
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the electrochemical reactions of nitrogen-containing organic compounds
Babak Kaboudin
 *,
Milad Behroozi
and
Sepideh Sadighi
*,
Milad Behroozi
and
Sepideh Sadighi
Department of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran. E-mail: Kaboudin@iasbs.ac.ir; kaboudin@gmail.com
First published on 26th October 2022
Abstract
The electrochemical reaction of amines, nitriles, amides, nitroaromatics, and imines has been proven to be a valuable method for the synthesis of various nitrogen-containing organic compounds. Synthetic uses of electrochemical methods for organic transformations of amines, nitriles, imines, and amides to heterocylic compounds and coupling products are discussed. This review aims to demonstrate the ongoing application of electrosynthesis in the preparation of various classes of organic compounds. Furthermore, to address the recent collective articles, this review also describes and summarizes manuscripts on the electrochemical reactions of amines, nitriles, amides, and imines from 2015 until today.
 Babak Kaboudin | Babak Kaboudin was born in 1970 in Iran. He studied chemistry at Isfahan University (Iran), where he obtained his bachelor's degree in chemistry (1992). In the same year, he went to Shiraz University (Iran) and earned his master's degree in 1994. Then, he joined Professor Hashem Sharghi's group at the Shiraz Institute for Organic Synthesis, where he completed his PhD thesis in September 1998. Subsequently, he started his independent research at IASBS (Iran), where he was promoted to full professor in 2007. His research interests include organophosphorus compounds, electrosynthesis, heterocycle synthesis, and the catalytic application of nanomaterials. |
 Milad Behroozi | Milad Behroozi was born in Maragheh, Iran. He received his bachelor's degree in 2019 from the University of Maragheh, and in 2020, he joined Prof. Kaboudin's research group. Currently, he is an MSc student of organic chemistry at the Institute for Advanced Studies in Basic Sciences (IASBS). His research focuses on organic electrosynthesis. |
 Sepideh Sadighi | Sepideh Sadighi obtained her bachelor's degree at Maragheh University in 2020. She joined Prof. Kaboudin's research group at the Institute for Advanced Studies in Basic Sciences (IASBS). Currently, she is an MSc student of organic chemistry. Her current research interest is organic electrochemical synthesis. |
1. Introduction
During the last two decades, there has been a great deal of research to develop greener synthetic methods and chemical processes.1 Designing chemical reaction methods that eliminate the use of catalysts (metal or organic catalysts) is an important and interesting approach that is applicable to all chemistry aspects. Among the novel methods for the synthesis of various organic compounds, electrochemical reactions have several advantages due to reduced environmental pollution and prevention of side reactions, which sometimes lead to failed reactions, and they also reduce the risks to human health.2–10 In electrochemical reactions, the main role of the surface of an electrode is in electron transfer, which leads to common reactive intermediates (carbocations, carbanions, radicals, and radical ions) via diffusion of the substrates from the reaction mixture to the electrode.11 Due to the higher reactivity of the intermediates and their higher concentration on the surface of the electrode, the electrosynthesis of organic compounds is highly selective as compared to the usual chemical reaction where intermediates are uniformly spread over the reaction medium.12The use of electrochemistry continues to this day and produces millions of tons of valuable chemicals. Furthermore, the electrochemical reactions are ‘green’ processes due to the use of electric current in place of stoichiometric oxidants or reductants. However, despite the aforementioned advantages, this technology is not used widely by organic chemists due to the complex reaction setup (potentiostat, divided/undivided cell, electrode composition, and electrolyte experiment type) and the misconception that product separation is difficult because only aqueous solvents may be employed. Additionally, there is no standard instrument for the electrosynthesis of organic compounds, and in many of the recent reports, homebuilt equipment was used. Thus, reports on the synthesis of organic compounds using electricity are few. The aim of this review article is to address the aforementioned difficulties by presenting the reported research works on the use of electricity in organic synthesis.
Organic transformations using electricity can be classified based on the nature of the electron transfer process. Although the catalytic processes at the surface of electrodes can provide useful properties in terms of selectivity and reactivity, a direct transformation at inert electrodes is very applicable, cost-effective, and environmentally benign. It is challenging to optimize reaction parameters, and the appropriate cell design is required for the electrosynthesis of organic compounds. Electrosynthesis is usually carried out via galvanostatic potentiostatic reactions. While the system setup is simple in galvanostatic reactions, a higher selectivity is achieved in potentiostatic electrolysis processes.
Nitrogen is the most important element in nature, and nitrogen-containing organic compounds are of considerable synthetic interest due to their unique bioactivities. They are also the main building blocks of living organisms with important roles in nature. Nitrogen atoms can form part of simple functional groups such as amines, imines, nitriles, amides, and carbamates or complex heterocyclic systems due to varying degrees of substitution and the oxidation of nitrogen. Furthermore, from a medicinal chemistry point of view, the nitrogen atom is a very common element in a large class of active pharmaceutical components existing in heterocyclic or acyclic molecules. Electrochemical reactions of functional groups containing amines, imines, and nitriles are highly powerful strategies for the synthesis of valuable organic compounds. This review aims to demonstrate the ongoing application of electrosynthesis in the preparation of various classes of nitrogen-containing organic compounds. Furthermore, to address the recent collective articles, this review describes and summarizes manuscripts on the electrochemical reactions of amines, nitriles, imines, and amides from 2015 until today.
2. Electrochemical reactions of amines
In 1834, Faraday reported the application of the electrical current in organic synthesis.13 The key species in electrochemical reactions are radical and cation intermediates. The efficiency and selectivity of electrosynthesis of organic compounds is controlled by the reaction conditions, including the current density, the temperature, the concentration, the pH value, the solvent, the electrolyte, and the electrodes.The electrochemical reactions of amines (primary, secondary, and tertiary) have been widely investigated. Various important organic materials are prepared by the electrochemical reactions of amines. This part of the review describes the electrochemical studies of primary, secondary, and tertiary amines in organic transformation since 2015. The presented reactions are selected examples involving typical and interesting substrates, with particular attention to representative reaction mechanisms.
2.1 Primary amines
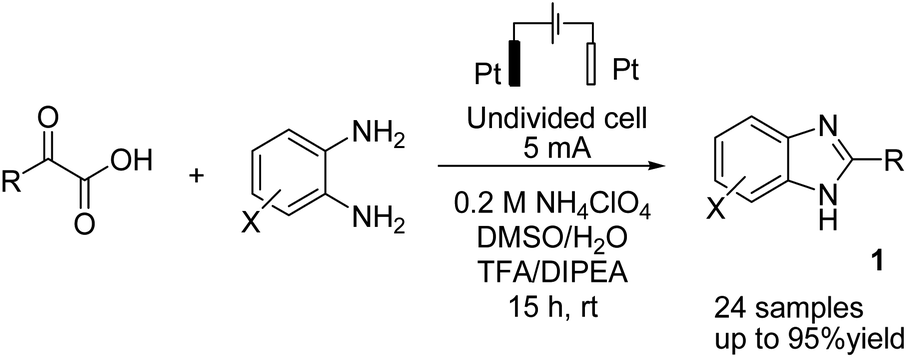
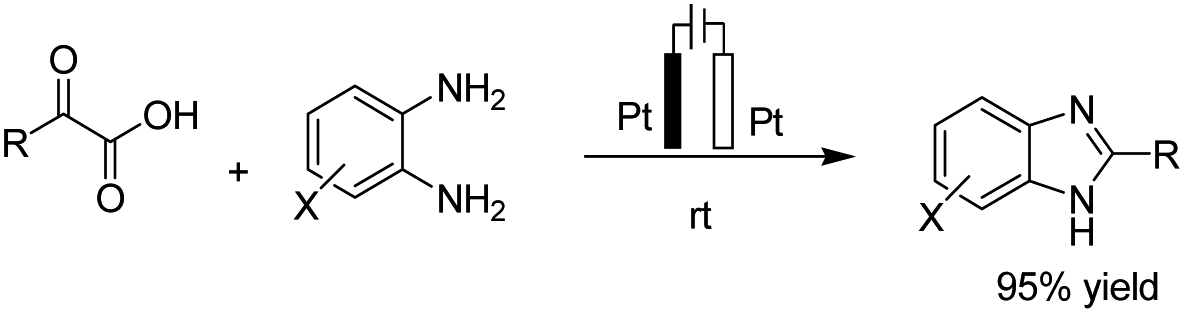
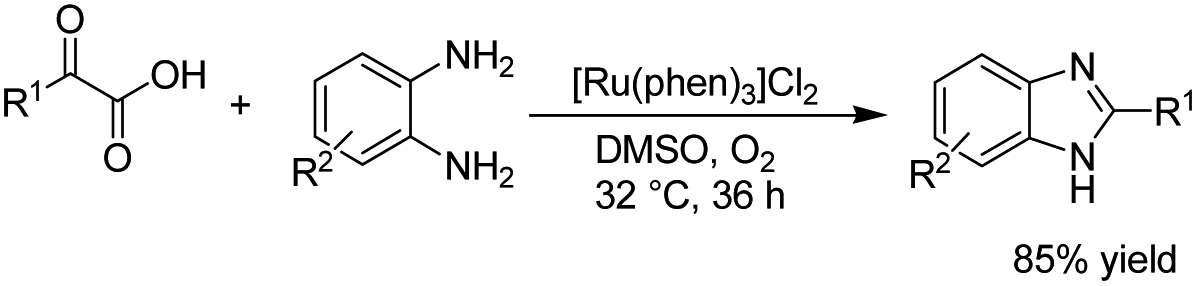
There is relatively less interest in the preparation of benzimidazoles from α-keto acids. Only a photocatalyzed de-carboxylation of α-keto acids with amines to form amides and benzazoles has been reported by Lei et al. under visible light irradiation.14a However, in 2016, Huang et al. reported the electrochemical synthesis of benzimidazoles from the decarboxylative C–N coupling of α-keto acids with ortho-phenylenediamines. The reaction proceeded via anodic oxidation similar to a Kolbe-type reaction.14b The reaction was carried out in an undivided cell, and various conditions were examined to increase the reaction efficiency. The most optimal conditions were obtained with platinum electrodes as the anode and cathode in dimethyl sulfoxide (DMSO)/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) at constant current and at room temperature (Scheme 1).
:3, v/v) at constant current and at room temperature (Scheme 1).
 | ||
| Scheme 1 Electrochemical synthesis of benzimidazoles. | ||
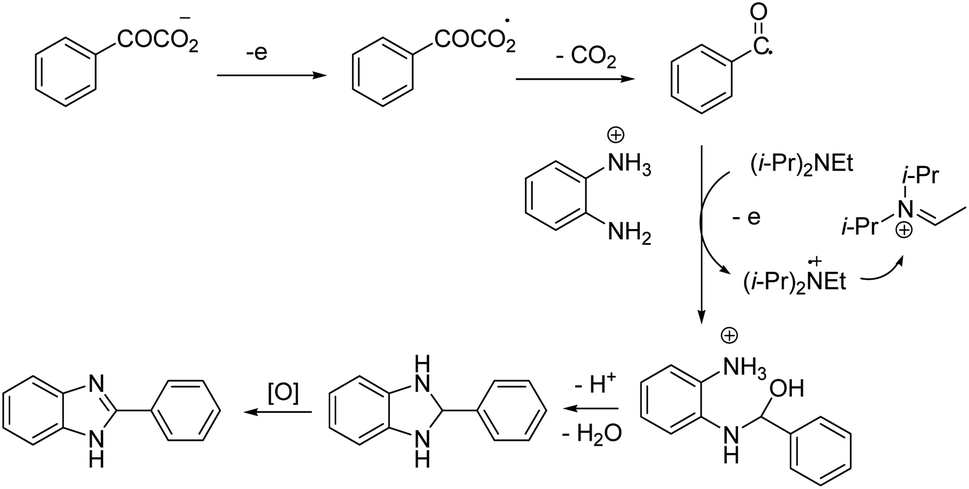
The reaction proceeded in the presence of a mixture of trifluoroacetic acid (TFA, 1 equiv.) and N,N-diisopropylethylamine (DIPEA, 2 equiv.). There is a critical and important role played by DIPEA in this reaction. According to the proposed mechanism (Scheme 2), an acyl radical forms in the anode via a similar Kolbe-type reaction, from α-keto acid anion that then undergoes a coupling with protonated diamine. In this reaction, DIPEA follows a hydrogen atom transfer rule to afford the coupling product. In the final step, dehydrogenation proceeds in the presence of O2 (Scheme 2).
 | ||
| Scheme 2 Proposed mechanism of electrochemical synthesis of benzimidazoles. | ||
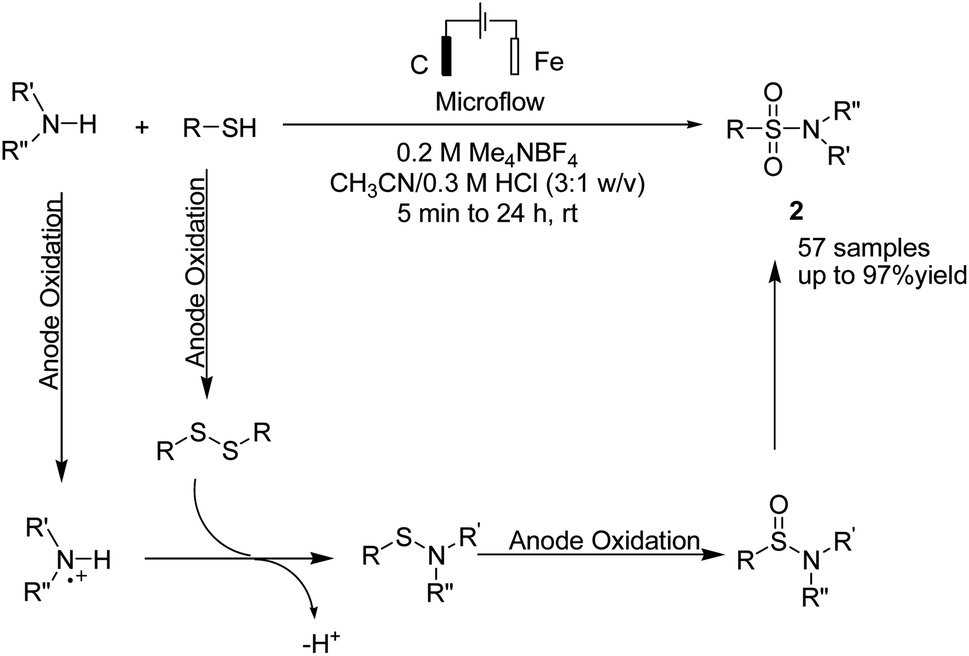
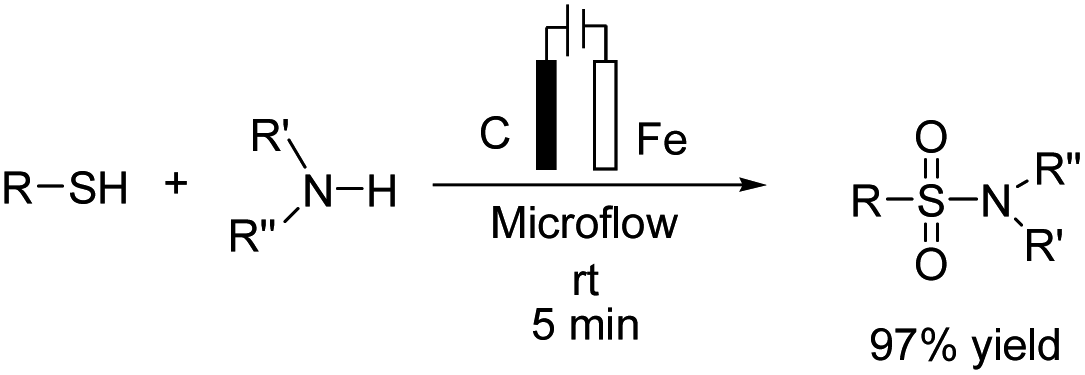
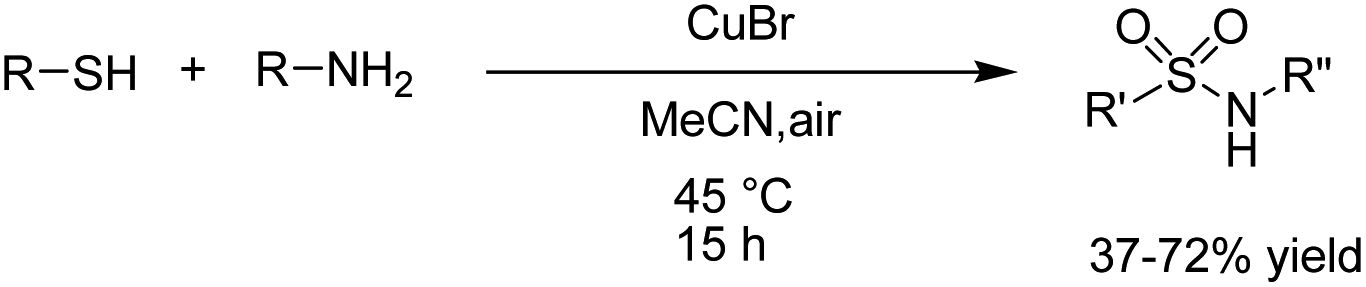
A one-pot procedure for the synthesis of sulfonamides via direct use of commodity chemicals such as thiols and amines is important for transformation. However, a suitable transformation would require two steps, including an S–N bond formation and a subsequent oxidation of the sulfur atom. The development of novel techniques for this transformation would be particularly useful, given the broad availability and the low cost of these starting materials. In 2019, the electrochemical synthesis of sulfonamides from the simple reaction of amines with thiols was reported by Noël et al.15 The reaction proceeded through the oxidative coupling between two readily available and inexpensive chemicals with a broad substrate scope and functional group compatibility. The synthesis of sulfonamides was carried out using this method in the absence of any oxidant or catalysts. In this reaction, hydrogen gas is formed only as a byproduct in the cathode (Scheme 3). Mechanistic studies showed that the thiol substrate is completely converted to the corresponding disulfide via anodic oxidation followed by coupling with amine to yield the corresponding sulfonamide via two oxidation steps in the anode.
 | ||
| Scheme 3 Electrochemical synthesis of sulfonamides. | ||
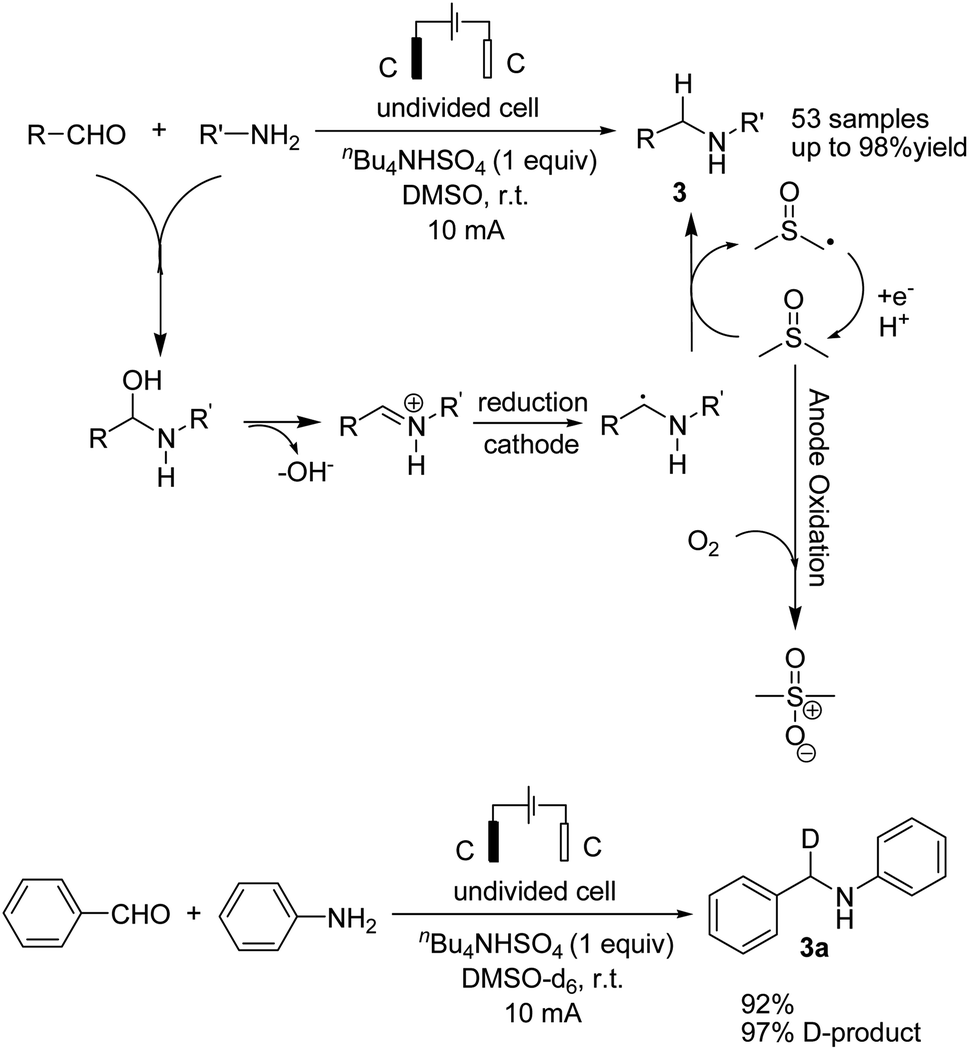
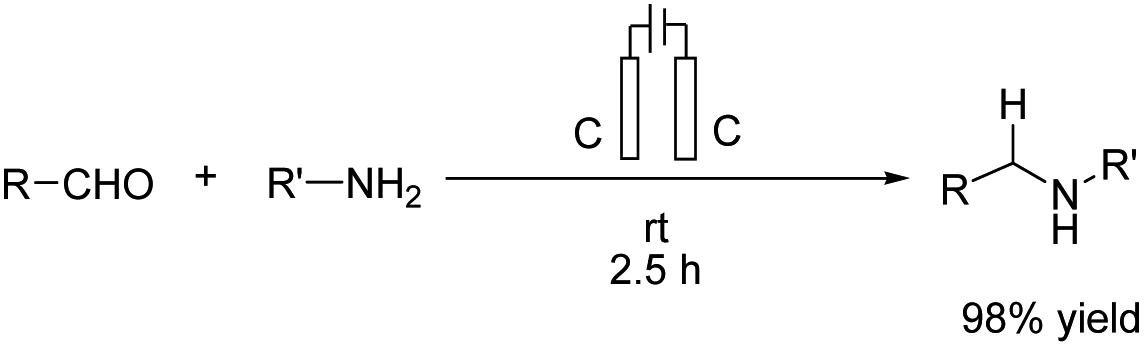
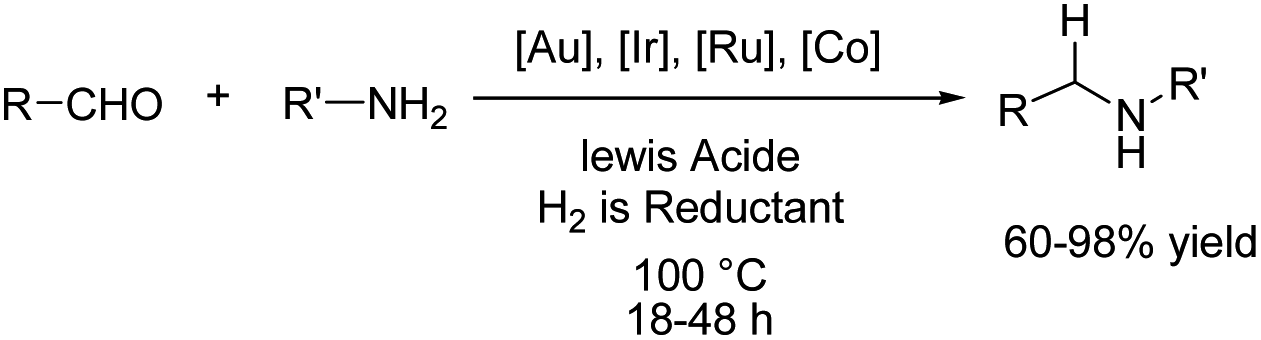
Reductive amination is considered to be one of the most versatile and efficient methods for the synthesis of amines. For efficient and highly selective reductive amination, there have been great efforts to find a suitable method. High-pressure molecular hydrogen in the presence of a transition metal has been widely utilized as a reductant instead of using stoichiometric amounts of NaBH4 and NaBH3CN as strong reductants. In 2020, Huang et al. reported the synthesis of secondary amines via an electrochemical reduction reaction of aldehyde with amine.16 The experimental results showed that the reaction proceeds very well in an undivided cell at a constant current of 10 mA using nBu4NHSO4 as the electrolyte in DMSO. Mechanistic studies showed that DMSO has a reductant role. In a control experiment, a deuterium-labelled secondary amine was obtained in the presence of DMSO-d6 through the formation of a C–D bond (Scheme 4).
 | ||
| Scheme 4 Electrochemical synthesis of secondary amines. | ||
An electrochemical transition metal and peroxide-free oxidative multicomponent cascade dehydrogenative [2 + 2 + 1] annulations of ketones and amines for the synthesis of imidazoles were reported by He et al.17 The reaction proceeded via the formation of α-iodo ketone from the reaction of aryl methyl ketone with iodine, followed by nucleophilic attack of the amine to the C–I bond to form α-amino ketone. Finally, condensation of the α-amino ketone with benzylamine, cyclization, and aromatization via an oxidative dehydrogenation reaction gave product 4 (Scheme 5).
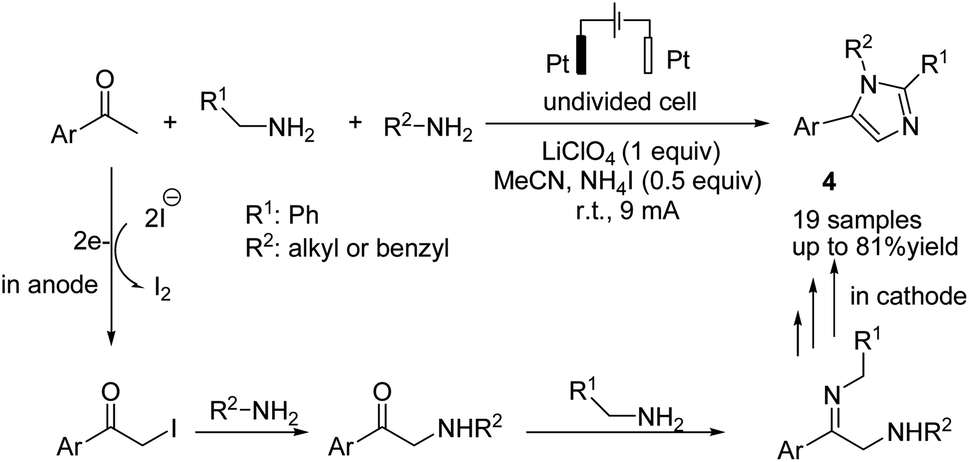 | ||
| Scheme 5 Electrochemical synthesis of imidazoles. | ||
Lei and co-workers reported the gram-scale synthesis of poly-substituted pyrroles via an electrochemical oxidative annulation from amines with carbonyl compounds in an undivided cell.18 By this method, various β-substituted and tetra-substituted pyrroles were obtained via the reaction of amines with aryl acetaldehydes and alkyl ketones, respectively (Scheme 6). The reaction proceeded by the formation and homo coupling of radicals at the anode via single-electron-transfer (SET) oxidation of imine, followed by intramolecular nucleophilic attack and cyclization to form the desired product.
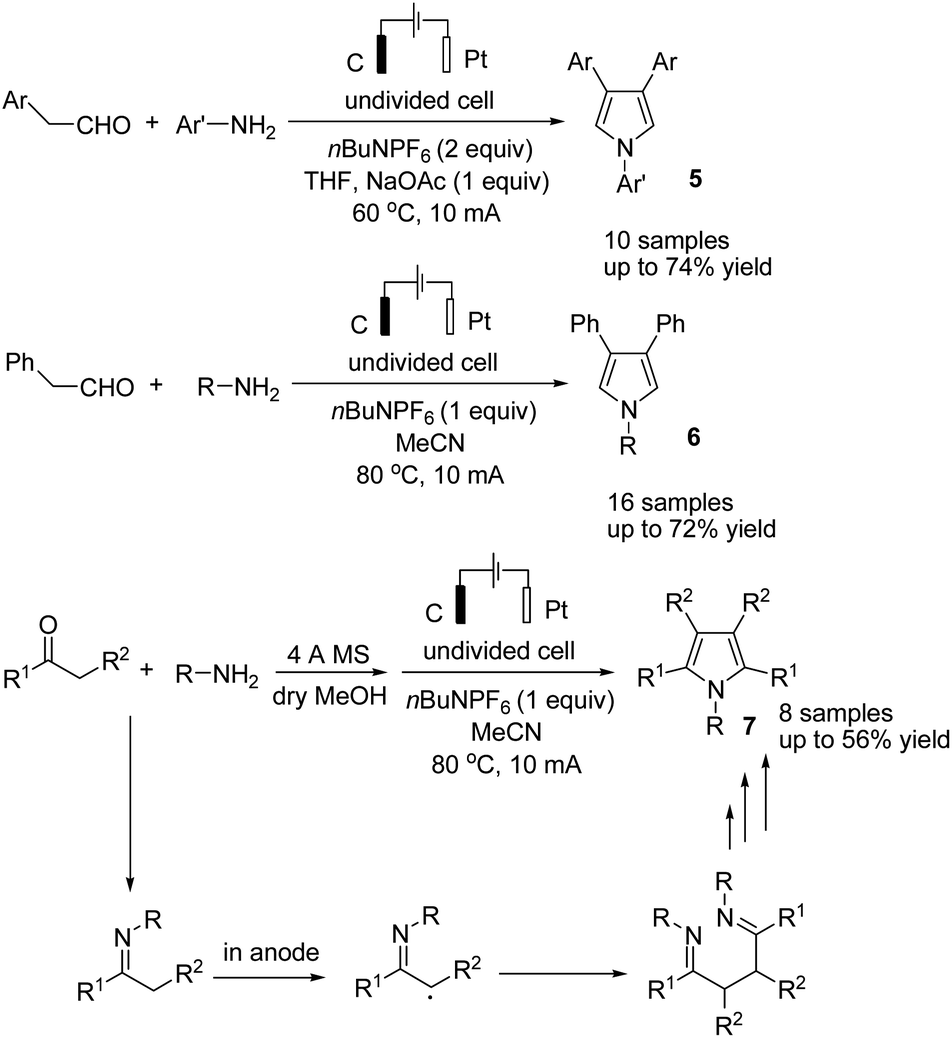 | ||
| Scheme 6 Electrochemical synthesis of polysubstituted pyrroles. | ||
Wang et al. reported the electrochemical synthesis of quinazolines via a C(sp3)–H amination/C–N cleavage by anodic oxidation under aqueous conditions (Scheme 7).19 Studies showed that iminium ion was formed via the loss of two electrons and one proton of tetramethyl ethylene diamine (TMEDA) at the anode, with ammonia generated from the electrolyte at the cathode (Scheme 7).
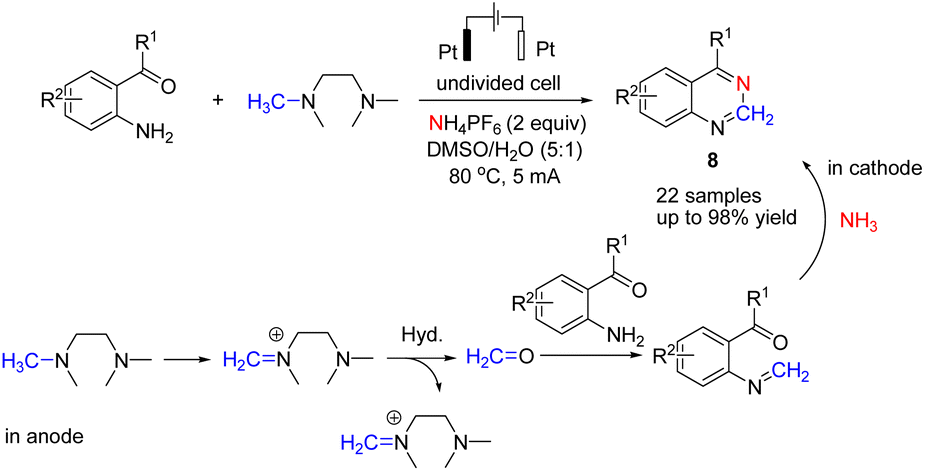 | ||
| Scheme 7 Electrochemical synthesis of quinazolines. | ||
In 2019, Huang et al. reported the electrochemical synthesis of 3-bromoimidazo[1,2-a]pyridines from 2-aminopyridines and α-bromo ketones in a simple undivided cell without any external oxidant through a domino condensation/bromination sequence.20 The reaction proceeded by a simple condensation of 2-aminopyridines with α-bromoketones, followed by bromination, which resulted in anode oxidation of the bromide anion to yield the target molecule 9 (Scheme 8).
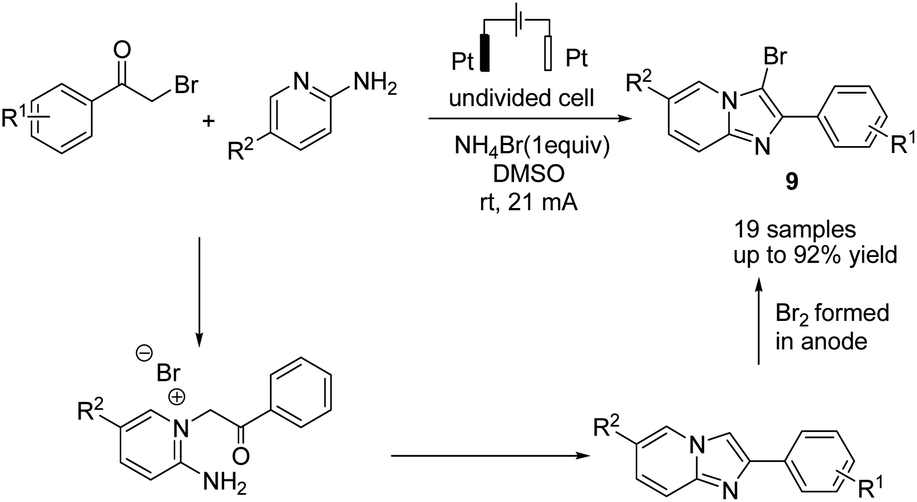 | ||
| Scheme 8 Electrochemical synthesis of imidazopyridines. | ||
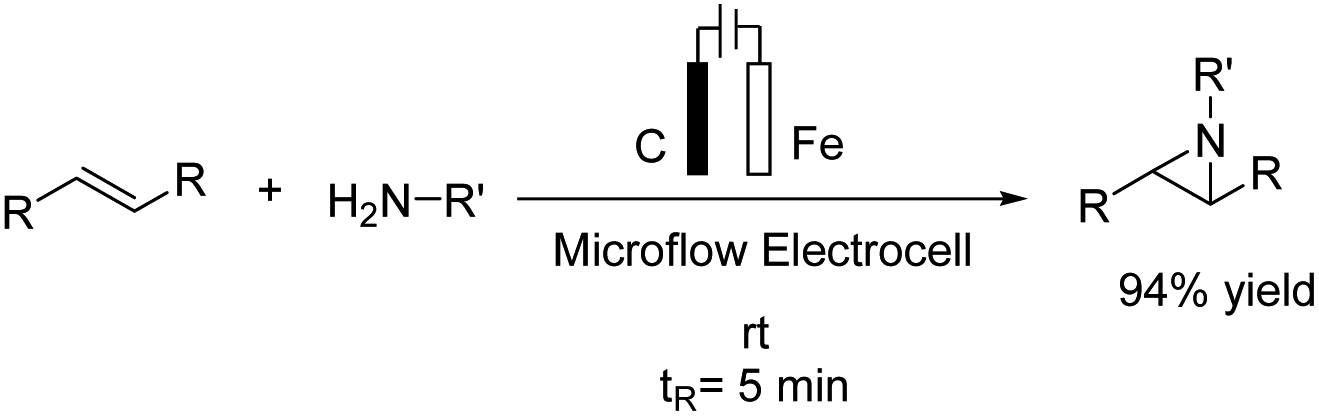
An electrochemical aziridination of internal alkenes with primary amines via an oxidative coupling between alkenes and primary alkyl amines in an electrochemical flow reactor was reported by Noël et al.21 Further investigations and density functional theory (DFT) calculations showed that the alkene was oxidized in the anode and subsequently reacted with the amine to yield the corresponding aziridine (Scheme 9). In another attempt, hydrogen generated at the cathode was used in a second reactor to reduce the aziridine to the corresponding hydroaminated product.
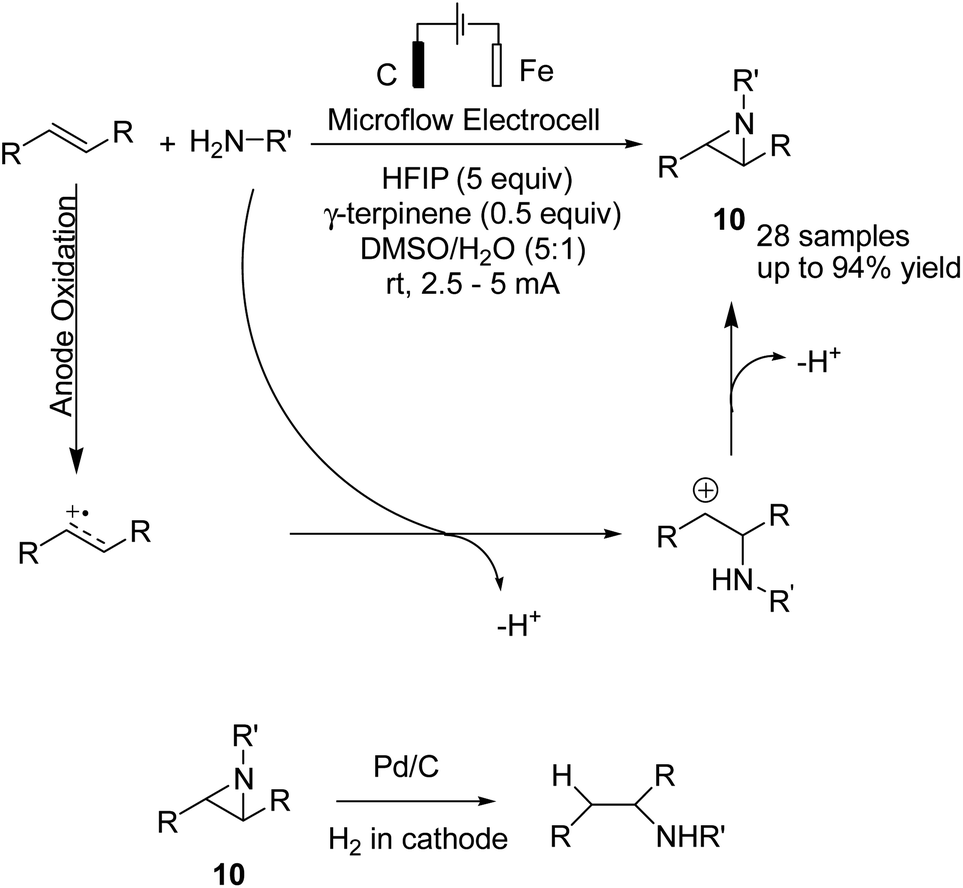 | ||
| Scheme 9 Electrochemical synthesis of azirdines. | ||
2.2 Secondary amines
In 2018, Mei et al. reported the copper-catalyzed electrochemical C–H amination of arenes with secondary amines at room temperature using undivided electrochemical cells.22 The n-butyl ammonium iodide played a crucial role as a redox mediator for this transformation. Mechanistic studies including kinetic profiles, isotope effects, cyclic voltammetry analysis, and radical inhibition experiments showed that the reaction proceeded via a single-electron-transfer (SET) with a high valent Cu(III) species. In this process, the Cu(II) complex was oxidized by iodine radicals (generated at the anode) to form Cu(III) species (Scheme 10).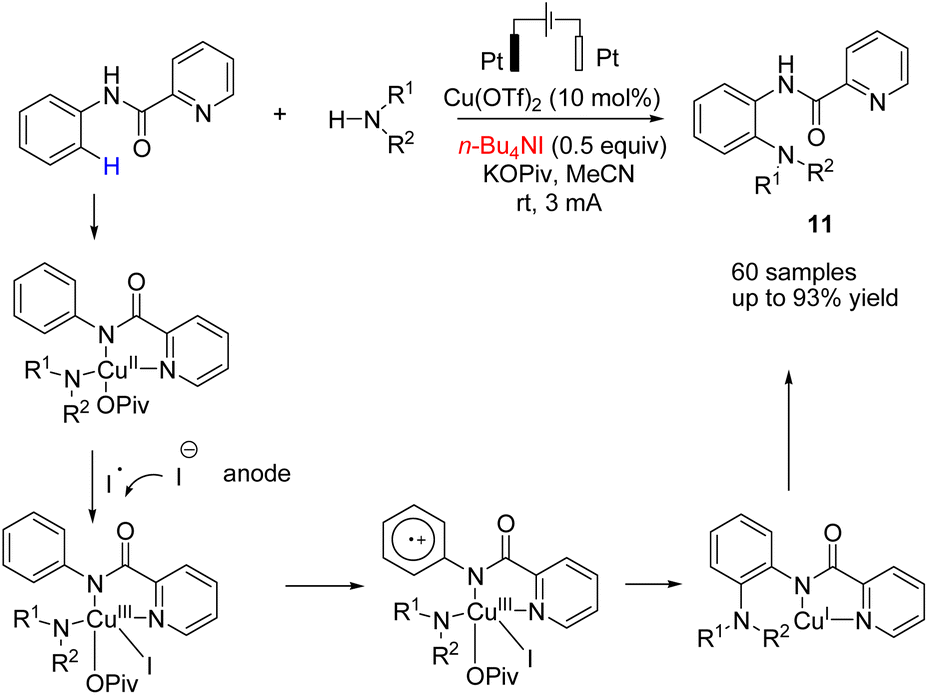 | ||
| Scheme 10 Electrochemical copper-catalyzed amination of arenes with secondary amines. | ||
Metal-free electrosynthesis of phosphinic amides via oxidative cross-coupling of secondary amines with diarylphospine oxides has been reported by Wang et al. in 2017.23 Mechanistic studies showed that the reaction proceeded via iodide ion oxidation into an iodine radical at the anode surface, which reacted with diarylphospine oxide to generate a P–I intermediate (Scheme 11). The amine nucleophile was easily reacted with the P–I intermediate, yielding the final product 12. At the cathode, the ethoxide anion and hydrogen molecule are produced through the reduction of ethanol.
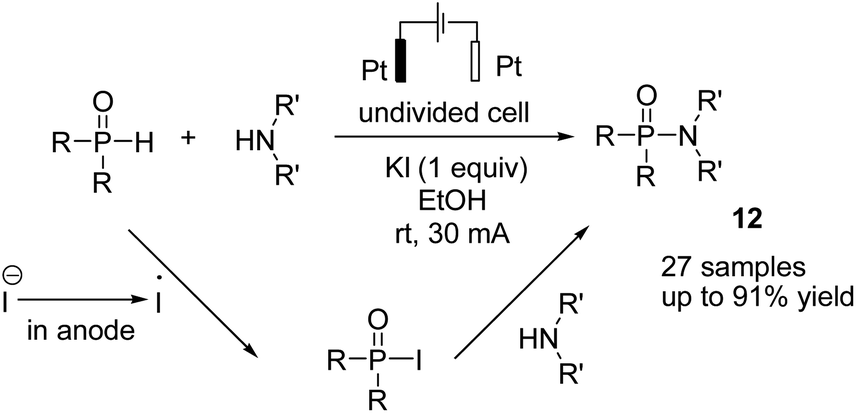 | ||
| Scheme 11 Electrochemical synthesis of phosphinic amides. | ||
In 2018, Huang et al. reported an electrochemical N-formylation of amines with glyoxylic acid via a decarboxylative process in the presence of copper acetate as an active oxidant.24 The mechanistic studies showed that the high valent copper was generated by anodic oxidation (see the detailed mechanism in Scheme 12). Glyoxylic acid was first converted into carboxylate ion by cesium carbonate, followed by condensation with the aniline to form an imine intermediate (NiCl2 was proposed to act as a Lewis acid to promote the imine condensation). The intermediate was oxidized by cupric acetate, followed by decarboxylation to generate the N-formylation product.
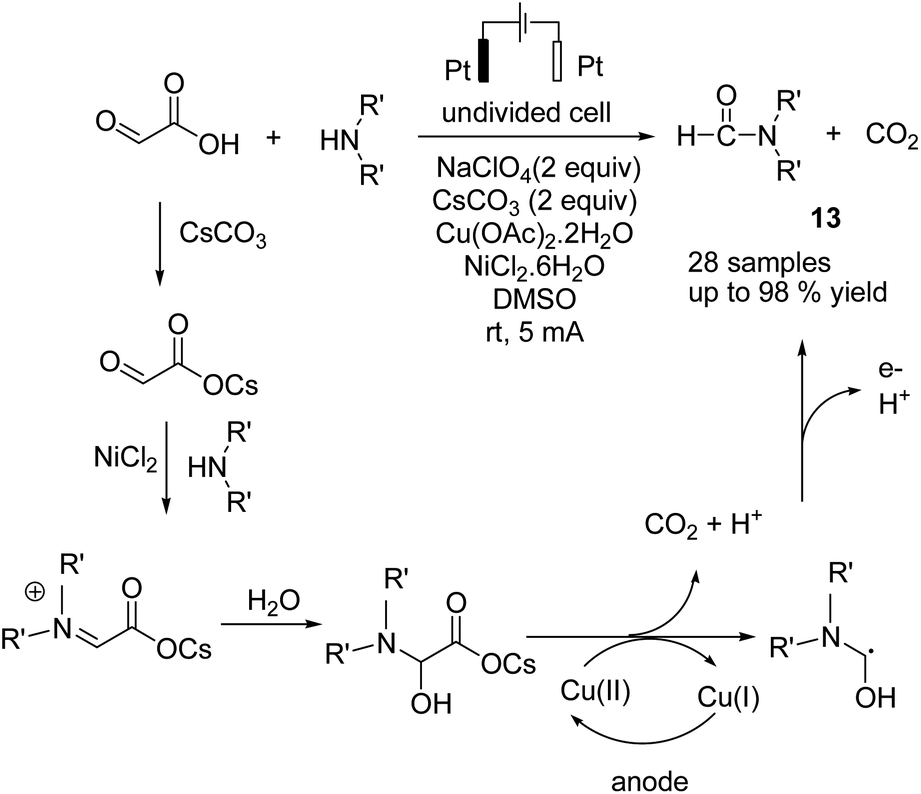 | ||
| Scheme 12 Electrochemical N-formylation of amines. | ||
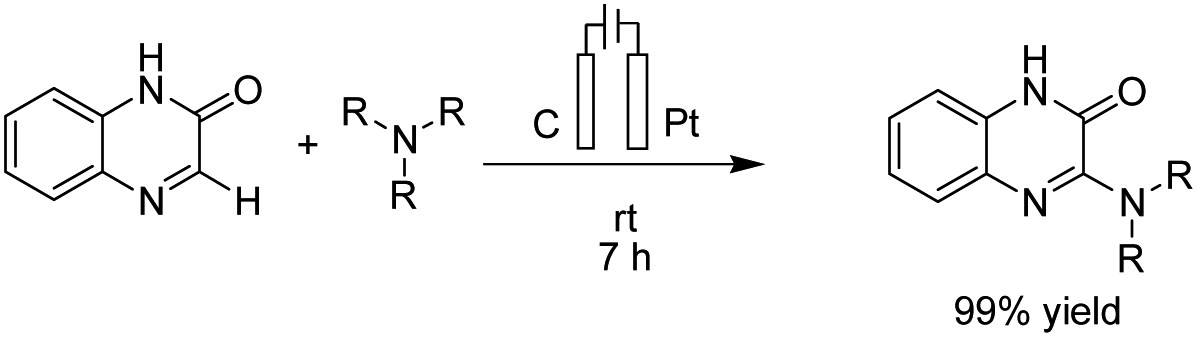
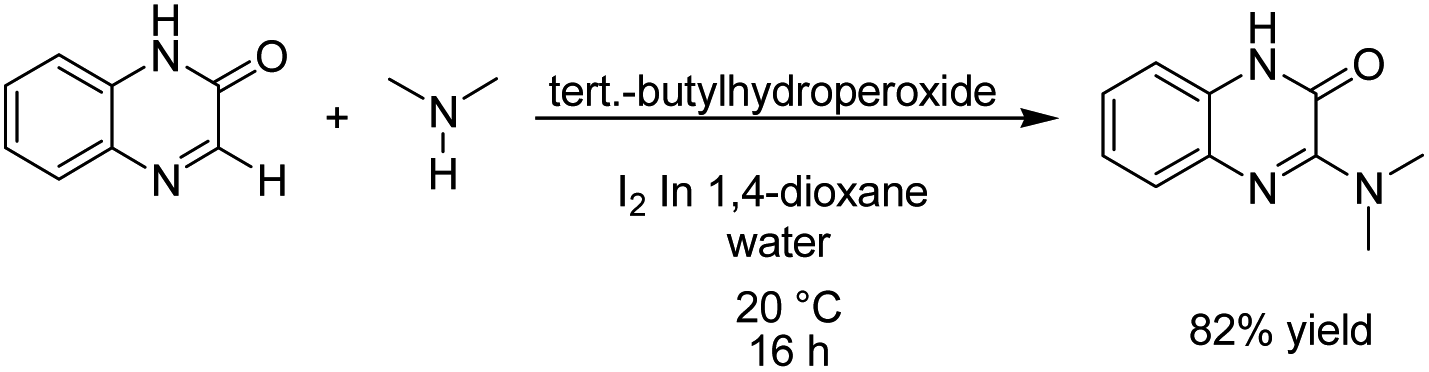
Zeng et al. reported an electrochemical dehydrogenative transition metal-free cross-coupling of quinoxalin-2(1H)-ones with secondary amines for the synthesis of 3-aminoquinoxalinones.25 It was assumed that the reaction proceeded through nucleophilic addition of the substrate amine to protonated quinoxaline-2(1H)-one (Scheme 13), followed by further anodic oxidation and deprotonation, yielding the desired products 14. Molecular hydrogen was produced at the cathode surface.
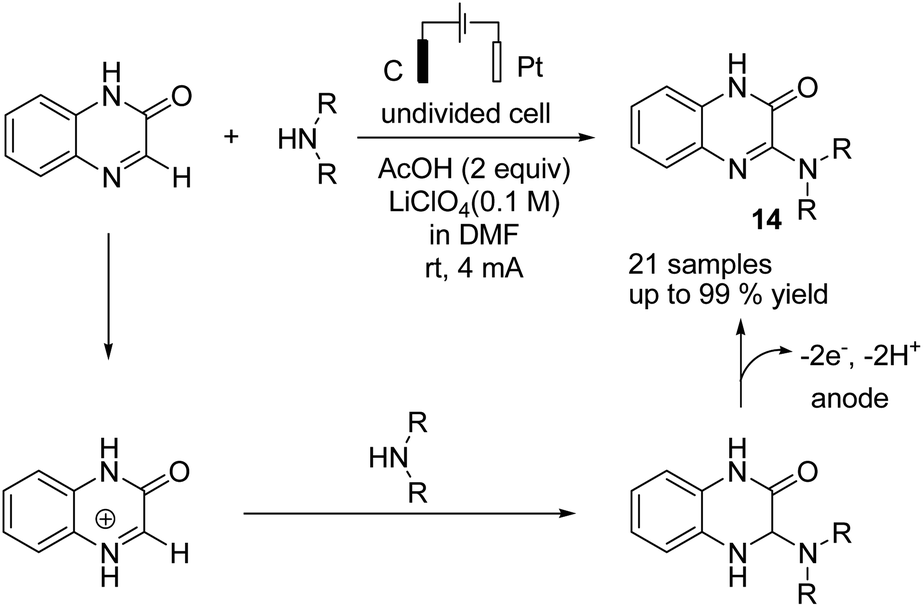 | ||
| Scheme 13 Electrochemical synthesis of 3-aminoquinoxalinones. | ||
In 2019, Ding et al. reported synthesis of amino phosphonates by an electrochemical C–H phosphonylation of unprotected secondary amines through metal-free and exogenous oxidant-free conditions.26 Mechanistic investigations revealed that an amine compound was oxidized at the anode electrode, giving an imine intermediate, which was attacked by phosphite catalyzed by sodium acetate, yielding the final product, aminophosphonate 15. Hydrogen evolution occurred at the cathode, and an acetate anion was regenerated (Scheme 14).
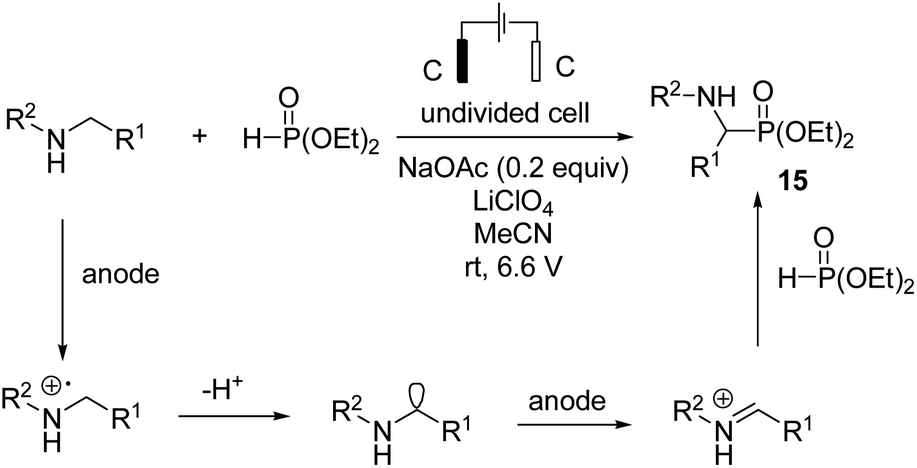 | ||
| Scheme 14 Electrochemical synthesis of 1-aminophosphonates. | ||
2.3 Tertiary amines
Electrochemical coupling of arylsulfonyl hydrazides with tertiary amines for the synthesis of amidovinyl sulfones under mild electrochemical conditions has been reported by Kim and Lee.27 The reaction was carried out using an acid, in a solution of n-Bu4NBF4 in DMSO in undivided cells with graphite–platinum electrodes under a constant current. Based on the experiments, it was assumed that the tertiary amine was activated and converted to a radical cation, and then, a C–H bond at the alpha position of the tertiary amine was oxidized to yield an iminium ion intermediate. However, arylsulfonyl hydrazide was oxidized and transformed into an arylsulfonyl radical at the anode. Finally, the arylsulfonyl radical reacted with the enamine (from the reaction of imine intermediate and acetate anion) to give the amidovinyl sulfone. Air or an oxygen atmosphere was required as an oxidant for this process (Scheme 15).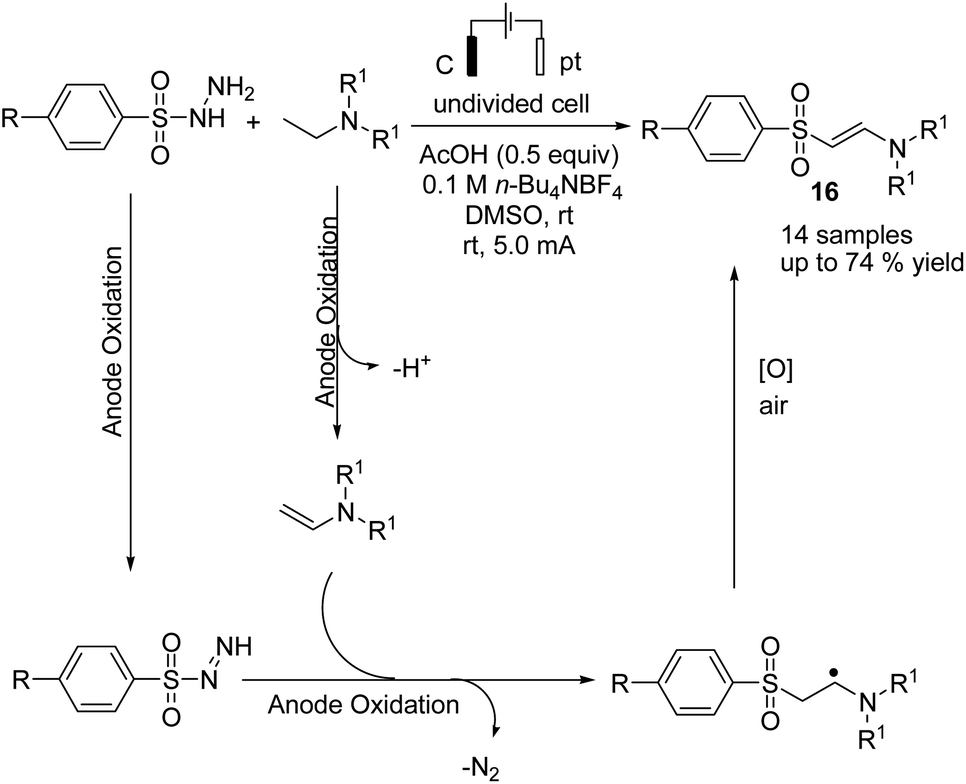 | ||
| Scheme 15 Electrochemical synthesis of amidovinyl sulfone. | ||
Luo et al. reported the catalytic asymmetric electrochemical C–H functionalization of simple ketones with tetrahydroisoquinolines in the presence of chiral primary amine catalysts for the synthesis of C1-alkylated tetrahydroisoquinolines in high yields and with excellent enantioselectivities.28 The reaction proceeded via an electrochemical oxidation of tetrahydroisoquinolines to the corresponding iminium ion intermediate form, followed by reaction with the enamine intermediate to yield C1-alkylated tetrahydroisoquinolines (Scheme 16).
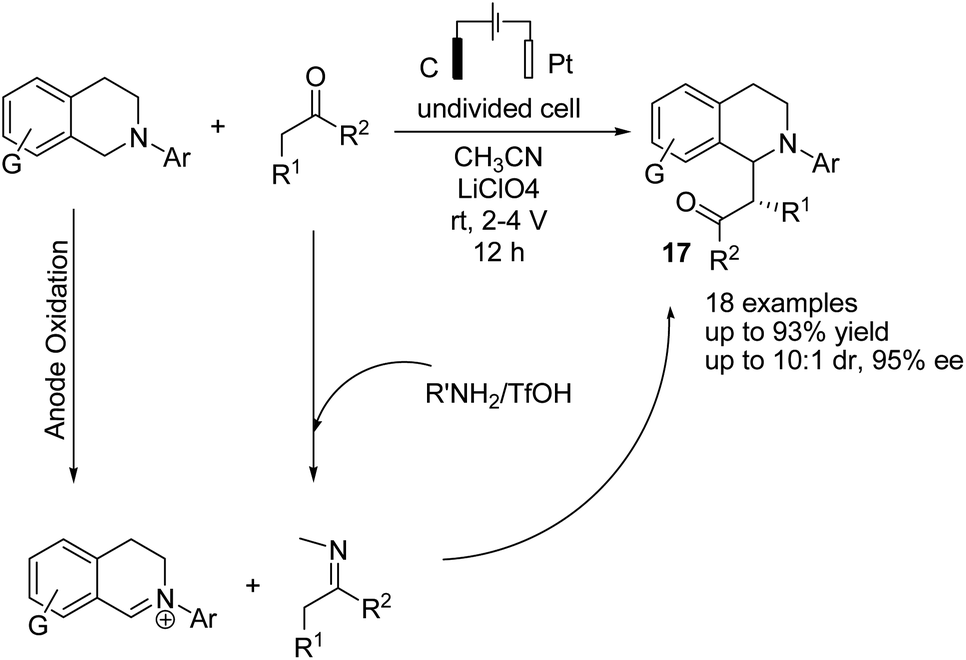 | ||
| Scheme 16 Electrochemical C–H functionalization of simple ketones with tetrahydroisoquinolines. | ||
A novel electrochemical strategy for the asymmetric oxidative cross-coupling of tetrahydroisoquinolines with alkynes was reported by Mei et al. in the presence of copper catalysis and 2,2,4,4-tetramethylpiperidine N-oxide (TEMPO).29 TEMPO is used as a co-catalyst to decrease the oxidation potential of the reaction. The reaction proceeded via the electrochemical oxidation of tetrahydroisoquinolines to the corresponding iminium ion intermediate form, followed by reaction with a copper acetylide intermediate (including chiral bisoxazoline ligand) to yield highly C1-alkynylated tetrahydroisoquinolines with up to 97% enantiomeric excess (ee) (Scheme 17).
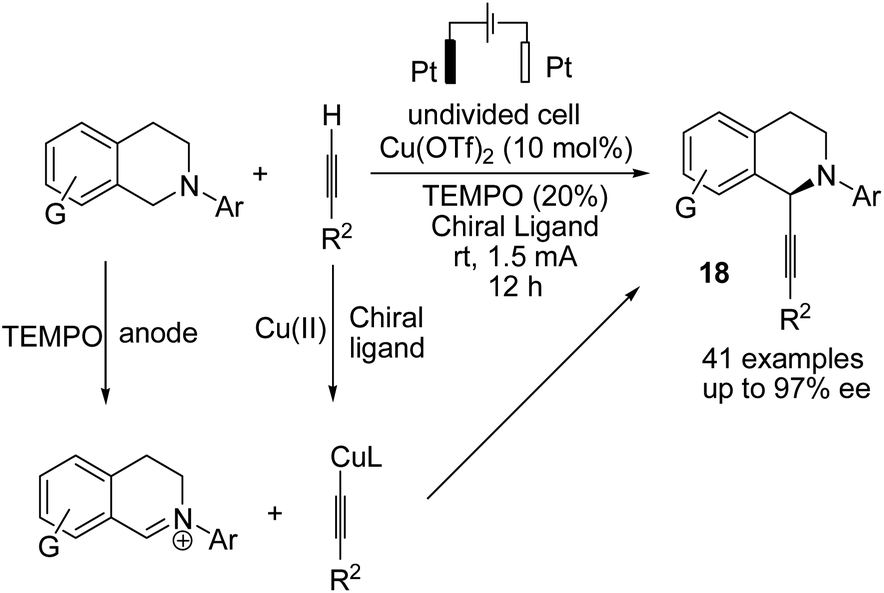 | ||
| Scheme 17 Electrochemical synthesis of amidovinyl sulfone. | ||
In 2019, Li et al. reported the electrochemical synthesis of polycyclic N-heterocycles under oxidant-free conditions via oxidation/[3 + 2] cycloaddition/aromatization cascade.30 The reaction proceeded via the anodic oxidation of NHPI to form phthalimide N-oxyl (PINO) and the cathodic reduction of MeOH to H2 and methoxide. The azomethine ylide was formed with hydrogen abstraction of tetrahydroisoquinoline acetate with the assistance of PINO, followed by reaction with N-methylmaleimide as the dipolarophile to yield product 19 via a [3 + 2] cycloaddition (Scheme 18).
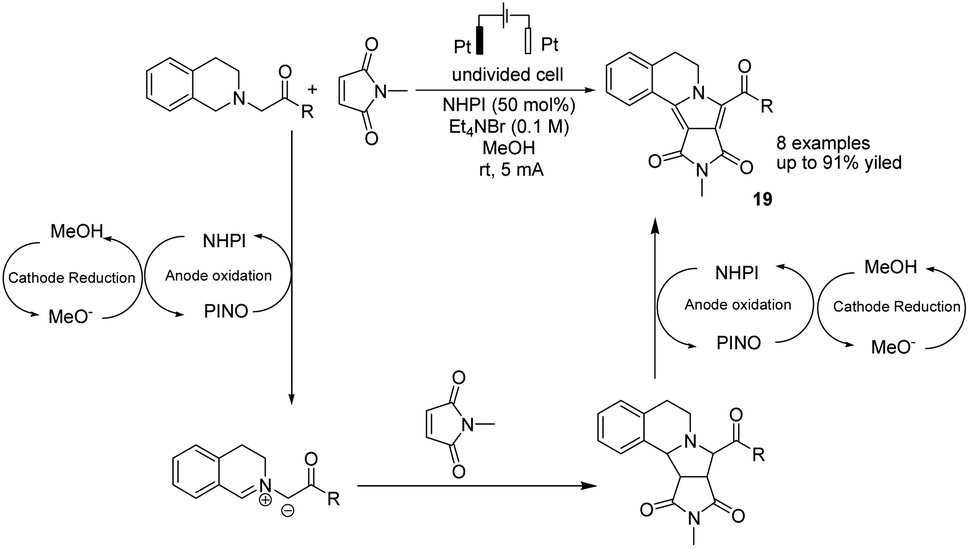 | ||
| Scheme 18 Electrochemical synthesis of polycyclic N-heterocycles. | ||
3. Electrochemical reactions of nitriles
In 2021, Findlater et al. reported the electrochemical arylation of aldehydes, ketones, and alcohols with 1,4-dicyanobenzene or para-substituted electron-withdrawing cyanobenzene.31 The mechanistic investigations revealed that benzylic alcohol undergoes an oxidation process at the anode surface, which results in the corresponding benzaldehyde. The nucleophilic addition between benzaldehyde and anion radical arising from 1,4-dicyanobenzene at the cathode surface is the key step toward arylation product 20 (Scheme 19). In this reaction, the cyanide leaving group was trapped by valeraldehyde.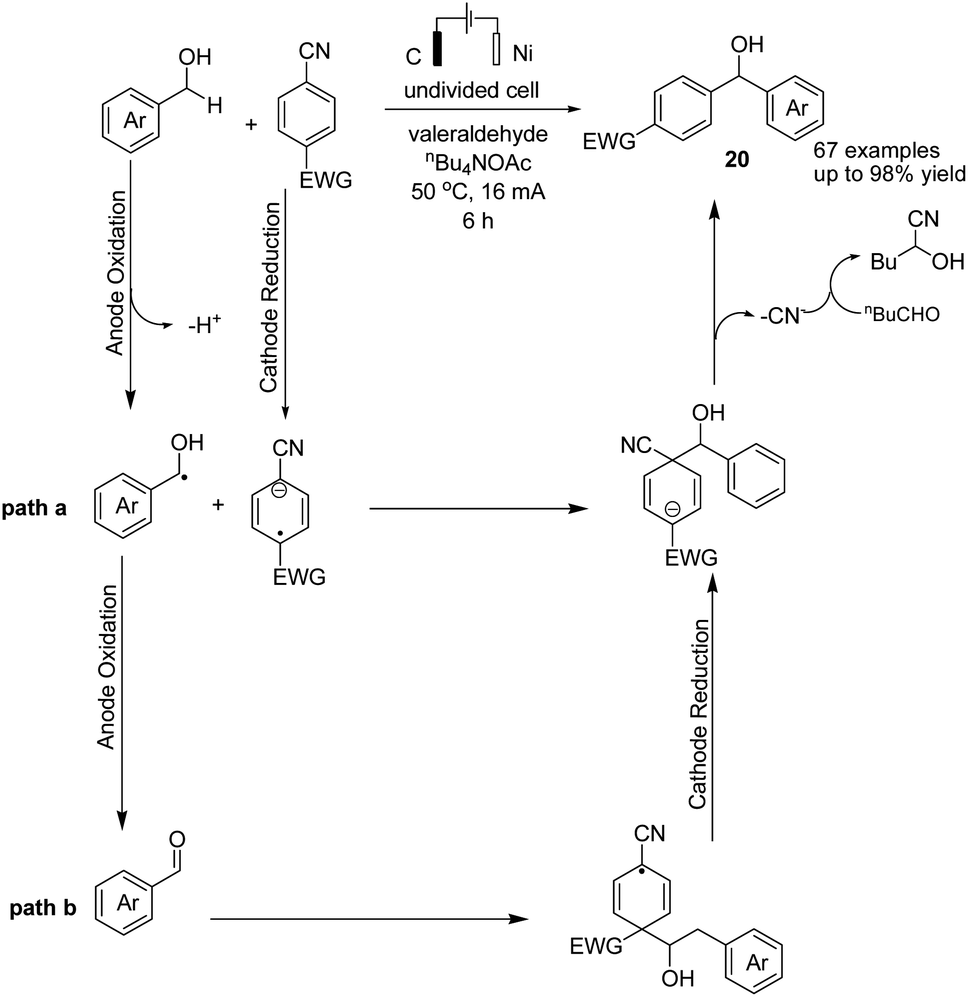 | ||
| Scheme 19 Electrochemical arylation of aldehydes and alcohols. | ||
Singh et al. reported the electrochemical synthesis of 3,5-disubstituted triazoles from nitriles and hydrazides.32 The conversion proceeded via the reaction of an amide radical cation (formed in the anode by an anodic oxidation of an amide) with hydrazide. The intermediate then converted to the final product 21 by cyclization and cathodic reduction, which subsequently underwent a proton shift dehydration to afford the desired product (Scheme 20).
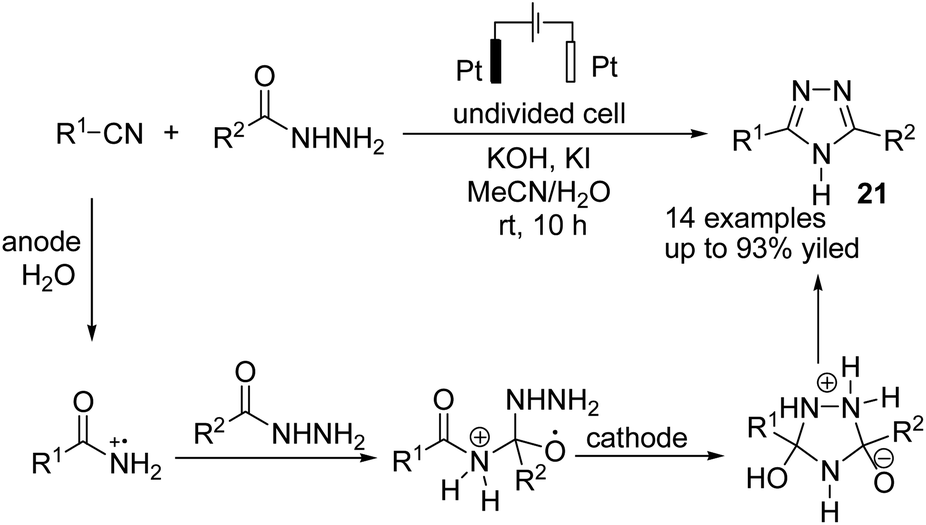 | ||
| Scheme 20 Electrochemical synthesis of 3,5-disubstituted triazoles. | ||
In 2019 Ye et al. reported an electrochemical TEMPO-catalyzed direct arylation of tertiary amines with benzonitrile derivatives via α-amino C(sp3)–H bond formation.33 The reaction proceeded via the anodic conversion of TEMPO to TEMPO+, which reversibly oxidized the tertiary arylamine to TEMPO and amino radical in the presence of 2,6-lutidine. In the next step, a coupling of amino radical with anodic formed anion radical, which underwent subsequent elimination of cyanide anion and aromatization to give the final product 22 (Scheme 21).
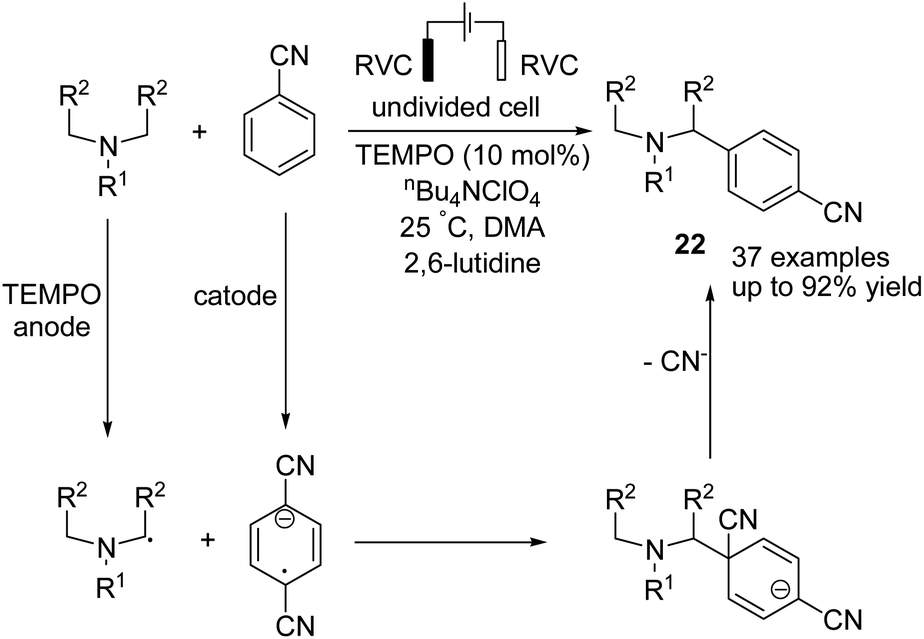 | ||
| Scheme 21 Electrochemical arylation of tertiary amines. | ||
In 2019 Wang, Yuan, and Li et al. reported an electrochemical oxidative C sp3–H/S–H cross-coupling of acetonitrile with thiols for the synthesis of tetrasubstituted olefins.34 Mechanistic investigations revealed that the reaction proceeded via one hydrogen atom abstraction of acetonitrile to yield the corresponding radical form by iodine radical in the anode. Sequential radical addition to another acetonitrile molecule was followed by hydrogen atom transfer from RSH to yield product 23 (Scheme 22).
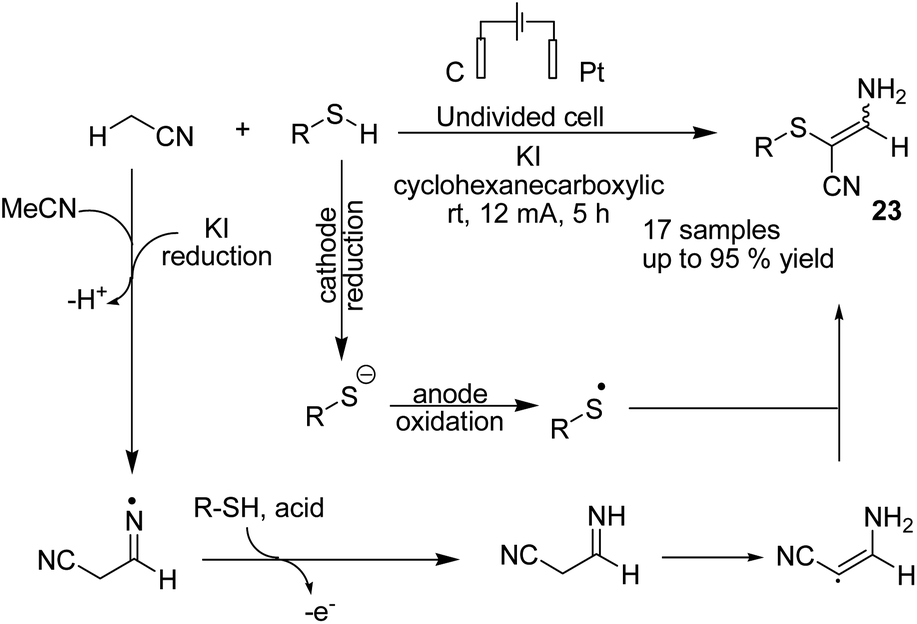 | ||
| Scheme 22 Electrochemical oxidative C sp3–H/S–H cross-coupling reaction. | ||
4. Electrochemical reactions of imines
In 2020, Lehnherr, Rovis, and co-workers reported the synthesis of hindered primary and secondary amine 24 via an electrochemical reaction of bench top-stable iminium salts with cyanoheteroarenes.35 According to the reported method, a wide variety of substituted heterocycles (pyridine, pyrimidine, pyrazine, purine, azaindole) has been utilized in the cross-coupling reaction, including those substituted with a halide, trifluoromethyl, ester, amide, or ether group, a heterocycle, or an unprotected alcohol or alkyne. The mechanistic studies based on DFT data, as well as cyclic voltammetry and NMR spectroscopy, showed that the reaction proceeded via a bi-radical cross-coupling of α-amino radicals and radicals derived from cyanoheteroarenes (Scheme 23).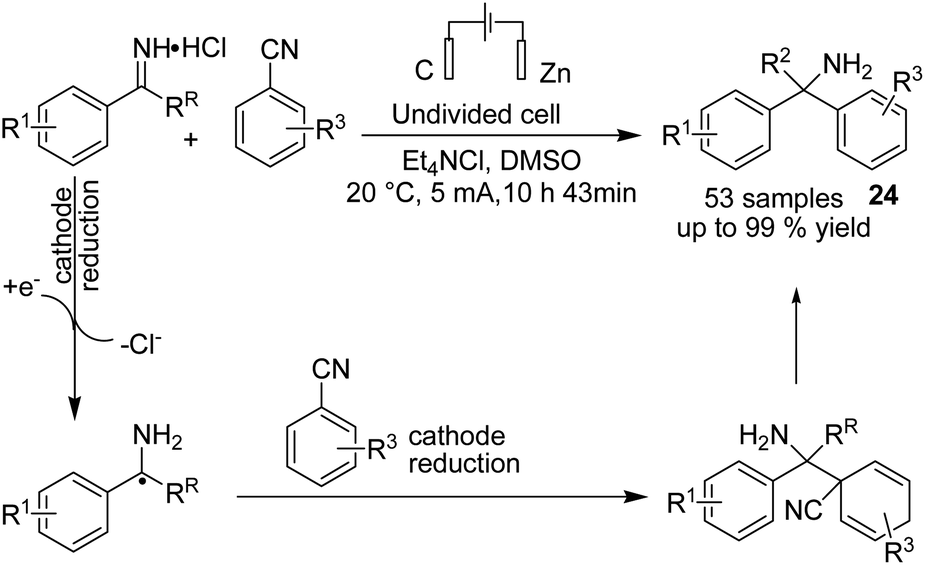 | ||
| Scheme 23 Electrochemical synthesis hindered the primary and secondary amine. | ||
The electrochemical synthesis of 1,2,4-triazole-fused heterocycles 25 via an intramolecular dehydrogenative C–N cross coupling reaction was developed by Zhang et al. in 2018.36 By this method, valuable 1,2,4-triazolo[4,3-a]pyridines and related heterocyclic compounds were efficiently synthesized from commercially available aliphatic or (hetero)aromatic aldehydes and 2-hydrazinopyridine. The reaction proceeded via the condensation of 2-hydrazinopyridine and aldehyde and gave the hydrazone, which subsequently underwent deprotonation by hydroxide generated from the cathodic reduction of water to produce a nitrogen ion intermediate. The final product was obtained by a single-electron transfer (SET) oxidation followed by intramolecular radical addition, anodic oxidation, and deprotonation (Scheme 24).
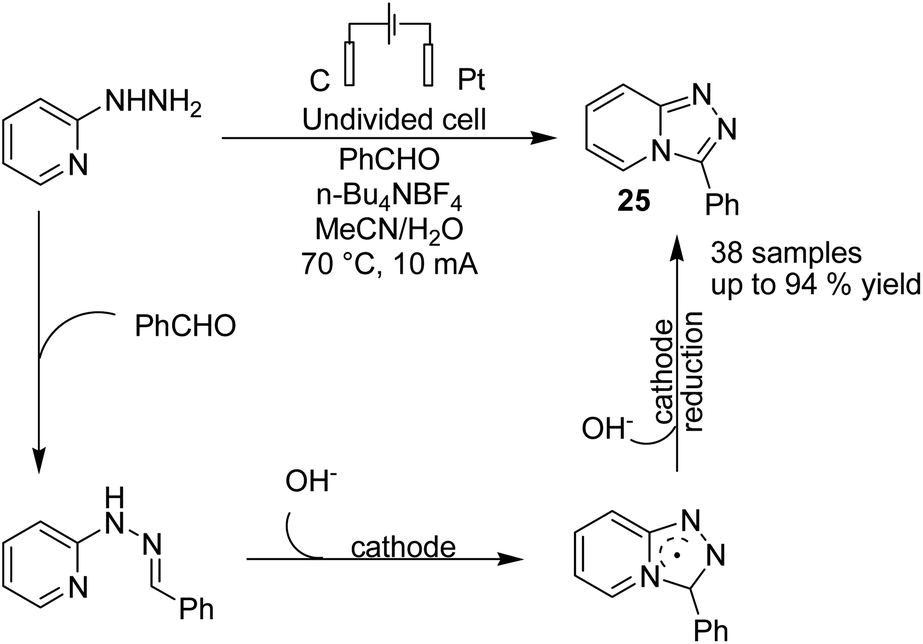 | ||
| Scheme 24 Electrochemical synthesis of 1,2,4-triazole-fused heterocycles. | ||
In 2016, Xu et al. reported the electrochemical formation of amidinyl radical (through the anodic cleavage of N–H bonds) for functionalization of the aromatic C–H bond. The resulting nitrogen radicals underwent cyclizations with arenes, followed by re-aromatization, to yield functionalized tetracyclic benzimidazoles 26 (Scheme 25).37
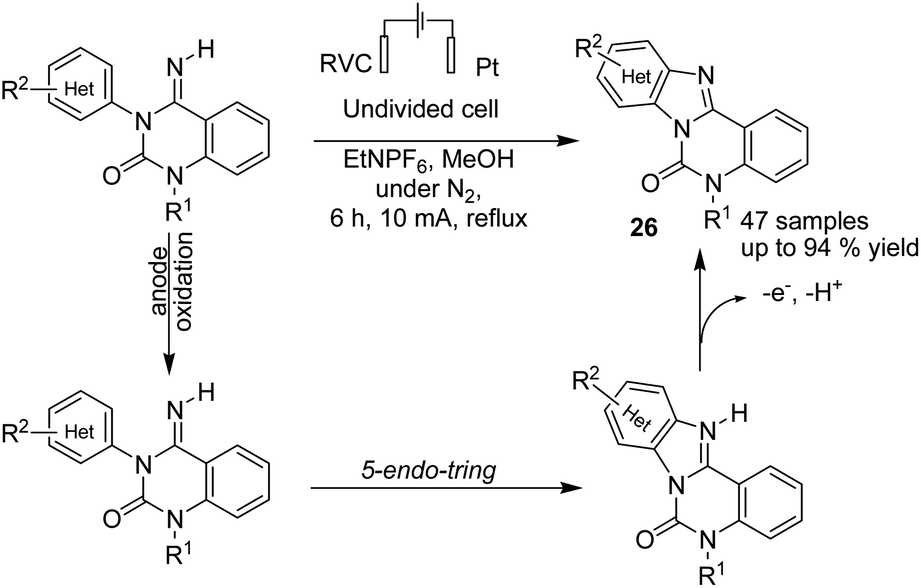 | ||
| Scheme 25 Electrochemical synthesis hindered the primary and secondary amine. | ||
5. Electrochemical reactions of amides
Lie and co-workers in 2019 reported electrochemical dehydrogenative aryl C–H/N–H cross-coupling of aromatic C–H compounds with N–H of sulfonimides.38 The reaction proceeded via an electrochemical oxidation-induced intermolecular cross-coupling with high regioselectivity through the N-radical addition pathway under external-oxidant-free and catalyst-free conditions. The cyclic voltammetry mechanistic study indicated that N-centered imidyl radicals are initially generated, and subsequently, radical addition to the aromatic C–H compound furnished a new C–N bond. The radical species underwent further oxidation to furnish a carbon cation intermediate, which finally was aromatized to provide the aryl C(sp2)–H imidation product 27 (Scheme 26).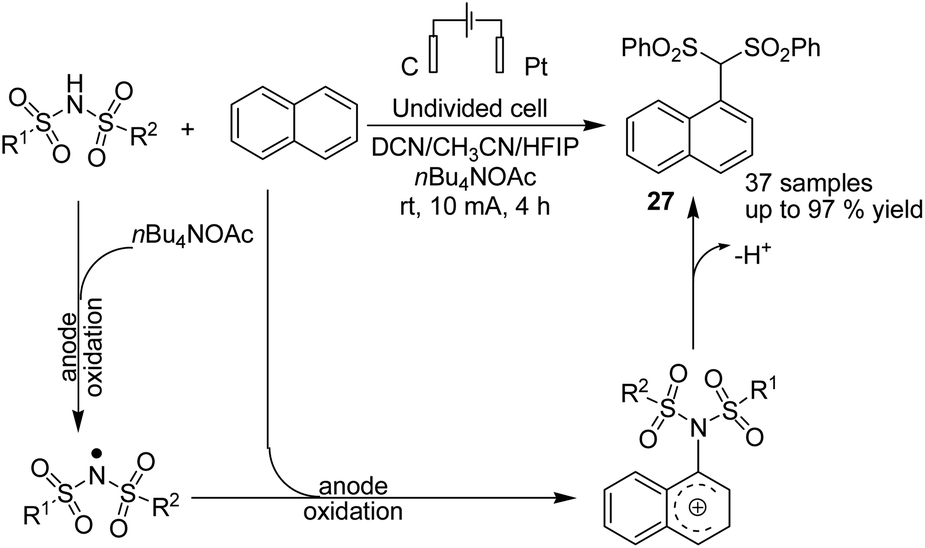 | ||
| Scheme 26 Electrochemical dehydrogenative aryl C–H/N–H cross-coupling reactions. | ||
In 2018, Ahmed et al. reported the efficient electrosynthesis of thiazolidin-2-imines via oxysulfurization of thiourea-tethered terminal alkenes.39 The reported method was the first electrochemical cyclisation to access thiazolidin-2-imines. The reaction was carried out via electrolysis of N-allylic thioureas to generate radical intermediates of nitrogen and sulfur that subsequently cyclised via oxysulfurisation of terminal alkenes to give thiazolidin-2-imines 28 with satisfactory to high yields (Scheme 27). Later, they also studied the above process in the presence of TEMPO, and the results showed that product 29 was obtained in satisfactory to high yield.40
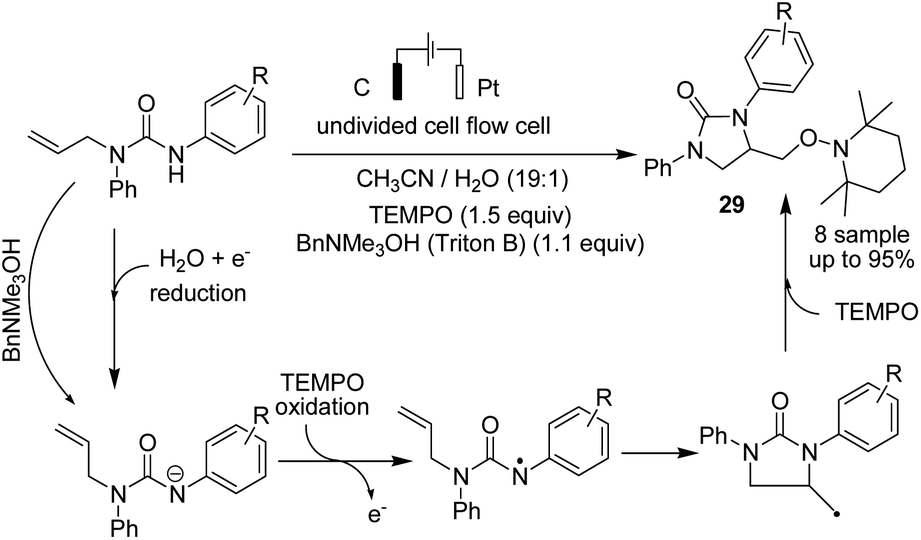 | ||
| Scheme 27 Electrosynthesis of thiazolidin-2-imines 28 and trapped product 29 in the presence of TEMPO. | ||
Waldvogel et al. in 2019 reported the electrochemical synthesis of 2-oxazolines 30 via the fluorocyclization of allylcarboxamides by a hypervalent iodine mediator.41 The process proceeded via anodic oxidation of the iodoarene to the activated hypervalent iodine. ArIF2 was attacked by the nucleophilic double bond in the amide compound to form the iodonium species. Subsequently, the three-membered heterocycle was opened by the carbonyl, and finally, the intermediate was converted into product after an SN2-type substitution reaction (Scheme 28). In another report, this group studied the electrochemical fluorocyclization of N-propargylamides for the synthesis of oxazoles.42 This reaction also proceeded via hypervalent ArIF2 generation by direct electrochemical oxidation of iodoarene ArI in Et3N·5HF, and it mediated the fluorocyclization of N-propargylamides to 5-fluoromethyl-2-oxazoles 31 (Scheme 29).
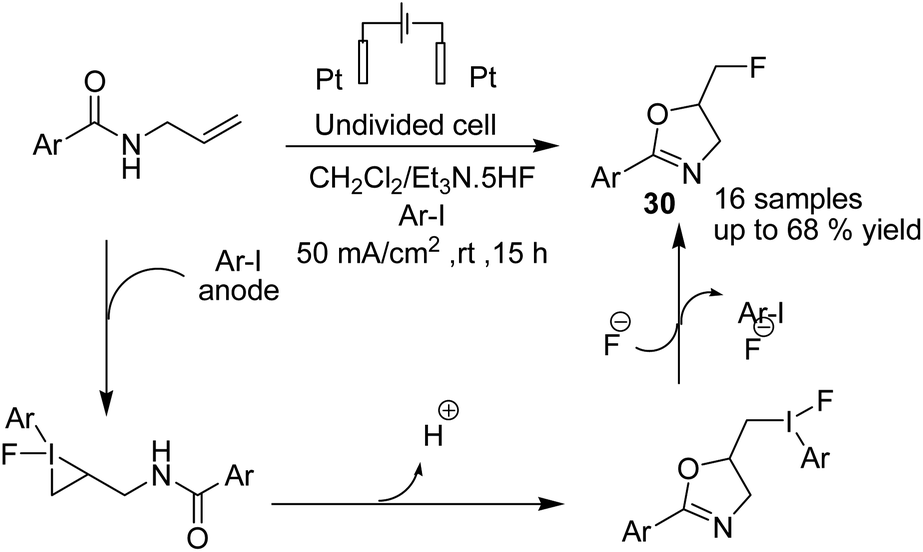 | ||
| Scheme 28 Electrosynthesis of 2-oxazolines 30 by in situ generation of ArIF2. | ||
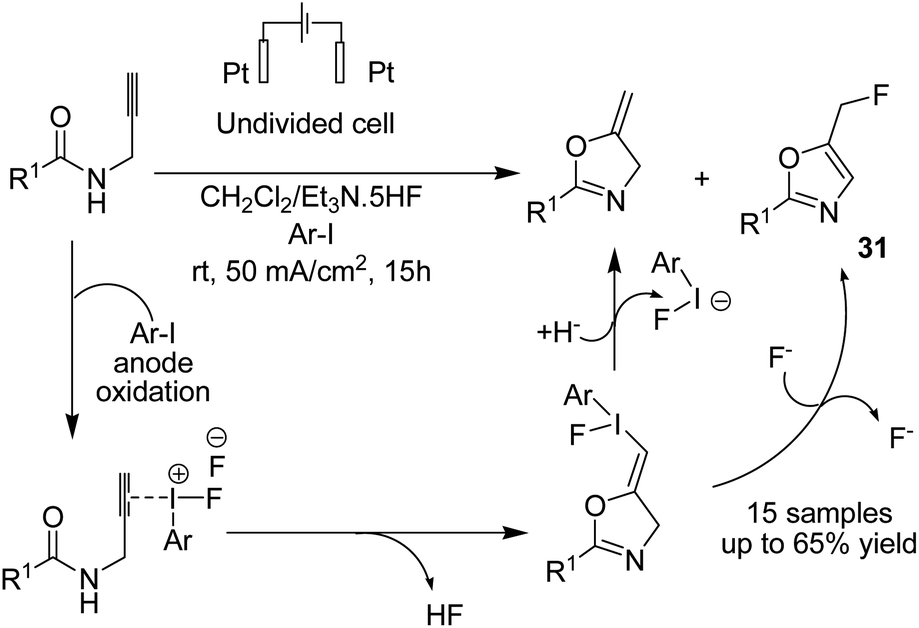 | ||
| Scheme 29 Electrosynthesis of 5-fluoromethyl-2-oxazoles 31 by in situ generation of ArIF2. | ||
In 2018, Xu et al. reported the electrochemical synthesis of 7-membered carbocycles through cascade 5-exo-trig/7-endo-trig radical cyclization of carbamate containing a disubstituted cis-alkene and a monosubstituted alkene in the presence of Cp2Fe (Scheme 30).43 A 5-membered ring was initially formed with trans-disposition of the radical centre, and finally, the 6-heptenyl radical underwent regioselective 7-endo cyclization. The reaction proceeded via transfer of one electron from Cp2Fe to the anode to afford Cp2Fe+. The methoxide base anion generated at the cathode deprotonated the substrate to give its conjugate base. A formed radical via oxidation of conjugated base by Cp2Fe+ through single-electron transfer (SET) underwent stereoselective 5-exo-trig cyclization to give carbon-centred radical species. Finally, the formed radical underwent 7-endo-trig cyclization with the remaining terminal alkene to give the bicyclic radical intermediate, and the reduction of radical via H-atom transfer afforded the final 7-membered ring product 32 (Scheme 30).
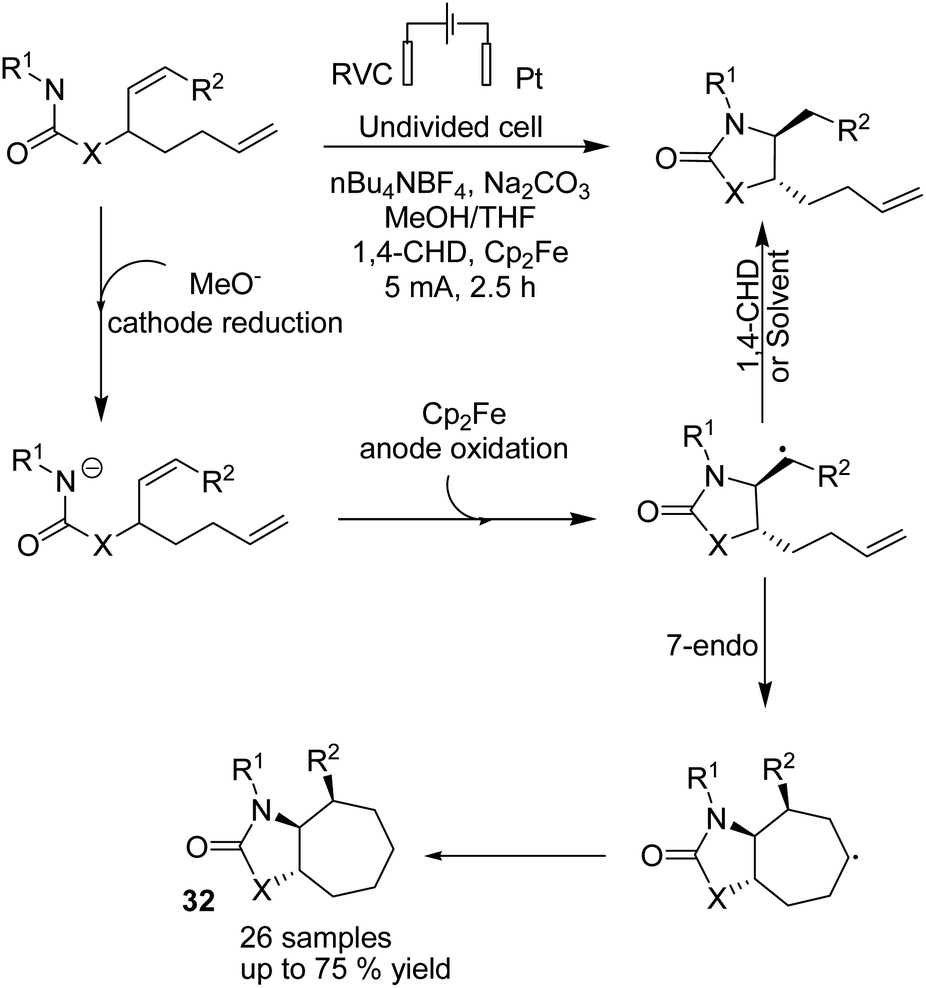 | ||
| Scheme 30 Electrochemical synthesis of 7-membered carbocycles. | ||
In 2019, Ackermann et al. reported an electrochemical position-, regio-, and chemo-selective ruthenium-catalyzed alkyne annulation by C–H/N–H activation of aryl carbamates.44 The mechanistic studies showed that the reaction proceeded via a plausible catalytic cycle to commence by a facile organometallic C–H activation (Scheme 31). A generated seven-membered ruthena(II) cycle from the insertion and migration of alkyne rapidly underwent reductive elimination to product 33. The ruthenium(0) sandwich reoxidized in the anode.
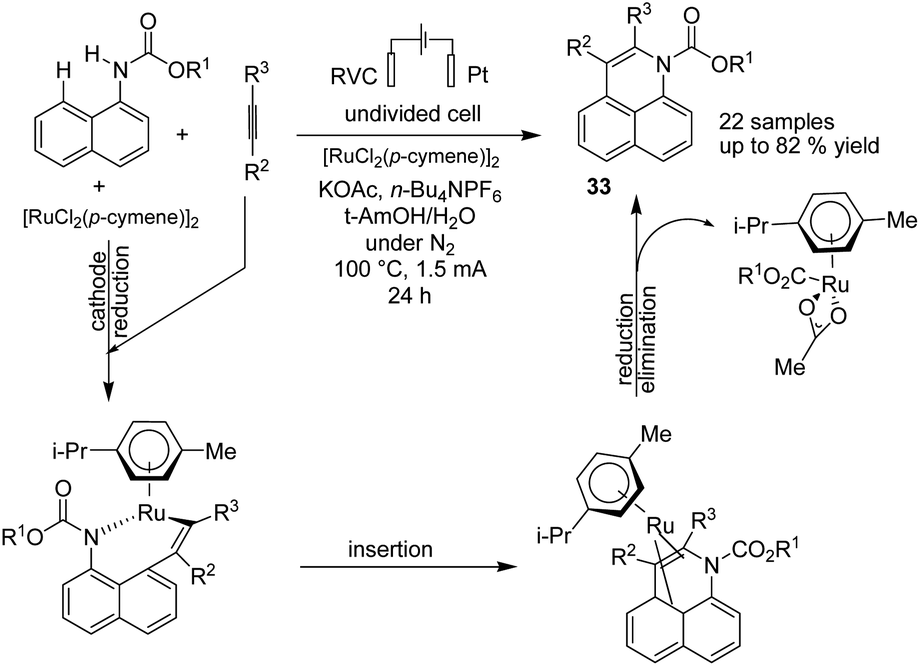 | ||
| Scheme 31 Electrochemical selective ruthenium-catalyzed alkyne annulations by C–H/N–H activation of aryl carbamates. | ||
Hu and Yi reported a formal aza-wacker cyclization via oxidative amination of crotyl N-arylcarbamates in the presence of a Cu catalyst for the synthesis of a wide range of 5-membered N-heterocycles including oxazolidinone, imidazolidinone, thiazolidinone, pyrrolidinone, and isoindolinone.45 The transformation of secondary and primary alkyl radical intermediates into alkenes was carried out in the presence of Cu catalyst. The mechanistic studies showed that the crotyl N-arylcarbamate associates with the base to give a product, which is oxidized at the anode to give an amidyl radical. The radical underwent 5-exo-trig cyclization to afford the alkyl a radical, which was captured by Cu(II) to generate a formal Cu(III) alkyl intermediate, and subsequently, product 34 was formed via a reductive elimination process (Scheme 32).
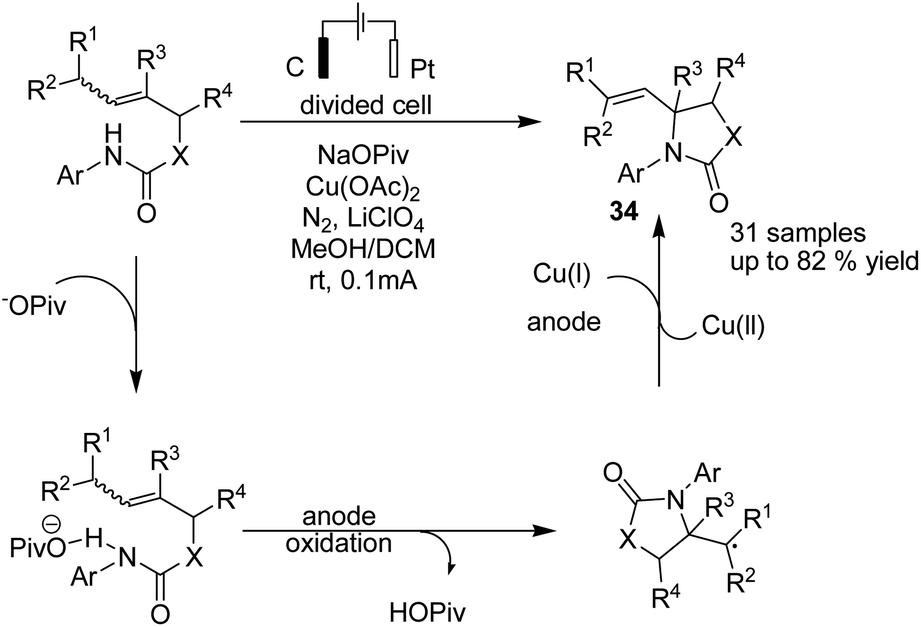 | ||
| Scheme 32 Electrochemical aza-wacker cyclization of crotyl N-arylcarbamates. | ||
In the other study,46 Ackermann et al. reported cobaltaelectro-catalyzed C–H/N–H activation with carbon monoxide or isocyanides (Scheme 33). The reaction proceeded via a plausible catalytic cycle of initiation of the cobalt(II) pre-catalyst by anodic oxidation to form the catalytically competent cobalt(III). In the next step, carboxylate-assisted C–H activation and subsequent migratory insertion gave rise to the six-membered cobalta(III) cycle, from which products 35 and 35′ formed via reductive elimination (the catalytically active cobalt(III) carboxylate complex is regenerated by anodic oxidation).
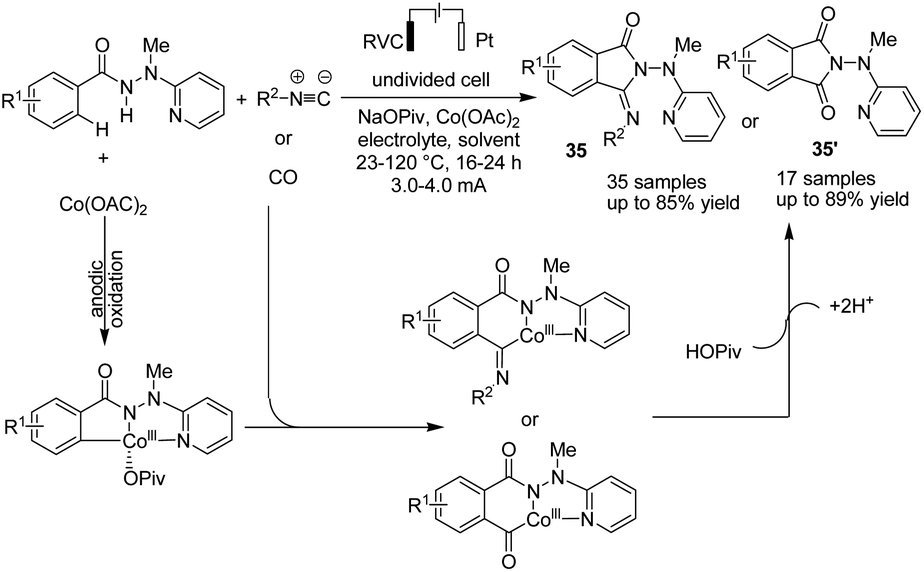 | ||
| Scheme 33 Electrochemical cobaltaelectro-catalyzed C–H/N–H activation with carbon monoxide or isocyanides. | ||
An electrochemical reductive radical Smiles rearrangement for C–N bond formation of compound 36 was reported by Guo et al. in 2019.47 The process proceeded via amidyl radical generation from the cleavage of the N–O bond of compound 36 under reductive electrolytic conditions, which played a crucial role in this transformation. The mechanistic studies showed that a single-electron transfer reduction in the cathode generated the radical amidyl intermediate, which subsequently underwent a radical Smiles rearrangement to form an O-centred radical intermediate, from which product 37 formed by cathodic reduction and protonation (Scheme 34).
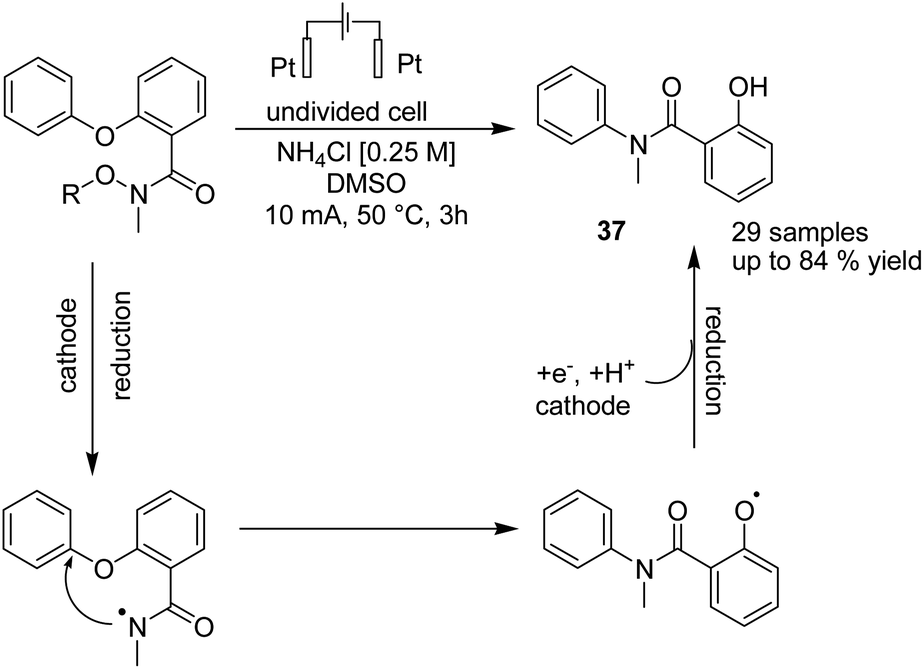 | ||
| Scheme 34 Electrochemical reductive radical smiles rearrangement. | ||
6. Electrochemical reactions of other organic nitrogen sources
Waldvogel and Rodrigo in 2019 reported an electrochemical reduction of aromatic and heteroaromatic nitrones to amines.48 The reduction reaction needs four electrons, whereby two electrons yield the imine, and the other two electrons are used for the reduction of the imine double bond to the corresponding amine (Scheme 35).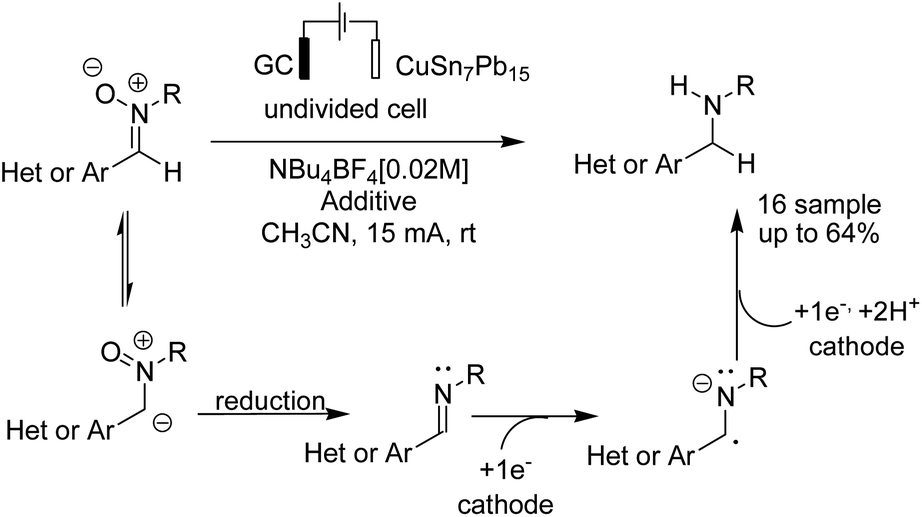 | ||
| Scheme 35 Electrochemical reduction of aromatic and heteroaromatic nitrones to amines. | ||
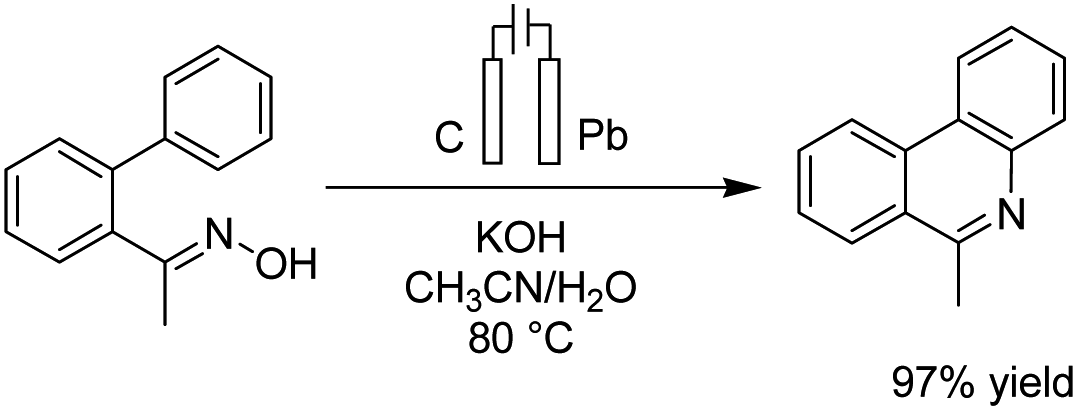
Xu et al. reported the electrochemical C–H functionalization of biaryl ketoximes for the synthesis of polycyclic N-heteroaromatic compounds and their corresponding N-oxides in the presence of TEMPO and Pt as a cathode.49 The electrosynthesis proceeded via the anodic oxidation of TEMPO into TEMPO+, which then reacted with oxime to afford an iminoxyl radical. N-Cyclization of the iminoxyl radical onto the phenyl ring, followed by re-aromatization, yields N-oxide product 38 (Scheme 36).
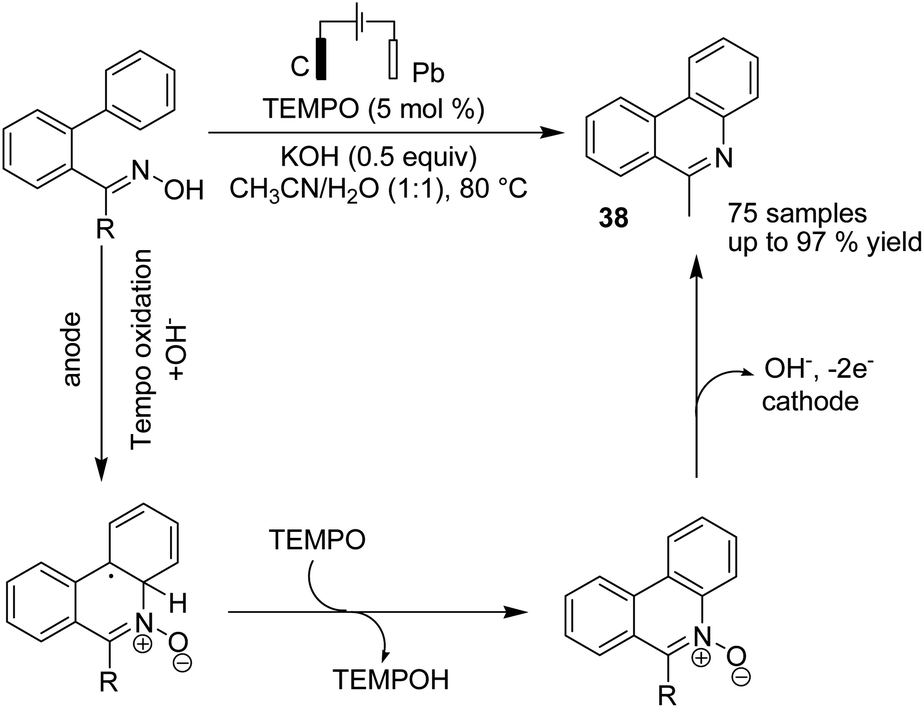 | ||
| Scheme 36 Electrochemical C–H functionalization of biaryl ketoximes for the synthesis of polycyclic N-heteroaromatic compounds. | ||
In 2018, the Waldvogel research team reported a novel electrosynthesis method for the synthesis of 2,1-benzisoxazoles and quinoline N-oxides from nitro aromatic compounds.50 The reaction proceeded via a cathodic reduction of the nitro moiety, and subsequent intramolecular cyclization afforded different substituted 2,1-benzisoxazoles and quinoline N-oxides (Scheme 37).
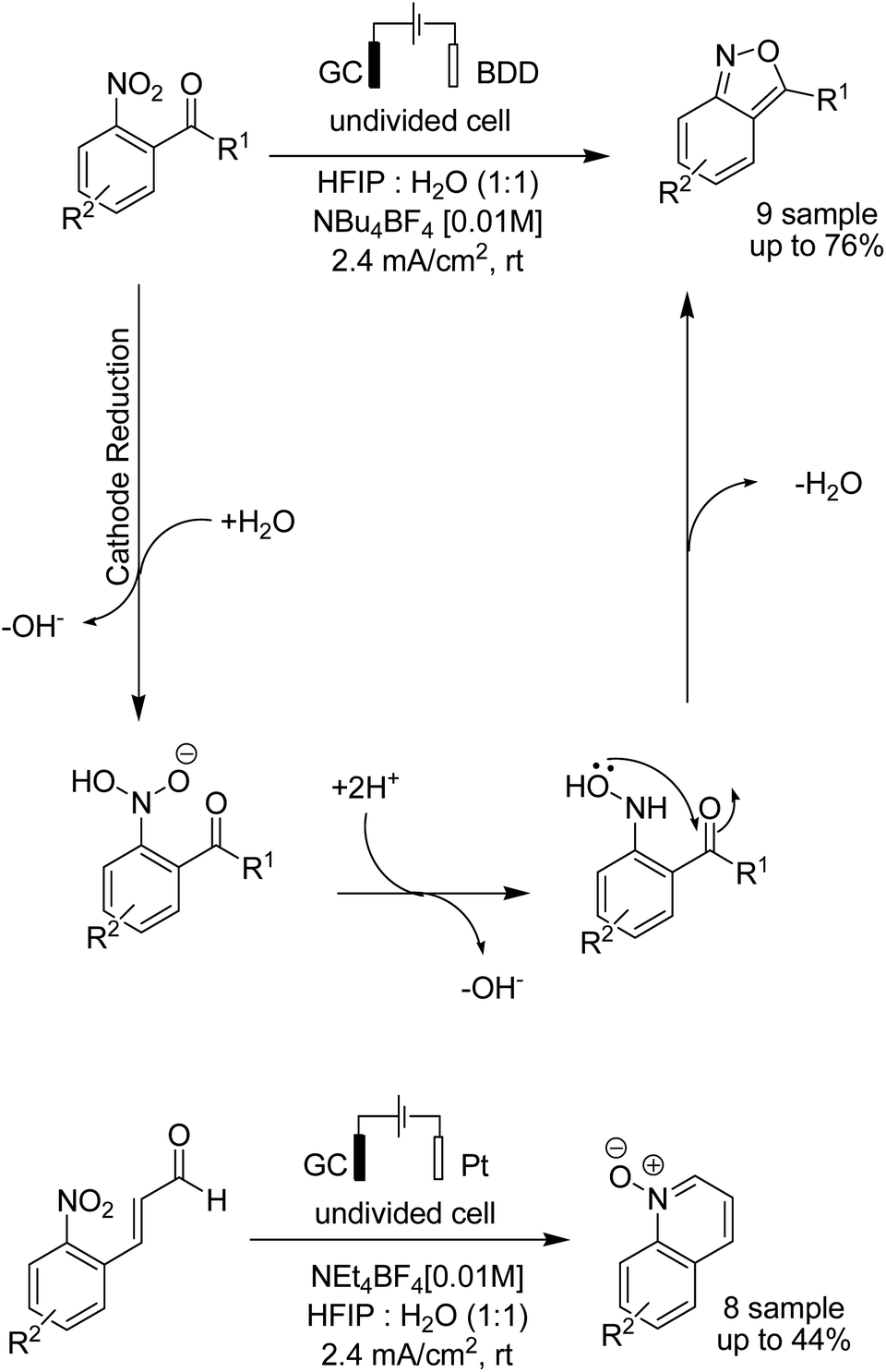 | ||
| Scheme 37 Electrochemical synthesis of 2,1-benzisoxazoles and quinoline N-oxides. | ||
In another report, Waldvogel and Hartmer reported the electrosynthesis of nitriles from oximes.51 The reaction proceeded via anodic oxidation of the nitrile to nitrile oxide, and subsequent cathodic reduction of nitrile oxide afforded the nitrile compound (Scheme 38).
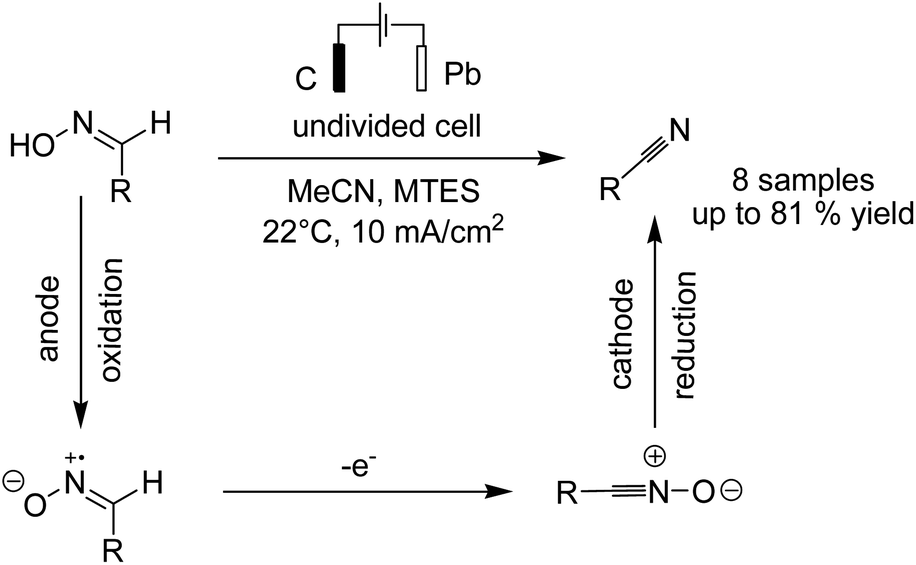 | ||
| Scheme 38 Electrochemical conversion of oximes to nitriles. | ||
7. Summary and outlook
Electrosynthesis is an efficient and applicable method in organic transformation. The application of this method in organic synthesis is interesting and attractive. In this review, the electrochemical reaction of amines, nitriles, amides, and imines for the synthesis of various heterocyclic compounds and coupling products have been summarized and discussed. The developments in the application of electrosynthesis as an efficient method for organic transformations are impressive, and hopefully can initiate further evolution in this area.This review is an introduction to an area that will inspire others to try electrochemical reactions for new organic transformations. The examples outlined in this review represent some of the organic transformations of nitrogen-containing compounds that assist scientists in trying to solve various problems in organic transformations by electrochemistry (Table 1). Electrosynthesis methods assist us in performing organic reactions in a simple manner, and there should be no reason to use stoichiometric amount of reagents to accomplish simple conversions, such as the conversion of alcohols to aldehydes, when the reaction can be efficiently carried out using electrochemistry. There is great potential in the chemoselectivity of the electrosynthesis method for various organic transformations, and especially their applications in the total synthesis of natural products. Although it is interesting that the reactions can be run using simple homemade equipment, we believe that the use of simple and standard instruments requires the development of electrosynthesis methods in organic transformations by scientists. We also believe that the development of electrosynthesis of organic compounds depends on mechanistic insights into electro–organic reactions. Our research group has recently entered this research field,52–54 and we hope that electrochemistry will soon become a routine technique in modern organic chemistry laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support by the Institute for Advanced Studies in Basic Sciences (IASBS).References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed.
- H. Lund, J. Electrochem. Soc., 2002, 149, S21–S33 CrossRef CAS.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zibes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- Y. Yuan and A. Lie, Nat. Commun., 2020, 11, 802 CrossRef CAS.
- T. Wirtanen, E. Rodrigo and S. R. Waldvogel, Adv. Synth. Catal., 2020, 362, 2088–2101 CrossRef.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- J. L. Rockl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef.
- M. Faraday, Ann. Phys. Chem., 1834, 109, 433–451 CrossRef.
- (a) J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi and Y. A.-W. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS; (b) H. B. Wang and J. M. Huang, Adv. Synth. Catal., 2016, 358, 1975–2198 CrossRef CAS.
- G. Laudadio, E. Barmpoutsis, C. Schotten, L. Struik, S. Govaerts, D. L. Browne and T. Noël, J. Am. Chem. Soc., 2019, 141, 5664–5668 CrossRef CAS PubMed.
- H. Hong, Z. Zou, G. Liang, S. Pu, J. Hu, L. Chen, Z. Zhu, Y. Li and Y. Huang, Org. Biomol. Chem., 2020, 18, 5832 RSC.
- L. Zeng, J. Li, J. Gao, X. Huang, W. Wang, X. Zheng, L. Gu, G. Li, S. Zhang and Y. He, Green Chem., 2020, 22, 3416 RSC.
- X. Gao, P. Wang, Q. Wang, J. Chen and A. Lei, Green Chem., 2019, 21, 4941 RSC.
- Z. Zhou, K. Hu, J. Wang, Z. Li, Y. Zhang, Z. Zha and Z. Wang, ACS Omega, 2020, 5, 31963–31973 CrossRef CAS PubMed.
- W. Q. Jian, H. B. Wang, K. S. Du, W. Q. Zhong and J. M. Huang, ChemElectroChem, 2019, 6, 2733 CrossRef CAS.
- M. Ošeka, G. Laudadio, N. P. van Leest, M. Dyga, A. D. A. Bartolomeu, L. J. Gooßen, B. de Bruin, K. T. de Oliveira and T. Noël, Chem, 2021, 7, 255–266 Search PubMed.
- Q. –L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS.
- Y. Wang, P. Qian, J.-H. Su, Y. Li, M. Bi, Z. Zha and Z. Wang, Green Chem., 2017, 19, 4769–4773 RSC.
- D.-Z. Lin and J.-M. Huang, Org. Lett., 2018, 20, 2112–2115 CrossRef CAS.
- K.-J. Li, K. Xu, Y.-G. Liu, C.-C. Zeng and B.-G. Sun, Adv. Synth. Catal., 2019, 361, 1033–1041 CrossRef CAS.
- M. Huang, J. Dai, X. Cheng and M. Ding, Org. Lett., 2019, 21, 7759–7762 CrossRef CAS.
- H.-S. Kim and S. Lee, Eur. J. Org. Chem., 2019, 2019, 6951–6955 CrossRef CAS.
- N. Fu, L. Li, Q. Yang and S. Luo, Org. Lett., 2017, 19, 2122–2125 CrossRef CAS.
- P.-S. Gao, X.-J. Wang, Z. –H. Wang, C. Zhang, B. Sun, Z. –H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254–15259 CrossRef CAS.
- Q. Wang, T. Yuan, Q. Liu, Y. Xu, G. Xie, X. Lv, S. Ding, X. Wang and C. Li, Chem. Commun., 2019, 55, 8398–8401 RSC.
- S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong, G. Li and M. Findlater, Angew. Chem., Int. Ed., 2021, 60, 7275–7282 CrossRef CAS PubMed.
- M. Singh, L. K. Sharma, R. Dubey, M. K. Patel, V. Prakash and R. K. P. Singh, ChemistrySelect, 2020, 5, 3847–3849 CrossRef CAS.
- Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548–16553 CrossRef CAS PubMed.
- F. Lu, Z. Yang, T. Wang, T. Wang, Y. Zhang, Y. Yuan and A. Lei, Chin. J. Chem., 2019, 37, 547–551 CrossRef CAS.
- D. Lehnherr, Y.-H. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478 CrossRef CAS PubMed.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732 RSC.
- H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587–592 CrossRef CAS PubMed.
- X. Hu, G. Zhang, L. Nie, T. Kong and A. Lie, Nat. Commun., 2019, 10, 5467–5476 CrossRef.
- M. Islam, B. M. Kariuki, Z. Shafiq, T. Wirth and N. Ahmed, Eur. J. Org. Chem., 2019, 2019, 1371–1376 CrossRef CAS.
- N. Ahmed and A. Vgenopouloua, SynOpen, 2019, 3, 46–48 CrossRef CAS.
- J. D. Haupt, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 242–245 CrossRef CAS PubMed.
- J. D. Herszman, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 7893–7896 CrossRef CAS PubMed.
- H. Long, J. Song and H.-C. Xu, Org. Chem. Front., 2018, 5, 3129–3132 RSC.
- R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC.
- X. Yi and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 4700–4704 CrossRef CAS PubMed.
- S. C. Sau, R. Mei, J. Struwe and L. Ackermann, ChemSusChem, 2019, 12, 3023–3027 CrossRef CAS PubMed.
- X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 10–13 CrossRef CAS PubMed.
- E. Rodrigo and S. R. Waldvogel, Chem. Sci., 2019, 10, 2044–2047 RSC.
- H.-B. Zhao, P. Xu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 15153 CrossRef CAS PubMed.
- E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255–12258 RSC.
- M. F. Hartmer and S. R. Waldvogel, Chem. Commun., 2015, 51, 16346 RSC.
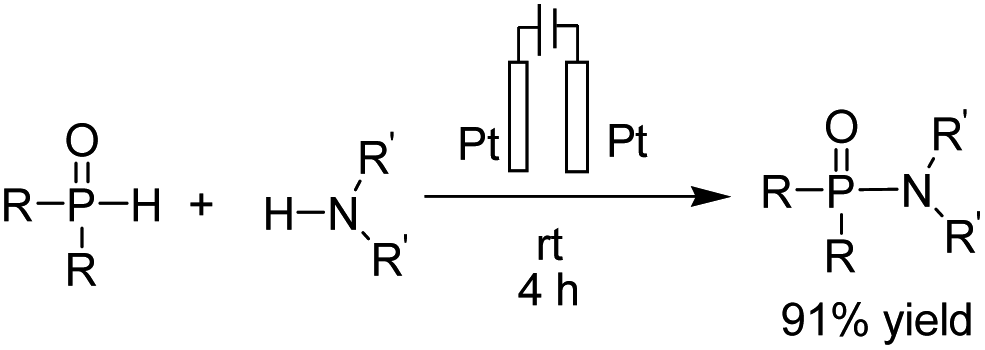
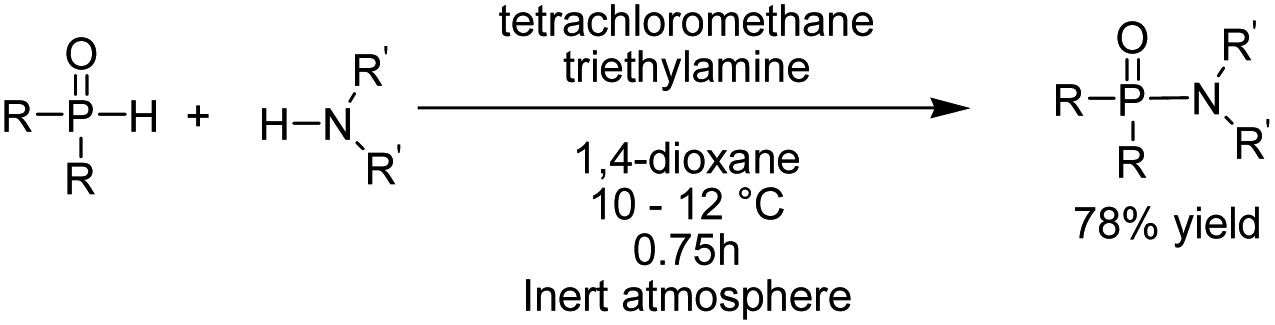
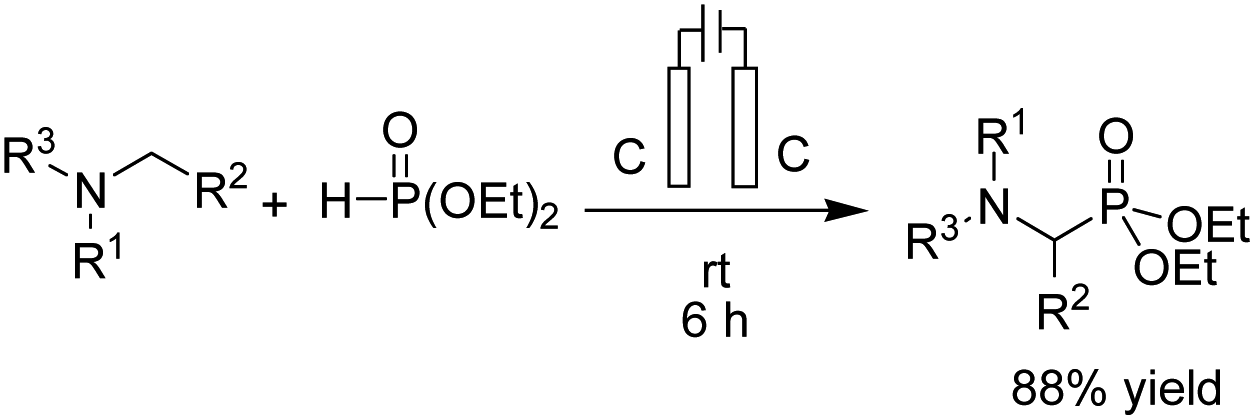
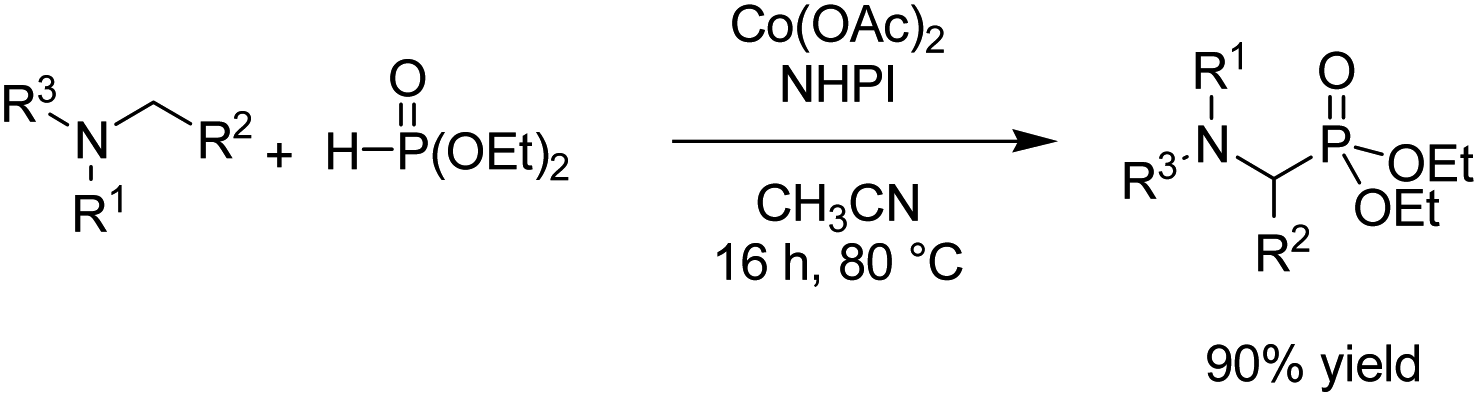
- B. Kaboudin, L. Behrouzi, F. Kazemi, M. M. Najafpour and H. Aoyama, ACS Omega, 2020, 5, 17947–17954 CrossRef CAS PubMed.
- L. Behrouzi, R. Bagheri, M. R. Mohammadi, Z. Song, P. Chernev, H. Dau, M. M. Najafpour and B. Kaboudin, Sci. Rep., 2020, 10, 19378 CrossRef CAS PubMed.
- L. Behrouzi, R. Bagheri, Z. Song, F. Kazemi, B. Kaboudin and M. M. Najafpour, Mater. Res. Express, 2019, 6, 125607 CrossRef CAS.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS PubMed.
- B. Du, Y. Wang, W. Sha, P. Qian, H. Mei, J. Han and Y. Pa, Asian J. Org. Chem., 2017, 6, 153–156 CrossRef CAS.
- A. Lator, Q. G. Gaillard, D. S. Mérel, J. F. Lohier, S. Gaillard, A. Poater and J. L. Renaud, J. Org. Chem., 2019, 84, 6813–6829 CrossRef CAS PubMed.
- Ueno, Y. Ikeda and E. Shirakawa, Eur. J. Org. Chem., 2017, 28, 4188–4193 CrossRef.
- Y. Liu, L. Lu, H. Zhou, F. Xu, C. Ma, Z. Huang and J. X. S. Xu, RSC Adv., 2019, 9, 34671–34676 RSC.
- S. K. Y. Leung, W. M. Tsui, J. S. Huang, C. M. Che and J. L. L. N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629–21664 CrossRef CAS PubMed.
- H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055–4059 CrossRef CAS PubMed.
- N. I. Ivanova, P. A. Volkov, L. I. Larina, N. K. Gusarovaand and B. A. Trofimov, Chem. Heterocycl. Compd., 2012, 47, 1384–1389 CrossRef CAS.
- A. Gupta, M. S. Eshmukhand and N. Jain, J. Org. Chem., 2017, 82, 4784–4792 CrossRef CAS PubMed.
- B. Lin, S. Shi, R. Lin, Y. Cui, M. Fang, G. Tangand and Y. Zhao, J. Org. Chem., 2018, 83, 6754–6761 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |