G. Barclay, H. M. Hegab, S. R. Clarke and M. Ginic-Markovic, Adv. Mater. Interfaces, 2017, 4, 1601192 CrossRef.
- Y. Zhu, D. Liu, X. Wang, Y. He, W. Luan, F. Qi and J. Ding, J. Mater. Chem. B, 2019, 7, 2019–2031 RSC.
- X. Wang, X. Peng, P. Yue, H. Qi, J. Liu, L. Li, C. Guo, H. Xie, X. Zhou and X. Yu, Mater. Sci. Eng., C, 2020, 109, 110544 CrossRef CAS.
- E. H. Backes, E. M. Fernandes, G. S. Diogo, C. F. Marques, T. H. Silva, L. C. Costa, F. R. Passador, R. L. Reis and L. A. Pessan, Biomater. Adv., 2021, 122, 111928 CrossRef CAS.
- F. Zhang, J. Yang, Y. Zuo, K. Li, Z. Mao, X. Jin, S. Zhang, H. Gao and Y. Cui, Mater. Des., 2022, 216, 110558 CrossRef CAS.
- C. G. Pitt, F. I. Chasalow, Y. M. Hibionada, D. M. Klimas and A. Schindler, J. Appl. Polym. Sci., 1981, 26, 3779–3787 CrossRef CAS.
- C. Wu, P. Han, X. Liu, M. Xu, T. Tian, J. Chang and Y. Xiao, Acta Biomater., 2014, 10, 428–438 CrossRef CAS PubMed.
- Y. W. Chen, Y. F. Shen, C. C. Ho, J. Yu, Y. H. A. Wu, K. Wang, C. T. Shih and M. Y. Shie, Mater. Sci. Eng., C, 2018, 91, 679–687 CrossRef CAS.
- V. Ball, D. D. Frari, V. Toniazzo and D. Ruch, J. Colloid Interface Sci., 2012, 386, 366–372 CrossRef CAS PubMed.
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS.
- N. F. D. Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. d’Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef.
- N. F. D. Vecchia, A. Luchini, A. Napolitano, G. D’Errico, G. Vitiello, N. Szekely, M. d’Ischia and L. Paduano, Langmuir, 2014, 30, 9811–9818 CrossRef.
- L. Yang, S. L. Phua, J. K. H. Teo, C. L. Toh, S. K. Lau, J. Ma and X. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3026–3032 CrossRef CAS.
- H. Suo, J. Zhang, M. Xu and L. Wang, Mater. Sci. Eng., C, 2021, 123, 111963 CrossRef CAS PubMed.
- Y. H. Lin, Y. C. Chiu, Y. F. Shen, Y. H. A. Wu and M. Y. Shie, J. Mater. Sci.: Mater. Med., 2017, 29, 11 CrossRef.
- D. Xiang, E. Harkin-Jones and D. Linton, RSC Adv., 2014, 4, 44130–44140 RSC.
- S. Kumar, M. R. Ramesh, M. Doddamani, S. M. Rangappa and S. Siengchin, Polym. Test., 2022, 114, 107703 CrossRef CAS.
- G. He, Z. Yang, L. Pan, J. Zhang, S. Liu and Q.-L. Yan, J. Mater. Chem. A, 2017, 5, 13499–13510 RSC.
- K. S. Jack, S. Velayudhan, P. Luckman, M. Trau, L. Grøndahl and J. Cooper-White, Acta Biomater., 2009, 5, 2657–2667 CrossRef CAS PubMed.
- A. A. Soufivand, N. Abolfathi, S. A. Hashemi and S. J. Lee, Addit. Manuf., 2020, 33, 101181 Search PubMed.
- Y. H. Lin, K. X. Lee, C. C. Ho, M. J. Fang, T. Y. Kuo and M. Y. Shie, Biomater. Adv., 2022, 135, 112660 CrossRef.
- C. X. F. Lam, D. W. Hutmacher, J. Schantz, M. A. Woodruff and S. H. Teoh, J. Biomed. Mater. Res., Part A, 2009, 90A, 906–919 CrossRef CAS.
- C. Y. Chien and W. B. Tsai, ACS Appl. Mater. Interfaces, 2013, 5, 6975–6983 CrossRef CAS.
- A. R. Bastos, F. R. Maia, J. M. Oliveira, R. L. Reis and V. M. Correlo, Mater. Sci. Eng., C, 2021, 129, 112413 CrossRef CAS PubMed.
- C. Hou, Y. Liu, W. Xu, X. Lu, L. Guo, Y. Liu, S. Tian, B. Liu, J. Zhang and C. Wen, Biomater. Adv., 2022, 139, 213018 CrossRef CAS.
- R. Batul, T. Tamanna, A. Khaliq and A. Yu, Biomater. Sci., 2017, 5, 1204–1229 RSC.
- S. K. Ghorai, A. Dutta, T. Roy, P. G. Ray, D. Ganguly, M. Ashokkumar, S. Dhara and S. Chattopadhyay, ACS Appl. Mater. Interfaces, 2022, 14, 28455–28475 CrossRef CAS.
- A. H. Aghajanian, A. Bigham, A. Sanati, A. Kefayat, M. R. Salamat, M. Sattary and M. Rafienia, Biomater. Adv., 2022, 137, 212809 CrossRef CAS PubMed.
- Z. Lyu, Y. Zhao, S. Huo, F. Wang, X. Meng, Z. Yuan, T. Long and Y. Wang, Mater. Des., 2022, 222, 111069 CrossRef CAS.
- Z. Liu, S. Qu, X. Zheng, X. Xiong, R. Fu, K. Tang, Z. Zhong and J. Weng, Mater. Sci. Eng., C, 2014, 44, 44–51 CrossRef CAS PubMed.
- Y. Sun, Y. Li, Y. Zhang, T. Wang, K. Lin and J. Liu, Biomater. Adv., 2021, 131, 112482 CrossRef CAS.
- S. Cheng, J. Ke, M. Yao, H. Shao, J. Zhou, M. Wang, X. Ji, G. Zhong, F. Peng, L. Ma and Y. Zhang, J. Mater. Sci. Technol., 2021, 69, 106–118 CrossRef CAS.
- C. T. Kao, Y. J. Chen, H. Y. Ng, K. X. Lee, T. H. Huang, T. F. Lin and T. T. Hsu, Materials, 2018, 11, 1664 CrossRef.
- X. Zhou, X. Cheng, D. Xing, Q. Ge, Y. Li, X. Luan, N. Gu and Y. Qian, Mater. Des., 2021, 198, 109300 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |