
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic defluorinative ketyl–olefin coupling by halogen-atom transfer†
Peter
Bellotti‡
 a,
Huan-Ming
Huang‡
ab,
Teresa
Faber
a,
Ranjini
Laskar
a and
Frank
Glorius
*a
a,
Huan-Ming
Huang‡
ab,
Teresa
Faber
a,
Ranjini
Laskar
a and
Frank
Glorius
*a
aWestfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 36, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
bSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: huanghm@shanghaitech.edu.cn
First published on 10th June 2022
Abstract
Ketyl–olefin coupling reactions stand as one of the fundamental chemical transformations in synthetic chemistry and have been widely employed in the generation of complex molecular architectures and natural product synthesis. However, catalytic ketyl–olefin coupling, until the recent development of photoredox chemistry and electrosynthesis through single-electron transfer mechanisms, has remained largely undeveloped. Herein, we describe a new approach to achieve catalytic ketyl–olefin coupling reactions by a halogen-atom transfer mechanism, which provides innovative and efficient access to various gem-difluorohomoallylic alcohols under mild conditions with broad substrate scope. Preliminary mechanistic experimental and computational studies demonstrate that this radical-to-polar crossover transformation could be achieved by sequentially orchestrated Lewis acid activation, halogen-atom transfer, radical addition, single-electron reduction and β-fluoro elimination.
Introduction
Developing catalytic chemical transformations that bypass stoichiometric—and often harsh—reagents stands as a pillar principle of green chemistry.1 Across the plethora of fundamental functional groups, carbonyl groups arguably rank amongst the most useful synthetic building blocks to efficiently construct alcohol derivatives.2 Extending the repertoire of the well-established Grignard addition3 and Nozaki–Hiyama–Kishi (NHK) reaction,4 the carbonyl group has also been employed to generate ketyl radicals through single-electron transfer (SET).5 Since their early discovery by Corey and Pyne,6 ketyl–olefin coupling reactions have surged to become a popular means of constructing alcohol derivatives through carbon–carbon bond formation, despite mandating stoichiometric amounts of reductant and harsh conditions.7 Spurred by Corey,8a catalytic ketyl–olefin coupling reactions have been developed by different research groups all over the world through visible light photocatalysis,8b–g electrosynthesis8h or radical relay strategies8i–l (Scheme 1A).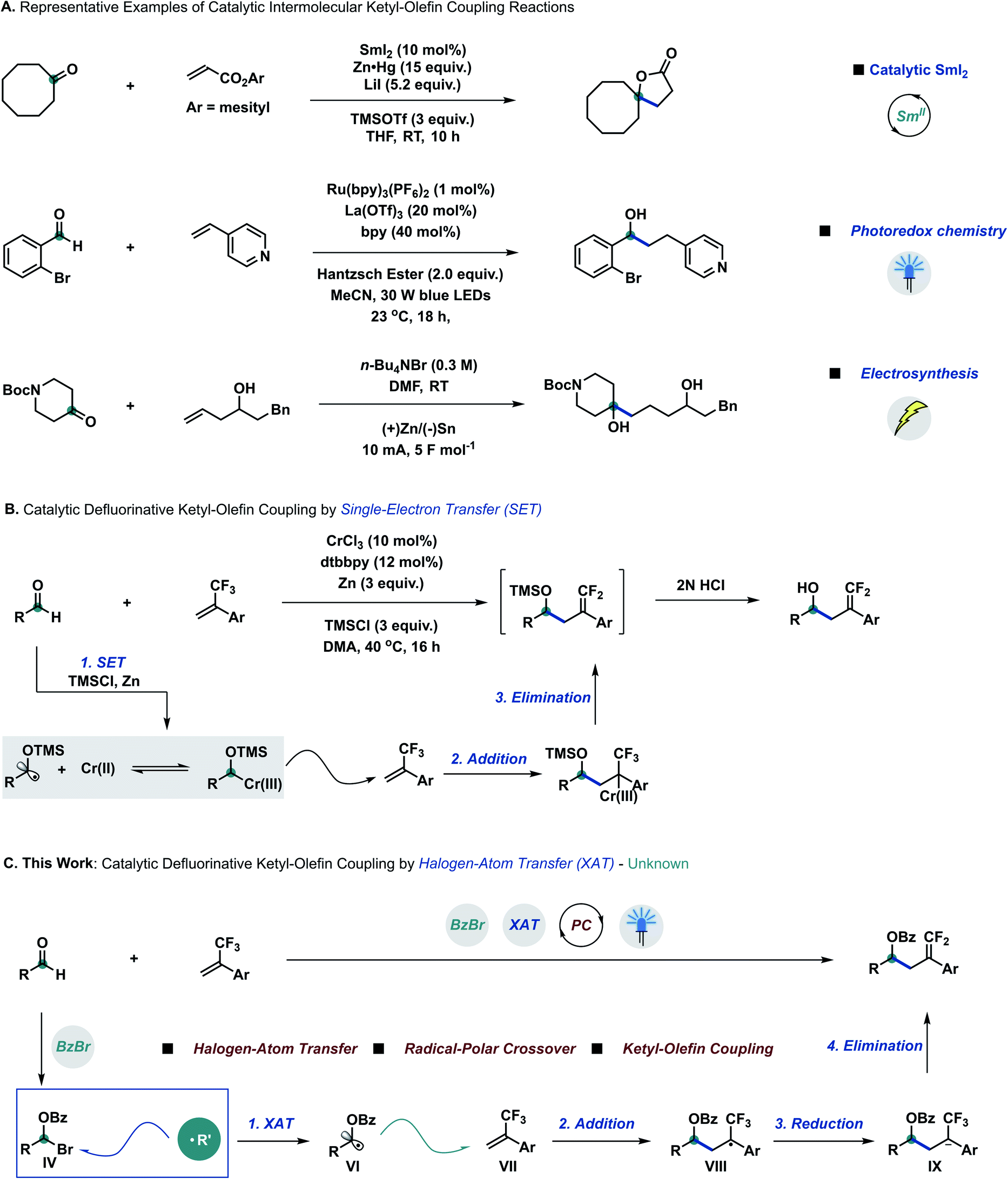 | ||
| Scheme 1 State-of-the-art of ketyl–olefin coupling reactions and our design approach enabled by halogen-atom transfer. (A) Selected examples of intermolecular ketyl–olefin coupling by means of catalytic SmI2, photoredox catalysis and electrosynthesis. (B) Report of defluorinative ketyl–olefin coupling via single-electron transfer. (C) Our strategy to achieve the mild defluorinative ketyl–olefin coupling via halogen-atom transfer. bpy = 2,2′-bipyridine; DMA = N,N-dimethylacetamide; DMF = N,N-dimethylformamide; dtbbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl; PC = photocatalysis; THF = tetrahydrofuran; TMS = trimethylsilyl; RT = room temperature; XAT = halogen-atom abstraction. | ||
Radical-to-polar crossover reactions—which intertwine single- and two-electron chemistry—have become an emerging synthetic tool to overcome the intrinsic limitations of traditional radical and polar chemistry,9 especially due to the rapid development of photoredox chemistry.10 Organofluorine motifs, owing to their unique reactivity, stability, and biological properties, have gained preeminent importance as building blocks in medicinal and agricultural chemistry.11a α-Trifluoromethylstyrene derivatives are versatile synthetic intermediates for the construction of gem-difluoroalkene compounds, attainable through radical-to-polar crossover manifolds under mild conditions.11b–i Due to the high reduction potential of aliphatic aldehydes,12 defluorinative ketyl–olefin coupling was only achieved very recently through single-electron transfer by using catalytic amounts of chromium (Wang),13a iron (Wang)13b and nickel (Montgomery)13c with stoichiometric reducing metals and additives (Scheme 1B).
Halogen-atom transfer (XAT) can be leveraged in synthetic chemistry to efficiently generate carbon centred radical species from organohalides.14 MacMillan15a,b and other research groups15c–e have recently merged the concept of XAT into metallaphotoredox chemistry.15f More recently, Leonori16a,b and Doyle16c discovered that aminoalkyl radicals could be used as a new type of halogen-atom abstracting reagent to form carbon based radical species from alkyl and aryl halides, to be further employed in related arylation,16d amination,16e hydroxymethylation16f and elimination16g processes. We questioned whether aldehydes could be directly employed as radical precursors to achieve carbon based radical intermediates through a XAT mechanism, prospectively extending the library of radical precursors besides overcoming the limitation of high reduction potentials. According to this hypothesis, we proposed that α-bromo alkyl intermediate IV could be formed in situ by reacting aldehydes and benzoyl bromide with catalytic amounts of Lewis acid.17 Pioneering work by Nagib and co-workers demonstrated that α-oxy halides can productively generate α-oxy radicals upon XAT using manganese metallaradicals in an overall atom-transfer catalytic cycle.17d,e We posited that α-oxy bromide IV could react with suitable XAT reagents to generate α-oxy radical species VI,14 to be trapped by α-trifluoromethylstyrene derivatives VII to form radical intermediates VIII.11b–i After sequential single-electron reduction by a suitable photocatalyst and β-fluoro elimination, the final ketyl–olefin coupling products could be obtained (Scheme 1C).
Results and discussion
Reaction design and optimization
Based on our concept and previous studies, hexanal 1a and α-trifluoromethylstyrene 2a were investigated to explore the catalytic defluorinative ketyl–olefin coupling reaction (Table 1). Pleasingly, coupling product 3 could be obtained in 74% isolated yield with 5 mol% 4CzIPN, 1.5 equiv. of (TMS)3SiOH and 4 equiv. of KOAc under visible light conditions (entry 1). Several bases were screened, indicating KOAc to be the most suitable base (entries 2–7). Highly oxidizing iridium based photocatalysts PC2 and PC3 were also investigated, but delivered lower yields (entries 8 and 9). When commercially available (TMS)3SiH was used in our system, only 53% yield of 3 was obtained (entry 10). Several control experiments also indicated that a photocatalyst, silane reagent, base and visible light are necessary to achieve the ketyl–olefin coupling (entry 11). Inspired by the work of Leonori16a,b,d–g and Doyle,16c we tested their standard conditions (entry 12) and other amines (entry 13).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yielda (%) |
| a Yields were determined by 1H NMR using dibromomethane as an internal standard. b Isolated yield in parentheses. c = concentration; DIPEA = N,N-diisopropylethylamine; I = light intensity; T = temperature; TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl. | ||
| 1 | None | 78 (74)b |
| 2 | Na2CO3 instead of KOAc | 65 |
| 3 | K2CO3 instead of KOAc | 70 |
| 4 | K3PO4 instead of KOAc | 71 |
| 5 | Na2HPO4 instead of KOAc | 57 |
| 6 | NaOAc instead of KOAc | 62 |
| 7 | NaHCO3 instead of KOAc | 53 |
| 8 | 1 mol% PC2 instead of 5 mol% PC1 | 64 |
| 9 | 1 mol% PC3 instead of 5 mol% PC1 | 28 |
| 10 | (TMS)3SiH instead of (TMS)3SiOH | 53 |
| 11 | Without 4CzIPN or (TMS)3SiOH or KOAc or light | 0 |
| 12 | Bn3N instead of (TMS)3SiOH, MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (9:1) :H2O (9:1) |
20 |
| 13 | Et3N or DIPEA instead of (TMS)3SiOH | 0 |
| 14 | With 2 equiv. of TEMPO | 0 |
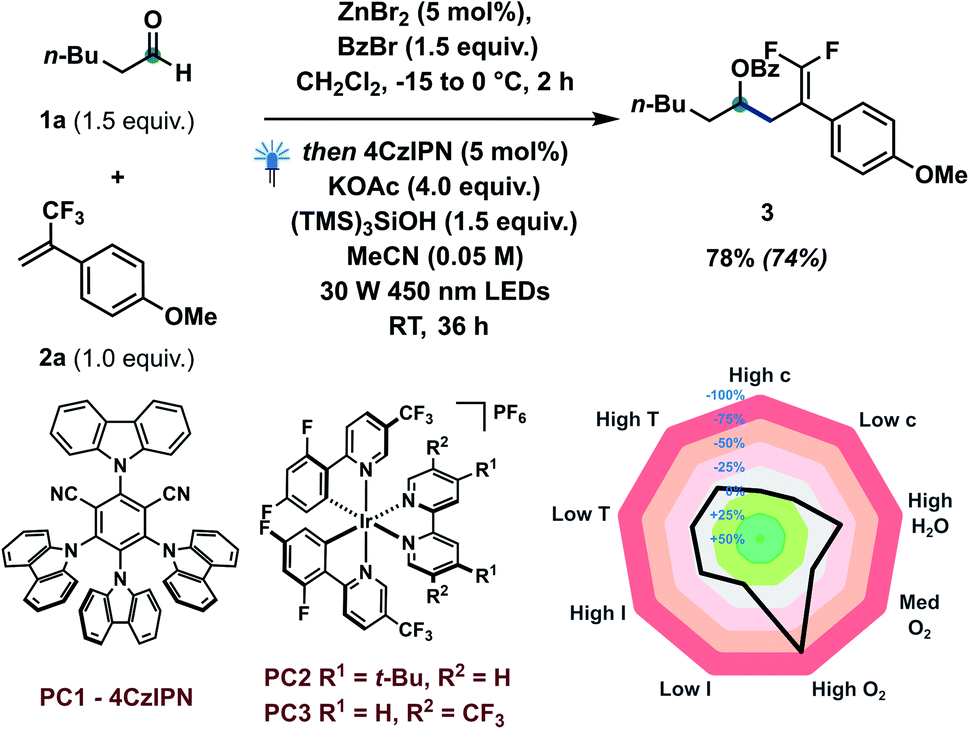
Unfortunately, only 20% yield was detected using tribenzylamine as a XAT reagent. We observed that a relatively low concentration (0.05 M) offered improved reaction yield, likely due to maximization of the MeCN:CH2Cl2 ratio. When two equivalents of TEMPO were added to the standard conditions, no desired product was generated, which indicated that the transformation may proceed through a radical mechanism (entry 14). Condition-based sensitivity screening18 was also applied in our current catalytic approach and showed that the reaction is sensitive to water, oxygen and lower reaction temperature.
Synthetic scope
With the optimized conditions in hand, we started to investigate the reaction scope of the catalytic defluorinative ketyl–olefin coupling reaction (Scheme 2). Firstly, different α-primary aldehydes were screened, and the corresponding coupling products 4–20 could be obtained in moderate to excellent yields. For example, paraformaldehyde (4), isopentyl aldehyde (5), and n-nonyl (6) and phenyl-substituted aliphatic aldehydes (7–13) were all well tolerated. Substituted phenylpropanal, functionalised with fluoro (8), ester (9, 10), methoxy (11) and phenyl (12) groups reacted equally well in 60–72% yield. Esters bearing gem-difluoro (17) and aliphatic chains (18) reacted in 46–58% yield. Aliphatic aldehydes bearing bromide (14), alkyne (15), imide (16), chloride (19) and alkene (20) substituents were all tolerated in the newly developed system. In particular, compounds derived from herbicide 2,4-D (19) and linoleic acid (20) testify to the appeal of the strategy towards the selective modification of complex scaffolds. Interestingly, a deuterated compound (13)—otherwise step-intensive to achieve—was also efficiently formed in 57% isolated yield. Furthermore, a variety of α-secondary aldehydes, both cyclic and acyclic, were also investigated (21–26). Cyclohexylcarbaldehyde (21) and small ring cyclobutylcarbaldehyde (22) reacted in moderate yields. An acyclic 2-methyl (24) substituted aldehyde delivered the corresponding difluoroalkene in 60% yield. Notably, aldehydes derived from non-steroidal anti-inflammatory drugs ibuprofen and naproxen were also tolerated, generating 25 and 26 in 57 and 31% isolated yield, respectively. Finally, sterically hindered tertiary aldehydes were also tested in this catalytic ketyl–olefin coupling. Remarkably, the corresponding coupling products (27–32) were formed in moderate to good yields. Pivalic (27) and adamantyl aldehyde (28) reacted to give the corresponding difluoroalkenes in 58 and 70% yield, respectively. α-Cyclic aldehydes featuring tetrahydrofuran (29), cyclopentyl (30) and cyclohexyl (31) substituents proved equally reactive and allowed for the generation of highly congested protected homoallylic alcohols. Pleasingly, gemfibrozil derivative 32 could also be obtained without interference of the electron-rich aryl ether. Benzaldehydes failed at delivering—under the optimized activation conditions—benzylic α-oxy bromides,17c therefore hampering their successful defluorinative ketyl–olefin coupling.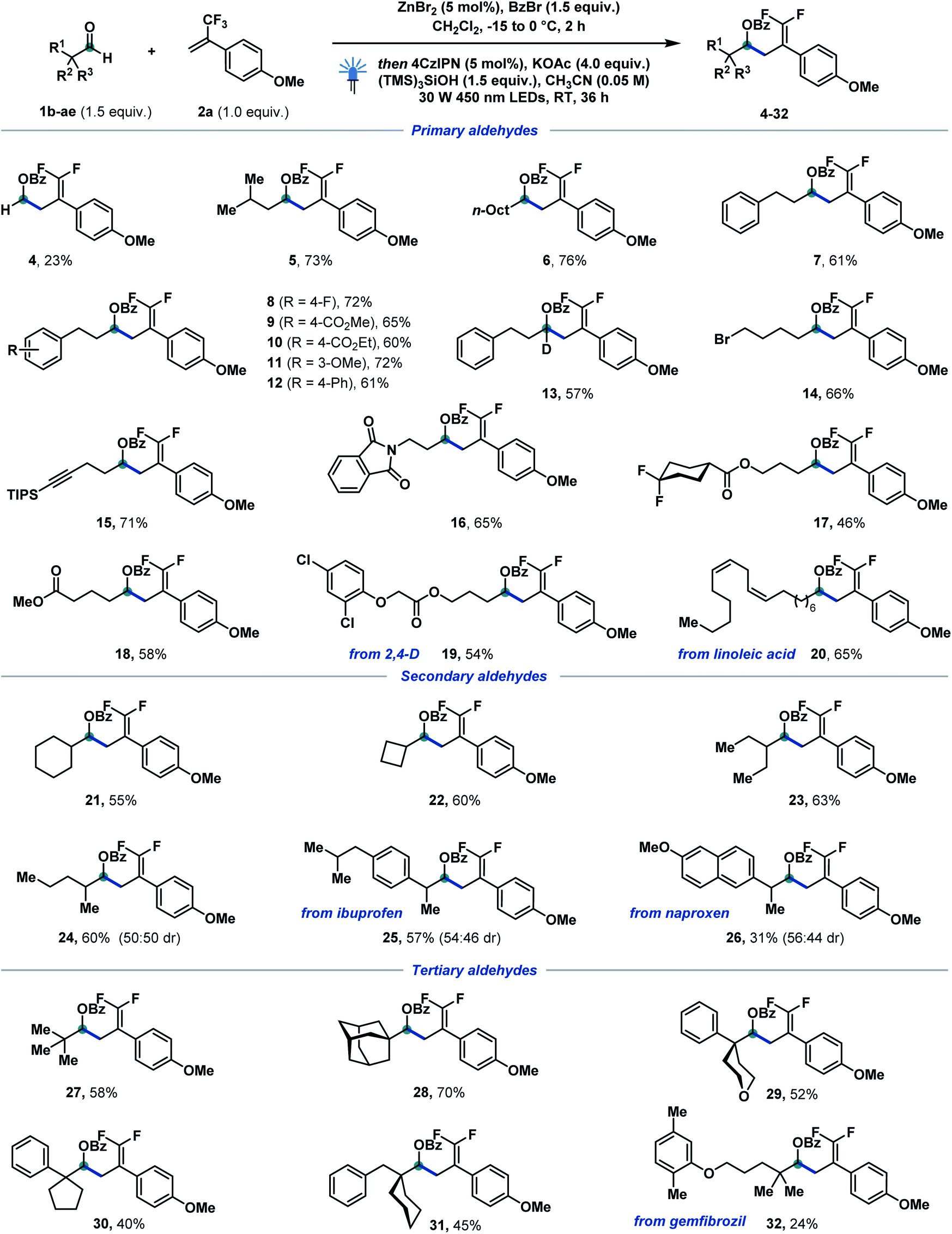 | ||
| Scheme 2 Reaction scope of α-primary, -secondary and -tertiary aldehydes. Reaction conditions: 1b–1ae (0.3 mmol, 1.5 equiv.), benzoyl bromide (0.3 mmol, 1.5 equiv.), ZnBr2 (0.01 mmol, 5 mol%), CH2Cl2 (1 M), −15 to 0 °C, 2 h, then 2a (0.2 mmol, 1.0 equiv.), 4CzIPN (0.01 mmol, 5 mol%), KOAc (0.8 mmol, 4.0 equiv.), (TMS)3SiOH (0.3 mmol, 1.5 equiv.), MeCN (0.05 M), 30 W 450 nm LEDs, RT, 36 h. For experimental details, see the ESI.† | ||
We then explored a variety of olefin derivatives to further investigate this catalytic protocol (Scheme 3). As shown below, a wide range of olefin derivatives could couple with hexanal 1a to generate the corresponding products (3, 33–46) in good to excellent yields. para-Substituted trifluoromethyl styrenes bearing methoxyl (3), phenoxyl (33), trifluoromethoxyl (34), thiomethyl (35), chloride (40), trifluoromethyl (36), trimethylsilyl (37), and hydroxyl (38) successfully delivered the corresponding products in moderate to good yields. Polysubstituted systems bearing 3,5-dimethoxy (39), 2-methoxy-4-chloro (40), 3,4-dimethoxy (41) and 3-fluoro-4-phenyl (42) substituents were well tolerated. 2-Naphthyl (43), indole (44), pyridine (45) and 1,3-benzodioxole (46) were successfully incorporated into the final product. α-Trifluoromethyl styrenes featuring electron-withdrawing groups afforded—as reported by Wang13a and Molander11d—substantial amounts of trifluoromethylated side products from competitive protonation of IX (for details see the ESI†).
 | ||
| Scheme 3 Reaction scope of α-trifluoromethylstyrene derivatives. Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), benzoyl bromide (0.3 mmol, 1.5 equiv.), ZnBr2 (0.01 mmol, 5 mol%), CH2Cl2 (1 M), −15 to 0 °C, 2 h, then 2b–2p (0.2 mmol, 1.0 equiv.), 4CzIPN (0.01 mmol, 5 mol%), KOAc (0.8 mmol, 4.0 equiv.), (TMS)3SiOH (0.3 mmol, 1.5 equiv.), MeCN (0.05 M), 30 W 450 nm LEDs, RT, 36 h. a The reaction was performed on a 0.1 mmol scale. b Aldehyde 1n was used instead of 1a. Boc = tert-butoxycarbonyl. For experimental details, see the ESI.† | ||
Mechanistic studies
Based on previous studies,17a–f α-bromo alkyl intermediate IV could be formed in situ upon reacting aldehydes and benzoyl bromide with a catalytic amount of zinc bromide. When cyclopropyl-containing alkene 2q was employed under the optimized condition, ring-opening product 47 was formed, suggesting formation of a benzyl radical as a reaction intermediate.19 Density functional theory (DFT) calculations at the CAM-B3LYP-D3/def-SVP, CPCM (MeCN)//CAM-B3LYP-D3/def2-TZVPP, and CPCM (MeCN) levels of theory were used to shed light on the following questions: (I) What are the mechanistic intricacies of the α-oxy radical VI formation phase? (II) What is the energetic profile of the reaction?According to literature reports, we postulated the generation of radical (TMS)3SiO˙ (II) from (TMS)3SiOH (I) to occur in order to initiate the subsequent reaction steps (Scheme 4). The transfer of one electron and one proton was calculated to be thermodynamically downhill with ΔG = −10.0 kcal mol−1. Considering a consecutive SET/deprotonation, we found direct oxidation of supersilanol I to be highly endergonic (ΔG = +18.8 kcal mol−1). The subsequent deprotonation is exergonic (ΔG = −28.8 kcal mol−1). Considering the opposite stepwise deprotonation/SET process, we found that acetate-mediated deprotonation is endergonic with ΔG = +20.1 kcal mol−1 and the following oxidation of the deprotonated supersilyl anion via SET is exergonic (ΔG = −30.1 kcal mol−1). Given that, in both stepwise scenarios, the first reaction step is significantly endergonic, a concerted oxidative PCET is deemed likely.
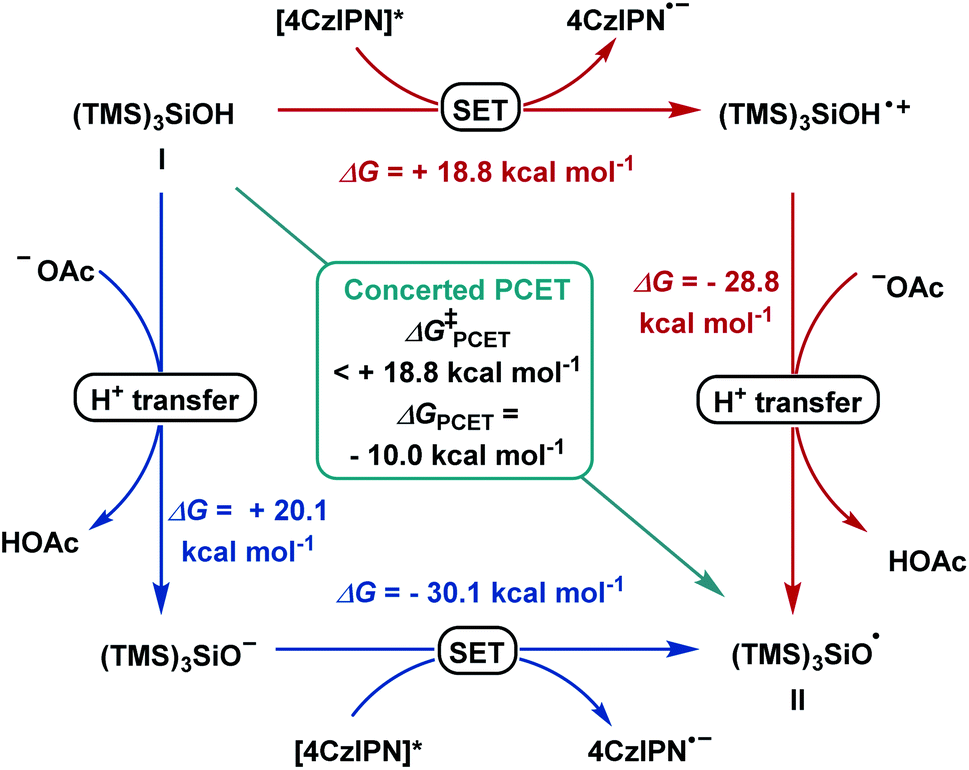 | ||
| Scheme 4 Reaction pathways for stepwise deprotonation/SET (blue), SET/deprotonation (red) and concerted PCET (green). | ||
Applying upper limit approximation, we propose the kinetic energy barrier of concerted PCET to be lower than 18.8 kcal mol−1 (ΔG‡ < +18.8 kcal mol−1 < +20.1 kcal mol−1). Next, the formation of a ketyl-type radical was further investigated (Scheme 5). Brook-type rearrangement to form silyl radical III has a small energy barrier (TS-1, ΔG‡ = +2.8 kcal mol−1) and it is exergonic by 43.9 kcal mol−1. Being both kinetically and thermodynamically favoured, Brook-type rearrangement is expected to happen before any bimolecular side reaction. The ensuing halogen atom transfer (XAT) from α-benzoate bromide IV to radical III was found to be thermodynamically favoured (ΔG = −28.8 kcal mol−1) and has to overcome a kinetic energy barrier of 12.4 kcal mol−1. This value agrees with reports by MacMillan and Houk for C–Br abstraction in 2-bromopropane by silyl radical III.20 For the addition of the ketyl-type radical to styrene VI, a kinetic energy barrier of +12.3 kcal mol−1 was found. The process was computed to be exergonic (ΔG = −18.1 kcal mol−1 for VI + VII → VIII). From trifluoroalkane radical VIII, single electron transfer from reduced 4CzIPN (Eox(4CzIPN/4CzIPN˙−) = −1.21 V vs. SCE)21 to give trifluoroalkane anion IX was found to be slightly exergonic (ΔG = −0.3 kcal mol−1 for VIII → IX). The subsequent E1cB-type fluoride elimination from methoxy substituted anion IX was significantly exergonic (ΔG = −16.2 kcal mol−1) and barrierless. Hence, this intramolecular process occurs immediately once intermediate IX is formed. Assuming that protonation occurs from supersilanol I, the process was found to be thermodynamically favoured compared to defluorination (ΔG = −35.5 kcal mol−1). However, given the barrierless E1cB-type process, we assume that the concentration of IX is so low that the bimolecular protonation—which has a first order dependence on the concentration of IX—is substantially suppressed. Considering the full energy profile, the ketyl–olefin coupling product proceeds via a sequence of halogen-atom abstraction, ketyl-type radical addition, SET and fluoride elimination. The rate limiting step features either halogen-atom abstraction (ΔG‡ = +12.4 kcal mol−1), radical addition to trifluoroalkene (ΔG‡ = +12.3 kcal mol−1) or PCET (ΔG‡ < +18.8 kcal mol−1).
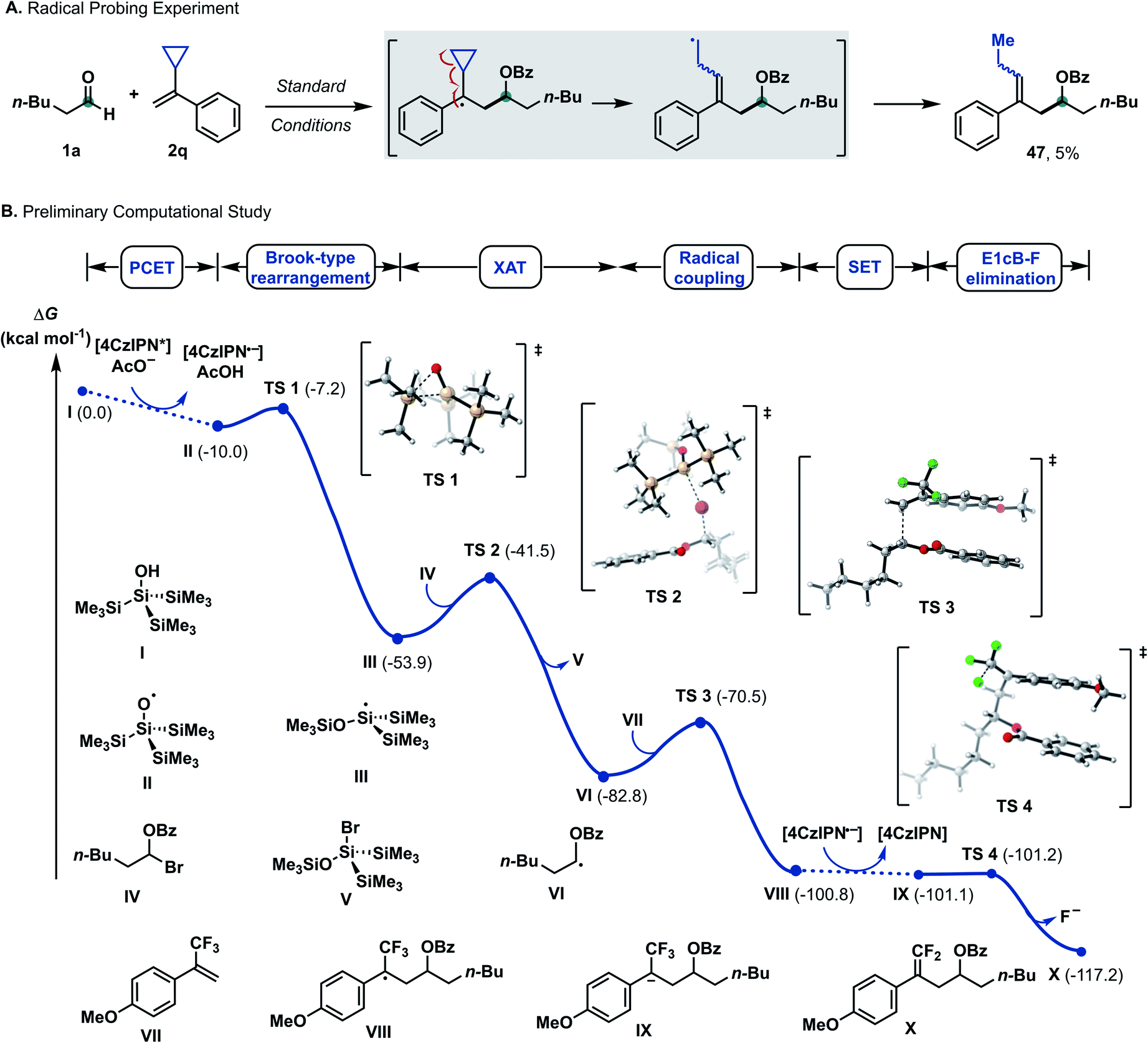 | ||
| Scheme 5 Selected mechanistic experiments and DFT calculations. Density functional theory calculations were performed at the CAM-B3LYP-D3/def-SVP, CPCM (MeCN)//CAM-B3LYP-D3/def2-TZVPP, and CPCM (MeCN) levels of theory. ΔG values are in kcal mol−1. For experimental details, see the ESI.† | ||
Conclusions
In summary, we successfully demonstrated the first example of catalytic defluorinative ketyl–olefin coupling reaction enabled by halogen-atom transfer. This radical-to-polar crossover methodology provides an alternative approach to achieve catalytic ketyl–olefin coupling reaction under visible light conditions, bypassing the need for (super)stoichiometric metal reductants. This newly developed method demonstrates that aliphatic aldehydes could be applied in the halogen-atom transfer induced radical chemistry and that in situ formed α-oxy bromides can serve as ketyl-radical surrogates to harness umpolung reactivity. Based on this concept, we expect that this approach will not only be applied in the synthesis of organofluoride compounds but also inspire related coupling reactions and radical carbonyl chemistry.Data availability
Experimental and computational data are provided in the ESI.† Raw NMR data (JCAMP and .mnova files) for literature unknown compounds and xyz coordinates from DFT calculations have been deposited at Zenodo, under the Creative Commons Attribution 4.0 International license: https://doi.org/10.5281/zenodo.6533181.Author contributions
H.-M. H., P. B. and F. G. conceptualized the research, H.-M. H., P. B., T. F. and R. L. performed the investigation, and T. F. performed the density functional theory investigation. H.-M. H., P. B. and F. G. wrote the manuscript with contributions from all authors, and F. G. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Alexander von Humboldt Foundation and the startup funding from ShanghaiTech University (H.-M. H.) and the Deutsche Forschungsgemeinschaft (Leibniz Award, SBF 858). We thank Philipp Pflüger (University of Münster) for support with the computational study.Notes and references
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- (a) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (b) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS PubMed.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- (a) A. Fürstner, Chem. Rev., 1999, 99, 991–1045 CrossRef PubMed; (b) H.-M. Huang, P. Bellotti and F. Glorius, Acc. Chem. Res., 2022, 55, 1135–1147 CrossRef CAS PubMed.
- (a) G. A. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307–338 CrossRef CAS PubMed; (b) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed; (c) Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC; (d) Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CAS; (e) K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC.
- E. J. Corey and S. G. Pyne, Tetrahedron Lett., 1983, 24, 2821–2824 CrossRef CAS.
- (a) S. Fukuzawa, A. Nakanishi, T. Fujinami and S. Sakai, J. Chem. Soc., Chem. Commun., 1986, 624 RSC; (b) G. A. Molander and C. Kenny, Tetrahedron Lett., 1987, 28, 4367–4370 CrossRef CAS; (c) A. L. Beckwith and D. H. Roberts, J. Am. Chem. Soc., 1986, 108, 5893–5901 CrossRef CAS PubMed; (d) E. J. Enholm and G. Prasad, Tetrahedron Lett., 1989, 30, 4939–4942 CrossRef; (e) D. S. Hays and G. C. Fu, J. Org. Chem., 1996, 61, 4–5 CrossRef CAS; (f) R. E. Estévez, J. L. Oller-Lopez, R. Robles, C. R. Melgarejo, A. Gansäuer, J. M. Cuerva and J. E. Oltra, Org. Lett., 2006, 8, 5433–5436 CrossRef PubMed.
- (a) E. J. Corey and G. Z. Zheng, Tetrahedron Lett., 1997, 38, 2045–2048 CrossRef CAS , for recent elegant examples of photoredox catalysis, see:; (b) H. Seo and T. F. Jamison, Org. Lett., 2019, 21, 10159–10163 CrossRef CAS PubMed; (c) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed; (d) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed; (e) K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed; (f) E. Fava, M. Nakajima, A. L. P. Nguyen and M. Rueping, J. Org. Chem., 2016, 81, 6959–6964 CrossRef CAS PubMed; (g) J.-Y. Gu, W. Zhang, S. R. Jackson, Y.-H. He and Z. Guan, Chem. Commun., 2020, 56, 13441–13444 RSC , for a recent elegant example of electrosynthesis, see:; (h) P. Hu, B. K. Peters, C. A. Malapit, J. C. Vantourout, P. Wang, J. Li, L. Mele, P. G. Echeverria, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 20979–20986 CrossRef CAS PubMed , for a recent elegant review and examples of radical relay catalysis, see:; (i) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC; (j) H.-M. Huang, J. J. W. McDouall and D. J. Procter, Nat. Catal., 2019, 2, 211–218 CrossRef CAS; (k) H.-M. Huang, Q. He and D. J. Procter, Synlett, 2019, 1, 2–7 Search PubMed; (l) S. Agasti, N. A. Beattie, J. J. W. McDouall and D. J. Procter, J. Am. Chem. Soc., 2021, 143, 3655–3661 CrossRef CAS PubMed.
- For recent reviews on radical-to-polar crossover reactions, see: (a) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed; (b) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- For selected reviews on photoredox catalysis, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS; (d) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (e) D. C. Fabry and M. Rueping, Acc. Chem. Res., 2016, 49, 1969–1979 CrossRef CAS PubMed; (f) K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242–252 CrossRef CAS PubMed; (g) E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed; (h) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (i) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (j) Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (c) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC; (d) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed; (e) W.-J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS PubMed; (f) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245–9251 CrossRef CAS; (g) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809–814 RSC; (h) Y. Liu, X. Tao, Y. Mao, X. Yuan, J. Qiu, L. Kong, S. Ni, K. Guo, Y. Wang and Y. Pan, Nat. Commun., 2021, 12, 6745 CrossRef CAS PubMed; (i) C. Zhu, Z.-Y. Liu, L. Tang, H. Zhang, Y.-F. Zhang, P. J. Walsh and C. Feng, Nat. Commun., 2020, 11, 4860 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- Very recently, Wang and Montgomery described several elegant examples of ketyl–olefin couplings through the SET mechanism, see: (a) C. Zhang, Z. Lin, Y. Zhu and C. Wang, J. Am. Chem. Soc., 2021, 143, 11602–11610 CrossRef CAS PubMed; (b) C. Zhang, L. Wang, H. Shi, Z. Lin and C. Wang, Org. Lett., 2022, 24, 3211–3216 CrossRef CAS PubMed; (c) J. Xiao and J. Montgomery, ACS Catal., 2022, 12, 2463–2471 CrossRef CAS.
- (a) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (b) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- (a) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed; (b) N. W. Dow, A. Cabré and D. W. C. MacMillan, Chem, 2021, 7, 1827–1842 CrossRef CAS PubMed; (c) R. J. Wiles, J. P. Phelan and G. A. Molander, Chem. Commun., 2019, 55, 7599–7602 RSC; (d) S. M. Hell, C. F. Meyer, G. Laudadio, A. Misale, M. C. Willis, T. Noël, A. A. Trabanco and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 720–725 CrossRef CAS PubMed; (e) J. H. Blackwell, R. Kumar and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 1598–1609 CrossRef CAS PubMed; (f) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- For recent elegant examples, see: (a) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (b) X.-S. Zhou, D.-M. Yan and J.-R. Chen, Chem, 2020, 6, 823–825 CrossRef CAS; (c) R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS PubMed; (d) T. Constantin, F. Juliá, N. S. Sheikh and D. Leonori, Chem. Sci., 2020, 11, 12822–12828 RSC; (e) B. Górski, A. L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef; (f) L. Caiger, C. Sinton, T. Constantin, J. J. Douglas, N. S. Sheikh, F. Juliá and D. Leonori, Chem. Sci., 2021, 12, 10448–10454 RSC; (g) H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed.
- (a) L. H. Ulich and R. Adams, J. Am. Chem. Soc., 1921, 43, 660–667 CrossRef CAS; (b) Z.-P. Yang and G. C. Fu, J. Am. Chem. Soc., 2020, 142, 5870–5875 CrossRef CAS PubMed; (c) H.-M. Huang, P. Bellotti, J. E. Erchinger, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2022, 144, 1899–1909 CrossRef CAS PubMed , Nagib and co-workers described two elegant examples, see:; (d) L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225–229 CrossRef CAS PubMed; (e) S. M. Rafferty, J. E. Rutherford, L. Zhang, L. Wang and D. A. Nagib, J. Am. Chem. Soc., 2021, 143, 5622–5628 CrossRef CAS PubMed; (f) H.-M. Huang, P. Bellotti, S. Kim, X. Zhang and F. Glorius, Nat. Synth., 2022 DOI:10.1038/s44160-022-00085-6.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- G. H. Lovett, S. Chen, X.-S. Xue, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 20031–20036 CrossRef CAS PubMed.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02732a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |