
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore–shell Fe3O4@CoFe2O4 nanoparticles as high-performance anode catalysts for enhanced oxygen evolution reaction†
Lisa
Royer
ab,
Iryna
Makarchuk
b,
Simon
Hettler
 cd,
Raul
Arenal
cde,
Tristan
Asset
a,
Benjamin
Rotonnelli
a,
Antoine
Bonnefont
f,
Elena
Savinova
a and
Benoit P.
Pichon
*bg
cd,
Raul
Arenal
cde,
Tristan
Asset
a,
Benjamin
Rotonnelli
a,
Antoine
Bonnefont
f,
Elena
Savinova
a and
Benoit P.
Pichon
*bg
aICPEES, UMR 7515 CNRS-ECPM-Université de Strasbourg, 25, rue Becquerel, F 67087, Strasbourg Cedex 2, France
bUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, F-67000, Strasbourg, France. E-mail: benoit.pichon@unistra.fr
cInstituto de Nanociencia y Materiales de Aragon (INMA), CSIC-Universidad de Zaragoza, Calle Pedro Cerbuna 12, 50009, Zaragoza, Spain
dLaboratorio de Microscopías Avanzadas (LMA), Universidad de Zaragoza, Calle Mariano Esquillor, 50018, Zaragoza, Spain
eARAID Foundation, 50018, Zaragoza, Spain
fInstitut de Chimie, UMR 7177, CNRS-Université de Strasbourg, 4 rue Blaise Pascal, CS 90032, 67081, Strasbourg Cedex, France
gInstitut Universitaire de France, 5 rue Descartes, 75015, Paris, France
First published on 14th June 2023
Abstract
Water electrolysis is a promising and environmentally friendly means for renewable energy storage. Recent progress in the development of anion exchange membranes (AEMs) has provided new perspectives for high-performance anode catalysts based on transition metal oxides (TMOs) for the sluggish anodic oxygen evolution reaction (OER). Here, we report on core–shell nanoparticles (Fe3O4@CoFe2O4) which allow combining an electrocatalytic shell (CoFe2O4) with a conductive core (Fe3O4). Such an original approach significantly minimizes critical Co content in the catalyst and avoids addition of unstable conductive carbon black. The core–shell nanoparticles outperform Co(1−x)Fe(2+x)O4 nanoparticles and show an exceptional OER activity per Co unit mass (2800 A gcobalt−1 at 1.65 V vs. RHE) suggesting synergistic interaction between the core and the shell. Along with the core–shell structure, the size of the Fe3O4 core is a critical parameter, with a large conductive Fe3O4 core being beneficial for OER enhancement.
Water electrolysis provides an environmentally friendly way to store renewable energies through H2 production. While proton exchange membrane water electrolysis is well suited to renewable energy storage, it relies on the utilization of scarce and expensive iridium (Ir) to accelerate the sluggish kinetics of the anodic oxygen evolution reaction (OER). Thanks to the recent progress in the development of anion-exchange membranes (AEMs), transition metal oxides (TMOs) appear as promising substitutes for noble metals.1–4 Various approaches have been proposed to enhance their OER activity, notably varying the composition, particle size, and defect concentration, or, recently, forming core–shell structures.5–9
Owing to their composition-dependent and widely tunable properties, TMOs with spinel structures appear very attractive for various energy-related applications. While Co3O4 and CoOOH thin films deposited on an Au(111) substrate have demonstrated significant surface-weighted OER activity,10 core–shell nanoparticles (NPs) combining a gold core and a TMO shell reached 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 A gTM−1 at 1.65 V vs. RHE. According to the authors, such a high OER activity of TMOs may result from an electronic effect of the gold core that favors the oxidation of the TM up to the 4+ state.11,12 The use of core–shell nanoparticles might be an efficient way to reduce the amount of expensive and rare electrocatalysts by circumscribing them to a thin shell,13,14 but the exclusive use of cost-effective and abundant materials is the next challenge for the worldwide dissemination of water electrolysis.
000 A gTM−1 at 1.65 V vs. RHE. According to the authors, such a high OER activity of TMOs may result from an electronic effect of the gold core that favors the oxidation of the TM up to the 4+ state.11,12 The use of core–shell nanoparticles might be an efficient way to reduce the amount of expensive and rare electrocatalysts by circumscribing them to a thin shell,13,14 but the exclusive use of cost-effective and abundant materials is the next challenge for the worldwide dissemination of water electrolysis.
TMO NPs are of significant interest since the catalytic activity can be greatly enhanced by adapting their size and shape, i.e. the active surface area. The valence of the metallic cations and their chemical environment are also critical to enhancing the OER activity.3,15,16 Beyond the fine control of these parameters, recent advances in nanoparticle synthesis in liquid media avoid aggregation, thus also favoring high surface area, i.e. enhanced mass-weighted catalytic activity. Consequently, the challenge lies in controlling the formation and the chemical structure of NPs in order to systematically study their electrochemical properties.
Although most TMOs are insulating, magnetite (Fe3O4) is conductive due to electron hopping between Fe(II) and Fe(III).17 Therefore, it may be considered a promising support material to avoid carbon black, which is often added to circumvent the lack of conductivity of TMO catalysts, but is unstable under anode operation conditions. Furthermore, iron cations in TMO phases may result in synergetic effects with the surrounding atoms, thus enhancing the OER.18,19 Hence, nanostructures combining Fe3O4 as a conductive core and a TMO as a catalyst shell would be promising high performance noble metal-free OER catalysts.
Here we report an original approach to design a new type of carbon-free, noble-metal-free nanostructured material for the OER. The core–shell structure of Fe3O4@CoFe2O4 NPs allows combining a conductive magnetite core and a catalytically active cobalt ferrite shell. Co being classified as a critical raw material by the European Union, such a design of NPs with a core–shell structure efficiently contributes to reducing the amount of Co used for the OER. Such a cost-effective material resulted in an excellent OER activity per unit mass of Co. The fine control of the core size and the shell thickness led us to investigate the influence of the core–shell structure on the OER activity which is markedly enhanced by a larger Fe3O4 core.
Core–shell NPs were synthesized by the thermal decomposition of metal complexes in high-temperature-boiling solvents (around 300 °C) as we recently reported20 (see the ESI† for details). Two different approaches were applied: (i) the seed-mediated growth of a CoFe2O4 shell onto the surface of pristine Fe3−δO4 nanoparticles which led to samples CS-1, CS-3, and CS-4. (ii) The diffusion of cobalt cations into vacancies at the surface of partially oxidized pristine Fe3−δO4 nanoparticles which led to sample CS-2.21 The core size was varied by adjusting the experimental conditions upon synthesis of Fe3–δO4 nanoparticles as we reported earlier.22,23 The shell thickness was modified by adjusting the amount of Co and Fe metal complexes with a Co/Fe molar ratio of 0.5. An increase in the amount of Co and Fe complexes by 2.5 from CS-3 to CS-4 resulted in a two-fold shell volume increase which agrees with the larger size of pristine Fe3–δO4 nanoparticles used for CS-4. The transmission electron microscopy (TEM) images showed faceted-spherical shapes and narrow size distributions both for pristine and for core–shell NPs (Fig. 1). Granulometry measurements showed stable colloidal suspensions of non-aggregated NPs thanks to the efficient coating of oleic acid which was added to the reaction medium (Fig. S3†).
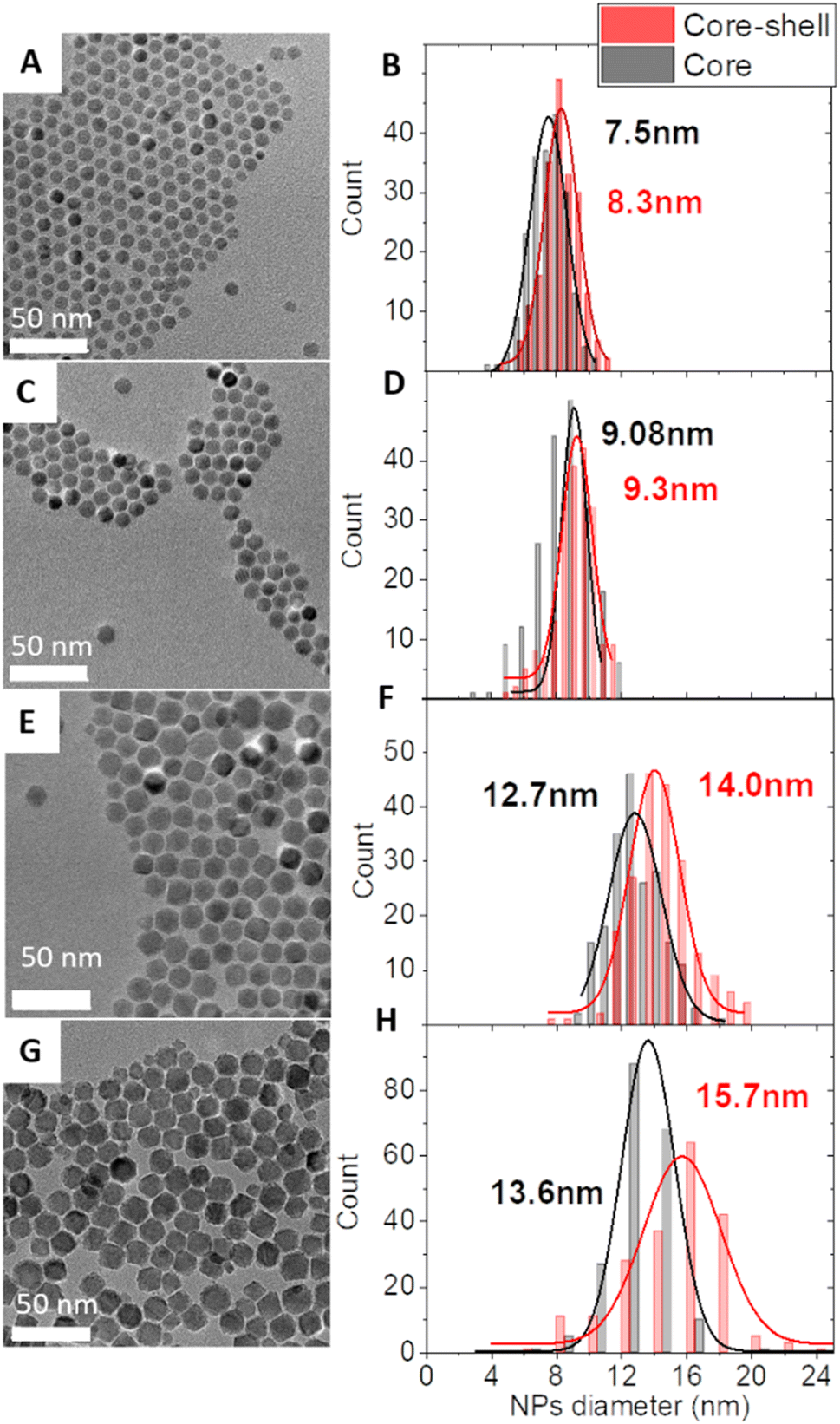 | ||
| Fig. 1 TEM micrographs of core–shell NPs: CS-1 (A), CS-2 (C), CS-3 (E), and CS-4 (G) and the corresponding size distributions (B, D, F and H) of the core–shell NPs and their pristine core NPs. | ||
Energy-dispersive X-ray (EDX) spectroscopy confirmed the presence of Co between 12 and 19 at% with respect to Fe in core–shell NPs (Table S1†). For CS-1, CS-2, and CS-3, these values are much higher than those corresponding to the size variation between pristine and core–shell NPs (Table S1†) which is indicative of the diffusion of cobalt in the Fe3−δO4 core as we have reported earlier.24 X-ray diffraction (XRD) patterns recorded for core–shell NPs display peaks that are all indexed to the spinel crystal structure (Fig. S4†). Although Fe3O4 and CoFe2O4 phases cannot be distinguished because of similar cell parameters (8.396 Å and 8.3919 Å, respectively),20 cell parameters of core–shell NPs are relatively close to those of Fe3O4 (Table S2†). This is ascribed to the low extent of the surface oxidation of pristine nanoparticles which are protected by the CoFe2O4 shell when exposed to air. The high crystallinity was confirmed by lattice fringes all-across core–shell NPs as observed in scanning transmission electron microscopy (STEM) micrographs recorded in the high angle annular dark field (HAADF) mode (Fig. S6†).
The local composition of these NPs was further investigated by electron energy-loss spectroscopy (EELS) analysis in STEM (Fig. 2 and S6†). Spatially resolved EELS maps show the homogeneous distribution of O in NPs. In contrast, the Co content is the highest at the edge while the Fe content is at its lowest. Cross-section profiles show that the Co fraction reaches a maximum value of 15–20% with respect to Fe and O on the edge while it quickly goes down to 5% (CS-2 and CS-4) and 2% (CS-3) in the center of the NP, in agreement with a much thinner shell than the core size. Considering stoichiometric CoFe2O4 (14% at. Co) and the resolution of the measurement (5 Å), Co is certainly distributed as a graded concentration (Co1+xFe2−xO4) which decreases from the NP surface to the center. This result is supported by the variation of the Co content over a longer distance from the surface to the center than the size variation usually ascribed to the shell thickness calculated from TEM micrographs. A slight increase in the O content can also be observed at the edges of nanoparticles which can be correlated to some cationic vacancies and surface defects as usually observed for such nanoparticles.
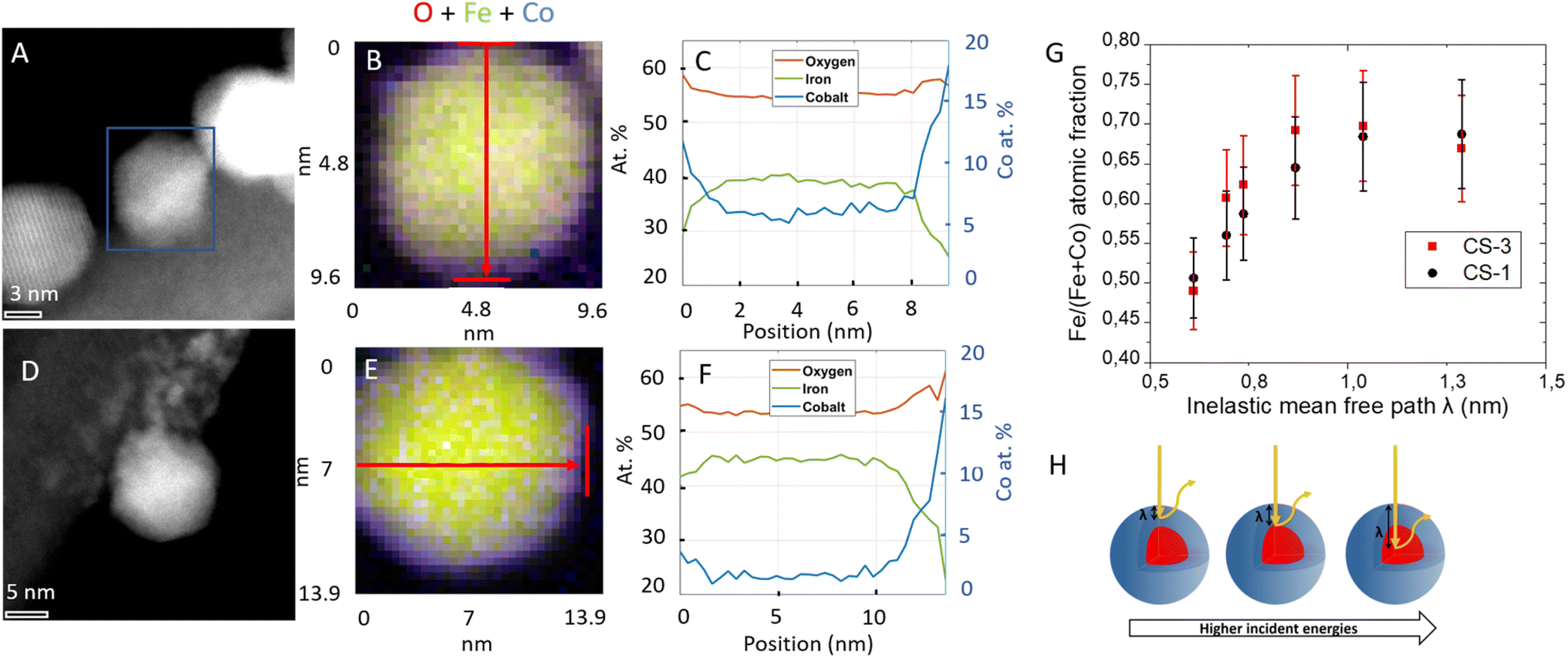 | ||
| Fig. 2 Scanning transmission electron microscopy (STEM) high angle annular dark field (HAADF) images of CS-2 (A–C) and CS-3 (D–F). Spatially resolved electron energy loss spectroscopy (EELS) analyses (B and E) showing the spatial distributions of oxygen O (red), iron Fe (green) and cobalt Co (blue). Quantitative cross-section profiles (C and F) recorded along the arrows depicted in (B and E) respectively. The left y-scale refers to percentages of Fe, Co and O atoms. (G) Fe atomic fraction (i.e. the ratio of Fe/(Co + Fe)) calculated from the depth profiling analysis performed for different incident energies on CS-1 and CS-3, the photon energy of the X-ray source was varied from 850 to 1350 eV in order to analyse Co 2p and Fe 2p peaks. Schematic illustration of the nanoparticle depth (H) probed as a function of the inelastic mean free path (λ). | ||
Complementary information on the average near-surface composition of NPs was obtained by performing X-ray photoelectron spectroscopy (XPS) measurements. The variation of the photon energy from 850 to 1350 eV allowed changing the inelastic mean-free path of the photoelectron emission (λ) from 0.6 to 1.3 nm thus allowing different NP depths to be probed. Considering that 68% of the emitted photoelectrons arise from a thickness λ and 99% emerge from 3λ,25,26 the Fe atomic fraction was calculated as the concentration of Fe normalized to the sum of Fe and Co from the area of Co 2p and Fe 2p peaks which were recorded for CS-1 and CS-3 (Fig. 2G). The Fe atomic fraction at small photon energies (hence short λ) is characteristic of the shell, but then gradually increases, since the contribution of the Fe3O4 core to the XP spectra increases with the photon energy. It is interesting to note that the Fe atomic fraction (Fe/(Fe + Co)) in the particle shell (∼0.5) is higher than stoichiometry (0.66 for CoFe2O4), which may be ascribed to the decomposition of Co stearate which happens at a higher temperature than that of Fe stearate.27 According to the La Mer theory, iron oxide starts growing before cobalt oxide at the surface of pristine NPs. This results in a gradient in the Co concentration in the shell. Nevertheless, both decomposition temperatures (of the iron and cobalt stearate) are sufficiently close to avoid phase segregation since no CoO phase was observed in the XRD patterns24 (Fig. S4†).
The electrochemical properties of core–shell NPs were studied in order to determine their OER activity and compare it to the activity of Co ferrite (Co(1−x)Fe(2+x)O4) NPs (for characterization see the ESI†). Cyclic voltammograms (CVs) were recorded after deposition of a thin layer of NPs on a glassy carbon (GC) electrode. To accurately determine the intrinsic electrocatalytic activity and avoid artefacts arising from charge and mass transport effects associated with thick NP layers (see e.g. ref. 28) the NP loading was set between ∼1 and 4 μg cm−2 (for more details regarding the influence of the NP loading on the apparent activity the reader is referred to ref. 29). In order to compare our results with the literature, current–potential curves were normalized in three different ways: to the mass of metal oxide, to the mass of cobalt (Co being considered a ‘strategic’ element), and to the NP surface area (estimated from the TEM mean diameter), see Fig. 3A. Considering the low NP loading on the electrode, the mass- and surface-weighted OER activities were measured at a relatively high potential of 1.65 V vs. RHE (Fig. 3D–F). While all core–shell NPs exhibit excellent OER activities (much exceeding the 28 A goxyde−1 activity of the Fe3O4 NPs of 10 nm), the largest CS-4 and CS-3 NPs display Co-mass weighted-activities (2300 and 2800 A gcobalt−1, respectively) which are twice those of CS-1 and CS-2 (about 1300 A gcobalt−1). This trend is even clearer when considering the surface-weighted activity which has increased 3–4-fold. It is noteworthy that both the mass- and the surface-weighted OER activity of the best (CS-3) core–shell NPs is significantly higher than that of the Co(1−x)Fe(2+x)O4 ferrite NPs synthesized through a similar protocol (see the ESI†). Hence, the OER activity of NPs unambiguously arises not only from the cobalt ferrite shell but also from the combination of Fe3O4 and CoFe2O4 into a core–shell structure. The difference in activity between CS-1 and CS-2 vs. CS-3 and CS-4 likely originates from the core size (7.5–9.1 vs. 12.7–13.6 nm). A larger core is likely to be essential for efficient OER since it results in a higher fraction of conductive magnetite.30,31 This hypothesis is supported by the low activity of Fe3O4@CoO, consisting of a 4.5 nm core.32 Indeed, our recent operando soft X-ray absorption spectroscopy data suggest that the synergy results from cooperative redox interaction between Fe(II) from the core and Co(II) from the shell.33 Additionally, we also showed that the OER activity of such Fe3O4@CoFe2O4 NPs is significantly enhanced by a thicker CoFe2O4 shell although the resistivity of the NP layer increases.29 It seems that the insulating properties of Co ferrite are compensated for by a larger number of active sites on the NP surface. It is worth noting that the Co-weighted activity of the most active Fe3O4@CoFe2O4 core–shell NPs (CS-3) greatly outperforms that of TMO catalysts, either with uniform or core–shell structures, which were reported in the literature (Table S4†). Indeed only CoOx(OH)y NPs34 and core–shell NPs with a gold core6,8 (Au@CoFeOx and Au@NiCo2S4) display higher activity although they are unstable when mixed with carbon black under the OER operation.
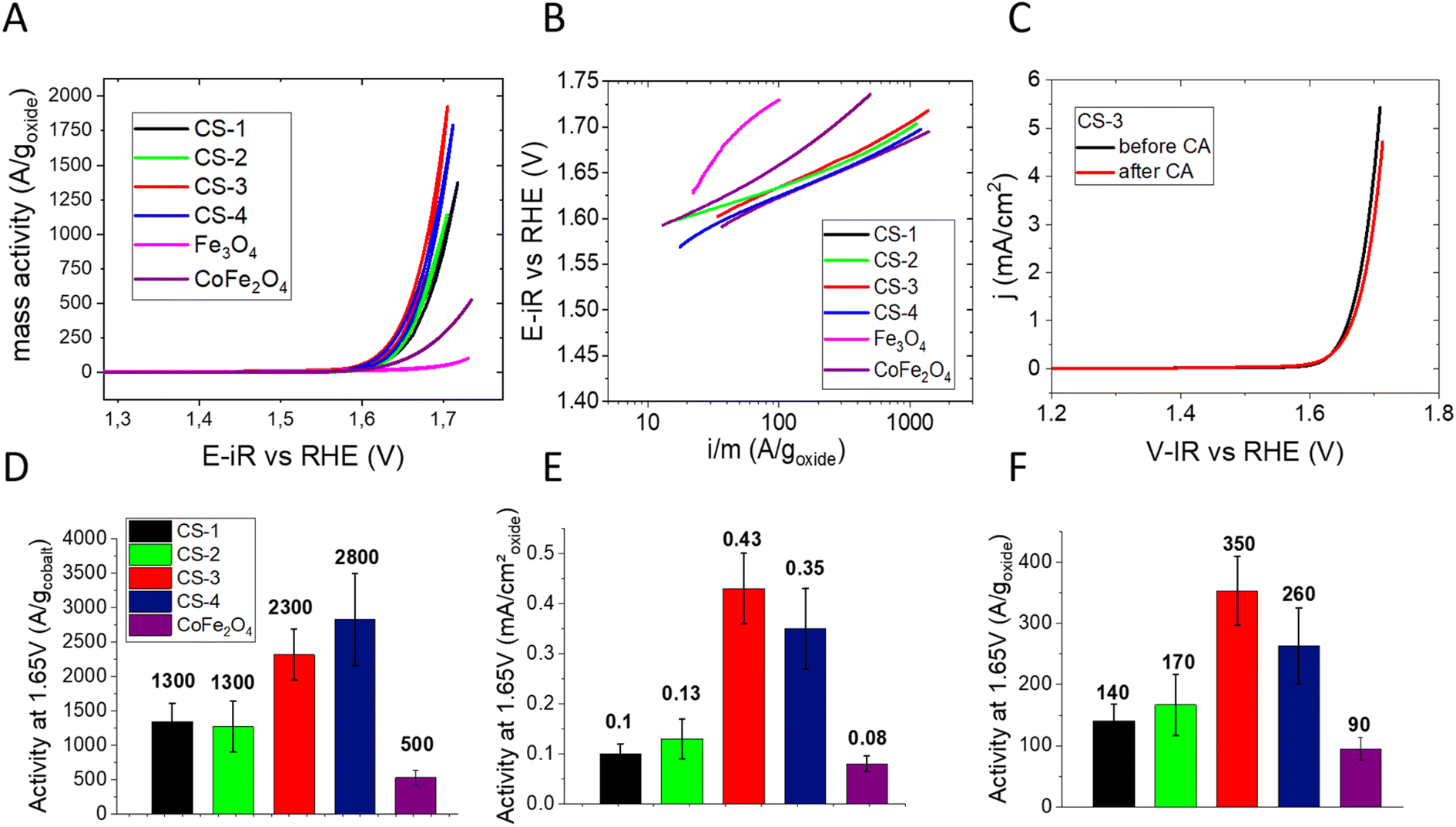 | ||
| Fig. 3 OER activity in 0.1 M NaOH: (A) cyclic voltammetry measurements performed on CS-1, CS-2, CS-3, CS-4 and Co(1−x)Fe(2+x)O4 nanoparticles deposited on a glassy carbon electrode (oxide loading between ∼1 and 4 μg cm−2, see the ESI†), scan rate 10 mV s−1. The electrode potential is ohmic drop-corrected, and the current is divided by the oxide mass. (B) Tafel slopes of these NPs (in mV decade−1). (C) Positive scans of CVs acquired for CS-3 NPs before (black) and after chronoamperometry measurements at 1.65 V vs. RHE for 3 hours. NP loading: 1.3 μg cm−2 and scan rate: 10 mV s−1. Panels (D)–(F) compare OER activity of core–shell and Co(1−x)Fe(2+x)O4 NPs at 1.65 V vs. RHE (extracted from CVs) per gram of cobalt (D), per cm2 of oxide (E) and per gram of oxide. | ||
With Tafel slopes being often used for mechanistic interpretations, one should note that they are often affected by ohmic losses in catalytic layers.29 Indeed, Tafel slopes increase with the electrode potential (Fig. 3B), notably for the Co(1−x)Fe(2+x)O4 NPs. The Tafel slopes were extracted from the anodic scans of CVs at two electrode potentials: 1.62 (where the OER just started) and 1.67 V vs. RHE (where the OER fully began). At the lower potential, all core–shell NPs display Tafel slopes in the range of 50 to 65 mV decade−1 which is slightly less than that for Co(1−x)Fe(2+x)O4 and much lower than those of the OER-inactive Fe3O4 NPs (Table S3†). The increase in the Tafel slope at higher potentials may be related to the ohmic resistance across the catalytic layer.29 At the higher potential, the Tafel slope of the Co(1−x)Fe(2+x)O4 particles exceeds the one for core–shell NPs, which is in agreement with the lower conductivity of the cobalt ferrite NPs. Finally, the medium-term stability of core–shell CS-3 NPs was studied using chronoamperometry at 1.65 V vs. RHE. Fig. 3C shows that anodic CV scan after 3 hours of polarization is within the reproducibility limit (±10%).
To sum up, Fe3O4@CoFe2O4 core–shell NPs with a narrow size distribution and uniform shape were successfully synthesized through two alternative approaches (diffusion and crystal growth). They present excellent OER activities per unit mass of Co (from 1300 to 2800 A gcobalt−1) which greatly outperform the OER activities of transition metal oxide nanoparticles reported in the literature. Along with the core–shell structure, the size of the Fe3O4 conductive core and the thickness of the Co-containing shell are critical parameters for efficient OER activity. Thus, synthesizing TMO based core–shell nanostructures with fine control of the chemical structure is a viable approach for high performance alkaline OER electrocatalysts.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This project was financially supported by the Foundation for Frontier Research in Chemistry. We thank the Helmholtz-Zentrum Berlin für Materialien und Energie for the allocation of synchrotron radiation beamtime. The authors acknowledge Juan Velasco Velez for his help on the synchrotron beamline as well as Vinavadini Ramnarain, Barbara Freis and Théo Lucante for the TEM images. STEM and EELS measurements have been performed at the Laboratorio de Microscopías Avanzadas (LMA) at the Universidad de Zaragoza (Spain). I. M. acknowledges the funding from IdEx Université de Strasbourg. R. A. and S. H. acknowledge the funding from the Spanish MICINN (project grant PID2019-104739GB-100/AEI/10.13039/501100011033), from the Government of Aragon (project DGA E13-20R) and from the European Union H2020 program “ESTEEM3” (823717).References
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- M.-I. Jamesh and X. Sun, J. Power Sources, 2018, 400, 31–68 CrossRef CAS.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- S.-C. Chou, K.-C. Tso, Y.-C. Hsieh, B.-Y. Sun, J.-F. Lee and P.-W. Wu, Materials, 2020, 13(12), 2703 CrossRef CAS PubMed.
- A. L. Strickler, M. Escudero-Escribano and T. F. Jaramillo, Nano Lett., 2017, 17, 6040–6046 CrossRef CAS PubMed.
- Z. Luo, S. Martí-Sànchez, R. Nafria, G. Joshua, M. de la Mata, P. Guardia, C. Flox, C. Martínez-Boubeta, K. Simeonidis, J. Llorca, J. R. Morante, J. Arbiol, M. Ibáñez and A. Cabot, ACS Appl. Mater. Interfaces, 2016, 8, 29461–29469 CrossRef CAS PubMed.
- Y. Lv, S. Duan, Y. Zhu, P. Yin and R. Wang, Nanomaterials, 2020, 10, 611 CrossRef CAS PubMed.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- F. Reikowski, F. Maroun, I. Pacheco, T. Wiegmann, P. Allongue, J. Stettner and O. M. Magnussen, ACS Catal., 2019, 9, 3811–3821 CrossRef CAS.
- Y. Gorlin, C.-J. Chung, J. D. Benck, D. Nordlund, L. Seitz, T.-C. Weng, D. Sokaras, B. M. Clemens and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 4920–4926 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- L. Gloag, T. M. Benedetti, S. Cheong, R. F. Webster, C. E. Marjo, J. J. Gooding and R. D. Tilley, Nanoscale, 2018, 10, 15173–15177 RSC.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- D. Guo, H. Kang, P. Wei, Y. Yang, Z. Hao, Q. Zhang and L. Liu, CrystEngComm, 2020, 22, 4317–4323 RSC.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- A. I. Dmitriev, S. I. Alekseev and S. A. Kostyuchenko, Inorg. Mater., 2019, 55, 576–581 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- M. Li, Y. Gu, Y. Chang, X. Gu, J. Tian, X. Wu and L. Feng, Chem. Eng. J., 2021, 425, 130686 CrossRef CAS.
- K. Sartori, A. Musat, F. Choueikani, J.-M. Grenèche, S. Hettler, P. Bencok, S. Begin-Colin, P. Steadman, R. Arenal and B. P. Pichon, ACS Appl. Mater. Interfaces, 2021, 13, 16784–16800 CrossRef CAS PubMed.
- X. Liu, B. P. Pichon, C. Ulhaq, C. Lefèvre, J.-M. Grenèche, D. Bégin and S. Bégin-Colin, Chem. Mater., 2015, 27, 4073–4081 CrossRef CAS.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- K. Sartori, D. Gailly, C. Bouillet, J.-M. Grenèche, P. Dueñas-Ramirez, S. Begin-Colin, F. Choueikani and B. P. Pichon, Cryst. Growth Des., 2020, 20, 1572–1582 CrossRef CAS.
- K. Sartori, G. Cotin, C. Bouillet, V. Halté, S. Bégin-Colin, F. Choueikani and B. P. Pichon, Nanoscale, 2019, 11, 12946–12958 RSC.
- B. R. Strohmeier, Surf. Interface Anal., 1990, 15, 51–56 CrossRef CAS.
- T. A. Carlson and G. E. McGuire, J. Electron Spectrosc. Relat. Phenom., 1972, 1, 161–168 CrossRef CAS.
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Grenèche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halté, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063–5073 CrossRef CAS.
- D. Y. Chung, S. Park, P. P. Lopes, V. R. Stamenkovic, Y.-E. Sung, N. M. Markovic and D. Strmcnik, ACS Catal., 2020, 10, 4990–4996 CrossRef CAS.
- L. Royer, J. Guehl, M. Zilbermann, T. Dintzer, C. Leuvrey, B. P. Pichon, E. Savinova and A. Bonnefont, Electrochim. Acta, 2023, 446, 141981 CrossRef CAS.
- M. Pauly, J.-F. Dayen, D. Golubev, J.-B. Beaufrand, B. P. Pichon, B. Doudin and S. Bégin-Colin, Small, 2012, 8, 108–115 CrossRef CAS PubMed.
- G. Rydzek, D. Toulemon, A. Garofalo, C. Leuvrey, J.-F. Dayen, D. Felder-Flesch, P. Schaaf, L. Jierry, S. Begin-Colin, B. P. Pichon and F. Boulmedais, Small, 2015, 11, 4638–4642 CrossRef CAS PubMed.
- L. Zhou, B. Deng, Z. Jiang and Z.-J. Jiang, Chem. Commun., 2019, 55, 525–528 RSC.
- L. Royer, A. Bonnefont, T. Asset, B. Rotonnelli, J.-J. Velasco-Vélez, S. Holdcroft, S. Hettler, R. Arenal, B. Pichon and E. Savinova, ACS Catal., 2023, 13, 280–286 CrossRef CAS.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Nanoparticle synthesis, TEM images, XRD patterns, cyclic voltammetry curves, impedance measurements, EELS and STEM HAADF micrographs. See DOI: https://doi.org/10.1039/d3se00130j |
| This journal is © The Royal Society of Chemistry 2023 |