
Verteporfin-loaded supramolecular micelles for enhanced cisplatin-based chemotherapy via autophagy inhibition†
Junwei
Ye‡
ab,
Bo
Yu‡
c,
Haitao
Hu
ab,
Dongfang
Zhou
 d,
Qiao
Jin
*ce,
Jian
Ji
c and
Zhe
Tang
*abf
d,
Qiao
Jin
*ce,
Jian
Ji
c and
Zhe
Tang
*abf
aDepartment of Surgery, The Fourth Affiliated Hospital, School of Medicine, Zhejiang University, Yiwu, 322000, China. E-mail: 8xi@zju.edu.cn
bInternational Institutes of Medicine, The Fourth Affiliated Hospital, School of Medicine, Zhejiang University, Yiwu, 322000, China
cMOE Key Laboratory of Macromolecule Synthesis and Functionalization of Ministry of Education, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: jinqiao@zju.edu.cn
dSchool of Pharmaceutical Sciences, Southern Medical University, Guangzhou, 510515, China
eCancer Center of Zhejiang University, Hangzhou, 310006, China
fDepartment of Surgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, 310000, China
First published on 5th January 2022
Abstract
Cisplatin (CDDP) is one of the most successful chemotherapeutic agents for cancer therapy. However, CDDP can activate pro-survival autophagy, which inhibits the therapeutic efficacy of CDDP. Herein, autophagy inhibitor verteporfin (VTPF) is integrated into CDDP-conjugated micelles to address this issue. The CDDP-conjugated micelles are prepared by host–guest interaction of zwitterionic poly(2-(methacryloyloxy)ethyl phosphorylcholine)-co-poly(2-(methacryloyloxy)ethyl adamantane-1-carboxylate) (P(MPC-co-MAd)) and CDDP conjugated β-cyclodextrin (CD-CDDP). VTPF is then physically encapsulated into the supramolecular micelles by hydrophobic interaction. Due to the zwitterionic corona of the supramolecular micelles, the micelles are stable in different media. CDDP and VTPF could be released in a reductive environment. CDDP-activated autophagy could be inhibited by VTPF, which is fully characterized by western blot, fluorescence imaging, and transmission electron microscopy (TEM). Moreover, the outstanding therapeutic efficacy of CDDP and VTPF co-loaded micelles is validated both in vitro and in vivo. This research not only provides a new strategy to fabricate CDDP delivery systems by supramolecular self-assembly, but also presents an innovative way to enhance cisplatin-based chemotherapy via autophagy inhibition.
Introduction
cis-Diamminedichloridoplatinum(II) (cisplatin, CDDP), which is called “The Penicillin of Cancer”, is one of the most widely used chemotherapeutic drugs in cancer therapy in the world.1,2 Due to its highly effective and broad-spectrum antineoplastic activity, CDDP is used in the first-line treatment of small cell lung cancer, ovarian cancer, bladder cancer, prostate cancer, and so on. It is known that CDDP exerts its antineoplastic effects by forming intra-strand and inter-strand DNA cross-linking. However, the clinical performance of CDDP is strongly limited by its multi-drug resistance and serious adverse effects.3,4 The clinical outcome of CDDP-based chemotherapy indicates that CDDP shows serious dose-dependent toxicities, such as nephrotoxicity, myelosuppression, ototoxicity, and neurotoxicity. The very low water solubility is another obstacle for the clinical use of CDDP. The development of innovative drug delivery systems is therefore urgently needed to improve the therapeutic performance of CDDP in the clinic.5–7Autophagy is a very important physiological process in balancing sources of energy via a self-digesting procedure.8 In general, autophagy is considered as a self-preservation manner. If cancer cells are exposed to chemotherapeutic agents, reactive oxygen species (ROS), or hyperthermia, autophagy will be activated to inhibit the apoptosis of cancer cells. Therefore, the therapeutic efficacy of many anticancer strategies including chemotherapy, photodynamic therapy (PDT), and photothermal therapy (PTT) can be significantly inhibited by pro-survival autophagy.9–13 CDDP-based chemotherapy can also increase the autophagy level, leading to reduced cell apoptosis. The inhibition of autophagy might be an effective way to improve the therapeutic performance of CDDP.
In recent years, emerging nano drug delivery systems were well-developed as powerful tools to improve the therapeutic efficacy and reduce adverse effects of chemotherapeutic agents.14–18 Various platinum (Pt)-based drug delivery systems were reported.19–22 For example, NC-6004 (Nanoplatin™), a kind of CDDP-incorporating polymeric micelles composed of poly(ethylene glycol)–poly(α-glutamic acid) (PEG–PGA), could sustainably release CDDP and be selectively accumulated in tumor tissues, reaching the Phase III stage of clinical trials.23 Tobacco mosaic virus, glyco-decorated tobacco mosaic virus, and tobacco mosaic virus coat protein were used as promising vectors for CDDP delivery.24–26 CDDP-loaded gelatin nanoparticles camouflaged with cancer cell membrane were reported for personalized cancer therapy.27 Targeted drug delivery systems and biomimetic technologies were also fabricated to achieve efficient delivery of CDDP.28 Supramolecular assembly might be another very important tool to fabricate novel vectors for CDDP delivery although it is rarely reported. CDDP conjugated prodrug micelles are especially promising since they can achieve simultaneous long blood circulation, selective tumour accumulation, and minimal drug leakage.29,30 Moreover, CDDP-based prodrug micelles can also be used to encapsulate other chemotherapeutic agents for combination chemotherapy, which is an effective way to overcome drug resistance and improve the therapeutic efficacy.31,32 As a typical example, we fabricated nitric oxide (NO) loaded CDDP prodrug nanoassemblies, which could enhance the therapeutic efficacy of CDDP by peroxynitrite anion-mediated down-regulation of glutathione reductase and xeroderma pigmentosum group A (XPA).33
Due to the unique zwitterionic chemical structures, zwitterionic polymers with balanced positive and negative charges show great potential in designing various biomaterials.34–39 Phosphorylcholine (PC), carboxybetaine (CB), and sulfobetaine (SB) are three typical representatives of zwitterions, which show excellent stealth properties, low immunogenicity, and reduced accelerated blood clearance (ABC) phenomenon.40–44 Importantly, zwitterions even show much better properties than poly(ethylene glycol) (PEG) in many aspects. For example, zwitterionic micelles showed much longer blood circulation time than PEGylated micelles.45,46 Therefore, zwitterions are particularly promising in fabricating drug nanocarriers with outstanding physiological stability and biocompatibility.47
In this research, we fabricated CDDP and autophagy inhibitor verteporfin (VTPF) co-loaded zwitterionic supramolecular micelles by host–guest interaction for enhanced cancer chemotherapy (Scheme 1). CDDP conjugated micelles were firstly prepared by host–guest interaction between CDDP conjugated β-cyclodextrin (CD–CDDP) and poly(2-(methacryloyloxy)ethyl phosphorylcholine)-co-poly(2-(methacryloyloxy)ethyl adamantane-1-carboxylate) (P(MPC-co-MAd)). VTPF was then loaded into CDDP-conjugated micelles via hydrophobic interaction. CDDP and VTPF were expected to be released in an intracellular reductive environment. CDDP could induce cell apoptosis, accompanied by increased autophagy. The integration of VTPF into CDDP-conjugated micelles was expected to inhibit CDDP-triggered autophagy, leading to enhanced cancer chemotherapy.
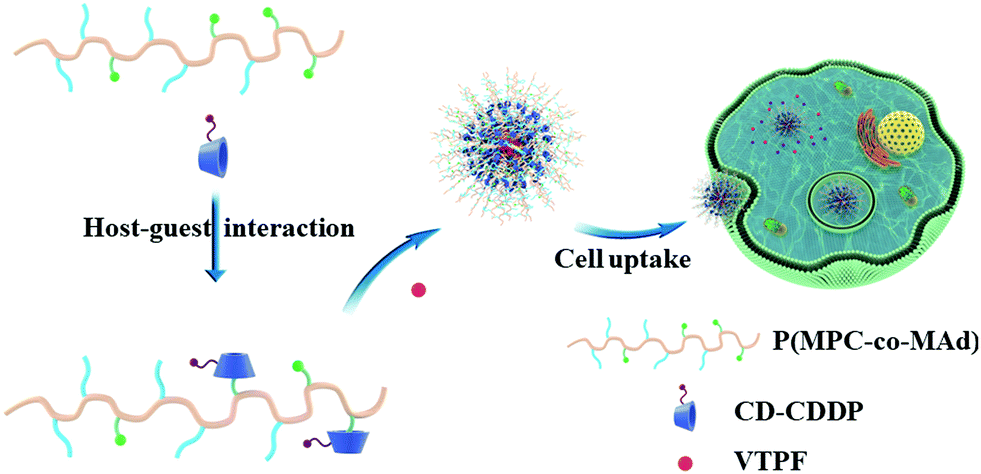 | ||
| Scheme 1 Schematic illustration of the self-assembly of CDDP and autophagy inhibitor VTPF co-loaded zwitterionic supramolecular micelles. | ||
Experimental section
Synthesis of 2-(methacryloyloxy)ethyl adamantane-1-carboxylate (MAd)
2-(Methacryloyloxy)ethyl adamantane-1-carboxylate (MAd) was synthesized by the reaction of acyl chloride and hydroxyl groups similar to our previous research.48 HEMA (3.6 mL, 0.03 mmol), triethylamine (4.2 mL, 0.03 mol), and 80 mL of anhydrous dichloromethane (CH2Cl2) were added in a three-necked flask. The flask was exposed in an ice water bath. 6 g of 1-adamantanecarbonyl chloride (0.03 mol) in 20 mL CH2Cl2 was added into the flask dropwise under an Ar atmosphere. The ice water bath was then removed and the solution was stirred overnight. After 24 h, the solution was filtered off and then extracted by distilled water twice. The solution was then concentrated by a rotary evaporator. The crude product was further purified by silica gel column chromatography. Light yellow liquid was finally obtained with a yield of 54%. Eluent: mixture of petroleum ether and ethyl acetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Synthesis of zwitterionic copolymer P(MPC-co-MAd)
P(MPC-co-MAd) was synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization. Briefly, azodiisobutyronitrile (AIBN, 3.7 mg, 22.5 μmol), 4-cyanopentanoic acid dithiobenzoate (25 mg, 90 μmol), MAd (788 mg, 2.7 mmol), and MPC (1.6 g, 5.4 mmol) were added in a Schlenk tube. 5 mL of methanol and 5 mL of N,N-dimethylformamide (DMF) were then added. The solution was then degassed by three freeze–pump–thaw cycles. After that, the pink solution was stirred at 60 °C. After 24 h, the reaction was terminated by exposing the solution to air. The solution was then precipitated into excess tetrahydrofuran (THF, 200 mL). The solid was collected and then dried under vacuum. Yield: 87%.Preparation of drug-loaded micelles
VTPF loaded micelles (VTPF MCs) were prepared by a typical cosolvent method. Briefly, 3 mg of VTPF and 20 mg of P(MPC-co-MAd) were dissolved in 4 mL mixed solvents of dimethyl sulfoxide (DMSO) and methanol (1:1). After the solution was stirred for 30 min, 4 mL of distilled water was added dropwise. After stirring for 1 h, the solution was dialyzed against distilled water for 2 days. Water was exchanged every 6 h. VTPF MCs were stored at 4 °C prior to use.
CDDP conjugated micelles (CDDP MCs) were prepared by host–guest interaction between β-cyclodextrin (β-CD) and adamantane (Ad). 15 mg of CDDP conjugated β-cyclodextrin (CD–CDDP) and 10 mg of P(MPC-co-MAd) were dissolved in 4 mL mixed solvents of DMSO and methanol (1:1). After the solution was stirred for 30 min, 4 mL of distilled water was added dropwise. After stirring for 1 h, the solution was dialyzed against distilled water for 2 days. Water was exchanged every 6 h. CDDP MCs were stored at 4 °C prior to use. In order to prepare CDDP and VTPF co-loaded micelles (CDDP/VTPF MCs), 3 mg of VTPF was added during the preparation of CDDP MCs.
The loading content of VTPF in the micelles was determined by UV-vis spectrophotometry after dissolving the micelles in DMSO. VTPF has a characteristic absorption at 692 nm. The loading content of CDDP in the micelles was determined by inductively coupled plasma-mass spectrometry (ICP-MS).
Reduction-sensitive drug release
2 mL of CDDP/VTPFMCs (2 mg mL−1) were added into a dialysis bag (3500 Da) by a pipette. The dialysis bag was immersed in 20 mL phosphate buffer saline (PBS) with or without 5 mM ascorbic acid. At the predetermined time intervals, 0.5 mL of external solution was collected to determine the concentration of CDDP and VTPF. 0.5 mL of fresh PBS with or without 5 mM ascorbic acid was supplemented. The release of CDDP and VTPF was determined by ICP-MS and UV-vis spectrophotometry, respectively.In vitro cell viability assay
The cytotoxicity of blank P(MPC-co-MAd) micelles was evaluated by cell counting kit-8 (CCK-8) assay using 200 μL of NIH-3T3 cells or LM3 cells in culture media (3 × 104 cells per milliliter). After cells were incubated for 24 h, different concentrations of P(MPC-co-MAd) were added. The cells were incubated for another 48 h and washed three times using PBS. 10 μL of CCK-8 was then added to each well. After 2 h incubation, the absorption value of each well was determined by a microplate reader at 450 nm. Data were expressed as average ± SD (n = 4).The cytotoxicity of CDDP MCs, VTPF MCs, and CDDP/VTPF MCs was evaluated by CCK-8 assay as well using 200 μL of LM3 cells in culture media (3 × 104 cells per milliliter). After LM3 cells were incubated for 24 h, different concentrations of CDDP MCs, VTPF MCs, and CDDP/VTPF MCs were added. The cells were incubated for 48 h and washed three times using PBS. 10 μL of CCK-8 was then added to each well. After 2 h incubation, the absorption value of each well was determined by a microplate reader at 450 nm. Data were expressed as average ± SD (n = 4).
In vitro detection of autophagy
The in vitro autophagy after different treatments was evaluated by western blot assay, monodansylcadaverine (MDC) staining, LC3 dot formation, and TEM images of autophagosomes. The detailed experimental procedure is presented in the ESI.†In vivo detection of autophagy
The in vivo autophagy after different treatments was measured by western blot assay. LM3 tumor bearing nude mice received 100 μL of saline, CDDP (5 mg kg−1), CDDP MCs (5 mg kg−1), VTPF MCs (6.1 mg kg−1), CDDP/VTPF MCs (5 mg kg−1 CDDP and 6.1 mg kg−1 VTPF), or a mixture of VTPF MCs and CDDP (5 mg kg−1 CDDP and 6.1 mg kg−1 VTPF). The mice were sacrificed after 24 h and the tumor tissues were harvested. The collected tumor tissues were then minced and homogenized in protein lysate buffer. The levels of LC3 and p62 were measured by western blot assay as mentioned above.In vivo tumor growth inhibition on a subcutaneous hepatoma model
The antitumor experiment was carried on a subcutaneous hepatoma model. 100 μL of LM3 cells (4 × 107 cells per milliliter) was subcutaneously administrated in the right flank region of nude mice. After the tumor was grown to about 150 mm3, the nude mice were randomly divided into five groups (four mice in every group). 100 μL of saline, CDDP (5 mg kg−1), CDDP MCs (5 mg kg−1), VTPF MCs (6.1 mg kg−1), or CDDP/VTPF MCs (5 mg kg−1 CDDP and 6.1 mg kg−1 VTPF) were injected into the tumor-bearing nude mice via the tail vein on day 0, day 3, and day 6, respectively. The tumor volume and body weight of each nude mouse were recorded regularly. The mice were sacrificed on day 21. The main organs (heart, liver, spleen, lung, kidney) and tumor tissues were harvested. The tumor tissues of each group were weighed and photographed. Subsequently, tumor tissues were fixed in 4% formaldehyde for histological analysis. Hematoxylin and eosin (H&E) staining, Ki67, and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assays were employed to further confirm the proliferation and apoptosis after treatments. The main organs including the heart, liver, spleen, lungs, and kidneys were fixed in 4% formaldehyde for H&E staining. The tumor volume was calculated according to the following equation:| Volume = (tumor length) × (tumor width)2/2. |
Results and discussion
Synthesis and characterization of P(MPC-co-MAd)
The synthetic route for the synthesis of MAd and P(MPC-co-MAd) is illustrated in Scheme S1 (ESI†). MAd was obtained by the reaction of 2-hydroxyethyl methacrylate (HEMA) and 1-adamantanecarbonyl chloride. The 1H NMR spectrum of MAd in Fig. S1 (ESI†) proved the successful synthesis of MAd. All the peaks and their integrals matched well with the chemical structure of MAd. P(MPC-co-MAd) was synthesized by RAFT polymerization of MPC and MAd using 4-cyanopentanoic acid dithiobenzoate as the chain transfer agent (CTA). The 1H NMR result of P(MPC-co-MAd) is shown in Fig. 1. By comparing the integrals of the characteristic peak of CTA (δ 7.46, δ 7.62, δ 7.89), the MPC segment (δ 3.71, δ 4.02), and the MAd segment (δ 4.18, 4.27), the polymerization degrees of MPC and MAd were calculated to be 55 and 27, respectively. Hence, the molecular weight of P(MPC-co-MAd) was about 24200 Da. Since P(MPC-co-MAd) can only be molecularly dissolved in mixed solvents, it is very difficult to determine the molecular weight and molecular weight distribution by gel permeation chromatography (GPC). CDDP conjugated β-cyclodextrin (CD–CDDP) was synthesized according to our previous publication (Scheme S2, ESI†).33 CD–CDDP showed strong host–guest interaction with adamantane. The binding constant of CD–CDDP and adamantane was 2.06 × 105 M−1 detected by isothermal titration calorimetry (ITC).33
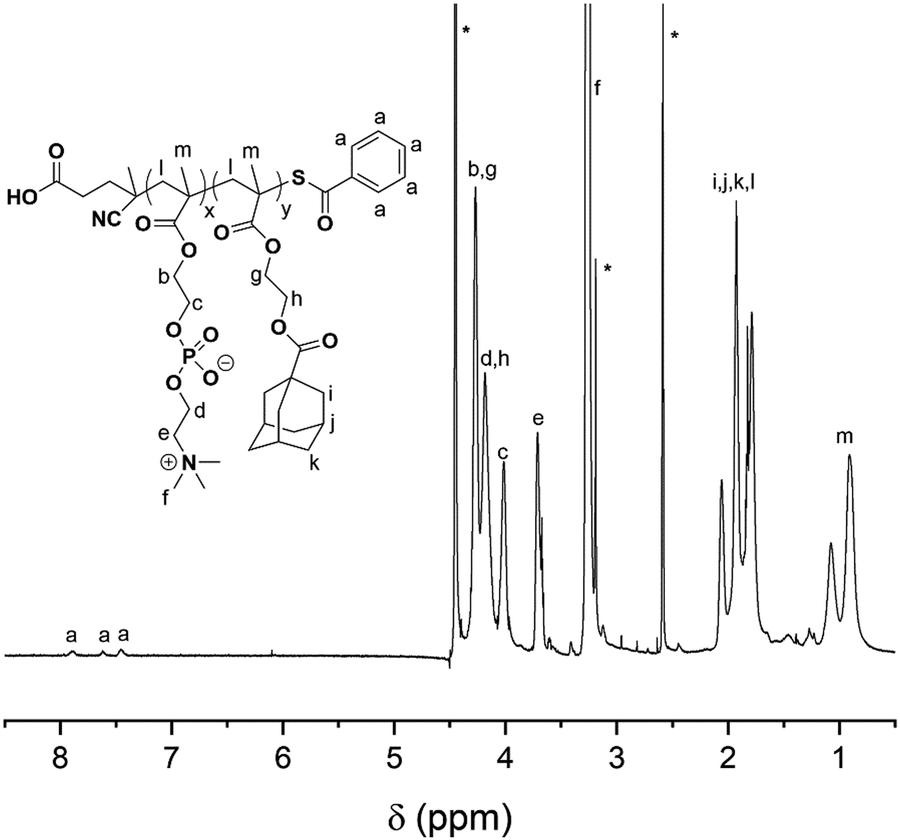 | ||
| Fig. 1 1H NMR spectrum of P(MPC-co-MAd) in the mixed solvent of CD3OD and DMSO-d6. | ||
Preparation of drug-loaded micelles
CDDP conjugated micelles (CDDP MCs) were prepared by host–guest interaction of CD–CDDP and P(MPC-co-MAd). The intensity-averaged hydrodynamic diameter of CDDP MCs was 124.6 nm with a polydispersity index (PDI) of 0.18, measured by dynamic light scattering (DLS) (Fig. 2A). Meanwhile, the TEM image in Fig. 2B indicated that CDDP MCs showed a spherical morphology with a diameter of 103.4 nm. The relatively small size of CDDP MCs determined by TEM might be ascribed to the shrinkage of the micelles after drying. The DLS and TEM results confirmed the successful preparation of zwitterionic supramolecular micelles.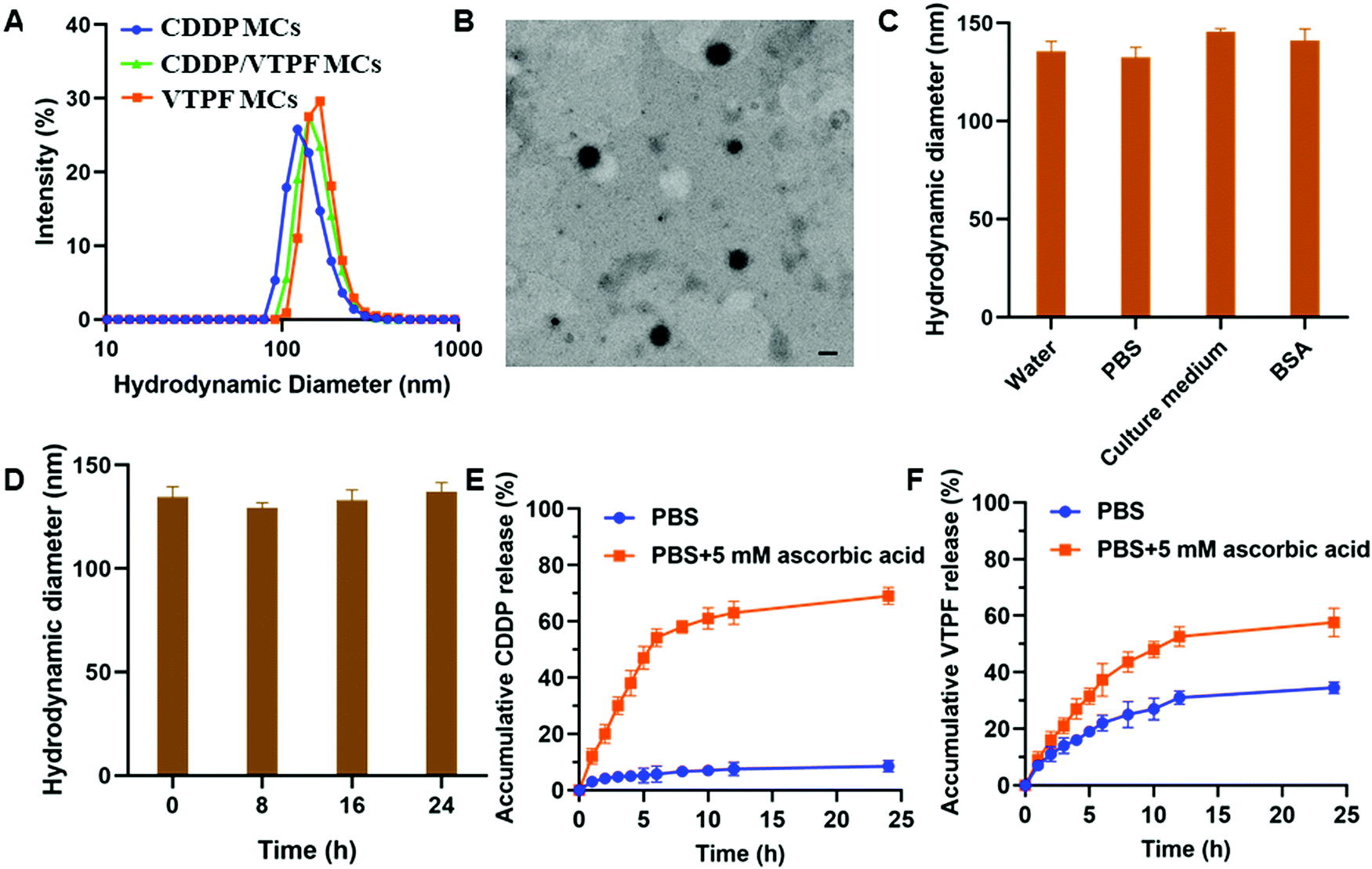 | ||
| Fig. 2 (A) The intensity-averaged hydrodynamic diameter of CDDP MCs, VTPF MCs, and CDDP/VTPF MCs. (B) The TEM image of CDDP MCs. Scale bar: 100 μm. (C) The hydrodynamic diameter of CDDP/VTPF MCs in different media, including water, PBS, cell culture medium, and 2 mg mL−1 BSA. (D) The hydrodynamic diameter of CDDP/VTPF MCs after incubating in PBS for different times. (E) The in vitro release of CDDP in PBS (pH 7.4) with or without 5 mM ascorbic acid. (F) The in vitro release of VTPF in PBS (pH 7.4) with or without 5 mM ascorbic acid. | ||
CDDP and VTPF co-loaded micelles (CDDP/VTPF MCs) were prepared by physical encapsulation of VTPF in CDDP MCs. The intensity-averaged hydrodynamic diameter of the CDDP/VTPF MCs was 139.1 nm, slightly larger than that of CDDP MCs. Meanwhile, VTPF loaded micelles (VTPF MCs) were prepared by physical encapsulation of VTPF into P(MPC-co-MAd) micelles via hydrophobic interaction. The loading content of CDDP in CDDP MCs was about 7.6% measured by ICP-MS. The loading content of VTPF in VTPF MCs was about 9.3% measured by UV-vis spectrophotometry. The loading contents of CDDP and VTPF in CDDP/VTPF MCs were 7.4% and 8.9%, respectively. The mole ratio of CDDP and VTPF in CDDP/VTPF MCs was about 2:1. According to the loading contents of CDDP, 11 adamantane molecules in one polymer chain combined with CD–CDDP, which implied that 16 adamantane molecules in one polymer chain were uncapped with CD–CDDP. The uncapped adamantane in P(MPC-co-MAd) was very hydrophobic, which could provide the driving force to form supramolecular micelles by hydrophobic interaction. Hydrophobic VTPF can then be encapsulated into CDDP MCs to obtain CDDP/VTPF MCs. The stability of the supramolecular micelles is very important for drug delivery. As shown in Fig. 2C, the hydrodynamic diameter of CDDP/VTPF MCs in different media including water, PBS, cell culture medium, and 2 mg mL−1 bovine serum albumin (BSA) didn’t change much. Meanwhile, CDDP/VTPF MCs exhibited excellent stability in PBS with negligible size change after 24 h (Fig. 2D). These results proved the excellent stability of CDDP/VTPF MCs, which is a prerequisite for drug delivery.
In vitro drug release
As an ideal drug delivery system, drugs should be retained in the micelles during circulation with minimal drug leakage. However, drugs should be released rapidly after being internalized by cancer cells. In this research, CDDP was conjugated to β-cyclodextrin (β-CD) by reduction-sensitive linkage, obtaining inert Pt(IV)–β-CD conjugates (CD–CDDP). The inert CD–CDDP could be reduced to cytotoxic Pt(II) by intracellular reductive compounds, such as ascorbic acid, glutathione (GSH), and cysteine, leading to the release of CDDP. The released CDDP can bind with DNA to induce cell apoptosis. The release of CDDP and VTPF from CDDP/VTPF MCs was then investigated in the presence or absence of 5 mM ascorbic acid, which simulated the intracellular reductive environment and extracellular physiological environment. As shown in Fig. 2E, the release of CDDP was very slow in PBS since CDDP was chemically conjugated to the micelles. Only less than 10% of CDDP could be released from CDDP/VTPF MCs after 24 h. However, the release of CDDP could be dramatically accelerated if CDDP/VTPF MCs were incubated with 5 mM ascorbic acid, leading to more than 50% release of CDDP after 6 h. At the same time, the release of CDDP in 5 mM ascorbic acid could make the micellar core more hydrophilic, which is helpful for the release of VTPF. Therefore, the release of VTPF could also be accelerated in 5 mM ascorbic acid (Fig. 2F).After CDDP was released from CDDP MCs, the hydrophobic micellar core would be more hydrophilic. However, because of the presence of uncapped hydrophobic adamantane, CDDP MCs would not be disassembled after drug release. The hydrodynamic diameter of CD–CDDP NPs reduced from 124.6 nm to 96.8 nm (Fig. S2, ESI†), which might be attributed to the decrease of hydrophobic segments after drug release.
Investigation of autophagy in vitro
Autophagy is known to play a pro-survival role in chemotherapy, which can inhibit cell apoptosis. In order to address this issue, the autophagy inhibitor VTPF was integrated into CDDP-conjugated micelles for enhanced cancer chemotherapy. The autophagy level was then investigated after different treatments. The autophagy-related proteins including LC3 and p62 were detected at first by western blot to investigate the changes of autophagic flux after different treatments. LC3-I localizes in the cytosol and LC3-II is generally found in the autophagosomes. LC3-I will conjugate with phosphatidylethanolamine to form LC3-II in the autophagy process. Furthermore, p62 is the substrate protein of autophagy, which will be specifically degraded during autophagy. As shown in Fig. 3A–C, compared to the PBS treated control group, VTPF MCs could lead to a 31.1% reduction of the LC3-II/LC3-I ratio and a 14.2% increase of p62 level, which indicated that VTPF could effectively inhibit cellular autophagy. Meanwhile, CDDP MCs could result in a 3-fold increase of LC3-II/LC3-I ratio as well as a 63% decrease of p62 expression, which indicated that CDDP could significantly increase autophagy of LM3. However, compared to CDDP MC treated LM3 cells, the autophagy level of CDDP/VTPF MC treated cells was greatly reduced, accompanied by a significantly reduced LC3-II/LC3-I ratio and much higher p62 expression.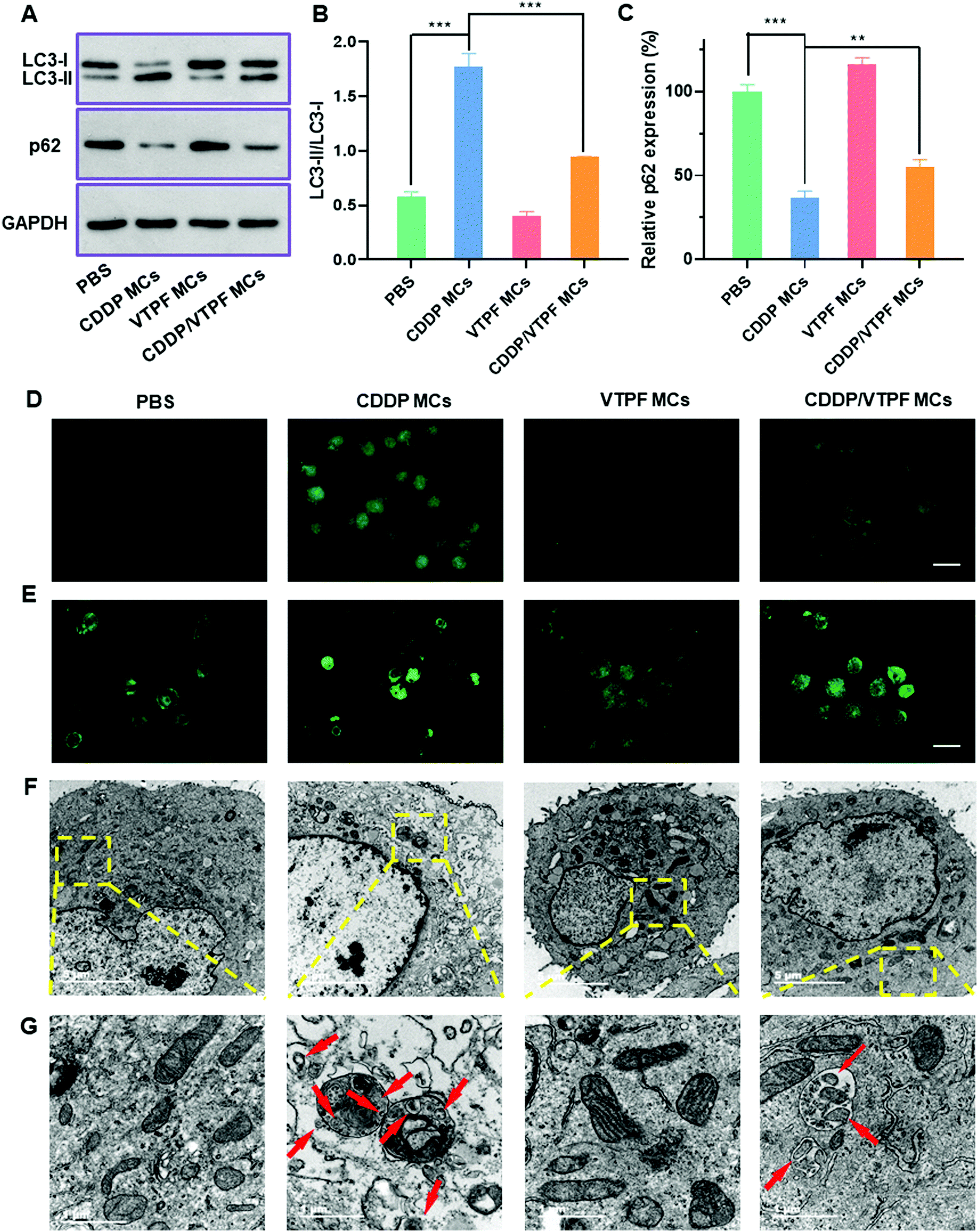 | ||
| Fig. 3 Study of autophagy after LM3 cells were treated with different drug-loaded micelles in vitro. (A) Western blot results of LC3-I, LC3-II, and p62 levels after LM3 cells were treated with CDDP MCs, VTPF MCs, or CDDP/VTPF MCs. (B) Quantitative analysis of the LC3-II/LC3-I ratio after different treatments. (C) Quantitative analysis of relative p62 level after LM3 cells were treated with different drug-loaded micelles. (D) Immunofluorescent images of LC3 punctate dots after LM3 cells were treated with CDDP MCs, VTPF MCs, or CDDP/VTPF MCs. Scale bar: 50 μm. (E) Fluorescent images of MDC staining after LM3 cells were treated with CDDP MCs, VTPF MCs, or CDDP/VTPF MCs. Scale bar: 50 μm. (F) TEM images of LM3 cells after being treated with CDDP MCs, VTPF MCs, or CDDP/VTPF MCs to observe the formation of autophagosomes. Scale bar: 5 μm. (G) The magnified TEM images of (F). Typical autophagosomes were labelled with red arrows. Scale bar: 1 μm. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
The formation of LC3 punctate dots was further visually observed by immunofluorescent imaging (Fig. 3D). Compared to the untreated PBS group, CDDP MC treated LM3 cells exhibited much stronger green fluorescence of LC3-II punctate dots, implying the activation of autophagy by CDDP MCs. Furthermore, if LM3 cells were treated with CDDP/VTPF MCs, the green fluorescence of LC3-II punctate dots was much weaker than that in CDDP MC treated cells, which indicated that CDDP-triggered autophagy could be effectively inhibited by VTPF. In the process of autophagy, acidic vesicular autophagosomes will be produced. A monodansylcadaverine (MDC) staining assay was used to observe the formation of acidic autophagosomes (Fig. 3E). As expected, much stronger fluorescence was observed in CDDP MC treated LM3 cells than that in the untreated PBS group, indicating the generation of more autophagosomes. However, if VTPF was integrated into CDDP MCs, the CDDP-activated autophagy could be efficiently inhibited, leading to much weaker fluorescence of acidic autophagosomes, which was in accordance with western blot and LC3 immunofluorescent imaging results. Furthermore, the formation of autophagosomes was directly observed by TEM. As shown in Fig. 3F and G, autophagosomes almost cannot be observed in PBS treated LM3 cells. However, a lot of autophagosomes could be observed in CDDP MC treated LM3 cells clearly, which again indicated the activation of autophagy by CDDP-based chemotherapy. Moreover, the quantity of autophagosomes was greatly reduced if VTPF was loaded into CDDP MCs, implying the efficient inhibition of autophagy by VTPF. Therefore, VTPF exhibited strong autophagy inhibition ability, which might enhance the therapeutic efficacy of CDDP-based chemotherapy.
In vitro cytotoxicity studies
Since CDDP-activated autophagy could be inhibited by VTPF, we further investigated if the cytotoxicity of CDDP could be enhanced by VTPF. At first, the biocompatibility of the zwitterionic blank P(MPC-co-MAd) micelles was studied by CCK-8 assay. As depicted in Fig. S3 (ESI†), the blank P(MPC-co-MAd) micelles didn’t show any cytotoxicity against NIH-3T3 and LM3 cells even when the concentration was as high as 1 mg mL−1, which indicated that zwitterions are good candidates to fabricate biocompatible micelles. The cytotoxicity of the drug-loaded micelles was then studied by CCK-8 assay after incubating with LM3 cells for 48 h. CDDP MCs exhibited obvious cytotoxicity against LM3 cells with a half-maximal inhibitory concentration (IC50) of 3.59 μM (Fig. 4). Free CDDP exhibited stronger cytotoxicity than CDDP MCs against LM3 cells with IC50 of 3.34 μM, which might be ascribed to the fast internalization and binding with DNA. Although VTPF MCs alone didn’t inhibit cell proliferation with more than 95% cell viability, CDDP/VTPF MCs exhibited much stronger cytotoxicity than CDDP MCs with IC50 of 3.01 μM, implying a strong synergistic effect of CDDP and VTPF in inhibiting the proliferation of LM3 cells. The remarkable cytotoxicity of CDDP/VTPF MCs might be ascribed to the inhibition of CDDP-activated autophagy by VTPF, which enhanced the cytotoxicity of CDDP.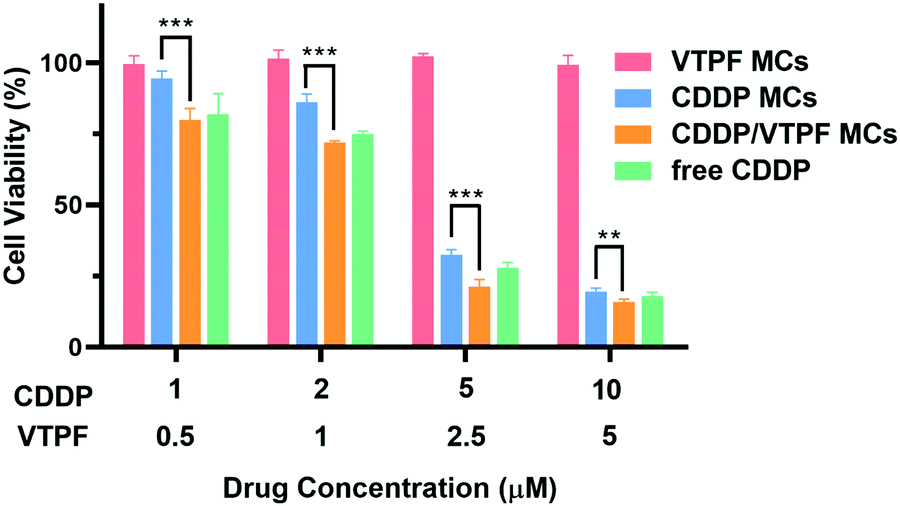 | ||
| Fig. 4 Viability curves of LM3 cells after treatment with different concentrations of CDDP MCs, VTPF MCs, or CDDP/VTPF MCs for 48 h (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001. | ||
In vivo autophagy inhibition by VTPF
Since VTPF could effectively inhibit CDDP-induced autophagy in vitro, it is interesting to know if VTPF could inhibit autophagy in vivo. After nude mice were received with different formulations, the LC3-II/LC3-I ratio and p62 level of the tumor tissues were detected by western blot. As shown in Fig. 5, the nude mice that received CDDP MCs exhibited a significantly higher autophagy level because of the increase of LC3-II/LC3-I ratio and decrease of p62 level. Specifically, the LC3-II/LC3-I ratio in the CDDP MCs treated group was 10-fold higher than that of the saline treated group. Meanwhile, the p62 level in the CDDP MC treated group reduced to 34.9% that of the saline group. These results indicated that CDDP MCs can induce cellular autophagy in vivo. Because autophagy is known to inhibit the apoptosis of cancer cells in CDDP-based chemotherapy, we intended to inhibit CDDP-induced autophagy by the integration of autophagy inhibitor VTPF. As expected, compared to the CDDP MC treated group, the LC3-II/LC3-I ratio in the CDDP/VTPF MC treated group reduced to as low as 21.1%, accompanied by the increase of the p62 level, which implied that the introduction of VTPF into CDDP MCs could effectively inhibit CDDP-induced autophagy. At the same time, the autophagy level in the VTPF MCs and CDDP treated group was also relatively low. Therefore, the integration of VTPF in CDDP-based chemotherapy could effectively inhibit cellular autophagy in vivo, which is beneficial for enhanced CDDP-based cancer therapy.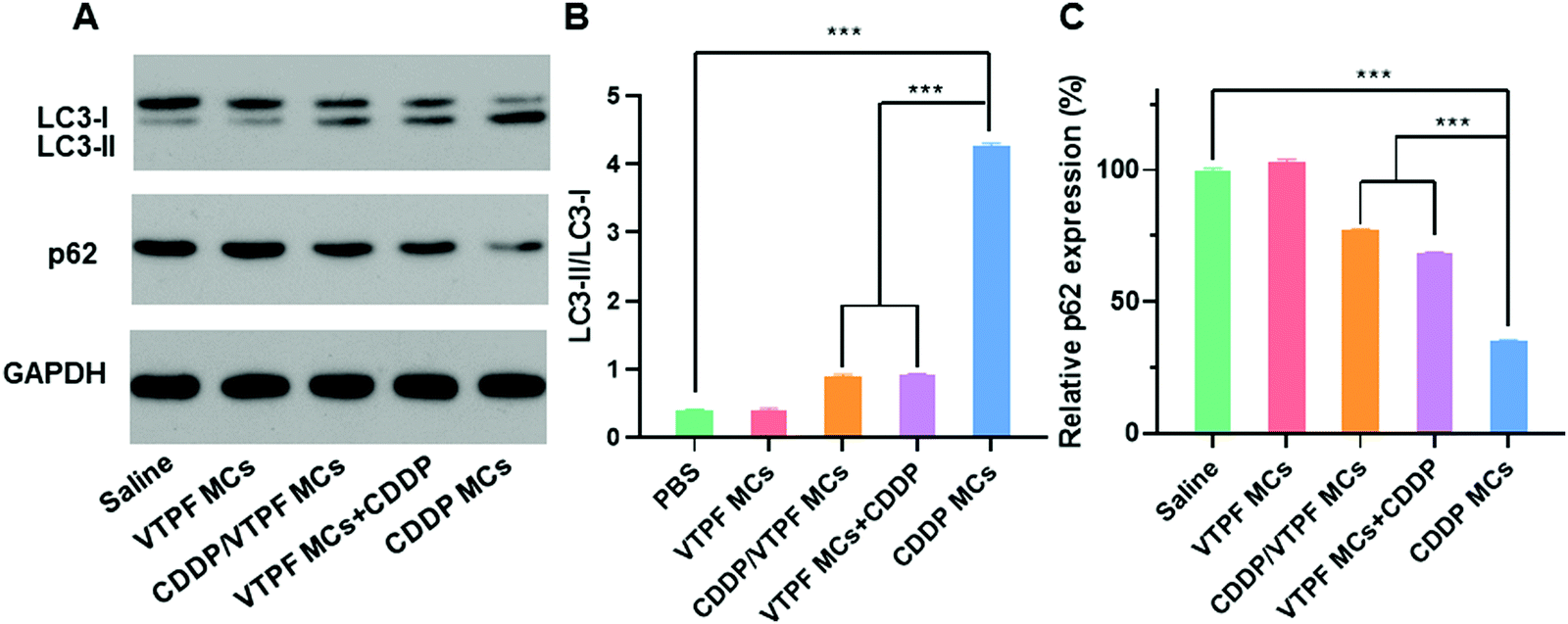 | ||
| Fig. 5 Study of autophagy after LM3 tumor-bearing nude mice were treated with different formulations in vivo. (A) Western blot results of LC3-I, LC3-II, and p62 levels after the mice were treated with saline, CDDP MCs, VTPF MCs, CDDP/VTPF MCs or a mixture of VTPF MCs and CDDP. (B) Quantitative analysis of LC3-II/LC3-I ratio after different treatments. (C) Quantitative analysis of relative p62 level after different treatments. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
In vivo antitumor evaluation
Encouraged by the excellent autophagy inhibition ability of VTPF, the in vivo antitumor efficacy of the zwitterionic supramolecular micelles was evaluated on a subcutaneous hepatoma xenograft model. The LM3 tumor-bearing nude mice were randomly divided into 5 groups and treated with different formulations (saline, CDDP, CDDP MCs, VTPF MCs, and CDDP/VTPF MCs). The therapeutic performance of different treatments was then evaluated by tracking the changes of tumor volume. As shown in Fig. 6A, CDDP MCs showed a moderate antitumor effect with about 5-fold increase of the tumor volume. VTPF MCs almost cannot inhibit tumor growth, with as high as an 8-fold increase of the tumor volume after 21 days, which was similar to the saline treated group. Interestingly, CDDP/VTPF MCs exhibited much better antitumor activity than CDDP MCs. A 2.8-fold increase of the tumor volume was observed after the mice were treated with CDDP/VTPF MCs. The much better antitumor activity of CDDP/VTPF MCs might be attributed to the inhibition of autophagy by VPTF. After 21 days post-injection, the mice were sacrificed and the tumor tissues were harvested and photographed. The digital photographs of the tumors after different treatments intuitively confirmed the excellent antitumor activity of the CDDP/VTPF MCs (Fig. 6B). The average tumor weights were 1.41, 0.27, 0.58, 1.35, and 0.18 g for the mice treated with saline, free CDDP, CDDP MCs, VTPF MCs, and CDDP/VTPF MCs, respectively. It should be noted that free CDDP exhibited much better antitumor activity than CDDP MCs. CDDP MCs should penetrate into the tumor tissues and then release CDDP to kill cancer cells. Compared to free CDDP, the tumor penetration of CDDP MCs and release of CDDP might be a problem, which can influence the therapeutic efficacy of CDDP. Therefore, it is possible that free CDDP exhibited better antitumor activity than CDDP MCs, which was also found in the previous literature.32,33,49 The therapeutic efficacy of CDDP MCs could be improved by introducing targeting ligands into CDDP MCs. However, free CDDP resulted in serious side effects. An obvious decrease of body weight was observed after the mice were injected with free CDDP (Fig. 6C). Furthermore, protein-like precipitates were observed in the gaps of the renal tubules in the H&E image of the kidney section for the mice treated with free CDDP, which was not observed in other groups, implying severe nephrotoxicity of free CDDP (Fig. 7). Therefore, the severe side effects of cisplatin could be effectively reduced after it was integrated into supramolecular micelles, which implied that the maximum-tolerated dose (MTD) of cisplatin could be increased in the form of CDDP MCs.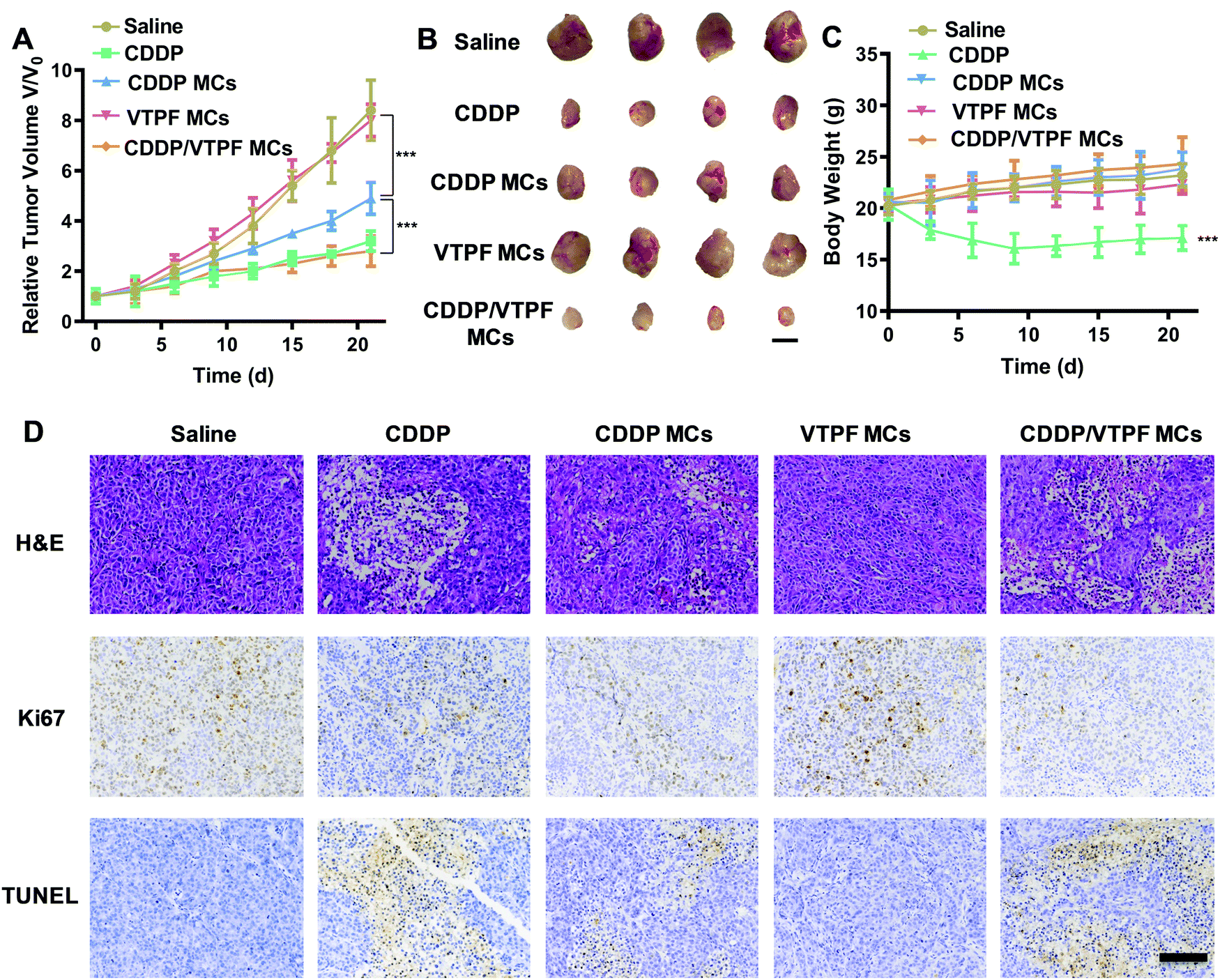 | ||
| Fig. 6 In vivo antitumor evaluation on a subcutaneous hepatoma xenograft model. The mice were treated with saline, CDDP, CDDP MCs, VTPF MCs, or CDDP/VTPF MCs, respectively. (A) Tumor growth inhibition curves after different treatments (n = 4). (B) Photograph of the harvested tumor tissues after different treatments. (C) The change of bodyweights of the mice after different treatments (n = 4). (D) Immunohistochemical analysis of the tumor sections using H&E, Ki67, and TUNEL after different treatments. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
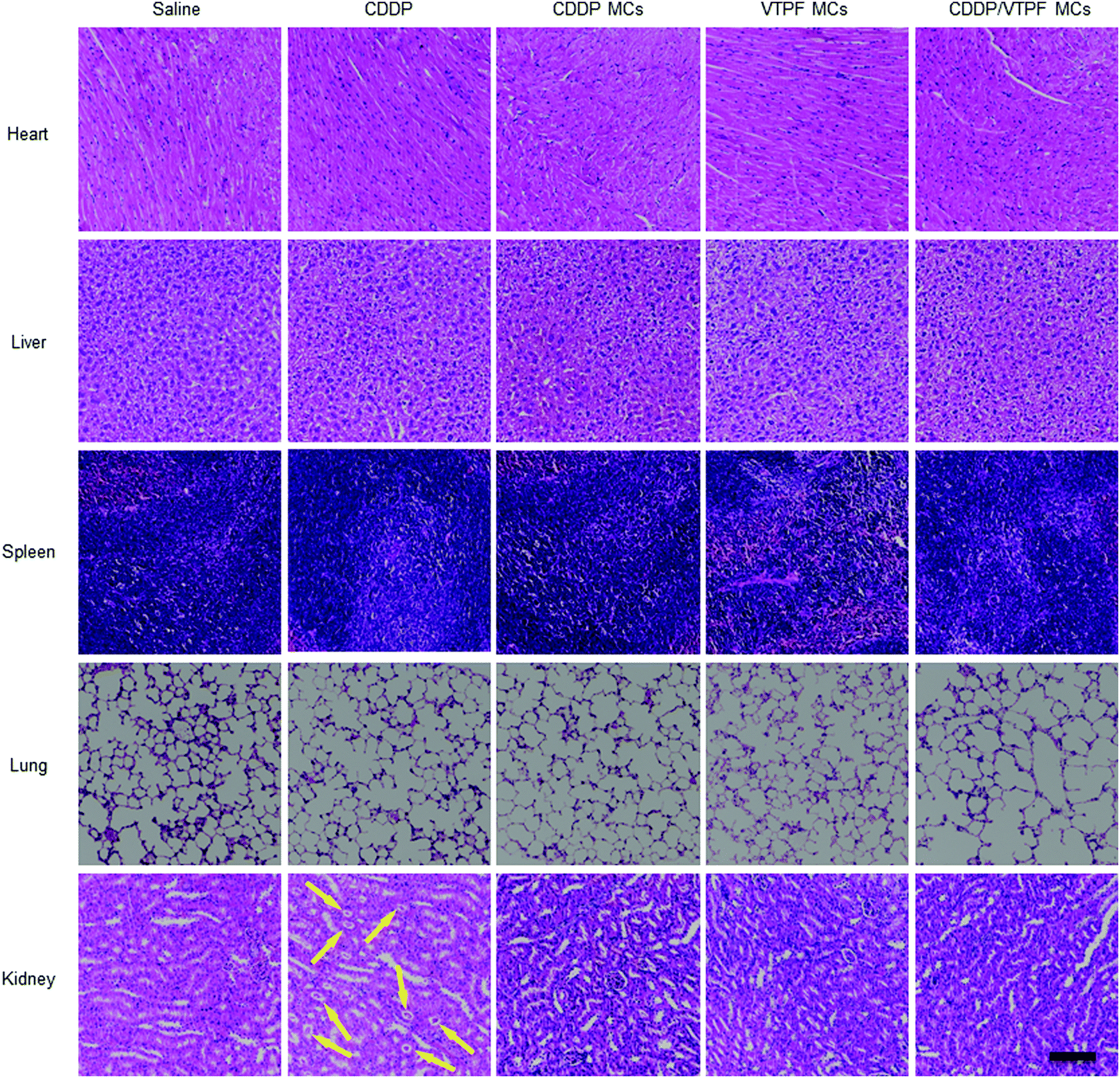 | ||
| Fig. 7 H&E staining of major organs (heart, liver, spleen, lung, kidney) harvested from tumor-bearing nude mice after different treatments. The protein-like precipitate in the H&E staining of the kidneys was labelled with yellow arrows. Scale bar: 100 μm. | ||
In order to further obtain insight into the antitumor activity of the supramolecular drug-loaded micelles, histologic sections of the tumor tissues after different treatments were then analyzed by immunohistochemistry, including H&E staining, Ki67 assay, and TUNEL assay (Fig. 6D). The immunohistochemistry results were in accordance with the tumor growth inhibition curves. CDDP/VTPF MCs gave rise to the highest apoptosis and lowest proliferation levels in tumor tissues compared to the control groups. These results together further indicated the excellent antitumor capability of CDDP/VTPF MCs.
Finally, the biosafety of the zwitterionic supramolecular micelles was evaluated. The bodyweight of the mice was almost not changed after being treated with CDDP MCs, VTPF MCs, or CDDP/VTPF MCs, indicating relatively low side effects (Fig. 6C). Meanwhile, the H&E staining of the major organs including the heart, liver, spleen, lungs, and kidneys indicated that the zwitterionic micelles didn’t show histopathological toxicity, implying low systematic toxicity (Fig. 7).
Conclusion
In summary, CDDP and VTPF co-loaded zwitterionic supramolecular micelles (CDDP/VTPF MCs) were successfully prepared by host–guest interactions. CDDP/VTPF MCs showed excellent stability in different media. Both CDDP and VTPF could be effectively released from CDDP/VTPF MCs in a reductive environment. After being incubated with LM3 cells, CDDP could activate autophagy to protect cells from apoptosis. However, the CDDP activated autophagy could be effectively inhibited by VTPF, which was confirmed by western blot, fluorescent images, and TEM images. The inhibition of autophagy by VTPF effectively enhanced the cytotoxicity of CDDP. The outstanding antitumor activity of CDDP/VTPF MCs was proved by a subcutaneous hepatocarcinoma model on nude mice. Meanwhile, CDDP/VTPF MCs exhibited excellent biosafety. This research provided a new strategy to enhance Pt-based chemotherapy via autophagy inhibition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the key project of the Natural Science Foundation of Zhejiang Province (LZ20H160002), the key research and development project of Zhejiang Province (2021C03048), and the National Natural Science Foundation of China (52022090) is gratefully acknowledged.References
- H. H. Xiao, L. S. Yan, E. M. Dempsey, W. T. Song, R. G. Qi, W. L. Li, Y. B. Huang, X. B. Jing, D. F. Zhou, J. X. Ding and X. S. Chen, Prog. Polym. Sci., 2018, 87, 70–106 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- H. Maeda and M. Khatami, Clin. Transl. Med., 2018, 7, 20 Search PubMed.
- S. Manohar and N. Leung, J. Nephrol., 2018, 31, 15–25 CrossRef CAS PubMed.
- R. J. Browning, P. J. T. Reardon, M. Parhizkar, R. B. Pedley, M. Edirisinghe, J. C. Knowles and E. Stride, ACS Nano, 2017, 11, 8560–8578 CrossRef CAS PubMed.
- D. F. Zhou, H. H. Xiao, F. B. Meng, X. Y. Li, Y. X. Li, X. B. Jing and Y. B. Huang, Adv. Healthcare Mater., 2013, 2, 822–827 CrossRef CAS PubMed.
- D. S. Wei, Y. J. Yu, Y. Huang, Y. M. Jiang, Y. Zhao, Z. X. Nie, F. Y. Wang, W. Ma, Z. Q. Yu, Y. Y. Huang, X. D. Zhang, Z. Q. Liu, X. C. Zhang and H. H. Xiao, ACS Nano, 2021, 15, 5428–5438 CrossRef CAS PubMed.
- D. Glick, S. Barth and K. F. Macleod, J. Pathol., 2010, 221, 3–12 CrossRef CAS PubMed.
- J. J. Reiners, P. Agostinis, K. Berg, N. L. Oleinick and D. Kessel, Autophagy, 2010, 6, 7–18 CrossRef CAS PubMed.
- Z. J. Zhou, Y. Yan, K. W. Hu, Y. Zou, Y. W. Li, R. Ma, Q. Zhang and Y. Y. Cheng, Biomaterials, 2017, 141, 116–124 CrossRef CAS PubMed.
- W. Yu, Y. T. Wang, J. Zhu, L. B. Jin, B. Liu, K. S. Xia, J. J. Wang, J. Q. Gao, C. Z. Liang and H. M. Tao, Biomaterials, 2019, 192, 128–139 CrossRef CAS PubMed.
- X. Sui, R. Chen, Z. Wang, Z. Huang, N. Kong, M. Zhang, W. Han, F. Lou, J. Yang, Q. Zhang, X. Wang, C. He and H. Pan, Cell Death Dis., 2013, 4, 12 Search PubMed.
- M. L. Chen, D. Yang, Y. Sun, T. Liu, W. H. Wang, J. T. Fu, Q. Q. Wang, X. Q. Bai, G. L. Quan, X. Pan and C. B. Wu, ACS Nano, 2021, 15, 3387–3401 CrossRef CAS PubMed.
- H. J. Han, Y. Hou, X. H. Chen, P. S. Zhang, M. X. Kang, Q. Jin, J. Ji and M. Y. Gao, J. Am. Chem. Soc., 2020, 142, 4944–4954 CrossRef CAS PubMed.
- Q. Jin, Y. Y. Deng, X. H. Chen and J. Ji, ACS Nano, 2019, 13, 954–977 CAS.
- P. Tan, H. Cai, Q. Wei, X. D. Tang, Q. F. Zhang, M. Kopytynski, J. X. Yang, Y. Yi, H. Zhang, Q. Y. Gong, Z. W. Gu, R. J. Chen and K. Luo, Biomaterials, 2021, 277, 13 Search PubMed.
- X. Y. Wong, A. Sena-Torralba, R. Alvarez-Diduk, K. Muthoosamy and A. Merkoci, ACS Nano, 2020, 14, 2585–2627 CrossRef CAS PubMed.
- X. H. Chen, H. Q. Gao, Y. Y. Deng, Q. Jin, J. Ji and D. Ding, ACS Nano, 2020, 14, 5121–5134 CrossRef CAS PubMed.
- P. Xie, Y. S. Wang, D. S. Wei, L. P. Zhang, B. Zhang, H. H. Xiao, H. Q. Song and X. Z. Mao, J. Mater. Chem. B, 2021, 9, 5173–5194 RSC.
- D. S. Wei, Y. J. Yu, X. C. Zhang, Y. H. Wang, H. Chen, Y. Zhao, F. Y. Wang, G. H. Rong, W. W. Wang, X. Kang, J. Cai, Z. H. Wang, J. Y. Yin, M. Hanif, Y. B. Sun, G. F. Zha, L. X. Li, G. H. Nie and H. H. Xiao, ACS Nano, 2020, 14, 16984–16996 CrossRef CAS PubMed.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed.
- Y. J. Yu, Q. Xu, S. S. He, H. J. Xiong, Q. F. Zhang, W. G. Xu, V. Ricotta, L. Bai, Q. Zhang, Z. Q. Yu, J. X. Ding, H. H. Xiao and D. F. Zhou, Coord. Chem. Rev., 2019, 387, 154–179 CrossRef CAS.
- V. Subbiah, J. E. Grilley-Olson, A. J. Combest, N. Sharma, R. H. Tran, L. Bobe, A. Osada, K. Takahashi, J. Balkissoon, A. Camp, A. Masada, D. J. Reitsma and L. A. Bazhenova, Clin. Cancer Res., 2018, 24, 43–51 CrossRef CAS PubMed.
- X. Liu, B. Liu, S. Gao, Z. Wang, Y. Tian, M. Wu, S. Jiang and Z. Niu, J. Mater. Chem. B, 2017, 5, 2078–2085 RSC.
- S. Gao, X. Liu, Z. Wang, S. Jiang, M. Wu, Y. Tian and Z. Niu, Nanoscale, 2018, 10, 11732–11736 RSC.
- C. E. Franke, A. E. Czapar, R. B. Patel and N. F. Steinmetz, Mol. Pharmaceutics, 2018, 15, 2922–2931 CrossRef CAS PubMed.
- L. Rao, G. T. Yu, Q. F. Meng, L. L. Bu, R. Tian, L. S. Lin, H. Deng, W. Yang, M. Zan, J. Ding, A. Li, H. Xiao, Z. J. Sun, W. Liu and X. Chen, Adv. Funct. Mater., 2019, 29, 1905671 CrossRef CAS.
- L. Chen, W. Hong, W. Ren, T. Xu, Z. Qian and Z. He, Signal Transduct. Target. Ther., 2021, 6, 225 CrossRef PubMed.
- J. Ren, X. Shu, Y. Wang, D. Wang, G. Wu, X. Zhang, Q. Jin, J. Liu, Z. Wu, Z. Xu, C. Z. Li and H. Li, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.10.052.
- L. N. Wang, Z. J. Liu, S. M. He, S. S. He and Y. P. Wang, J. Mater. Chem. B, 2021, 9, 4587–4595 RSC.
- Q. F. Zhang, G. Z. Kuang, S. S. He, H. T. Lu, Y. L. Cheng, D. F. Zhou and Y. B. Huang, Nano Lett., 2020, 20, 3039–3049 CrossRef CAS PubMed.
- Y. W. Cong, H. H. Xiao, H. J. Xiong, Z. G. Wang, J. X. Ding, C. Li, X. S. Chen, X. J. Liang, D. F. Zhou and Y. B. Huang, Adv. Mater., 2018, 30, 1706220 CrossRef PubMed.
- Y. Y. Deng, Y. P. Wang, F. Jia, W. F. Liu, D. F. Zhou, Q. Jin and J. Ji, ACS Nano, 2021, 15, 8663–8675 CrossRef CAS PubMed.
- X. Zhang, L. Li, W. Peng, X. H. Dong, Y. H. Gu, Z. Z. Ma, D. L. Gan and P. S. Liu, J. Mater. Chem. B, 2021, 9, 4169–4177 RSC.
- K. T. Huang, P. S. Hsieh, L. G. Dai and C. J. Huang, J. Mater. Chem. B, 2020, 8, 7390–7402 RSC.
- T. L. Zhang, Z. G. Lu, X. Y. Wang, J. Shen, J. Z. Wang, Y. W. Niu, Z. B. Xiao and X. Zhang, Chin. Chem. Lett., 2021, 32, 573–576 CrossRef CAS.
- H. Zhang, Y. N. Zhu, Y. Li, X. Y. Qi, J. Yang, H. S. Qi, Q. S. Li, Y. M. Ma, Y. Zhang, X. Zhang and L. Zhang, Adv. Funct. Mater., 2021, 31, 9 Search PubMed.
- H. Nakano, S. Kakinoki and Y. Iwasaki, Colloids Surf., B, 2021, 205, 8 CrossRef PubMed.
- G. Q. Zheng, S. Liu, J. Q. Zha, P. Zhang, X. W. Xu, Y. T. Chen and S. Y. Jiang, Langmuir, 2019, 35, 1858–1863 CrossRef CAS PubMed.
- Y. L. Liu, D. Zhang, B. P. Ren, X. Gong, L. J. Xu, Z. Q. Feng, Y. Chang, Y. He and J. Zheng, J. Mater. Chem. B, 2020, 8, 3814–3828 RSC.
- Y. M. Zhao, Y. Y. Deng, Z. Tang, Q. Jin and J. Ji, Langmuir, 2019, 35, 1919–1926 CrossRef CAS PubMed.
- T. Bai, J. Q. Li, A. Sinclair, S. Imren, F. Merriam, F. Sun, M. B. O'Kelly, C. Nourigat, P. Jain, J. J. Delrow, R. S. Basom, H. C. Hung, P. Zhang, B. W. Li, S. Heimfeld, S. Y. Jiang and C. Delaney, Nat. Med., 2019, 25, 1566–1575 CrossRef CAS PubMed.
- Y. Y. Deng, F. Jia, S. Y. Chen, Z. D. Shen, Q. Jin, G. S. Fu and J. Ji, Biomaterials, 2018, 187, 55–65 CrossRef CAS PubMed.
- M. Kaneko, M. Ishikawa, S. Nakanishi and K. Ishihara, ACS Macro Lett., 2021, 10, 926–932 CrossRef CAS.
- H. B. Wang, X. S. Liu, Y. Wang, Y. J. Chen, Q. Jin and J. Ji, J. Mater. Chem. B, 2015, 3, 3297–3305 RSC.
- W. Yang, S. J. Liu, T. Bai, A. J. Keefe, L. Zhang, J. R. Ella-Menye, Y. T. Li and S. Y. Jiang, Nano Today, 2014, 9, 10–16 CrossRef CAS.
- Y. Y. Deng, P. Y. Song, X. H. Chen, Y. Huang, L. J. Hong, Q. Jin and J. Ji, ACS Nano, 2020, 14, 9711–9727 CrossRef CAS PubMed.
- Y. Chen, Y. Wang, H. Wang, F. Jia, T. Cai, J. Ji and Q. Jin, Polymer, 2016, 97, 449–455 CrossRef CAS.
- S. He, C. Li, Q. Zhang, J. Ding, X. J. Liang, X. Chen, H. Xiao, X. Chen, D. Zhou and Y. Huang, ACS Nano, 2018, 12, 7272–7281 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02583j |
| ‡ J. Ye and B. Yu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |