
Identifying the optimal oxidation state of Cu for electrocatalytic reduction of CO2 to C2+ products†
Liang
Xu
a,
Jiaqi
Feng
a,
Limin
Wu
ab,
Xinning
Song
 ab,
Xingxing
Tan
ab,
Libing
Zhang
ab,
Xiaodong
Ma
ab,
Shunhan
Jia
ab,
Juan
Du
c,
Aibing
Chen
c,
Xiaofu
Sun
*ab and
Buxing
Han
*abd
ab,
Xingxing
Tan
ab,
Libing
Zhang
ab,
Xiaodong
Ma
ab,
Shunhan
Jia
ab,
Juan
Du
c,
Aibing
Chen
c,
Xiaofu
Sun
*ab and
Buxing
Han
*abd
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: sunxiaofu@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Chemical and Pharmaceutical Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China
dShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 26th January 2023
Abstract
The electrocatalytic CO2 reduction reaction (CO2RR) to C2+ products is of great importance. It is known that the co-operation of Cu1+ and Cu0 in the catalysts can yield a high faradaic efficiency (FE). However, it is very difficult to figure out the optimal ratio of Cu1+ and Cu0 because Cu1+ can be reduced to Cu0 during CO2RR. To solve this problem and identify the optimal oxidation state of Cu, herein we propose a strategy to prepare Cu catalysts with different oxidation states, which could be stabilized by the pulsed electrolysis method during CO2RR. On the basis of this method, we have studied the effect of the oxidation state of Cu on CO2RR to form C2+ products. It has been found that the Cu catalyst with an oxidation state of +0.41 is the most efficient in our reaction system, and the FE of C2+ products is 70.3% in an H-type cell. This work provides a precise method to identify the optimal oxidation state of the catalysts that are not stable in the reaction.
The electrocatalytic CO2 reduction reaction (CO2RR) to form value-added carbon-based chemicals and fuels by utilizing renewable electricity is a promising technology to mitigate CO2 emissions, fulfil the anthropogenic carbon cycle, and store excess renewable electricity as chemical energy.1–7 Among the products that can be generated from CO2RR, C2+ products are the most desirable due to their high energy densities and industrial value as chemical feedstocks.8–12 However, their selectivity and activity are severely limited by multistep hydrogenation and the sluggish kinetics of C–C coupling steps. To date, copper is known to be the most efficient electrocatalyst for selectively converting CO2 to C2+ products.13–16 The synergism of Cu1+ and Cu0 sites in copper catalysts has been verified to achieve high faradaic efficiency (FE) for CO2-to-C2+ products.17–20 However, previous research has shown that Cu1+ species are reduced to Cu0 in the reaction.21–23 Therefore, it is challenging to confirm the optimal Cu oxidation state for efficient electrocatalytic CO2RR.
Various strategies have been used to tune the Cu electron structure, such as space confinement,24,25 the synthesis of alloys,26 doping heteroatoms,27,28 and organic ligand modification.29,30 Among these, organic ligand modification (e.g. carboxylate and imidazole) has been reported to stabilize metal centers with appropriate oxidation states, and thus affects the intermediate adsorption during CO2RR.30–36 Meanwhile, the Cu-based catalyst prepared by modifying the carboxylate ligand showed unique electrochemical CO2 reduction selectivity toward C2+ products.37–39 This has made the method to be viewed as a good candidate for constructing Cu1+ catalytic sites to promote the formation of C2+ products. However, the content of Cu1+ drops dramatically during the potentiostatic electrolysis, resulting in the change of CO2RR catalytic activity.
Pulsed potential electrolysis has emerged as a simple and effective method to increase the reaction durability and improve the product selectivity in CO2RR via tuning the surface architecture, oxidation state, surface adsorbate coverage and local pH.40–44 Meanwhile, the pulsed electrochemical method is also a simple and quick method to prepare various materials, such as metals, alloys, metal chalcogenides and porous materials.45–49 Recently, based on the pulsed electrolysis method, our group proposed the “in situ periodic regeneration of catalyst (PR-C)” strategy to give long-term high efficiency of CO2 electroreduction to generate C2+ products over the Cu catalyst by applying a positive potential pulse for a short time periodically in the halide-containing electrolyte.50 At the same time, we also found that the Cu catalyst could be in situ regenerated to maintain the stability of the oxidation state of Cu via the pulsed potential electrocatalytic CO2RR.
Identifying the optimal oxidation state of Cu in CO2RR to form C2+ products is of importance from both fundamental and practical points of views. Herein, we designed and prepared several CuxCyOz catalysts with different Cu oxidation states using the pulsed electrochemical method. The oxidation state of Cu was stabilized by the pulsed potential in CO2RR, and the optimal oxidation state of Cu for producing C2+ products was figured out. It was found that the catalyst with an average Cu valence state of 0.41 was most efficient, and the FE of C2+ products could reach 70.3% with a current density of 24.1 mA cm−2 at −1.0 V versus the reversible hydrogen electrode (RHE).
As illustrated in Fig. 1A, the catalysts were prepared via a pulsed electrochemical method in 0.1 M KHCO3 aqueous electrolyte containing 0.1 M potassium benzenedicarboxylate (K2BDC). A typical H-type cell with three-electrode configuration was used in this work, which included a Cu foil working electrode, a Pt gauze counter electrode, and an Ag/AgCl reference electrode. Cu2+ ions were generated at the anode potential (Ea = 1.25 V vs. RHE), and they interacted with negatively charged carboxylate ligands to form the Cu complex. The Cu complex was then reduced to CuxCyOz at the cathode potential (Ec = −1.0 V vs. RHE). Anodic pulses (ta) of 3 s followed by cathodic pulses (tc) of 5 s were applied in this work. Such pulses were repeated for 80 cycles.
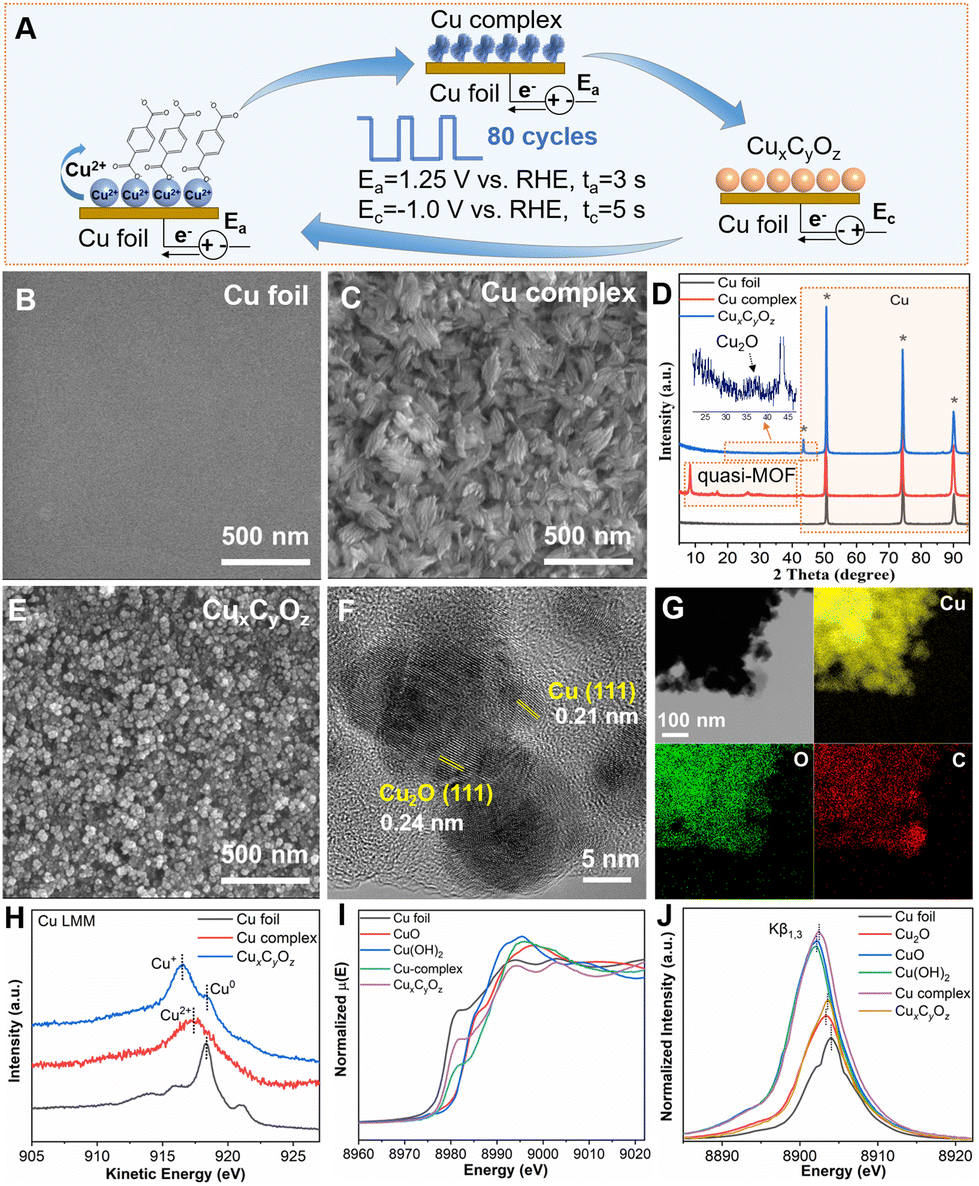 | ||
| Fig. 1 (A) Schematic diagram of the preparation of the CuxCyOz catalyst. SEM images of (B) Cu foil, (C) Cu complex, and (E) CuxCyOz. (D) XRD patterns of different samples. (F) HR-TEM image and (G) elemental mappings images of CuxCyOz. (H) The quasi in situ XPS signals of Cu Auger LMM spectra for the Cu foil, Cu complex, and CuxCyOz. (I) XANES spectra and (J) XES spectra of different samples. | ||
The scanning electron microscopy (SEM) image showed that the untreated Cu foil had a smooth surface (Fig. 1B). The Cu complex has a leaf-like structure generated by the stacking of lamellae due to the application of Ea (Fig. 1C). The X-ray diffraction (XRD) patterns in Fig. 1D confirmed the presence of a crystalline quasi-metal–organic framework (MOF).39 After that, the electrochemical reconstruction51–53 was performed by reducing the Cu complex in an electrolyte to form the CuxCyOz catalyst. The leaf-like structure was converted into uniform nanoparticles (Fig. 1E). The high-resolution transmission electron microscopy (HR-TEM) image (Fig. 1F) confirmed that both metallic Cu and Cu2O crystal lattices existed in the CuxCyOz catalyst, where 0.21 nm and 0.24 nm belong to Cu(111) and Cu2O(111), respectively.54,55 This can also be confirmed from the XRD patterns (Fig. 1D). The corresponding energy-dispersive X-ray spectroscopy (EDS) results showed that the atomic ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C:O was approximately 4:2:1 (Table S1†). The EDS for elemental mapping (Fig. 1G) showed that the Cu, C and O elements were distributed homogeneously throughout the entire architectures.
:C:O was approximately 4:2:1 (Table S1†). The EDS for elemental mapping (Fig. 1G) showed that the Cu, C and O elements were distributed homogeneously throughout the entire architectures.
The surface analysis of different samples was performed by quasi in situ X-ray photoelectron spectroscopy (XPS) (Fig. S1† and Fig. 1H). The Cu 2p XPS spectra and Cu Auger L3M45M45 transition indicated that Cu2+ was the major species in the Cu complex, while Cu0 and Cu1+ species existed in CuxCyOz. Furthermore, the detailed structural information of Cu was investigated by in situ X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES). As shown in Fig. 1I, Cu K-edge X-ray absorption near-edge spectroscopy (XANES) of the Cu complex exhibited an edge profile similar to that of CuO or Cu(OH)2 in the range from 8960 to 9020 eV, while the spectrum of CuxCyOz showed a close absorption edge with the Cu foil. These observations indicated that Cu in CuxCyOz has a lower oxidation state compared with that in the Cu complex. Both the Cu complex and CuxCyOz presented a characteristic Cu–Cu peak at 2.3 Å and a Cu–O peak at around 1.5 Å in the extended X-ray absorption fine-structure (EXAFS) spectra (Fig. S2†). However, the intensity of the Cu–O peak decreased and the intensity of the Cu–Cu peak increased in CuxCyOz compared to those of the Cu complex, suggesting that the oxidized copper in the Cu complex was partially reduced during the formation of CuxCyOz. Fig. 1J shows the Cu Kβ1,3 XES spectra of the Cu complex and CuxCyOz. When comparing the spectra with those of the reference samples, the in situ spectra for the Cu complex lay in between those for the Cu2+ and Cu+ references, whereas in the case of CuxCyOz, the spectra were in between those for Cu1+ and Cu0. Both the XAS and XES results indicated the co-existence of Cu1+ and Cu0 species in CuxCyOz, which is in line with the XPS data.
It is worth noting that application of different Ea values would affect the oxidation state of Cu in the CuxCyOz catalyst. As shown in Fig. 2A, we prepared a series of catalysts by changing the Ea. The SEM images are shown in Fig. S3,† and their morphologies were quite similar. The impact of the applied Ea on the Cu oxidation state was further investigated using XANES (Fig. 2B). The absorption edges of all the samples reside between those of Cu0 and Cu1+. We also acquired the oxidation state of Cu as a function of the Cu K-edge energy shift (Fig. 2C). The detailed calculation method for quantifying the oxidation average valence state of Cu is discussed in the ESI (Table S2†). The average valence of Cu increased gradually with the increase of the applied Ea, which was +0.20, +0.41, +0.47 and +0.59, when the applied Ea was 1.0 V, 1.25 V, 1.4 V, and 1.6 V vs. RHE, respectively. For making a clear distinction, these CuxCyOz catalysts with different Cu oxidation states are denoted as CuxCyOz(0.20), CuxCyOz(0.41), CuxCyOz(0.47), and CuxCyOz(0.59).
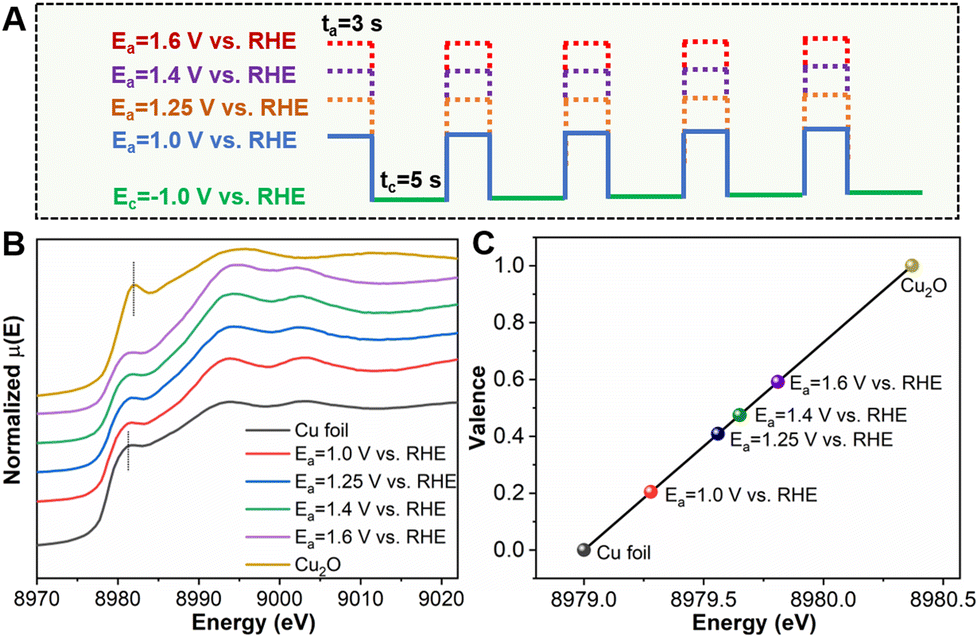 | ||
| Fig. 2 (A) Schematic diagram of the preparation of the CuxCyOz catalyst under different conditions (Ea = 1.0 V, 1.25 V, 1.4 V, and 1.6 V vs. RHE, ta = 3 s, Ec = −1.0 V vs. RHE, tc = 5 s). (B) XANES spectra at the Cu K-edge under different conditions. (C) Average oxidation state of Cu in CuxCyOz under different conditions from Cu K-edge XANES. | ||
Next, quasi in situ XPS and Auger LMM transition measurements were performed to characterize the composition and structure changes of the catalysts during the CO2RR in different routes. In route (1), pulsed electrolysis (Ea = 1.25 V vs. RHE, ta = 3 s; Ec = −1.0 V vs. RHE, tc = 50 s) was applied in 0.1 M KHCO3–K2BDC electrolyte. The same electrolysis conditions as applied for the preparation of CuxCyOz catalyst were followed for the CO2RR, except that the time of tc was extended. In route (2), potentiostatic electrolysis was applied in 0.1 M KHCO3–K2BDC electrolyte with Ec = −1.0 V vs. RHE. As shown in Fig. 3A and B, the content of Cu1+ could be maintained unchanged in route (1), while it would be decreased gradually in route (2). To further investigate the changes in the Cu oxidation state, operando XAS at the Cu K-edge was carried out. The Cu K-edge XANES spectra of CuxCyOz(0.41) used in route (1) for different times exhibited an edge profile similar to that of the catalyst before the reaction, while close to the absorption edge of the Cu foil when reacted in route (2) (Fig. 3C and D). On the other hand, the intensity of Cu–Cu coordination (2.3 Å) of CuxCyOz(0.41) in route (1) was basically unchanged compared to that of the catalyst before the reaction, but it gradually increased in route (2) in the Fourier transform (FT) of the EXAFS spectra in R space (Fig. S4†). The results demonstrated that the Cu oxidation state can be maintained by the in situ regeneration of Cu+ during the CO2RR in route (1), while it declined gradually to tend to Cu0 during the CO2RR in route (2).
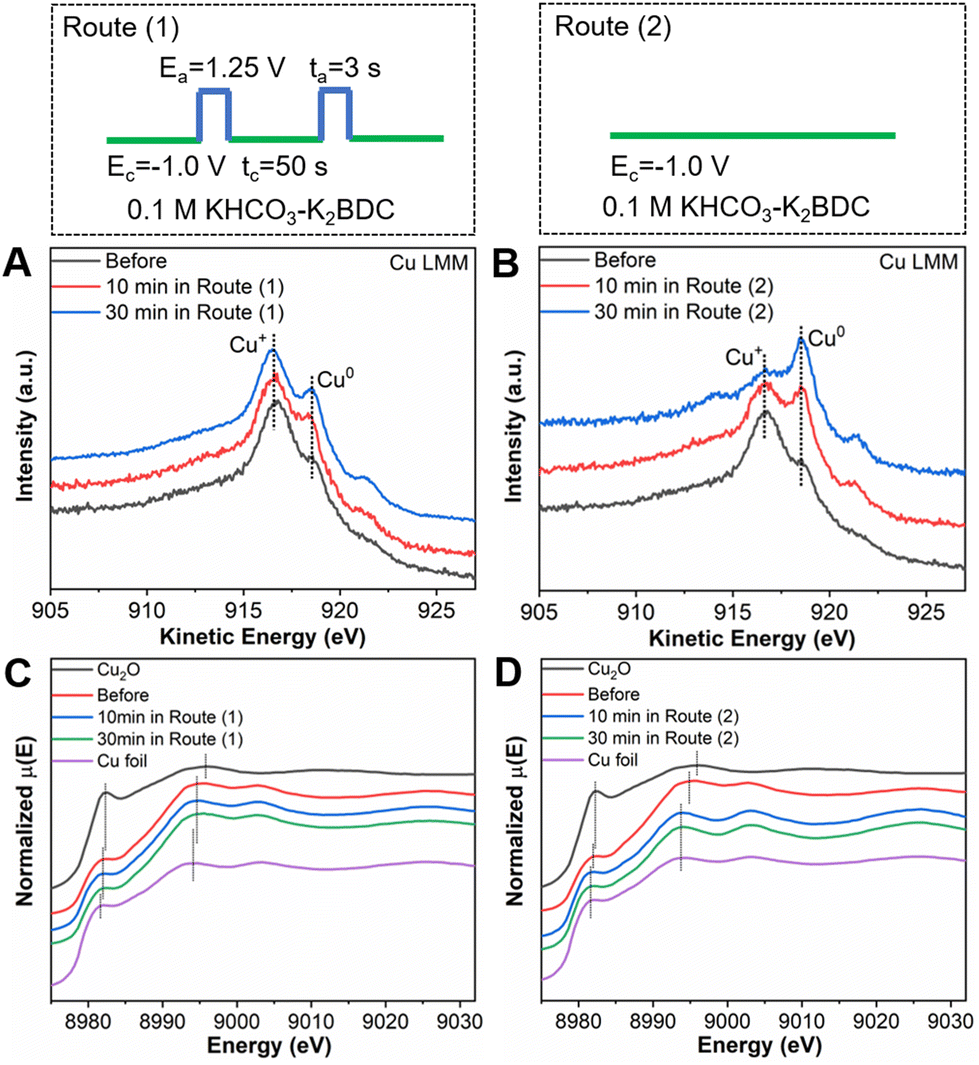 | ||
| Fig. 3 The quasi in situ Auger LMM spectra of Cu for CuxCyOz(0.41) reacted in (A) route (1) and (B) route (2) for different times. The in situ XANES spectra of CuxCyOz(0.41) reacted in (C) route (1) and (D) route (2) for different times. | ||
The CO2RR performances of CuxCyOz catalysts were then investigated in an H-type cell under different electrolysis conditions for 2 h. We considered the effect of K+ concentration on the CO2RR performance, and found that the increase of the K+ concentration in the electrolyte did not promote the formation of C2+ products (Fig. S5†). We also carried out the electrolysis experiments under a N2 atmosphere (without CO2). No carbon-based reduction product could be detected, indicating that CO2 was the carbon source in this work. For CuxCyOz(0.41), it had a higher current density at −0.9 to −1.25 V vs. RHE in route (1) than that in route (2) (Fig. 4A and B). Route (1) also showed a significant difference in the distributions of CO2RR products compared with route (2). C1 products (CO, formate and methane), C2+ products (ethylene, ethanol and n-propanol) and H2 can be detected by 1H nuclear magnetic resonance (NMR) spectroscopy and gas chromatography (GC). The FE for C2+ products in route (1) was much higher than that in route (2). In route (1), the CuxCyOz(0.41) catalyst exhibits a volcano-shaped dependence of total FE for C2+ products at different Ec values, and the maximum FE (C2+ products) could reach up to 70.3% at −1.0 V vs. RHE with a current density of 24.1 mA cm−2, while the highest FE for C2+ products was only 46.6% in route (2). The catalyst also showed lower CO2RR performance to form C2+ products in routes (3) and (4) in 0.1 M KHCO3 (Fig. S6†).
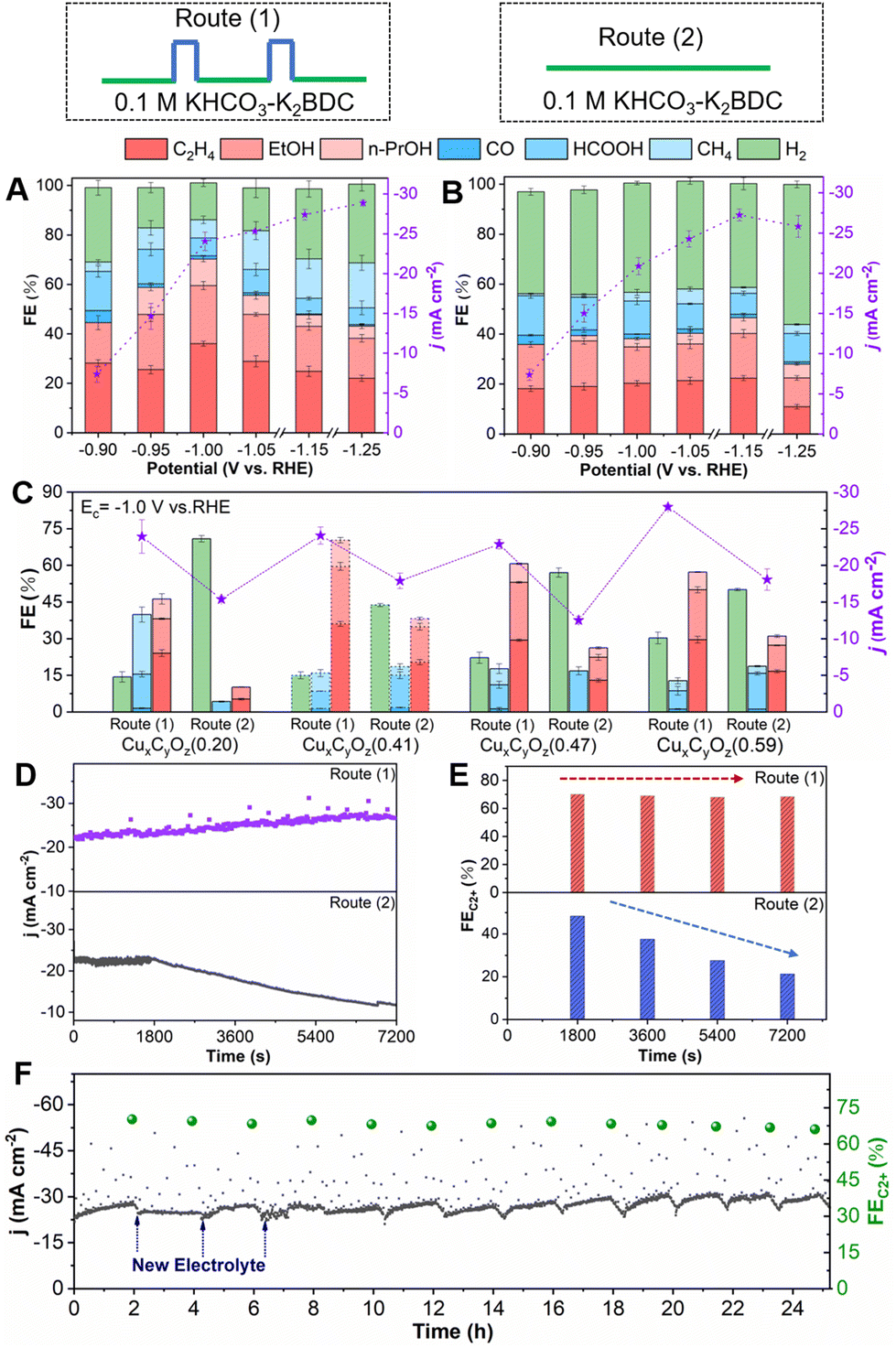 | ||
| Fig. 4 CO2RR product distribution and current density (j) of CuxCyOz(0.41) in (A) route (1) and (B) route (2). (C) CO2RR product distribution and current density of CuxCyOz with different oxidation states of Cu. (D and E) Dependence of the current density (j) and FE of CuxCyOz(0.41) on time in route (1) and route (2). (F) Long-term stability of CO2 pulsed electroreduction over CuxCyOz(0.41). | ||
Furthermore, the CO2RR performance of other CuxCyOz catalysts was also evaluated. The CuxCyOz(0.41) catalyst exhibited an optimal CO2-to-C2+ product performance with the average Cu valence state of 0.41 under an Ea of 1.25 V (Fig. 4C). In the control experiments, we optimized the duration of the pulse (ta and tc) and the concentration of the electrolyte. The detailed discussion is shown in the ESI (Fig. S7–S12†). The final optimized reaction conditions are Ea = 1.25 V vs. RHE, ta = 3 s, Ec = −1.0 V vs. RHE, tc = 50 s, and the FE of C2+ products was the highest in the electrolyte containing 0.1 M K2BDC.
Based on the above analysis, some important observations are summarized below.
(1) The pulsed electrolysis can effectively inhibit the hydrogen evolution reaction (HER) and promote the formation of C2+ products.
(2) The addition of K2BDC in the electrolyte did not contribute to the formation of C2+ products by comparing to that in routes (2) and (4) under potentiostatic electrolysis conditions (Fig. 4B and Fig. S6B†), while the addition of K2BDC in the electrolyte combined with pulsed electrolysis can achieve the in situ regeneration of the CuxCyOz catalyst to stabilize the oxidation state of Cu during the pulsed CO2RR, leading to the improved CO2-to-C2+ performance.
(3) The oxidation state of Cu in CuxCyOz catalysts could regulate the CO2-to-C2+ product performance. The optimal oxidation state of Cu in CuxCyOz catalysts was +0.41 corresponding to the best CO2-to-C2+ performance.
(4) The SEM images show that there was no obvious change after different reaction times in route (1) and route (2) (Fig. S13†), suggesting that the K2BDC in the electrolyte did not change the morphology of the catalyst, which also confirmed that the change in CO2RR performance did not originate from the variation of the catalyst morphology.
The stability was crucial for the application of CO2RR. First, the current density of CO2RR and the FE of C2+ products, which depend on the reaction time within 2 h, were investigated. Obviously, the current density and FE of C2+ products over the CuxCyOz(0.41) catalyst did not change significantly with time in route (1), while both of them decreased continuously in route (2) (Fig. 4D and E). The CuxCyOz catalysts with the other oxidation states of Cu also show a similar phenomenon (Fig. S14†). Furthermore, consecutive cycles were carried out to determine the long-term CO2RR stability of the CuxCyOz(0.41) catalyst in route (1). The current density and FE of C2+ products did not change notably over 25 hours in route (1) (Fig. 4F). The results demonstrated that CuxCyOz(0.41) exhibited outstanding catalytic activity and stability toward CO2RR in route (1) due to the fact that Cu in the catalyst has an optimal oxidation state and can be maintained by the in situ regeneration of Cu during the reaction.
Conclusions
In summary, the CuxCyOz catalysts with different Cu oxidation states have been synthesized via the pulsed electrochemical method. The oxidation state of Cu can be stabilized by the pulsed anode potential in CO2RR, which allows us to study the effect of the oxidation state of Cu on the performance of the catalysts more precisely. It is found that the FEs of C2+ products depend strongly on the oxidation state of Cu. The catalyst with a Cu oxidation state of +0.41 yields the highest C2+ FE of 70.3% with a current density of 24.1 mA cm−2 in an H-type cell. This work provides a precise method to identify the optimal oxidation state of the catalysts. This method is specifically favorable for studying the catalysts that are not stable during the electrochemical reaction due to the reduction of the active species. Obviously, it is also useful for designing efficient catalysts with a suitable oxidation state for CO2RR.Author contributions
L. X., X. F. S. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. L. X. performed the whole experiments. J. Q. F., L. M. W., X. N. S. and X. X. T. performed the analysis of experimental data. L. B. Z., X. D. M. and S. H. J. conducted a part of the characterization study. J. D. and A. B. C. participated in discussions. X. F. S. and B. X. H. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (22002172, 22203099 and 22121002), the Beijing Natural Science Foundation (J210020), the S&T Program of Hebei (B2021208074), the Chinese Academy of Sciences (QYZDY-SSW-SLH013) and the Photon Science Center for Carbon Neutrality. The X-ray absorption spectroscopy measurements were performed at Beamline 4B9A at Beijing Synchrotron Radiation Facility (BSRF). The X-ray emission spectroscopy beam time was granted by Beamline 4W1B of BSRF, Institute of High Energy Physics, Chinese Academy of Sciences. The staff members of 4W1B are acknowledged for their support in measurements and data reduction.References
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC
.
- R. B. Song, W. Zhu, J. Fu, Y. Chen, L. Liu, J. R. Zhang, Y. Lin and J. J. Zhu, Adv. Mater., 2020, 32, 1903796 CAS
- D. Gao, P. Wei, H. Li, L. Lin, G. Wang and X. Bao, Acta Phys.-Chim. Sin., 2021, 37, 2009021 Search PubMed
- X. Song, L. Xu, X. Sun and B. Han, Sci. China: Chem., 2023, 66, 315–323 CrossRef CAS
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed
- J. H. Ye, T. Ju, H. Huang, L. L. Liao and D. G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed
- D. G. Yu and L. N. He, Green Chem., 2021, 23, 3499–3501 RSC
- H. L. Zhu, H. Y. Chen, Y. X. Han, Z. H. Zhao, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2022, 144, 13319–13326 CrossRef CAS PubMed
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed
- X. Yan, C. Chen, Y. Wu, Y. Chen, J. Zhang, R. Feng, J. Zhang and B. Han, Green Chem., 2022, 24, 1989–1994 RSC
- Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 33, 2005798 CrossRef CAS PubMed
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed
- Y. Zhang, P. Li, C. Zhao, G. Zhou, F. Zhou, Q. Zhang, C. Su and Y. Wu, Sci. Bull., 2022, 67, 1679–1687 CrossRef CAS PubMed
- C. Liu, J. Gong, Z. Gao, L. Xiao, G. Wang, J. Lu and L. Zhuang, Sci. China: Chem., 2021, 64, 1660–1678 CrossRef CAS
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CAS
- T. C. Chou, C. C. Chang, H. L. Yu, W. Y. Yu, C. L. Dong, J. J. Velasco-Vélez, C. H. Chuang, L. C. Chen, J. F. Lee, J. M. Chen and H. L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y. M. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205832 CAS
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS PubMed
- Z. Z. Wu, F. Y. Gao and M. R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed
- J. Y. Kim, D. Hong, J. C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G. D. Lee, D. H. Nam and Y. C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed
- F. Hu, L. Yang, Y. Jiang, C. Duan, X. Wang, L. Zeng, X. Lv, D. Duan, Q. Liu, T. Kong, J. Jiang, R. Long and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 26122–26127 CrossRef CAS PubMed
- W. He, I. Liberman, I. Rozenberg, R. Ifraemov and I. Hod, Angew. Chem., Int. Ed., 2020, 59, 8262–8269 CrossRef CAS PubMed
- Z. Yin, C. Yu, Z. Zhao, X. Guo, M. Shen, N. Li, M. Muzzio, J. Li, H. Liu, H. Lin, J. Yin, G. Lu, D. Su and S. Sun, Nano Lett., 2019, 19, 8658–8663 CrossRef CAS PubMed
- H. L. Zhu, J. R. Huang, X. W. Zhang, C. Wang, N. Y. Huang, P. Q. Liao and X. M. Chen, ACS Catal., 2021, 11, 11786–11792 CrossRef CAS
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding, Y. Cao, X. Duan, J. Wang, J. Yang and Y. Wu, Adv. Energy Mater., 2021, 11, 2101424 CrossRef CAS
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed
- L. Zhang, X. X. Li, Z. L. Lang, Y. Liu, J. Liu, L. Yuan, W. Y. Lu, Y. S. Xia, L. Z. Dong, D. Q. Yuan and Y. Q. Lan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed
- L. Hao, Q. Xia, Q. Zhang, J. Masa and Z. Sun, Chin. J. Catal., 2021, 42, 1903–1920 CrossRef CAS
- Y. Yang, Z. Zhang, Z. Zhang, C. Tang, X. Chang and L. Duan, Chin. J. Chem., 2021, 39, 1281–1287 CrossRef CAS
- Y. Wang, X. P. Zhang, H. Lei, K. Guo, G. Xu, L. Xie, X. Li, W. Zhang, U. P. Apfel and R. Cao, CCS Chem., 2022, 4, 2959–2967 CrossRef CAS
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed
- W. Zhang, C. Huang, J. Zhu, Q. Zhou, R. Yu, Y. Wang, P. An, J. Zhang, M. Qiu, L. Zhou, L. Mai, Z. Yi and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112116 CAS
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Joule, 2021, 5, 1987–2026 CrossRef CAS
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. R. Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. R. Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS
- J. Li, Y. Kuang, Y. Meng, X. Tian, W. H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C. S. Ku, C. Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS
- Y. T. Huang, H. Lee, W. D. Li and S. P. Feng, J. Power Sources, 2019, 435, 226801 CrossRef CAS
- Y. Wang, Z. Meng, H. Chen, T. Li, D. Zheng, Q. Xu, H. Wang, X. Y. Liu and W. Guo, J. Mater. Chem. C, 2019, 7, 1966–1973 RSC
- J. Chen, Y. Wang, J. Cao, L. Liao, Y. Liu, Y. Zhou, J. H. Ouyang, D. Jia, M. Wang, X. Li and Z. Li, Electrochim. Acta, 2020, 361, 137036 CrossRef CAS
- L. Xu, X. Ma, L. Wu, X. Tan, X. Song, Q. Zhu, C. Chen, Q. Qian, Z. Liu, X. Sun, S. Liu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202210375 CAS
- M. B. Kale, R. A. Borse, A. Gomaa Abdelkader Mohamed and Y. Wang, Adv. Funct. Mater., 2021, 31, 2101313 CrossRef CAS
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef PubMed
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed
- Y. Zhong, X. Kong, Z. Song, Y. Liu, L. Peng, L. Zhang, X. Luo, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 2554–2560 CrossRef CAS PubMed
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04368h |
| This journal is © The Royal Society of Chemistry 2023 |