
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting reduced-symmetry ligands with pyridyl and imidazole donors to construct a second-generation stimuli-responsive heterobimetallic [PdPtL4]4+ cage†
Aston C.
Pearcy
 a,
Lynn S.
Lisboa
a,
Dan
Preston
b,
Nick B.
Page
a,
Tristan
Lawrence
a,
L. James
Wright
c,
Christian G.
Hartinger
c and
James D.
Crowley
*a
a,
Lynn S.
Lisboa
a,
Dan
Preston
b,
Nick B.
Page
a,
Tristan
Lawrence
a,
L. James
Wright
c,
Christian G.
Hartinger
c and
James D.
Crowley
*a
aDepartment of Chemistry, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz
bResearch School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
cSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 14th July 2023
Abstract
A new sequential metalation strategy that enables the assembly of a new more robust reduced symmetry heterobimetallic [PdPtL4]4+ cage C is reported. By exploiting a low-symmetry ditopic ligand (L) that features imidazole and pyridine donor units we were able to selectively form a [Pt(L)4]2+ “open-cage” complex. When this was treated with Pd(II) ions the cage C assembled. 1H and DOSY nuclear magnetic resonance (NMR) spectroscopy and electrospray ionisation mass spectrometry (ESIMS) data were consistent with the quantitative formation of the cage and the heterobimetallic structure was confirmed by single crystal X-ray crystallography. The cage C was shown to bind anionic guest molecules. NMR studies suggested that these guests interacted with the cavity of the cage in a specific orientation and this was confirmed for the mesylate ion (MsO−)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C host–guest adduct using X-ray crystallography. In addition, the system was shown to be stimulus-responsive and could be opened and closed on demand when treated with appropriate stimuli. If a guest molecule was bound within the cage, the opening and closing was accompanied by the release and re-uptake of the guest molecule.
:C host–guest adduct using X-ray crystallography. In addition, the system was shown to be stimulus-responsive and could be opened and closed on demand when treated with appropriate stimuli. If a guest molecule was bound within the cage, the opening and closing was accompanied by the release and re-uptake of the guest molecule.
Introduction
High symmetry is generally a desirable property often associated with beauty. Many self-assembled architectures in Nature display high-symmetry (i.e. viral protein capsids, ferritin, and protein tetramers such as haemoglobin).1,2 Inspired by these systems, much of the early work in self-assembled metallosupramolecular architectures (MSAs) targeted highly symmetric assemblies.3–13 There are now many approaches that enable the straightforward generation of these high-symmetry metallo-architectures from simple building blocks. The resulting materials have been exploited for a range of applications, including encapsulation,14–16 separations,17 catalysis,18–21 environmental remediation,22 and biological activity.23,24While these are important applications and the results are impressive they almost universally exploit the cavities of the MSAs in a dissimilar way to active sites in enzymes. Unlike the cavities of these high-symmetry MSAs, enzyme active sites tend to be lower symmetry and this provides the basis for their high substrate selectivity. As such, there has been a recent move toward developing lower symmetry MSAs in an effort to further enhance the properties and applications of these materials. There have been three main approaches to reduced/lower symmetry MSAs, these are (1) combine a metal ion with two or more different ligands and selectively form hetero-ligated MSAs, (2) combine a metal ion and a lower symmetry ligand to selectively form a single MSA, or (3) generate heterometallic MSAs.25–29
M2L4 lantern-shaped cages (Fig. 1a)30–35 have been at the forefront of efforts to generate reduced symmetry MSAs from lower symmetry ligands. This is due to their relative simplicity, as they only contain four ligands and two metal ions. A range of heteroleptic [M2LaxLby] cages (where x = 3 or 2 and y = 1 or 2) have been developed exploiting geometric, steric, templating or supramolecular effects to provide selectivity.36–43 These approaches have also been extended to larger heteroleptic palladium(II) architectures.44–46 In addition to these heteroleptic systems, a series of reduced symmetry homoleptic [Pd2L4]4+, [Pd4L8]8+ and [Pd6L12]12+ cages have been synthesised using lower symmetry ligands.47–54 Reduced/lower symmetry MSAs have also been generated using a heterometallic approach.26 However, often the ligands used to generate heterometallic MSAs are bi- or tri-dentate donor systems in conjunction with a mono-dentate donor unit and exploit the chelate effect to provide metal ion selectivity.55,56 Generating [M1M2L4]-type heterometallic architectures that feature ditopic linker ligands with two monodentate donor units has proven to be much more difficult.
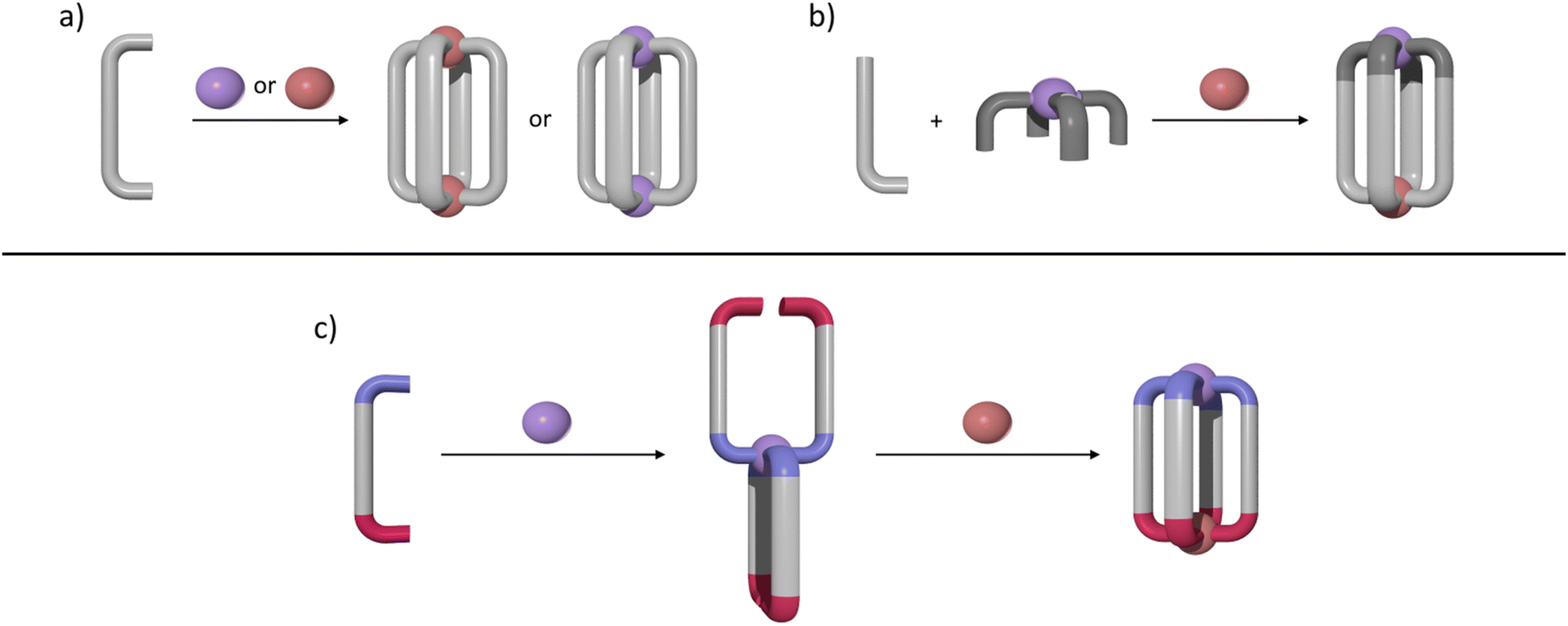 | ||
| Fig. 1 Cartoon representations of: (a) synthesis of homometallic high symmetry [M2L4]4+ cages where M = PdII or PtII; (b) synthesis of a low-symmetry heterobimetallic [PdPtL4]4+ cage using the subcomponent self-assembly approach; (c) formation of a heterobimetallic [PdPtL4]4+ cage from a low-symmetry ditopic ligand via a sequential complexation strategy. Colours: PdII brick, PtII purple, bis-monodentate ligands light or dark grey. Periwinkle blue or red ligand caps denote differing donor units. | ||
We have recently synthesised the first example of a low-symmetry heterobimetallic [PdPtL4]4+ cage.57 However, our approach used the subcomponent self-assembly method from a pre-formed inert platinum(II) tetrapyridylaldehyde complex, a substituted pyridylamine building block and a labile palladium(II) complex (Fig. 1b). The reversible formation of the dynamic covalent imine and labile palladium(II)–pyridyl bonds enabled the quantitative formation of the heterobimetallic [PdPtL4]4+ cage. While this method provides access to [PdPtL4]4+ architectures and has been exploited to generate related heterometallic double cavity cages,58 the presence of the water-labile imine functionality limits the potential applications of these systems. Herein, we report a new method for synthesis of more robust [PdPtL4]4+ architectures (Fig. 1c). Using a low-symmetry ditopic ligand that features imidazole and pyridine as two monodentate coordinating units with markedly different donor strengths, the platinum(II) and palladium(II) ions of the target [PdPtL4]4+ heterobimetallic cage can be introduced sequentially (Fig. 1c). As with our previously reported [PdPtL4]4+ cage, this architecture is stimuli-responsive and can be opened and closed reversibly with concurrent guest release and binding.59,60 Importantly, we also show that, unlike other low-symmetry assemblies, the new low-symmetry cage can bind anionic guests in a specific orientation, in a manner reminiscent of biologically occurring molecular containers such as enzymes.
Results and discussion
For the sequential complexation strategy (Fig. 1c) to be successful, we required a low-symmetry ditopic ligand that contained two monodentate coordinating units with different donor strengths. Due to the difference in pKa between imidazole (6.95)61 and pyridine (5.20)62 we reasoned that a ligand featuring those donor units might be appropriate. However, before committing to that ligand design we carried out some competition experiments with palladium(II) ions and 1-methylimidazole (meim) and 3-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]pyridine (3-pytri)63 (ESI, Sections 1 and 2†). Combining [Pd(CH3CN)4](BF4)2, meim and 3-pytri in a 1:4:4 ratio in CD3CN led to the quantitative formation of [Pd(meim)4](BF4)2. Similarly, reacting the preformed [Pd(3-pytri)4](BF4)2 complex with meim (4 eq.) led to the instantaneous displacement of the 3-pytri ligands and quantitative formation of the [Pd(meim)4](BF4)2 complex (ESI, Section 2, Fig. S21 and S22†). This strongly suggested that a ditopic ligand containing both imidazole pyridine donors would be a suitable candidate for selective, sequential hetero-dimetalation. As such we targeted the synthesis of L (Scheme 1 and ESI, Section 1, Scheme S1†).
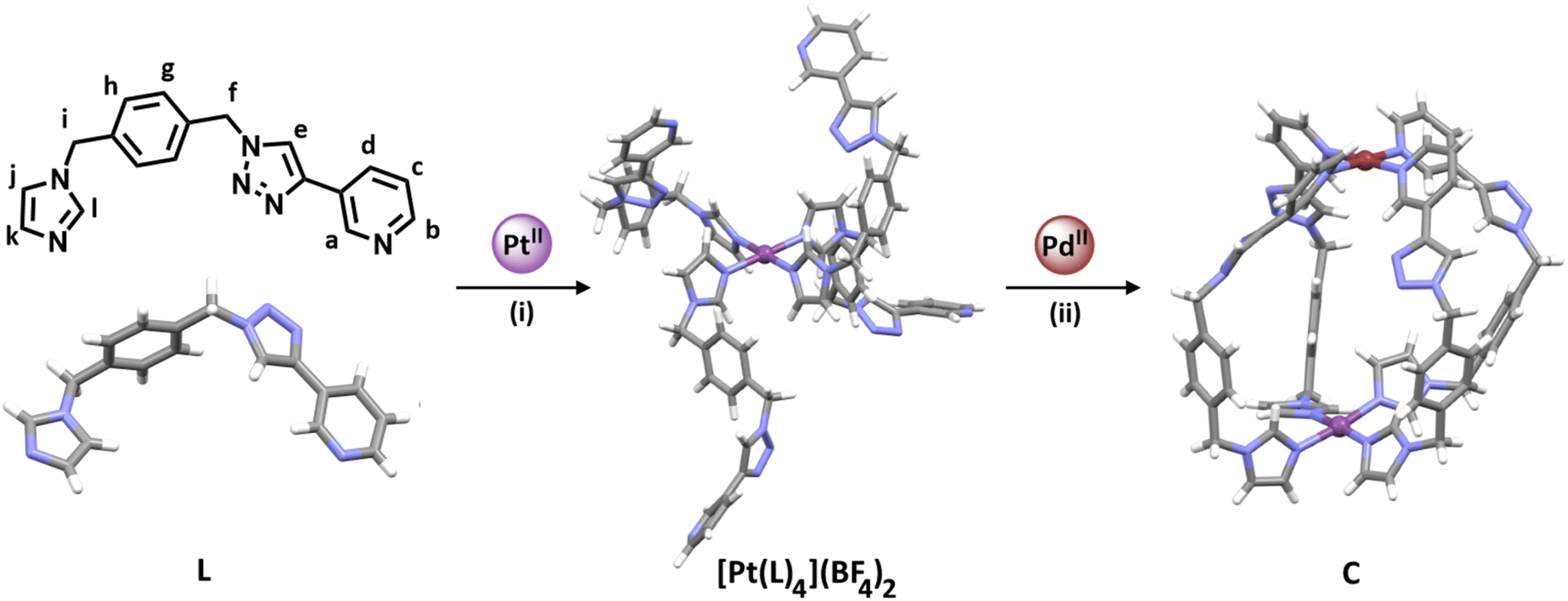 | ||
| Scheme 1 Synthesis of the platinum(II) complex [Pt(L)4](BF4)2, and the heterobimetallic low-symmetry cage (C): (i) L (4.1 eq.), AgBF4 (2.2 eq.), and [Pt(DMSO)2Cl2] (1 eq.), d6-DMSO, 60 °C, 7 days. (ii) [Pt(L)4](BF4)2 (1 eq.), [Pd(CH3CN)4](BF4)2 (1 eq.), CD3CN, RT, 10 min ([L] = 21.1 mM, [[Pt(L)4](BF4)2] = 5.27 mM). The stick model (MMFF) of [Pt(L)4](BF4)2 was generated using SPARTAN'18.66 Stick models of L and C are the molecular structures determined by X-ray diffraction analysis. Colours: PdII brick, PtII purple, N periwinkle blue, C grey, H white. Solvent molecules and counterions omitted for clarity. | ||
The synthesis of low-symmetry ligand L began with the desymmetrisation of commercially available α,α′-dibromo-p-xylene (ESI, Scheme S1†). α,α′-Dibromo-p-xylene (1 eq.) and NaN3 (1 eq.) were heated at 60 °C in acetonitrile for 24 h to give 1-(azidomethyl)-4-(bromomethyl)benzene64,65 in modest yields (isolated yields ranged from 27–45%). This mono-substituted intermediate (1 eq.) was then stirred with imidazole (1.1 eq.) and KOH (2 eq.) for 4 h at room temperature (RT) in acetonitrile to give 1-(4-(azidomethyl)benzyl)-1H-imidazole in good yield (77%). This azide building block (1 eq.) was then treated with 3-ethylnylpyridine (1.63 eq.) using standard copper-catalysed azide–alkyne cycloaddition (CuAAC) “click” conditions (CuSO4·5H2O (0.68 eq.) and sodium ascorbate (0.9 eq.) in 4:1 DMF:H2O at RT for 24 h) providing the low-symmetry ditopic imidazole pyridine ligand L in excellent yield (91%). L was characterised using 1H, 13C and DOSY nuclear magnetic resonance (NMR) spectroscopies, electrospray ionisation mass spectrometry (ESIMS) and the molecular structure was determined using X-ray crystallography (Scheme 1 and ESI, Section 1, Fig. S31†).
With the low-symmetry ditopic ligand L in hand, we attempted to selectively form the platinum(II) complex [Pt(L)4](BF4)2 in which each ditopic ligand is coordinated to platinum through the imidazole nitrogen donor only. L (4.1 eq.), AgBF4 (2.2 eq.), and [Pt(DMSO)2Cl2] (1 eq.) were combined in d6-DMSO at 60 °C in the absence of light and the reaction monitored using 1H NMR spectroscopy and ESIMS (ESI, Fig. S8–S10 and S22–S24†). After one day the resonances due to free ligand L had vanished and were replaced by two sets of shifted signals (ESI, Fig. S22 and S23†).
ESIMS data showed the presence of a major peak at m/z = 730.2721, which was consistent with the molecular formula [Pt(L)4]2+, suggesting the mono-PtII complex had formed. With continued heat over seven days the ratio of the two sets of signals slowly changed to a point where a 85:15 ratio67 was obtained.68 Addition of ethyl acetate to this mixture led to the precipitation of pure [Pt(L)4](BF4)2 in good isolated yield (68%). ESIMS of the isolated material in CH3CN again displayed a major peak at m/z = 730.2721 with a second smaller peak at m/z = 1546.5385 consistent with the formation of [Pt(L)4]2+ and [Pt(L)4](BF4)+ ions. The observed isotope patterns matched those calculated for the 2+ and 1+ ions, respectively (ESI, Fig. S10†). Comparison of the 1H NMR spectrum of the ligand L with the spectrum of the isolated colourless material showed that the chemical shifts of the pyridyl (Ha–d) and triazole (He) resonances remained essentially unchanged upon coordination to PtII, whereas the imidazole resonances (Hj,k and l) all moved significantly upon complex formation. This was consistent with the formation of a linkage isomer where all ligands are coordinated to the PtII ion via the imidazole donor units (Fig. 2 and ESI, Fig. S8†). Additionally, the 1H DOSY NMR spectra (CD3CN, 298 K) of L and [Pt(L)4](BF4)2 showed that all the proton resonances within the individual samples had the same diffusion coefficients (DL = 12.0 × 10−10 m2 s−1 and D[Pt(L)4](BF4)2 = 5.15 × 10−10 m2 s−1), suggesting formation of a single product (ESI, Section 4, Fig. S24–S30, Table S1†). The platinum(II) complex diffused more slowly than L consistent with the formation of a larger structure. While we have been unable to confirm the molecular structure via X-ray crystallography, the combined data suggest that the tetraimidazole linkage isomer of [Pt(L)4](BF4)2 (MMFF molecular model, Fig. 1) was formed.
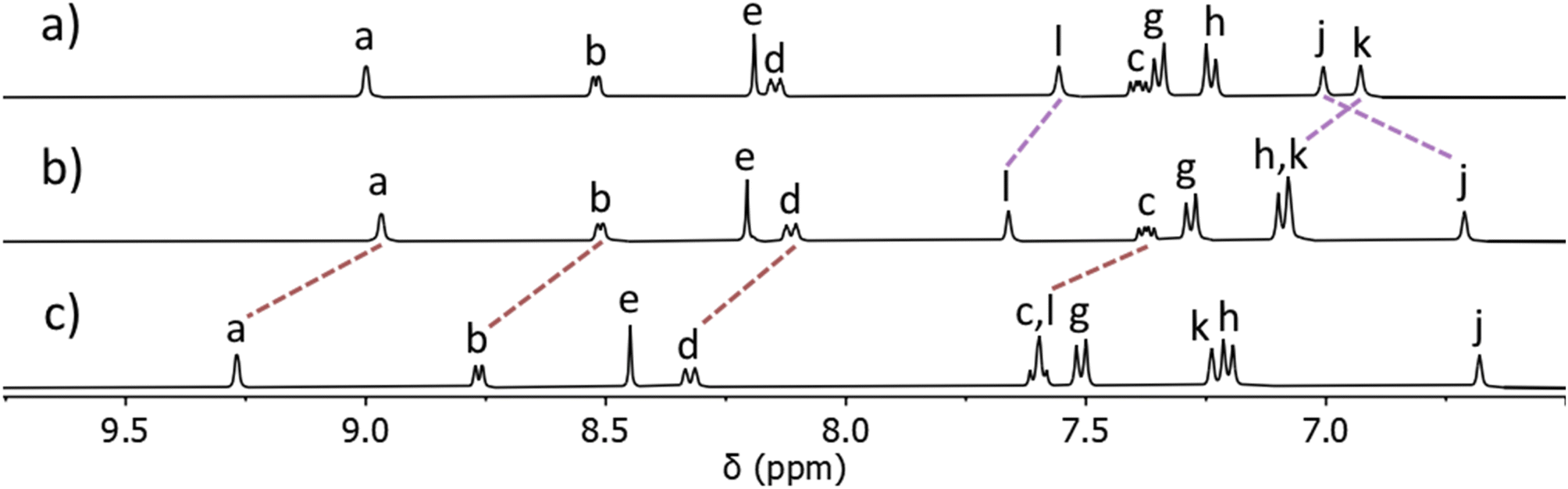 | ||
| Fig. 2 Stacked partial 1H NMR spectra (400 MHz, d6-DMSO, 298 K) of (a) ligand L, (b) the [Pt(L)4](BF4)2 complex, and (c) heterobimetallic cage C ([L] = 21.1 mM). Proton labels correspond to those shown in Fig. 1 and the ESI.† | ||
The isolated complex [Pt(L)4](BF4)2 (1 eq.) was then combined with [Pd(CH3CN)4](BF4)2 (1 eq.) in either CD3CN or d6-DMSO at room temperature and the reaction was monitored via1H NMR spectroscopy. Within 10 minutes, signals due to the free pyridyl unit (Ha–d) had completely vanished and were replaced by a new set of resonances shifted downfield (Δδ = 0.20–0.25 ppm, d6-DMSO, Fig. 2b and c). Large downfield shifts of the α-pyridyl protons (Ha and Hb) were observed, characteristic of coordination of the palladium(II) ion. Resonances due to platinum(II) coordinated imidazole units remained relatively unchanged (Fig. 2b and c). The new set of 1H NMR signals is distinct from those of both L and [Pt(L)4](BF4)2. The 1H DOSY NMR spectrum of the reaction mixture revealed that all the new resonances possess the same diffusion coefficient, suggesting the clean formation of a single product. The diffusion coefficient (DC = 5.54 × 10−10 m2 s−1) of the cage is different from that of the ligand L (DL = 12.0 × 10−10 m2 s−1) and similar to that of [Pt(L)4](BF4)2 (D[Pt(L)4](BF4)2 = 5.15 × 10−10 m2 s−1), consistent with the formation of a larger cage architecture. ESIMS data obtained from the reaction mixture also supported the formation of the [PdPt(L)4](BF4)4 heterobimetallic cage, C. The major peak at m/z = 391.6139 was consistent with the molecular formula [PdPt(L)4]4+ and the observed isotope pattern matched that calculated for this 4+ ion.
Other smaller peaks at m/z = 551.1534 and 870.2332, consistent with the [PdPt(L)4(BF4)]3+ and [PdPt(L)4(BF4)2]2+ ions, respectively, were also observed (ESI, Fig. S13†). The heterometallic low-symmetry cage C was isolated as a pale-yellow solid in 87% yield from acetonitrile solution by addition of excess ice-cold diethyl ether. The solid was dissolved in nitromethane and diethyl ether was vapour-diffused into the solution to provide colourless crystals of a suitable quality for single-crystal X-ray diffraction analysis. This unequivocally confirmed the formation of the heterobimetallic low-symmetry [PdPt(L)4]4+ cage (Fig. 3 and ESI, Section 5, Fig. S32†). The two metal ions are bridged by four ligands L adopting a syn arrangement. As indicated by the 1H NMR analysis, the platinum(II) ion is coordinated by the four imidazole units of the low-symmetry ligand and the palladium(II) ion is coordinated to the four pyridyl units. The nitrogen atoms of the 1,2,3-triazole moieties are not coordinated to either metal ion. The Pd⋯Pt distance (10.449 Å) is slightly smaller than that of the related imine-based heterobimetallic cage,57 and comparable to similar, more rigid dipyridyl homometallic cages,69–73 and other low-symmetry assemblies.74–76 The cavity of the cage C contains a molecule of nitromethane and a BF4− anion. Hydrogen bonding is observed between both the guest molecules with a variety of different units on the cage. The nitromethane molecule interacts with three of the four inwardly pointing imidazole protons. The BF4− ion interacts with all four α-pyridyl protons (as is often observed in related heterometallic cages) two inwardly pointing triazole protons, and one of the inwardly pointing imidazole protons (Fig. 3 and ESI, Section 5, Fig. S32†).
 | ||
| Fig. 3 Molecular structure of the heterobimetallic low-symmetry [PdPt(L)4]4+ cage C. A BF4− anion and a CH3NO2 molecule occupy the cavity of C. Colours: PdII brick, PtII purple, N periwinkle blue, C grey, H white, B pick, F green. Hydrogen atoms involved in H-bonding interactions (⋯) are shown. All other hydrogen atoms, most solvents of crystallisation and counterions have been omitted for clarity. | ||
Having successfully generated the second low-symmetry heterobimetallic [PdPt(L)4]4+ cage (C), we then examined if the architecture was a thermodynamic or kinetically metastable product. The cage C was initially heated in CD3CN solution at either 50 °C or 75 °C and the solution speciation was monitored using 1H NMR spectroscopy (ESI, Section 6, Fig. S35 and S36†). The 1H NMR spectra of C were unchanged after 4 days of heating in CD3CN (at either 50 or 75 °C) indicating that the dimetallic complex is robust under these conditions. Similar experiments were then carried out using d6-DMSO as the solvent. The cage C was unchanged after four days at 50 °C in d6-DMSO (Fig. S37†). When C was heated at 75 °C or 100 °C in d6-DMSO changes were observed (Fig. S38†). After 24 hours at 75 °C a new species can be observed. However, even after 4 days at that temperature in d6-DMSO, C remains the major (>90%) species present (Fig. S38†). At 100 °C in d6-DMSO the rearrangement/isomerisation process is faster. After 24 hours at 100 °C C is still the major (50%) compound in solution but several other species can be observed. As the solution is heated further the speciation continues to change and after 4 days only a small amount (10–15%) of C remains. The other species observed are presumed to be different [PdPt(L)4]4+ cage isomers. The combined results suggest that C is a kinetically metastable architecture. However, the complex is kinetically robust in both CD3CN and d6-DMSO over a range of temperatures.
The molecular structure of the low-symmetry heterometallic cage C confirmed that the cavity of the cage was accessible for guest molecules (Fig. 3). Therefore, we investigated the host–guest (HG) chemistry of the system with a wide variety of neutral organic and inorganic guest molecules (ESI, Fig. S41†). The HG interactions were examined using 1H NMR spectroscopy and ESIMS. In a series of experiments, one of the potential guest molecules (2 eq.) was combined with cage C (1 eq.) in CD3CN at 298 K and the 1H NMR spectrum acquired (ESI, Section 7, Fig. S42–S44†). The 1H NMR spectrum of the host–guest mixture was compared to the spectra of the “free” host and guest compounds (ESI, Fig. S42 and S43†). Disappointingly, no complexation-induced shifts (CIS) were observed for either the host or the guest resonances, suggesting that none of these neutral guests interacted with the cavity of cage C. Consistent with the 1H NMR spectra, the ESIMS data displayed no peaks due to HG complexes. Molecular models (SPARTAN'18,66 MMFF, see ESI, Section 7, Fig. S40†) suggest that there are no obvious steric interactions that would prevent host–guest formation for the majority of the HG pairs examined. The lack of neutral guest binding appears to be caused primarily by the BF4− counter-anions, outcompeting the neutral guests and preferentially occupying the cage cavity (Fig. 3 and ESI, Fig. S39†). This competitive anion binding behaviour has been observed before in related homometallic cages.77
Due to the lack of binding observed with neutral molecules, we turned to anionic sulfonate, organotrifluoroborate and phosphate ester guest molecules as these have been well established to bind within related homometallic cage systems.78–86 We examined methanesulfonate (MsO−), p-toluenesulfonate (TsO−), tolyltrifluoroborate (tolBF3−) and dimethylphosphate (dimephos−). All four of the anions displayed CIS consistent with guest binding (ESI, Section 7†). For example, upon titration of MsO− into an CD3CN solution of C, a large CIS of the Ha proton (Δδ = 0.55, with 2 eq. of MsO−) was observed (Fig. 4 and ESI, Fig. S50†). ESIMS data of the HG mixture also provided evidence for the formation of a C⊂MsO− host–guest adduct, and indicated a 1:1 C:MsO− ratio, with large peaks at m/z = 553.8112 ([C⊂MsO−]3+) and m/z = 874.2178 ([C⊂MsO−(BF4−)]2+) (Fig. 4 and ESI, Fig. S49†). Similar NMR and ESIMS data was obtained from CD3CN mixtures of C and TsO− (ESI, Fig. S45–S48†). Disappointingly, mixtures of C and either tolBF3− or dimephos− did not display any peaks due the HG adducts in the ESIMS data potentially suggesting that the HG interaction with those guests is weaker than those found with the sulfonates.
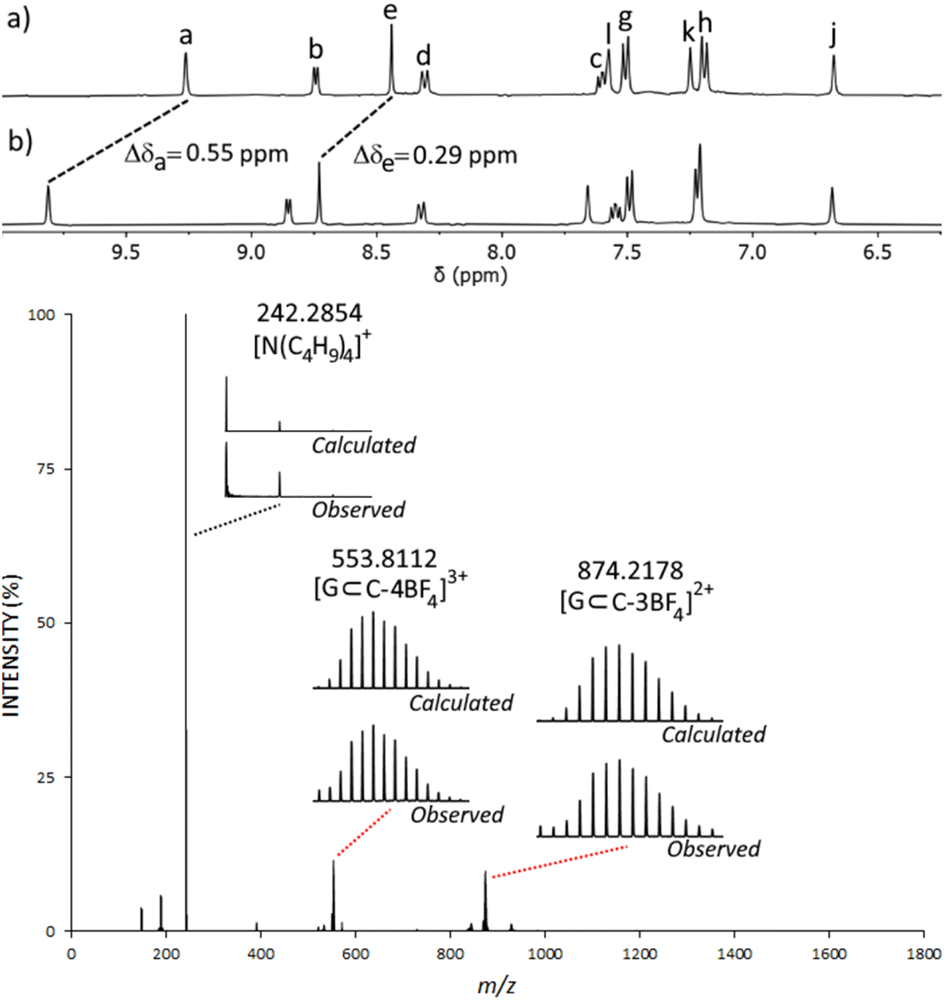 | ||
| Fig. 4 Top: stacked partial 1H NMR spectra (400 MHz, [D3]acetonitrile, 298 K) of (a) C and (b), C + MsO− (2 eq.) ([C] = 0.150 mM). Bottom: ESI-mass spectrum obtained from an acetonitrile solution of C (1 eq.) and MsO− (2 eq.). G refers to the MsO− guest molecule. | ||
The interactions of anionic guest molecules with cage C were examined further using 1H NMR spectroscopic titrations (CD3CN, 298 K) and the data was curve-fitted using bindfit87–89 to obtain the association constants (K, ESI, Section 7, Table S5†). Consistent with the ESIMS data, both sulfonate systems provided an excellent fit to a 1:1 binding model (KMsO− = 29400 ± 2700 M−1 and KTsO− = 17200 ± 1600 M−1). The smaller, more basic MsO− has a slightly stronger interaction with the cage C compared to the larger, less basic TsO−. The titration data with the tolBF3− guest also provided an excellent fit to a 1:1 binding model and the interaction was weaker (KtolBF3− = 2190 ± 40 M−1) than what had been found with the sulfonates. Interestingly, the NMR titration data from the interaction of C with dimephos− was consistent with a 1:2 HG interaction (K1(dimephos−) = 5870 ± 320 M−1, K2(dimephos−) = 1540 ± 70 M−1, ESI, Fig. S54–S57, Table S5†).
The lower symmetry of cage C means that there are potentially two different binding modes for the anionic guest: one where the anions bind at the [Pd(py)4]2+ end of the cage and the other where the guests interact with the [Pt(im)4]2+ end (ESI, Fig. S61†). The largest CIS observed in the 1H NMR data obtained from HG mixtures with the sulfonate and organotrifluoroborate anions were for the pyridyl (Ha) and triazole (He) protons of C (Fig. 4 and ESI, Section 7†). Little to no shifts were observed for the imidazole (Hj,k,l) resonances of C. The data for the C:dimephos− 1:2 HG complex was similar, large CIS were observed for the pyridyl (Ha) and triazole (He) protons of C at lower equivalents (0–3 eq.) of dimephos−. As more equivalents (5–42) of dimephos− are added to the mixture the Ha and He resonances stop shifting and the imidazole resonances (Hl and Hj) begin to move (ESI, Fig. S54†). The CIS data observed for the 1:1 HG complexes strongly suggest that the anions all interact selectively with the [Pd(py)4]2+ end of the cage as has been observed in related homometallic cages.78–82 The CIS data obtained for the 1:2 HG complex formed with dimephos− is also consistent with the first guest binding selectively at the [Pd(py)4]2+ end of the cage while the second anion then interacts with the [Pt(im)4]2+ end of C. Additional support for this HG orientation was obtained from X-ray crystallography (Fig. 5 and ESI, Section 7, Fig. S53†). Vapor diffusion of diethyl ether into an acetonitrile solution of the cage C and MsO− (2 eq.) provided colourless crystals suitable for single-crystal X-ray diffraction analysis. Determination of the molecular structure unequivocally confirmed the formation of the HG complex C⊂MsO− (Fig. 5 and ESI, Fig. S53†). The Pd–Pt distance (10.086 Å) in the HG complexes is slightly smaller than that of the parent cage (10.449 Å, vide infra). Importantly, the structure revealed that the MsO− guest molecule is interacting, through hydrogen bonding, with the [Pd(py)4]2+ motif of the heterometallic cage, consistent with the NMR data. The MsO− guest is rotationally disordered, but clearly forms hydrogen bonds to the endohedral α-pyridyl protons of the [Pd(py)4]2+ unit. The C⋯OS distances ranged from 3.12 to 3.26 Å, and the C–H⋯OS distances from 2.33 to 2.37 Å. Similar interactions have been observed in a range of related homometallic dipalladium cages.78–82,90,91 There are also interactions with the triazole protons consistent with the CIS observed in solution. We note that the BF4− counter anion in the X-ray structure of the “free” C (Fig. 3) is also interacting with the [Pd(py)4]2+ end of the cage providing additional solid-state support for the host–guest selectivity.
 | ||
| Fig. 5 Molecular structure of the C⊂MsO− host–guest adduct. The MsO− guest is rotationally disordered. Colours: PdII brick, PtII purple, N periwinkle blue, C grey, H white, O red, S orange. Hydrogen atoms involved in H-bonding interactions are shown. All other hydrogen atoms, most solvents of crystallisation and counterions have been omitted for clarity. | ||
Having established that C can act as a host for anionic guest molecules, we next examined the stimulus responsiveness of the architecture. We and others have shown that addition of the base N,N′-dimethylaminopyridine (DMAP) to a PdII architecture can remove the labile palladium(II) ions as the complex [Pd(DMAP)4]2+, and release free ligand.57,69,92 We have also shown that addition of DMAP to a PdII/PtII heterobimetallic architecture preferentially removes the more labile palladium(II) metal ion, leaving the coordination environment of the relatively inert platinum(II) metal ion untouched.57 This process is completely reversible and the addition of acid preferentially protonates the DMAP ligands, leading to reformation of the cage architecture. As we have used DMAP and p-toluenesulfonic acid (TsOH) as external stimuli previously, we chose to employ them in this new heterobimetallic system to examine if the cage could be reversibly opened and closed (ESI, Section 8, Scheme S2, Fig. S62–S65†).
DMAP (1–5 eq.) was titrated into a d6-DMSO solution of C and the reaction monitored via1H NMR spectroscopy (Fig. S64†). Signals attributable to C decreased in intensity, while a new set of signals that matched those of [Pt(L)4](BF4)2 grew in intensity after each addition of DMAP. Another set of signals due to a [Pd(DMAP)4]2+ complex also increased in intensity as more equivalents of DMAP were added. These observations indicate the successful opening of the cage. After 4 equivalents of DMAP had been added, the addition of one more equivalent of DMAP did not result in any further changes to the chemical shifts of the complex, and no signals were observed for free L. This suggests that an excess of DMAP under the conditions of this experiment was insufficient to extract the inert platinum(II) ion from the imidazole pocket of the [Pt(L)4](BF4)2 complex.
TsOH (5 eq.) was then added to the resulting [Pt(L)4](BF4)2:[Pd(DMAP)4]2+:DMAP mixture, and the reaction monitored via1H NMR spectroscopy (Fig. S64†). Resonances due to the [Pt(L)4](BF4)2 open complex steadily decreased in intensity, while a new set of signals that matched those of C grew in intensity after each addition of TsOH. Two more sets of resonances also steadily grew in intensity over the course of the experiment, assigned as protonated DMAP and deprotonated TsOH. These results are consistent with the reformation of C. Similar results are obtained when using methanesulfonic acid (MsOH) in the place of TsOH.
Having confirmed that C can open and close on demand, we then examined if this ability persists in the presence of a guest (Fig. 6 and ESI, Fig. S66 and S67†). The C⊂MsO− host–guest adduct (Fig. 6a) was formed by addition of MsO− (1 eq.) to a CD3CN solution of C (1 eq.). As detailed before large CIS are observed for Ha and He of the cage confirming the formation of the HG complex, additionally the methyl resonance of the bound MsO− anion (HMe = 2.01 ppm) is shifted upfield from “free” MsO− (HMe = 2.41 ppm, ESI, Fig. S66†). DMAP (4 eq.) was then added to the C⊂MsO− host–guest mixture and the reaction monitored using 1H NMR spectroscopy (Fig. 6b). Much like with “free” C, when DMAP was added to the HG complex the palladium(II) ions are removed from the cage and the [Pd(DMAP)4]2+ and [Pt(L)4]2+ complexes are formed (Fig. 6b). This was accompanied by release of the MsO− guest (HMeO = 2.43 ppm). Upon addition of methanesulfonic acid (MsOH, 4 eq.) to this mixture, reformation of C was observed along with protonation of the DMAP ligands (Fig. 6c). The reformation of C occurs with reuptake of MsO− guest molecules, with large CIS observed for Ha and He. The 1H NMR spectrum of the reformed host–guest adduct (Fig. 6c) is similar, but not exactly identical, to that of the original C⊂MsO− host–guest complex (Fig. 6a). This is because the solution now contains 5 equivalents of MsO− after the deprotonation of MsOH, which further shifts the resonances (Ha and He) of the host–guest adduct. Using TsOH in place of MsOH also provides similar results when switching the host–guest adduct open and closed, but the final HG spectrum (after the switching has been completed) is more complicated due to competition between MsO− (1 eq.) and TsO− (4 eq.) for the cavity of C (ESI, Fig. S67†).
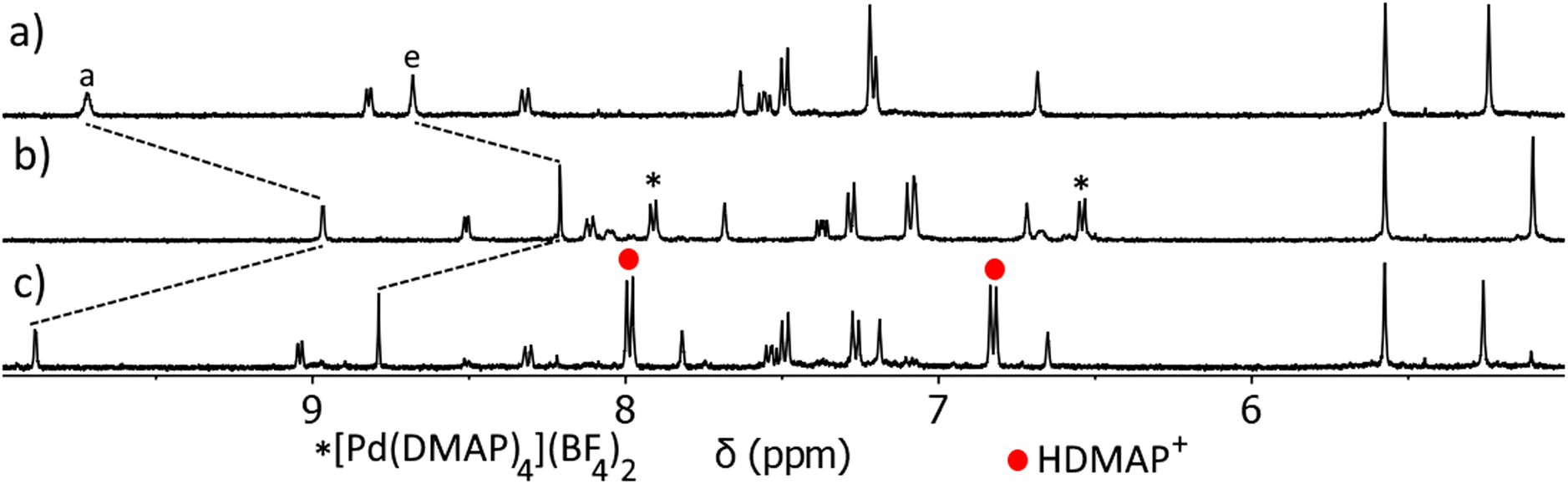 | ||
| Fig. 6 Stacked partial 1H NMR spectra (400 MHz, CD3CN, 298 K) of (a) C⊂MsO− at a 1:1 ratio ([C] = 1.00 mM), (b) [Pt(L)4](BF4)2 open complex after the addition of DMAP (4 eq.), (c) restored C⊂MsO− host–guest adduct after addition of MsOH (4.1 eq.). | ||
The results described above show that C retains all the key properties of the 1st generation imine-based heterobimetallic [PdPt(L)4]4+ cage.57 While this imine-based cage was stable in polar aprotic solvents (acetonitrile, nitromethane and DMSO) when it was exposed to water (5 or 50% in DMSO) the imines hydrolysed and the cage decomposed.93,94 To examine if the second generation heterobimetallic [PdPt(L)4]4+ cage C was more robust in aqueous conditions we carried out two stability experiments using 1H NMR spectroscopy (ESI, Fig. S68 and S69†). Initially, C was dissolved in a 19:1 d6-DMSO:D2O mixture and monitored over 21 days. There was no sign of any cage decomposition. A similar experiment in a 1:1 d6-DMSO:D2O mixture was then carried out. Again, no decomposition of the cage C was observed. These experiments indicate that the 2nd generation heterobimetallic [PdPt(L)4]4+ cage C is considerably more robust in aqueous conditions when compared to the parent imine-based cage.
Conclusions
We have developed a method that enables the formation of the low-symmetry heterobimetallic [PdPtL4]4+ cage C. By synthesising a low-symmetry ligand that features coordinating units of sufficiently different donor strengths, the individual donor units can be selectively coordinated to different metal ions in a stepwise fashion. This strategy enabled the formation of the complex [Pt(L)4](BF4)2, which can be transformed through the addition of palladium(II) ions to the heterobimetallic [PdPtL4]4+ cage in quantitative yield. Formation of the cage was examined with 1H and DOSY NMR spectroscopies as well as ESIMS, and unequivocally confirmed using X-ray crystallography. The cage was shown not bind any of the neutral guest molecules screened in the solvents used. However, the cage was able to strongly encapsulate a range of different anions (MsO−, TsO−, tolBF3− and dimephos−). Most importantly, solution NMR data and solid-state X-ray data from the host–guest complexes indicated that the anions interact orientationally selectively with the [Pd(py)4]2+ end of the cage C.Much like the first generation, imine-based [PdPtL4]4+ cage, C was shown to be stimuli-responsive. Upon addition of DMAP the cage was opened through the formation of the complexes [Pd(DMAP)4]2+, and it could be closed again by addition of MsOH or TsOH which protonated the DMAP ligands and released free palladium(II) ions back into the system resulting in spontaneous reformation of C. This same process could also be carried out on the C⊂MsO− host–guest adduct. In this case the guest molecule was released on deactivation of host–guest interactions when the cage was converted to the open platinum(II) complex. Reuptake of the guest molecule was then observed when the cage was reformed on addition of acid to release free PdII ions.
While the current cage C has limited guest binding capabilities, its stability in aqueous conditions suggests that more water soluble analogues should be able to take advantage of the hydrophobic effect and bind a wider range of neutral guest molecules.3 These new systems should provide access to switchable enzyme-like catalysts.71,95–99
Data availability
The data that support the findings of this study are available in the ESI† of this article. All experimental procedures, characterisation and titration data are available in the ESI.† Crystallographic data for the structures reported herein have been deposited as CCDC 2175042–2175046.Author contributions
AP, JC, CH, and LW conceived the idea. JC, CH, and LW obtained the funding. AP and JC analysed the data and wrote the manuscript. AP conducted the synthesis, host–guest chemistry and crystallography. TL carried out the model studies. NP assisted the ligand synthesis. LL assisted the collection of host–guest and crystallographic data. DP collected and analysed the DOSY NMR data. All authors provided feedback on the manuscript drafts and approved the submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ACP thanks the University of Otago and the John Edmond Trust for MSc scholarships. LSL thanks the University of Otago for a PhD scholarship. JDC thanks the University of Otago, Department of Chemistry and the MacDiarmid Institute for Advanced Materials and Nanotechnology for funding. All authors are grateful to the Marsden Fund for supporting this work through a grant (UOA1726).Notes and references
- D. L. Caulder and K. N. Raymond, J. Chem. Soc., Dalton Trans., 1999, 1185–1200 RSC.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- E. G. Percastegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS.
- N. B. Debata, D. Tripathy and H. S. Sahoo, Coord. Chem. Rev., 2019, 387, 273–298 CrossRef CAS.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- H. D. Mai, N. M. Tran and H. Yoo, Coord. Chem. Rev., 2019, 387, 180–198 CrossRef CAS.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- H. Wang, Y. Li, N. Li, A. Filosa and X. Li, Nat. Rev. Mater., 2021, 6, 145–167 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- L. Catti, R. Sumida and M. Yoshizawa, Coord. Chem. Rev., 2022, 460, 214460 CrossRef CAS.
- M. Yoshizawa and M. Yamashina, Chem. Lett., 2017, 46, 163–171 CrossRef CAS.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed.
- M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed.
- R. J. Hooley, Synlett, 2020, 31, 1448–1463 CrossRef CAS.
- E. G. Percastegui, Chem. Commun., 2022, 58, 5055–5071 RSC.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS.
- A. Pöthig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- M. Hardy and A. Lützen, Chem.–Eur. J., 2020, 26, 13332–13346 CrossRef CAS PubMed.
- J. Lewis and J. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS.
- D. Tripathy, N. B. Debata, K. C. Naik and H. S. Sahoo, Coord. Chem. Rev., 2022, 456, 214396 CrossRef CAS.
- C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed.
- D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295–3297 CrossRef CAS PubMed.
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- D. Bardhan and D. K. Chand, Chem.–Eur. J., 2019, 25, 12241–12269 CrossRef CAS PubMed.
- N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS.
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS PubMed.
- A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V. Schäfer and G. H. Clever, Chem.–Eur. J., 2018, 24, 12976–12982 CrossRef CAS.
- W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285–8289 CrossRef CAS PubMed.
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, A. Platzek, L. Schneider, K. Wu and G. H. Clever, Chem. Sci., 2022, 13, 1829–1834 RSC.
- J. A. Findlay, K. M. Patil, M. G. Gardiner, H. I. MacDermott-Opeskin, M. L. O'Mara, P. E. Kruger and D. Preston, Chem.–Asian J., 2022, 17, e202200093 CrossRef CAS PubMed.
- S. Sudan, R. J. Li, S. M. Jansze, A. Platzek, R. Rudolf, G. H. Clever, F. Fadaei-Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2021, 143, 1773–1778 CrossRef CAS PubMed.
- R. J. Li, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Chem.–Eur. J., 2021, 27, 9439–9445 CrossRef CAS PubMed.
- A. Tarzia, J. E. M. Lewis and K. E. Jelfs, Angew. Chem., Int. Ed., 2021, 60, 20879–20887 CrossRef CAS PubMed.
- J. E. M. Lewis, Chem.–Eur. J., 2021, 27, 4454–4460 CrossRef CAS PubMed.
- J. E. M. Lewis, A. Tarzia, A. J. P. White and K. E. Jelfs, Chem. Sci., 2020, 11, 677–683 RSC.
- S. S. Mishra, S. V. K. Kompella, S. Krishnaswamy, S. Balasubramanian and D. K. Chand, Inorg. Chem., 2020, 59, 12884–12894 CrossRef CAS PubMed.
- D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424–18428 CrossRef CAS PubMed.
- H. Yu, J. Li, C. Shan, T. Lu, X. Jiang, J. Shi, L. Wojtas, H. Zhang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 26523–26527 CrossRef CAS.
- T. Tsutsui, L. Catti, K. Yoza and M. Yoshizawa, Chem. Sci., 2020, 11, 8145–8150 RSC.
- R.-J. Li, A. Marcus, F. Fadaei-Tirani and K. Severin, Chem. Commun., 2021, 57, 10023–10026 RSC.
- F. Li and L. F. Lindoy, Aust. J. Chem., 2019, 72, 731–741 CrossRef CAS.
- L. Li, D. J. Fanna, N. D. Shepherd, L. F. Lindoy and F. Li, J. Inclusion Phenom. Macrocyclic Chem., 2015, 82, 3–12 CrossRef CAS.
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101–11107 CrossRef CAS.
- L. S. Lisboa, D. Preston, C. J. McAdam, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2022, 61, e202201700 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- T. Y. Kim, R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Eur. J., 2018, 24, 14878–14890 CrossRef CAS PubMed.
- H. Walba and R. W. Isensee, J. Org. Chem., 1961, 26, 2789–2791 CrossRef CAS.
- T. M. Krygowski, H. Szatyłowicz and J. E. Zachara, J. Org. Chem., 2005, 70, 8859–8865 CrossRef CAS.
- T. R. Chan and V. V. Fokin, QSAR Comb. Sci., 2007, 26, 1274–1279 CrossRef CAS.
- T. Zhao, V. M. Lynch and J. L. Sessler, Org. Biomol. Chem., 2022, 20, 980–983 RSC.
- Y. Qu, X. Du, K. Cheng, Y. Zang, L. Xu, K.-i. Shinohara, M. Teraguchi, T. Kaneko and T. Aoki, ACS Mater. Lett., 2020, 2, 1121–1128 CrossRef CAS.
- SPARTAN'18, Wavefunction, Inc Search PubMed.
- We presume that the minor species observed the 1H NMR spectra is a different linkage isomer of [Pt(L)4](BF4)2.
- Repeating the reaction under the same conditions but using CD3CN as the solvent results in a very similar outcome, with a mixture of products obtained even after seven days (ESI†).
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- R. A. S. Vasdev, J. A. Findlay, A. L. Garden and J. D. Crowley, Chem. Commun., 2019, 55, 7506–7509 RSC.
- V. Marti-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef CAS.
- N. Kishi, Z. Li, Y. Sei, M. Akita, K. Yoza, J. S. Siegel and M. Yoshizawa, Chem.–Eur. J., 2013, 19, 6313–6320 CrossRef CAS PubMed.
- Y.-H. Li, Y. Zhang, Y.-M. Legrand, A. van der Lee, J.-J. Jiang, C.-X. Chen, C.-Y. Su and M. Barboiu, Dalton Trans., 2017, 46, 15204–15207 RSC.
- D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424–18428 CrossRef CAS PubMed.
- H. Yu, J. Li, C. Shan, T. Lu, X. Jiang, J. Shi, L. Wojtas, H. Zhang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 26523–26527 CrossRef CAS PubMed.
- J. E. M. Lewis, Chem.–Eur. J., 2021, 27, 4454–4460 CrossRef CAS PubMed.
- D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed.
- R. A. S. Vasdev, J. A. Findlay, A. L. Garden and J. D. Crowley, Chem. Commun., 2019, 55, 7506–7509 RSC.
- I. Regeni, B. Chen, M. Frank, A. Baksi, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2021, 60, 5673–5678 CrossRef CAS PubMed.
- R. J. Li, J. Tessarolo, H. Lee and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3865–3873 CrossRef CAS PubMed.
- R.-J. Li, J. J. Holstein, W. G. Hiller, J. Andréasson and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 2097–2103 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef CAS PubMed.
- K. Yazaki, Y. Sei, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2016, 22, 17557–17561 CrossRef CAS PubMed.
- E. G. Sheetz, Z. Zhang, A. Marogil, M. Che, M. Pink, V. Carta, K. Raghavachari and A. H. Flood, Chem.–Eur. J., 2022, 28, e202201584 CrossRef CAS PubMed.
- A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS PubMed.
- S. Sudan, F. Fadaei-Tirani, R. Scopelliti, K. E. Ebbert, G. H. Clever and K. Severin, Angew. Chem., Int. Ed., 2022, 61, e202201823 CrossRef CAS PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- BindFit, https://supramolecular.org/ Search PubMed.
- J. E. M. Lewis and J. D. Crowley, Supramol. Chem., 2014, 26, 173–181 CrossRef CAS.
- D. Preston, K. M. Patil, A. T. O'Neil, R. A. S. Vasdev, J. A. Kitchen and P. E. Kruger, Inorg. Chem. Front., 2020, 7, 2990–3001 RSC.
- J. J. Henkelis, J. Fisher, S. L. Warriner and M. J. Hardie, Chem.–Eur. J., 2014, 20, 4117–4125 CrossRef CAS PubMed.
- L. S. Lisboa, Synthesis of Heterometallic Supramolecular Cages, Thesis, Doctor of Philosophy, University of Otago, 2022, Retrieved from http://hdl.handle.net/10523/12653 Search PubMed.
- L. S. Lisboa, M. Riisom, H. J. Dunne, D. Preston, S. M. F. Jamieson, L. J. Wright, C. G. Hartinger and J. D. Crowley, Dalton Trans., 2022, 51, 18438–18445 RSC.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- J. Wang, T. A. Young, F. Duarte and P. J. Lusby, J. Am. Chem. Soc., 2020, 142(41), 17743–17750 CrossRef CAS.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang and Q. Xu, Coord. Chem. Rev., 2021, 430, 213656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2175042–2175046. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01354e |
| This journal is © The Royal Society of Chemistry 2023 |