
Visible light triggered controlled formation of rapidly self-healing hydrogels based on thiol–disulfide exchange†
Linlin
Wang‡
ab,
Qingchen
Cao‡
bc,
Xing
Wang
 *bc and
Decheng
Wu
*bd
*bc and
Decheng
Wu
*bd
aKey Laboratory of Special Functional Aggregated Materials (Shandong University), Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P. R. China
bBeijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wangxing@iccas.ac.cn; dcwu@iccas.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dDepartment of Biomedical Engineering, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China
First published on 18th March 2022
Abstract
The properties of stimuli-responsive hydrogels can be tailored under various external stimuli, but it is difficult to realize the customized adjustment of hydrogel properties since the crosslinking degree in the gelation process is intractable. Here, a visible light triggered thiol–disulfide exchange reaction was applied for constructing disulfide-crosslinked hydrogels from P(EMA-SS-PEG), a poly(ethylene glycol) grafted poly(ethyl methacrylate) derivative with a disulfide linkage as the grafting point. This photochemical method provides mild gelation conditions to handily regulate the morphology, mechanical properties, swelling ratio, and degradation rate of hydrogels by simply varying the irradiation time. Based on this strategy, these disulfide-crosslinked hydrogel coatings showed rapid self-healing in 10 min under ambient conditions, which was dependent on the width of the scratch, temperature, and humidity. Notably, spraying water on these coatings could significantly accelerate the self-healing process of large scratches (360 μm) at room temperature with a self-healing time of 1 hour, enabling the practical application of hydrogel coatings in a natural environment.
Introduction
Hydrogels are widely used in engineering and biomedical fields owing to their soft, rubbery texture and good biocompatibility.1,2 Stimuli-responsive hydrogels, whose properties can be handily and readily tailored under various external stimuli, such as pH,3 temperature,4 electricity,5 redox,6 and light,7,8 have attracted increased interest in recent years. Based on these features, self-healing hydrogels can enable the repair of localized physical damage, such as cracks or scratches, to effectively prevent catastrophic failures and extend their lifetime, and thus have been widely developed for diverse applications,9 such as coatings/films,10–13 biosensors,14–16 drug delivery systems,17–19 and wound healing.20–23 As the dynamic covalent bonds, disulfide bonds are particularly attractive for the construction of self-healing materials24,25 due to their multiple stimulatory responses to pH,26,27 light,28–30 heat,31,32 mechanical force,33 and redox potential.34,35 Furthermore, recent research demonstrates that the disulfide exchange reaction can proceed under ambient conditions to endow disulfide-crosslinked networks with rapid self-healability.36–38 For example, dual sulfide–disulfide crosslinked networks exhibited spontaneous self-healing ability within 0.5–30 min at room temperature,36 and disulfide-crosslinked hydrogel coatings showed good self-healing ability without any catalyst or external intervention.37 However, self-healing hydrogels based on disulfide exchange reactions are still rarely developed to possess rapid and room temperature self-healing properties without external stimuli. In addition, it is difficult to realize the customized adjustment of the hydrogel properties since controlling the real-time crosslinking degree in the gelation process is intractable once the crosslinking reaction is initiated. To surmount this challenge, we previously established the controlled pH-triggered and photochemical gelling system via the thiol–disulfide exchange reaction for the customized manipulation of the structures and properties of hydrogels.39–41 Unfortunately, harsh pH environments or UV light exposure was employed to trigger the reported crosslinking process, which is not favorable for practical applications, such as in the engineering and biomedical areas.Besides the advantages of a remote trigger, tunable intensity and wavelength, “turn-off” effect (the light-triggered reaction could be stopped and restarted as needed by turning off or turning on the irradiation light), and spatiotemporal precision,42 visible light is the biosafe irritation to fabricate hydrogels.43,44 Herein, we develop the visible light triggered thiol–disulfide exchange reaction for the real-time and in situ control of the gelling process, which not only avoids harsh pH and UV exposure conditions but also allows regulation of the structures and properties of hydrogels. Meanwhile, the self-healing properties of the hydrogel coating on microcracks were investigated, demonstrating its rapid self-healing properties without any stimuli. The self-healing behavior of hydrogel coatings was adjustable by controlling humidity and temperature. Moreover, spraying water on coatings can remarkably accelerate the self-healing process of hydrogel coatings to satisfy the demand in the practical environment.
Experimental section
Materials and characterization
4-Cyanopentanoic acid dithiobenzoate (CPADB, 98%, J&KChemical), 2,2′-azobis(2-methylpropionitrile) (AIBN, 99%, Energy Chemical), N,N-dimethylacetamide (DMAc, 99%, Energy Chemical), eosin Y disodium salt (EY, 98%, J&K Chemical), 2-mercaptoethylamine hydrochloride (MCA, 99%, Energy Chemical) and dithiothreitol (DTT, 99%, J&K Chemical) were used as received. α-Methoxy-ω-thiol-poly(ethyleneglycol) (PEG-SH) was obtained from poly(ethyleneglycol)methylether (PEG-OH, average Mn = 750) according to the previous literature.45 Azodiisobutyronitrile (AIBN) was recrystallized twice from ethanol. 2-(Pyridin-2-yl-disulfanyl)ethylacrylate (EMA-SS-Py) was synthesized according to the previous literature,46 and the structure was characterized by the 1H NMR spectrum (Fig. S1, ESI†). Other reagents were commercially purchased from Beijing Chem. Co. and used without further purification. The coating substrates of round glass coverslips were modified to be more hydrophilic with piranha solution (H2O2/H2SO4 = 1/3),47 which is a very corrosive chemical. Please be particularly careful and take necessary protective measures to ensure the safety if using the piranha solution. 1H NMR spectra in CDCl3 were obtained on a BrukerDRX-400 spectrometer using tetramethylsilane (TMS) as the internal reference for chemical shifts (δ, ppm). Molecular weight was determined from gel permeation chromatography (GPC) implemented on a Waters 1525 binary HPLC pump connected to a Waters 2414 refractive index detector with THF as the eluent at a flow rate of 1.0 mL min−1.Synthesis of P(EMA-SS-Py)
A 25 mL Schlenk flask was equipped with EMA-SS-Py (200 mg, 0.78 mmol), CPADB (4.4 mg, 0.016 mmol), AIBN (0.5 mg, 0.003 mmol) and DMAc (2 mL). Three freeze–pump–thaw cycles were performed with an argon inflow into the reaction vessel in the final cycle and sealed to eliminate oxygen. The Schlenk flask was then placed in a preheated oil bath at 70 °C for 6 h and the monomer conversion rate was controlled at 82%, which was determined by 1H NMR spectra. After being precipitated twice in ether and dried in a vacuum, the pink solid powder was obtained in 90% yield and characterized by 1H NMR spectroscopy (Fig. S2, ESI†). The molecular weight was calculated using the formula| Mn = M(monomer) × [monomer]0 × conversion/[RAFT agent]0. |
Synthesis of P(EMA-SS-PEG)
0.5 mL of acetic acid was added into the P(EMA-SS-Py) (0.20 g, 5.54 μmol, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) solution in CH2Cl2 (15 mL) and then purged with nitrogen for 30 min. PEG-SH (1.2 g, 1.6 mmol, Mn = 750 g mol−1) in CH2Cl2 (10 mL) was added dropwise into the mixed solution and stirred for 3 days at room temperature. The reaction mixture was concentrated by rotary evaporation, and then further purified by ultrafiltration (MWCO 3500). A viscous liquid was obtained in 95% yield after freeze-drying.
700) solution in CH2Cl2 (15 mL) and then purged with nitrogen for 30 min. PEG-SH (1.2 g, 1.6 mmol, Mn = 750 g mol−1) in CH2Cl2 (10 mL) was added dropwise into the mixed solution and stirred for 3 days at room temperature. The reaction mixture was concentrated by rotary evaporation, and then further purified by ultrafiltration (MWCO 3500). A viscous liquid was obtained in 95% yield after freeze-drying.
Preparation of hydrogels
A polymer precursor solution containing P(EMA-SS-PEG) (20 mg), MCA (1.0 mg) and EY (0.4 mg) was prepared in deionized water (200 μL). The hydrogel precursor solution was then irradiated under a CEL-HXUV300 Xenon Light Source (50 W, 400–780 nm) with a gentle rotation by a mechanical agitator to ensure homogeneity. Stable hydrogels were obtained with visible light irradiation at a predetermined time.FT-IR characterization
Hydrogels with various irradiation times of 6 h, 12 h, and 24 h were prepared. These samples were washed several times with deionized water to remove the soluble substance, and then freeze-dried. The pure polymer P(EMA-SS-PEG) was used as the control sample. Test samples were obtained by the potassium bromide tableting technique, and then measured on a Bruker TENSOR-27 spectrometer in the frequency range 4000–400 cm−1 at a resolution of 2 cm−1 with a total of 32 scans.Morphology characterization
Hydrogels with various irradiation times (2 h, 6 h, 12 h and 24 h) were prepared. These samples were washed by deionized water to remove the soluble substance, and then freeze-dried. Pieces of the lyophilized hydrogel were carefully stuck onto a conducting resin with a double-sided adhesive. The morphology of hydrogels was then studied by the field emission scanning electron microscopy (SEM) at an acceleration voltage of 5 kV on a JSM-6700F microscope (JEOL, Japan). The samples were sputter-coated with a thin layer of Pt for 90 s to make the sample conductive before testing.Rheological measurements
Hydrogels with various visible light exposure time (1 h, 2 h, 4 h, 6 h, 12 h and 24 h) were placed between two plates of the Thermo Haake Rheometer equipped with cone-parallel plate geometry (35 mm of diameter) at a gap of 0.052 mm. An amplitude sweep (G′measured as a function of strain) was first performed in order to confirm that measurements were conducted within the linear viscoelastic regime. Oscillatory frequency sweeps were then performed at 25 °C with a constant strain of 0.05% in the frequency of 100–0.1 rad s−1.Swelling properties of hydrogels
Five samples with different visible light exposure time (2 h, 4 h, 6 h, 12 h and 24 h) were prepared for measurement. The samples were immersed into deionized water (200 mL) and incubated at 25 °C until the equilibrium of swelling had been reached. The swollen hydrogels were weighed after the excess of water on the surfaces was absorbed with a filter paper carefully. The swelling ratio (%) was calculated using the following equation:| SW = (Ws − Wd)/Wd × 100% |
Degradation of hydrogels
For degradation tests, hydrogels with the irradiation time of 2 h and 24 h were weighed and recorded as the original weight (Woriginal). After being incubated in 10 mL PBS solution containing 10 mM DTT at 25 °C for a predetermined time, hydrogels were washed by deionized water several times and then freeze-dried to get the solid residues. The residues were weighed and recorded as the weight of remained hydrogels (Wresidue) after degradation. The relative weight (%) of the remained hydrogels was calculated using the following equation:| RW = Wresidue/Woriginal × 100% |
Fabrication of hydrogel coatings and contact angle test
The hydrogel precursor solution of P(EMA-SS-PEG), MCA and EY was spread out onto the hydrophilic glass wafer (diameter = 5 mm) and irradiated under the CEL-HXUV300 Xenon Light Source (50 W, 400–780 nm) for 0 and 10 min to obtain the coatings of precursor and hydrogels, respectively. Hydrogel coating was immersed in water for 10 s to remove the soluble substance. After air-drying, samples were measured using a Kruss DSA100 contact angle measurement instrument with a Sanyo camera, and the standard water drop size was 2 μL. The contact angle test was conducted five times at different points of the coatings.Self-healing assays
40 μL of hydrogel precursor solution of P(EMA-SS-PEG), MCA and EY was spread onto the hydrophilic glass wafer (diameter = 5 mm) and irradiated under a CEL-HXUV300 Xenon Light Source (50 W, 400–780 nm) for 10 min to obtain the hydrogel coatings. Then micro-scratch with a certain width was created by using a razor blade. The scratch depth was controlled the same as the depth of the corresponding hydrogel coating. Unless otherwise stated, self-healing tests were all conducted in the constant temperature and humidity chamber. The self-healing behaviors of hydrogel coatings on glass substrates were investigated using an inverted fluorescence microscope Olympus IX71 with the mode of natural light. The area of the scratch before (Ab) and after healing (Af) were calculated by the measuring software of this inverted fluorescence microscope. The healing efficiency (%) was calculated using the following equation:| HE = (Ab − Af)/Ab ×100% |
Results and discussion
In our previous work, UV irradiation replaced pH to trigger the thiol–disulfide exchange reaction for constructing disulfide-crosslinked hydrogels, which not only avoids a strong alkali environment but also provides precise spatiotemporal control of structures and properties of hydrogels. However, the UV irradiation can damage cellular proteins and DNA.42,43 Considering biosafety issues, we thus employed the mild visible light as an attractive alternative to effectively reshuffle disulfide bonds and construct the biodegradable hydrogels.For visible light triggered formation of hydrogels, we prepared the comb-like polymer P(EMA-SS-PEG) via acid-catalyzed thiol–disulfide exchange reaction of P(EMA-SS-Py) with PEG-SH according to our previous work.41 The obtained P(EMA-SS-PEG) was characterized by 1H NMR spectroscopy (Fig. S3, ESI†), which showed no evidence of residual pyridyl disulfide, suggesting quantitative PEG grafting and the calculated molecular weight to be around 37600. The GPC trace of P(EMA-SSPEG) (Fig. S4, ESI†) exhibited the Mn of 42400 with a polydispersity index (PDI) of 1.57. The obtained polymer possessed good water solubility due to the hydrophilic PEG side chains.
When a 10 wt% P(EMA-SS-PEG) solution with photoinitiator EY and cysteamine hydrochloride (MCA) was exposed to visible light irradiation for 1 h, a stable hydrogel formed and retained the shape when it was tilted (Fig. 1A).48 To avoid the alkaline environment that may trigger the thiol–disulfide exchange, cysteamine hydrochloride instead of cysteamine was used as the active thiol in the formation of visible light triggered disulfide-crosslinked hydrogels. As the gelation system was almost neutral, it was compatible with physiological pH. Further irradiation caused the increased crosslinking degree of hydrogels with contraction of the gel structure and discharge of water. In addition, the colour of hydrogels became light with time prolonging. Therefore, visible light is effective to trigger the formation of hydrogels.
 | ||
| Fig. 1 (A) Images of hydrogel formation under visible light irradiation. (B) The schematic for formation of the crosslinked network. | ||
To further confirm the formation of chemical crosslinking network (Fig. 1B), the gelation process was monitored via FT-IR measurements. Before testing, the hydrogels with 6, 12 and 24 h irradiation were firstly washed several times with deionized water to remove the soluble substance, followed by lyophilization to obtain insoluble residue. As the amount of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups remained constant in the gelation process, the relative intensity of other peaks could quantitatively reflect the number variation of the corresponding functional groups. During the gelation, the EMA group remained constant while the PEG segment released from the system. The decreased PEG chains caused the decline of the total characteristic peak intensity of C–H stretching and C–O–C stretching in the IR spectra. As shown in Fig. 2A, the characteristic peak of C–O–C stretching (1114 cm−1) with reference to PEG chains and the relative peak intensity decreased after 24 h irradiation, indicating the decreased contents of PEG chains in the crosslinked network. Meanwhile, the decreased relative peak intensity of C–H stretching (2879 cm−1) after 24 h irradiation could further prove the decreased PEG contents in the gelation process. Both the reduced characteristic peak of C–O–C stretching and C–H stretching strongly suggested our proposed formation mechanism of a crosslinked network as indicated in Fig. 1B. Under initial irradiation of visible light, the thiyl radical transferred from MCA to polymer side chains via thiol–disulfide exchange reactions, and then initiated the radical-centered disulfide exchange reactions between intermolecular chains to form the crosslinking networks. The photoinitiated radical reaction could be stopped and restarted as needed by turning off or turning on visible light. Thus, various crosslinking degrees of hydrogels could be obtained by simply altering the radiation time. Thus, similar to the UV-triggered hydrogels,41 visible light could also trigger thiol–disulfide exchange reaction and cause the rearrangement of disulfide bonds in the gelation system, accompanied by detachment of PEG side chains and the formation of disulfide bond crosslinking points. Meanwhile, the relative peak intensity of C–O–C and C–H stretching decreased gradually with the extension of irradiation time (Fig. 2B and C), demonstrating the controlled crosslinking degree of hydrogels by simply altering their radiation time. In addition, the appeared peak of N–H stretching (3426 cm−1) from the amino group demonstrated the transfer of thiyl radicals from MCA to polymer chains. Since hydrogels cannot be formed without addition of MCA, we inferred that the thiyl radical of MCA transferred from the photoinitiator system was required to initiate radical-centered disulfide exchange reactions.49
O groups remained constant in the gelation process, the relative intensity of other peaks could quantitatively reflect the number variation of the corresponding functional groups. During the gelation, the EMA group remained constant while the PEG segment released from the system. The decreased PEG chains caused the decline of the total characteristic peak intensity of C–H stretching and C–O–C stretching in the IR spectra. As shown in Fig. 2A, the characteristic peak of C–O–C stretching (1114 cm−1) with reference to PEG chains and the relative peak intensity decreased after 24 h irradiation, indicating the decreased contents of PEG chains in the crosslinked network. Meanwhile, the decreased relative peak intensity of C–H stretching (2879 cm−1) after 24 h irradiation could further prove the decreased PEG contents in the gelation process. Both the reduced characteristic peak of C–O–C stretching and C–H stretching strongly suggested our proposed formation mechanism of a crosslinked network as indicated in Fig. 1B. Under initial irradiation of visible light, the thiyl radical transferred from MCA to polymer side chains via thiol–disulfide exchange reactions, and then initiated the radical-centered disulfide exchange reactions between intermolecular chains to form the crosslinking networks. The photoinitiated radical reaction could be stopped and restarted as needed by turning off or turning on visible light. Thus, various crosslinking degrees of hydrogels could be obtained by simply altering the radiation time. Thus, similar to the UV-triggered hydrogels,41 visible light could also trigger thiol–disulfide exchange reaction and cause the rearrangement of disulfide bonds in the gelation system, accompanied by detachment of PEG side chains and the formation of disulfide bond crosslinking points. Meanwhile, the relative peak intensity of C–O–C and C–H stretching decreased gradually with the extension of irradiation time (Fig. 2B and C), demonstrating the controlled crosslinking degree of hydrogels by simply altering their radiation time. In addition, the appeared peak of N–H stretching (3426 cm−1) from the amino group demonstrated the transfer of thiyl radicals from MCA to polymer chains. Since hydrogels cannot be formed without addition of MCA, we inferred that the thiyl radical of MCA transferred from the photoinitiator system was required to initiate radical-centered disulfide exchange reactions.49
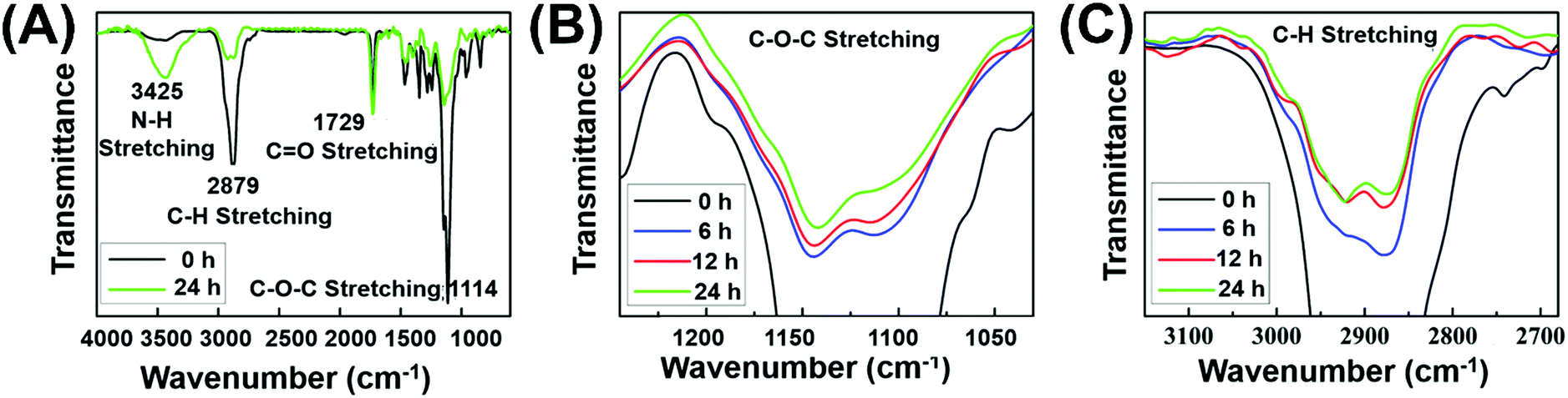 | ||
| Fig. 2 IR spectra of (A) P(EMA-SS-PEG) and its hydrogel with 24 h irradiation; (B) C–O–C stretching and (C) C–H stretching of P(EMA-SS-PEG) (0 h) and its hydrogels with irradiation of 6, 12 and 24 h. The peak intensity of CO stretching was constant and served as a reference value. | ||
In view of the temporal control of the photochemical reaction, we could handily tune the crosslinking degree of hydrogels by simply varying the irradiation time, and thereby regulate their morphology, mechanical properties, swelling ratio, and degradation rate. As shown in Fig. 3A, the initial hydrogel after 1 h gelation possessed a smooth structure. However, increasing gelation time tended to form inhomogeneous hydrogels with the lager pores of microstructures. Based on this observation, it was proposed that intramolecular thiol–disulfide exchange reaction occurred with irradiation time, which limited the formation of a crosslinked network between polymer backbones and led to the formation of microstructures as reflected in the appearance of some nanoaggregates. The increased elastic moduli (G′) and decreased swelling ratios of hydrogels (Fig. 3B and C) demonstrated that increasing gelation time and crosslinking degree could enhance the mechanical strength and reduce swelling ability. Notably, 24 h irradiation of hydrogels possessed the mechanical strength with the G′ value of up to 0.04 MPa, which could be comparable to the UV-triggered hydrogel in our previous work. This result proved that visible light could effectively trigger thiol–disulfide exchange and tailor the formation of hydrogels.
 | ||
| Fig. 3 (A) SEM images of hydrogels obtained after visible light irradiation of 2, 6, 12 and 24 h (scale bar = 1 μm). (B) Elastic moduli (G′) of hydrogels with irradiation times of 1, 2, 4, 6, 12 and 24 h. (C) Swelling ratios of hydrogels with irradiation times of 2, 4, 6, 12 and 24 h. (D) Degradation curves of the hydrogels with the irradiation times of 2 and 24 h after immersing in the 1 × PBS solution containing 0 and 10 mM of DTT, respectively. (Inset: the degradation curves of the hydrogels with 2 h). | ||
The hydrogels could be quickly decomposed due to the redox response of disulfide bonds, and their degradation rate was obviously different by changing the irradiation time. As reflected in Fig. 3D, the loose hydrogel with 2 h irradiation totally disappeared within 4 h after immersing into reductive dithiothreitol (DTT) solutions, while the degradation time was up to 5 days for the hydrogel with 24 h irradiation. Therefore, the morphology, mechanical properties, swelling ratio, and degradation behaviors of hydrogels could also be well regulated by simply changing the irradiation time. Meanwhile, this visible light triggered thiol–disulfide exchange reaction can be tailored under mild gelation conditions and avoids harsh pH and UV exposure conditions compared to our previous studies.39–41
Healing of the microcracks is of great importance in improving the stability and durability of materials in long-term applications, owing to the inevitable occurrence of microcracks that can lead to the devastation and failure of materials.50,51 In this work, the self-healing properties of the disulfide-crosslinked hydrogel coatings upon micro-scratching through the reformation of disulfide linkages were investigated in various environments. The preparation procedures of hydrogel coatings on glass slide surfaces are shown in Fig. 4A. Under visible light irradiation, the hydrogel precursor solution of P (EMA-SS-PEG), EY and MCA could form hydrogel coatings in situ. The hydrophilic property of hydrogel coating was studied and the result is summarized in Fig. 4B. The result showed that the hydrogel coating could remain stable after immersing in water, proving the formation of a crosslinked network. In addition, compared to the contact angle of 29.1 °C for the coating of hydrogel precursor solution, the contact angle of hydrogel coating with 10 min irradiation slightly increased to 35.4 °C due to the loss of some hydrophilic PEG side chains, further testifying the formation of hydrogels.
 | ||
| Fig. 4 (A) Schematic for the fabrication process of hydrogel coatings. (B) Pictures of water droplets on the coating of hydrogel precursor solution and the hydrogel coating with 10 min irradiation by marking the corresponding contact angles values. | ||
The self-healing ability of the resulting hydrogel coatings with microscopic scratches (about 25 μm in width) was investigated in ambient environments by changing their thickness and crosslinking degrees, and the detailed data are summarized in Table 1, Fig. S5 (ESI†) and Fig. 5A. For sample 1#–3#, the self-healing time was reduced evidently with the coating thickness increasing from 25.2 μm to 66.5 μm. For example, sample 1# could not completely self-healed even after 2 days (Fig. 5A-i), while only 10 min was required for sample 3# to fully repair the damage (Fig. 5A-k). The reason may ascribe to that thick coating was conducive to the material flows between scratches, thus resulting in rapid self-healing behavior. By changing the gelation time, hydrogel coatings with different crosslinking degrees demonstrated the obvious distinguished self-healing properties (Table 1). As shown in Fig. 5A, compared to 3#, the scratch of 4# was partially healed even after 20 h when prolonging the gelation time to 20 min. As crosslinking degree increased, the crosslinked network became compact to reduce the chain mobility of disulfide exchange in the self-healing process, resulting in the low healing rate for 4#. Meanwhile, the thin coating with a longer irradiation time was not beneficial to the material flows, which affected the self-healing ability. Therefore, the healing process of hydrogel coatings could occur spontaneously at ambient temperature without any external stimulus and the healing properties could be well regulated by the thickness and crosslinking degree of hydrogels.
| Sample | Amount of solution | Irradiation time | Coating thickness | Self-healing time |
|---|---|---|---|---|
| 1# | 20 μL | 10 min | 25.2 μm | >2 d |
| 2# | 30 μL | 10 min | 50.6 μm | 30 min |
| 3# | 40 μL | 10 min | 66.5 μm | 10 min |
| 4# | 40 μL | 20 min | 55.0 μm | >20 h |
 | ||
| Fig. 5 (A) Microscope images of self-healing in ambient environments for hydrogel coatings with the corresponding experiment parameters in Table 1 (Scratch width: about 25 μm, scale bar: 100 μm). (B) Healing curves of hydrogel coatings (3# sample in Table 1) with different scratch widths of 11.7, 36.2 and 119.4 μm. | ||
To investigate the effect of scratch width on self-healing kinetics, the healing efficiency of hydrogel coatings with three different scratch widths was estimated based on the decrease in the area of the scratches over time (Fig. 5B). For the three samples, all the damaged areas were all fully healed in 2 h with the healing efficiency of 100%, indicating the rapid healing behaviors at ambient temperature. The healing time was calculated to be 5 min for the 6.4 μm wide scratch, 30 min for the 26.2 μm wide scratch, and 2 h for 119.3 μm wide scratch, suggesting the dependence of self-healing kinetics on the dimension of scratch.
The self-healing properties of hydrogel coatings with a scratch width of about 180 μm at various temperatures were monitored by using an optical microscope, and the healing time was estimated based on the complete disappearance of the scratches. Fig. 6A shows the self-healing process of hydrogel coatings in the constant temperature and humidity chamber of 40% relative humidity (RH). The healing time of hydrogel coatings obviously reduced from 10 h to 2 h with the temperature increasing from 20 to 35 °C, suggesting the powerful temperature dependence of self-healing behaviour.
 | ||
| Fig. 6 (A) Microscope images of self-healing for hydrogel coatings under the temperature conditions of 20 °C, 25 °C, 30 °C, and 35 °C (scratch width: about 180 μm, humidity: 40% RH, scale bar: 100 μm). (B) Healing curves of hydrogel coatings with various scratch widths in different temperature conditions. | ||
At 20 °C, the scratch was healed partially after 6 h and completely healed at 48 h. In comparison, the scratch at 35 °C completely disappeared within 2 h, returning the surface with a uniform coating. At high temperature, the molecular chains were more extended, which was conducive to the mobility of the network between scratches. Meanwhile, the high temperature enabled the disulfide exchange reaction of the self-healing process. Both the molecular effect and rapid disulfide exchange reaction between scratches may lead to the rapid healing behaviour.
In addition, the self-healing properties of hydrogel coatings with the scratch width of about 270 μm (Fig. S6, ESI†) and 360 μm (Fig. S7, ESI†) at 20–35 °C are evaluated and summarized in Fig. 6B. In different scratch widths, the healing time possessed the same trend of variability with temperature increase, further confirming the temperature dependence of self-healing behaviors. At the same temperature, the scratch with a larger width size demonstrated the slow healing behavior, due to the long process of material flows. In particular, the impact of scratch width on the healing time becomes increasingly pronounced with lower temperatures. As a result, the high temperature and the small size of scratches are advantageous to the rapid healing of hydrogel coatings.
The effect of humidity on the self-healing properties of hydrogel coatings was further studied, and the temperature was kept at 25 °C in the constant temperature and humidity chamber during the healing process. As shown in Fig. 7A, the complete self-healing occurred at 20% RH within 24 h on the 180 μm wide scratch, while at 40% RH, only 6 h was required for full healing. Upon further increasing the humidity to 80% RH, the self-healing time reduced to 2 h, indicating the great influence of humidity on self-healing behaviors. The self-healing properties of hydrogel coatings with the scratch width of about 90 μm (Fig. S8, ESI†) and 270 μm (Fig. S9, ESI†) at 20 °C were also studied, as reflected in Fig. 7B. At the low humidity of 20% RH and 40% RH, the scratch width had a significant effect on the healing time. In particular, at 20% RH, the healing time greatly increased from 3 h to 24 h when the scratch width increased from 90 μm to 180 μm, and the scratch of 270 μm was only partially healed within 24 h hours. In comparison, the healing time hardly changed upon increasing the scratch width at the high humidity of 60% RH and 80% RH. This may be because high humidity enhanced the swelling behaviors of the hydrogel coatings, which led to the large enough volume changes to accelerate the material flows between scratches and obtain the rapid self-healing behaviors.
 | ||
| Fig. 7 (A) Microscope images of self-healing for hydrogel coatings under the humidity conditions of 20%, 40%, 60% and 80% (scratch width: about 180 μm, temperature: 25 °C, scale bar: 100 μm). (B) Healing curves of hydrogel coatings with various scratch widths in different humidity conditions. | ||
To meet the practical demand of self-healing coatings in low temperature and humidification environments, spraying water on the coating was used to accelerate the self-healing process. As shown in Fig. 8A, coatings with a scratch width of 270 μm could not be fully healed even after 48 h under the conditions of 25 °C and 20% RH (Fig. 8A-b). Upon spraying water 5 times and then healing for 1 h, the coatings obviously started healing (Fig. 8A-c), demonstrating that humidification could significantly promote the healing process. After repeating the process, a completely healed coating with a smooth surface was observed in Fig. 8A-d, indicating that enough humidity was needed for the full repair. Upon spraying water 10 times, the coatings with a scratch width of 360 μm could be completely healed in 1 h (Fig. 8B), demonstrating that spraying water was an effective method to obtain the rapid self-healing properties. To simplify the steps, a trace amount of water was dripped onto the scratch to accelerate the self-healing process, while an uneven surface was formed due to the excessive swelling (Fig. 8C). Therefore, spraying water was a suitable way for self-healing coatings to rapidly heal in low temperature and dry environments.
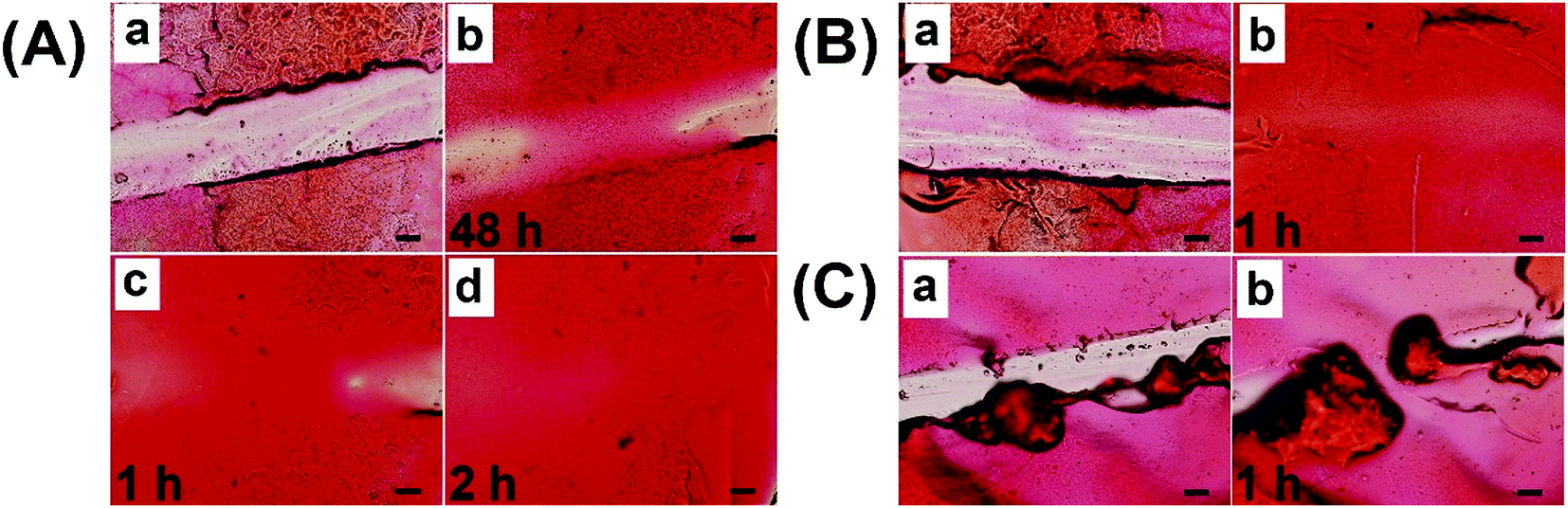 | ||
| Fig. 8 (A) Microscope images of self-healing for hydrogel coatings with scratch width of about 270 μm (scale bar: 100 μm): a-before healing, b-healing for 48 h, c-spraying water for 5 times and then healing 1 h, d-spraying water for 10 times and then healing 2 h. (B) Microscope images of self-healing for hydrogel coatings with scratch width of about 360 μm: a-before healing, b-spraying water 10 times and healing 1 h. (C) Microscope images of self-healing for hydrogel coatings with a scratch width of about 180 μm: a-before healing, b-trace water added and then healing 1 h. Self-healing behaviors were conducted in the temperature and humidity chamber of 25 °C and 20% RH. | ||
Based on the observed healing behavior, a detailed self-healing mechanism of hydrogel coatings was proposed. After cutting the hydrogel coating with a certain scratch width, the coating flowed back to the centre of the scratch due to the surface tension driven viscoelastic restoration.36,52 When the scratch surfaces contacted each other, disulfide exchange reaction occurred to regenerate chemical bonds between scratch edges and eventually healed the damage.53,54
Conclusions
In summary, we demonstrated an effective strategy for the construction of disulfide-crosslinked hydrogels with adjustable properties via the visible-triggered thiol–disulfide exchange reaction under mild gelation conditions. The disulfide-crosslinked hydrogel coatings possessed a rapid spontaneous self-healing behavior based on disulfide–disulfide exchange reactions under mild conditions, with a favorable dependence on the width of the scratch, temperature, and humidity. Importantly, spray humidification could obviously accelerate the self-healing process of large scratches at low temperature and humidity, which enabled the practical application of hydrogel coatings in a natural environment.Author contributions
Linlin Wang and Qingchen Cao contributed equally. Linlin Wang: conceptualization, methodology, writing-original draft and visualization. Qingchen Cao: methodology, writing-original draft and visualization. Xing Wang: supervision, writing-review and editing. Decheng Wu: conceptualization, writing-review and editing, supervision, funding acquisition. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21935011, 51973226 and 21725403) and the Youth Innovation Promotion Association CAS (No. 2019031).References
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed.
- J. Su, S. Lu, S. Jiang, B. Li, B. Liu, Q. Sun, J. Li, F. Wang and Y. Wei, Adv. Mater., 2021, 33, 2100619 CrossRef CAS PubMed.
- A. Bhat, J. M. Amanor-Boadu and A. Guiseppi-Elie, ACS Sens., 2020, 5, 500–509 CrossRef CAS PubMed.
- S. Huang, J. Shen, N. Li and M. Ye, J. Appl. Polym. Sci., 2015, 132, 41530 CrossRef.
- X. Ying, Y. Wang, J. Liang, J. Yue, C. Xu, L. Lu, Z. Xu, J. Gao, Y. Du and Z. Chen, Angew. Chem., Int. Ed., 2014, 53, 12436–12440 CAS.
- L. Liu, N. Wang, Y. Han, Y. Li and W. Liu, Macromol. Rapid Commun., 2014, 35, 344–349 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- N. Tao, D. Zhang, X. Li, D. Lou, X. Sun, C. Wei, J. Li, J. Yang and Y. N. Liu, Chem. Sci., 2019, 10, 10765–10771 RSC.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- M. Abdolah Zadeh, S. van der Zwaag and S. J. Garcia, ACS Appl. Mater. Interfaces, 2016, 8, 4126–4136 CrossRef CAS PubMed.
- K. Chen, S. Zhou and L. Wu, ACS Nano, 2016, 10, 1386–1394 CrossRef CAS PubMed.
- Q. Yang, P. Wang, C. Zhao, W. Wang, J. Yang and Q. Liu, Macromol. Rapid Commun., 2017, 38, 1600741 CrossRef PubMed.
- M. Diba, S. Spaans, K. Ning, B. D. Ippel, F. Yang, B. Loomans, P. Y. Dankers and S. C. Leeuwenburgh, Adv. Mater. Interfaces, 2018, 5, 1800118 CrossRef.
- Y. Yao, Y. Wang, Z. Li and H. Li, Langmuir, 2015, 31, 12736–12741 CrossRef CAS PubMed.
- G. Ge, Y. Lu, X. Qu, W. Zhao, Y. Ren, W. Wang, Q. Wang, W. Huang and X. Dong, ACS Nano, 2020, 14, 218–228 CrossRef CAS PubMed.
- Z. Liang, J. Zhang, C. Wu, X. Hu, Y. Lu, G. Wang, F. Yu, X. Zhang and Y. Wang, Biosens. Bioelectron., 2020, 155, 112105 CrossRef CAS PubMed.
- S. Merino, C. Martín, K. Kostarelos, M. Prato and E. Vázquez, ACS Nano, 2015, 9, 4686–4697 CrossRef CAS PubMed.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- M. Chen, J. Tian, Y. Liu, H. Cao, R. Li, J. Wang, J. Wu and Q. Zhang, Chem. Eng. J., 2019, 373, 413–424 CrossRef CAS.
- F. Sun, Y. Bu, Y. Chen, F. Yang, J. Yu and D. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 9132–9140 CrossRef CAS PubMed.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS PubMed.
- W. Huang, Y. Wang, Z. Huang, X. Wang, L. Chen, Y. Zhang and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 41076–41088 CrossRef CAS PubMed.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- P. Casuso, I. Odriozola, A. Pérez-San Vicente, I. Loinaz, G. Cabañero, H. J. Grande and D. Dupin, Biomacromolecules, 2015, 16, 3552–3561 CrossRef CAS PubMed.
- X. Wang, P. Y. Gao, Y. Y. Yang, H. X. Guo and D. C. Wu, Nat. Commun., 2018, 9, 2772 CrossRef PubMed.
- X. Du, J. Li, A. Welle, L. Li, W. Feng and P. A. Levkin, Adv. Mater., 2015, 27, 4997–5001 CrossRef CAS PubMed.
- Z. Y. Deng, S. A. Yuan, R. X. Xu, H. J. Liang and S. Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 8896–8900 CrossRef CAS PubMed.
- Z. Q. Yu, J. T. Sun, C. Y. Pan and C. Y. Hong, Chem. Commun., 2012, 48, 5623–5625 RSC.
- X. Wu, J. Li, G. Li, L. Ling, G. Zhang, R. Sun and C. P. Wong, J. Appl. Polym. Sci., 2018, 135, 46532 CrossRef.
- A. P. Wiita, S. R. K. Ainavarapu, H. H. Huang and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7222–7227 CrossRef CAS PubMed.
- L. Jia, D. Cui, J. Bignon, A. Di Cicco, J. Wdzieczak-Bakala, J. Liu and M. H. Li, Biomacromolecules, 2014, 15, 2206–2217 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- S. Y. An, S. M. Noh, J. H. Nam and J. K. Oh, Macromol. Rapid Commun., 2015, 36, 1255–1260 CrossRef CAS PubMed.
- W. J. Yang, X. Tao, T. Zhao, L. Weng, E. T. Kang and L. Wang, Polym. Chem., 2015, 6, 7027–7035 RSC.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef PubMed.
- D. C. Wu, X. J. Loh, Y. L. Wu, C. L. Lay and Y. Liu, J. Am. Chem. Soc., 2010, 132, 15140–15143 CrossRef CAS PubMed.
- J. Zhang, F. Yang, H. Shen and D. Wu, ACS Macro Lett., 2012, 1, 1295–1299 CrossRef CAS.
- L. Wang, L. Li, X. Wang, D. Huang, F. Yang, H. Shen, Z. Li and D. Wu, Polym. Chem., 2016, 7, 1429–1438 RSC.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- A. Fu, K. Gwon, M. Kim, G. Tae and J. A. Kornfield, Biomacromolecules, 2015, 16, 497–506 CrossRef CAS PubMed.
- Z. Wang, H. Kumar, Z. Tian, X. Jin, J. F. Holzman, F. Menard and K. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 26859–26869 CrossRef CAS PubMed.
- X. Wang, D. Li, F. Yang, H. Shen, Z. Li and D. Wu, Polym. Chem., 2013, 4, 4596–4600 RSC.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS PubMed.
- L. Li, X. X. Deng, Z. L. Li, F. S. Du and Z.-C. Li, Macromolecules, 2014, 47, 4660–4667 CrossRef CAS.
- G. M. Scheutz, J. L. Rowell, S. T. Ellison, J. B. Garrison, T. E. Angelini and B. S. Sumerlin, Macromolecules, 2020, 53, 4038–4046 CrossRef CAS.
- O. Valdes-Aguilera, C.-P. Pathak, J. Shi, D. Watson and D.-C. Neckers, Macromolecules, 1992, 25, 541–547 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- X. Y. Dou, X. Wang and D. C. Wu, Acta Polym. Sin., 2019, 50, 429–441 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01698a |
| ‡ These authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2022 |