
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurved graphene nanoribbons derived from tetrahydropyrene-based polyphenylenes via one-pot K-region oxidation and Scholl cyclization†
Sebastian
Obermann
 a,
Wenhao
Zheng
b,
Jason
Melidonie
a,
Steffen
Böckmann
c,
Silvio
Osella
d,
Nicolás
Arisnabarreta
e,
L. Andrés
Guerrero-León
a,
Felix
Hennersdorf
f,
David
Beljonne
g,
Jan J.
Weigand
f,
Mischa
Bonn
b,
Steven
De Feyter
e,
Michael Ryan
Hansen
c,
Hai I.
Wang
b,
Ji
Ma
*ah and
Xinliang
Feng
*ah
a,
Wenhao
Zheng
b,
Jason
Melidonie
a,
Steffen
Böckmann
c,
Silvio
Osella
d,
Nicolás
Arisnabarreta
e,
L. Andrés
Guerrero-León
a,
Felix
Hennersdorf
f,
David
Beljonne
g,
Jan J.
Weigand
f,
Mischa
Bonn
b,
Steven
De Feyter
e,
Michael Ryan
Hansen
c,
Hai I.
Wang
b,
Ji
Ma
*ah and
Xinliang
Feng
*ah
aCenter for Advancing Electronics Dresden (cfaed), Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, D-01069 Dresden, Germany. E-mail: xinliang.feng@tu-dresden.de
bMax-Planck-Institute for Polymer Research, D-55128 Mainz, Germany
cInstitute of Physical Chemistry, Westfählische Wilhelms-Universität (WWU) Münster, D-48149 Münster, Germany
dChemical and Biological Systems Simulation Lab, Centre of New Technologies University of Warsaw, Banacha 2C, Warsaw 02-097, Poland
eDepartment of Chemistry, Division of Molecular Imaging and Photonics, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
fChair of Inorganic Molecular Chemistry, Technische Universität Dresden, Dresden, Germany
gLaboratory for Chemistry of Novel Materials, Materials Research Institute, University of Mons, Mons 7000, Belgium
hMax Planck Institute of Microstructure Physics, Weinberg 2, 06120 Halle, Germany. E-mail: ji.ma@mpi-halle.mpg.de
First published on 27th July 2023
Abstract
Precise synthesis of graphene nanoribbons (GNRs) is of great interest to chemists and materials scientists because of their unique opto-electronic properties and potential applications in carbon-based nanoelectronics and spintronics. In addition to the tunable edge structure and width, introducing curvature in GNRs is a powerful structural feature for their chemi-physical property modification. Here, we report an efficient solution synthesis of the first pyrene-based GNR (PyGNR) with curved geometry via one-pot K-region oxidation and Scholl cyclization of its corresponding well-soluble tetrahydropyrene-based polyphenylene precursor. The efficient A2B2-type Suzuki polymerization and subsequent Scholl reaction furnishes up to ∼35 nm long curved GNRs bearing cove- and armchair-edges. The construction of model compound 1, as a cutout of PyGNR, from a tetrahydropyrene-based oligophenylene precursor proves the concept and efficiency of the one-pot K-region oxidation and Scholl cyclization, which is clearly revealed by single crystal X-ray diffraction analysis. The structure and optical properties of PyGNR are investigated by Raman, FT-IR, solid-state NMR, STM and UV-Vis analysis with the support of DFT calculations. PyGNR exhibits a narrow optical bandgap of ∼1.4 eV derived from a Tauc plot, qualifying as a low-bandgap GNR. Moreover, THz spectroscopy on PyGNR estimates its macroscopic charge mobility μ as ∼3.6 cm2 V−1 s−1, outperforming several other curved GNRs reported via conventional Scholl reaction.
Introduction
Structurally well-defined graphene nanoribbons (GNRs) are constantly developing materials, amassing more and more attention due to their unique opto-electronic properties1–3 and potential to be integrated into future nanoelectronics and spintronics.3–9 The physicochemical properties of atomically precise GNRs highly depend on their edge structures and widths.6,7,10–13 In the well-investigated armchair-edged GNRs (AGNR), three different families of ribbons can be distinguished as a function of carbon atoms along the ribbon width, exhibiting semiconducting properties.6,14–21 On the other hand, zigzag-edged GNRs (ZGNR) possess edge states and metallic character, but could only be synthesized on surfaces to date, drastically limiting their applicability.2,6,22–24 In stark contrast to largely uncontrolled top-down methods, the mentioned examples were achieved by bottom-up synthesis, an approach also rendering mixed edges possible. For instance, mixing armchair edge with zigzag edge can create intriguing physical phenomena, as recently exemplified by the topological quantum phase in GNRs.7,25–27 Aside from the edge type, curvature (or non-planarity) is another powerful structural feature for tuning the properties of GNRs,28,29 such as liquid-phase dispersibility and charge transport behavior.30–32 A common synthetic pathway to achieve curvature in GNRs is to introduce the structural motif of small [n]helicene units on its periphery, such as [4]helicene or [5]helicene,33–35 creating the cove edge12,36–38 or fjord edge39 respectively. For example, the regular incorporation of cove subunits by elimination of a single carbon atom along the opposite ZGNR edges can produce a series of curved GNRs with periodic cove-zigzag edge structure, providing GNRs with well-tunable band structures and effective masses (Fig. 1a).11 In the context of this structural modification of ZGNRs towards cove-edge topologies, it raises the synthetic question if cove-edges can also be accessed from the parent design of AGNRs (Fig. 1b).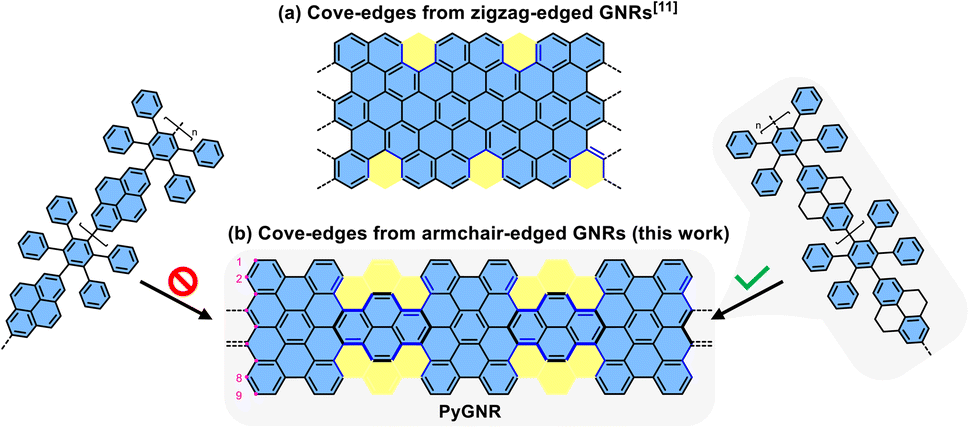 | ||
| Fig. 1 (a) One example of curved GNRs with periodic cove-zigzag edges; (b) curved GNRs obtained by incorporation of cove units along the 9-AGNR edges, reported in this work. | ||
Pyrene (2) is one of the widely used building blocks in material science.40 Its biphenyl-like structure with two double bond characteristics, called the K-region (Scheme 1), distinguishes pyrene from other polycyclic aromatic hydrocarbons (PAHs).40,41 Moreover, its full-zigzag edge structure renders it a versatile motif for constructing highly conjugated π-systems with tailored edge topologies and functions. Although it is possible to functionalize pyrene on every position by various synthetic methods,42–44 its incorporation into large PAHs (or nanographenes) via Scholl reaction of pyrene-based dendritic oligophenylene precursors is often unsuccessful due to its high reactivity and rich electron density.45,46 Therefore, the expansion of pyrene-based precursors toward the synthesis of GNRs remains a synthetic challenge.
 | ||
| Scheme 1 Reoxidation of 4,5,9,10-tetrahydropyrene (3) and synthetic route to model compound 1. Reagents and conditions: (a) (i) RANEY®-nickel, EtOAc, rt, 48 h, (ii) Pd/C (10%), EtOAc, H2, 60 °C, 24 h; (b) FeCl3 (10 eq.), MeNO2, CH2Cl2, rt, 5 min; (c) (BPin)2, 4,4′-di-tert-butyl-2,2′-bipyridine, [Ir(OMe)(COD)]2, THF, 80 °C, overnight; (d) 5, 6, Pd(dppf)Cl2·CH2Cl2, K3PO4, PhMe/H2O, 115 °C, overnight; (e) (i) SnCl2, EtOH/EtOAc, reflux, overnight, (ii) NBS, CHCl3, rt, 10 min; (f) 6, Pd(dppf)Cl2, K3PO4, PhMe/H2O, 115 °C, 48 h; (g) CHBr3, isoamylnitrite, 100 °C, overnight; (h) Pd(PPh3)4, K2CO3, PhMe/H2O, 115 °C, 48 h; (i) DDQ, MeSO3H, CH2Cl2, rt, 30 min, then DDQ, TfOH, rt, 3 min. | ||
Herein, we demonstrate an efficient solution synthesis of a novel curved pyrene-based GNR (PyGNR) bearing cove- and armchair-edges from the predesigned tetrahydropyrene-based polyphenylene precursor P1. It is worth mentioning that when trying to obtain PyGNR from pyrene-embedded polyphenylene directly (Fig. 1b), only short and insoluble oligomers could be obtained due to the poor solubility of the pyrene bearing polymers. To overcome this obstacle, the unsaturated polymer P1 is first obtained by A2B2-Suzuki polymerization of 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4,5,9,10-tetrahydro-pyrene (4) and alkyl-chain equipped tetraphenyl-substituted 1,4-diiodobenzene (12). Subsequently, one-pot oxidation of the tetrahydropyrene K-regions towards pyrene moieties together with the Scholl reaction, enables the desired formation of PyGNR. The resultant PyGNR reaches a length of up to ∼35 nm according to the gel-permeation chromatography (GPC) analysis of P1. As a cutout of PyGNR, model compound 1 was successfully synthesized to evaluate the efficiency and feasibility of the one-pot cyclodehydrogenation process. The structure and optical properties of PyGNR were well investigated by solid-state NMR, FT-IR, Raman, STM imaging and UV-Vis spectroscopy. Due to the unique edge structure, a narrow optical bandgap of ∼1.4 eV was determined by a Tauc-plot (ESI Fig. S2†), slightly below the value of 1.75 eV predicted by DFT. Furthermore, time-resolved terahertz spectroscopy reveals a macroscopic charge mobility of ∼3.6 cm2 V−1 s−1, outperforming several other examples37,39 of curved GNRs obtained by conventional Scholl reactions.
Results and discussion
Synthesis of model compound 1
The synthesis of model compound 1 is illustrated in Scheme 1. Considering the solubility issue of 1, mesitylene side groups were installed instead of the more common tert-butyl groups. First, 4,5,9,10-tetrahydropyrene (3) was obtained from the commercially available pyrene (2) after desulfurization with RANEY®-nickel and hydrogenation under pressure in the presence of Pd/C among over-hydrogenated products in 31% yield (Scheme 1). Then, iridium-catalyzed borylation42 was used to furnish compound 4 in 73% yield. Slightly modifying the synthesis route of Lungerich et al.47 allowed us to synthesize compound 10 starting from 1,2,3-tribromo-5-nitrobenzene (5, see ESI†). A Suzuki coupling reaction between 5 and 6 afforded compound 7 in 69% yield, followed by a two-step sequence of reduction and bromination to furnish compound 8 in 60% yield. Another Suzuki coupling of 8 and 6 gave compound 9 in 77% yield. In a Sandmeyer-type reaction, aniline 9 was brominated to compound 10 with a yield of 47%. Finally, building blocks 4 and 10 were cross-coupled by the Suzuki reaction, providing the oligophenylene precursor 11 in a low yield of 35% due to the steric hindrance of the inner mesitylene units in 11. For the cyclodehydrogenation of 11, we initially examined the classic Scholl conditions by use of FeCl3 or triflic acid with DDQ in CH2Cl2, but failed to obtain a defined product (ESI Chapter 1.2.12†). We then turned to a milder approach using methylsulfonic acid, and DDQ, which furnished a time-stable MALDI-TOF signal 12 proton masses above the desired signal, indicating a partially closed intermediate (ESI Fig. S32†).After adding the stronger triflic acid to the reaction mixture, the bright red compound 1 was obtained in 60% yield after only three minutes of additional reaction time.
The chemical identity of 1 was confirmed by high-resolution mass spectroscopy and NMR spectroscopy, whose key changes for compounds 11 and 1 are illustrated in Fig. 2, respectively. The MALDI-TOF spectra show a mass difference of 28 proton masses. This implies that in addition to the cyclodehydrogenation of 24 protons, 4 additional hydrogens were removed from the tetrahydropyrene moiety to reestablish the pyrene K-region. In addition to the reduction of NMR signals in the case of compound 1, the total amount of protons in the aliphatic region was reduced from 98 to 90 (ESI Fig. S19–S21†), further confirming the successful conversion of the tetrahydropyrene moieties. The anti π–π-stacking properties of the mesitylene side groups enabled NMR spectroscopy of 1 with a sufficient resolution to assign the protons using two-dimensional COSY- and NOESY-NMR spectroscopy (ESI Fig. S22 and S23†). Furthermore, a shift in the downfield due to the aromatic ring currents was observed, especially for the protons a and b (proton labels seen in Scheme 1), resulting from their location on the cove edge.
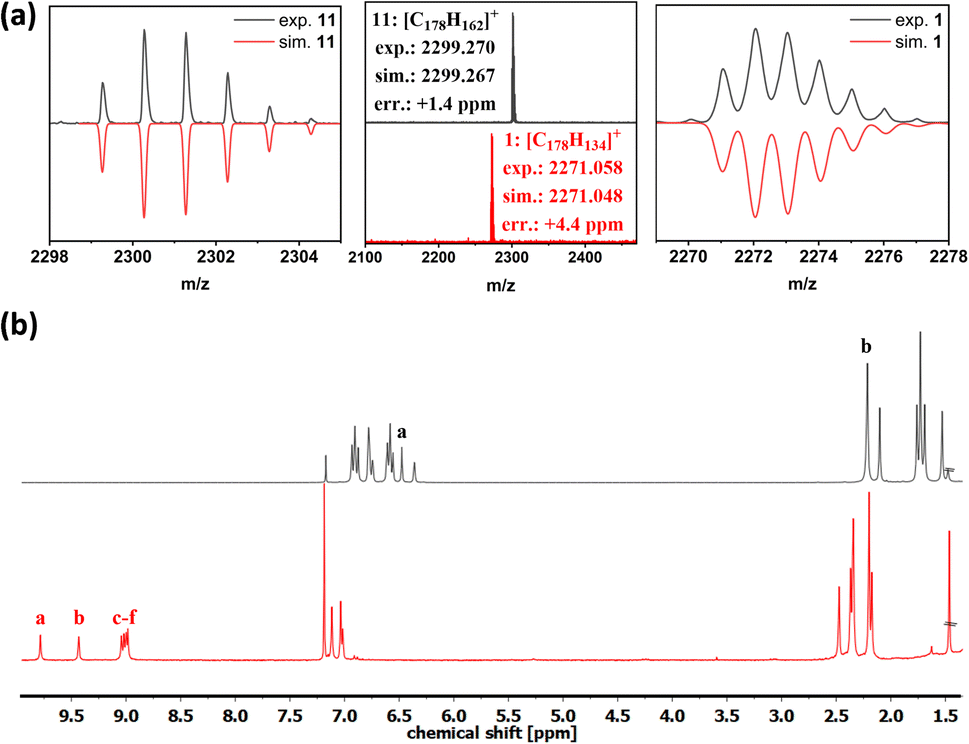 | ||
| Fig. 2 (a) MALDI-TOF-MS of compounds 11 and 1. (b) 1H-NMR spectra of compound 11 (top) and compound 1 (bottom), recorded in CDCl3 at 300 MHz (proton labels see Scheme 1). | ||
Crystallographic analysis of compounds 11 and 1
Sufficiently large crystals for X-ray crystallographic analysis were obtained for 11 and 1, respectively (Fig. 3). In the case of 11, a suitable crystal for single crystal X-ray diffraction analysis (SCXRD) was grown from 11 dissolved in CH2Cl2 by slow evaporation of the solvent. SCXRD reveals an unconjugated phenylic structure combined with twisted sp3-bonds of the central tetrahydropyrene unit. The sp3-carbon single bonds are revealed by the bond lengths of 1.57 Å and 1.58 Å, respectively. Compound 11 crystallizes in the triclinic space group P1. In the case of 1, a single crystal was obtained by slow diffusion of methanol in its CS2-solution. The SCXRD analysis reveals 1 as a curved π-conjugated molecule featuring four [4]helicene subunits. The hexa-peri-hexabenzocoronene (HBC) units have been fully fused and share a pyrene unit in the center, revealing the successful conversion of the tetrahydropyrene moieties back to a fully sp2-hybridized pyrene core and supporting the proposed one-pot K-region oxidation and Scholl cyclization. The cove edges between the pyrene K-region and the HBC moieties possess an average torsion angle of 21°, resulting in curvature of the molecule. In contrast to compound 11, compound 1 crystallizes in the monoclinic space group P21/c. The bond lengths in the K-regions of compound 1 are significantly shorter, about 1.35 Å (1.337 Å and 1.361 Å, respectively), matching with the value for pyrene (2) reported by the literature and indicating double bond character.48 In the solid packing, the crystal layers of 1 are slightly slipped with the π-surfaces facing towards each other, forming π–π-stacks (Fig. 3c). | ||
| Fig. 3 X-ray crystallographic structure (ORTEP drawing at the 50% probability level) of (a) 11 and (b) 1 (top and side view). (c) Crystal-packing of 1. Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
Synthesis of PyGNR
After confirming the model reaction formed the desired compound 1, we further extended the synthesis towards PyGNR based on tetrahydropyrene monomer 4. Early attempts of this A2B2 Suzuki polymerization with 2,7-diborylated pyrene gave only oligomers according to linear mode MALDI-TOF MS measurements, which remained insoluble and, therefore, unavailable for further solution processing (ESI Fig. S34†). When these oligomers were exposed to Scholl-reaction conditions as a suspension, partial cyclization and decomposition were observed. In consequence, the 4,5,9,10-tetrahydropyrene containing polymer P1 (Scheme 2) was designed. The monomer 12,15 substituted with 3,7-dimethyloctyl chains, was chosen as the coupling partner for the diborylated pyrene derivative 4 to increase the processibility of the GNR later. In contrast to pyrene (2), when switching to the tetrahydropyrene derivative 4, soluble polymers were obtained when refluxing the monomers, Pd(PPh3)4, K2CO3 in toluene and H2O for 48 hours. Linear mode MALDI-TOF MS of the crude material revealed groups of signals up to m/z ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, spaced by the repetition unit of 1145 g mol−1 (ESI Fig. S33†). After washing with HCl/MeOH, filtration and subsequent dissolution and filtration in chloroform, the crude polymer P1 was obtained after removing the chloroform in vacuo. P1 was then divided into 4 fractions by recycling GPC and further analyzed by analytical GPC against polystyrene standards. Our analysis reveals an averaged molar mass of Mn ∼ 24000 Da and a narrow polydispersity index (PDI) of 1.2 for the heaviest (10 wt%) crude polymers (ESI Fig. S36–S38†), significantly enhancing the molecular weight compared to other GNRs reported via the Suzuki polymerization method.15,39 The protons could be assigned to their positions in P1 by 1H-NMR spectroscopy (ESI Fig. S30†). Due to P1's excellent solubility, even 13C spectra could be recorded effortlessly (ESI Fig. S31†). Finally, the Scholl reaction of the first fraction of P1 with FeCl3 (7 equiv./H) as the Lewis acid and oxidant in CH2Cl2 for 7 days yielded the PyGNR. The resultant PyGNR possesses combined cove and armchair edges with an estimated average length of ∼35 nm based on the Mn of the corresponding P1.
000, spaced by the repetition unit of 1145 g mol−1 (ESI Fig. S33†). After washing with HCl/MeOH, filtration and subsequent dissolution and filtration in chloroform, the crude polymer P1 was obtained after removing the chloroform in vacuo. P1 was then divided into 4 fractions by recycling GPC and further analyzed by analytical GPC against polystyrene standards. Our analysis reveals an averaged molar mass of Mn ∼ 24000 Da and a narrow polydispersity index (PDI) of 1.2 for the heaviest (10 wt%) crude polymers (ESI Fig. S36–S38†), significantly enhancing the molecular weight compared to other GNRs reported via the Suzuki polymerization method.15,39 The protons could be assigned to their positions in P1 by 1H-NMR spectroscopy (ESI Fig. S30†). Due to P1's excellent solubility, even 13C spectra could be recorded effortlessly (ESI Fig. S31†). Finally, the Scholl reaction of the first fraction of P1 with FeCl3 (7 equiv./H) as the Lewis acid and oxidant in CH2Cl2 for 7 days yielded the PyGNR. The resultant PyGNR possesses combined cove and armchair edges with an estimated average length of ∼35 nm based on the Mn of the corresponding P1.
 | ||
| Scheme 2 Synthetic route to PyGNR. Reagents and conditions: (a) 4 (stoichiometric), 12, Pd(PPh3)4, K2CO3, Aliquat 336, PhMe/H2O, 110 °C, 48 h; (b) FeCl3, MeNO2, CH2Cl2, rt, 7 d. Inset is the optimized geometry of PyGNR, aliphatic side chains omitted for clarity. | ||
Structural characterizations of PyGNR
The structures of precursor P1 and PyGNR were then characterized by a combination of solid-state 1H and 13C{1H} MAS NMR (1H decoupled 13C magic angle spinning NMR) at a high spinning speed of 62.5 kHz (Fig. 4a and b), FT-IR and Raman (Fig. 4c and d). While the precursor P1 shows well-defined signals in the 1H MAS NMR spectrum (Fig. 4a) for both the aromatic and aliphatic protons centered around 0.9 ppm and 6.6 ppm, respectively, broader lines are observed for PyGNR along with a downfield shift of the aromatic protons to 8.9 ppm. This can be attributed to a more rigid structure and extended π-conjugation of PyGNR compared to P1. Deconvolution of the quantitative 1H MAS NMR spectra shows a lower relative content of aromatic protons in PyGNR compared to P1 as a result of the graphitization and matches the expected relative intensities within the expected error margins. The graphitization process can also be confirmed by the 13C{1H} MAS NMR spectra that were acquired with a short recycle delay of 3.5 s (Fig. 4b). Under the chosen experimental conditions, the 13C signal of the side chains only shows low intensity. In addition, the structural evolution from P1 to PyGNR is also supported by 2D solid-state NMR experiments (see ESI Chapter 9†).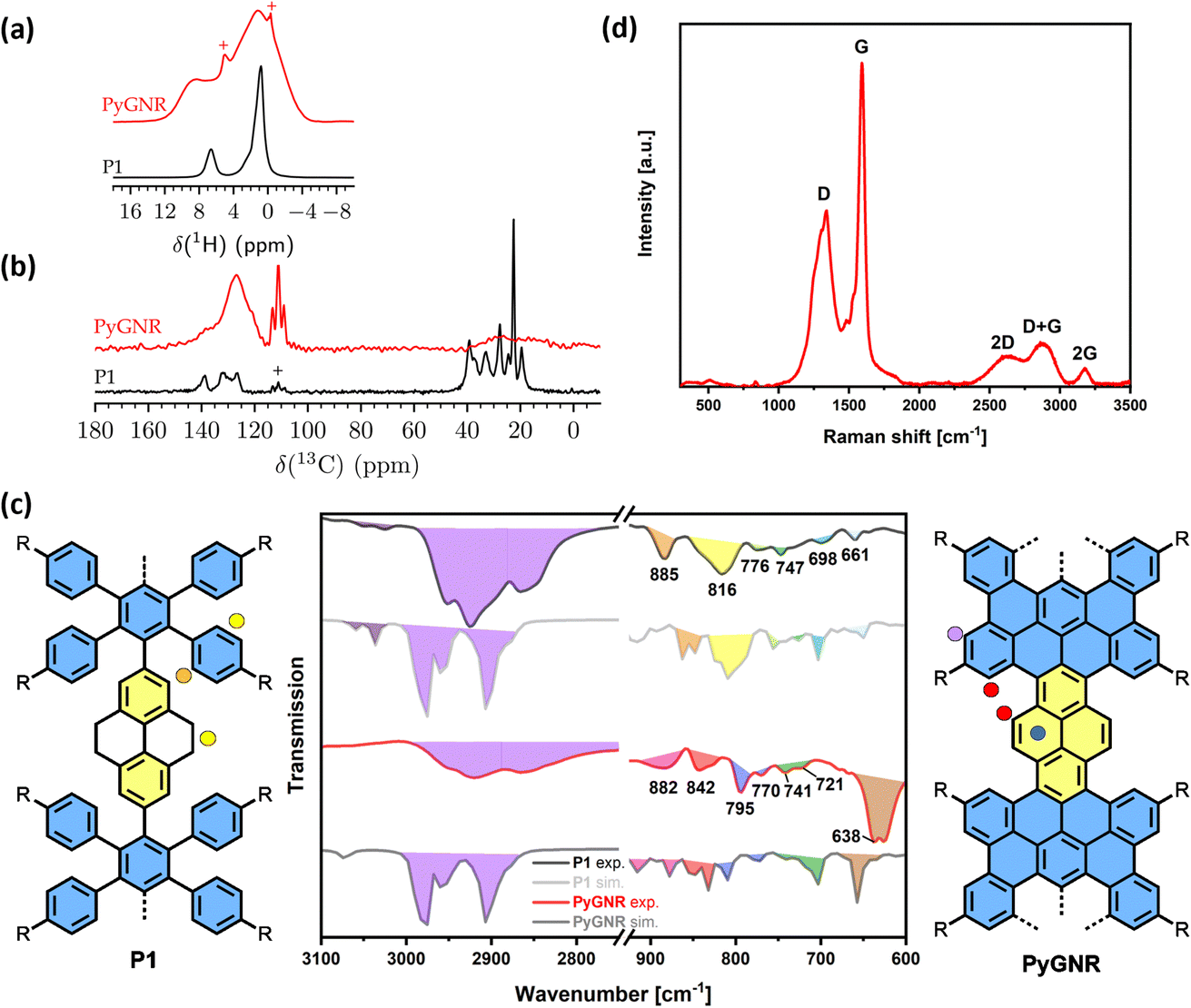 | ||
| Fig. 4 (a) Quantitative 1H MAS NMR spectra of P1 and PyGNR with 64 s recycle delay. Signals of remaining solvent are marked with +; (b) 13C{1H} MAS NMR spectra with a short recycle delay of 3.5 s and 25 kHz of low-power CW decoupling. The signal of PTFE spacer material is marked with +. All spectra were acquired at a static field of 11.74 T and a spinning frequency of 62.5 kHz. (c) Measured and calculated (at the DFT HSE/6-31G(d) level) FT-IR spectra of P1 and PyGNR. In the fingerprint region (900–600 cm−1), the calculated IR intensity was scaled by a factor 10; (d) Raman spectra of PyGNR, recorded at 514 nm. | ||
The FT-IR spectra of P1 (Fig. 4c) exhibit typical vibrational modes for its structural elements. A more detailed discussion can be found in Chapter 10 of the ESI.† To assist in the identification of the corresponding vibrations, DFT calculations on the HSE/6-31G(d) level of theory were performed. The peak at 885 cm−1, which was identified as the SOLO-mode (CH-wagging of an aromatic proton without adjacent protons, orange)49 of the tetrahydropyrene moiety, confirms once more the presence of unsaturated bonds in P1. The FT-IR spectrum of PyGNR was then compared to P1. The abundant aromatic CH-stretchings in the range of 3020–3060 cm−1 were diminished in the graphitized ribbon due to the cyclodehydrogenation. The CH-stretchings from the aliphatic side chains from 2750 to 3000 cm−1 were retained. Further, characteristic vibrations were investigated in the fingerprint region and identified by comparison to the HSE/6-31G(d) prediction. First, the SOLO-modes can be found around 882 cm−1 (pink), a further indication of successful graphitization. Interestingly, PyGNRs CH-wagging of the cove SOLO-mode and pyrene DUO moieties (CH-wagging of two adjacent aromatic protons) can be found in a joint vibration mode around 840 cm−1 (red), demonstrating the successful conversion of the tetrahydro-moieties into pyrene K-regions. This claim is further supported by the ring vibrations of the pyrene K-region, which can be attributed to the newly emerged signals around 790–770 cm−1 (blue), absent in P1. Last, the broad signal at 640 cm−1 (brown) provides another strong indication of successful graphitization, as the DFT-prediction assigns these modes as in plane aromatic CC-scaffold vibrations. The Raman spectrum of PyGNR (Fig. 4d) shows two strong D and G bands at ∼1340 and ∼1590 cm−1. Furthermore, double resonance peaks can be found at ∼2605, ∼2880 and ∼3170 cm−1, which can be assigned as the 2D, D + G and 2G bands. The peak positions are in line with other bottom-up solution-synthesized GNRs reported in the literature.13,21,37 As expected from the low twist in the cove regions observed from the crystal structure of compound 1 (21°), PyGNR shows only mediocre dispersibility and processability in polar organic solvents. However, imaging techniques such as STM vastly favor planar structures and usually fail to yield results for strongly curved geometries. Fig. 5 shows the results obtained by the STM characterization of PyGNR at the HOPG/heptanoic acid interface. Large scale (Fig. 5a) and high resolution (Fig. 5b) STM images show the presence of a clear stripe patterned structure observed in isolated areas of the HOPG surface. Line profile analysis performed in graphite calibrated STM images (Fig. 5c) confirms the presence of parallel rows comprising a periodicity of 3.3 (±0.2) nm which matches the expected periodicity from molecular modelling (Fig. 5d). This distance indicates that PyGNR is adsorbed without alkyl chain interdigitation, but instead, next to one another50 as depicted in Fig. 5d. The stability obtained during the STM measurements suggests high on-surface adsorption energies due to strong vdW-interactions between the PyGNR core and the graphitic substrate. This allowed the acquisition of multiple STM images (see ESI Fig. S51†).
 | ||
| Fig. 5 Large scale (a) and high resolution (b) STM images of PyGNR self-assembled at the heptanoic acid/HOPG interface; (c) graphite-calibrated representative line-profile measured along the red arrow direction in STM image of panel (a); (d) molecular model of PyGNR showing packing without alkyl chain interdigitation. Ubias = −0.4 V and Iset = 0.05 nA. | ||
In contrast, the low twist of PyGNR seems to be sufficient to resolve the π–π-interactions observed in planar GNRs,15 as additional investigations by TEM, SEM and powder XRD revealed an amorphous structure of the material (see ESI Chapters 11–13†). Subsequent to SEM imaging, EDX confirmed the complete removal of residual iron ions (see ESI Chapter 11†).
Optical property and THz spectroscopy study of PyGNR
The UV-Vis absorption spectra were recorded in CHCl3 solutions. An NMP dispersion of PyGNR that was allowed to settle overnight was used (Fig. 6a). Due to its lack of conjugation, polymer P1 (dark grey) possesses only high energy absorption bands starting with a first peak at 338 nm, which agrees well with the polymer fragment 11 (light gray), exhibiting peaks at similar wavelengths (326 nm and 267 nm). Significantly red-shifted to 559 nm due to its full conjugation, the first absorption of 1 (blue) can be found. Two distinct absorption peaks of 1 are visible at 437 nm and 367 nm. | ||
| Fig. 6 (a) Absorption spectra of 1, 11, P1 (CHCl3), and PyGNR (NMP) as well as DFT-predicted 1 and PyGNR; (b) time-resolved terahertz photoconductivity of PyGNR following photoexcitation. The absorbed photon fluence is ∼1 mJ cm−2. (c) The frequency-resolved THz complex conductivity measured ∼1.2 ps after photoexcitation. The solid lines are fits to the Drude–Smith model. | ||
Compared to its short fragment 1, the absorption spectrum of PyGNR (purple) is further red-shifted and displays an absorption peak with a maximum at ∼680 nm as well as a shoulder at 485 nm, showing strong proof for graphitization compared to the high energy absorptions of P1. The optical bandgap was determined as 1.4 eV by a Tauc-plot (ESI Fig. S2†), qualifying as a low bandgap GNR. The experimental result is in good agreement with the DFT-prediction of 1.75 eV. In addition, the simulated TD-DFT curves for 1 (light blue) and PyGNR (light purple) match well with the experimental spectra.
To investigate the charge transport properties of PyGNR, we conducted optical pump-THz probe (OPTP) spectroscopy measurements (see the Experimental details in the ESI Chapter 8†).51,52Fig. 6b shows the time-resolved complex photoconductivity of PyGNR dispersed in 1,2,4-trichlorobenzene, following above-gap optical excitation (here, 1.55 eV laser pulses). The rapid sub-picosecond rise of the real photoconductivity indicates charge carrier generation, and the following rapid decay can be assigned to the carrier trapping and/or the formation of bound electron–hole states (i.e., excitons) due to the strong quantum confinement and the reduced dielectric screening effect.52,53 The exciton formation is featured by a large imaginary conductivity and a small real conductivity at the later time scale. To further study the free charge carriers' transport properties, we measured the conductivity spectrum around the peak photoconductivity (∼1.2 ps after photoexcitation), as shown in Fig. 6c (for Experimental details, see ESI Chapter 8†). The phenomenological Drude–Smith (DS) model was applied to describe the frequency-resolved complex conductivity (for details, see ESI†).54,55 The DS model considers the charge scattering occurring via preferential backscattering due to conjugation or structural deformation in materials. A parameter c is introduced to characterize the backscattering probability, ranging from 0 (isotropic scattering) to −1 (100% backscattering). From the results, we obtained an effective charge scattering time τ = 38 ± 1 fs and c = −0.970 ± 0.003. Furthermore, by considering a reduced charge carrier effective mass m* = 0.55m0 from DFT (considering contributions from both electrons and holes; see the ESI† for details), we estimate the macroscopic charge mobility  to be ∼3.6 cm2 V−1 s−1, outperforming similar examples of curved GNRs in the recent literature, such as hybrid cove- and gulf-edged GNRs,37 and a 9AGNR based fjord-edged GNR.39
to be ∼3.6 cm2 V−1 s−1, outperforming similar examples of curved GNRs in the recent literature, such as hybrid cove- and gulf-edged GNRs,37 and a 9AGNR based fjord-edged GNR.39
Conclusion
In summary, we demonstrate the efficient synthesis of the first curved pyrene-based GNR (PyGNR) by introducing cove units along 9-AGNR edges. The synthetic strategy to obtain PyGNR was carried out in a previously unreported strategy, regenerating the K-regions of the pyrene moieties in one step during the cyclodehydrogenation of a well-soluble unsaturated polyphenylene precursor P1. Proving the structural concept of the ribbon and the proposed one-pot oxidation, model compound 1 was also synthesized. The obtained PyGNR was fully characterized by solid-state NMR, FT-IR, Raman, STM and UV-Vis spectroscopy, also supported by the DFT spectra simulations. THz study of PyGNR revealed a macroscopic charge mobility of ∼3.6 cm2 V−1 s−1, outperforming similar GNRs reported by conventional Scholl-cyclizations.37,39 Furthermore, PyGNR is the first GNR with accessible pyrene K-regions, rendering further functionalization, e.g., by oxidation to diketones, possible.43 Strategic use of tetrahydro-derivatives demonstrates the feasibility of obtaining novel GNRs in one step from sp3-C containing polymer precursors and opens up possibilities for other GNRs suffering from polymer solubility issues or dehydrogenation problems.Data availability
Experimental and computational data can be found in the ESI.† The cif-files of the crystal structures can be found alongside the ESI.†Author contributions
J. Ma and X. Feng supervised the project. S. Obermann, J. Melidonie and L. A. Guerrero-León synthesized and characterized the compounds. W. Zheng, M. Bonn and H. I. Wang measured and interpreted the THz measurements. S. Böckmann and M. R. Hansen measured and interpreted the solid-state NMR. S. Osella and D. Beljonne performed the DFT calculations. N. Arisnabarreta and S. De Feyter carried out and analyzed the solid-liquid STM imaging. F. Hennersdorf and J. J. Weig and measured and refined the single crystal structures. S. Obermann, J. Ma and X. Feng co-wrote the paper with contributions from all co-authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was financially supported by the EU Graphene Flagship (Graphene Core 3, 881603), ERC Consolidator Grant (T2DCP, 819698), H2020-MSCA-ITN (ULTIMATE, No. 813036), the Center for Advancing Electronics Dresden (cfaed), H2020-EU.1.2.2.-FET Proactive Grant (LIGHT-CAP, 101017821), the DFG-SNSF Joint Switzerland-German Research Project (EnhanTopo, No. 429265950) and the Research Foundation–Flanders (FWO) (grant G.0A41.20N, 12ZS623N), KU Leuven Internal Funds (grants C14/18/06 and C14/19/079). The authors gratefully acknowledge the GWK support for funding this project by providing computing time through the Center for Information Services and HPC (ZIH) at TU Dresden. The computational resources in Mons are supported by the FNRS “Consortium des Equipments de Calcul Intensif–CECI” program Grant No. 2.5020.11 and by the Walloon Region (ZENOBE Tier-1 supercomputer, under grant 1117545). D. B. is a research director at Belgian F.R.S./FNRS. We also thank the Polish National Science Centre for funding (grant no. UMO-2020-39-I-ST4-01446). The computation was carried out with the support of the Interdisciplinary Center for Mathematical and Computational Modeling at the University of Warsaw (ICM UW) under grants no. (G83-28 and GB80-24). The authors also express their gratitude to Markus Göbel for the Raman measurement, Frank Drescher for the ESI-measurements, Enrique Caldera Cruz for the analytical GPC measurements and Tobias Nickel for the MALDI-TOF measurements. N. A. acknowledges K. S. Mali for fruitful discussions.References
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS PubMed.
- P. Ruffieux, S. Wang, B. Yang, C. Sanchez-Sanchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489–492 CrossRef CAS PubMed.
- W. Niu, J. Ma, P. Soltani, W. Zheng, F. Liu, A. A. Popov, J. J. Weigand, H. Komber, E. Poliani, C. Casiraghi, J. Droste, M. R. Hansen, S. Osella, D. Beljonne, M. Bonn, H. I. Wang, X. Feng, J. Liu and Y. Mai, J. Am. Chem. Soc., 2020, 142, 18293–18298 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1231 CrossRef CAS.
- V. Saraswat, R. M. Jacobberger and M. S. Arnold, ACS Nano, 2021, 15, 3674–3708 CrossRef CAS PubMed.
- O. v. Yazyev, Acc. Chem. Res., 2013, 46, 2319–2328 CrossRef CAS PubMed.
- D. J. Rizzo, G. Veber, T. Cao, C. Bronner, T. Chen, F. Zhao, H. Rodriguez, S. G. Louie, M. F. Crommie and F. R. Fischer, Nature, 2018, 560, 204–208 CrossRef CAS PubMed.
- Y. Huang, W. T. Dou, F. Xu, H. B. Ru, Q. Gong, D. Wu, D. Yan, H. Tian, X. P. He, Y. Mai and X. Feng, Angew. Chem., Int. Ed., 2018, 57, 3366–3371 CrossRef CAS PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- Y. F. Zhang, Y. Zhang, G. Li, J. Lu, Y. Que, H. Chen, R. Berger, X. Feng, K. Müllen, X. Lin, Y. Y. Zhang, S. Du, S. T. Pantelides and H. J. Gao, Nano Res., 2017, 10, 3377–3384 CrossRef CAS.
- X. Wang, J. Ma, W. Zheng, S. Osella, N. Arisnabarreta, J. Droste, G. Serra, O. Ivasenko, A. Lucotti, D. Beljonne, M. Bonn, X. Liu, M. R. Hansen, M. Tommasini, S. De Feyter, J. Liu, H. I. Wang and X. Feng, J. Am. Chem. Soc., 2022, 144, 228–235 CrossRef CAS PubMed.
- J. Liu, B. W. Li, Y. Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097–6103 CrossRef CAS PubMed.
- M. G. Schwab, A. Narita, Y. Hernandez, T. Balandina, K. S. Mali, S. de Feyter, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 18169–18172 CrossRef CAS.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS PubMed.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- R. S. Jordan, Y. L. Li, C. W. Lin, R. D. McCurdy, J. B. Lin, J. L. Brosmer, K. L. Marsh, S. I. Khan, K. N. Houk, R. B. Kaner and Y. Rubin, J. Am. Chem. Soc., 2017, 139, 15878–15890 CrossRef CAS PubMed.
- L. Talirz, H. Söde, T. Dumslaff, S. Wang, J. R. Sanchez-Valencia, J. Liu, P. Shinde, C. A. Pignedoli, L. Liang, V. Meunier, N. C. Plumb, M. Shi, X. Feng, A. Narita, K. Müllen, R. Fasel and P. Ruffieux, ACS Nano, 2017, 11, 1380–1388 CrossRef CAS PubMed.
- K. Sun, P. Ji, J. Zhang, J. Wang, X. Li, X. Xu, H. Zhang and L. Chi, Small, 2019, 15, 1804526 CrossRef.
- K. Y. Yoon and G. Dong, Mater. Chem. Front., 2020, 4, 29–45 RSC.
- G. Li, K. Y. Yoon, X. Zhong, J. Wang, R. Zhang, J. R. Guest, J. Wen, X. Y. Zhu and G. Dong, Nat. Commun., 2018, 9, 1687 CrossRef PubMed.
- S. Wang, L. Talirz, C. A. Pignedoli, X. Feng, K. Müllen, R. Fasel and P. Ruffieux, Nat. Commun., 2016, 7, 11507 CrossRef CAS PubMed.
- P. P. Shinde, J. Liu, T. Dienel, O. Gröning, T. Dumslaff, M. Mühlinghaus, A. Narita, K. Müllen, C. A. Pignedoli, R. Fasel, P. Ruffieux and D. Passerone, Carbon, 2021, 175, 50–59 CrossRef CAS.
- P. P. Shinde, O. Gröning, S. Wang, P. Ruffieux, C. A. Pignedoli, R. Fasel and D. Passerone, Carbon, 2017, 124, 123–132 CrossRef CAS.
- Y. L. Lee, F. Zhao, T. Cao, J. Ihm and S. G. Louie, Nano Lett., 2018, 18, 7247–7253 CrossRef CAS PubMed.
- T. Cao, F. Zhao and S. G. Louie, Phys. Rev. Lett., 2017, 119, 076401 CrossRef PubMed.
- O. Gröning, S. Wang, X. Yao, C. A. Pignedoli, G. Borin Barin, C. Daniels, A. Cupo, V. Meunier, X. Feng, A. Narita, K. Müllen, P. Ruffieux and R. Fasel, Nature, 2018, 560, 209–213 CrossRef PubMed.
- Y. Hu, P. Xie, M. de Corato, A. Ruini, S. Zhao, F. Meggendorfer, L. A. Straasø, L. Rondin, P. Simon, J. Li, J. J. Finley, M. R. Hansen, J. S. Lauret, E. Molinari, X. Feng, J. v. Barth, C. A. Palma, D. Prezzi, K. Müllen and A. Narita, J. Am. Chem. Soc., 2018, 140, 7803–7809 CrossRef CAS PubMed.
- W. Niu, J. Ma and X. Feng, Acc. Chem. Res., 2022, 55, 3322–3333 CrossRef CAS PubMed.
- N. J. Schuster, L. A. Joyce, D. W. Paley, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2020, 142, 7066–7074 CrossRef CAS PubMed.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- I. Ivanov, Y. Hu, S. Osella, U. Beser, H. I. Wang, D. Beljonne, A. Narita, K. Müllen, D. Turchinovich and M. Bonn, J. Am. Chem. Soc., 2017, 139, 7982–7988 CrossRef CAS PubMed.
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS.
- T. J. Sisto, Y. Zhong, B. Zhang, M. T. Trinh, K. Miyata, X. Zhong, X. Y. Zhu, M. L. Steigerwald, F. Ng and C. Nuckolls, J. Am. Chem. Soc., 2017, 139, 5648–5651 CrossRef CAS PubMed.
- L. Yang, J. Ma, W. Zheng, S. Osella, J. Droste, H. Komber, K. Liu, S. Böckmann, D. Beljonne, M. R. Hansen, M. Bonn, H. I. Wang, J. Liu and X. Feng, Adv. Sci., 2022, 9, 2200708 CrossRef CAS.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS PubMed.
- X. Yao, W. Zheng, S. Osella, Z. Qiu, S. Fu, D. Schollmeyer, B. Müller, D. Beljonne, M. Bonn, H. I. Wang, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 5654–5658 CrossRef CAS.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- S. Sasaki, S. Suzuki, K. Igawa, K. Morokuma and G. Konishi, J. Org. Chem., 2017, 82, 6865–6873 CrossRef CAS PubMed.
- L. Ji, K. Fucke, S. K. Bose and T. B. Marder, J. Org. Chem., 2015, 80, 661–665 CrossRef CAS PubMed.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- X. Feng, J.-Y. Hu, C. Redshaw and T. Yamato, Chem.–Eur. J., 2016, 22, 11898–11916 CrossRef CAS.
- N. Ponugoti and V. Parthasarathy, Chem.–Eur. J., 2022, 28, e202103530 CAS.
- F. Liu, X. Shen, Y. Wu, L. Bai, H. Zhao and X. Ba, Tetrahedron Lett., 2016, 57, 4157–4161 CrossRef CAS.
- D. Lungerich, D. Reger, H. Hölzel, R. Riedel, M. M. J. C. Martin, F. Hampel and N. Jux, Angew. Chem., 2016, 128, 5692–5696 CrossRef.
- C. S. Frampton, K. S. Knight, N. Shankland and K. Shankland, J. Mol. Struct., 2000, 520, 29–32 CrossRef CAS.
- M. Tommasini, A. Lucotti, M. Alfè, A. Ciajolo and G. Zerbi, Spectrochim. Acta, Part A, 2016, 152, 134–148 CrossRef CAS PubMed.
- A. Narita, I. A. Verzhbitskiy, W. Frederickx, K. S. Mali, S. Alkaersig Jensen, M. R. Hansen, M. Bonn, S. De Feyter, C. Casiraghi, X. Feng and K. Müllen, ACS Nano, 2014, 8(11), 11622–11630 CrossRef CAS PubMed.
- R. Ulbricht, E. Hendry, J. Shan, T. F. Heinz and M. Bonn, Rev. Mod. Phys., 2011, 83, 543–586 CrossRef CAS.
- A. Tries, S. Osella, P. Zhang, F. Xu, C. Ramanan, M. Kläui, Y. Mai, D. Beljonne and H. I. Wang, Nano Lett., 2020, 20, 2993–3002 CrossRef CAS PubMed.
- G. Soavi, S. Dal Conte, C. Manzoni, D. Viola, A. Narita, Y. Hu, X. Feng, U. Hohenester, E. Molinari, D. Prezzi, K. Müllen and G. Cerullo, Nat. Commun., 2016, 7, 11010 CrossRef CAS PubMed.
- S. A. Jensen, R. Ulbricht, A. Narita, X. Feng, K. Müllen, T. Hertel, D. Turchinovich and M. Bonn, Nano Lett., 2013, 13, 5925–5930 CrossRef CAS PubMed.
- T. L. Cocker, D. Baillie, M. Buruma, L. V. Titova, R. D. Sydora, F. Marsiglio and F. A. Hegmann, Phys. Rev. B, 2017, 96, 205439 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on synthetic procedures, NMR spectroscopy, HR MALDI-TOF mass, X-ray crystallography, GPC, THz-spectroscopy, solid-state NMR and DFT calculations (PDF). Crystal data for compound 1 and 10 (CIF). CCDC 2226715 and 2226716. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02824k |
| This journal is © The Royal Society of Chemistry 2023 |