
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMediator-free NADH photochemical regeneration with the aid of the amino acid L-cysteine†
Alberto
Bianco
 a,
Mirko
Zaffagnini
b and
Giacomo
Bergamini
*a
a,
Mirko
Zaffagnini
b and
Giacomo
Bergamini
*a
aDipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: giacomo.bergamini@unibo.it
bDipartimento di Farmacia e Biotecnologie, Alma Mater Studiorum – Università di Bologna, Via Irnerio 42, 40126 Bologna, Italy
First published on 24th August 2022
Abstract
A strongly photoreducing ruthenium complex has been used to regenerate nicotinamide cofactor with only visible light and L-cysteine. The photoinduced processes have been deeply characterized, and the emission of NADH has been employed for the assessment of selectivity towards the enzymatically active photoregenerated cofactor.
Enzyme catalysis, thanks to its high activity and selectivity, has been exploited in the fields of pharmaceuticals, chemical synthesis, and biodegradations.1,2 Moreover, in recent years, enzymes that can generate molecular hydrogen or reduce CO2 into solar fuels have attracted a lot of attention as a potential candidate to fight against the energy crisis and global warming.3–5
A quarter of all enzymes are represented by oxidoreductases, most of which depends on the coenzyme the coenzyme β-Nicotinamide Adenine Dinucleotide (NAD+) and its phosphate (NAD(P)+);1,6–8 but due to the high cost of the reduced forms (€ 190 per gram for NADH, € 1520 per gram for NADPH, Roche Diagnostic GmbH) that are consumed stoichiometrically during the process, the use of these enzymes is very restricted and not sustainable for artificial applications. The employment of cofactors in further industrial application can be pushed only with a simple, effective and sustainable method for NAD(P)H regeneration.1,6–10
In the last decade, enzymatic, chemical, electrochemical and photochemical approaches for regenerating cofactors were proposed7 and, thanks to its inspiration from natural photosynthesis, the photochemical approach is attracting increasing interest.11–13
Most examples on photoregeneration reported in the literature rely on a mediator, such as rhodium complex [Cp*Rh(bpy)(H2O)]2+, whose role is to selectively transfer an hydride to NAD+, whereas the role of the photocatalyst is only to regenerate the mediator.7,14 Moreover, triethanolamine (TEOA) has been extensively applied as the electron donor in the regeneration of cofactors.7,14 However, few years ago Grzelczak et al. reported a new role of TEOA in the photocatalytic regeneration of cofactor molecules.15 In this study, the authors demonstrated that an oxidation product of TEOA, glycolaldehyde, is able to directly reduce NAD+. This finding creates a point of attention in identifying the mechanisms underlying photocatalytic processes where TEOA is used as a scavenger.
In the present paper, we propose a new photocatalytic method that avoids the use of an electron mediator and employs a biomolecule that has the double function of the source of electrons and the hydrogen atom donor.
In order to overcome the high monoelectronic reduction potential of NAD+ (ENAD+/NAD˙ = −1.18 V vs. SCE),16 a chromophore with remarkably reducing and long-living excited state is needed, along with the desired properties of water-solubility, high photostability, and capability to absorb visible light.
To this aim, we selected the luminescent metal complex mono(bipyridyl)-tetracyanoruthenate [Ru(bpy)(CN)4]2− (Fig. 1) owing to its peculiar photophysical and photochemical properties. The absorption and emission spectra of this complex have been thoroughly investigated in water and other polar solvents.17–19 The low-energy absorption band and the weak luminescence band exhibited in the visible region (Fig. 1) are related to metal-to-ligand (bpy) charge-transfer (MLCT) excited states (spin-allowed states for the absorption bands, the lowest spin-forbidden state for the emission band).
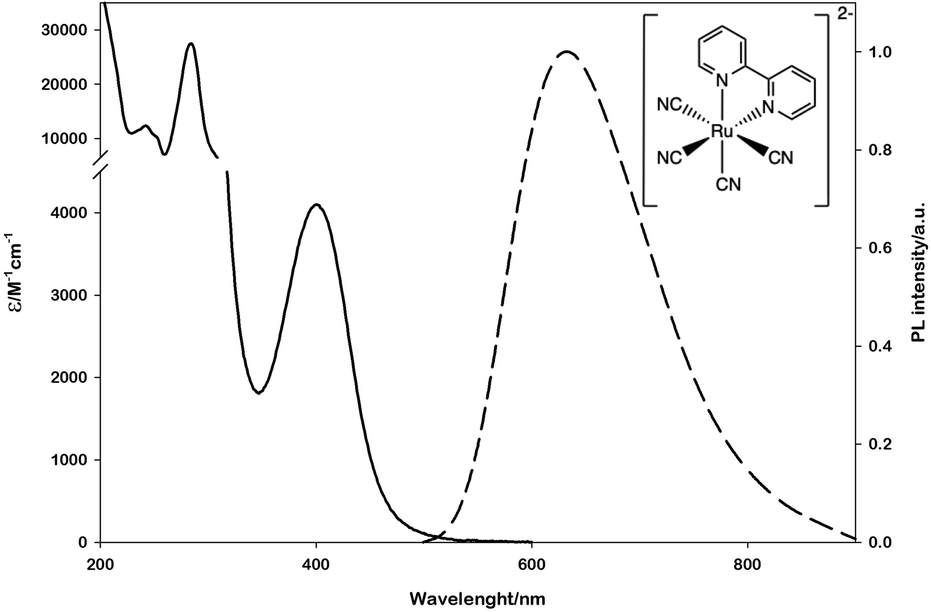 | ||
| Fig. 1 Absorption (solid line) and emission (dashed line) spectra of [Ru(bpy)(CN)4]2− in water. The emission spectrum is obtained upon excitation at 400 nm. | ||
The emission lifetime recorded in water is τ = 101 ns in deaerated solution and τ = 91 ns in air-equilibrated solution,17 demonstrating the limited quenching effect by molecular oxygen and the potential of this complex to be employed as a photocatalyst. Moreover, within the approximation of the thermally equilibrated excited state, the calculated excited-state potentials in dimethylformamide (EP+/P* = - 1.83 V vs. SCE and EP*/P- = + 0.08 V vs. SCE, Scheme S1†)17 indicate that *[Ru(bpy)(CN)4]2− is a powerful reductant and a rather poor oxidant.
In water only the oxidation potential of the ground state can be measured due to the very high reduction potential (Scheme S1†). [Ru(bpy)(CN)4]−/*[Ru(bpy)(CN)4]2− (EP+/P* = −1.6 V vs. SCE in water)17,19 is thermodynamically sufficient for the high direct monoelectronic reduction of NAD+ (ENAD+/NAD˙ = −1.18 V vs. SCE),16 which can then follow different deactivation pathways (Scheme 1).
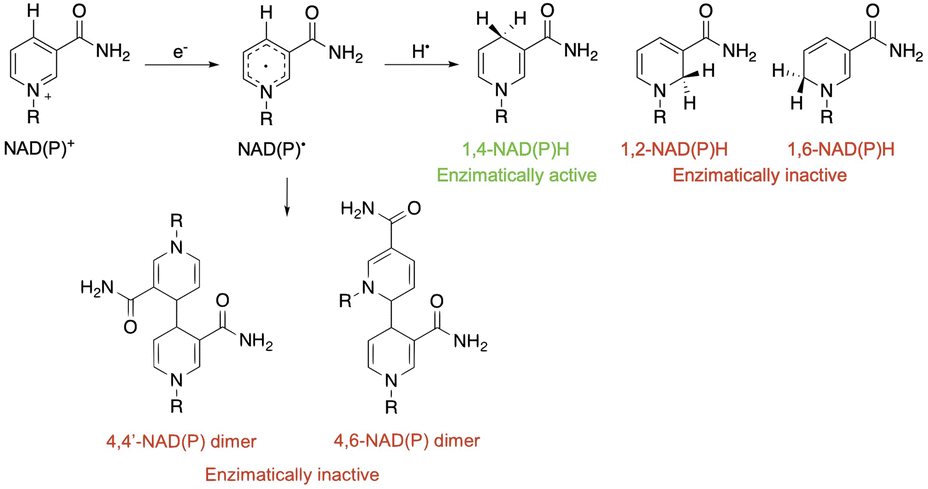 | ||
| Scheme 1 Possible deactivation pathways of the NAD˙ radical. | ||
After NAD˙ formation, several products can be generated but only 1,4-NADH is the desired enzymatically active cofactor.20,21 Particular attention should be paid to the NAD2 dimer that has an UV-Vis absorption band at ca. 340 nm, very similar to that of 1,4-NADH, and therefore may interfere with the correct estimation of the concentration of the regenerated cofactor. In order to prevent NAD˙ radical dimerization, the system needs a fast hydrogen atom donor, which has to be water-soluble at a high concentration and preferably optically transparent above 300 nm. Furthermore, to close the photocatalytic cycle, a sacrificial electron donor is required to reduce the oxidized ruthenium complex. In the present study we tested L-cysteine, a proteinogenic amino acid that can act both as a hydrogen atom donor and sacrificial electron donor at once, as shown in Scheme 2.
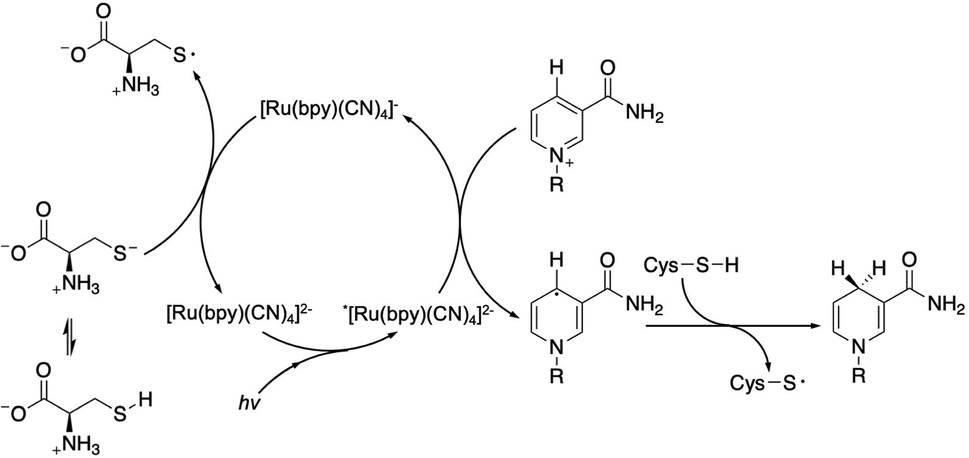 | ||
| Scheme 2 Proposed mechanism of the photocatalytic cycle for NAD˙ radical formation and its quenching by L-cysteine with the evolution of 1,4-NADH. | ||
To prove the proposed mechanism, we first studied the photoinduced processes. To address the deactivation pathways of *[Ru(bpy)(CN)4]2−, the weak emission of the complex (Φ = 0.0068)17 can be monitored in order to prove the interaction between its excited state and the other reactants. An aqueous solution of ruthenium was titrated with increasing amounts of NAD+ and it was observed that the emission spectra (Fig. S1†) and the lifetime decays decrease linearly with the oxidized cofactor concentration (Fig. 2).
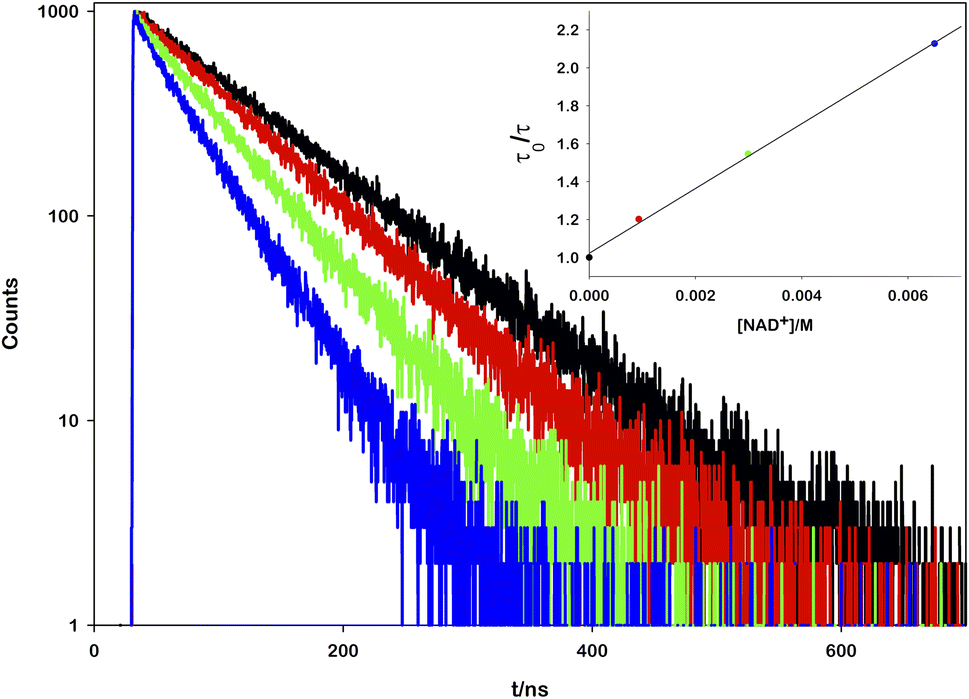 | ||
| Fig. 2 Emission lifetime decays of [Ru(bpy)(CN)4]2− in air-equilibrated aqueous solution (black), [NAD+] = 0.93 mM (red), [NAD+] = 2.99 mM (green), [NAD+] = 6.5 mM (blue). The inset shows the Stern–Volmer plot with linear fitting (R2 = 0.9998). | ||
From the mono-exponential behaviour of lifetime decays, we can infer a dynamic quenching of the ruthenium excited state by NAD+ and, using the Stern–Volmer equation, it was possible to calculate a quenching constant equal to 1.88 × 109 M−1 s−1 (see the ESI for details†).
On the other hand, no relevant quenching mechanism has been observed upon additions of L-cysteine (up to ca. 0.1 M, see Fig. S2†), as expected by the low oxidation potential (ECySH˙+/CySH = −0.57 V vs. SCE).22 These results prove that the first step of the photocatalytic cycle is the oxidative quenching with the formation of NAD˙ and [Ru(bpy)(CN)4]2−. An experiment of continuous irradiation of a solution containing [Ru(bpy)(CN)4]2− (0.25 mM), NAD+ (2.0 mM), L-cysteine (0.1 M), and 30 minutes purging with N2 shows the preservation of the ruthenium complex (confirmed by absorption spectra, Fig. S3†) demonstrating the closing of the photocatalytic cycle by L-cysteine. Moreover, it has been proved that neither the excited state nor the oxidized ruthenium complex [Ru(bpy)(CN)4]− (EP+/P = + 0.78 V vs. SCE in water)19 is able to oxidize NADH (ENADH˙+/NADH = −0.81 V vs. SCE),23,24 avoiding the consumption of the reduced photoproduct.
To prove the ability of L-cysteine (BDES–H = 360.3 kJ mol−1)25 to act also as a hydrogen atom donor in the proposed cycle (Scheme 2), we carried out a comparison test with TEOA (BDEO–H = 441.0 kJ mol−1),25 irradiating two samples that differ only in the electron donor.
In this experiment, 2 mL of aqueous reaction mixture, composed of [Ru(bpy)(CN)4]2− (0.25 mM), NAD+ (2.0 mM) and Tris·HCl buffer (0.1 M) were placed in two quartz gas-tight cuvettes (1 cm path length), the electron donor (TEOA or L-cysteine, 0.1 M) was added to each sample and the pH was adjusted to 7.2 by the addition of 2 M NaOH for the slightly acidic sample containing L-cysteine and 2 M HCl for the alkaline containing TEOA. Then, the mixtures were purged with N2 for 30 minutes. The solutions have been irradiated with a 405 nm high-power LED (see the ESI†), recording the absorption and emission spectra in order to follow the variations in the NADH characteristic spectral regions. The concentration of the regenerated cofactor is usually measured by monitoring the UV-Vis absorbance at 340 nm. It is worth noting that also some enzymatically inactive products (1,6-NADH isomer 345 nm and NAD2 dimer at 340 nm, Scheme 1) have an absorption contribution in this region thus making it difficult to evaluate the ratio of the different forms.26 Spectral changes reported in Fig. 3 show the formation of a photoproduct that absorbs in the same spectral region of NADH but, as mentioned before, the absorption is ambiguous. Another photophysical method to follow the cofactor regeneration is monitoring through the emission band centred at 470 nm, which is an exclusive property of the 1,4-NADH (Fig. S4†). This method has been extensively used in enzyme function in vitro and in vivo27,28 but, surprisingly, scarcely used in cofactor photoregeneration. Recording the absorption, emission, and excitation spectra of a mixture of all the possible non dimeric 1,x-NADH isomers (Scheme 1) obtained by chemical reduction of NAD+ with NaBH4,29 it is possible to assess which is the emissive form (Fig. S5,† comparison with commercial 1,4-NADH, from Roche Diagnostic GmbH, product code 10107735001). The results confirm that the combined monitoring of the absorption and emission gives an unambiguous characterization of the enzymatically active cofactor. Ratiometric evaluation of absorption and emission signals opens the possibility of employing steady-state photophysical measurements to evaluate the ratio between the active 1,4-NADH and the dimer obtained via radical coupling.
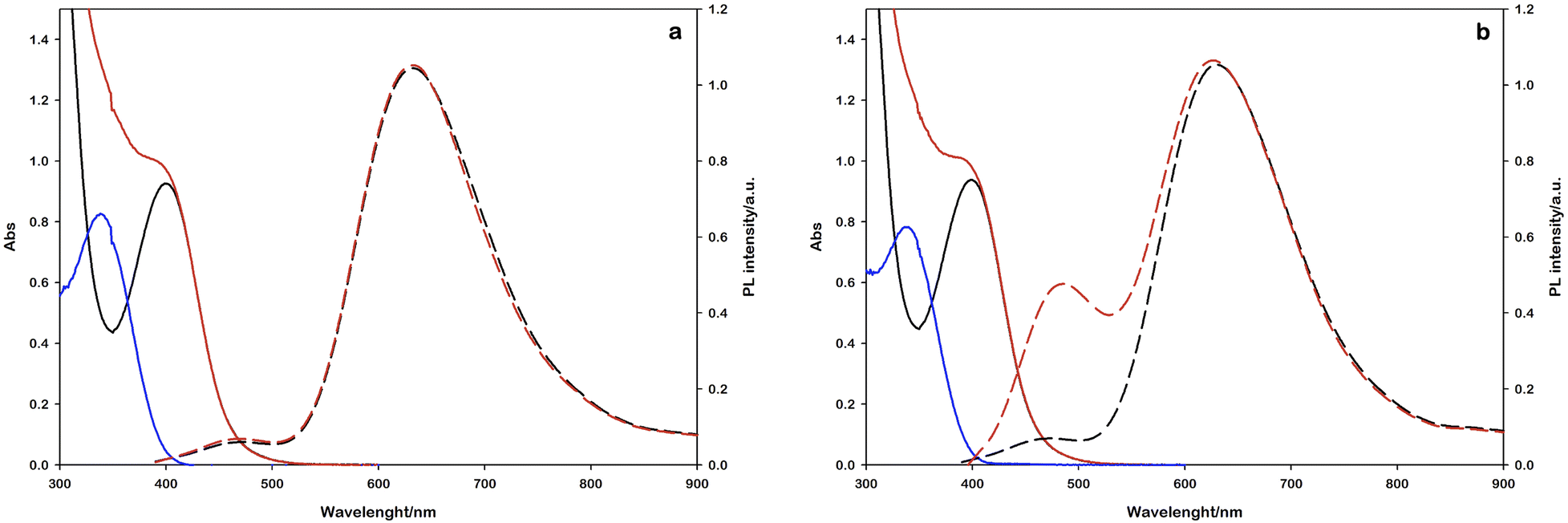 | ||
| Fig. 3 Absorption (solid lines) and emission (dashed lines) spectral changes observed upon 405 nm irradiation using TEOA (panel a) or L-cysteine (panel b) as the electron donor. Black lines represent the starting reaction mixtures and red lines after 90 minute irradiation. The difference absorption spectra are also reported (blue lines). Emission spectra are obtained upon 340 nm excitation. | ||
In this direction, the emission arising at 470 nm with L-cysteine (Fig. 3b) is ascribable to the enzymatically active 1,4-NADH, proving the desired product formation in contrast to the sample with TEOA.
The same experiments were performed under air equilibrated conditions, leading to no spectral changes (Fig. S6†) and this behaviour can be ascribed to the quenching of the NAD˙ radical by molecular oxygen.30
Performing photoregeneration tests with different irradiation setups (different wavelengths and photon fluxes), we noticed that decreasing the irradiance the rate of photoregeneration obviously decreases, but the percentage of 1,4-NADH in the photoproduct increases. In these experiments, equivalent aqueous solutions were prepared ([Ru(bpy)(CN)4]2− (0.25 mM), NAD+ (2.0 mM), L-cysteine (0.1 M), Tris·HCl buffer (0.1 M), pH = 7.2, N2 purging for 30 minutes) and irradiated with different sources (Fig. S9†) at different distances, measuring the spectral irradiance with a diode array detector (see the ESI†). The data obtained show that with similar ΔA at 340 nm, higher emissions at 470 nm were recorded in low irradiance experiments (Fig. 4). This behaviour is ascribable to the higher NAD˙ concentration generated per time unit with more powerful sources, which promotes the radical–radical dimerization; thus, NAD2 was almost solely obtained, which absorbs in the same spectral region of 1,4-NADH but exhibits no emission.31
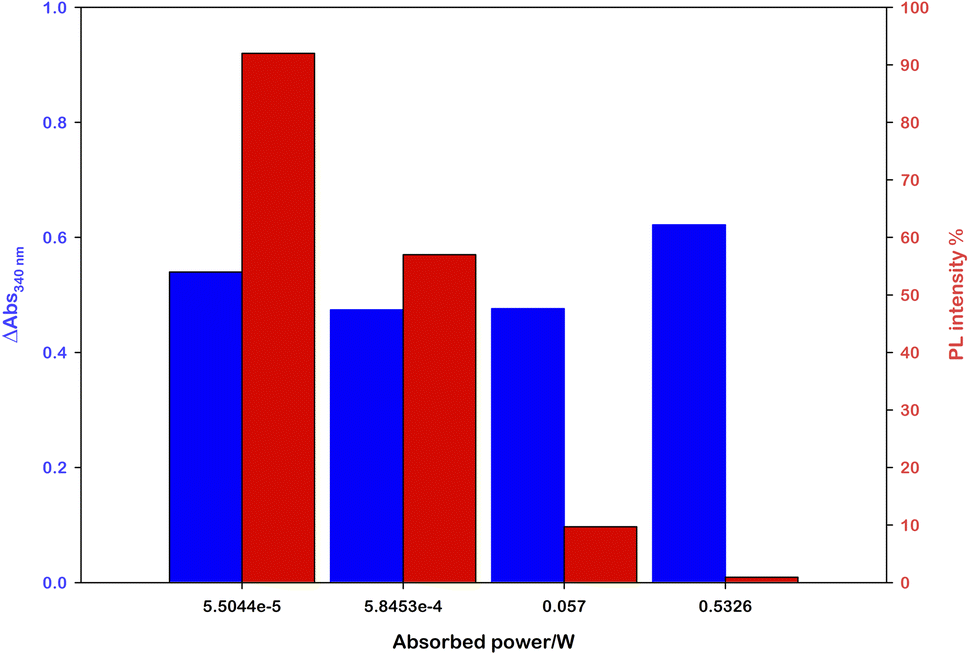 | ||
| Fig. 4 Effect of the light source power on the 1,4-NADH emission signal at similar absorbance variations; the percentage refers to ruthenium emission used as an internal standard. | ||
The most straightforward method to validate the regenerated cofactor is by an enzyme assay.32 This is normally performed using a substrate that can be reduced by the enzyme in combination with NADH and monitoring the disappearance of the absorption band centred at 340 nm. To obtain a photophysical clear signal, the substrate and the product of the enzymatic reaction must not absorb light in this spectral region. For this reason, we selected plant alcohol dehydrogenase 1 (ADH-1) enzyme that consumes NADH to convert acetaldehyde into ethanol and NAD+.
Enzymatic assays were performed on each obtained photoproduct and the first evidence was that the samples with no photoluminescence signal at 470 nm showed completely no enzyme activity and the absorption band at 340 nm remained unchanged. Among the samples showing emission at 470 nm, the enzymatic activity followed a linear correlation with the emission intensity, thus confirming that the emission is the unequivocal signal of the enzymatically active species. Optimizing the experimental setup and irradiation conditions, the best result was achieved irradiating a typical solution ([Ru(bpy)(CN)4]2− (0.25 mM), NAD+ (2.0 mM), L-cysteine (0.1 M), Tris·HCl buffer (0.1 M), pH = 7.2, N2 purging for 30 minutes) with a commercial white lamp for 8 hours (see the ESI, Fig. S9†). The following addition of acetaldehyde (15 mM) and ADH-1 (0.1 μM) leads to a strong decrease in absorption (around 65% of the photoproduct) and an almost total disappearance of the 1,4-NADH emission signal (Fig. 5). HPLC analyses performed on this sample confirm the production of 1,4-NADH (blue vs. yellow curve, Fig. S7†) and the total consumption after the enzymatic reaction (blue vs. green curve, Fig. S7†). We also performed HPLC studies to prove the stability of the photocatalyst (Fig. S8†).
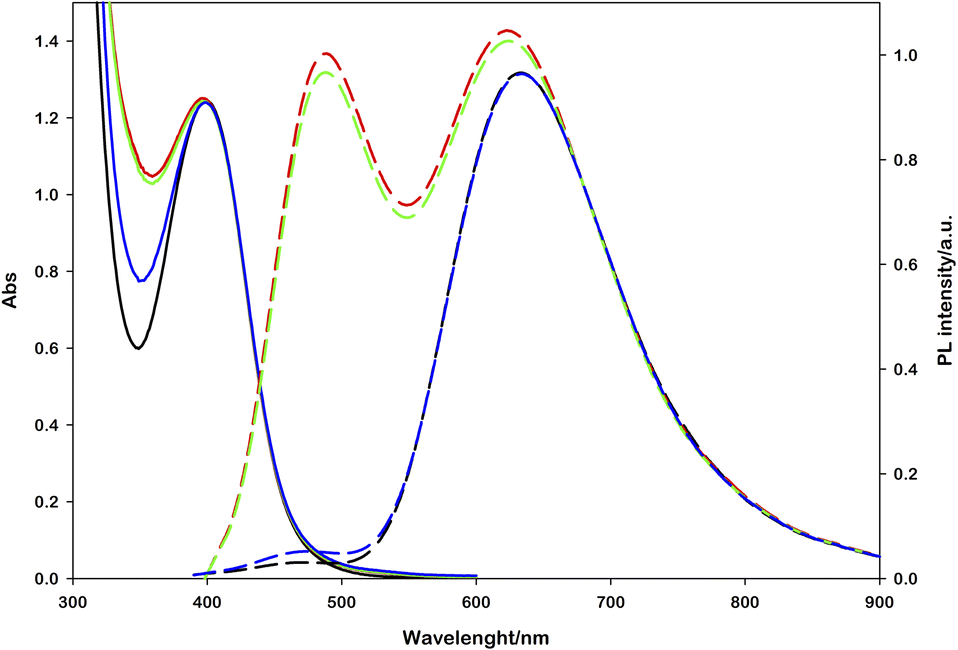 | ||
| Fig. 5 Absorption (solid lines) and emission (dashed lines) spectral variations observed upon white lamp irradiation and subsequent enzyme assay. Black lines represent the starting reaction mixture, red lines after irradiation (15 hours), green lines upon 15 mM acetaldehyde addition and blue lines after 0.1 μM ADH-1 addition. Emission spectra are obtained upon 340 nm excitation. | ||
Conclusions
In summary, a water-soluble and highly photostable ruthenium complex ([Ru(bpy)(CN)4]2−) has been successfully employed for the direct photochemical monoreduction of NAD+. The high reduction potential of the Ru complex is able to generate NAD˙ under visible light irradiation followed by a fast hydrogen atom transfer from the L-cysteine that prevents dimerization to the inactive NAD2 dimer. We proposed the distinctive emission at 470 nm of 1,4-NADH as a straightforward tool to investigate the formation of the enzymatically active cofactor. This approach could accelerate the use of visible light in the regeneration of natural cofactors thus opening new horizons in the use of enzymatic reactions in industrial applications.Author contributions
A. B. designed and carried out the experiments, and wrote the first draft of the paper. M. Z. provided the enzyme and supervised the enzymatic experiments. G. B. conceived, designed and supervised the research, and wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Bologna for financial support.Notes and references
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50, 8003–8049 RSC.
- T. Katagiri and Y. Amao, Green Chem., 2020, 22, 6682–6713 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- Z. Jiang, B. Wang, J. C. Yu, J. Wang, T. An, H. Zhao, H. Li, S. Yuan and P. K. Wong, Nano Energy, 2018, 46, 234–240 CrossRef CAS.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- L. Sellés Vidal, C. L. Kelly, P. M. Mordaka and J. T. Heap, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 327–347 CrossRef PubMed.
- Y. Wang, G. Xiao, Y. Zhao, S. Wang, Y. Jin, Z. Wang and H. Su, Nanotechnology, 2021, 32, 485703 CrossRef CAS.
- T. Katagiri and Y. Amao, New J. Chem., 2021, 45, 15748–15752 RSC.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS.
- F. F. Özgen, M. E. Runda and S. Schmidt, ChemBioChem, 2021, 22, 790–806 CrossRef PubMed.
- Y. Zhang, Y. Zhao, R. Li and J. Liu, Sol. RRL, 2021, 5, 2000339 CrossRef CAS.
- A. K. Mengele, D. Weixler, S. Amthor, B. J. Eikmanns, G. M. Seibold and S. Rau, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202114842.
- K. Kinastowska, J. Liu, J. M. Tobin, Y. Rakovich, F. Vilela, Z. Xu, W. Bartkowiak and M. Grzelczak, Appl. Catal., B, 2019, 243, 686–692 CrossRef CAS.
- J. A. Farrington, E. J. Land and A. J. Swallow, Biochim. Biophys. Acta, Bioenerg., 1980, 590, 273–276 CrossRef CAS.
- C. A. Bignozzi, C. Chiorboli, M. T. Indelli, M. A. Rampi Scandola, G. Varani and F. Scandola, J. Am. Chem. Soc., 1986, 108, 7872–7873 CrossRef CAS PubMed.
- M. T. Indelli, C. A. Bignozzi, A. Marconi and F. Scandola, in Photochemistry and Photophysics of Coordination Compounds, ed. H. Yersin and A. Vogler, Springer Berlin Heidelberg, Berlin, Heidelberg, 1987, pp. 159–164 Search PubMed.
- M. D. Ward, Coord. Chem. Rev., 2006, 250, 3128–3141 CrossRef CAS.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CrossRef CAS PubMed.
- T. Saba, J. W. H. Burnett, J. Li, P. N. Kechagiopoulos and X. Wang, Chem. Commun., 2020, 56, 1231–1234 RSC.
- T. A. Enache and A. M. Oliveira-Brett, Bioelectrochemistry, 2011, 81, 46–52 CrossRef CAS PubMed.
- B. W. Carlson, L. L. Miller, P. Neta and J. Grodkowski, J. Am. Chem. Soc., 1984, 106, 7233–7239 CrossRef CAS.
- R. Ruppert and E. Steckhan, J. Chem. Soc., Perkin Trans. 2, 1989, 811–814 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- J. W. H. Burnett, R. F. Howe and X. Wang, Trends Chem., 2020, 2, 488–492 CrossRef CAS.
- A. Mayevsky and G. G. Rogatsky, Am. J. Physiol.: Cell Physiol., 2007, 292, C615–C640 CrossRef CAS PubMed.
- P. M. Schaefer, S. Kalinina, A. Rueck, C. A. F. von Arnim and B. von Einem, Cytometry, Part A, 2019, 95, 34–46 CrossRef.
- B. A. Beaupre, M. R. Hoag, J. Roman, F. H. Försterling and G. R. Moran, Biochemistry, 2015, 54, 795–806 CrossRef CAS PubMed.
- S. Fukuzumi and T. Tanaka, in Photoinduced Electron Transfer Part C. Photoinduced Electron Transfer Reactions: Organic Substrates, ed. M. A. Fox and M. Chanon, Elsevier, 1988, pp. 578–635 Search PubMed.
- S. S. Chan, T. M. Nordlund, H. Frauenfelder, J. E. Harrison and I. C. Gunsalus, J. Biol. Chem., 1975, 250, 716–719 CrossRef CAS.
- W. Jones, J. W. H. Burnett, J. Shi, R. F. Howe and X. Wang, Joule, 2020, 4, 2055–2059 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2se01045c |
| This journal is © The Royal Society of Chemistry 2022 |