
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction of haloacetonitrile-associated risk by adjustment of distribution system pH†
Kevin
Stewart
ab,
Dong
An
cd and
David
Hanigan
 *a
*a
aDepartment of Civil and Environmental Engineering, University of Nevada, Reno, NV 89557-0258, USA. E-mail: dhanigan@unr.edu
bNow with Keller Associates, Reno, NV 89502, USA
cDepartment of Environmental Science & Engineering, Fudan University, Shanghai 200238, China
dShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
First published on 16th August 2023
Abstract
Haloacetonitriles (HANs) and haloacetamides (HAMs) have been scrutinized recently due to their relative toxicity compared to regulated carbonaceous disinfection byproducts (DBPs). Rates of HAN hydrolysis to HAMs and HAMs to haloacetic acids (HAAs) increase with pH and the ranked toxicity of these groups is HAN > HAM > HAA. Thus, intentional hydrolysis during distribution of drinking water would result in reduced exposure to HANs and HAMs, and subsequently lower toxicological risk. To evaluate the potential for such a hydrolysis scheme to reduce the toxicological burden of delivered water, we examined the formation and degradation of HANs and HAMs in conventionally treated surface water and in Milli-Q water spiked with two natural organic matter isolates and free chlorine at varying pH. HAN concentrations in finished drinking water at pH 6, 7.5, and 9 at the end of the 5-day experimental period were 12.5, 7.1, and 1.9 μg L−1, respectively, equivalent to a 77% (pH 6 to 9) and 45% (pH 7.5 to 9) decrease in summed calculated toxicity from the disinfection by-products measured. Similar conclusions followed from experiments with natural organic matter isolates. In samples from the full-scale treatment plant and experiments conducted with isolates, the potential to exceed THM and HAA5 regulatory limits was increased while driving HANs, an unregulated but highly toxic class of DBPs, to lower toxicity hydrolysis products. We find that increasing distribution system pH is an effective method to reduce HAN-associated toxicity, although potential tradeoffs in distribution system corrosion, scale stability, and the potential to violate THM and HAA regulations should be carefully evaluated prior to implementation.
Water impactHaloacetonitriles contribute substantially to the overall toxicity of disinfected drinking water. We demonstrate that increasing pH in the distribution system to levels feasible at scale reduces the risk associated with haloacetonitriles by hydrolyzing them to less toxic products. |
Introduction
Total trihalomethanes (THMs or THM4, the sum of chloroform, bromodichloromethane, dibromochloromethane, and bromoform) and five haloacetic acids (HAA5, sum of mono-, di-, and trichloroacetic acid, and mono-, and dibromoacetic acid) are currently regulated in drinking water by the United States Environmental Protection Agency at 80 μg L−1 and 60 μg L−1 respectively.1 The US EPA is currently considering whether to regulate disinfection byproducts (DBPs) further, and as of November 2022 the Contaminant Candidate List 5 has been published and includes dichloroacetonitrile (DCAN), and dibromoacetonitrile (DBAN) as possible candidates for regulation, likely in-part due to recent research suggesting they contribute substantially to the total risk pool of known DBPs with measured toxic potency.2–4Although haloacetonitriles (HANs) generally account for less than 2% of total organic halides (TOX) by mass in chlorinated waters,5 their contribution to calculated toxicity (weighting DBP concentrations by their Chinese Hamster Ovary (CHO) cell lethality, often referred to as the “summed calculated toxicity” or “calculated additive toxicity”) compared to currently known or regulated DBPs, suggests that they are the main drivers of DBP-associated toxicity.6,7 There are many limitations to such a method of comparing calculated additive toxicity, as demonstrated by McKenna et al.,8 but overall the current understanding by scientists and engineers is that HANs contribute a substantial portion of the toxicity in a disinfected drinking water of the DBPs that have measured toxicity.9
Treatment methods which have been proposed for the control of HANs in drinking water include pre-ozonation,10 UV photolysis, persulfate, UV/H2O2, UV/persulfate,11 various strategies utilizing nanomaterial catalysts,12,13 point-of-use heat treatment,14 and natural organic matter (NOM) removal.15 However, these treatment methods generally have high capital costs and are not easily implementable at existing drinking water treatment plants. Installing UV-AOP or ozonation equipment for treatment of DBPs at a drinking water treatment plant, for example, would only destroy DBPs formed upon chlorination and disinfection in the treatment plant clearwell. With a chlorine residual required in the distribution system, DBPs continue to form as water is distributed to customers. Treatment units that only eliminate DBPs at the treatment plant are unlikely to be an effective solution for reducing HAN levels at the tap. Further, many treatment plants are already equipped with the ability to increase pH via addition of caustic soda or soda ash upon distribution. More easily implementable treatment strategies that reduce HAN levels by the time water reaches consumers would be beneficial in meeting potential future regulatory requirements for HANs.
Multiple kinetic studies have been published in recent years demonstrating hydrolysis and chlorination of HANs to haloacetamides (HAMs), and HAMs to HAAs.16–18 Higher pH leads to increased hydrolysis rates through nucleophilic attack by hydroxide at the nitrile and carbonyl carbons of HANs and HAMs, respectively. Similarly, greater chlorine residual increases chlorination rate through nucleophilic attack. Notably, this sequential pathway is in order of decreasing cytotoxicity (Fig. 1, excepting bromoacetamide (BAM) which is slightly higher in cytotoxicity than bromoacetonitrile (BAN)).18
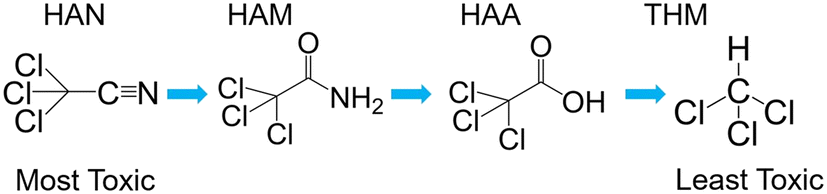 | ||
| Fig. 1 Example reaction scheme showing hydrolysis of trichloroacetonitrile to chloroform. Note that the initial reaction is substantially faster than the following two. | ||
We propose that increasing distribution system pH will take advantage of the cascading toxicity profile of the base-catalyzed hydrolysis products, producing a straightforward, viable, and easily implementable treatment method for lowering HAN-associated toxicity in delivered drinking water. To demonstrate this, a distribution system was simulated by holding treated drinking water, or Milli-Q water spiked with NOM isolates, spiking sodium hypochlorite at varying pH, and measuring DBPs. Measured DBPs were multiplied by their respective CHO LC50s and summed the summed values were compared across three different pH representative of distributions systems delivering water at low, high, and neutral pH.
Materials and methods
Sample collection
Finished drinking water was collected in October 2021 from a conventional drinking water treatment plant in Reno, NV utilizing coagulation, flocculation, sedimentation, filtration, and free chlorine disinfection. The sample was stored on ice and immediately transported to the laboratories at the University of Nevada, Reno. General water quality characteristics for the finished drinking water were analyzed within two hours of sample collection and are provided in Table SI-1.† Suwannee River NOM (SRNOM, 2R101N) and Upper Mississippi River NOM (UMRNOM, 1R110N) were purchased from the International Humic Substances Society.Experimental methods
To choose an appropriate buffer for the hydrolysis experiments, several buffer systems were tested at pH 6, 7.5, and 9. A 10 mM phosphate buffer was chosen for the HAN hydrolysis experiments as no noticeable change in pH was observed after 24- and 48-hours following addition of the buffers to finished drinking water, as well as the NOM isolate samples. To understand the effect of phosphate buffer strength on HAN degradation kinetics, Milli-Q water was buffered to pH 8 at three different buffer concentrations: 1 mM, 10 mM, and 100 mM, and 10 μg L−1 of trichloroacetonitrile (TCAN), chloroacetonitrile (CAN), dibromoacetonitrile (DBAN), and BAN were spiked. Samples were collected immediately after spiking and after 24-hours, and HANs were extracted and quantified. No noticeable change in degradation kinetics was observed over the varying buffer concentrations, indicating that phosphate does not catalyze the reaction via phosphorolysis, agreeing well with experiments conducted by others who compared HAN hydrolysis kinetics in both phosphate and carbonate buffered solutions.19 Therefore, a buffer concentration of 10 mM was used in all experiments.The finished drinking water sample had a free chlorine residual of 1.7 mg Cl2 L−1 which was not further supplemented. Samples were buffered using a 10 mM phosphate buffer at three different pH: 6, 7.5, and 9, confirmed using a pH probe. For experiments with NOM, 3 mg C L−1 was spiked of either SRNOM or UMRNOM. The samples were buffered the same as the drinking water samples and sodium hypochlorite stock solution was added, resulting in an initial concentration of 9 mg Cl2 L−1. While the chlorine concentration employed is likely higher than what is typical for drinking water treatment facilities, it was used here to confirm trends in DBP formation and hydrolysis seen in the full-scale treated samples at higher DBP concentrations (i.e., worst case scenario). To produce the initial data points, chlorine was quenched within one hour after chlorination at the treatment plant for the drinking water sample or immediately after the chlorine spike in the NOM tests. Treatment groups were partitioned to headspace-free 70 mL reaction vials for the allotted reaction times (up to 5 days) at room temperature (22 °C). The reaction times for this study included 0-, 1-, 16-, 24-, 30-, 48-, and 120-hours at pH 6 and 7.5. At pH 9, a 72-hour sample was taken instead of a 120-hour sample. Variable sampling intervals were used to capture differences in hydrolysis kinetics at different pH. 36% of the data was produced via experimental triplicates (initial, 1-hour, and 24-hour samples). After the planned reaction duration, free chlorine was quenched with 50 μL of 0.5 M ascorbic acid. Ascorbic acid was chosen to minimize the degradation/reduction of DBPs.20,21 Experiments were also conducted to ensure negligible DBP degradation over a one-week storage period. No noticeable change in DBP concentration was observed when calibration standards were spiked, stored for one week at 5 °C, and extracted following the analytical methods described below. Samples were then acidified using 150 μL of 12 N HCl to preserve DBPs and stored at 5 °C for up to two weeks before extraction, although most samples were extracted within one week.
Statistical methods
Statistical analyses including ANOVA22 and Pearson correlations were conducted in Graphpad Prism v.9.5.1 and Microsoft Excel. Normality was assumed for Pearson correlation analysis as error in the data is derived in random experimental and analytical error, which is non-biased.Analytical methods
Four THMs, six HANs, and six HAMs were extracted and quantified using liquid–liquid extraction (LLE) with methyl tert-butyl ether (MTBE, Fisher HPLC grade) and gas chromatography with a HP-5 column (Agilent, 30 m length, 0.32 mm diameter, 0.25 μm film) with electron capture detection (GC-ECD, Agilent 8860). The method was similar to EPA Method 551. Specific oven programs can be found in the supplemental information in Text SI-1.† In brief, 30 mL of sample was pipetted into a pre-ashed 40 mL vial. 3 mL of MTBE containing 1 μg mL−1 of 1,2-dibromopropane and 10 grams of granular anhydrous sodium sulfate (Fisher, 99%) were added. Samples were shaken at 250 rpm for 30 minutes using a mechanical shaker. Approximately 1 mL of the upper organic layer of each extract was transferred to a GC vial and stored at −20 °C for up to 14 days until GC-ECD analysis.Nine HAAs were extracted using LLE, esterified, and quantified using GC-ECD with a DB-1 column (Agilent, 30 m length, 0.25 mm diameter, 1 μm film) similar to EPA Method 552. Specific oven programs can be found in Text SI-2.† Briefly, 30 mL of sample was transferred to a pre-ashed 40 mL vial. 20 μL of 20 μg mL−1 2-bromobutyric acid (surrogate standard), 1.5 mL concentrated sulfuric acid, 3 mL of MTBE containing 1 μg mL−1 1,2,3-trichloropropane (internal standard), and 10 grams of anhydrous sodium sulfate were then added to each vial. Samples were then shaken at 250 rpm for 30 minutes using a mechanical shake table. 1 mL of MTBE extract was then transferred to a pre-ashed 20 mL vial containing 2 mL of acidic methanol (5% H2SO4). The extract and acidic methanol mixture was placed in a water bath at 50 °C for 2 hours. Upon completion of the esterification, vials were cooled for approximately 5 minutes, and 5 mL of saturated sodium bicarbonate solution (96 g L−1) was added. 1 mL of MTBE was then added to each sample and vials were mechanically shaken for 2 minutes. 1 mL of organic extract was transferred to GC vials and stored at −20 °C for up to 14 days until GC-ECD analysis.
A free chlorine solution (∼5%) was diluted to ∼2 mg L−1 and standardized using a Hach DR 6000 spectrophotometer with the N,N-diethyl-1,4 phenylenediamine (DPD) reagent method.23 Total organic carbon (TOC) was quantified using a Shimadzu TOC analyzer using standard methods.24 pH was measured with a Fischer Scientific Accumet 13-620-299B pH probe. Error associated with summed calculated toxicity includes error propagated from experimental/analytical uncertainty from DBP formation experiments and their measurement, along with an assumed relative standard error of 12% for published LC50 values.8,25 CHO LC50 values for compounds measured in this study are tabulated in Table SI-2.†
Reagents
EPA 501/601 2000 μg mL−1 trihalomethanes calibration mix containing chloroform (99.9%), bromodichloromethane (BDCM, 97.2%), dibromochloromethane (DBCM, 96.5%), and bromoform (96.1%) and EPA 552.2 2000 μg mL−1 haloacetic acids mix (HAA9) containing chloroacetic acid (CAA, 98.1%), dichloroacetic acid (DCAA, 98.5%), bromoacetic acid (BAA, 98.1%), trichloroacetic acid (TCAA, 99.9%), bromochloroacetic acid (BCAA, 98.3%), bromodichloroacetic acid (BDCAA, 99.6%), dibromoacetic acid (DBAA, 100%), chlorodibromoacetic acid (CDBAA, 98%), and tribromoacetic acid (TBAA, 99.7%) were purchased from Sigma Aldrich. 5000 μg mL−1 disinfection byproduct standard containing bromochloroacetonitrile (BCAN), dibromoacetonitrile (DBAN), dichloroacetonitrile (DCAN), and trichloroacetonitrile (TCAN) was purchased from Agilent. Bromoacetonitrile (BAN, 97%), chloroacetonitrile (CAN, 98%), chloroacetamide (CAM, 98%), bromoacetamide (BAM, 98%), dichloroacetamide (DCAM, 98%), trichloroacetamide (TCAM, 98%), 2-bromobutyric acid (99%), 1,2-dibromopropane (98%), and 1,2,3-trichloropropane (98%) were purchased from Sigma Aldrich. Dibromoacetamide (DBAM, 99%), and bromochloroacetamide (BCAM, 99%) were purchased from Toronto Research Chemicals.Results and discussion
DBP formation at circumneutral pH
We initially focused on finished drinking water collected from a regional drinking water treatment plant in Reno, NV, and confirmed the conclusions using Milli-Q water spiked with NOM and sodium hypochlorite, buffered at varying pH. Fig. 2 contains example data of the formation of individual DBPs in the finished drinking water sample at circumneutral pH, stacked by common functional group. Data for other pH are provided in Fig. SI-1 and SI-2.† At pH 7.5, THMs, HAAs, HAMs, and HANs were formed at detectable concentrations at all pH tested. The greatest species by mass for each DBP class was chloroform, BCAA or BDCAA (temporally dependent), DCAM, and TCAN, and this was similar over time. THMs, HAAs, and HAMs formed continuously throughout the experiment but began to plateau at the longest time point measured (120-hour). HANs, however, formed rapidly and did not substantially increase in concentration after the initial measurement. HAN concentrations reached a maximum of 10.2 μg L−1, and HAMs reached a maximum concentration of 4.3 μg L−1. THMs reached a high of 81.7 μg L−1, HAA5 reached a maximum of 29 μg L−1, and HAA9 reached a maximum concentration of 101.6 μg L−1, reflecting the substantial contribution of BCAA and BDCAA, which are included in HAA9 but not in HAA5. Notably, in this finished drinking water sample, the concentration of THMs exceeded the U.S. EPA maximum contaminant level (MCL), although this duration of chlorine contact may not represent (and was not intended to represent) the distribution system from which it was taken. Overall, at circumneutral pH, HAAs were present at the highest concentration by mass, followed by THMs, HANs, and HAMs.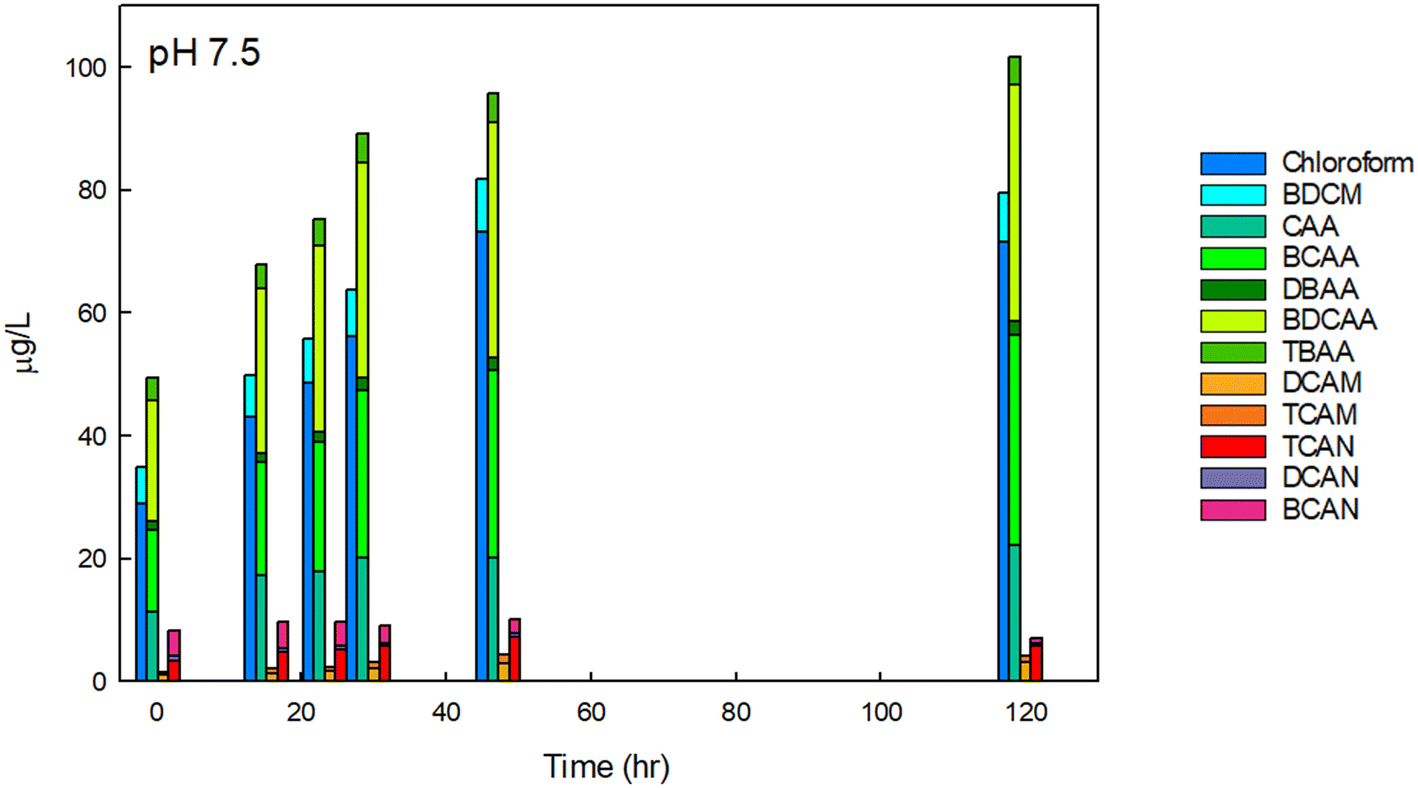 | ||
| Fig. 2 DBPs in finished drinking water at pH 7.5 at varying reaction durations. DBPs which were not present or present below the detection limits are not shown. The initial free chlorine concentration was 1.7 mg Cl2 L−1. Data for 0-, 1-, and 24-hour time intervals were conducted in experimental triplicate and the mean is shown, others are from single experiments. For experiments conducted in replicate, the coefficient of variation for the summation of THMs was always less than 0.03, always less than 0.17 for HAAs, 0.22 for HAMs, and 0.03 for HANs, accounting for error propagation of the individual DBPs measured in the replicates. The concentrations of each species measured are provided in Table SI-3.† | ||
Effect of pH on DBP formation and degradation
Comparing formation/degradation of DBPs vs. pH, we found a generally positive correlation between pH and formation of THMs and HAMs (Fig. 3), but pH and total HANs were negatively correlated (Pearson correlation coefficients for pH vs. DBP classes are presented in Table SI-6†). HAAs tended to be minimally impacted by pH with differences in formation across the three pH being generally <10%. At shorter reaction periods, HAN formation was similar across the range of pH of the experiment; in all three pH groups, the 1-hour samples had HAN concentrations of approximately 8 μg L−1. However, after the first sampling point, HAN concentrations diverged from this trend, with concentrations at higher pH substantially lower than those at lower pH. This is likely because the degradation rate exceeded the formation rate in the pH 9 sample, although no such formation rate constants are available. BCAN concentrations exceeded DCAN concentrations at lower pH and shorter time intervals, but the trend reversed at increased pH and longer time intervals (both for the disinfected drinking water and for SRNOM and UMRNOM, see section on NOM isolates), potentially explaining some of the variability in published literature regarding which species dominates.26 Using all data points from the time series at each pH combined with ANOVA, we found that pH had a statistically significant impact on formation of HAMs (p = 2 × 10−3) and HANs (p = 2 × 10−4), but the effects were weaker for THMs (p = 0.09) and not statistically significant for HAAs (p = 0.66). Finally, the greatest concentration of THMs measured was at pH 9, equal to 84.1 μg L−1, respectively. Again, this could potentially violate the EPA MCL assuming a chlorine concentration and residence time that are similar to these bench-scale experiments. HAA5 concentrations did not exceed the respective MCL at any pH.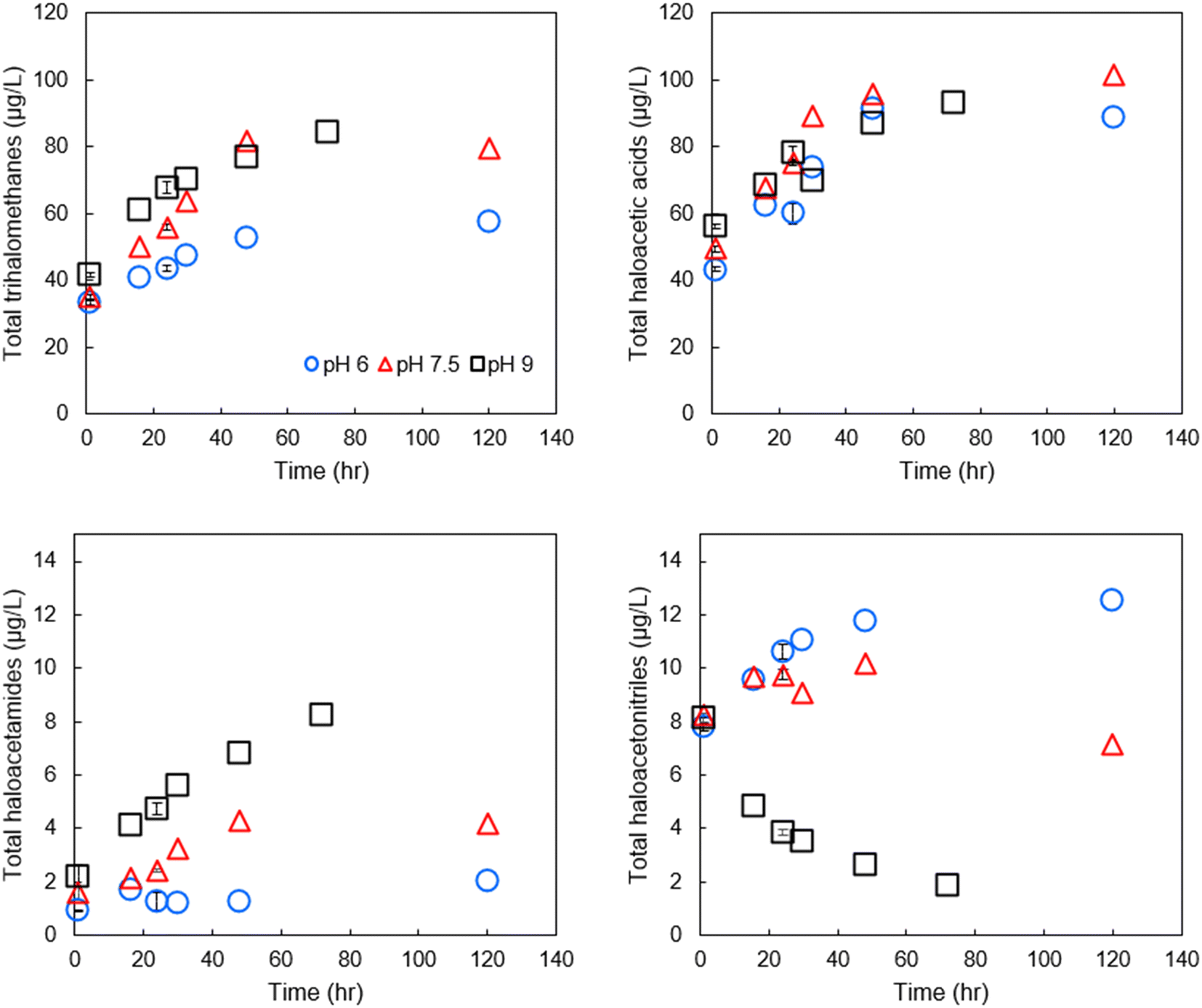 | ||
| Fig. 3 DBP formation across time by DBP class at pH 6, 7.5, and 9 in finished drinking water. Variable sampling intervals were used to capture differences in hydrolysis at varying pH. Error bars represent one standard deviation of experiments conducted in triplicate. | ||
Although HAMs have been reported to have basic hydrolysis rate constants on the same order of magnitude as their HAN counterparts,27,28 HAMs were not, on net, degraded in this study. Rather, HAMs were maximized at the highest pH tested. It is not clear why lower pH resulted in lower concentration of HAMs compared to more basic pH, but one possibility is that HAM concentrations are substantially influenced by hydrolysis of HANs to HAMs. Comparing concentrations of HANs at the 48-hour sampling between the pH 6 and pH 9 samples demonstrates a loss of 61 nM HAN (28 nM TCAN, 33 nM BCAN, no loss of DCAN). The documented hydrolysis mechanism suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HAN to HAM molar relationship18 and thus, the hydrolysis of HANs represents a corresponding formation 61 nm of HAMs, or 70% of the observed difference between HAM concentrations at pH 6 and 9. Thus, the observed lack of HAM hydrolysis is likely due to the competing mechanisms of HAM hydrolysis and HAM formation from HAN hydrolysis. Note that although this roughly accounts for HAM formation from hydrolysis of HANs, this comparison does not account for differences in formation kinetics of HAMs at varying pH from reactions between chlorine and organic matter. While the chemical kinetic model and rate constants proposed by Yu and Reckhow18 are useful for predicting hydrolysis of HANs in waters with fixed starting concentrations, a model incorporating both formation kinetics from NOM and hydrolysis kinetics may be necessary for quantitative predictions of HAN, HAM, HAA, and THM concentrations in drinking water treatment and distribution systems. Measurements of formation kinetics could potentially be estimated from the difference between observed formation rates here and published hydrolysis rate constants, although the results would likely have a substantial amount of propagated error and would be specific to the single chlorine dose employed.
:1 HAN to HAM molar relationship18 and thus, the hydrolysis of HANs represents a corresponding formation 61 nm of HAMs, or 70% of the observed difference between HAM concentrations at pH 6 and 9. Thus, the observed lack of HAM hydrolysis is likely due to the competing mechanisms of HAM hydrolysis and HAM formation from HAN hydrolysis. Note that although this roughly accounts for HAM formation from hydrolysis of HANs, this comparison does not account for differences in formation kinetics of HAMs at varying pH from reactions between chlorine and organic matter. While the chemical kinetic model and rate constants proposed by Yu and Reckhow18 are useful for predicting hydrolysis of HANs in waters with fixed starting concentrations, a model incorporating both formation kinetics from NOM and hydrolysis kinetics may be necessary for quantitative predictions of HAN, HAM, HAA, and THM concentrations in drinking water treatment and distribution systems. Measurements of formation kinetics could potentially be estimated from the difference between observed formation rates here and published hydrolysis rate constants, although the results would likely have a substantial amount of propagated error and would be specific to the single chlorine dose employed.
Impact of pH on calculated toxicity
To assess the relative impact of pH on DBP-associated toxicity, individual DBP concentrations were divided by their measured cytotoxicity and the results summed (i.e., summed or calculated additive toxicity, Fig. 4). At pH 6, calculated toxicity increased throughout the experimental period. At pH 7.5, calculated toxicity increased for the first 16 to 24 hours and then decreased throughout the remainder of the experimental period. At pH 9 there was a rapid decrease in calculated toxicity, reaching a minimum approximately 24 hours after treatment.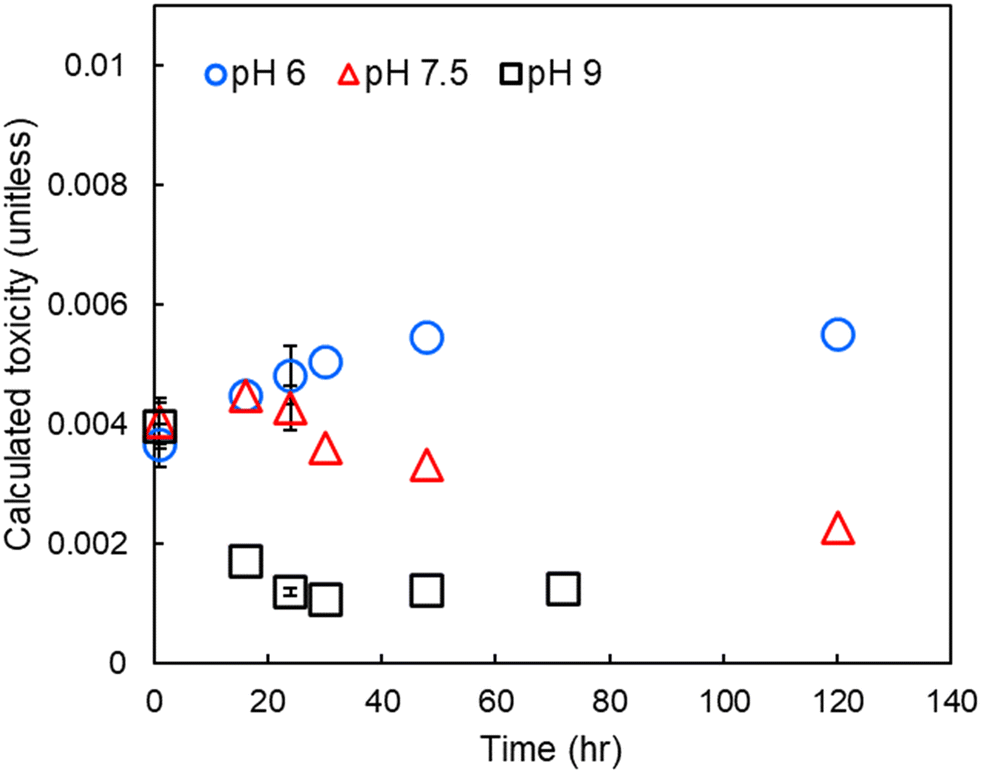 | ||
| Fig. 4 Calculated toxicity in finished drinking water at pH 6, 7.5 and 9. Errors bars show one standard deviation of experimental triplicates, including propagated error from both experimental/analytical uncertainty and an assumed 12% relative standard error in the LC50.8,25 | ||
To understand which DBPs drove the observed changes in calculated toxicity at varying pH, we show the contribution to calculated toxicity by DBP class in Fig. 5. HANs were the primary drivers of DBP-associated toxicity at pH 6 and 7.5 in finished drinking water, accounting for as much as 85% of calculated toxicity, despite accounting for ≤9% of the total mass of DBPs measured. At pH 9 HANs were rapidly hydrolyzed to HAMs and HAAs, with HANs accounting for approximately 82% of the initial calculated toxicity and decreasing to 10% of calculated toxicity after 72 hours. At pH 6 HAAs contributed the second greatest to calculated toxicity at all reaction times. At pH 7.5 HAAs contributed the second greatest to calculated toxicity at reaction times less than 48 hours and dominated in samples with longer reaction times. HAAs accounted for up to 58% of total DBPs by mass and 15% to 82% of calculated toxicity.
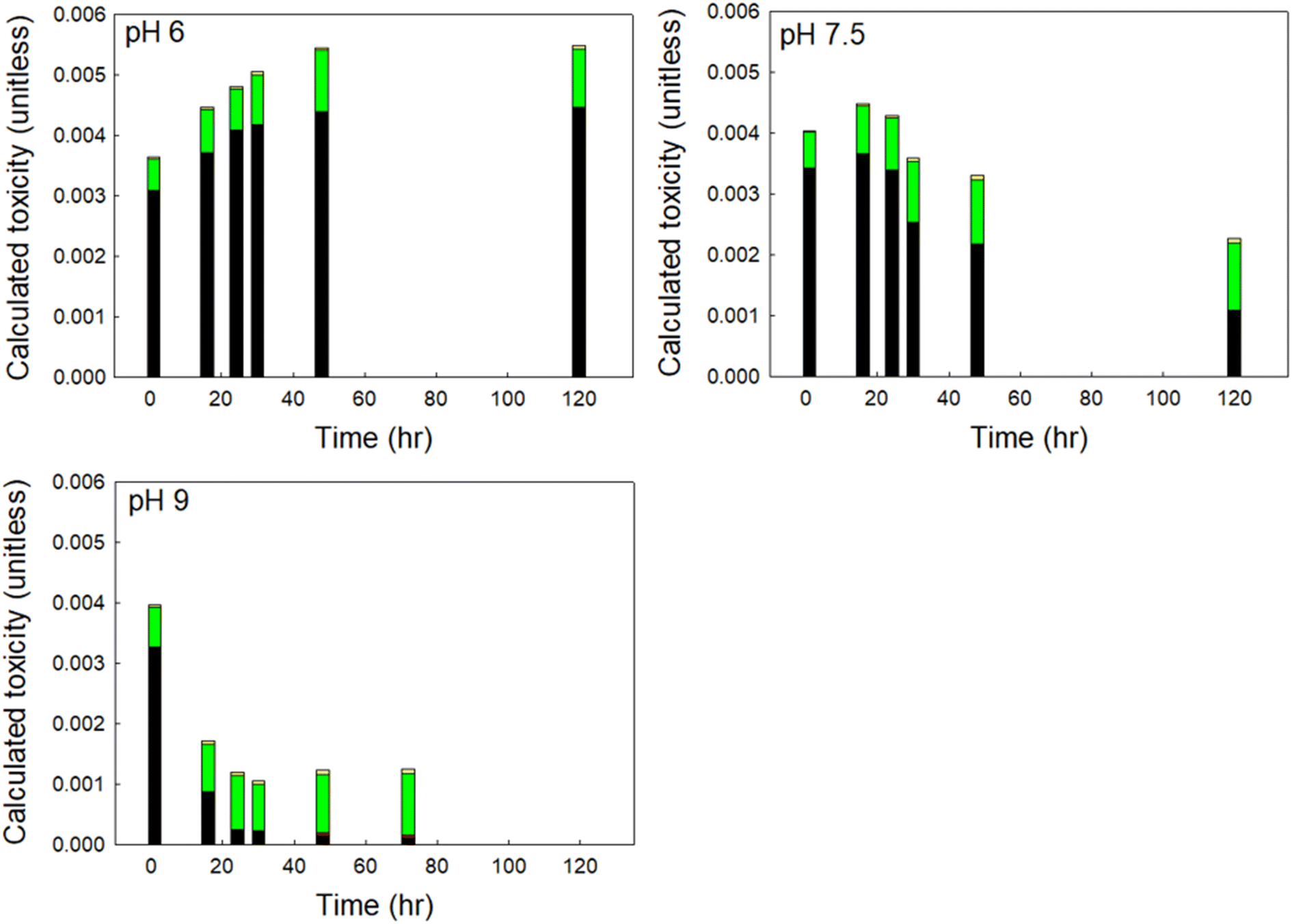 | ||
| Fig. 5 Contribution to calculated toxicity of each DBP class in a sample of finished drinking water at pH 6, 7.5, and 9. The coefficient of variation was always less than 0.1. | ||
Although HAMs may be up to an order of magnitude more toxic than similarly substituted HAAs, HAMs accounted for only 0.1% to 2.6% of calculated toxicity, attributable to their relatively low concentration formed at all pH (≤4% of total DBPs by mass). Similarly, THMs accounted for up to 47% of total DBPs by mass, but only contributed 0.7% to 5.6% to calculated toxicity across all time steps, representative of their relatively low toxicity. Overall, HANs represented the greatest contribution to DBP-associated toxicity immediately following chlorine exposure at all pH. At circumneutral and acidic pH, their contribution was relatively unchanged as the overall calculated toxicity of the sample increased. But at more alkaline pH, the concentration of HANs decreased rapidly, and the associated contribution to calculated toxicity was reduced correspondingly.
Confirmation of conclusions with NOM isolates
To verify trends in DBP formation and calculated toxicity across varying NOM sources, experiments were conducted with two different IHSS NOM sources spiked to pH buffered Milli-Q water at 3 mg C L−1. The spiked NOM was exposed to an initial addition of 9 mg Cl2 L−1 free chlorine. In Fig. 6 we show calculated toxicity over time from these experiments. Plots similar to Fig. 4 and 5, showing formation over time and species contribution to toxicity, are provided in the ESI† (Fig. SI-3 through SI-6). Tabulated concentrations of DBPs measured for SRNOM and UMRNOM are provided in Tables SI-4 and SI-5.†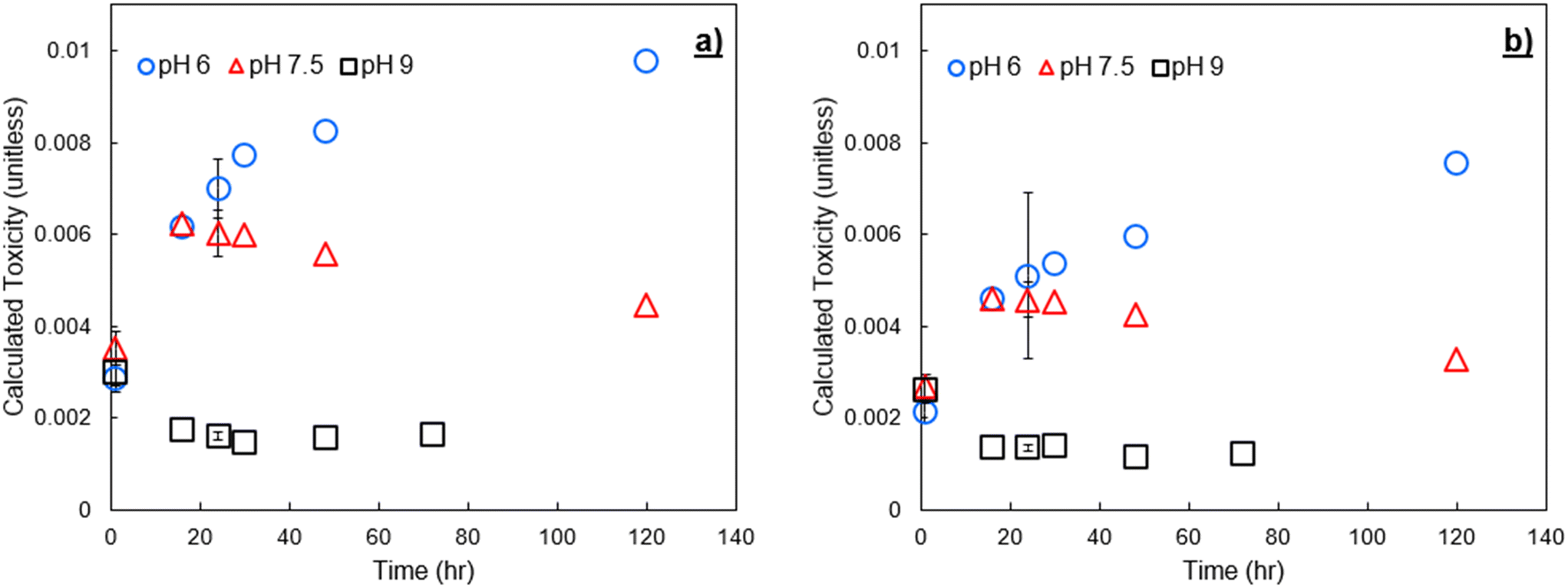 | ||
| Fig. 6 Calculated toxicity from formed DBPs at pH 6, 7.5, and 9 after chlorination of a) Suwannee River NOM, and b) upper Mississippi River NOM. The chlorine dose was 9 mg Cl2 L−1. Error bars show one standard deviation of experimental triplicates including propagated error from both experimental/analytical uncertainty and an assumed 12% relative standard error in the LC50 values.8,25 | ||
DBP-associated calculated toxicity from chlorination of NOM isolates had similar trends to that of finished drinking water, rapidly decreasing at pH 9 due to HAN degradation. In all experiments, calculated toxicity at pH 7.5 increased for the first 16 to 24 hours following initial chlorine contact, then declined steadily throughout the remaining sampling period. pH 6 resulted in the greatest calculated toxicity at all time points due to the increased stability of HANs at low pH (i.e., decreased hydrolysis). pH 9 resulted in decreased toxicity for all time points beyond the initial sampling. The EPA MCL for THMs was also exceeded with both SRNOM and UMRNOM, and basic pH exacerbated the issue. These samples have not been subject to traditional drinking water treatment methods and have been spiked with relatively high concentrations of chlorine and are therefore not representative of drinking water samples or subject to the MCL, but it is notable that in multiple experiments with both spiked NOM and conventionally treated surface water, there was an inverse relationship between calculated toxicity and the potential to violate MCLs for regulated DBPs.
Conclusions, implications, and limitations
HANs form rapidly (i.e., hours) following chlorine addition, and pH is a crucial factor in determining subsequent persistence in treated drinking waters. HANs are stable at reduced pH but relatively unstable at alkaline pH, thus increasing distribution system pH will lead to degradation of these compounds through based catalyzed hydrolysis, and reduced exposure. At pH 9 in finished drinking water and samples containing NOM isolates, we demonstrated the rapid destruction of HANs resulting in a subsequent decrease in calculated toxicity. In any full-scale application of such an approach intended to improve water quality and protect public health from DBP exposure, consideration must be given to the potential for pipe scaling and pipe corrosion when adjusting distribution pH. Correspondingly, we also demonstrated that at longer distribution system residence times, pH at or above 7.5 is likely to cause HAN hydrolysis to supersede formation. In other words, any pH at or above 7.5 will likely improve calculated toxicity, and pH as high as 9 is not likely to be required. While the hydrolysis kinetics are independent of individual water chemistry, water chemistry is likely to be a factor in formation kinetics and individual site investigations may be required to determine the pH which balances distribution system impacts with HAN formation and hydrolysis. Additionally, the potential to violate THM and HAA5 MCLs was increased while driving HANs, an unregulated but highly toxic class of DBPs, to lower toxicity hydrolysis products. Thus, the current regulations are somewhat at odds with delivering water that is higher quality, as measured by calculated toxicity. Finally, only the compounds which were measured are used to make the presented conclusions. Other known and unknown DBPs are likely to be present in any disinfected water and were not included, and this may bias the conclusions. This bias is inherent to the approach, present in all efforts to use to calculated toxicity to make engineering and public health decisions,8 and any decisions made based on conclusions from such an approach should strongly consider the limitations and the potential for unintended consequences, including formation of other currently unknown DBPs, possibly of higher toxicity than those which were investigated in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by the National Science Foundation CBET #1804255 and OIA #2229313.References
- Agency USEP, National Primary Drinking Water Regulations, 2022 Search PubMed.
- US EPA, Contaminant Candidate List 5, 2022 Search PubMed.
- S. S. Lau, X. Wei, K. Bokenkamp, E. D. Wagner, M. J. Plewa and W. A. Mitch, Assessing Additivity of Cytotoxicity Associated with Disinfection Byproducts in Potable Reuse and Conventional Drinking Waters, Environ. Sci. Technol., 2020, 54(9), 5729–5736 CrossRef CAS PubMed.
- S. W. Krasner, T. C. F. Lee, P. Westerhoff, N. Fischer, D. Hanigan and T. Karanfil, et al., Granular Activated Carbon Treatment May Result in Higher Predicted Genotoxicity in the Presence of Bromide, Environ. Sci. Technol., 2016, 50(17), 9583–9591 CrossRef CAS PubMed.
- S. D. Richardson, A. D. Thruston, S. W. Krasner, H. S. Weinberg, R. J. Miltner and K. M. Schenck, et al., Integrated Disinfection By-Products Mixtures Research: Comprehensive Characterization of Water Concentrates Prepared from Chlorinated and Ozonated/Postchlorinated Drinking Water, J. Toxicol. Environ. Health, Part A, 2008, 71(17), 1165–1186 CrossRef CAS PubMed.
- X. Wei, M. Yang, Q. Zhu, E. D. Wagner and M. J. Plewa, Comparative Quantitative Toxicology and QSAR Modeling of the Haloacetonitriles: Forcing Agents of Water Disinfection Byproduct Toxicity, Environ. Sci. Technol., 2020, 54(14), 8909–8918 CrossRef CAS PubMed.
- J. M. Allen, M. J. Plewa, E. D. Wagner, X. Wei, K. Bokenkamp and K. Hur, et al., Drivers of Disinfection Byproduct Cytotoxicity in U.S. Drinking Water: Should Other DBPs Be Considered for Regulation?, Environ. Sci. Technol., 2022, 56(1), 392–402 CrossRef CAS PubMed.
- E. McKenna, K. A. Thompson, L. Taylor-Edmonds, D. L. McCurry and D. Hanigan, Summation of disinfection by-product CHO cell relative toxicity indices: sampling bias, uncertainty, and a path forward, Environ. Sci.: Processes Impacts, 2020, 22(3), 708–718 RSC.
- E. D. Wagner and M. J. Plewa, CHO cell cytotoxicity and genotoxicity analyses of disinfection by-products: An updated review, J. Environ. Sci., 2017, 58, 64–76 CrossRef CAS PubMed.
- X. Yang, C. Shang, Q. Shen, B. Chen, P. Westerhoff, J. Peng and W. Guo, Nitrogen origins and the role of ozonation in the formation of haloacetonitriles and halonitromethanes in chlorine water treatment, Environ. Sci. Technol., 2012, 46(23), 12832–12838 CrossRef CAS PubMed.
- S. Hou, L. Ling, C. Shang, Y. Guan and J. Fang, Degradation kinetics and pathways of haloacetonitriles by the UV/persulfate process, Chem. Eng. J., 2017, 320, 478–484 CrossRef CAS.
- J. J. Molnar, J. R. Agbaba, B. D. Dalmacija, M. T. Klašnja, M. B. Dalmacija and M. M. Kragulj, A comparative study of the effects of ozonation and TiO2-catalyzed ozonation on the selected chlorine disinfection by-product precursor content and structure, Sci. Total Environ., 2012, 425, 169–175 CrossRef CAS PubMed.
- X. Chang, X. Yao, N. Ding, X. Yin, Q. Zheng, S. Lu, D. Shuai and Y. Sun, Photocatalytic degradation of trihalomethanes and haloacetonitriles on graphitic carbon nitride under visible light irradiation, Sci. Total Environ., 2019, 682, 200–207 CrossRef CAS PubMed.
- W. Shi, L. Wang and B. Chen, Kinetics, mechanisms, and influencing factors on the treatment of haloacetonitriles (HANs) in water by two household heating devices, Chemosphere, 2017, 172, 278–285 CrossRef CAS PubMed.
- S. E. Barrett, S. W. Krasner and G. L. Amy, Natural Organic Matter and Disinfection By-Products: Characterization and Control in Drinking Water-An Overview, ACS Symp. Ser., 2000, 761, 2–14 CrossRef CAS.
- G. Ersan, M. S. Ersan, A. Kanan and T. Karanfil, Predictive modeling of haloacetonitriles under uniform formation conditions, Water Res., 2021, 201, 117322 CrossRef CAS PubMed.
- V. Glezer, B. Harris, N. Tal, B. Iosefzon and O. Lev, Hydrolysis of haloacetonitriles: Linear free energy relationship, Kinetics and products, Water Res., 1999, 33(8), 1938–1948 CrossRef CAS.
- Y. Yu and D. A. Reckhow, Kinetic Analysis of Haloacetonitrile Stability in Drinking Waters, Environ. Sci. Technol., 2015, 49(18), 11028–11036 CrossRef CAS PubMed.
- D. Reckhow and R. Yu, Personal communication, 2022 Search PubMed.
- S. Ding, W. Chu, S. W. Krasner, Y. Yu, C. Fang, B. Xu and N. Gao, The stability of chlorinated, brominated, and iodinated haloacetamides in drinking water, Water Res., 2018, 142, 490–500 CrossRef CAS PubMed.
- N. Moore, S. Ebrahimi, Y. Zhu, C. Wang, R. Hofmann and S. Andrews, A comparison of sodium sulfite, ammonium chloride, and ascorbic acid for quenching chlorine prior to disinfection byproduct analysis, Water Supply, 2021, 21(5), 2313–2323 CrossRef CAS.
- R. Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org. 2019.
- Hach, Chlorine, Free and Total, High Range, DOC3165301449, 2022 Search PubMed.
- American Water Works Association, 5310 TOTAL ORGANIC CARBON, Standard Methods For the Examination of Water and Wastewater, 2022 Search PubMed.
- E. S. Peterson, S. Johnson, S. Shiokari, Y. Yu, S. M. Cook and R. S. Summers, Impacts of carbon-based advanced treatment processes on disinfection byproduct formation and speciation for potable reuse, Water Res., 2022, 220 Search PubMed.
- T. Bond, M. R. Templeton, N. H. Mokhtar Kamal, N. Graham and R. Kanda, Nitrogenous disinfection byproducts in English drinking water supply systems: Occurrence, bromine substitution and correlation analysis, Water Res., 2015, 85, 85–94 CrossRef CAS PubMed.
- S. Ding, W. Chu, S. W. Krasner, Y. Yu, C. Fang and B. Xu, et al., The stability of chlorinated, brominated, and iodinated haloacetamides in drinking water, Water Res., 2018, 142, 490–500 CrossRef CAS PubMed.
- G. Hua and D. A. Reckhow, Effect of alkaline pH on the stability of halogenated DBPs, J. - Am. Water Works Assoc., 2012, 104(2), E107–E120 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00230f |
| This journal is © The Royal Society of Chemistry 2023 |