
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic turnover of CO2 under visible light by [Re(CO)3(1-(1,10) phenanthroline-5-(4-nitro-naphthalimide))Cl] in tandem with the sacrificial donor BIH†
Alyssa Spear a,
Robson L. Schuarcab,
Jesse Q. Bondb,
Timothy M. Kortera,
Jon Zubieta*a and
Robert P. Doyle*a
a,
Robson L. Schuarcab,
Jesse Q. Bondb,
Timothy M. Kortera,
Jon Zubieta*a and
Robert P. Doyle*a
aDepartment of Chemistry, Syracuse University, Syracuse, NY 13244, USA. E-mail: jazubiet@syr.edu; rpdoyle@syr.edu
bDepartment of Biomedical and Chemical Engineering, Syracuse University, Syracuse, NY 13244, USA
First published on 10th February 2022
Abstract
Optimized photocatalytic conversion of CO2 requires new potent catalysts that can absorb visible light. The photocatalytic reduction of CO2 using rhenium(I) has been demonstrated but suffers from low turnover. Herein, we describe a [Re(CO)3(1-(1,10)phenanthroline-5-(4-nitro-naphthalimide))Cl] photocatalyst, which when combined with the sacrificial donor 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole, results in selective production of formic acid and a high turnover number of 533 and turnover frequency of 356 h−1. Single-crystal X-ray diffraction and DFT studies are also discussed.
1. Introduction
Increasing levels of atmospheric and anthropogenic CO2 require the development of new technologies and strategies in the pursuit of sustainable energy and climate preservation.1–3 Homogenous mononuclear rhenium(I) complexes have been explored as reductive catalysts for the conversion of CO2 to fuels and feedstocks, primarily CO.4,5 While this subset of catalysts has been tuned for high turnover numbers (TN, [CO2 reduction product]/[catalyst]), notably as high as 350,6 rarely is selective CO2 to formic acid conversion reported. Formate and formic acid are attractive products due to ease of transportation and storage, and high energy densities, and as favourable candidates for hydrogen storage.7–9 Any reports of CO2 to formate reduction by mononuclear rhenium(I) complexes are typically in trace amounts and hence there is an unmet need to tune such complexes towards formate/formic acid selectivity if they are to provide a viable option for CO2 conversion to more diverse products, especially under visible light.Previously, we have reported on the synthesis, characterization, electrochemistry, and catalytic activity of a [Re(CO)3(1-(1,10)-phenanthroline-5-(4-nitro-naphthalimide)Cl] complex (1). 1 consists of a rhenium(I) tricarbonyl core with a naphthalimide based photosensitizing ligand, (1,10)-phenanthroline-5-(4-nitro-naphthalimide) (5-PAN), for the visible light photoreduction of CO2 to formate (Fig. 1).10 A sacrificial electron donor, triethylamine (TEA), was required for photocatalysis.7 The catalytic activity of 1 increased with a concomitant increase in TEA, and no CO2 conversion was obtained in the absence of light (450–460 nm). Catalytic saturation was reached at 0.87 mmol of HCOO−, corresponding to a TN ([HCOO–]/[1]) of 48.10
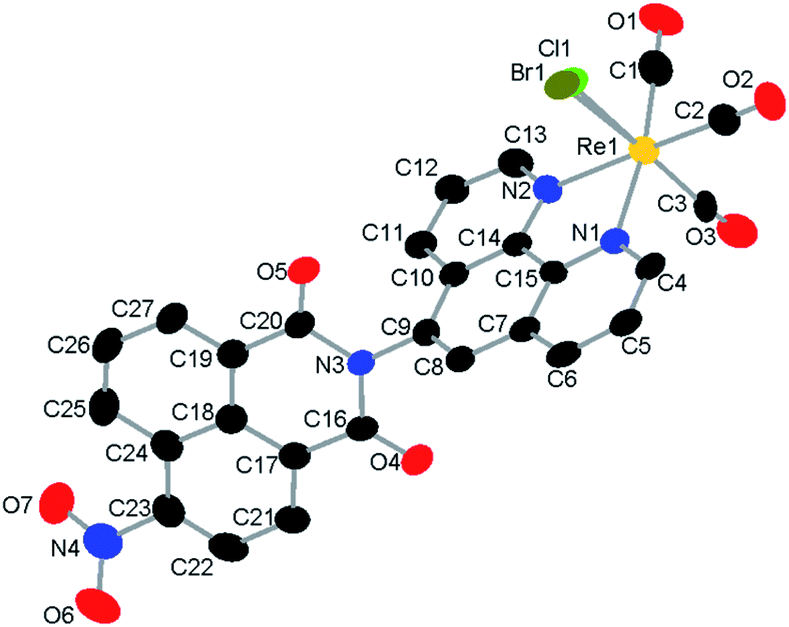 | ||
Fig. 1 Single-crystal X-ray diffraction structure of a mixture of 1 and 2. H-atoms and toluene solvent molecule are omitted for clarity. The halido ligand site is modelled as a disorder of Br![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl, 0.80:0.20. Structure deposited as CCDC # 2086581. :Cl, 0.80:0.20. Structure deposited as CCDC # 2086581. | ||
In the present research, we aim to discern a mechanism for the system and improve the photocatalytic ability of 1. We synthesized the bromide analogue, [Re(CO)3(1-(1,10)-phenanthroline-5-(4-nitro-naphthalimide)Br] (2), to evaluate the effect of the halide. Moreover, we obtained crystals of combined 1 and 2 which allowed X-ray diffraction studies and subsequent DFT calculations reported herein. To further drive CO2 reduction, we replaced TEA with a 2-electron proton donor in the form of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d] imidazole (BIH), which has been shown to significantly quench the excited state of rhenium(I) photocatalysts thereby yielding better turnover.11,12 Consequently, we have combined BIH with 1 and 2 in the presence of blue light (450–460 nm) to produce formate in high selectivity over CO.
2. Experimental
2.1. Materials and methods
CDCl3, CD3OD, deuterated DMSO, Re(CO)5(X) (X = Cl, Br), 2-phenylbenzimidazole, iodomethane, sodium borohydride, sulfuric acid, and diethyl ether were purchased through Sigma Aldrich (Milwaukee, WI, USA). Sodium hydroxide was purchased through Fisher Scientific (Agawam, MA, USA). Methanol (MeOH), ethanol, and dichloromethane (DCM) were purchased through Sigma Aldrich (Milwaukee, WI, USA) and dried over 4 Å molecular sieves (Sigma-Aldrich) in-house to remove water. Barium carbonate (13C, 98%+) was purchased from Cambridge Isotope Laboratories, Inc (Tewksbury, MA, USA). 1 M H2SO4 was prepared in-house. A 12 W LED blue light (450–460 nm) was purchased from Abi, USA (Vernon, CA, USA).1H/13C NMR were conducted on a Bruker Avance III HD 400 MHz instrument and calibrated to the residual CDCl3 signal at 7.24 ppm, D1 = 5s for 13C NMR. FT-IR was carried out on a Nicolet Infrared Spectrophotometer utilizing potassium bromide (KBr) disks. Crystals of [Re(CO)3(1-(1,10)phenanthroline-5-(4-nitro-naphthalimide))X] (X = Cl or Br) suitable for single crystal X-ray diffraction studies were only obtained by combining 1 and 2 (1:4 by mass) in MeOH/DCM (1:9 v/v) and gently layering toluene on top, then sealing the glass vial with a cap and parafilm. Gas chromatography (GC) was conducted on an HP-6890 instrument with a ShinCarbon packed column (Restek, PA, USA), a Flame Ionization Detector (FID), and a methanizer. The FID was calibrated with the standards 1% CO in helium, 1% CO2 in helium, and 0.5% CH4 in helium (Airgas). All injections were done with a calibrated 1 mL syringe (Agilent). Ion chromatography was conducted by Intertek Pharmaceuticals (Whitehouse, NJ, USA).
2.2. Synthetic procedures
1 was prepared as previously reported.10 2 and BIH were prepared as described below. Re(CO)5Br was refluxed in water for 24 h to produce [Re(CO)3(H2O)3Br].13 The ligand 5-PAN was prepared as previously reported.10 [Re(CO)3(H2O)3Br] (0.26 mmol, 105 mg) and 5-PAN (0.26 mmol, 110 mg) were combined in 30 mL MeOH/DCM (1:9 v/v), put under argon, and stirred for 1.5 hours. The solution volume was reduced to half in vacuo and diethyl ether was added to precipitate the product. The mixture was centrifuged, and the ether decanted. The yellow solid noted was collected and dried under vacuum overnight giving 68% yield based on rhenium.
The synthesis of BIH was modified from literature procedures.14,15 2-Phenylbenzimidazole (3 g) was mixed with methyl iodide (8 g) in 15 mL of anhydrous MeOH containing NaOH (0.64 g) in a pressure tube. The tube was purged with argon then sealed and heated at 110 °C with stirring for 3 days. After cooling to room temperature, crystals formed. The crude intermediate was filtered and washed with MeOH and dried in vacuo. The intermediate was dissolved in 80 mL of anhydrous MeOH and NaBH4 (1.5 g) was slowly added and stirred for 1 hour under argon. The solvent was then evaporated, and the product was recrystallized in hot ethanol/water 5:1 to give white crystals of BIH in 75% yield.
2.3. Density functional theory
The electronic structure of the metal complex was simulated with Gaussian 16 Rev C.01 using the B3LYP density functional and the def2-TZVP basis set (all-electron basis functions for atoms except Re, which utilized an effective-core potential). The starting geometry of the complex was taken from the experimental crystallographic structure and the structure was then fully geometry optimized. Solvent effects were included in all calculations using the Polarizable Continuum Model (nonequilibrium for the excited states) with DCM as the specified solvent. Vertical electronic transitions were calculated with time-dependent DFT based on the Tamm-Dancoff approximation (TDA) and included 100 states (singlets and triplets) to explore the possible transitions.2.4. Typical photocatalysis experiments
The following is a typical experiment using 1. In each sample tested, 0–110.3 equivalences of BIH (0–2 mmol), were premixed in MeOH/DCM (1:9 v/v) for 10 min, after which, 13.5 mg (0.018 mmol) of 1 was added. All samples were kept at a 5 mL total reaction volume, irradiated with blue light positioned 1 inch from the reaction vessel and at a 21° angle of incident light. The solution was bubbled with 13CO2 evolved in situ with 0.500 g Ba13CO3 and 1 M H2SO4 drip for a reaction time of 1.5 hours. For each trial, the drip rate was set such that the Schlenk flask containing Ba13CO3 was full at the end of 1.5 hours and the H2SO4 was well in excess. All catalytic experiments were conducted at least in triplicate under blue light irradiation.
At the end of each trial, liquid products were quantified via 13C NMR. 400 μL of the reaction solution and 250 μL of CDCl3 were added to an NMR tube and wrapped in aluminium foil until it was ready to be loaded into the NMR. The carbon peaks for BIH and its oxidative product, BI+, at ∼33 ppm were integrated and set to 1. The H13COO− peak at ∼160 ppm was integrated and quantified with a calibration curve (S12†).
For the detection of gaseous products, 1 and BIH were dissolved in MeOH/DCM (1:9 v/v) in a vial with a stir bar and septum cap. The vial was purged with CO2 for 20 min, then irradiated with blue light for 30 min. A headspace sample was taken with a 1 mL airtight syringe and analysed by GC.
3. Results and discussion
3.1. Crystal structure
We sought to obtain a solid-state structure of the catalytic species to facilitate DFT studies. Single-crystal X-ray diffraction analysis revealed that the catalyst crystalizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the unit cell, one catalyst and one toluene solvate molecule (S8†). Diffraction quality crystals were only obtained from a mix of 1 and 2; thus, the six-coordinate, octahedral rhenium center includes a disordered halido ligand comprised of Br:Cl, 0.80:0.20. The CO ligands are facially coordinated, and the ligand, 5-PAN, is coordinated to the rhenium center in a bidentate fashion at N1 and N2 (Fig. 1). Detailed description of the structure is included in ESI†.
with two molecules in the unit cell, one catalyst and one toluene solvate molecule (S8†). Diffraction quality crystals were only obtained from a mix of 1 and 2; thus, the six-coordinate, octahedral rhenium center includes a disordered halido ligand comprised of Br:Cl, 0.80:0.20. The CO ligands are facially coordinated, and the ligand, 5-PAN, is coordinated to the rhenium center in a bidentate fashion at N1 and N2 (Fig. 1). Detailed description of the structure is included in ESI†.
3.2. Density functional theory
The full geometry optimization yielded a final structure that was in good agreement with the X-ray diffraction results described herein for 1.The TDA simulation yielded a predicted a UV/VIS spectrum that matched very well with the experimental data7 (S9†), indicating the high quality of the calculated orbital energies and participating states. Of particular interest to the photocatalytic behaviour are the four transitions with non-zero oscillator strength in the 400 to 500 nm region. These singlet transitions are detailed in Table 1 and the strongest transition, HOMO−1 → LUMO+1, is visualised in Fig. 2. Other transitions are visualised in the ESI (S10)†.
| Excitation wavelength (nm) | Oscillator strength (f) | Main participating orbitals and percentage contributions |
|---|---|---|
| 470.63 | 0.0016 | HOMO → LUMO+1 (99%) |
| 446.59 | 0.0680 | HOMO−1 → LUMO+1 (90%) |
| HOMO → LUMO+2 (8%) | ||
| 426.62 | 0.0345 | HOMO−1 → LUMO+1 (8%) |
| HOMO → LUMO+2 (90%) | ||
| 416.46 | 0.0172 | HOMO−1 → LUMO+2 (98%) |
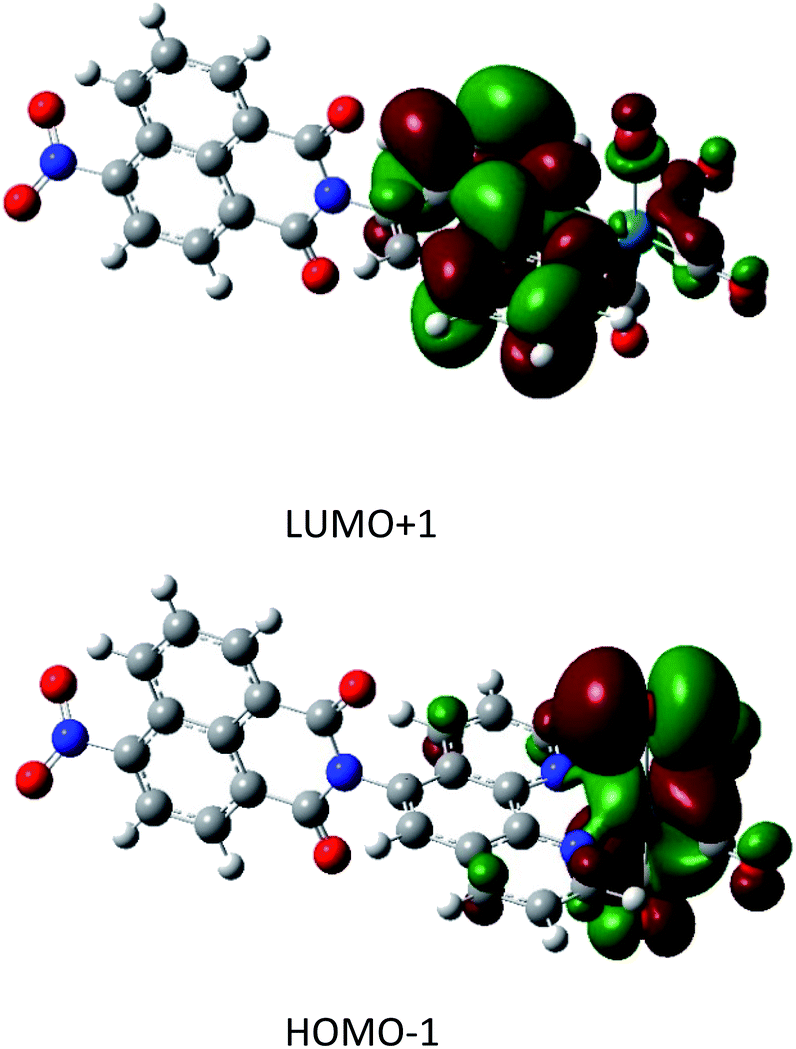 | ||
| Fig. 2 Orbital density plots for HOMO−1 → LUMO+1. The trend in these excitations is a migration of electron density from the rhenium atom to the ligand. | ||
3.3. Homogenous photocatalysis
Previously reported10 cyclic voltammetry of 1 indicated sufficient potentials of −0.95 V and −1.18 V for the two-electron reduction of CO2 to formate (−0.53 V).4 Thus, 1 was evaluated for catalytic reduction of CO2 in the presence and absence of blue light (450–460 nm) utilizing 13C NMR tracking of H13COO− (typical 13C NMR included in S11†).It should be noted that at higher concentrations of BIH (≥1 mmol), the limit of solubility in MeOH/DCM (1:9 v/v) in the presence of catalyst was reached, requiring that the BIH be dissolved in the solvent system prior to addition of the catalyst. Without this order of dissolution, the catalytic production of H13COO− at concentrations of BIH ≥ 1 mmol, was highly variable (S12†) as undissolved BIH would be misrepresented in the calibration curve (S13†).
The production of H13COO− correlates with the addition of excess BIH and blue light in an exponential trend with R2 = 0.9932 (Fig. 3). A plateau at 9.48 mmol indicated the maximum H13COO− produced and catalytic saturation. The maximum TN achieved was 533, and turnover frequency (TOF = TN/reaction time) 356 h−1 at ∼110 equivalences of BIH, where the concentration of 1 was 3.62 mM in 5 mL of MeOH/DCM (1:9 v/v). 2 was also tested for photocatalytic activity and showed no functional difference from that of 1, which is in contradiction to some literature reports which have seen halide degradation kinetics differences.16,17 No formate was detected in the absence of catalyst, BIH, or 13CO2 and a negligible amount was detected in the absence of irradiation. The calculated TN of 533 not only surpasses our previously reported 1/TEA system tenfold but introduces a standard for selective turnover of CO2 to formate via a mononuclear rhenium(I) catalyst.
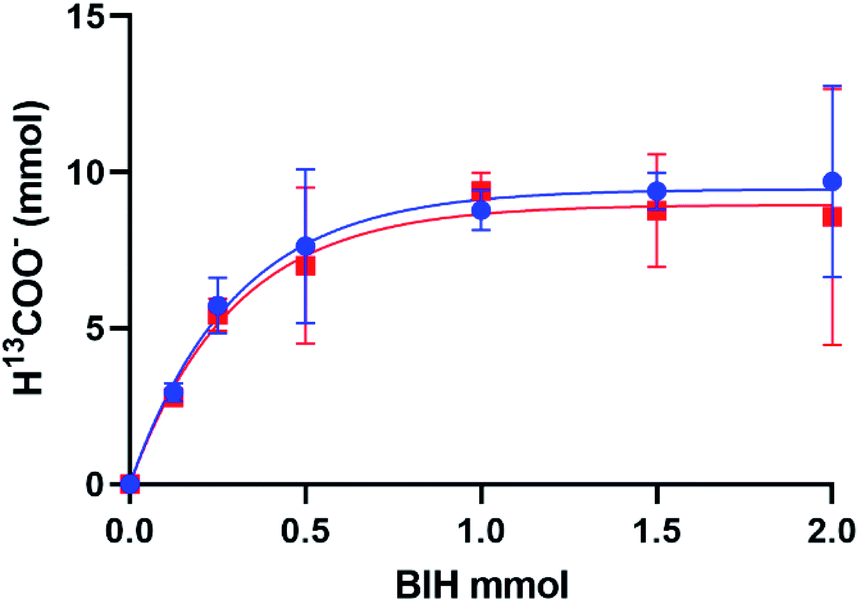 | ||
| Fig. 3 A plot of H13COO− (mmol) vs. BIH (mmol) upon blue light (450–460 nm) irradiation of 1 (blue circles, R2 = 0.9932) and 2 (red squares, R2 = 0.9864). H13COO− increased with an exponential curve fit and reached a plateau at y = 9.45 and 8.96 mmol H13COO−, respectively. | ||
The production of HCO3− has also been reported appearing on 13C NMR at ∼158 ppm.18 Ion chromatography was therefore conducted and confirmed the identity of formic acid, and lack of carbonate (S14†). Gaseous products of photocatalysis were also analysed by GC. Trace quantities of CO were detected at a maximum production of 820 nmol CO corresponding to a TN of 0.04 and a TOF of 0.09 h−1 (S15†).
3.4. Solvent effects
The solvent system MeOH/DCM (1:9 v/v) was chosen because it best solubilized all of the reaction components, however MeOH has been reported to enhance the catalytic activity of rhenium diimine complexes by increasing the current in electrolysis experiments.19 To reveal the effect of MeOH in photocatalysis, experiments were done in the presence of deuterated methanol (along with DMF at typical 1:9 v/v ratio used before). When DCM was used as the lone solvent, no formate was produced as evidenced by the loss of the ∼160 ppm peak in the 13C NMR and the subsequent emersion of a weak peak at ∼156 ppm with an integration of 0.6. When MeOH was replaced with CD3OD (CD3OD/DCM 1:9 v/v), the ∼160 ppm peak was reduced to an integration of 0.9 (S16†).
In DMF alone, a common solvent used in rhenium photocatalysis, the formate peak was also not seen in the 13C NMR and a peak at ∼156 ppm was seen with an integration of 7.6. It should be noted that the aldehyde carbon of DMF has a peak at 161 ppm, therefore a formate peak could be occluded. Nonetheless, the absence of MeOH resulted in the loss of the signature formate peak at 160 ppm and subsequent appearance of a peak slightly upfield at 156 ppm with greatly diminished integration. When experiments were conducted in MeOH/DCM with no CO2, no carbon signal was observed above 150 ppm, thus the source of the carbon is indeed CO2 with no degradation of MeOH itself. MeOH is likely necessary for catalysis as a major proton source.
3.5. Photocatalytic mechanism
A variety of factors including solvent, sacrificial donor, and halide ligand can affect the catalytic activity of rhenium diimine complexes. TLC analysis of the reaction mixture before and after irradiation showed no obvious degradation of the catalyst while BI+ did separate from BIH (S17†), which was also indicated by NMR. With the addition of BIH, compared to TEA, TN was dramatically increased possibly due to the increase in reductive power and efficient formation of the one-electron reduced catalytic species.20 When TEMPO was added in equimolar concentrations to BIH, the integration of formate increased, suggesting TEMPO acting as a hydride donor. These observations allude to a hydride mechanism.A possible mechanism is described (Fig. 4). After reductive quenching of the photoexcited catalyst by BIH, the halide dissociates, and the active catalyst is produced. Literature descriptions of formate generation then propose the formation of a metal hydride intermediate.4,21–23 Insertion of CO2 into the M−H bond then produces the intermediate. This process is in competition with H2 generation, which cannot be detected as H2 is used a co-feed gas for the methanizer during GC analysis.
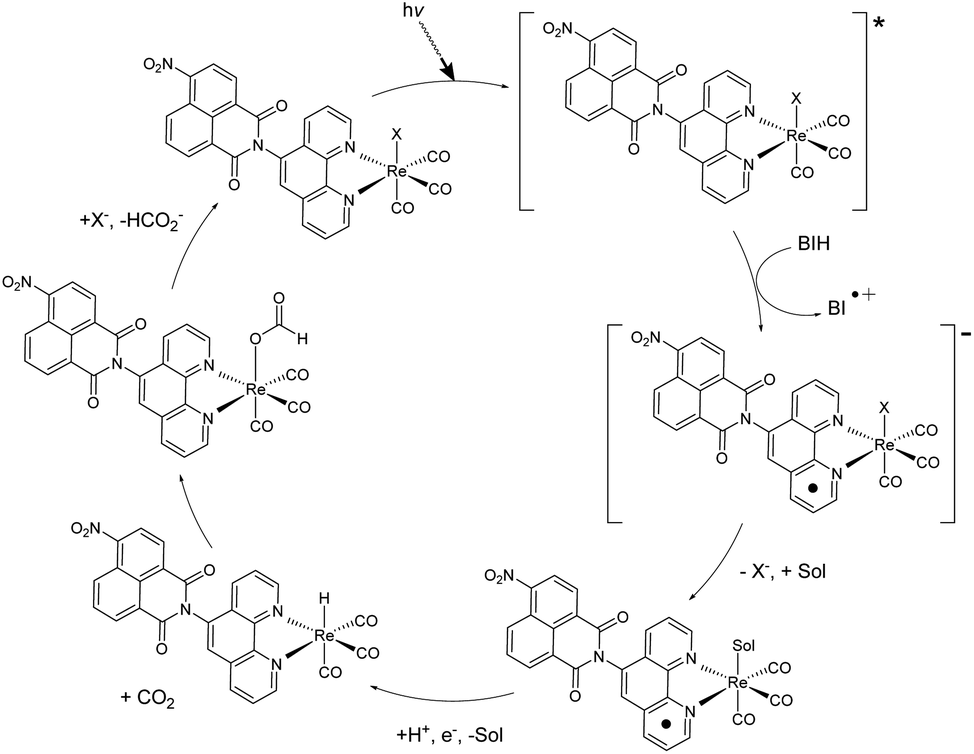 | ||
| Fig. 4 Proposed hydride mechanism of CO2 reduction to formate via 1 or 2. X = Cl or Br, Sol = DCM or MeOH. | ||
In control experiments in the absence of light, formate was still detected, reaching a maximum amount of 3.20 mmol (R2 = 0.9037) corresponding to a TN of 176 at 110 equivalents of BIH to 1. To explain the cause of this activity, we noted a literature report of BIH forming a radical cation in the presence of atmospheric O2 without irradiation.24 Under blue light irradiation, BIH alone, in MeOH/DCM (1:9 v/v), was able to reduce CO2 to formate with a low turnover of 5.7 (TN = [formate]/[BIH]). Given the sizable difference in TN from 5.7 to 533, we conclude that any BIH-only mechanism is overridden in the presence of 1 by the hydride mechanism.
The hydride mechanism shown in Fig. 4 only accounts for one BIH per catalytic cycle, whereas the TN is 533 with 110 equivalences of BIH. This suggests multiple mechanisms or components are contributing to the formation of formate herein. Notably, the role of methanol is underappreciated in the proposed mechanism and may account for the enhanced TN.
4. Conclusion
We have reported an extraordinarily improved photocatalytic TN and TOF of a rhenium(I) naphthalimide complex in the presence of the sacrificial electron and proton donor, BIH. The crystal structure of [Re(CO)3(1-(1,10)phenanthroline-5-(4-nitro-naphthalimide)) X] (X = Cl or Br) was also reported, indicating facial coordination of three CO ligands, coordination of a halide in the solid state, and bidentate coordination of the naphthalimide-based photosensitizing ligand. The generated H13COO− increased with excess BIH until a plateau was reached at 9.45 mmol of H13COOH. The corresponding TN at 110 equivalents of BIH to 1 is 533, a tenfold improvement to catalysis using 1 with the sacrificial donor TEA10 and marked selectivity over CO production, which was observed in nanomole quantities. MeOH/DCM was not only used for its ability to solvate the catalyst, but to optimize the production of H13COO−; DCM or DMF lead to diminished H13COO− signal. While the addition of MeOH improved catalytic activity, the source of the carbon was CO2. To date, this TN is the only reported value for a rhenium(I) mononuclear homogeneous photocatalyst for the selective photoreduction of CO2 to formate.1 is an exciting complex to apply towards CO2 reduction as the field is developing new methods of synthesizing valuable chemicals while maintaining sustainable and mild conditions. Notably, at similar reduction potentials to those reported for 1,10 electrosynthesis of methylamine from CO2 and NO3− was recently reported25 and will be explored with 1/BIH. Further mechanistic studies are needed to elaborate on the complex mechanism at play, especially the role of MeOH. The complex itself is an interesting avenue for future investigation in terms of development through derivatization of the bidentate naphthalimide ligand and optimization of the solvent system to better harness the quenching capabilities of BIH.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Archer, J. Geophys. Res., 2005, 110, C09S05 CrossRef.
- E. T. Sundquist, Quat. Sci. Rev., 1991, 10, 283–296 CrossRef.
- G. Shaffer, S. M. Olsen and J. O. P. Pedersen, Nat. Geosci., 2009, 2, 105–109 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- L. Q. Qiu, K. H. Chen, Z. W. Yang and L. N. He, Green Chem., 2020, 22, 8614–8622 RSC.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Nat. Catal., 2019, 2(8), 702–708 CrossRef CAS.
- D. R. Case, A. Spear, A. F. Henwood, M. Nanao, S. Dampf, T. M. Korter, T. Gunnlaugsson, J. Zubieta and R. P. Doyle, Dalton Trans., 2021, 50, 3479–3486 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Chem.–Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed.
- N. Lazarova, S. James, J. Babich and J. Zubieta, Inorg. Chem. Commun., 2004, 7, 1023–1026 CrossRef CAS.
- I. S. H. Lee, E. H. Jeoung and M. M. Kreevoy, J. Am. Chem. Soc., 1997, 119, 2722–2728 CrossRef CAS.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- J. Hawecker, J. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Chem. Commun., 1983, 536–538 RSC.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- L. Rotundo, E. Azzi, A. Deagostino, C. Garino, L. Nencini, E. Priola, P. Quagliotto, R. Rocca, R. Gobetto and C. Nervi, Front. Chem., 2019, 7, 417 CrossRef CAS PubMed.
- L. Rotundo, D. C. Grills, R. Gobetto, E. Priola, C. Nervi, D. E. Polyansky and E. Fujita, ChemPhotoChem, 2021, 5, 526–537 CrossRef CAS.
- N. D. Loewen, T. V. Neelakantan and L. A. Berben, Acc. Chem. Res., 2017, 50, 2362–2370 CrossRef CAS PubMed.
- D. B. Cluff, A. Arnold, J. C. Fettinger and L. A. Berben, Organometallics, 2018, 38, 1230–1235 CrossRef.
- D. W. Cunningham and J. Y. Yang, Chem. Commun., 2020, 56, 12965–12968 RSC.
- E. Hasegawa, S. Nakamura, K. Oomori, T. Tanaka, H. Iwamoto and K. Wakamatsu, J. Org. Chem., 2021, 86, 47 CrossRef PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the X-ray determinations, bond lengths and angles. CCDC 2086581. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08261b |
| This journal is © The Royal Society of Chemistry 2022 |