
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysine methyltransferase inhibitors: where we are now
Alessandra
Feoli†
,
Monica
Viviano†
,
Alessandra
Cipriano
,
Ciro
Milite
,
Sabrina
Castellano
and
Gianluca
Sbardella
 *
*
Department of Pharmacy, Epigenetic Med Chem Lab, University of Salerno, via Giovanni Paolo II 132, I-84084 Fisciano, SA, Italy. E-mail: gsbardella@unisa.it; Fax: +39-089-96-9602; Tel: +39-089-96-9770
First published on 13th December 2021
Abstract
Protein lysine methyltransferases constitute a large family of epigenetic writers that catalyse the transfer of a methyl group from the cofactor S-adenosyl-L-methionine to histone- and non-histone-specific substrates. Alterations in the expression and activity of these proteins have been linked to the genesis and progress of several diseases, including cancer, neurological disorders, and growing defects, hence they represent interesting targets for new therapeutic approaches. Over the past two decades, the identification of modulators of lysine methyltransferases has increased tremendously, clarifying the role of these proteins in different physio-pathological states. The aim of this review is to furnish an updated outlook about the protein lysine methyltransferases disclosed modulators, reporting their potency, their mechanism of action and their eventual use in clinical and preclinical studies.
 Alessandra Feoli | Alessandra Feoli received her Master of Science degree (MSc) in Pharmaceutical Chemistry in 2011 at the University of Salerno. In 2012, she spent 3 months at LMU (Munich) in the laboratory of Prof. Axel Imhof for a COST action. In 2016 she obtained her PhD in Pharmaceutical Sciences at the University of Salerno and, during her PhD program, she spent 6 months at NanoTemper Technologies (Munich) for improving her knowledge of MST and nanoDSF. Since 2017, she has been carrying out post-doctoral studies in the laboratory of Prof. Gianluca Sbardella at the University of Salerno, where she works on biochemical and biophysical techniques. |
 Monica Viviano | Monica Viviano received her Pharmaceutical Chemistry degree in 2009 at the University of Salerno. During her PhD, she spent 6 months in the Kappe Laboratory at Karl Franz University (Austria) to expand her knowledge in MW and continuous-flow techniques. In 2013, she obtained her PhD in Pharmaceutical Sciences at the University of Salerno, where she was a post-doc working on small-molecule modulators of epigenetic targets. Since October 2021, she has been an assistant professor at the Department of Pharmacy of the same university, focusing on advanced procedures for the synthesis and biological evaluation of small molecules of biological interest. |
 Alessandra Cipriano | Alessandra Cipriano received her Master of Science degree (MSc) in Pharmaceutical Chemistry and Technologies in 2016 at the University of Salerno. During her PhD program, she spent 6 months in the laboratory of Prof. Ciulli (University of Dundee, Scotland, UK) to expand her knowledge in the PROTACTM field. In 2020, she obtained her PhD in Drug Discovery and Development at the University of Salerno, where she is currently a post-doctoral researcher working on the design and synthesis of modulators of epigenetic targets. Her main research interests are the development of advanced procedures for the synthesis of small molecules that targeting epigenetic and non-epigenetic targets. |
 Ciro Milite | Ciro Milite received his Master of Science degree (MSc) in Pharmaceutical Chemistry in 2007 at the University of Salerno where he obtained his PhD in Medicinal Chemistry in 2011. During his PhD, he spent a period at the “SCRIPPS Research Institute” at the Barbas Laboratory to expand his knowledge in the field of organocatalyzed enantioselective chemical reactions. In 2018 he gained a position as assistant professor at the Department of Pharmacy, University of Salerno and, since 2021, he has been an associate professor. His research activity is focused on epigenetic drug discovery, spanning synthetic strategies, chemical biology, and biophysical techniques. |
 Sabrina Castellano | Sabrina Castellano obtained her Medicinal Chemistry degree in 1995 from the University of Trieste (IT) and her PhD at the University of Milan (IT). In 1999, she got a Postdoctoral Fellowship from the University of Trieste and in 2001 a position as an assistant professor. She has spent two years as a visiting scientist in Oyun Kwon's group at UCLA (USA). Since 2014 she has been an associate Professor at the University of Salerno (IT). In 2018, she obtained the National Scientific Habilitation to become full professor in medicinal chemistry. She has published more than 70 scientific peer-reviewed papers and holds one patent. Her main research interests are the broad area of medicinal chemistry, covering the design of small-molecule modulators of biological targets, the development of advanced procedures for the synthesis of compounds to be used in drug-discovery projects, and the application of modern biophysical approaches to study the different aspects of protein–ligand interactions. |
 Gianluca Sbardella | Gianluca Sbardella graduated in Pharmaceutical Chemistry and Technology at the University of Rome “La Sapienza” (Italy) in 1993 and received his PhD from the same university in 1997. In 2001 he joined the Department of Pharmacy of the University of Salerno, where since 2016 he has been Full Professor in Medicinal Chemistry. During his career, Gianluca was a Research Fellow at the Drug Chemistry Center of CNR (Italian National Research Center) in 1997, a Research Fellow at the Istituto Superiore di Sanità (ISS) – the Italian National Institute of Health – in 1998–1999, a postdoctoral fellow at the University of Rome “La Sapienza” in 2000, and a research associate at the University of Siena in 2000–2001. In 2004 he spent a sabbatical period as a visiting professor at the Department of Chemistry & Biochemistry of UCLA. After early interest in the discovery of antiviral drugs, since 2000 his research has regarded most aspects of epigenetic drug discovery, spanning synthetic strategies, medicinal chemistry, chemical biology, and biophysical techniques. He is currently the Past President of the Medicinal Chemistry Division of the Italian Chemical Society (Divisione di Chimica Farmaceutica della Società Chimica Italiana). |
Introduction
Despite evidence of histone methylation being reported in the 1960s,1 the first enzyme that is able to catalyse this modification was only identified forty years later.2,3From the chemical point of view, histone methyltransferases (HMTs) catalyse, using S-adenosyl-L-methionine (SAM, AdoMet) as a cofactor, the introduction of a methyl mark to the arginine or lysine residues of target proteins. Lysine methyltransferases (KMTs) are responsible for the mono-, di- and trimethylation of N ε-lysine in histone and non-histone substrates (Fig. 1).
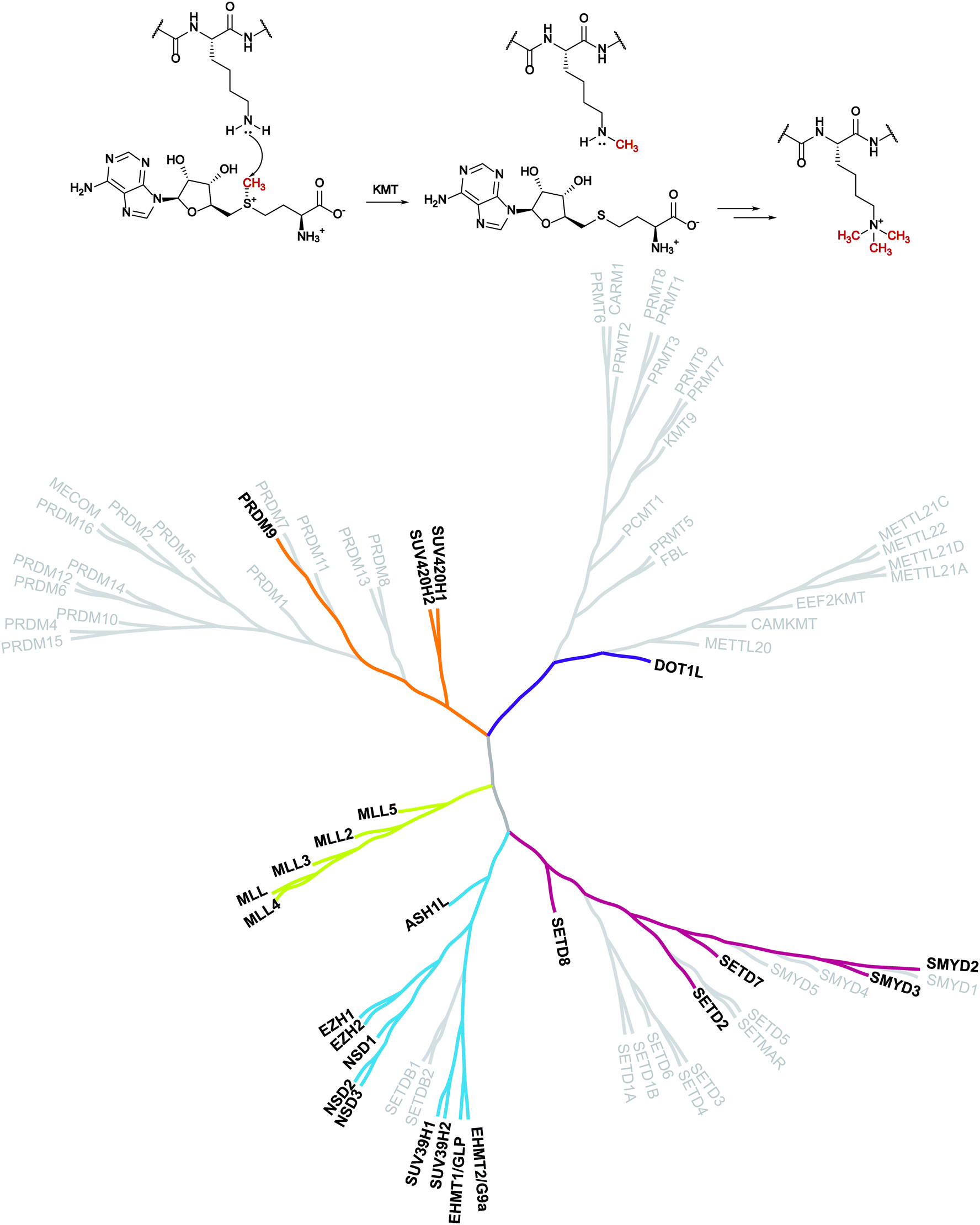 | ||
| Fig. 1 Lysine methylation catalysed by lysine methyltransferases and the KMT phylogenetic tree. In bold are depicted the proteins that are the object of this review for which inhibitors are known. | ||
The introduction of this methyl mark is a dynamic process that controls transcriptional regulation and gene expression and it has a pivotal role in cell fate determination. As a result, these proteins have been related to several disease conditions such as cancer and neurodegeneration.4–8 These findings make KMTs an appealing target for therapeutic intervention and have encouraged the identification of modulators of these enzymes.
The first group of KMTs includes around 55 members, which share a common evolutionarily conserved SET [Su(var),9 enhancer of zeste,10 trithorax11] domain that is responsible for the catalytic activity. The SET domain is approximately 130 amino acids long and the central region (i-SET) is preceded and followed by n-SET and c-SET domains, which interact with each other and are required for methyltransferase activity.12,13
The second group of KMTs is composed of non-SET-containing proteins that transfer the methyl group, exploiting a catalytic portion which looks like that of DNA and protein arginine methyltransferases (DNMTs and PRMTs).14
In the past decade, several reviews regarding these proteins have been published, some of them focusing on structural features of the proteins, some on their involvement in different pathologies, and others reporting the currently available inhibitors for some of the proteins.15–23
In this review, we report an updated overview of the current literature regarding selected KMTs for which high-quality chemical probes have been reported. For each of these proteins, we provide a summary of their biological functions, including the implications in pathological states, and describe the corresponding inhibitors on the basis of their mechanism of action. For selected inhibitors (at least one for each protein), structural insights into the binding mode are also provided.
DOT1L
The protein DOT1 was identified for the first time in a yeast screen (Saccharomyces cerevisiae) performed for searching genes that, when overexpressed, disrupt telomeric silencing.24 Homolog genes of DOT1 (DOT1-like or DOT1L) present 88% homology in their amino acid sequence and they have been found in different species such as mammals, drosophila, and protozoa.25 DOT1L (disruptor of telomeric silencing 1-like) is the most studied non-SET-containing methyltransferase that is responsible for the mono-, di- and trimethylation of lysine 79 of histone H3 (H3K79) in yeasts and mammals, where it is associated with transcriptionally active genes. DOT1L represents the sole epigenetic protein responsible for this modification; indeed, the knockout of its gene in different species causes the complete loss of H3K79 methylation.26–28 DOT1L catalyses the transfer of a methyl group from the cofactor SAM to the H3 substrate thanks to the presence of an AdoMet binding motif, which resembles that of DNMTs and PRMT1.14DOT1L can methylate histone H3 only in the presence of DNA, while no activity has been detected on free histone H3, meaning that only parental histones will be methylated by this protein.26
DOT1L is involved in different processes, including physiological as well as pathological ones. From the physiological point of view, this protein has a key role in transcriptional activation, promoting the formation of heterochromatin. In addition, it is involved in embryonic development, in the regulation of haematopoiesis, in cell cycle progression and in DNA repair after damage.27
Moreover, DOT1L together with other proteins such as AF10, AF17, AF9, ENL, Skp1, TRRAP and β-catenin, forms the multimeric complex “DotCom”, which has a specific enzymatic activity as it can catalyse H3K79 di- and trimethylation.29 Considering that three of these proteins (AF9, ENL and AF10) are fusion partners with the mixed-lineage leukaemia (MLL) genes, there is a strict link between their chromatin-reading effect and DOT1L activity in leukemogenesis.30 Indeed, it has been demonstrated that aberrant transcriptional activation via H3K79 methylation by DOT1L is linked to the insurgence of MLL, a subtype of childhood leukaemia that derives from oncogenic chromosomal rearrangements of the MLL genes.31 Thus, although the expression of DOT1L is not modified in leukaemia, its altered methyltransferase activity is a consequence of the chromosomal translocations observed in MLL. However, the precise mechanism by which DOT1L contributes to the gene-activation process has not been fully elucidated and it requires further investigation.
DOT1L inhibitors
Because of the importance of the protein DOT1L in many physiological and pathological processes, different inhibitors of this protein have been developed over the years and they can be classified, based on their structure, into two main classes: nucleoside inhibitors (Fig. 2) and non-nucleoside inhibitors (Fig. 4).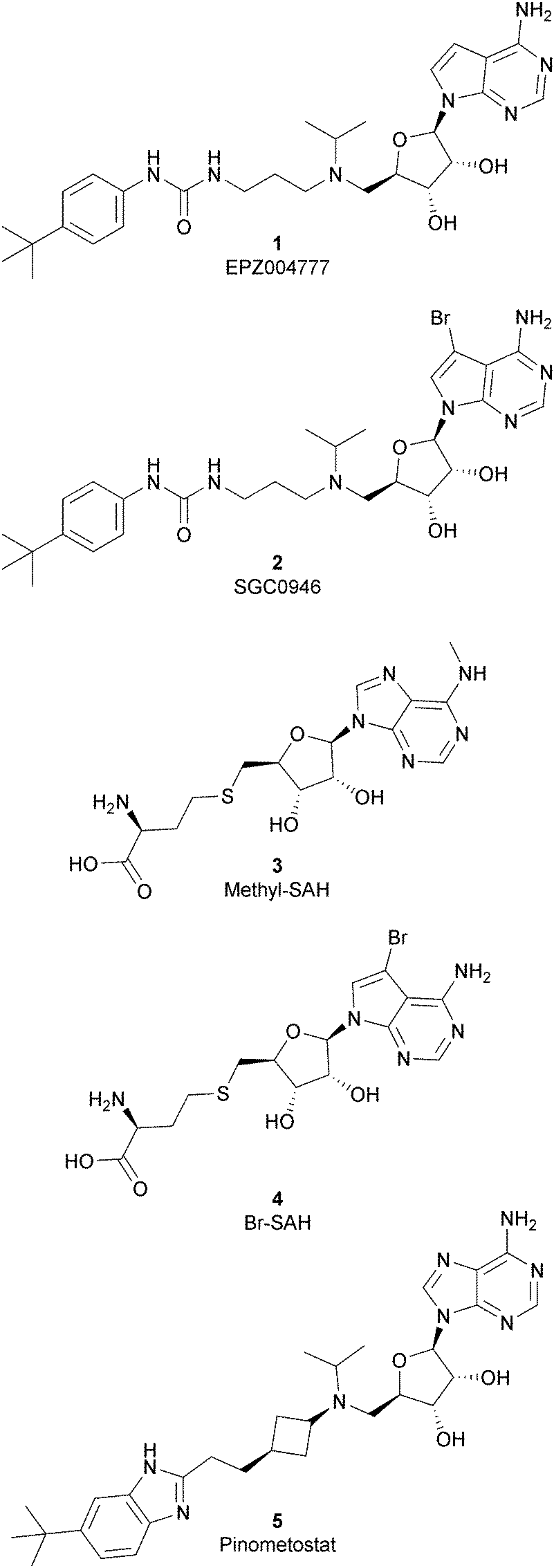 | ||
| Fig. 2 Nucleoside inhibitors of DOT1L. | ||
Nucleoside inhibitors
As the DOT1L mechanism of action is based on the presence of an AdoMet binding motif, early efforts to find inhibitors were focused on the design of small molecules that are able to occupy the same binding pocket of the cofactor SAM. Therefore, the first class of DOT1L inhibitors is characterized by structural features that mimic the SAM molecule. 1 (EPZ004777) was the first SAM competitor able to strongly inhibit DOT1L (IC50 of 0.4 nM), showing in vitro and in vivo antiproliferative activity in MLL-re-arranged leukemic cells (MV4-11).In addition, selectivity assays against a panel of 9 other protein methyltransferases (PMTs) showed no inhibition at concentrations up to 50 μM.32 Despite being one of the strongest DOT1L inhibitors, 1 cannot be used in therapy due to its low half-life, poor pharmacological properties, and side effects. Moreover, even if the etiology of the effect is not clear, a significant inhibition of this enzyme by 1 was observed to be responsible, in a 14 day treatment, for the increasing number of white blood cells, neutrophils, monocytes and lymphocytes in mice.32,33
Starting from 1, different analogues were synthesized, such as 2 (SGC0946), characterized by a bromide on the adenosine ring, which was more active than the parent compound, showing an IC50 of 0.3 nM in a cell-free assay, and an IC50 of 8.8 nM and 2.6 nM in MCF10a and A431 cells, respectively.34 Several compounds based on the SAH (S-adenosylhomocysteine) structure also belong to the first class of DOT1L inhibitors: some of them were identified by alkylation of the primary amine of the adenine ring of SAH (methyl-SAH, 3)35 or by adding a bromide on the adenosine ring (BrSAH, 4), but these modifications led to a deep decrease in DOT1L inhibition and selectivity with respect to the parent compound.36
In 2013, 5 (pinometostat) was identified as a potent (Ki ≤ 80 pM) and selective DOT1L inhibitor, showing a 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold selectivity over other PMTs. It exhibits strong inhibitory activity towards DOT1L in cells and is able to inhibit MV4-11 proliferation with an IC50 value of 3.5 nM.37 However, this molecule has low oral bioavailability in mouse, rat, and dog;38 therefore, it is necessary to carry out intraperitoneal administration. A representation of the binding mode of compound 5 obtained using the LigPlot+ program39 is depicted in Fig. 3. The X-ray crystal structure of the inhibitor in complex with DOT1L (PDB 4HRA) shows that 5 is able to occupy the SAM binding site and to induce the opening of a hydrophobic pocket beyond the amino acid portion of SAM. Specifically, the adenosine moiety binds in a similar manner as in SAM structures. The positively charged 5′-amino group is responsible for a hydrogen bond with the carbonyl moiety of Gly163. On the other hand, the benzimidazole nitrogen engages with the carboxylate side chain of Asp161.
000-fold selectivity over other PMTs. It exhibits strong inhibitory activity towards DOT1L in cells and is able to inhibit MV4-11 proliferation with an IC50 value of 3.5 nM.37 However, this molecule has low oral bioavailability in mouse, rat, and dog;38 therefore, it is necessary to carry out intraperitoneal administration. A representation of the binding mode of compound 5 obtained using the LigPlot+ program39 is depicted in Fig. 3. The X-ray crystal structure of the inhibitor in complex with DOT1L (PDB 4HRA) shows that 5 is able to occupy the SAM binding site and to induce the opening of a hydrophobic pocket beyond the amino acid portion of SAM. Specifically, the adenosine moiety binds in a similar manner as in SAM structures. The positively charged 5′-amino group is responsible for a hydrogen bond with the carbonyl moiety of Gly163. On the other hand, the benzimidazole nitrogen engages with the carboxylate side chain of Asp161.
 | ||
| Fig. 3 Flat representation (generated using LigPlot+ from PDB ID 4HRA) showing contacts between compound 5 and active-site residues in DOT1L. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
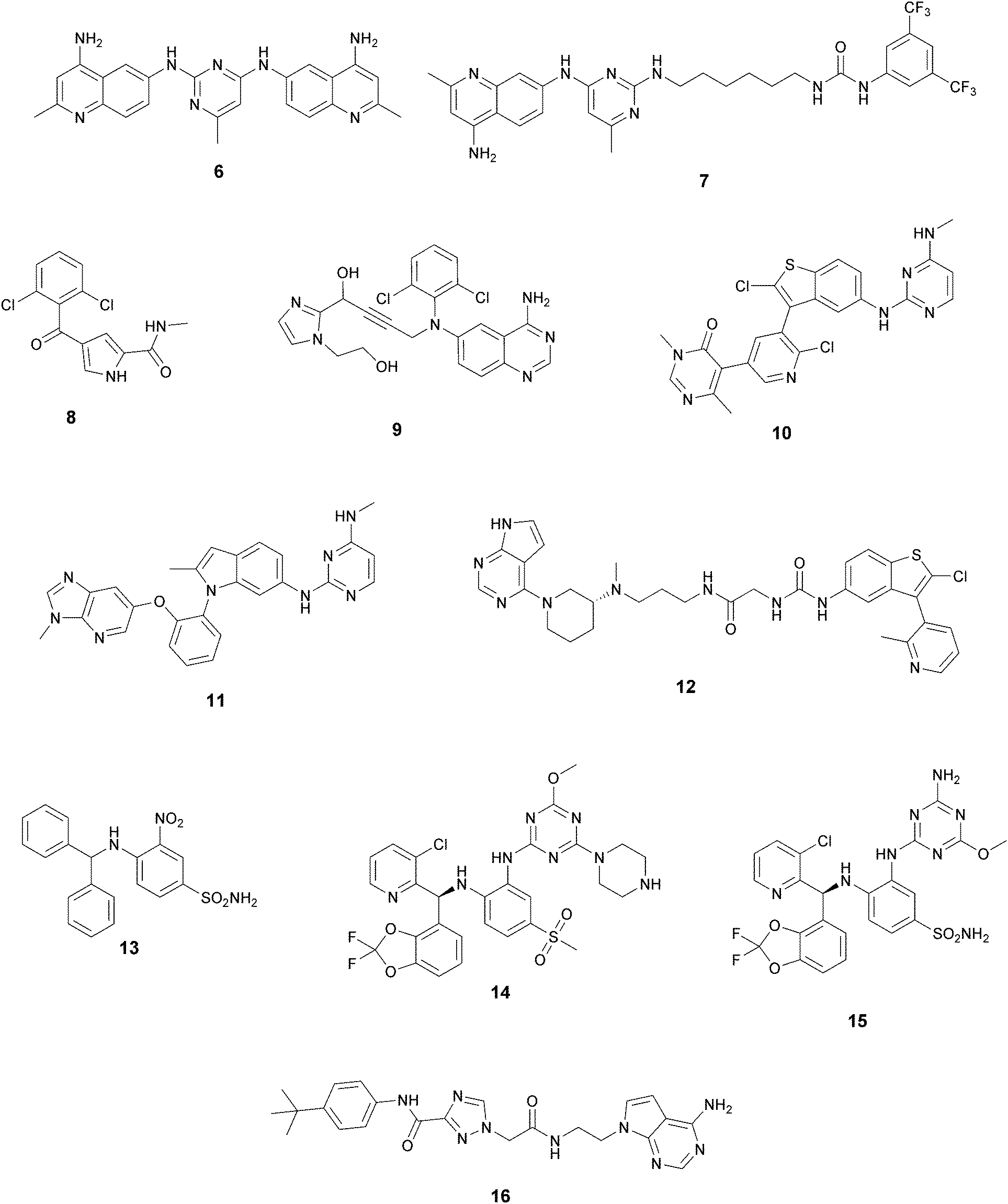 | ||
| Fig. 4 Non-nucleoside inhibitors of DOT1L. | ||
Similar to other SAM-mimicking inhibitors, 5 has a low half-life and the effects are visible only after more than 7 days of treatment. However, preclinical experiments evidenced clear tumour regression and little or no re-growth for more than 30 days after cessation of treatment with the compound. 5 was subjected to phase I clinical trials against relapsed or refractory acute myeloid leukaemia in children (NCT02141828)40 and adults (NCT01684150),41 and two different clinical trials are now evaluating the combination of 5 with azacytidine, cytarabine and daunorubicin (NCT03724084;42 NCT0370129543) to improve cell killing and tolerability in vivo.
Non-nucleoside inhibitors
In recent years, several approaches to discover new DOT1L inhibitors that overcome the pharmacokinetic (PK) limitations of SAM-related compounds have been developed. Luo and co-workers identified the first non-nucleoside DOT1L inhibitor through structure-based virtual screening (SBVS):44 the bisaminoquinoline 6 showed high inhibitory activity towards DOT1L, but its poor cell permeability was responsible for the weak activity against MV4-11 proliferation. In order to improve the permeability, the same research group developed compound 7, which showed strong DOT1L inhibition (IC50 of 3.55 μM) and could effectively down-regulate both the level of H3K79 methylation and the overexpression of leukemogenic genes in MV4-11, via qRT-PCR and western blot studies.45In 2016, a fragment-based screening (FBS) approach led to the identification of the weak inhibitor 8 (IC50 of 320 μM), whose co-crystallization with DOT1L and structure-based ligand optimization yielded 9 with nanomolar activity (IC50 of 14 nM). X-ray crystallography studies confirmed that these new SAM-competitive compounds are not able to interact within the SAM binding site, but with an induced binding pocket adjacent to the binding site of the cofactor: upon the binding of compound 9, a conformational change of the enzyme did not allow anymore the binding of the SAM cofactor that is fundamental for the activity of DOT1L.46 Exploiting again the FBS approach, the same research group identified compounds 10 and 11 as potent and selective DOT1L inhibitors, where both interact with the same binding pocket and had IC50 values of 1.4 and 0.4 nM, respectively.47 Cellular assays showed that these compounds are able to significantly reduce dimethylated H3K79 levels (IC50 of 23 and 16 nM, respectively) and can inhibit the proliferation of MV4-11 leukaemia cells in the nanomolar range, whilst maintaining good PK properties, a moderate half-life and good oral bioavailability.
In 2017, Mobits and co-workers used a combination of FBS, virtual screening (VS) and fragment-linking approaches, which led to the discovery of compound 12. This derivative was obtained by connecting two different fragments: a non-nucleoside fragment, which mimics the key interactions of SAM bound to DOT1L, and an induced back pocket binder. This new inhibitor showed a Ki in the very low picomolar range and a favourable selectivity profile against a panel of 22 KMTs and PRMTs with no inhibitory activity up to 50 μM. Moreover, in a head-to-head comparison in cellular assays, 12 worked equal to or better than 5, suppressing H3K79 dimethylation (IC50 of 3 nM) both in HeLa and Molm-13 cells and inhibiting proliferation of MV4-11 cells.48
The binding interactions of compound 12 with the enzyme are represented in Fig. 5. The X-ray crystal structure of the inhibitor bound to the enzyme (PDB 5MW4) confirmed the expected binding mode: the NH of the pyrrolopyrimidine core serves as a hydrogen-bond donor, interacting with the side chain of Asp222, while the adjacent nitrogen acts as a hydrogen-bond acceptor for the backbone NH of Phe223. An interaction of the pyridine nitrogen with Ser140 is also observed.
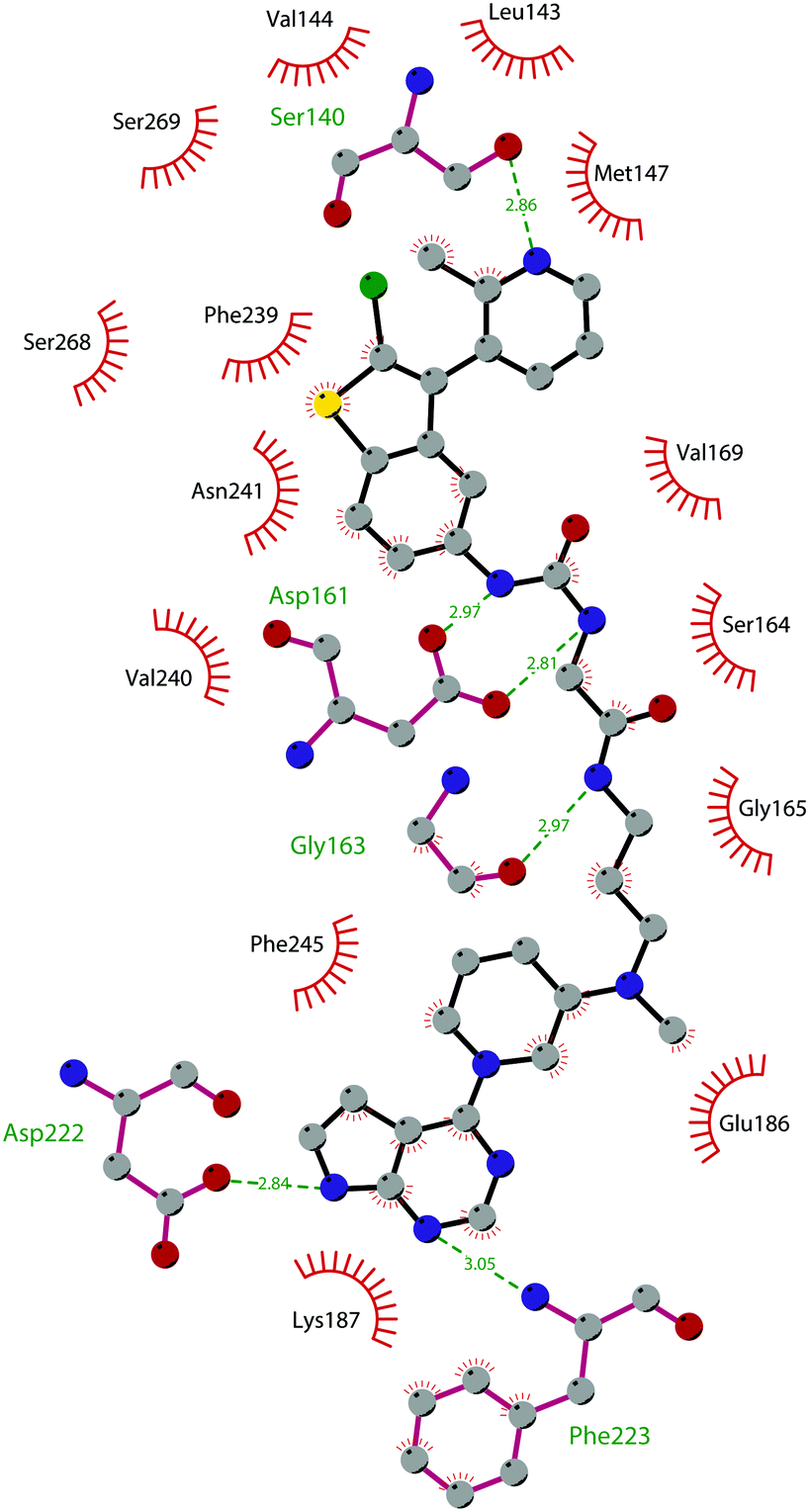 | ||
| Fig. 5 Flat representation (generated using LigPlot+ from PDB ID 5MW4) showing contacts between compound 12 and active-site residues in DOT1L. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
Two years later, Stauffer and co-workers exploited the structure-guided optimization of a high-throughput screening (HTS) hit (13, IC50 of 8.5 μM) to obtain new DOT1L inhibitors (14 and 15) with sub-nanomolar potency (IC50 of 0.11 nM and 0.17 nM, respectively). Moreover, selectivity testing for these compounds against a selected panel of histone methyltransferases showed no inhibition below 5 μM. Despite the good results, a limited efficacy in a mouse tumour xenograft model was obtained, illustrating the difficulty of reaching the good level of DOT1L inhibition in vivo required to effectively suppress MLL-fusion tumour growth.49
More recently, Gibbons and co-workers applied a VS strategy on a nucleoside-focused library of 1200 compounds, adenosine-based as well as small molecules with ‘nucleoside-like’ scaffolds, which is different from the DOT1L clinical candidates and other reported inhibitors. Using structure-based design and molecular modelling studies, compound 16 was identified, showing an IC50 of 1.0 μM and very modest activity against DNMT1, PRMT3, PRMT5 and PRMT8, with selectivity for DOT1L of 250, 130, 175, and >250-fold, respectively. This compound exhibited dose-dependent cellular activity against the proliferation of murine MLL-AF9 transformed cells, accompanied by a dose-dependent decrease of H3K79 dimethylation and induction of cell differentiation. Although this new inhibitor is only modestly potent, it represents a new chemical probe with a unique non-nucleoside scaffold that is able to bind and compete with the SAM-binding site of DOT1L, which could be the starting point for further optimizations.50
G9A/GLP
G9a and the related G9a-like protein (GLP) are two proteins with a homology of around 80% in the sequence of their catalytic domain. G9a and GLP are reported also as euchromatin histone lysine methyltransferases (EHMTs), a family of proteins that regulate gene transcription. G9a is also known as lysine methyltransferase 1C or euchromatic histone lysine N-methyltransferase 2 (EHMT2) while GLP is reported as lysine methyltransferase 1D or euchromatic histone methyltransferase 1 (EHMT1).51,52 G9a and GLP are mainly responsible for the mono- and dimethylation of H3K9, but they are also able to catalyse the methylation of other histone substrates (H3K27, H3K56 and H1),53–57 as well as non-histone proteins, including p53,58 sirtuin59 and G9a itself. In particular, autocatalytic G9a methylation represents a mechanism of gene silencing.60Dimethylated H3K9 is associated with repressed euchromatic regions of the genome, meaning that G9a and GLP repress euchromatic gene transcription through H3K9 dimethylation of specific target genes. In addition, both G9a and GLP have been found to interact with several transcriptionally repressive protein complexes.61
From the structural point of view, G9a and GLP are characterized by the presence of a C-terminal SET domain, a large N-terminal region for a nuclear localization signal (with a glutamate-rich region and a cysteine-rich ring finger-like domain) and a middle domain characterized by 6 ankyrin repeats as reader domains.62,63
G9a and GLP can exist as homomeric and heteromeric complexes. The heteromeric complex is functional in vivo so it was initially believed that this complex was necessary for the catalytic activity. Subsequent studies suggested that G9a and GLP proteins have independent physiological functions.64
G9a and GLP are widely expressed in several organs and tissues, as they have multiple biological roles. Mainly, they are linked with both facultative and constitutive heterochromatin formation and transcriptional repression. They are also involved in the development of the brain, as G9a knockout led to embryonic lethality caused by growth defects.52,65
On the other hand, aberrant activity and expression of G9a and GLP is linked to the development of diseases, mainly cancer.66 Several pieces of experimental evidence correlate G9a and GLP up-regulation with metastatic disease and poor prognosis in various types of human cancer. Consistently, knockdown of G9a reduces tumour cell growth in in vitro experiments and the invasiveness of cancer.67 In addition, dysregulation of G9a and GLP is also associated with inflammatory and neurodegenerative disorders.68,69
G9A/GLP inhibitors
Considering the crucial role of G9a and GLP in many biological processes, over the past decade several G9a and GLP inhibitors have been developed. Based on their mode of action, these compounds can be categorized into two different group: SAM-competitive inhibitors (Fig. 6) and substrate-competitive inhibitors (Fig. 7).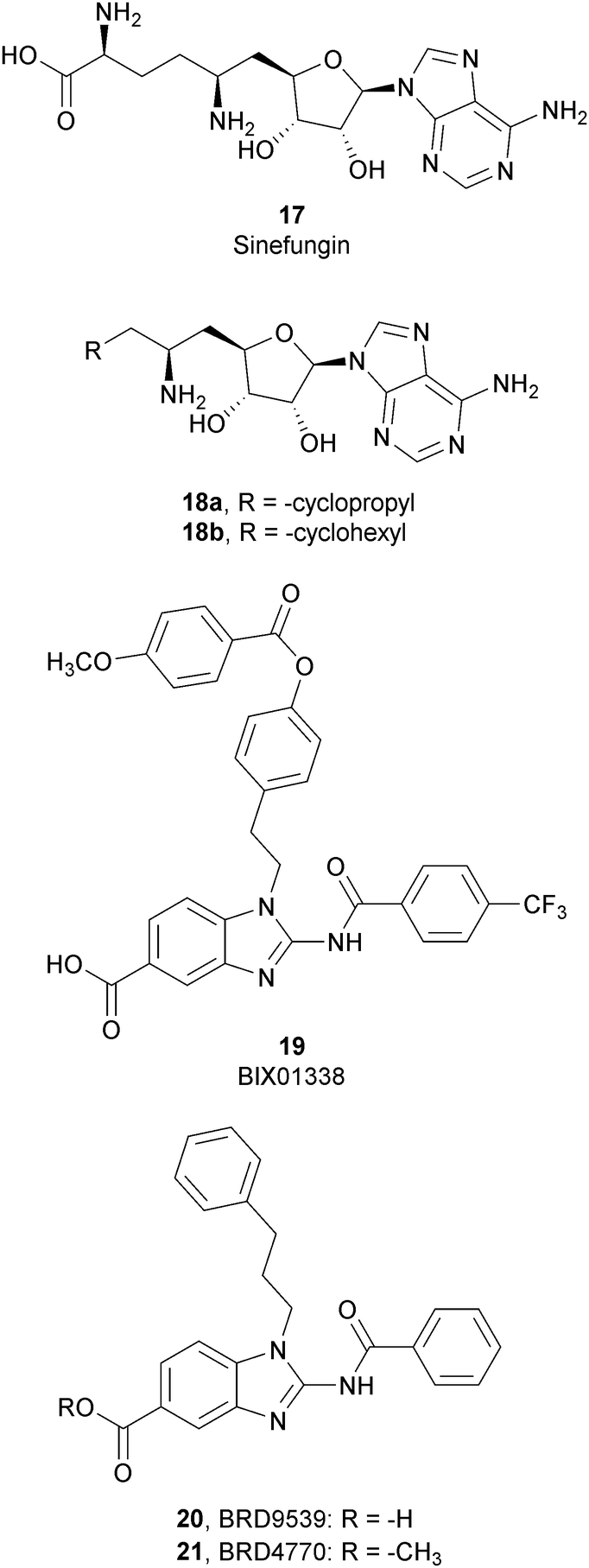 | ||
| Fig. 6 G9a SAM-competitive inhibitors. | ||
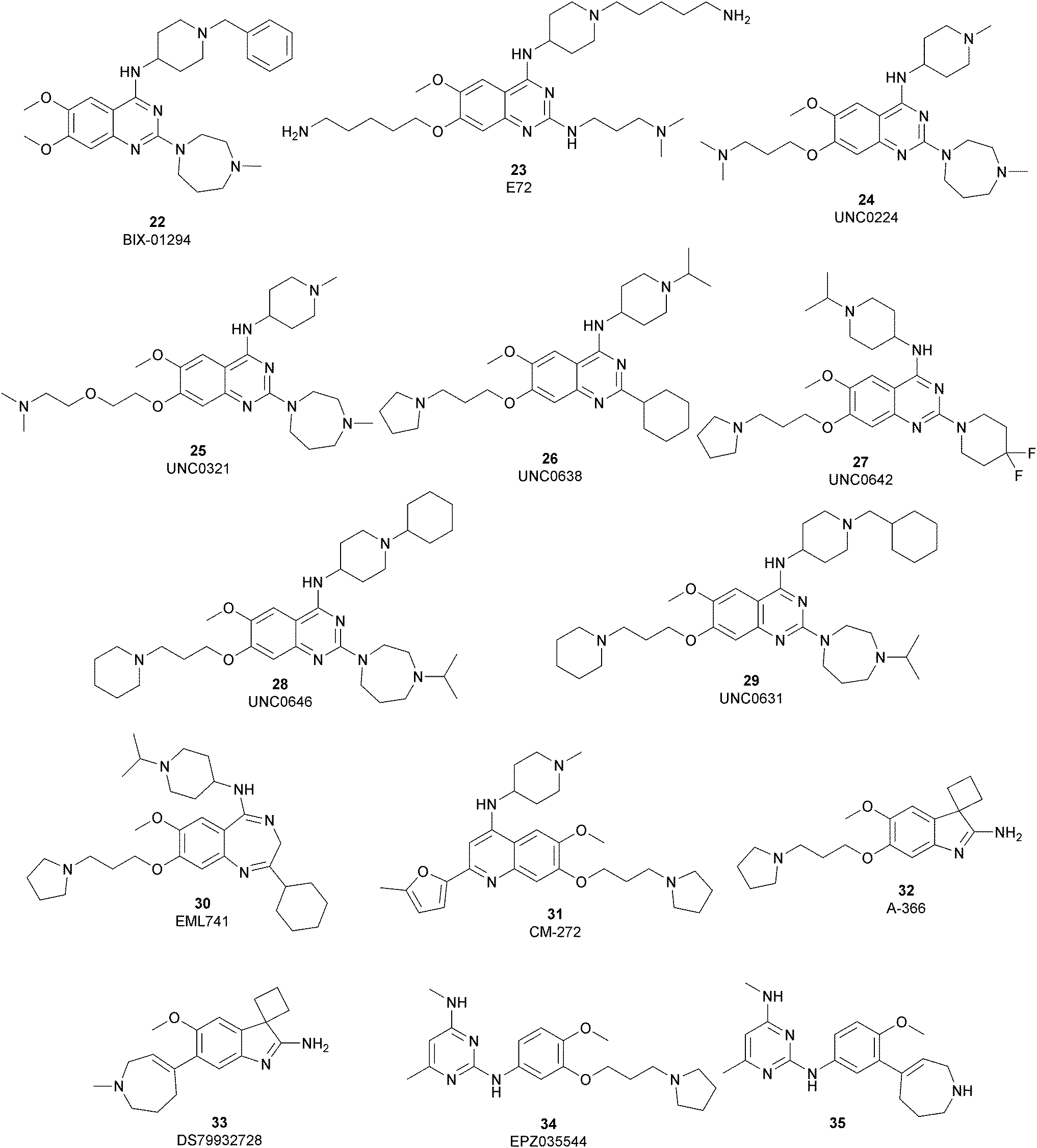 | ||
| Fig. 7 G9a substrate-competitive inhibitors. | ||
SAM-competitive inhibitors
SAM-competitive inhibitors bind the cofactor SAM binding pocket of G9a, preventing histone methylation and, consequently, G9a activity. Among these compounds, 17 (sinefungin), a natural product structurally correlated to SAM, inhibits GLP/G9a with an IC50 of 28.4 and 30.1 μM, respectively, although it displays no selectivity over a wide range of methyltransferases.Modifications at the amino acid moiety led to the identification of a series of cycloalkyl-substituted analogues such as 18a,b. These compounds showed a comparable or better potency than sinefungin (IC50 of 21.8 and 39.6 μM for 18a and IC50 of 1.5 and 1.6 μM for 18b on GLP and G9a, respectively). Although the selectivity was evaluated on a limited number of methyltransferases, these compounds appear to be more selective than sinefungin, i.e., 18a showed no inhibition on PRMT1 and 18b showed a weak inhibition of DNMT1 and no activity on PRMT1 and SET7/9.70,71
Starting from compound 19 (BIX01338), identified via HTS as a good G9a inhibitor (IC50 of 4.7 μM) but with poor selectivity and cellular activity, other SAM-mimetic benzimidazoles were designed, leading to the identification of 20 (BRD9539).72,73 This compound has an IC50 value of 6.3 μM against G9a but is inactive in cell-based assays, probably due to its low cell permeability. Conversely, the methyl ester analogue 21 (BRD4770) displayed a higher cellular activity, although its reduced biochemical potency compared to 20. It was hypothesized that 20 is the potentially active form of 21 in cells, considering that the methyl ester group could be easily hydrolysed in cells acting as a “prodrug”. 21 reduced cellular levels of di- and trimethylated H3K9 with low cellular toxicity and inhibited both anchorage-dependent and -independent proliferation in the pancreatic cancer cell line PANC-1. Despite their interesting profiles, these compounds suffer from low selectivity, considering that 20 also inhibits PRC2 activity, with a similar potency to G9a inhibition as well as NSD2, even if the effects were observed only at 40 μM.73
The low selectivity is one of the major drawbacks of this class of inhibitors; consequently, SAM-competitive inhibitors have not been further investigated.
Substrate-competitive inhibitors
The most potent, selective, and widely used chemical probes of G9a/GLP are substrate-competitive inhibitors. These compounds specifically occupy the substrate site in G9a, gaining a better selectivity against the protein target over SAM-competitive inhibitors. In 2007, via the HTS of approximately 125000 compounds, Kubicek and co-workers identified the quinazoline 22 (BIX-01294) as a G9a/GLP inhibitor. 22 inhibits G9a and GLP (IC50 of 1.9 μM and 0.7 μM, respectively) in an uncompetitive manner with SAM. 22 reduces dimethylated H3K9 levels in different G9a target genes, promoting autophagy-dependent cell death and reducing cell proliferation in different types of cancer cell lines, such as colorectal, breast, bladder cancer and glioma cells.74–76 Insertion of the 5-aminopentyloxy moiety on 22 to mimic the lysine side chain, yielded 23 (E72), which retains a similar activity to 22 but displays a very low cytotoxicity.77 The 2,4-diamino-6,7-dimethoxyquinazoline core was identified as the key pharmacophore, and structure–activity relationship (SAR) investigations allowed the identification of several derivatives. 24 (UNC0224) is an efficient and selective G9a/GLP inhibitor (IC50 of 15 nM and 20 nM, respectively), whose X-ray crystal structure in complex with G9a showed that the 7-dimethylaminopropoxy group is responsible for important interactions with the lysine-binding channel of this enzyme.78
In the quinazoline scaffold, replacement of 7-dimethylaminopropoxy group with an ethoxyethyl chain resulted in 25 (UNC0321) (Ki of 63 pM), which is the first reported G9a inhibitor with picomolar potency, although this had a lower cellular activity compared with 22.79 Despite their high in vitro potency, the cellular activity of the above mentioned quinazoline-based compounds is limited, considering that the concentration required to produce functional effects is close to the toxic concentration and that they are characterized by poor druglikeness properties.
In order to overcome these drawbacks, further optimization of the quinazoline class of inhibitors led to the discovery of 26 (UNC0638), a cell-permeable G9a/GLP inhibitor (IC50 < 15 nM and 19 nM, respectively) with high selectivity over a wide range of other lysine and arginine methyltransferases, as well as over non-epigenetic targets. The X-ray crystal structure of 26 bound to the protein and the SAH (PDB 3RJW) provided structural information about the binding mode, confirming its substrate-competitive mode of action. As depicted in Fig. 8A, the compound engages the substrate binding pocket, inserting the 7-(3-pyrrolidin-1-yl)propoxy side chain into the lysine-binding groove. Moreover, the hydrogen of the amino group at the 4-position of the quinazoline ring establishes an important hydrogen bond with Asp1083, as well as the nitrogen of the quinazoline being hydrogen bonded with Asp1088.
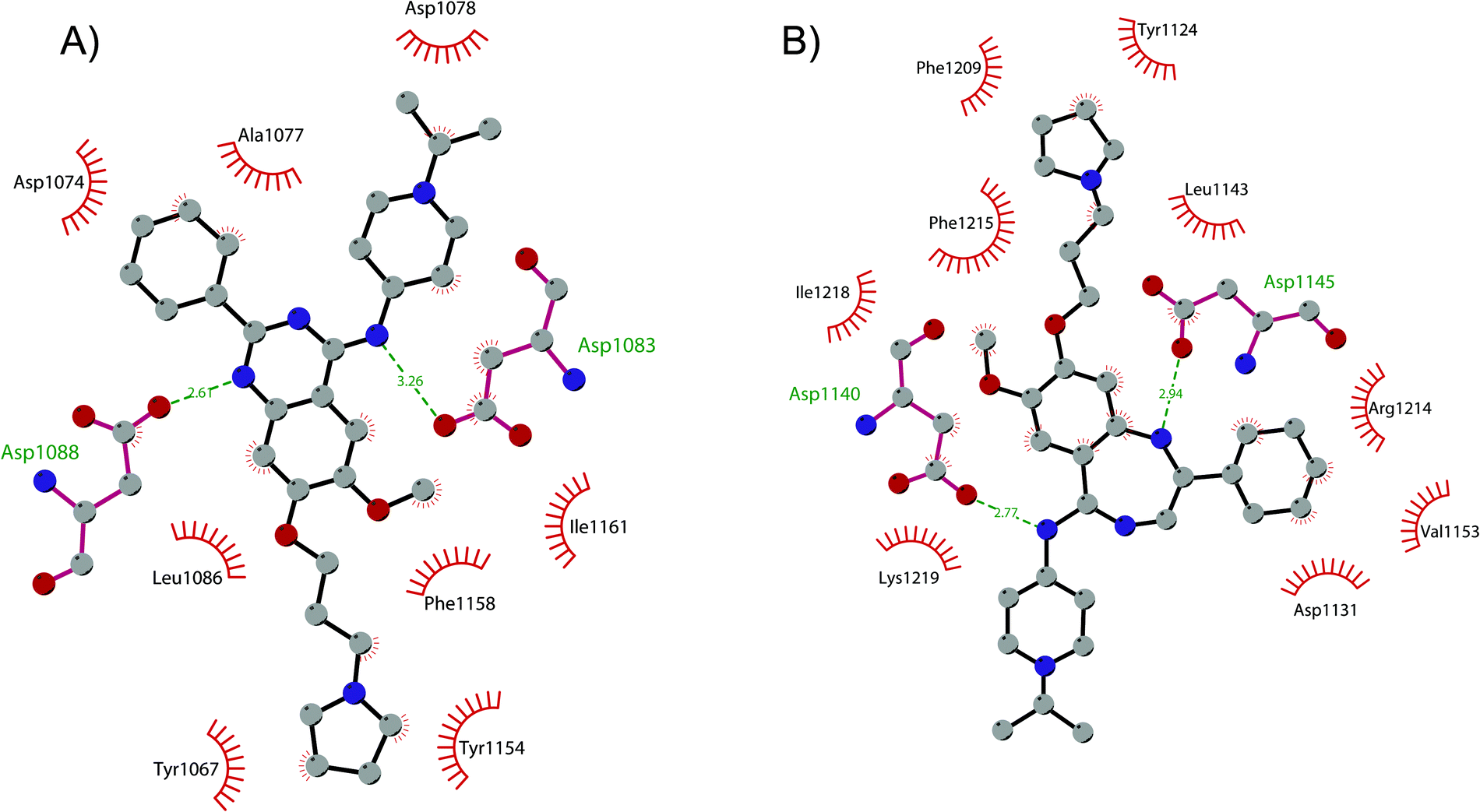 | ||
| Fig. 8 Flat representation (generated using LigPlot+) showing contacts between (A) compound 26 and active-site residues in G9a (from PDB ID 3RJW) and (B) compound 30 and active-site residues in GLP (from PDB ID 6MBP). Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compounds. | ||
26 potently reduces levels of dimethylated H3K9 in a variety of cell lines and with a lower cellular toxicity than the previously reported compounds. However, the poor PK properties limited its employment in animal studies.80 The hypothesis that the cyclohexyl group at position 2 of the quinazoline core could be related to the poor metabolic stability, and supported its replacement with a 4,4-difluoropiperidine moiety, yielding 27 (UNC0642). This new derivative maintains the high in vitro potency (IC50 < 2.5 nM) and cellular activity and with a lower toxicity than the parent compounds but also exhibits improved in vivo PK properties.81 Other related compounds are 28 (UNC0646) and 29 (UNC0631), potent G9a inhibitors (IC50 of 6 and 4 nM, respectively) with excellent potency in a variety of cell lines.82
Apart from the quinazoline derivatives, a limited number of G9a/GLP inhibitors have been identified. Exploiting the scaffold-hopping approach, the quinazoline moiety of 25 was replaced with a benzodiazepine scaffold, resulting in the identification of 30 (EML741). This derivative showed high in vitro (IC50 of 23 nM) and cellular potency, improved selectivity against other methyltransferases and low cellular toxicity. Notably, 30 is also able to inhibit DNMT1, synergizing with G9a inhibition in reducing cellular proliferation. The major interactions of 30 with the target protein are depicted in Fig. 8B. The co-crystal structure of the compound in complex with the catalytic SET domain of GLP in the presence of SAH (PDB 6MBP) showed that the inhibitor and the cofactor are located in two opposite binding sites. Specifically, 30 is bound to the acidic surface of the substrate-binding pocket, inserting the pyrrolidine moiety into the lysine-binding channel. The benzodiazepine scaffold is lodged with Arg1214, Ile1218, and Leu1143 forming van der Waals interactions. Also, the amino group at the 4-position and the protonated nitrogen of the N-isopropylpiperidine group are hydrogen bonded with two negatively charged residues, Asp1140 and Asp1145.83
Starting from the quinazoline scaffold, its bioisosteric replacement with the quinoline one led to 31 (CM-272), a dual small-molecule inhibitor that is able to target simultaneously the methyltransferase activity of G9a and DNMTs. In fact, G9a interacts with DNMT1 to coordinate DNA and histone methylation, promoting transcriptional silencing of the target genes. The dual small-molecule 31 inhibits cellular proliferation, promotes apoptosis, and prolongs the survival of haematological neoplasia xenogeneic models, offering a novel approach for treating several human cancers with poor prognoses.84 Through a library screen of a subset of compounds, the spiroindole 32 (A-366) was identified as a potent and selective inhibitor (IC50 of 3.3 nM and 38 nM for G9a and GLP, respectively).85 The compound occupies the lysine-binding site of G9a and is peptide-competitive and SAM-uncompetitive. Therefore, it is not very surprising that, two years after its identification as a G9a/GLP inhibitor, compound 32 was also reported as a sub-micromolar inhibitor of the epigenetic methyl reader protein Spindlin1 (IC50 of 186.3 nM), which is also able to engage the target in cells (although only at 100 μM).86,87 In fact, the capability of the compound to bind to the same pocket of the lysine substrate of the methyltransferase makes it also able to bind to the reader protein that recognizes the methylation reaction product.
The introduction of a tetrahydroazepine in 32 led to the identification of 33 (DS79932728), which retained the potency on G9a (IC50 of 4.50 nM and 33.9 nM for G9a and GLP, respectively), showed good metabolic stability, and the capability to induce γ-globin in monkeys, with interesting implications in the study of β-thalassemia.88
Beyond the well-characterized quinazoline structures, interest in the identification of new valuable chemical scaffolds as G9a inhibitors led to consideration of the diaminopyrimidine core of 34 (EPZ035544), a reported G9a/GLP inhibitor (IC50 of 16.9 and 185 nM, respectively) with low PK profile, as an optimizable novel template. Insertion of the tetrahydroazepine group as a side chain, used successfully in the design of 33, allowed the identification of 35, with increased potency on G9a/GLP (IC50 of 4.3 and 32.3 nM, respectively) compared with 34 and an improved PK profile in mice (the oral bioavailability increased up to 40%).89
SUV39H1 and SUV39H2
The suppressors of variegation 3–9 homolog 1 (SUV39H1, also known as KMT1A) and 2 (SUV39H2, also known as KMT1B) are members of the SUV39 subfamily of KMTs and are localized in the nucleus. They contain a highly conserved C-terminal SET domain that is responsible for the di- and trimethylation of H3K9, which is associated with gene silencing and heterochromatin formation, while in the N-terminal region there is a chromodomain that is fundamental for the catalytic activity.90,91 SUV39H2 has a homology sequence of 59% with SUV39H1; however, it differs from this last protein by the presence of a basic histone H1-like N-terminus.92SUV39H1 preferentially methylates the monomethylated H3K9, exploiting the binding of the chromodomain to the methylated residue, although it remains unclear if SUV39H2 prefers the methylated or unmethylated substrate H3.93,94 In addition, SUV39H2 methylates histone H2AX on Lys134, which is a critical modification for the production of phosphorylated H2AX (γ-H2AX) in cancer cells.95
SUV39H1 and SUV39H2 play an essential role in several biological mechanisms, such as transcriptional repression and epigenetic silencing in euchromatin, heterochromatin organization, the regulation of telomere length in mammalian cells, and embryonic development.96,97
Knockout of both SUV39H1 and SUV39H2 can lead to abnormally long telomeres, due to the lack of their enzymatic activities of targeting heterochromatin,97 and it causes prenatal lethality, accompanied by a global reduction in trimethylated H3K9,98 as well as a reduction of hematopoietic stem cell function.99
Despite sharing the same enzymatic activity, SUV39H1 and SUV39H2 might play different roles in cancer, probably due to their different substrate preference. In fact, SUV39H1 is considered as a tumour-suppressor gene because of its cell-proliferation-suppressing activity, while for SUV39H2 a tumour-promoting activity has been reported.100 In particular, alterations as well as an overexpression of SUV39H2 can be found in different cancers, indicating that SUV39H2 is involved in carcinogenesis, invasion, and metastasis of different kinds of solid tumours and haematological malignancies.85,101 Dysregulation of SUV39H2 has also been reported in several other diseases, such as hereditary nasal parakeratosis, which is caused by a mutation of SUV39H2 in dogs.102 Moreover, it has been reported that alterations in the regulation of SUV39H2 are also associated with autoimmune diabetes103 and steatohepatitis in mice.104
SUV39H1 and SUV39H2 inhibitors
As discussed above, SUV39H1 and SUV39H2 have emerged as critical proteins involved in the insurgence of several pathological states, mainly cancer. In spite of great efforts having been spent toward the development of modulators of both these proteins, only three inhibitors have been reported so far (Fig. 9).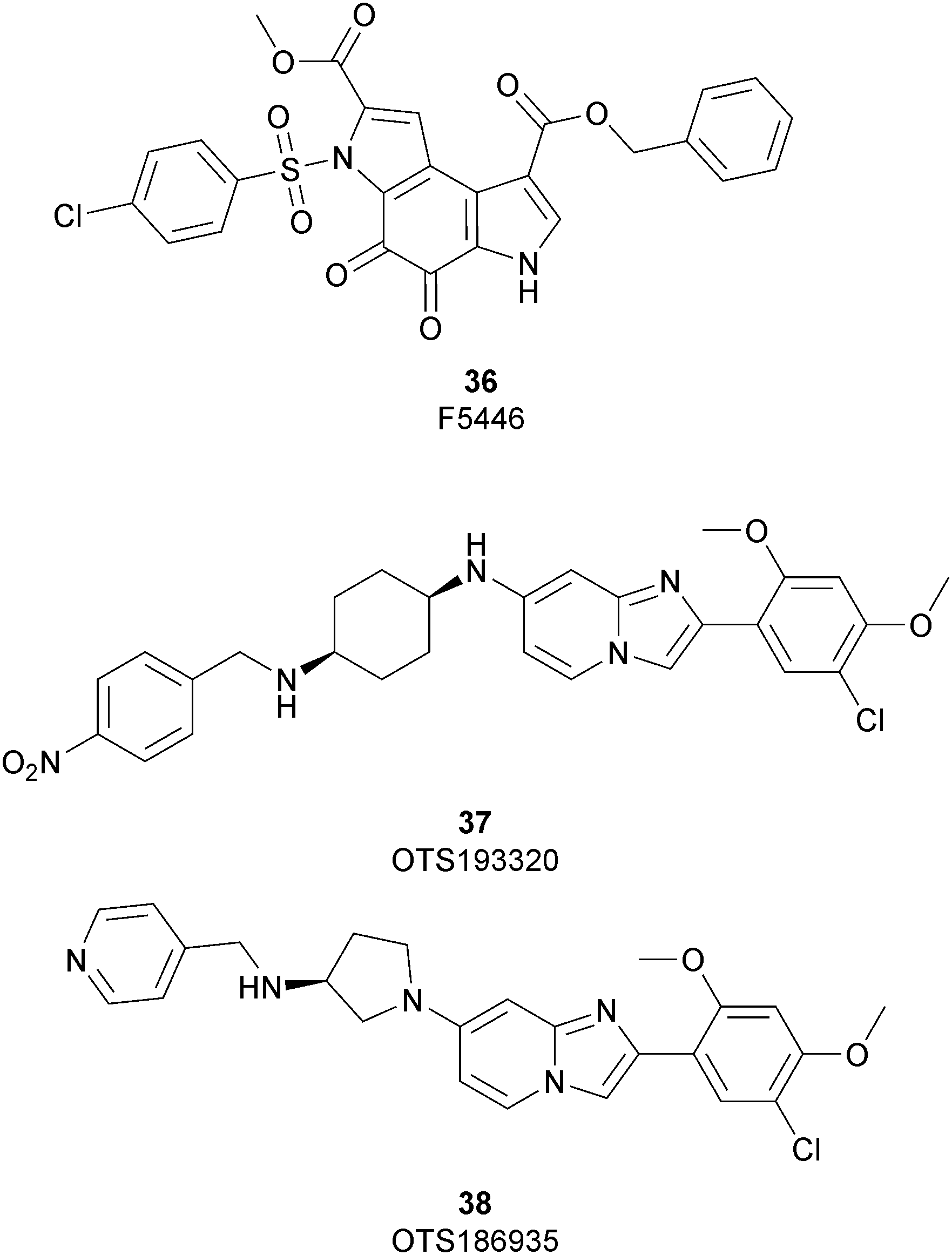 | ||
| Fig. 9 SUV39H1 and SUV39H2 inhibitors. | ||
Until recently, the fungal epipolythiodioxopiperazine metabolites chaetocin105 and verticillin A106 were the sole SUV39H1 inhibitors described; however, although potent (an IC50 value of 0.6 μM was reported for chaetocin), both compounds also inhibit several other histone methyltransferases and are non-specific redox and covalently active compounds.105–107
In 2019 a virtual screening campaign performed on the SET domain of human SUV39H1, followed by an in vitro validation, led to the identification of a few hits, including a 4,5-dioxo-6H-pyrrolo(3,2-e)indole-2,8-dicarboxylate derivative. Subsequent structural optimization studies yielded compound 36 (F5446), which was found to inhibit SUV39H1 with an IC50 of 0.496 μM and decreased the deposition level of the repressive H3K9me3 mark at the promoter regions of the genes coding for the major proteins used by CD8+ cytotoxic T lymphocytes (CTLs) to induce tumour cell apoptosis, thus increasing their expression.108 Compound 36 also showed low toxicity in mice models, and in vivo studies showed that it suppresses established tumour growth in a CD8+ CTL-dependent manner. Subsequent studies also showed that the inhibition of SUV39H1 by 36 effectively suppresses human colon carcinoma xenograft growth in vivo.109 In 2018 the screening of a panel of commercially available compounds enabled the identification of some imidazo[1,2-a]pyridine compounds with weak SUV39H2 methyltransferase inhibitory activity, with IC50 values of <10 μM. A subsequent SAR-based drug optimization study yielded 37 (OTS193320), a potent SUV39H2 inhibitor (IC50 of 22.2 nM) that displays a growth-suppressive outcome of SUV39H2-positive A549 lung cancer cells (IC50 of 0.38 μM). In in vitro studies, this compound decreased global H3K9 trimethylation levels in breast cancer cells and activated apoptotic cell death. Furthermore, when combined with doxorubicin, 37 reduced γ-H2AX levels as well as cancer cell viability compared with a single agent administration. Further optimization allowed the identification of 38 (OTS186935) (IC50 of 6.49 nM), which is able to significantly inhibit tumour growth without any detectable toxicity in mouse xenograft models, exploiting MDA-MB-231 breast cancer cells and A549 lung cancer cells.110 In order to show the binding interactions between 38 and the active-site residues of SUV39H2, a flat representation using LigPlot+ has been prepared (Fig. 10). The deposited X-ray crystal structure of the methyltransferase domain of the enzyme bound to 38 (PDB 6P0R) revealed the importance of the nitrogen of the imidazo[1,2-a]pyridine scaffold for the interaction with Thr303, responsible of the correct allocation of the molecule inside the binding site.
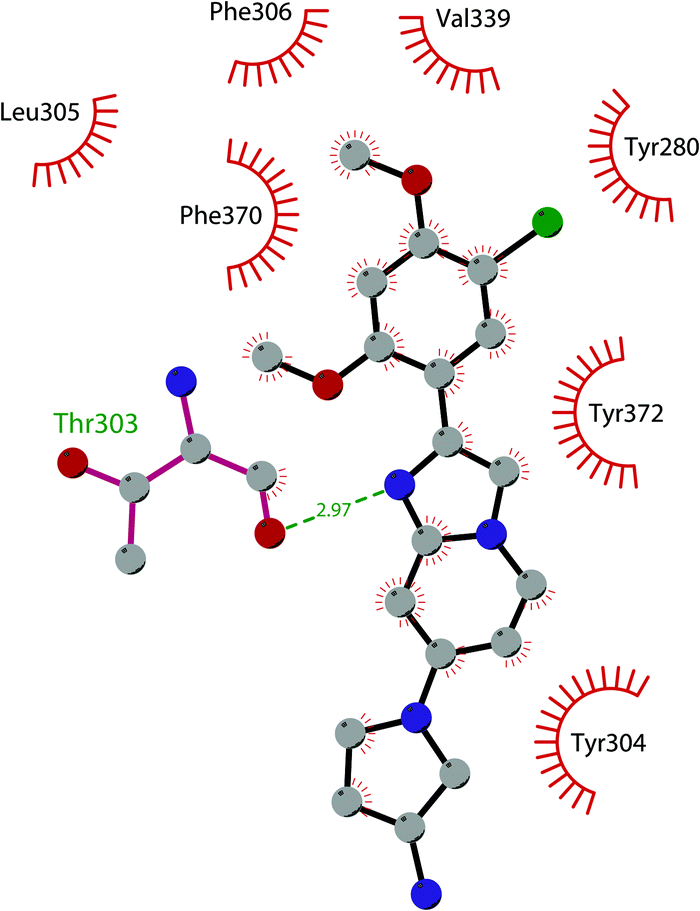 | ||
| Fig. 10 Flat representation (generated using LigPlot+ from PDB ID 6P0R) showing contacts between compound 38 and active-site residues in SUV39H2. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
However, it should be noted that these compounds were not well characterized by the authors in terms of their selectivity against other PMTs and, consequently, caution should be exercised when considering their use as chemical probes. Nevertheless, to the best of our knowledge they are the most potent SUV39H2 inhibitors reported so far.
EZH1 and EZH2
E(Z) is one of the founding members of the SET domain family of methyltransferase proteins.111 Enhancer of zeste homologs 1 and 2 (EZH1 and EZH2) are two homolog proteins belonging to Polycomb Repressive Complex 2 (PRC2), a family of proteins involved in the maintenance of gene silencing thanks to the introduction of post-translational modifications on histones and chromatin condensation.112 PRC2 is composed of four core components: EZH1 and EZH2, suppressor of zeste 12 (SUZ12), retinoblastoma suppressor associated protein 46/48 (RBAP46/48) and embryonic ectoderm development (EED). EZH1 and EZH2 represent the catalytic subunit and they are responsible for lysine mono-, di- and trimethylation of H3K27 thanks to their SET domain,113 while the other core subunits behave as regulators of the methyltransferase activity of EZH1 and EZH2. However, it remains unclear if they possess other functions independently from EZH1 and EZH2.114–116 Among all of these, of particular importance is the EED subunit, a WD40-repeat-containing protein that recognizes, thanks to the presence of an aromatic cage, the methyl marks deposited by EZH proteins, and this interaction causes a conformational change in the structure of these methyltransferases, which increases their catalytic activity. This mechanism allows the propagation of H3K27 trimethylation on nucleosomes, contributing to the maintenance of the chromatin in a repressive form.116From the functional point of view, the PRC2 complex is recruited to non-methylated CpG islands (CGIs) and trimethylates H3K27, with the subsequent recruitment of canonical PRC1 via chromodomain proteins (CBX), a subunit of PRC1 that binds to these trimethylated residues. In turn, PRC1 mono-ubiquitylates histone H2A at K119, inducing the condensation of chromatin.111,117,118
Despite sharing the same target genes, EZH1 and EZH2 are differently expressed in tissues and have distinct functions. While EZH1 is ubiquitously expressed, EZH2 expression is found in proliferating tissues. In addition, EZH1 exhibits low levels of histone lysine methyltransferase activity while EZH2 performs this modification robustly. Regarding the condensation of chromatin, EZH1 represses transcription and compacts chromatin more efficiently than EZH2; nevertheless, EZH1 and EZH2 have a complementary role, as EZH2 knockdown resulted in EZH1 upregulation.112
EZH1 and EZH2 are responsible for the regulation of gene expression during development and haematopoiesis, therefore mutations and abnormal expression of these proteins are reported in various types of cancer, mainly haematological malignancies, breast, and prostate cancer. Specifically, overexpression of both EZH1 and EZH2 is associated with tumour aggressiveness, poor prognosis, and relapses.119–122
EZH1 and EZH2 inhibitors
The development of small-molecule inhibitors of EZH1/EZH2 has been an intense area of research. So far, several EZH1/EZH2 inhibitors have been developed and some of them have been entered in clinical trials alone or in combination with other therapies for a variety of tumours. Considering that EZH1/EZH2 are the catalytic subunit of the PRC2 complex, different drug-design strategies have been explored, targeting both the catalytic domain as well as other complex components such as the EED subunit. Consequently, inhibitors can be categorized into two different classes: inhibitors of the EZH1/EZH2 catalytic domain (Fig. 11) and inhibitors of the EED subunit (Fig. 13). Moreover, the development of degraders (Fig. 18) that are able to induce protein degradation of the PRC2 complex will be discussed. | ||
| Fig. 11 Inhibitors of the EZH1/2 catalytic domain. | ||
Inhibitors of the EZH1/EZH2 catalytic domain
The first class of inhibitors developed comprises small molecules targeting the catalytic SET domain EZH1/EZH2. It is worth noting that EZH1 and EZH2 share 96% sequence identity within their catalytic SET domain and 76% sequence identity overall, making the identification of selective ligands challenging. Despite these difficulties, most of the compounds identified have been shown to be more active towards EZH2 over EZH1, while some molecules targeted both proteins, acting as dual inhibitors. The benefits of selective or dual EZH1/EHZ2 inhibition are still under discussion. Notwithstanding the good effects of compounds with a greater activity on EZH2 versus EZH1, good results can also be achieved with dual EZH1/EZH2 inhibitors considering that EZH1, despite the lower methyltransferases activity compared with EZH2, plays a minor role in balancing the deletion of EZH2.122,123From a structural point of view, the pyridone–benzamide core is a highly optimized scaffold for the binding to EZH2 in a SAM-competitive mode. In fact, in 2012 Epizyme Inc. identified 39 (EPZ005687) as an EZH2 inhibitor (Ki of 24 nM), displaying 500-fold selectivity over 15 other PMTs and 50-fold selectivity over the closely related enzyme EZH1. This compound selectively inhibits H3K27 methylation in EZH2 wild type and Y641- or A677-mutant lymphoma cells as well as other tumour cell lines, such as breast and prostate cancers.124 The optimization process of 39 led to the identification of 40 (EPZ6438 or tazemetostat), with an increased potency (Ki of 2.5 nM) and a better PK profile compared with the parent compound. 40 is more than 35-fold more selective towards EZH2 over EZH1125 and it selectively killed wild type as well as human lymphoma cell lines harbouring point mutations in the EZH2 catalytic domain, inhibiting H3K27 trimethylation.12640 is orally available and, in 2020, the US FDA approved it, and it was marketed as Tazverik®, the first EZH2 inhibitor for the treatment of metastatic or locally advanced epithelioid sarcoma in adults and paediatric patients older than 16 years. In the same year, the FDA conferred accelerated approval to 40 for patients with relapsed or refractory follicular lymphoma, who have received at least two preceding systemic therapies and whose tumours have an EZH2 mutation or have no satisfactory alternative treatment options. In addition, 40 was also evaluated in other clinical trials in combination with other therapeutic agents (NCT04179864; NCT01897571; and NCT03854474).127–129
In 2015, Epizyme Inc. developed 41 (EPZ011989), a selective and orally available inhibitor of EZH2, structurally related to 40. In particular, the central aromatic core was differently decorated: besides the pyridone scaffold, the pyran substituent of 40 was replaced with a N-(2-methoxyethyl)-N-methylcyclohexanamine and the morpholine was connected through an acetylene linker instead of a benzene moiety. 41 maintains its activity both on mutant and wild-type EZH2 (Ki of <3 nM) with more than 3000-fold selectivity over the other tested HMTs and more than 15-fold selectivity for EZH2 over EZH1. 41 significantly reduced tumour growth in a mouse xenograft model of human B cell lymphoma.130
Maintaining the pyridone-amidic moiety, which is crucial for the EZH2 binding, Lu and co-workers exploited a scaffold-hopping approach on the benzene rings of compound 40, obtaining a series of benzofuran-based EZH2 inhibitors. Among them, 42 (EBI-2511) displayed good enzymatic (IC50 of 4.0 nM) as well as cellular activity, showing a reduction of trimethylated H3K27 levels in a dose-dependent manner both in Pfeiffer cells (IC50 of 6.0 nM) and WSU-DLCL2 cells (IC50 of 55 nM). In a Pfeiffer xenograft mouse model, 42 showed a dose-dependent inhibition of the tumour growth, resulting in a good reduction in tumour size with no changes in body weight. Notably, 42 showed a superior antitumor efficacy compared with 40 at the same dose, suggesting that it might accomplish a similar efficacy but at lower dosages in clinical studies.131
Through HTS, in 2012 Glaxo Smith Kline USA (GSK) disclosed 43 (GSK126) a selective inhibitor of both wild-type and mutant EZH2 (Kappi of 0.5–3 nM).132 Suppressing EZH2 activity, 43 significantly decreased the growth of EZH2-mutant diffuse large B-cell lymphoma (DLBCL) in xenograft mice upon intravenous administration.13343 entered in a phase I clinical trial for different types of solid tumours and lymphoma but showed insufficient clinical activity (NCT02082977).134 It was suggested that the activity of EZH2 inhibitors is drastically affected by the tumour microenvironment, as confirmed by the antitumor efficacy of 43 when administered to immunodeficient but not to immunocompetent hosts.135 Liu and co-workers were able to solve the crystal structure of human PRC2 bound to 43 (PDB 5WG6).136 The main interactions are depicted in Fig. 12. The pyridone moiety is embedded in a binding pocket formed by residues of Trp624, Arg685, and Phe686. In particular, the interactions of the pyridone group confirmed the SAM-competitive mode of action of this compound. In fact, the amide nitrogen and the carbonyl oxygen establish distorted hydrogen bonds with both the carbonyl oxygen and the amide nitrogen of Trp624 of EZH2, thus preventing binding to the cofactor SAM. In addition, the Tyr111 is hydrogen bonded with the carbonyl oxygen between the indole group and the pyridone ring. Interestingly, the linked pyridine and piperazine rings of 43 protrude above the gate to the solvent.
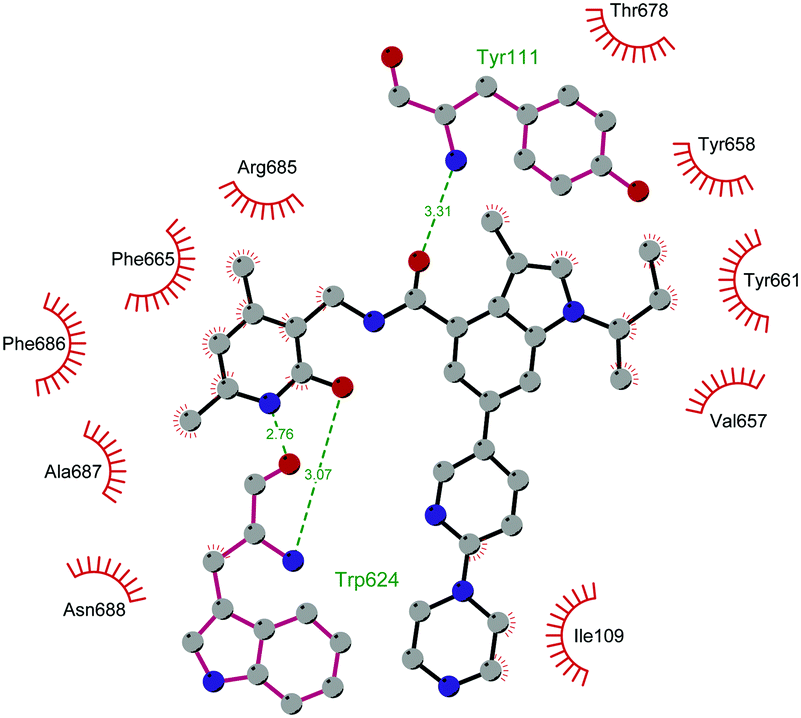 | ||
| Fig. 12 Flat representation (generated using LigPlot+ from PDB ID 5WG6) showing contacts between compound 43 and human PRC2. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
 | ||
| Fig. 13 Inhibitors of the EED subunit. | ||
In 2012, compounds 44 (GSK343) and 45 (EI1) were also identified. Specifically, GSK developed 44 as an efficient (EZH2 Kappi of 1.2 nM), selective, and cell-active EZH2 inhibitor. Despite its great potential as an in vitro tool to explore EZH2 biology, 44 is not suitable for in vivo studies, considering the high clearance in rat PK studies.137 At the same time, Novartis exploited HTS, followed by structural optimization, and obtained derivative 45, which is able to inhibit the methyltransferases activity of EZH2 with more than 10000-fold selectivity over other methyltransferases and 90-fold selectivity over EZH1. 45 showed good cellular activity, decreasing H3K27 methylation without disrupting the chromatin binding of PRC2. 45 is equally active against both wild-type and Y641-mutant DLBCL cells, reducing proliferation, causing cell cycle arrest, and apoptosis.138
In 2016, Constellation Pharmaceuticals published a series of inhibitors characterized by the presence of an indole ring, identifying 46 (CPI-1205) as a compound with a modest selectivity for EZH2 over EZH1 (IC50 of 2 nM and 52 nM, respectively) compared with the high selectivity shown against 30 other histone or DNA methyltransferases.139 In a KARPAS-422 lymphoma xenograft model, 46 demonstrated significant tumour growth reduction upon 25 days of administration. The compound completed a phase I study in patients with B-cell lymphomas, (NCT02395601)140 showing anti-cancer activity and no significant toxicity. In a phase Ib study, 46 also produced positive results for patients with metastatic castration-resistant prostate cancer (mCRPC), demonstrating good tolerability, minimal safety issues and encouraging clinical activity. Currently, 46 is under investigation in a phase Ib/II trial to evaluate its effects on mCRPC in combination with enzalutamide or abiraterone/prednisone (NCT03480646).141 Another interesting aspect is that EZH2 inhibition in human T cells by 46 produced phenotypic and functional alterations of the regulatory T-cells and enhanced the cytotoxic activity of effector T-cells. It was speculated that modulation of EZH2 expression in T-cells can improved antitumor responses via anti-CTLA-4 therapy. Indeed, ipilimumab increased EZH2 expression in peripheral T-cells, suggesting a strong rationale for its combination trial with 46.142
Compound 47 (ZLD1039), featuring a cyclohexane group fused with the pharmacophoric pyridone moiety, inhibited EZH2 wild type as well as Y641F and A677G mutant (IC50 of 5.6, 15, and 4 nM, respectively). 47 reduced the global H3K27 methylation levels and re-established the expression tumour suppressors that are responsible for the higher survival of patients with breast cancer. In three different breast tumour xenograft models (MCF-7, MDA-MB-231, and 4T1 cell lines), 47 decreased tumour growth and metastasis, endorsing the role of EZH2 in breast cancer progression.143
In 2018, the Pfizer Global Research group optimized a previous series of lactam-derived EZH2 inhibitors, developing the oxetane-containing compound 48 (PF-06821497), which exhibited remarkable potency (Ki less than 0.1 nM) and pharmaceutical properties as well as robust tumour growth inhibition in vivo.14448 entered a phase I clinical trial for the treatment of adults with relapsed/refractory small cell lung cancer, castration-resistant prostate cancer and follicular lymphoma (NCT03460977).145
Regarding dual inhibitors, 49 (UNC1999) was reported by researchers at the University of North Carolina as dual EZH1 and EZH2 inhibitor (IC50 of 45 and <10 nM, respectively). This compound reduced trimethylated H3K27 levels in cells and selectively killed DLBCL cells carrying the EZH2-Y641 mutations together with inhibition in growth of MV4-11 cells.146,147
Other orally bioavailable EZH1/EZH2 dual inhibitors are 50 ((R)-OR-S1) and 51 ((R)-OR-S2), which strongly and selectively inhibited methyltransferase activity of both EZH2 and EZH1 (IC50 of 8.4 and 7.4 nM and 2.5 and 10 nM for EZH2/EZH1, respectively). Compared with EZH2 selective inhibitors, these compounds showed a remarkable antitumor efficacy in vitro as well as in vivo against DLBCL cells with EZH2 mutation. These dual inhibitors displayed a strong efficacy both in haematological and solid cancer panel assays, suggesting that both EZH2 and EZH1 are implicated in some types of cancer and their simultaneous inhibition would be effective for targeting them. Accordingly, 51 inhibited the in vitro growth of MV4-11 cells more effectively than 49, which is probably due to the weaker enzymatic activity of 49 on EZH1.148
Inhibitors of the EED subunit
Besides inhibitors of the EZH1/2 catalytic subunit, other components of the PRC2 complex have been targeted to inhibit the methyltransferase activity. This strategy could prevent some recently emerged issues, such as the gain-of-function mutations of EZH2 resulting in EZH2-inhibitor-resistant cancer cells lines. Considering the role of EED in enhancing the catalytic efficiency of the PRC2 complex, inhibition of the EED interaction with the trimethylated histone substrate was explored as a strategy to modulate the enzymatic activity of the complex. So far, several inhibitors targeting the EED subunit have been reported.In 2017, 52 (EED226) and 53 (A-395) were identified as nanomolar inhibitors of EED and studies helped to clarify the effects on EED inhibition. Specifically, Novartis identified 52 as a potent allosteric binder of the PRC2 complex (IC50 of 23.4 nM). The high-resolution crystal structure of 52 bound to EED and to an EED binding domain (EBD) (PDB 5GSA) revealed that the compound binds the pocket responsible for the recognition of the trimethylated H3K27 substrate. Specifically, the compound is inserted into a deeper pocket that is created by a conformational change of the four aromatic residues which form the ‘aromatic cage’ of the binding site (Phe97, Tyr148, Trp364, and Tyr365), positioning the furan group on the inside and the methylsulfonic group towards the solvent. Fig. 14 depicts the major interactions of 52: the triazolopyrimidine core forms π–π interactions with Tyr148, Tyr365, and Phe97 residues while the furan group is stabilized by van der Waals and cation–π interactions with other residues of the binding pocket. Moreover, two hydrogen bonds stabilize the interactions of 52 inside the pocket: the amino group between the bicyclic and the furan moieties is hydrogen bonded with the side chain carbonyl of Asn194 while the bottom nitrogen of the triazolopyrimidine core establishes a hydrogen bond with the side chain of Lys211. Despite the moderate cell permeability, 52 led to a dose-dependent reduction of both di- and trimethylated H3K27 markers in G401 cells in addition to a strong proliferation inhibition in lymphoma cells carrying EZH2 mutations as well as cell lines with acquired resistance to SAM-competitive EZH2 inhibitors. Administration of 52 in mice using a subcutaneous xenograft model of Karpas422 showed a clear dose-dependent reduction of trimethylated H3K27 levels with a good tolerability. To better evaluate the effect of PRC2 inhibition in vivo, 52 was administered orally to mice for a longer period and a complete tumour regression was observed after 32 days. Furthermore, a combination of 52 and the EZH2 inhibitor 45 exhibited synergistic effects in suppressing cancer cell growth, suggesting a potential combination therapy.149
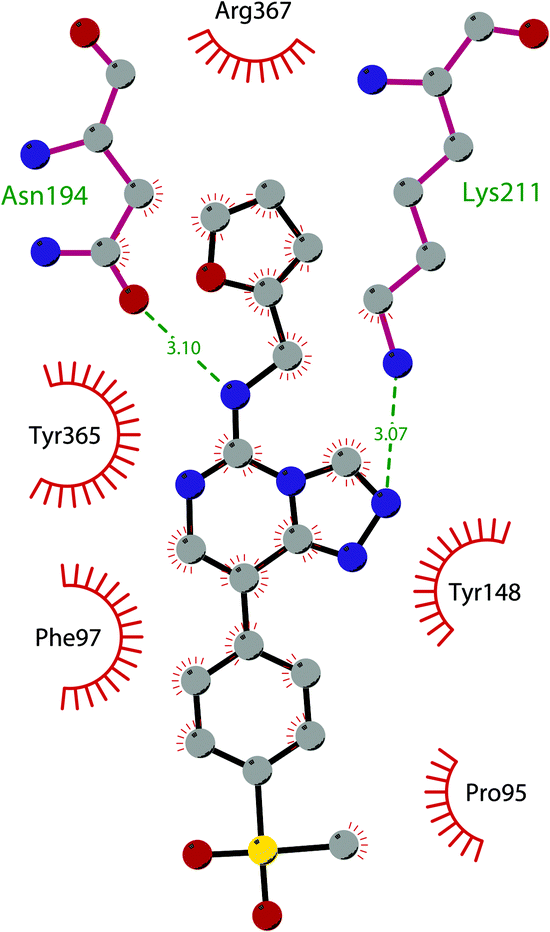 | ||
| Fig. 14 Flat representation (generated using LigPlot+ from PDB ID 5GSA) showing contacts between compound 52, EED, and an EZH2 peptide referred to as the EED binding domain (EBD). Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compound. | ||
In the same year, AbbVie reported the identification of 53 as a selective EED inhibitor with high in vitro potency (KD of 1.5 nM from surface plasmon resonance (SPR) assay and IC50 of 18 nM in a radioactivity-based assay). Structurally, 53 is a mixture of two diastereomers, epimeric at the indane stereocenter, and is used for biological evaluation without separation. The crystal structure of 53 with the EED WD40 domain showed that the compound competes with the histone substrate for binding to the aromatic cage within the WD40 repeat domain. Cellular studies showed that 53 is effective against PRC2 in human cancer cell lines that are sensitive to EZH2 inhibition, leading to phenotypic effects on H3K27 methylation that are comparable to inhibitors of the catalytic EZH1/EZH2 domain.
In a DLBCL Pfeiffer xenograft model, administration of 53 two times weekly for 5 weeks, showed a reduction of tumour growth of 84%. Moreover, 53 retains its effects on EZH2-inhibitor-resistant cell lines. Unfortunately, oral administration and systemic administration using an osmotic pump resulted in insufficient exposures and the only route to administer the compound was subcutaneously.150
In 2019, the pyrimidone-based compound 54 (BR-001) was identified, which is able to selectively bind to EED, competing with trimethylated H3K27 binding (IC50 of 4.5 nM). The compound exhibited a strong antiproliferative effect on Karpas 422 cells and anti-tumour efficacy in the CT26 syngeneic model. It has been reported that 54 acts both directly, leading to inactivation of PRC2, as well as modulating the tumour microenvironment promoting CD8+ T-cell tumour infiltration.151 Currently, 55 (MAK683), patented by Novartis as a new triazolo pyrimidine EED inhibitor, is in a phase I/II study in patients with advanced malignancies such as DLBCL, nasopharyngeal carcinoma or other advanced solid tumours for whom no further effective standard treatment is available (NCT02900651).152
EZH2 and EED degraders
Besides canonical single-site inhibitors, novel approaches have been exploited to target the PRC2 complex, developing chemical agents that are able to selectively induce protein degradation. Specifically, PROteolysis TArgeting Chimeras (PROTACTM) have been developed, linking a binder for the protein-of-interest and an E3 ligase ligand to induce ubiquitination, thereby achieving protein degradation via the proteosome. At the same time, hydrophobic tagging was exploited, attaching a hydrophobic group to a selected protein binder, leading the misfolding or unfolding of the target protein and its subsequent proteasome-degradation. Following the same strategies developed to obtain single-site inhibitors, EZH2 and EED degraders were developed, using selective warheads for both proteins (Fig. 15a and b, respectively).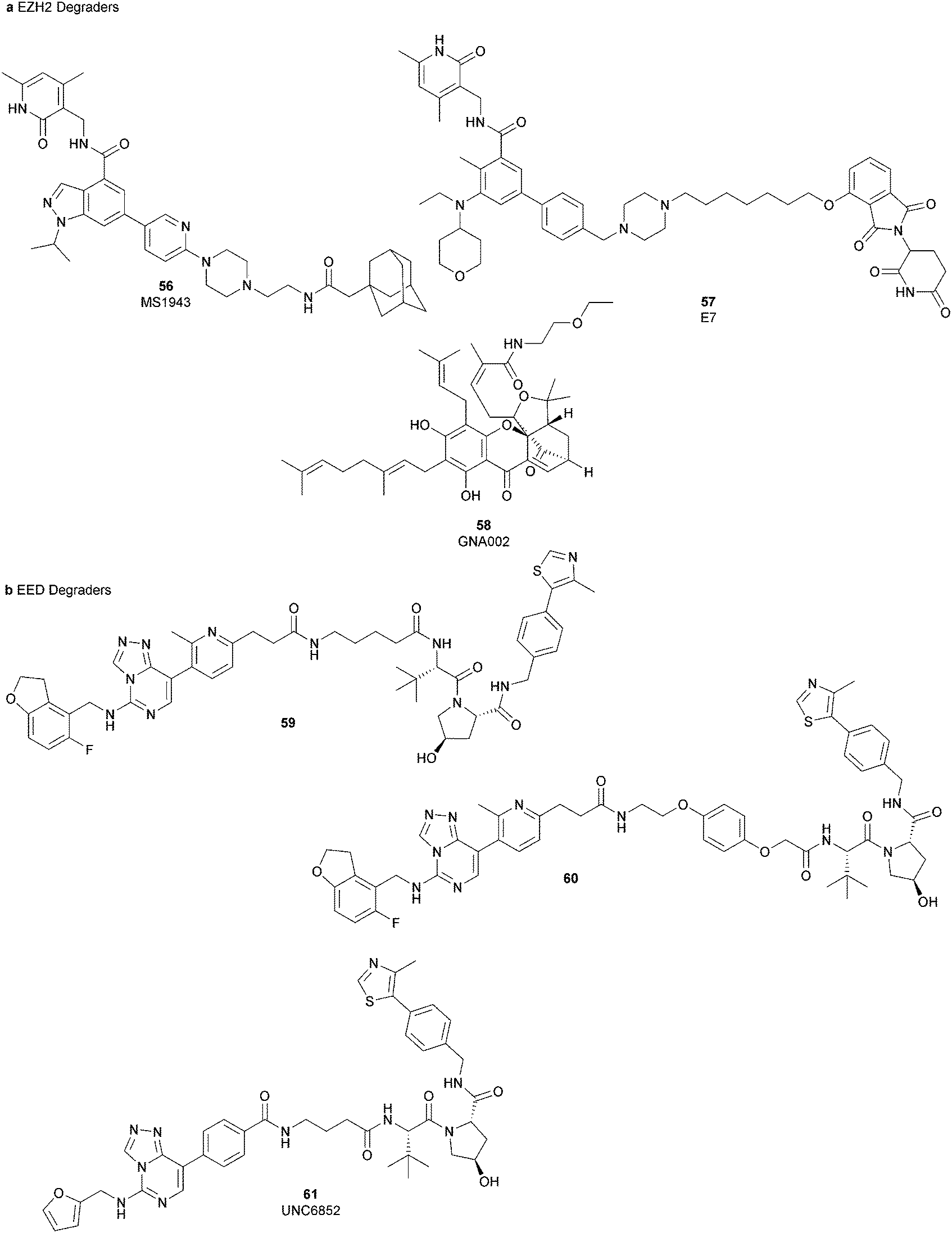 | ||
| Fig. 15 (a) EZH2 and (b) EED degraders. | ||
In 2018, the discovery of a first-in-class EZH2-selective degrader 56 (MS1943) was reported. This compound was designed by merging a non-covalent and selective inhibitor of EZH2 (IC50 of 12 nM and 2.5 μM for EZH2 and EZH1, respectively) that is structurally related to 44 to a bulky adamantyl group as a hydrophobic tag. Compound 56 preserved the potency (IC50 of 120 nM) and selectivity in inhibiting EZH2 activity (IC50 more than 10 μM for EZH1). In cancer and non-cancerous cell lines, 56 significantly reduced the EZH2 protein levels while it had a minimum or no effect in reducing the EZH1 protein levels. 56-induced EZH2 degradation resulted in significant cell growth inhibition and cytotoxic effects in multiple triple-negative breast cancer cell lines but not in normal cells. Moreover, 56 is able to completely suppress tumour growth in vivo with good tolerability, suggesting an alternative treatment for EZH2-dependent cancer.153 The PROTACTM57 (E7), formally obtained by connecting the protein binder 43 to 4-hydroxythalidomide, a well-studied ligand of the cereblon (CRBN) E3 ligase, through an alkyl linker, has been also reported. 57 showed high potency (IC50 of 2.7 nM) in reducing the EZH2 activity and good selectivity over EZH1 (IC50 of 179 nM), fully inhibiting the oncogenic activity of EZH2 with antiproliferative effects on EZH2-dependent tumours. Notably, the recruitment of E3 ubiquitin ligase resulted in the indiscriminate ubiquitination and degradation of all PRC2 components, including EZH2, EED, SUZ12, and RbAp48.154
Besides bifunctional degraders, the compound 58 (GNA002) is able to induce EZH2 degradation without featuring a hydrophobic tail or an E3 ligase binder. Specifically, the compound covalently binds the Cys668 residue located within the EZH2-SET-domain, leading to its ubiquitination and consequently its degradation by the protein quality control E3 ligase, CHIP. Moreover, as 58 covalently bound a specific cysteine of EZH2, it did not affect the activity of EZH1 or other examined SET domain-containing PMTs. The ability to significantly suppress tumour growth was investigated using both in vitro and in vivo assays in wild-type EZH2 solid tumours as well as in lymphomas harbouring EZH2 mutations. These encouraging results suggested an alternative approach for EZH2 degradation as an anticancer strategy.155
On the other hand, three bifunctional degraders were developed using a selective EED warhead. AstraZeneca researchers reported the identification of two EED-targeted PROTACsTM (59 and 60) obtained by merging an EED binder, structurally related to compound 55, and a well-characterized ligand of von Hippel–Lindau ligase (VHL) with two different types of linker. Both compounds displayed a comparable binding affinity for EED via an in vitro assay (pIC50 of 8.11 and 8.17 for 59 and 60, respectively). In cellular assays, these PROTACsTM induced rapid EED degradation in cells, inhibiting proliferation of the EZH2 mutant DLBCL cell line as well as the EZH2 wild-type rhabdoid cancer cell line. Unfortunately, no growth inhibition was observed in a limited number of tested EZH2-dependent cell lines, probably due to a cellular fraction of EZH2 that is resistant to PROTACTM-mediated degradation. Interestingly, these degraders also induce the degradation of EED associated proteins in the PRC2 complex, such as EZH2 and SUZ12. Considering that EED degradation is faster than for EZH2 and SUZ12, it was suggested that EED degradation destabilizes the PRC2 complex, triggering other components to the proteasome. Otherwise, it was speculated that formation of the EED–PROTACsTM–E3 ligase ternary complex could expose the lysine residues of EZH2 or SUZ12, which are consequently ubiquitinated.156 In the same year, 61 (UNC6852) was disclosed as an EED degrader, featuring a 52-derived ligand and a VHL binder. Even if the degrader showed a slight decrease in potency in the binding of WD40 of the EED protein compared with 52 (IC50 of 247 nM and 45 nM for 61 and 52 in the same assay, respectively), it is still able to bind the target protein. Like compounds 59 and 60, 61 also induces proteasomal degradation of all PRC2 components, probably due to the aforementioned mechanisms. 61 is able to prevent the histone methyltransferase activity of EZH2 in both HeLa cells and DLBCL cells that contain EZH2 gain-of-function mutations. Furthermore, 61 has anti-proliferative effects that are comparable to EZH2 and EED inhibitors in DB and Pfeiffer cells but it is substantially less toxic.157
SETD7
The SET domain containing lysine methyltransferase 7 (SETD7, also called SET7/9 or KMT7) was the first lysine methyltransferase identified to specifically monomethylate H3K4.158–160This mark furnishes a site for further modifications, but several studies reported a limited involvement of SETD7 in the di- and trimethylation of H3K4.161,162
SETD7 can also methylate many non-histone proteins (including p53, TAF10, ER, P65, STAT3, SOX2, pRb, SIRT1, DNMT1, SUV39H1 and FOXO34-14) and probably these are the main SETD7 substrates.143 Overall, the monomethylation of K372 of p53 represents a central regulatory event as it has a key role for p53 activation in human cell lines. SETD7 activity is triggered by DNA damage and induces the acetylation of lysine residues in the regulatory domain of p53 by different acetyltransferases (p300, CBP and TIP60). The acetylation prevents the ubiquitin-mediated degradation of p53 by the proteasome and, as a final outcome, K372 methylation extends the p53 life.163
On the other hand, SETD7 methylation of other substrates can be associated with protein degradation. For instance, human DNMT1 methylation at K1094 by SETD7 increases the turnover of the protein.164 In addition, DNMT1 can also be monomethylated at K142, leading to its ubiquitination and degradation.165
Considering the abundance of its substrates in cells, SETD7 is involved in the regulation of various pathways. It has been demonstrated that SETD7, in atherosclerosis and diabetes, acts as a coactivator of inflammatory genes that interact with NF-KB.166 Increasing evidence suggests that SETD7 also has a key role in the regulation of processes such as cell differentiation and proliferation. Indeed, many genes required for normal embryonic development frequently harbour the monomethylated H3K4 modification. SETD7 is also overexpressed in a variety of cancers, such as colorectal cancer167 and hepatocellular carcinoma,168 making this protein an interesting target for drug discovery.
SETD7 inhibitors
To better explore the regulatory role of SETD7, several attempts have been made to develop potent and selective SETD7 inhibitors. However, the number of selective compounds reported so far is still limited, and the majority of these have a weak inhibitory activity (Fig. 16). As for other methyltransferases, different derivatives have been designed, exploiting as a starting point the structure of the cofactor SAM. The replacement of the sulphur atom with a nitrogen and the introduction of different alkylamino chains that mimic the side chain of lysine yielded different derivatives.169 Among these, in an ELISA assay, 62 (DAAM-3) showed approximately 50% inhibition against SETD7 at 10 μM. The co-crystal structure of the SET domain in complex with 62 revealed that the compound binds the cofactor binding site, inserting the n-hexyl group within the lysine access channel.170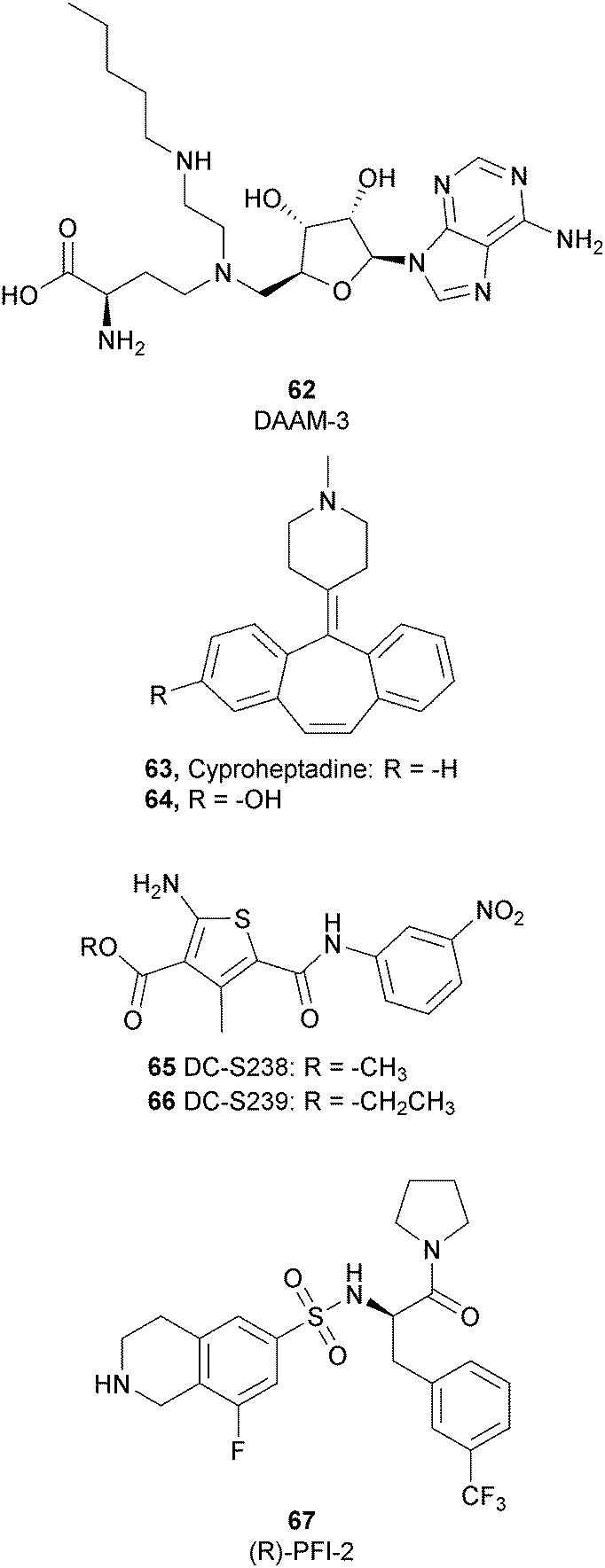 | ||
| Fig. 16 SETD7 inhibitors. | ||
An HTS campaign led to the identification of 63 (cyproheptadine), an antagonist of the histamine and serotonin receptor clinically approved as an antiallergy drug, as a SETD7 inhibitor (IC50 of 1 μM). The X-ray structure of SETD7 in complex with 63 revealed that the compound binds to the substrate-binding site of the enzyme, inducing conformational changes in the C-terminal region and in the amino acid residues involved in the formation of the channel for lysine access. According to SETD7 knockdown, 63 reduced the amount of ERα in a dose- and time-dependent manner, enhancing its degradation by the proteasome. This evidence highlights the critical role of SETD7 in the estrogen-dependent transactivation of ER target genes and its potential therapeutic use in diseases associated with upregulation of ERα such as breast cancer.171 Structural optimization of 63via the insertion of various functional groups on the aromatic rings of the dibenzocycloheptene moiety led to the identification of derivative 64, with an improved potency (IC50 of 0.41 μM).172
SAM competitive and selective inhibitors of SETD7 are 65 (DC-S238) and 66 (DC-S239) (IC50 of 4.88 μM and 4.59 μM, respectively). Compound 66 exhibited anti-proliferative effects on breast cancer and leukaemia cell lines MCF7, HL60 and MV4-11 cells but had no cellular activity against HCT116 (human colon cancer) and DHL4 (lymphoma) cells, suggesting that this derivative is more active towards breast cancer and leukaemia cells.173
So far, the most potent and widely used SETD7 inhibitor is 67 ((R)-PFI-2), which was identified via the HTS of approximately 150000 compounds of Pfizer's collection.174 This compound showed a high potency (IC50 of 2.0 nM) and is more than 1000-fold selective over a panel of 18 other methyltransferases and DNMT1. The X-ray crystal structure of the SETD7 catalytic domain bound to 67 revealed an uncommon mode of action, suggesting a substrate-competitive and SAM-dependent binding mode. This evidence was supported by the X-ray crystal structure of the compound bound to the target protein (PDB 4JLG). As depicted in Fig. 17, the pyrrolidine amide engages the pocket that is normally occupied by the target lysine residue, also making direct hydrophobic interactions with the leaving methyl group of SAM, preventing useful interactions of the protein with lysine substrate. Upon the binding of 67 with the protein, the post-SET loop (residues 336–349) adopts a distinctive conformation, making hydrophobic interactions with the trifluoromethyl group. Moreover, the ligand anchors the peptide-binding site, making several hydrogen bonds with different residues, including Gly336 of the post-SET loop Ser268 on the opposite side of the substrate-binding channel.
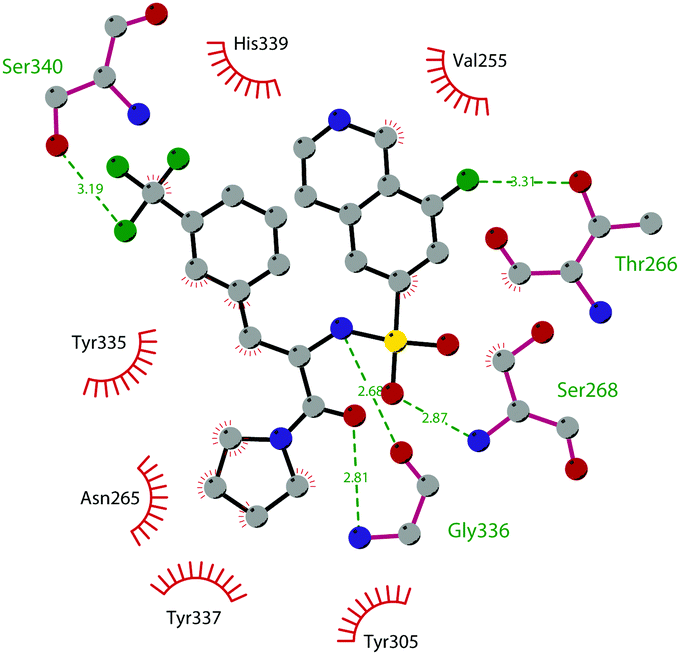 | ||
| Fig. 17 Flat representation (generated using LigPlot+ from PDB ID 4JLG) showing contacts between compound 67 and active-site residues in SETD7. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compound. | ||
The cellular activity of 67 highlighted the role of SETD7-dependent methylation in the control, subcellular localization, and function of the transcriptional coactivator Yes-associated protein (YAP). Treatment of 67 in high-density MCF7 cells resulted in a dose-dependent increase of the nuclear YAP and intensified the expression of its target genes, such as AREG and CYR61, indicating that YAP localization in this cell line requires persistent SETD7 activity. It is worthy of note that only 67 is active whereas its (S)-enantiomer is 500-fold less active. Molecular dynamics simulations helped to discriminate their different efficacy based on different bioactive energy barriers. This study provides helpful information for future structure-based drug development, revealing the impact of chirality on SETD7 inhibition.175
SMYD2
SMYD2 (SET and MYND Domain-Containing Protein 2, also called KMT3C) is a member of a subfamily of SET-domain-containing proteins with a particular domain architecture. This family of proteins is composed of a SET domain that is divided into two parts by a MYND domain/zinc finger motif, followed by a cysteine-rich post-SET domain.176 SMYD2 has been shown to methylate H3K36 even if it lacks specificity on the free histones. This protein is a relatively promiscuous enzyme as it is responsible for the methylation of many substrates (at least six non-histone substrates have been reported in cells),177 the best characterized of which is p53, where it shows a highly selective activity. SMYD2 methylates p53 at K370, a repressing modification to p53-mediated transcriptional regulation, and reduces p53-mediated apoptosis. It is of note that the methylation of K370 in p53 is inhibited by SET9-mediated methylation of K372, corroborating the regulatory crosstalk between these post-translational modifications.178Physiologically, SMYD2 is involved in the differentiation of embryonic stem cells and, differently from the other SMYD proteins that are equally expressed in somatic and pluripotent cells, it is expressed at high levels in somatic rather than in pluripotent cells.179 Moreover, it has been reported that SMYD2 is important for cardiac and skeletal function, although it is not essential for the development of the heart.180
From the pathological point of view, SMYD2 overexpression promotes oncogenic transformation, and high protein levels have been reported in various cancers such as gastric, breast, cervical, pancreatic and lymphoma.181–184
SMYD2 inhibitors
Since the structure description of SMYD2 that was specified in 2011,185 several pharmaceutical companies have focused research interest on this enzyme. Despite great efforts so far, no SMYD2 inhibitor has been subjected to a clinical-evaluation process, and all compounds reported (Fig. 18) are still at the research level.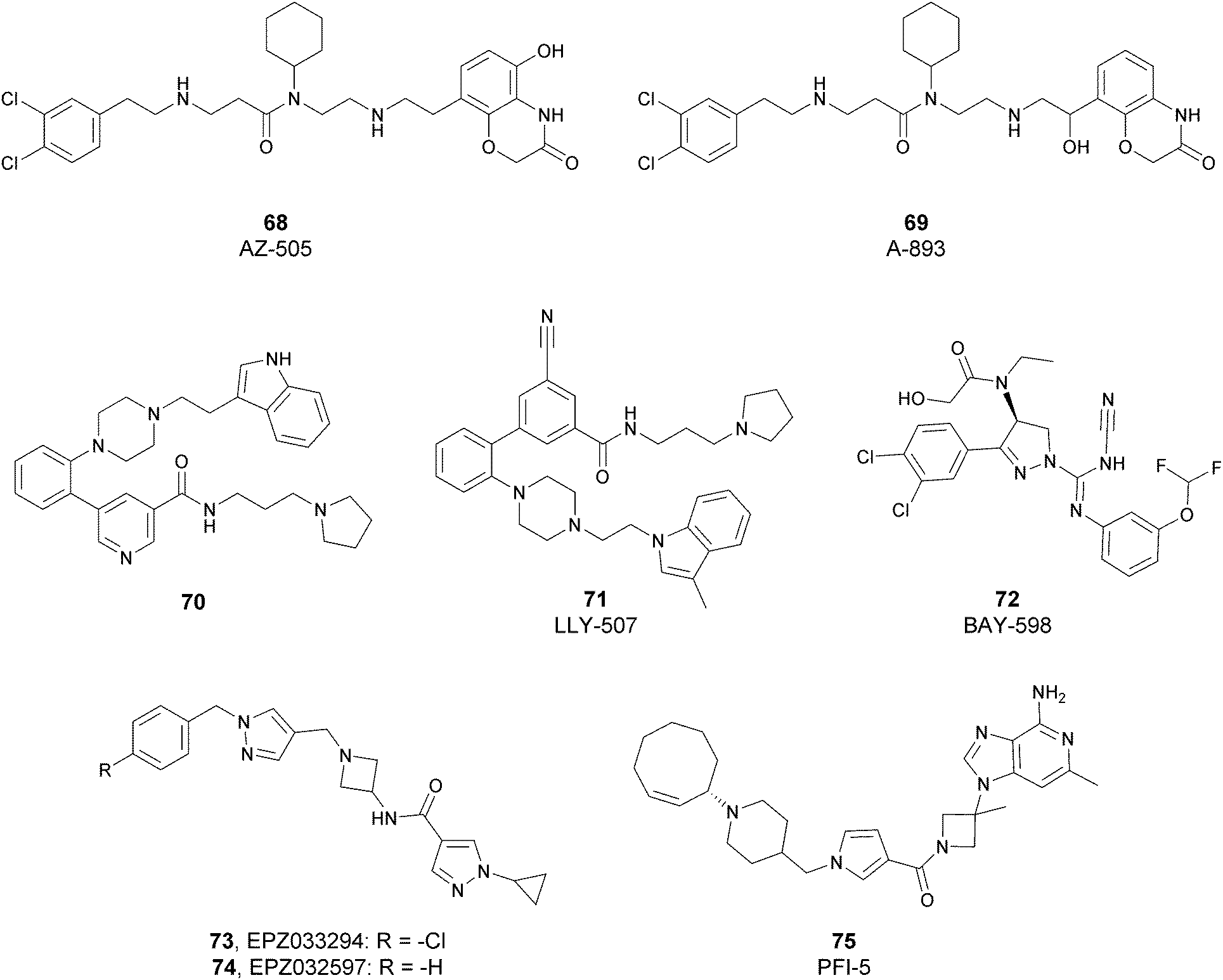 | ||
| Fig. 18 SMYD2 inhibitors. | ||
In 2011, within a large screening campaign, Ferguson and co-workers from AstraZeneca identified the first substrate-competitive and selective SMYD2 inhibitor 68 (AZ-505). This compound showed an inhibitory potency in the sub-micromolar range (IC50 of 0.12 μM) and the crystallographic structure of SMYD2 in the complex of the cofactor SAM and 68 confirmed the mechanism of action of the inhibitor, giving interesting guidelines for further optimization.185 Starting from 68, the formal shift of the hydroxyl group from position 5 of the benzoxazinone to the amino ethyl linker at position 8 resulted in an increased potency, as compound 69 (A-893) is 80-fold more active than the parent compound (IC50 of 2.8 nM). The co-crystal structure of SMYD2 with the inhibitor (PDB 4YND) highlighted the binding mode of this compound. As depicted in Fig. 19, the ether oxygen of the benzoxazinone accepts a hydrogen bond from the side-chain hydroxyl of Tyr258 of the lysine channel, and the ring amine maintains a water-bridged interaction. The configuration of the chiral carbon has only a marginal influence on the in vitro inhibitory activity (IC50 of 2.8 vs. 5.6 nM for (R)- and (S)-69, respectively). 69, as the parent compound, is a substrate-competitive inhibitor with a good selectivity for SMYD2 over a panel of 31 PMTs and can reduce p53 methylation levels by 42% in A549 lung cancer cells.186
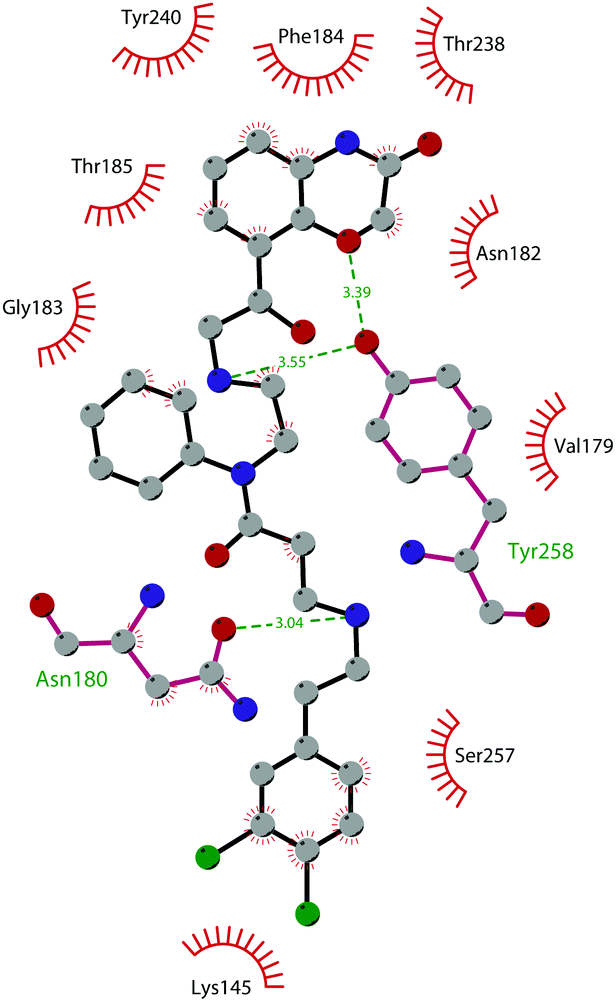 | ||
| Fig. 19 Flat representation (generated using LigPlot+ from PDB ID 4YND) showing contacts between compound 69 and active-site residues in SMYD2. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
In 2013, the AstraZeneca group performed an internal hit identification campaign using a combination of HTS and high-concentration fragment screening, which led to the discovery of different substrate-competitive chemical series of SMYD2 inhibitors.187,188 In order to improve the biochemical and cellular potency as well as the PK properties, scaffold hybridization techniques were used and compound 70 was developed. This new pyridyl inhibitor showed good selectivity towards SMYD2 over a focused panel of KMTs (i.e., EZH2, NSD1, G9a, and DOT1L), as well as an interesting potency both in vitro (IC50 of 17 nM) than in the U2OS cell line (EC50 of 1.02 μM).189
Again, the lead generation efforts by the AstraZeneca group paved the way for further studies that led to the discovery of novel SMYD2 inhibitors. In 2015, Nguyen and co-workers from Eli Lilly and Company, in collaboration with the SGC, designed the potent (IC50 < 15 nM) SMYD2 inhibitor 71 (LLY-507), which shows activity in biochemical as well as cell-based assays. This compound showed high selectivity towards SMYD2 (more than two-orders of magnitude on a panel of 25 different methyltransferases, including SMYD3, and other non-epigenetic targets), which is strictly related to the presence of the pyrrolidine group that is able to bind the lysine pocket of the peptide substrate channel of the enzyme. This substrate-competitive compound reduced p53 methylation at sub-micromolar concentrations (IC50 of 0.6 μM) in osteosarcoma (U2OS) and other cell lines and inhibited proliferation in cancer cells characterized by SMYD2 overexpression.190
In 2016, an optimization of HTS hits by Bayer Pharma resulted in the identification of 72 (BAY-598) as a potent (IC50 of 27 nM) substrate-competitive (Ki of 8 nM) SMYD2 inhibitor endowed with suitable PK properties. In this case, stereoselectivity played a significant role in binding to SMYD2, as the S-enantiomer is 50-fold more potent than the R-enantiomer (IC50 of 27 nM vs. 1.7 μM for (S)-72 and (R)-72, respectively). 72 exhibited more than 100-fold selectivity toward SMYD2 on a panel of 32 PMTs, including the related SMYD3 and SUV420H1/H2. The high inhibitory activity of 72 was confirmed in HEK293T (embryonic kidney) cells in which it decreased p53 methylation in a dose-dependent manner (IC50 of 58 nM) without altering the protein levels. Nevertheless, when tested on a variety of 240 cancer cell lines of different origin, the antiproliferative effect of this inhibitor was limited. In vivo studies on mice bearing subcutaneous tumour xenografts derived from the KYSE-150 cell line overexpressing SMYD2 confirmed the capability of this compound to reduce methylation upon oral administration. On the other hand, only mice groups treated with 72 in combination with doxorubicin exhibited a considerable decrease of tumour area with respect to monotherapy or control groups.191
In 2018, Thomenius and co-workers (Epizyme Inc.) described two novel SMYD2 inhibitors, 73 (EPZ033294) and 74 (EPZ032597), characterized by a very good potency (IC50 of 3.9 nM and 16 nM, respectively) against SMYD2 methyltransferase activity. Both compounds showed a 3-fold selectivity ratio toward SMYD2 over a panel of 15 PMTs, including SMYD3. The crystal structure of the ternary SAM–SMYD2–73 complex underlined the unique binding mode of 73, positioning the cyclopropyl pyrazole portion in the lysine groove and the hydrophobic tail into a new channel generated by the presence of the inhibitor itself.192 It is noteworthy that, due to its nanomolar inhibitory activity, 73 is the most potent and the first non-competitive SMYD2 inhibitor known so far.
In 2019, Taylor and co-workers, within a HTS campaign of a large number of SAM-mimicking compounds, identified the first SAM-competitive SMYD2 inhibitors. The structural optimization of a promising hit resulted in the development of 75 (PFI-5), characterized by the presence of a 3-deaza-adenine ring. Moreover, the amino acid side chain and the ribose of SAM were replaced with a linear string of three rings and an azetidine group, respectively. 75 showed a good capability to inhibit the methylation of p53 in MCF7 cells (IC50 of 1.3 μM). In addition, this inhibitor showed high selectivity towards SMYD2 (less than 20% inhibition against a panel of 33 PKMTs, PRMTs and DNMTs) when tested at 50 μM.193
SMYD3
SMYD3 (SET and MYND Domain-Containing 3, also known as KMT3E) is a protein that catalyses the methylation of H3K4.194 Structural analysis of the SMYD3 protein reveals the presence of a tetratricopeptide repeat (TPR) domain, which is responsible for the peculiar structure of the substrate-binding region and a deep and narrow pocket, which constitutes the C-terminal domain of SMYD3. Both the TPR and post-SET domains are of main importance for the methyltransferase activity.195SMYD3 is not able to methylate H3K4 on free histones or on nucleosomes; however, HSP90A can assist SMYD3 in the protein folding, and methylation can occur at H3K4. This interaction of HSP90A with SMYD3 takes place via the MYND domain and the TPR motif.196
Among the non-histone substrates, SMYD3 methylates K831 in VEGFR1, resulting in an enhancement of the kinase activity of the enzyme, which is strictly connected to the migration and invasion of cancer cells.197 In addition, SMYD3 plays a pivotal role in the regulation of Ras signalling in oncogenic processes through the methylation of MAP3K2.198
SMYD3 expression has been reported in different tissues such as the brain, kidney, and ovary with the highest expression in skeletal muscle and the thymus. Like SMYD2, SMYD3 is involved in embryonic development, as it has a key role in early embryonic lineage commitment and peri-implantation development in mice, thanks to the activation of specific genes.199
From the pathological side, SMYD3 is responsible for the transcriptional regulation of carcinogenesis and the development of human cancer.200,201 This protein activates the transcription of a set of downstream genes (oncogenes, cell adhesion genes, and signal transduction genes) and it is overexpressed in different human cancers (especially hepatocellular and colorectal carcinomas).202 However, the molecular mechanism by which SMYD3 regulates cancer pathways and its relationship to tumorigenesis in vivo are largely unknown.
SMYD3 inhibitors
As described above for SMYD2, an intense involvement of several pharmaceutical companies in the discovery of SMYD3-modulators resulted in the identification of a good number of validated SMYD3-selective inhibitors (Fig. 20), which can be employed as tools to investigate the effects of SMYD3 inhibition. | ||
| Fig. 20 SMYD3 inhibitors. | ||
As the result of in silico structure-based HTS, in 2015 Peserico and co-workers described 76 (BCI-121) as the first-in-class SMYD3 inhibitor that is able to significantly reduce selected methylation marks at 100 μM. This compound demonstrated a good potency both in biochemical (IC50 of 3 nM) and cellular (IC50 of 36 nM) assays, which also confirmed it to be orally bioavailable and suitable for in vivo analyses. The treatment of HCT116 and HT29 (Caucasian colon adenocarcinoma) cell lines with 76 at 100 μM caused a substantial antiproliferative effect after 72 h (54 and 46% inhibition of cell growth, respectively).203
In 2016, starting from visual inspection of the ternary SMYD3–MEKK2 peptide–SAH complex, Van Aller and co-workers designed bi-substrate inhibitors. A series of aza-SAH analogues was synthesized and tested as SMYD3 inhibitors, focusing on the identification of a better side-chain length for good inhibition. The most potent (IC50 of 130 nM) derivative was 77 (GSK2807), which is characterized by a propyl dimethylamine side chain, and is able to fit into the lysine channel anchoring the inhibitor in the pocket with a high degree of similarity with SAH-binding mode. The inhibition of methyltransferase activity by 77 was selective for SMYD3 toward a panel of 8 methyltransferases, and was 24-fold less active toward SMYD2. Despite these interesting data, the poor PK properties of this compound did not permit in vivo assays.204
In the same year, screening of the proprietary histone methyltransferase-biased library and subsequent hit optimization phases enabled Epizyme Inc. to develop 78 (EPZ031686) and 79 (EPZ030456). These derivatives showed a common oxindole scaffold, capable of making several interactions with amino acid residues in the lysine channel of SMYD3. 78, whose 4,4,4-trifluorobutyl chain is responsible for increasing of the lipophilicity of the molecule, showed strong activity both in biochemical (IC50 of 3 nM) and cellular (IC50 of 36 nM) assays. In the same way, the insertion of a sulphonamide portion allowed the identification of 79, which was characterized by a nanomolar inhibitory activity in biochemical (IC50 of 4 nM) and cellular (IC50 of 48 nM) assays. Both derivatives were selective SMYD3 inhibitors, even if the low solubility of 79 troubled its in vivo testing.205
Two years later, after a screening campaign on isoxazole sulphonamides, the same company identified 80 (EPZ028862), which was active both in biochemical (IC50 of 1.3 nM) and cellular (IC50 of 32 nM) assays with satisfactory PK properties for further in vivo studies using different animal models. This derivative was a selective SMYD3 inhibitor that is able to reduce methylation marks on MEKK2 in 293T cells co-transfected with SMYD3 and MEKK2. The crystal structure of 80 in complex with the protein (PDB 5V37) disclosed a binding mode similar to 79, which is probably due to the fact that both compounds are characterized by a common azabicyclo-octanyl moiety, as well as a sulphonamide group, and is able to involve different protein moieties. Moreover, as shown in Fig. 21, the acetamide motif is responsible for the interactions with both Thr184 and Tyr239 residues of the binding pocket. Despite the good activity profile, no antiproliferative activity was observed when tested in lung cancer cell lines, and no effect on cell growth was detected in a panel of other cancer cell lines.192
 | ||
| Fig. 21 Flat representation (generated using LigPlot+ from PDB ID 5V37) showing contacts between compound 80 and active-site residues in SMYD3. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
In 2018, a collaboration between SGC and Bayer allowed the identification of 81 (BAY-6035), a substrate-competitive SMYD3 inhibitor with nanomolar IC50 values both in vitro (88 nM) and in cell-based (70 nM) assays toward the methylation of MEKK2. 81 is commercially available as a chemical probe for SMYD3, even if the corresponding publication has not been yet released.
Starting from a HTS campaign of 503954 compounds, in 2019 Huang and co-workers identified the tetrahydroacridine derivative 82 (IC50 of 4.78 μM) as a reasonable chemical starting point for the development of new SMYD3 inhibitors. Subsequent SAR studies and further optimization resulted in the discovery of a new class of 4-chloroacridines that are able to covalently modify SMYD3 via a nucleophilic aromatic substitution reaction at position 4. Among them, compound 83 showed a high inhibitory potency towards the enzymatic activity of SMYD3 (IC50 of 11.7 nM), as well as antiproliferative activity against the HepG2 human liver cancer cell line in 3D cell culture.206
Recently, a new HTS within the GSK proprietary compounds collection resulted in the identification of compound 84, an isoxazole amide related to 80, which is characterized by a lower biochemical potency (IC50 of 5 μM vs. 1.3 nM for 84 and 80, respectively) but has an interesting ligand efficiency (LE of 0.35) and PK properties. Modifications of the HTS hit on the basis of the structure-guided SAR study resulted in the identification of compound 85, a potent (IC50 of 6.3 and 44 nM in biochemical and cellular MEKK2me assays, respectively), selective and orally bioavailable small-molecule inhibitor of SMYD3, and is considered a useful tool to explore the SMYD3 function in cancer as well as other disorders.207
Very recently, Talibov and co-workers identified 86 (diperodon) as an allosteric SMYD3 ligand. The R- and S-enantiomers showed a KD of 42 and 84 μM, respectively, and co-crystallization studies disclosed that both compounds were able to bind to an allosteric site in the C-terminal protein binding domain, representing a novel starting point for the design of new compounds that interact with a new druggable allosteric site.208
SETD2
SETD2 (also known as KMT3A and HYPB) was first isolated from human hemopoietic stem cells, and it represents a unique protein since it is the sole methyltransferase that is responsible for the trimethylation of H3K36.209 SETD2 has a high homology with the yeast protein SET2, which was previously identified in S. cerevisiae and currently SET2 is the sole nucleosomal H3K36 methyltransferase reported in yeast (responsible for all the methylation states of H3K36), where it acts as a transcriptional repressor.210 Besides H3K36, two non-histone substrates of SETD2 are mainly reported, which are α-tubulin (methylated at K40)211 and STAT1 (methylated at K525).212 Nevertheless, in a recent study, other additional non-histone peptide substrates were identified and nine of them showed strong methylation at the target lysine.213From the structural point of view, SETD2 is characterized by the presence of different functional domains: the methyltransferase activity domains (AWS, SET and post-SET domains), the protein-binding domains (WW (tryptophan–tryptophan), CC (coiled–coiled), SRI (Set2–Rpb1 interacting)) and other domains with a function that is still unknown.214,215
SETD2 is involved in different important cellular processes, such as transcriptional regulation, DNA damage repair and cytoskeletal remodelling. Regarding transcriptional regulation, SETD2, exploiting its SRI domain, binds to the hyperphosphorylated C terminal domain of RNA polymerase II, and it trimethylates H3K36 allowing the recruitment of reader proteins (MRG15, PTB) during transcriptional processes.216 SETD2 contributes in maintaining the microtubule stability via the methylation of α-tubulin, highlighting the cross-talk between the epigenome and the cytoskeleton.211
From the pathological point of view, it has been reported that mutations in the sequence or aberrations in the SETD2 activity are linked to renal cancer,217 glioma218 and leukaemia.219 In particular, these alterations can lead to tumorigenesis, the progression of cancer and unfavourable prognosis, even if the exact mechanism is still to be clarified.220–222
SETD2 inhibitors
So far, the only known inhibitors of SETD2 are sinefungin derivatives (Fig. 22). With the aim of identifying target-specific methyltransferase inhibitors, in 2012 Zheng and co-workers carried out screening against a panel of methyltransferases of N-alkyl sinefungin analogues as privileged small-molecule scaffolds. N-Propyl sinefungin (87, Pr-SNF) and N-benzyl sinefungin (88, Bn-SNF) were identified as SAM-competitive inhibitors of human SETD2-specific inhibitors with definite potency and selectivity. 87 and 88 exhibited an IC50 of 0.80 and 0.48 μM, respectively, a value that is 10-fold higher than that of sinefungin itself and displayed 2/200-fold and 10/100-fold preferences, respectively, for SETD2 over other examined methyltransferases. Specifically, 87 was shown to preferentially interact with SETD2 by matching the distinct transition-state features of SETD2's catalytically active conformer, locating its secondary amine at the substrate–cofactor interface and the N-alkyl chain in the lysine-binding pocket.223 | ||
| Fig. 22 SETD2 inhibitors. | ||
In fact, as depicted in Fig. 23, the SETD2–87 co-crystal structure (PDB 4FMU) shows that the N-propyl moiety of the inhibitor is buried in a hydrophobic binding pocket formed by Tyr1579, Tyr1605, Phe1664, and Tyr1666. On the other hand, a large number of hydrogen bonds are also detected, engaging the hydroxy groups of the ribose portion and His1603, Tyr1604 and Gln1676. Despite their specificity, the activity of 87 and 88 is influenced by the peptide substrate, and their poor membrane permeability prevented in cell or in vivo assays.
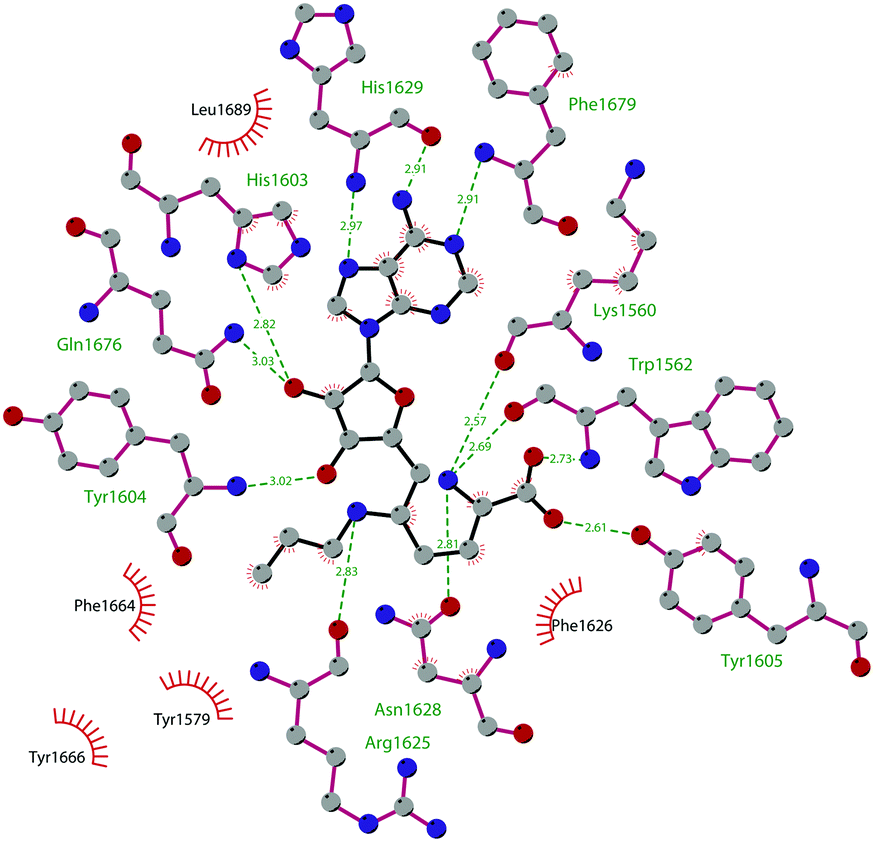 | ||
| Fig. 23 Flat representation (generated using LigPlot+ from PDB ID 4FMU) showing contacts between compound 87 and active-site residues in SETD2. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
SETD8
SET domain-containing lysine methyltransferase 8 (also called PR-SET7 or KMT5A) is a member of the SET domain-containing family and is characterized by a unique activity as it is the only reported mammalian protein responsible for the monomethylation of H4K20 in vivo. SETD8 preferentially monomethylates nucleosomes instead of histones, suggesting this protein is able to interact with other regions of the nucleosome as well as the H4 N-terminal region surrounding the residue H4K20.23 These multivalent interactions of SETD8 involve both the SET domain and a basic region N-terminal to the SET domain.224,225As with other lysine methyltransferases, SETD8 methylates not only histones but also other proteins, including p53, Numb and PCNA. Monomethylation of p53 K382 by SETD8 triggers the suppression of p53-dependent transcription activation in cancer cells.226 Numb is methylated by SETD8 on the K158 and K163 residues, and this methylation disrupts the complex Numb–p53–MDM2, which is responsible for the promotion of apoptosis. Hence, the methylation of Numb prevents apoptosis and finally results in the ubiquitination and degradation of p53.227 The monomethylation of K248 of proliferating cell nuclear antigen (PCNA) (PCNAK248me1) enhances its interaction with the flap endonuclease FEN1, promoting carcinogenesis by deregulating the PCNA expression.228
Several studies have provided evidence that SETD8 may be involved in many cellular processes, such as DNA replication, DNA damage response, transcription modulation, and cell cycle regulation as well as in carcinogenesis and cancer invasiveness.229–231 SETD8 is overexpressed in different types of cancer tissues including bladder cancer, non-small-cell and small-cell lung carcinoma, chronic myelogenous leukaemia, hepatocellular carcinoma, pancreatic and breast cancer.232,233
SETD8 inhibitors
To better elucidate the SETD8 biology as well as its effects in both normal and diseased cells, some inhibitors have been developed (Fig. 24), even if just a few of them displayed a certain degree of selectivity and/or cellular activity.22 | ||
| Fig. 24 SETD8 inhibitors. | ||
The screening of a library of marine natural products allowed the identification of the polyketide 89a (nahuoic acid A), a decalin polyketide isolated from cultures of Streptomyces sp. obtained from a marine sediment collected in Papua New Guinea. 89a inhibited SETD8 activity with an IC50 value of 6.5 μM, and with no significant effect on other tested enzymes. Cellular treatment with 89a led to a decrease in the levels of both mono- and trimethylated H4K20 without affecting other methylated histone marks.234,235 Exposure of U2OS cells to 89a and its prodrug version (nahuoic acid A pentaacetate, 89b), prepared to improve the cellular permeability, resulted in a modest decrease of cellular proliferation (IC50 of 65 μM and 39 μM, respectively). The same results were obtained when treating breast cancer cell lines such as SUM159 and MDA-MB-436.
In 2014, Luo and co-workers reported three small molecules (90–92) as SETD8 inhibitors, which were identified via the HTS of a library containing more than 5000 commercially available compounds. These derivatives differ in their mechanisms for SETD8 inhibition: 90 (SPS8I1, IC50 of 0.21 μM) turned out to be a substrate-dependent inhibitor, 91 (SPS8I2, IC50 of 0.5 μM) exhibited no substrate or SAM dependence, while 92 (SPS8I3, IC50 of 0.5 μM) was found to be both a substrate- and SAM-dependent inhibitor. In all cases, the quinonic motif, common to all inhibitors, interacts with active cysteine residues (Cys270) in SETD8 through an irreversible slow-onset process. According to SETD8 knockdown, these compounds produced a cell cycle arrest phenotype and reduced the monomethylation of H4K20. Unfortunately, their low selectivity against other protein methyltransferases and other cellular targets prevents their further development.236
In the same year, Jin and co-workers reported the identification of 93 (UNC0379) via a cross-screening of a library containing more than 150 quinazoline-based compounds, the same scaffold previously explored for G9a ligands. In a radioactive methyl transfer assay, 93 showed an IC50 value of 7.3 μM. This compound showed no appreciable activity on 14 other methyltransferases tested, with IC50 values above 100 μM also for G9a and GLP. By contrast, compound 93 inhibited PRC2 only at the two highest concentrations (50 and 100 μM), showing an IC50 value of more than 50 μM. A SAR study was undertaken on the 4-amino group of the quinazoline scaffold, showing that reducing the length of the alkyl linker between the two amino groups resulted in a lower potency. Moreover, it was shown that the basicity of the pyrrolidinyl group was critical for the SETD8 inhibitory activity. Regarding the mode of action, 93 acts as a substrate-competitive inhibitor, non-competing with the cofactor SAM, showing a good selectivity over other methyltransferases.237 Recently, it has been shown that the pharmacological inhibition of SETD8 by 93 led to cell-cycle defects and apoptosis in a p53-dependent or -independent manner. In particular, SETD8 inhibition in multiple myeloma (MM) cell lines induced by 93 led to cell death activating the p53 canonical pathway. However, 93-induced cytotoxicity in MM cells is not necessarily dependent on p53 activation but is mainly caused by cellular stress during G1 progression. Moreover, a low dose of 93 co-administered with the cytotoxic agent melphalan strongly increases DNA damage and overcomes drug resistance in both p53 wild-type and deficient MM cells. This evidence suggested that targeting SETD8 could be an alternative therapeutic strategy in high-risk patients with multiple myeloma independently of their mutational status for p53.238
In 2016, a higher potent inhibitor, 94 (MS2177), was identified by inserting an aminoethyl group as a side chain on 93. Indeed, the resulting compound 94 had an in vitro IC50 of 1.9 μM and a much lower affinity in isothermal titration calorimetry (ITC) experiments compared with 93 (KD of 1.3 μM vs. 18 μM). The compound was found to be competitive with the substrate but not with the co-factor SAM.239 In the same study, a potent covalent SETD8 inhibitor, 95 (MS453), was identified by inserting into compound 93 an acrylamide group as the electrophilic warhead, and which is responsible for the reaction with a surface-exposed Cys311 in the active site. The crystal structure of 95 in complex with SETD8 (PDB 5TH7) showed the formation of a homodimer in which each covalently modified Cys311 residue locates the quinazoline moiety to its binding site of the other complementary subunit. As illustrated in Fig. 25, the compound establishes a hydrogen bond between the secondary amino group in the 4-position and the carbonyl oxygen of Cys311 on the peptide backbone. Moreover, 95 makes solvent-mediated hydrogen bonds between the quinazoline N1 nitrogen and the carbonyl groups on the protein backbone, inserting the 2-pyrrolidine motif into a hydrophobic pocket induced by the binding of the inhibitor to the protein.
 | ||
| Fig. 25 Flat representation (generated using LigPlot+ from PDB ID 5TH7) showing contacts between compound 95 with active-site residues in SETD8. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compound. | ||
95 showed an IC50 value of 804 nM with a good selectivity over 28 other methyltransferases. Unfortunately, no significant reduction in the methylated H4K20 levels was observed from treating HeLa cells with the inhibitor (10 μM), probably due to its poor cellular permeability.239
SUV420H1 and SUV420H2
SUV420H1 (KMT5B) and SUV420H2 (KMT5C) are two highly homologous enzymes in mammals (65% sequence identity in the catalytic domain). Although both enzymes can use as a substrate the unmodified H4K20, they prefer the monomethylated H4K20 as a substrate in vivo and exploit the SETD8-generated monomethylated H4K20 to produce di- and trimethylated H4K20.240 In particular, it has been reported that, in mouse embryonic fibroblasts, SUV420H1 catalyses the formation of most of the dimethylated H4K20 while SUV420H2 is responsible for trimethylated H4K20. The catalytic domains of SUV420H1 and SUV420H2 consist of three subdomains: an N-terminal helical domain, a SET domain and a Zn-binding post-SET domain. SUV420H1 has a bigger N-domain than SUV420H2 due to a 22-residue insertion between the first and second helices.241SUV420H1 and SUV420H2 proteins play critical roles in the chromatin structure, genomic integrity and in the regulation of the cell cycle. Indeed, both these proteins are linked to transcriptional repression, DNA replication and repair.242,243 Therefore, the loss of both of them is accompanied with chromosomal aberrations, deficiencies in DNA double-strand break repair and reduction in telomere elongation, together with a decrease in proliferation rates and in cell cycle progression.230,243–245 SUV420H1 and SUV420H2 are pivotal regulators of normal embryonic development and in self-renewal or lineage-commitment of stem cell populations. Indeed, the loss of SUV420H1/2 results in perinatal lethality. Alterations in the expression and mutations of SUV420 enzymes are related to the insurgence of cancer. Regarding SUV420H2, it has been reported that in breast cancer and in osteosarcoma there is a reduction in the levels of trimethylated H4K20, together with a decrease in the expression of SUV420H2, which is associated with invasiveness and poor prognosis.246 However, in pancreatic cancer, SUV420H2 is overexpressed and it is associated with invasive cancer. In particular, it has been reported that this protein behaves as a regulator of epithelial/mesenchymal state control and its high levels promote the epithelial-to-mesenchymal transition, which is related to metastasis.247 Regarding SUV420H1, it has been reported that this protein is overexpressed and mutated in different cancers. On the basis of the type of mutation, different outcomes in cancer have been observed, where the protein can assume a tumour suppressor or a tumorigenic role.248 Hence, we can conclude that both SUV420 enzymes have multiple and divergent roles in cancer that need to be fully elucidated.
SUV420H1 and SUV420H2 inhibitors
Although the identification of modulators of these enzymes may be essential for understanding their function, so far only three inhibitors have been reported (Fig. 26). In 2017, Bromberg and co-workers developed a high-throughput homogeneous activity-based scintillation proximity assay with the aim of identifying SUV420 inhibitors. A library of approximately 470000 compounds was screened, and among them 96 (A-196) was described as a first-in-class chemical probe of both SUV420 enzymes and a good starting point for the development of new inhibitors. Co-crystallization and biochemical analyses confirmed that 96 is a substrate-competitive inhibitor of SUV420H1 and SUV420H2 with IC50 values of 25 and 144 nM, respectively. Examination of the crystal structure of 96 bound to SUV420H1 (PDB 5CPR) revealed that the cyclopentyl moiety is able to occupy a hydrophobic pocket made by Ser251, Val252, Tyr307 and Met253, which could be further explored. On the other hand, the pyridyl unit is located in a solvent-exposed region (Fig. 27). A further global decrease in di- and trimethylated H4K20 and a simultaneous increase in monomethylated H4K20 were found after treatment of the prostate adenocarcinoma cell line PC-3 or U2OS osteosarcoma cells with 96, without any toxic effects.
 | ||
| Fig. 26 SUV420H1 and SUV420H2 inhibitors. | ||
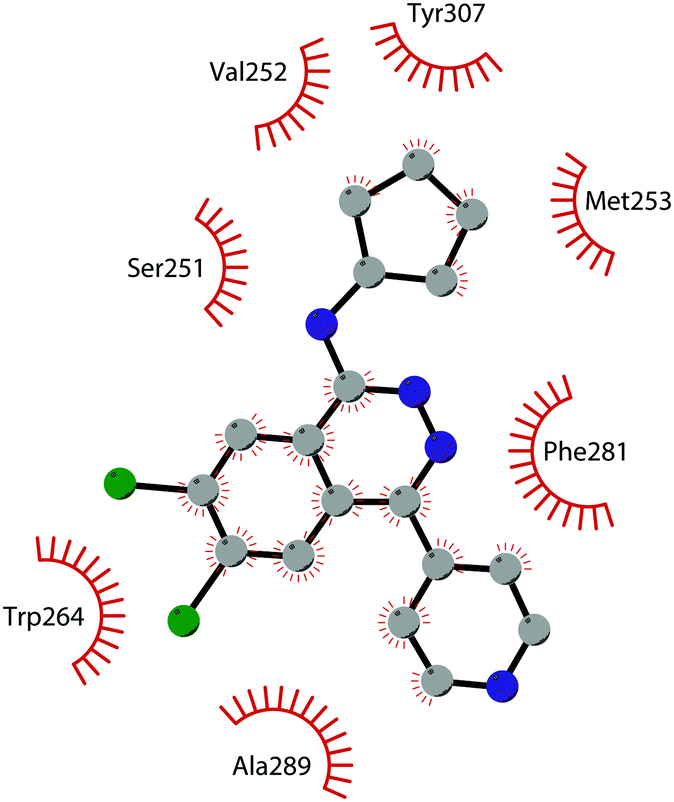 | ||
| Fig. 27 Flat representation (generated using LigPlot+ from PDB ID 5CPR) showing contacts between compound 96 and active-site residues in SUV420H1. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
Furthermore, this inhibitor does not induce resistance/compensation effects, as di- and trimethylated H4K20 remained low after 20 population doublings.249
Recently, a HT and SBVS combined with molecular dynamics simulation has been used by Zheng and co-workers in order to find novel potential SUV420H1 inhibitors. Starting from the Specs subset of the ZINC database, containing over 200000 compounds, 97 (ZINC08398384) and 98 (ZINC08439608) were selected and showed an inhibitory effect similar to the known inhibitor 96, effectively reducing the expression level of dimethylated H4K20 in U2OS (at 1.25 and 2.5 μM, respectively) and attenuating the migration of osteosarcoma cells in vitro.250
NSD1, NSD2, NSD3
The NSD (Nuclear receptor SET Domain-containing) family is a phylogenetically distinct family of methyltransferases comprised of NSD1, NSD2 (MMSET/WHSC1) and NSD3 (WHSC1L1), three proteins with pivotal roles in chromatin regulation. These proteins share the presence of a catalytic SET domain (with a high homology with the SETD2 protein) and multiple conserved domains, such as proline–tryptophan–tryptophan–proline (PWWP) and the plant homeodomain (PHD) zinc finger domains, which are involved in the interaction with DNA and other methylated histones.251Regarding their specific substrates, NSD proteins catalyse the methylation of several lysine marks on histones H3 and H4.252 In particular, NSD1 dimethylates H3K36 and H4K20, whereas NSD2 methylates H4K20, H3K4, H3K27 and H3K36, and NSD3 methylates H3K4 and H3K27.253,254
NSD proteins play essential roles in embryonic development, and it has been shown that the deletion of NSD1 protein causes problems in the development of post-implantation embryos.255 Moreover, the dysregulation of NSD proteins in human cells affects cell growth and differentiation and has been related to several diseases, such as mental retardation and developmental defects that are common symptoms of Sotos syndrome256,257 and Wolf–Hirschhorn syndrome.258,259 In addition, aberrant activity of NSD proteins, as well as their overexpression, has been linked to the insurgence of neoplastic diseases such as breast carcinoma, acute myeloid leukaemia, and multiple myeloma.260–262
NSD1, NSD2, NSD3 inhibitors
Although NSD proteins are appealing therapeutic targets, a limited number of small-molecule inhibitors have been described so far (Fig. 28). Based on the homology of the catalytic SET domain of the NSDs to that of G9a-like protein, in 2017 the G9a/GLP inhibitor 22 (Fig. 8) was tested on NSD proteins, showing an in vitro inhibitory activity on all these targets and lowering monomethylated H3K36 levels. Specifically, 22 revealed the highest inhibition on NSD2 (IC50 of 41 μM), followed by NSD3 (IC50 of 95 μM), and finally NSD1 (IC50 of 112 μM). This different activity has been associated with the diverse binding modes of the compound towards the catalytic portion of the three proteins. Specifically, 22 is able to shallowly bind into the lysine channel of NSD1; conversely, in NSD2, the diazepane side of the molecule fits into the lysine channel; in NSD3 the dimethoxy quinazoline group spreads deeply into the lysine channel, like in GLP. Furthermore, the better IC50 value observed against NSD2 is probably due to a tighter hydrogen-bond network in the NSD2 active site compared with NSD1 and NSD3.263 | ||
| Fig. 28 NSD1–3 inhibitors. | ||
Recently, an NMR fragment-based screening approach, followed by chemical optimization, resulted in the identification of 99 (BT5), the first-in-class irreversible small-molecule inhibitor of NSD1, whose electrophilic methyl-aziridine moiety is responsible for the covalent binding to the C2062 residue of the NSD1 SET domain. The crystal structure of NSD1 in complex with 99 disclosed a conformational modification in the autoinhibitory loop of the SET domain, leading to the formation of a unique, channel-like site that is suitable for targeting with small molecules. 99 inhibits NSD1 at low micromolar concentrations (IC50 of 5.8 μM upon 4 h incubation, which improved to IC50 of 1.4 μM after 16 h due to irreversible binding). NSD2 and NSD3 inhibition was also observed, although non-covalent and at higher concentrations. The cellular thermal shift assay confirmed that 99 is able to bind the NSD1 SET domain in cells, generating a global reduction of dimethylated H3K36 levels without perturbing trimethylated H3K36.264
The natural product sinefungin17 (Fig. 6) and the N-alkyl sinefungin derivative 87 (Fig. 22) have been identified as NSD2 inhibitors (IC50 of 26 μM and 3.3 μM, respectively), but inhibition data on NSD1 and NSD3 have not been reported.265 In 2019, de Luccio and co-workers by VS identified 100 (LEM-14). This inhibitor showed an in vitro IC50 of 132 μM against H3K36 methylation by NSD2, a very weak activity against NSD1, and no activity against NSD3, representing an interesting tool for studying the biology of NSD2.266
In 2021, a VS followed by a ligand-based scaffold-hopping approach, enabled the identification of 101, a small-molecule antagonist that blocks the protein–protein interaction between the N-terminal PWWP domain of NSD2 (PWWP1) and dimethylated H3K36 with a KD of 3.4 μM.267 The crystal structure of 101 in complex with NSD2 and subsequent molecular docking simulations enabled the development of the more potent 102 (MRT866), which is capable of binding NSD2–PWWP1 with a KD of 349 nM. Further structure-based optimization resulted in 103 (UNC6934), which showed potent binding to PWWP1 (KD of 91 nM).268 In order to better understand the binding mode of 103, a representation of the main interaction is shown in Fig. 29. The crystal structure of the inhibitor in complex with NSD2–PWWP1 (PDB 6XCG) shows how the cyclopropyl moiety is inserted in a hydrophobic pocket made of Val230 and Phe266. Moreover, the amidic NH is involved in a hydrogen bond with Gln321, while the carbonyl oxygen establishes a strong interaction with Ala270.
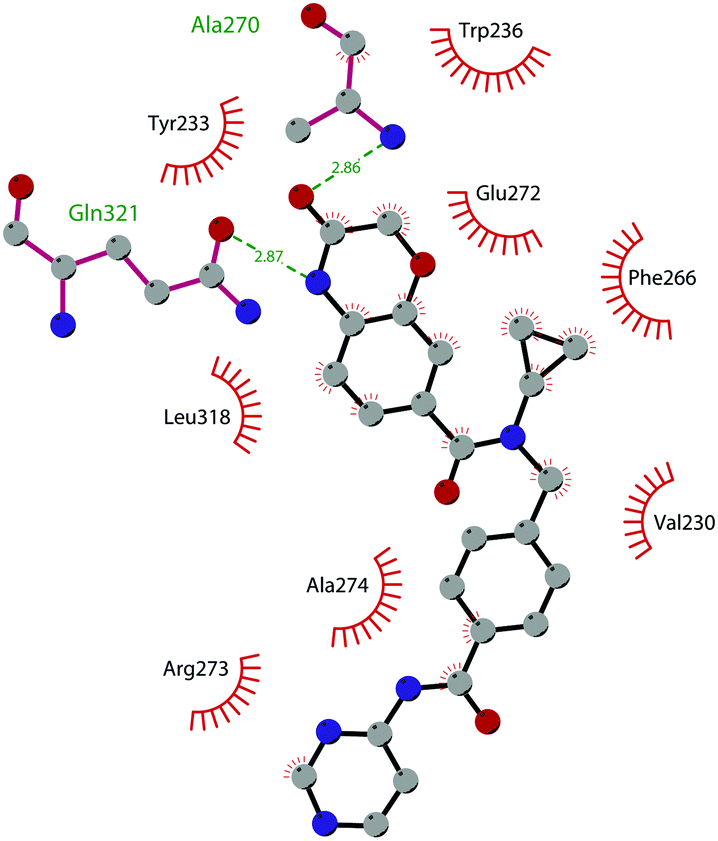 | ||
| Fig. 29 Flat representation (generated using LigPlot+ from PDB ID 6XCG) showing contacts between compound 103 and active-site residues in NSD2–PWWP1. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
In 2019 Mc Connell and co-workers reported the fragment-based virtual screening discovery of 104 (BI-9321), a strong and selective antagonist of the NSD3–PWWP1 domain. This compound showed the capability of targeting the methyl-lysine binding site of the PWWP1 domain with an IC50 of 203 nM and a KD of 166 nM. Moreover, 104 was able to downregulate Myc messenger RNA expression, as well as reduce cellular proliferation in the MOLM-13 human leukaemia cell line.269 The co-crystal structure of NSD3–PWWP1 in complex with 104 (PDB 6G2O) showed the importance of the aminic group in the binding mode of the inhibitor. In fact, as depicted in Fig. 30, this motif is responsible for a hydrogen bond with Glu318, while the pyridinic nitrogen is involved in an interaction with the Ser314 residue of the binding pocket.
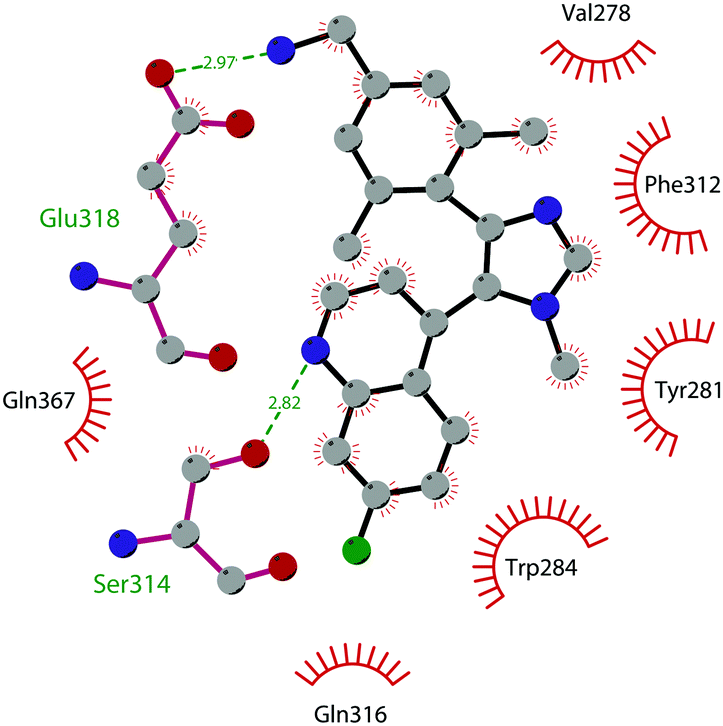 | ||
| Fig. 30 Flat representation (generated using LigPlot+ from PDB ID 6G2O) showing contacts between compound 104 and active-site residues in NSD3–PWWP1. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
MLL proteins
The family of lysine methyltransferase KMT2 is composed of SETD1A, SETD1B and four mixed-lineage leukaemia proteins (MLL 1,2,3, and 4) that play pivotal roles in the regulation of gene expression during embryogenesis and development. These proteins catalyse the methylation of H3K4,270 and they exist in nuclear complexes (human COMPASS), which include ASH2L, RbBP5, Cfp1, WDR5 and Dpy30. The components of the complex greatly enhance the enzymatic activity of MLL methyltransferases.271,272Structurally, MLL proteins are characterized by a peculiar domain architecture: in addition to the C-terminal SET domain, all the proteins of the family feature a different number of DNA-binding and protein–protein interaction domains, which are exploited for binding to specific interaction partners. Despite the structural homology in the organization of the domains, each MLL protein differs from the others on the basis of the type, number and position of each specific domain. In particular, there is high variability in the number of PHD finger domains: four for MLL1 and 2, eight for MLL3, and seven for MLL4.273 In addition, MLL1 and MLL2 display the CXXC zinc-finger domain for the interaction with unmethylated CpG islands, and this domain is absent in the other members of the family.274 The structural differences in MLL proteins are reflected in their enzymatic activity: for instance, while MLL1 and 2 are responsible for both the mono- and dimethylation of H3K4, proteins MLL3 and MLL4 have been reported as monomethyltransferases.275,276
Many studies have shown that alterations in the expression of MLL genes are linked to the insurgence of diseases. Beside the involvement of MLL proteins in the pathogenesis of leukaemia, for instance through the interaction with menin (which lead to myeloid transformation),277 a huge number of somatic mutations for MLL proteins have been identified in specific solid cancer types, mainly of the lung, endometrium, and large intestine.278–280
MLL protein inhibitors
A deeper understanding of the biological role and function of MLL proteins could be triggered by the discovery of potent chemical probes that selectively target them. So far, only a small SAM-based focused chemical library has been designed with the aim of directly targeting the active site of MLL proteins. Among them, compound 105 (TC-5115, Fig. 31) has emerged as the best candidate (IC50 of 16 nM). Its co-crystal structure revealed that the compound is positioned in the cofactor binding site, while the azetidine group induces the “open” conformation of the substrate binding pocket, thus allowing additional interactions between the inhibitor and the MLL SET domain.281 | ||
| Fig. 31 Structure of SAM-based MLLs inhibitor. | ||
On the other hand, efforts made to target protein–protein interactions in the MLL core complex have been more successful. Specifically, disruptors of WDR5–MLL (Fig. 32) as well as menin–MLL interactions (Fig. 34) have been developed.
 | ||
| Fig. 32 WDR5–MLL interaction disruptors. | ||
WDR5–MLL interaction disruptors
The central role of WDR5 in the maintenance of integrity and the methyltransferase activity of MLL complexes has suggested that disrupting the WDR5–MLL interaction is a possible strategy to develop inhibitors of MLL methyltransferases. The identification of a conserved arginine-containing motif within MLL, i.e., the WIN (WDR5-interacting) sequence, is responsible for the association with WDR5 and, consequently, is essential for the enzymatic activity of MLL1282 and represents a good starting point from which to design disruptors of WDR5–MLL interactions.Karatas and co-workers reported the design of peptidomimetics to disrupt WDR5–MLL interactions. Specifically, the identification of the minimum motif responsible for the high-affinity binding of MLL1 to WDR5 (–CO–ARA–NH–), led to the design of two 3-mer peptides (Ac–ARA–NH2 and Ac–ART–NH2) as efficient WDR5 binders with Ki values of 120 and 20 nM, respectively.283 Further extensive modifications enabled the identification of 106 (MM-102), a potent peptidomimetic compound (Ki of <1 nM) that mimics the WIN peptide for binding to WDR5. However, 106 exhibits a very modest cellular potency and poor metabolic stability in human microsomes.284 It was anticipated that the reduction of conformational flexibility could overcome these issues. Therefore, the linear structure of the parent compound was replaced with a macrocyclic equivalent, obtaining diverse compounds.285,286 Among them, derivative 107 (MM-589) binds to WDR5 with a very high affinity (Ki less than 1 nM), significantly inhibiting the MLL1 methyltransferase activity (IC50 of 12.7 nM). Moreover, 107 inhibits cell growth in the MOLM-13 and MV4-11 human AML cell lines that carry the MLL translocation with more than 30-fold selectivity over the HL-60 leukaemia cell line that lacks the MLL translocation.286 In a screening of commercially available compounds, 108 (WDR5-0103) was identified as a modest WDR5 binder (Kdis of 3.0 μM in a peptide-displacement assay, and KD of 0.45 μM). The compound efficiently antagonizes the WDR5–WIN peptide interaction, negatively affecting the catalytic activity of MLL complexes in vitro.287 The X-ray crystal structure supported the optimization of 108-based nitro analogues, facilitating the identification of 109 (WDR5-47), an improved WDR5 binder (Kdis of 0.3 μM).288 Replacement of the nitro group with the 4-piridinyl group led to 110 (W-23), which efficiently disrupted the MLL1–WDR5 interaction with an IC50 value of 104 nM.289
Further optimization yielded derivative 111 (WL-15), which better disrupted the interaction of MLL1–WDR5 (IC50 of 26.4 nM) and inhibiting the catalytic activity of the MLL1 complex (IC50 of 5.4 μM). Unfortunately, the poor cellular permeability limited its cellular activity in inhibiting the proliferation of leukaemia cell lines with or without the MLL1 fusion protein.290
Based on the N-(2-(4-methylpiperazin-1-yl)phenyl)acetamide scaffold, other derivatives were designed as disruptors of MLL1–WDR5 interactions with increased cellular activity. Specifically, 112 (DDO-2084) inhibited MLL1 methyltransferase activity both in vitro (IC50 of 1.65 μM) and in the MV4-11 cell line (IC50 of 17.7 μM), showing a down-regulation of the MLL-target genes expression level. Insertion of the 4-aminobutanamido group on 112 improved the inhibitory activity: compound 113 (DDO-2117) exhibited an IC50 of 0.19 μM and strongly inhibited the MLL1–WDR5 interaction (IC50 of 7.6 nM and KD of 13.6 nM).291
Additional optimization of the substituents in the above-mentioned scaffold led to the identification of 114 (OICR-9429), a compound that competitively disrupted the WDR5 interaction with the WIN motif of MLL (Kdisp of 64 nM) with good WDR5 affinity (KD of 30 and 93 nM in SPR and ITC assay, respectively). 114 is highly selective for WDR5 with no inhibition of 22 other human methyltransferases, 9 different WD40 domains, and other non-epigenetic targets. The crystal structure of WDR5 in complex with 114 helped to clarify its detailed binding mode (PDB 4QL1). The compound is inserted in an arginine-binding pocket of WDR5, precluding the interaction of the WIN peptide residue with MLL. Upon 114 binding, the protein undergoes conformational rearrangements to better accommodate the compound (Fig. 33). In particular, the Phe263 moved to lodge the N-methylpiperazine moiety in position of the WIN peptide guanidinium group, while the Phe133 and Phe149 residues moved to accommodate the opposite side of the molecule. Moreover, the piperazine, the amide linker and the pyridone group interact (directly or by water-mediated hydrogen bonds) with the surrounding residues of Cys261, Ser91 and Asp107, whereas the other side of the molecule interacts with a hydrophobic pocket in WDR5 formed by the Phe133, Tyr191 and Pro173 residues. Critical for compound binding is the interaction with Ser91. Notably, the morpholino ring is solvated, lacking a clear electron density, suggesting that it can assume different conformations. The efficacy and target selectivity of 114 were also confirmed in cells in which the oncogenic transcription factor p-30 drives cell growth in a WDR5–MLL-dependent manner. These findings offer an attractive strategy for targeting cancer depending on the oncogenic transcription factor mutations.292,293
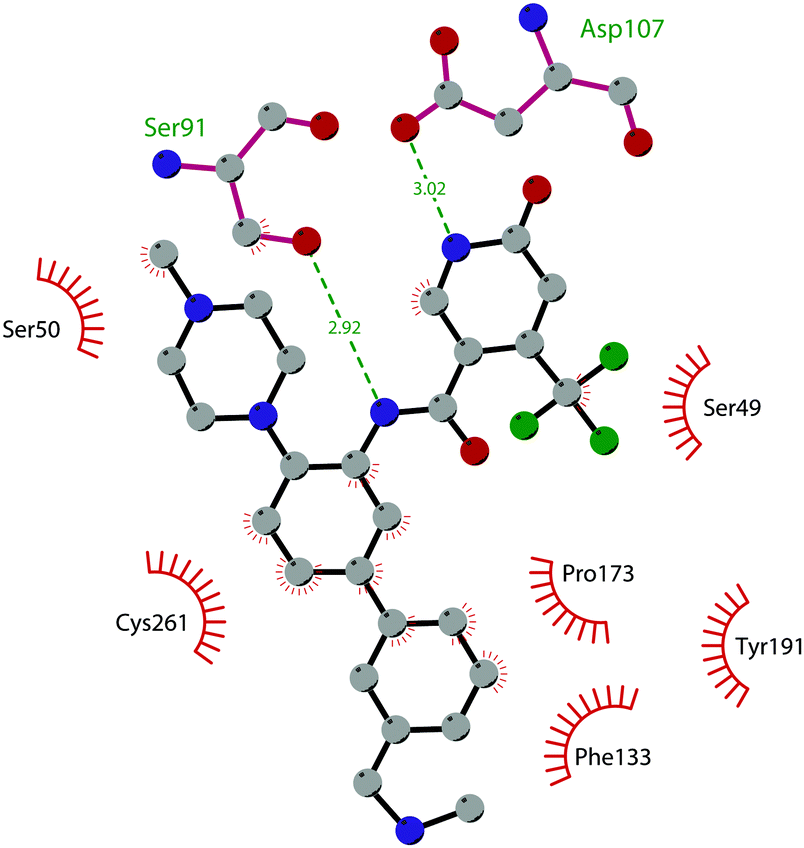 | ||
| Fig. 33 Flat representation (generated using LigPlot+ from PDB ID 4QL1) showing contacts between compound 114 and active-site residues in WDR5. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compound. | ||
 | ||
| Fig. 34 MLL–menin interaction disruptors. | ||
In 2018, exploiting fragment-based methods and structure-based design, dihydro-pyrroloimidazole was disclosed as an effective WDR5 binder. Crystal structure analysis depicted the binding of this core to WDR5 in a pocket that normally accommodates the MLL1–peptide–R3765–guanidine side chain. The most potent inhibitor derivative, 115, displayed a high potency (Ki of 0.9 nM) causing the dose-dependent inhibition of H3K4-methylation activity and a moderate growth reduction in the MV4-11 cell line.294
Menin–MLL interaction disruptors
Another therapeutic strategy explored to develop new drugs for MLL leukaemia patients is via disruption of the protein–protein interactions between MLL and the oncogenic cofactor menin, which are required for the initiation of MLL-mediated leukemogenesis as well as for regulating the expression of target genes, such as HOXA9 and MEIS1 genes, essential for the leukemogenic activity of MLL fusion proteins. Menin binds with two different fragments of MLL: the high-affinity menin binding motif (MBM1 with a KD of 56 nM) and the low-affinity menin binding motif (MBM2 with a KD of 1 μM). Even if MBM2 contributes to binding to menin, deletion of the MBM1 motif is sufficient to abolish the activity of MLL oncoproteins.295 Therefore, small-molecule inhibitors have been developed that mainly target the MBM1 motif.From a structural point of view, most of the MLL–menin inhibitors developed feature a thienopyrimidine core structure.
HTS and further medicinal chemistry optimization guided the identification of the parent compound of this class, 116 (MI-2). It inhibited the the menin–MLL interaction with an IC50 of 446 nM, disrupting the menin–MLL interaction both in vitro and in human cells. The treatment of different MLL leukaemia cell lines with 116 resulted in different effects, such as the downregulation of HOXA9 and MEIS1 expression, inhibition of proliferation and induction of apoptosis and differentiation.296 These pioneering results highlighted the potentiality of targeting menin–MLL interactions in acute leukaemia associated with MLL translocations and prompted the synthesis of several derivatives featuring the thienopyrimidine scaffold.
117 (MI-2-2) showed an 8-fold improved binding affinity toward menin (KD of 22 nM) disrupting the MBM1–menin interaction with high affinity (IC50 of 46 nM) comparable to the fragment including both MBM1 and MBM2. In this way, the compound is able to fully dissociate the entire menin–MLL fusion protein complex.297 Despite the good cellular permeability, 117 showed modest activity in human MLL leukaemia cell lines and poor metabolic stability, limiting in vivo studies. Insertion of a cyano-indole ring connected to the thienopyrimidine scaffold via a piperidine linker furnished the compound 118 (MI-136), which maintained a good inhibitory activity and a strong binding affinity for menin (IC50 of 31 nM and KD of 24 nM, respectively).298 The introduction of a methyl and/or a methyl pyrazole substituent on the indole nitrogen yielded compounds 119 (MI-463) and 120 (MI-503) as efficient menin binders with low-nanomolar binding affinities (IC50 of 15.3 nM and 14.7 nM and KD of 9.9 nM and 9.3 nM, respectively). Treatment of human leukaemia cell lines, such as MV4-11 and MOLM-13 (which express the MLL-AF9 oncogene), with 120 resulted in a strong downregulation expression of homeobox genes (HOXA9, HOXA10, HOXA7, PBX3, and MEIS1) and other downstream targets of MLL fusion proteins. 119 and 120 displayed good metabolic stability and a PK profile in mice, and their in vivo oral administration resulted in a longer survival time in a mouse model of MLL leukaemia.298 Further SAR studies led to the identification of 121 (MI-538), which displayed a strong in vitro inhibition of the menin–MLL interaction and a high binding affinity for menin (IC50 of 21 nM and KD of 6.5 nM, respectively). Compared with 119 and 120, this compound revealed a significant improvement regarding the cellular activity, selectivity for MLL leukaemia cells, and PK properties.299 Additional optimization of the substituent of the indole nitrogen led to an improvement in inhibitory activity over previously identified compounds. 122 (MI-1481) emerged as potent inhibitor of the menin–MLL interaction (IC50 of 3.6 nM). The improved inhibitory activity of 122 could be associated with a stronger interference, not only with the MBM1 motif but also with the MBM2 portion of MLL, resulting in a better displacement of the bivalent MLL peptide from its binding to menin. On the other hand, the increased polarity of 122 negatively affected the oral bioavailability of this compound.300 The introduction of an amino group in the thienopyrimidine scaffold, combined with optimization of the pattern of substituents on the indole ring, furnished 123 (MI-3454). This compound exhibited a sub-nanomolar inhibitory activity (IC50 of 0.51 nM), efficiently disrupting the interaction of menin with an MLL fragment. 123 has a high oral bioavailability and its administration in mice significantly induces a complete remission or regression of leukaemia in different models, including patient-derived xenografts (PDXs).301
Iterative structure-based drug design of a piperazinyl pyrimidine fragment initially identified subsequently yielded 124 (VTP50469), a potent, highly selective, and orally bioavailable inhibitor. Structurally, the compound maintains the pyrimidine core, while the piperazine was replaced with a spirocyclic moiety decorated with a cyclohexyl sulfonamide group. The binding of 124 into the menin–MLL binding pocket was highlighted via the co-crystal structure (PDB 6PKC). In Fig. 35 are summarized the key binding features, including the hydrogen bond with Tyr276 and Trp341 and cation–π interactions with Tyr319 and Tyr323 side chains. In cellular studies, 124 inhibited proliferation and produced apoptosis of leukaemia cell lines harbouring MLL rearrangements, with no effect on the cell lines lacking them. Moreover, the treatment of PDX models developed from patients with different types of leukaemia with 124 resulted in impressive reductions of leukaemia. Interestingly, 124 is able to displace menin from chromatin genome-wide also decreasing the DOT1L chromatin occupancy, probably causing its destabilization.302
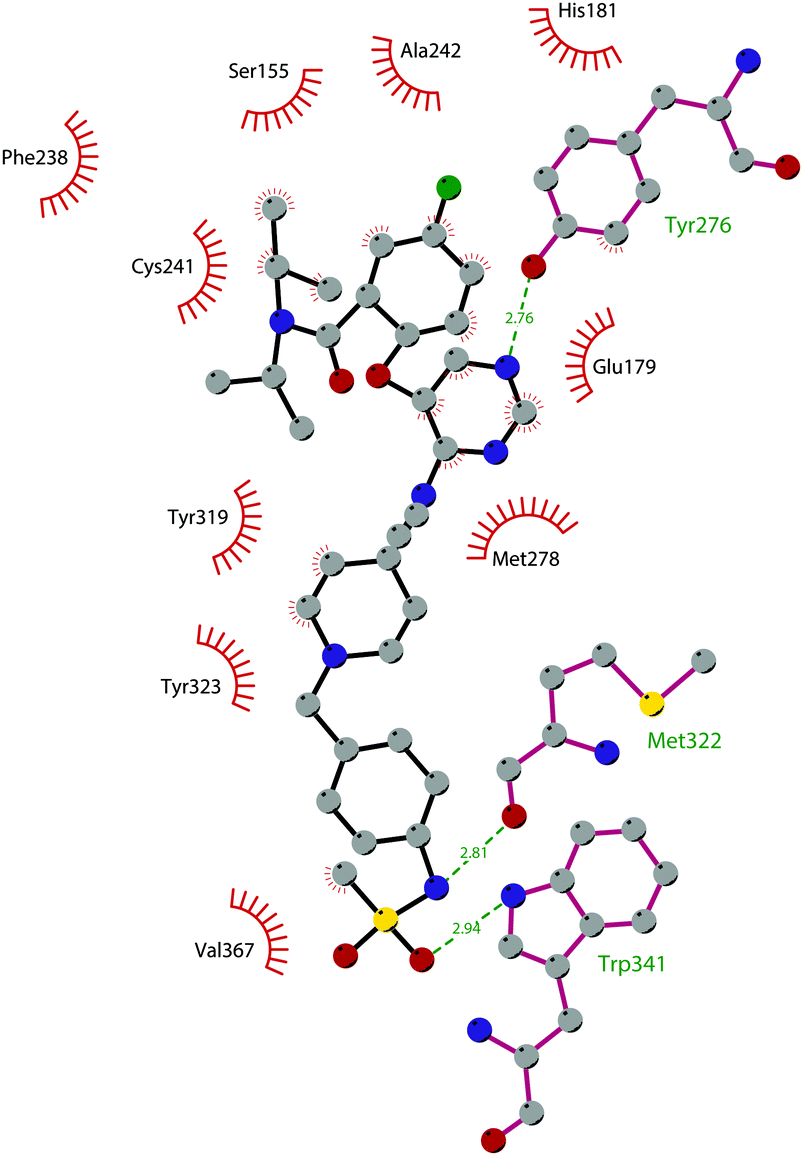 | ||
| Fig. 35 Flat representation (generated using LigPlot+ from PDB ID 6PKC) showing contacts between compound 124 and the active-site residues in menin. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with compounds. | ||
Aiming to identify new menin–MLL inhibitors with diverse chemical scaffolds, the Grembecka group identified a novel class of compounds starting from the HTS of approximately 288000 small molecules. 125 (MIV-6R), an aminomethylpiperidine-based compound, displayed a high binding affinity for menin and high potency in disrupting menin–MLL interactions (KD of 85 nM and IC50 of 56 nM). The compound efficiently induced the hematopoietic differentiation in MLL leukaemia cells and inhibited their proliferation, reducing the expression level of key MLL–fusion protein target genes.303 However, the primary amino group could affect the metabolic stability of 125. In order to improve this issue, some modifications were applied, leading to the identification of 126 (M-470). Specifically, the primary amino group was replaced with a metabolically stable nitrile group and the cyclopentyl ring was functionalized with a reverse carbamate, inserting a fluorine into the phenyl ring to improve the binding affinity. Moreover, the linker was constrained into a 3-methylazetidine group and the nitrile group in position 6 was replaced with a cyclobutylsulfonyl group.304 These modifications improved the inhibitory activity (IC50 of 7 nM), the metabolic stability and, above all, offered the chance to design a covalent inhibitor. Indeed, the insertion of an acrylamide group as a Michael acceptor on the 4-membered ring allowed the identification of 127, a covalent binder of menin (IC50 of 3 nM), which strongly inhibited cell growth in MV4-11 cells. To improve the solubility, a (dimethylamino)-methyl group was attached to the acrylamide group, obtaining 128 (M-525), which showed a potent antiproliferative effect in a variety of human acute leukaemia cell lines, mostly harbouring MLL fusions. The co-crystal structure of 128 in complex with menin revealed that the compound forms a covalent bond with the sulphur atom of the C329 residue through its acrylamide group.304
Further optimization of 128 led to the identification of 129 (M-808), a covalent menin inhibitor that is able to potently inhibit cell growth in MV4-11 and MOLM-13 cell lines harbouring MLL fusion (IC50 of 1 and 4 nM, respectively). Notably, the compound demonstrated strong antitumor effects in the MV4-11 xenograft tumour model in mice with severe combined immunodeficiency, and its intravenous administration (25 mg kg−1 every day, three times per week) resulted in 97% tumour growth inhibition.305
PRDM9
PRDM9 is a member of the PRDM (PRDI-BF1 (positive regulatory domain I-binding factor 1) and RIZ1 (retinoblastoma protein-interacting zinc finger gene 1)) family, a group of 19 methyltransferases that are involved in the epigenetic regulation of gene expression.306 A common feature of all these proteins is the presence of several protein–protein and protein–DNA binding domains and an N-terminal PR domain, which is structurally and functionally related to the SET domain.307However, despite the high homology with the SET domain, only a few PRDM proteins (PRDM2, PRDM7 and PRDM9) have been characterized by their methyltransferase activity, while the others are mainly involved in recruiting other writers through protein–protein interactions.308
PRDM9 catalyses the trimethylation of H3K4 and H3K36 on nucleosome substrates; however; thanks to the presence of its specific protein–protein domains, it interacts with other enzymes, leading to genetic homologous recombination. In addition, PRDM9 is considered an important enzyme in the meiotic prophase.309 Physiologically, PRDM9 is expressed only in germ cells in the gonads and testes and the absence of PRDM9 in mice is associated with sterility for the male as well as the female phenotype. Alterations in the expression of the protein and also the presence of PRMD9 mutations has been related to the insurgence of tumours such as head and neck squamous cell carcinoma, renal cell cancer and leukaemia.310–313
PRDM9 inhibitors
Despite the increasing interest in the PRDM subfamily of enzymes, so far only one PRDM9 inhibitor has been reported (Fig. 36). In 2019, a radioactivity-based methyltransferase assay was used to screen a library of 7500 compounds, enabling the identification of 39 hits that are able to inhibit PRDM9 with IC50 values ranging from 4 to 30 μM. After an extensive SAR campaign, 130 (MRK-740) emerged as a potent (IC50 of 80 nM) and selective PRDM9 inhibitor. The crystal structure of the PRDM9 catalytic domain–SAM–130 ternary complex (PDB 6NM4) enabled the confirmation that this compound acts as a SAM-dependent substrate-competitive inhibitor. | ||
| Fig. 36 PRDM9 inhibitor. | ||
A representation of the intermolecular contacts is reported in Fig. 37, revealing a large number of hydrophobic interactions between the PRDM9 side chains and 130. In addition, the pyridine ring is responsible for the π-stacking interactions with Trp356. On the other hand, the pyridine nitrogen is assumed to be protonated and forms a long-range electrostatic interaction with the side chain of Asp324. In cellular assays, 130 showed the direct inhibition of H3K4 methylation at endogenous PRDM9 target loci, representing the first-in-class PRDM subfamily chemical probe.314
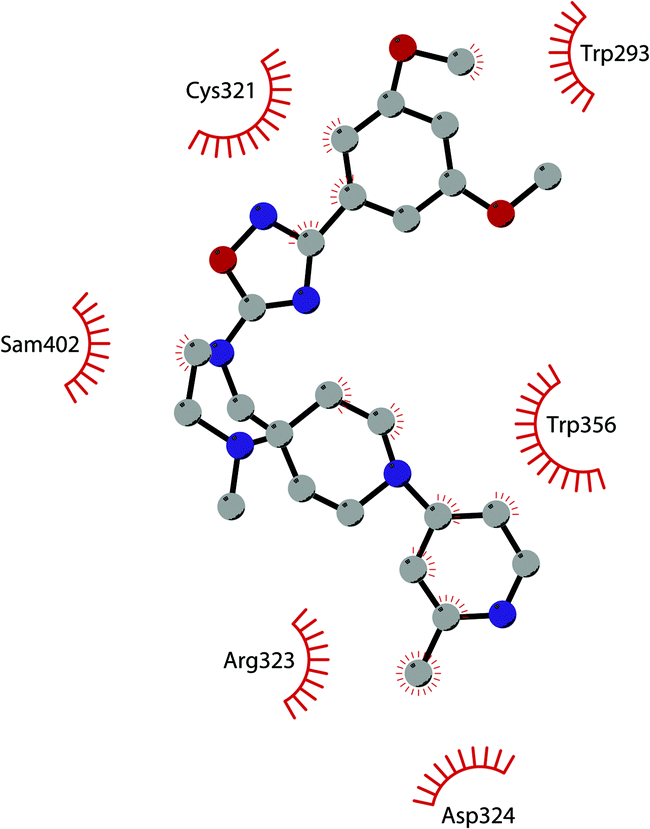 | ||
| Fig. 37 Flat representation (generated using LigPlot+ from PDB ID 6NM4) showing contacts between compound 130 and active-site residues in PRDM9. Hydrogen bonds are shown as dashed lines. The spoked arcs represent protein residues making non-bonded (hydrophobic) contacts with the compound. | ||
ASH1L
The protein Ash1 (absent, small, or homeotic disc 1) was first identified in Drosophila as a member of the trithorax group of proteins,315 which act as major regulators in embryonic development and whose function counteracts the polycomb group of proteins that are responsible for gene silencing. The mammalian homolog of Ash1, named ASH1L (ASH1-like, also known as KMT2H), is a methyltransferase that catalyses the dimethylation of H3K36. This protein is localized in the promoter-proximal coding regions of active genes, suggesting a role as an epigenetic activator.316From the structural point of view, ASH1L contains a SET domain that is located in the middle of the sequence, followed by a post-SET domain, a bromodomain, a bromo-adjacent homology (BAH) domain and a PHD zinc finger domain.317
Functionally, ASH1L interacts with proteins MRG15 and NURF55, constituting the ASH1 complex. In particular, the interaction with MRG15 significantly promotes the catalytic activity of ASH1L, probably by inducing a conformational change that abolishes the block of the auto-inhibitory loop of ASH1L.318
ASH1L is widely expressed mostly in the brain as it plays an important role in the development and in the function of the nervous system. As a matter of fact, alterations of the ASH1L gene are related to the development of neurological disorders such as Tourette syndrome, autism spectrum disorder, and intellectual disability during development.319 In addition, ASH1L is overexpressed in breast cancer,184 in hepatocellular carcinoma320 and in anaplastic thyroid cancer,321 although the role in the biology of these tumours has still not been fully disclosed.
ASH1L inhibitors
The involvement of ASH1L in several cancer types makes this protein an interesting therapeutic target. Unfortunately, the identification of selective inhibitors has been challenging, mainly due to the limited access to the active site of the protein, considering that the catalytic SET domain adopts an inactive conformation with an autoinhibitory loop. Very recently, Grembecka and co-workers reported the identification of the first-in-class ASH1L inhibitors. The NMR-based screening of approximately 1600 fragments from an internal library, followed by medicinal chemistry optimization and structure-based design, enabled the identification of thioamide-containing compounds, of which derivative 131 (AS-99, Fig. 38) was the best candidate with an IC50 of 0.79 μM and a KD of 0.89 μM. | ||
| Fig. 38 ASH1L inhibitor. | ||
This inhibitor was found to be highly selective towards ASH1L, with no inhibition observed at 50 μM on a panel of 20 HMTs as well as on a panel of representative kinases. Interestingly, structural studies revealed that the thioamide-containing derivatives allosterically inhibited the enzymatic activity of the protein, inserting into a pocket that flanks the autoinhibitory loop in the SET domain and stabilizing this conformation. Compound 131 is able to reduce dimethylated levels of H3K36 in MLL leukaemia cells, inhibiting their proliferation and inducing their differentiation and apoptosis. Moreover, this compound downregulates the expression of MLL fusion target genes and reduces the leukaemia burden in a systemic model of MLL leukaemia. This evidence endorses 131 as a valuable chemical probe to further investigation of ASH1L biology.322
Conclusions
In the past 20 years, a great abundance of studies has shown the importance of KMTs in a huge number of physiological processes and in the development of several diseases. These findings have led to the discovery of a plethora of modulators with the aim of fully elucidating the biological role of these proteins as well as establishing them as targets for new therapeutic approaches. In this review, we have reported the so-far identified modulators of KMTs. Among them, one compound has been recently approved by the FDA, some of them are in advanced phases of preclinical studies, and others have only recently been identified and for them a thorough characterization is still needed before they can enter the clinic. Nevertheless, there is a huge number of proteins, endowed with a clear role in several pathological conditions, for which no compounds have been identified yet. Hence, despite the recent advancements in this field, there is still an urgent need to identify new potent and selective modulators of these epigenetic targets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Epigenetic Med Chem Lab's work on epigenetics has received funding from the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Ricerca di Interesse Nazionale (PRIN 20152TE5PK and PRIN 2017MT3993), from the University of Salerno (FARB grant), and from Regione Campania (Italy) grant “Combattere la resistenza tumorale: piattaforma integrata multidisciplinare per un approccio tecnologico innovativo alle oncoterapie—CAMPANIA ONCOTERAPIE” (project no. B61G18000470007).Notes and references
- K. Murray, Biochemistry, 1964, 3, 10–15 CrossRef CAS PubMed.
- D. Chen, H. Ma, H. Hong, S. S. Koh, S.-M. Huang, B. T. Schurter, D. W. Aswad and M. R. Stallcup, Science, 1999, 284, 2174–2177 CrossRef CAS PubMed.
- S. Rea, F. Eisenhaber, D. O'Carroll, B. D. Strahl, Z.-W. Sun, M. Schmid, S. Opravil, K. Mechtler, C. P. Ponting, C. D. Allis and T. Jenuwein, Nature, 2000, 406, 593–599 CrossRef CAS PubMed.
- R. Hamamoto, V. Saloura and Y. Nakamura, Nat. Rev. Cancer, 2015, 15, 110–124 CrossRef CAS PubMed.
- T. Vougiouklakis, B. J. Bernard, N. Nigam, K. Burkitt, Y. Nakamura and V. Saloura, Clin. Epigenet., 2020, 12, 146 CrossRef CAS PubMed.
- M. Urulangodi and A. Mohanty, J. Cell Commun. Signal, 2020, 14, 31–45 CrossRef PubMed.
- X. Wei, X. Yi, X.-H. Zhu and D.-S. Jiang, Clin. Epigenet., 2020, 12, 30 CrossRef CAS PubMed.
- E. M. Michalak, M. L. Burr, A. J. Bannister and M. A. Dawson, Nat. Rev. Mol. Cell Biol., 2019, 20, 573–589 CrossRef CAS PubMed.
- B. Tschiersch, A. Hofmann, V. Krauss, R. Dorn, G. Korge and G. Reuter, EMBO J., 1994, 13, 3822–3831 CrossRef CAS PubMed.
- R. S. Jones and W. M. Gelbart, Mol. Cell. Biol., 1993, 13, 6357–6366 CAS.
- M. J. Stassen, D. Bailey, S. Nelson, V. Chinwalla and P. J. Harte, Mech. Dev., 1995, 52, 209–223 CrossRef CAS PubMed.
- R. C. Trievel, B. M. Beach, L. M. Dirk, R. L. Houtz and J. H. Hurley, Cell, 2002, 111, 91–103 CrossRef CAS PubMed.
- M. Schapira, Curr. Chem. Genomics, 2011, 5, 85–94 CrossRef CAS PubMed.
- K. Sawada, Z. Yang, J. R. Horton, R. E. Collins, X. Zhang and X. Cheng, J. Biol. Chem., 2004, 279, 43296–43306 CrossRef CAS PubMed.
- P. A. Del Rizzo and R. C. Trievel, Epigenetics, 2011, 6, 1059–1067 CrossRef CAS PubMed.
- D. Husmann and O. Gozani, Nat. Struct. Mol. Biol., 2019, 26, 880–889 CrossRef CAS PubMed.
- E. M. Rowe, V. Xing and K. K. Biggar, Brain Res., 2019, 1707, 164–171 CrossRef CAS PubMed.
- K. P. Bhat, H. Ümit Kaniskan, J. Jin and O. Gozani, Nat. Rev. Drug Discovery, 2021, 20, 265–286 CrossRef CAS PubMed.
- C. Zagni, U. Chiacchio and A. Rescifina, Curr. Med. Chem., 2013, 20, 167–185 CrossRef CAS PubMed.
- R. A. Copeland, Philos. Trans. R. Soc., B, 2018, 373, 20170080 CrossRef PubMed.
- H. U. Kaniskan and J. Jin, ACS Chem. Biol., 2015, 10, 40–50 CrossRef CAS PubMed.
- C. Milite, A. Feoli, M. Viviano, D. Rescigno, A. Mai, S. Castellano and G. Sbardella, ChemMedChem, 2016, 11, 1680–1685 CrossRef CAS PubMed.
- C. Milite, A. Feoli, M. Viviano, D. Rescigno, A. Cianciulli, A. L. Balzano, A. Mai, S. Castellano and G. Sbardella, Clin. Epigenet., 2016, 8, 102 CrossRef PubMed.
- M. S. Singer, A. Kahana, A. J. Wolf, L. L. Meisinger, S. E. Peterson, C. Goggin, M. Mahowald and D. E. Gottschling, Genetics, 1998, 150, 613–632 CrossRef CAS PubMed.
- J. Min, Q. Feng, Z. Li, Y. Zhang and R. M. Xu, Cell, 2003, 112, 711–723 CrossRef CAS PubMed.
- F. van Leeuwen, P. R. Gafken and D. E. Gottschling, Cell, 2002, 109, 745–756 CrossRef CAS PubMed.
- B. Jones, H. Su, A. Bhat, H. Lei, J. Bajko, S. Hevi, G. A. Baltus, S. Kadam, H. Zhai, R. Valdez, S. Gonzalo, Y. Zhang, E. Li and T. Chen, PLoS Genet., 2008, 4, e1000190 CrossRef PubMed.
- G. A. Shanower, M. Muller, J. L. Blanton, V. Honti, H. Gyurkovics and P. Schedl, Genetics, 2005, 169, 173–184 CrossRef CAS PubMed.
- M. Mohan, H. M. Herz, Y. H. Takahashi, C. Lin, K. C. Lai, Y. Zhang, M. P. Washburn, L. Florens and A. Shilatifard, Genes Dev., 2010, 24, 574–589 CrossRef CAS PubMed.
- Y. Okada, Q. Feng, Y. Lin, Q. Jiang, Y. Li, V. M. Coffield, L. Su, G. Xu and Y. Zhang, Cell, 2005, 121, 167–178 CrossRef CAS PubMed.
- K. M. Bernt, N. Zhu, A. U. Sinha, S. Vempati, J. Faber, A. V. Krivtsov, Z. Feng, N. Punt, A. Daigle, L. Bullinger, R. M. Pollock, V. M. Richon, A. L. Kung and S. A. Armstrong, Cancer Cell, 2011, 20, 66–78 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, C. R. Majer, C. J. Sneeringer, J. Song, L. D. Johnston, M. P. Scott, J. J. Smith, Y. Xiao, L. Jin, K. W. Kuntz, R. Chesworth, M. P. Moyer, K. M. Bernt, J. C. Tseng, A. L. Kung, S. A. Armstrong, R. A. Copeland, V. M. Richon and R. M. Pollock, Cancer Cell, 2011, 20, 53–65 CrossRef CAS PubMed.
- A. T. Nguyen, J. He, O. Taranova and Y. Zhang, Cell Res., 2011, 21, 1370–1373 CrossRef CAS PubMed.
- S. S. Spurr, E. D. Bayle, W. Yu, F. Li, W. Tempel, M. Vedadi, M. Schapira and P. V. Fish, Bioorg. Med. Chem. Lett., 2016, 26, 4518–4522 CrossRef CAS PubMed.
- J. L. Anglin, L. Deng, Y. Yao, G. Cai, Z. Liu, H. Jiang, G. Cheng, P. Chen, S. Dong and Y. Song, J. Med. Chem., 2012, 55, 8066–8074 CrossRef CAS PubMed.
- W. Yu, D. Smil, F. Li, W. Tempel, O. Fedorov, K. T. Nguyen, Y. Bolshan, R. Al-Awar, S. Knapp, C. H. Arrowsmith, M. Vedadi, P. J. Brown and M. Schapira, Bioorg. Med. Chem., 2013, 21, 1787–1794 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, A. Basavapathruni, L. Jin, P. A. Boriack-Sjodin, C. J. Allain, C. R. Klaus, A. Raimondi, M. P. Scott, N. J. Waters, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon and R. M. Pollock, Blood, 2013, 122, 1017–1025 CrossRef CAS PubMed.
- A. Basavapathruni, E. J. Olhava, S. R. Daigle, C. A. Therkelsen, L. Jin, P. A. Boriack-Sjodin, C. J. Allain, C. R. Klaus, A. Raimondi, M. P. Scott, A. Dovletoglou, V. M. Richon, R. M. Pollock, R. A. Copeland, M. P. Moyer, R. Chesworth, P. G. Pearson and N. J. Waters, Biopharm. Drug Dispos., 2014, 35, 237–252 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- I. Epizyme and C. Corporation, A Phase 1 Dose Escalation and Expanded Cohort Study of EPZ-5676 in the Treatment of Pediatric Patients With Relapsed/Refractory Leukemias Bearing a Rearrangement of the MLL Gene, https://ClinicalTrials.gov/show/NCT02141828, 2014.
- I. Epizyme and Celgene, A Phase 1, Open-Label, Dose-Escalation & Expanded Cohort, Continuous IV Infusion, Multi-center Study of the Safety, Tolerability,PK & PD of EPZ-5676 in Treatment Relapsed/Refractory Patients With Leukemias Involving, https://ClinicalTrials.gov/show/NCT01684150, 2012.
- N. C. Institute, Pinometostat With Standard Chemotherapy in Treating Patients With Newly Diagnosed Acute Myeloid Leukemia and MLL Gene Rearrangement, https://ClinicalTrials.gov/show/NCT03724084, 2019.
- N. C. Institute, Pinometostat and Azacitidine in Treating Patients With Relapsed, Refractory, or Newly Diagnosed Acute Myeloid Leukemia With 11q23 Rearrangement, https://ClinicalTrials.gov/show/NCT03701295, 2019.
- S. Chen, L. Li, Y. Chen, J. Hu, J. Liu, Y.-C. Liu, R. Liu, Y. Zhang, F. Meng, K. Zhu, J. Lu, M. Zheng, K. Chen, J. Zhang, H. Jiang, Z. Yao and C. Luo, J. Chem. Inf. Model., 2016, 56, 527–534 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhang, J. Liu, L. Chen, L. Zhao, B. Li and W. Wang, Bioorg. Med. Chem. Lett., 2017, 27, 1584–1587 CrossRef CAS PubMed.
- C. Scheufler, H. Möbitz, C. Gaul, C. Ragot, C. Be, C. Fernández, K. S. Beyer, R. Tiedt and F. Stauffer, ACS Med. Chem. Lett., 2016, 7, 730–734 CrossRef CAS PubMed.
- C. Chen, H. Zhu, F. Stauffer, G. Caravatti, S. Vollmer, R. Machauer, P. Holzer, H. Mobitz, C. Scheufler, M. Klumpp, R. Tiedt, K. S. Beyer, K. Calkins, D. Guthy, M. Kiffe, J. Zhang and C. Gaul, ACS Med. Chem. Lett., 2016, 7, 735–740 CrossRef CAS PubMed.
- H. Möbitz, R. Machauer, P. Holzer, A. Vaupel, F. Stauffer, C. Ragot, G. Caravatti, C. Scheufler, C. Fernandez, U. Hommel, R. Tiedt, K. S. Beyer, C. Chen, H. Zhu and C. Gaul, ACS Med. Chem. Lett., 2017, 8, 338–343 CrossRef PubMed.
- F. Stauffer, A. Weiss, C. Scheufler, H. Mobitz, C. Ragot, K. S. Beyer, K. Calkins, D. Guthy, M. Kiffe, B. Van Eerdenbrugh, R. Tiedt and C. Gaul, ACS Med. Chem. Lett., 2019, 10, 1655–1660 CrossRef CAS PubMed.
- G. S. Gibbons, A. Chakraborty, S. M. Grigsby, A. C. Umeano, C. Liao, O. Moukha-Chafiq, V. Pathak, B. Mathew, Y. T. Lee, Y. Dou, S. C. Schurer, R. C. Reynolds, T. S. Snowden and Z. Nikolovska-Coleska, Eur. J. Med. Chem., 2020, 189, 112023 CrossRef CAS PubMed.
- M. Tachibana, K. Sugimoto, T. Fukushima and Y. Shinkai, J. Biol. Chem., 2001, 276, 25309–25317 CrossRef CAS PubMed.
- M. Tachibana, J. Ueda, M. Fukuda, N. Takeda, T. Ohta, H. Iwanari, T. Sakihama, T. Kodama, T. Hamakubo and Y. Shinkai, Genes Dev., 2005, 19, 815–826 CrossRef CAS PubMed.
- D. Patnaik, H. G. Chin, P. O. Estève, J. Benner, S. E. Jacobsen and S. Pradhan, J. Biol. Chem., 2004, 279, 53248–53258 CrossRef CAS PubMed.
- H. Wu, X. Chen, J. Xiong, Y. Li, H. Li, X. Ding, S. Liu, S. Chen, S. Gao and B. Zhu, Cell Res., 2011, 21, 365–367 CrossRef CAS PubMed.
- Y. Yu, C. Song, Q. Zhang, P. A. DiMaggio, B. A. Garcia, A. York, M. F. Carey and M. Grunstein, Mol. Cell, 2012, 46, 7–17 CrossRef CAS PubMed.
- P. Trojer, J. Zhang, M. Yonezawa, A. Schmidt, H. Zheng, T. Jenuwein and D. Reinberg, J. Biol. Chem., 2009, 284, 8395–8405 CrossRef CAS PubMed.
- T. Weiss, S. Hergeth, U. Zeissler, A. Izzo, P. Tropberger, B. M. Zee, M. Dundr, B. A. Garcia, S. Daujat and R. Schneider, Epigenet. Chromatin, 2010, 3, 7 CrossRef PubMed.
- J. Huang, J. Dorsey, S. Chuikov, L. Pérez-Burgos, X. Zhang, T. Jenuwein, D. Reinberg and S. L. Berger, J. Biol. Chem., 2010, 285, 9636–9641 CrossRef CAS PubMed.
- K. E. Moore, S. M. Carlson, N. D. Camp, P. Cheung, R. G. James, K. F. Chua, A. Wolf-Yadlin and O. Gozani, Mol. Cell, 2013, 50, 444–456 CrossRef CAS PubMed.
- S. C. Sampath, I. Marazzi, K. L. Yap, S. C. Sampath, A. N. Krutchinsky, I. Mecklenbräuker, A. Viale, E. Rudensky, M. M. Zhou, B. T. Chait and A. Tarakhovsky, Mol. Cell, 2007, 27, 596–608 CrossRef CAS PubMed.
- C. Mozzetta, J. Pontis, L. Fritsch, P. Robin, M. Portoso, C. Proux, R. Margueron and S. Ait-Si-Ali, Mol. Cell, 2014, 53, 277–289 CrossRef CAS PubMed.
- R. E. Collins, J. P. Northrop, J. R. Horton, D. Y. Lee, X. Zhang, M. R. Stallcup and X. Cheng, Nat. Struct. Mol. Biol., 2008, 15, 245–250 CrossRef CAS PubMed.
- H. Wu, J. Min, V. V. Lunin, T. Antoshenko, L. Dombrovski, H. Zeng, A. Allali-Hassani, V. Campagna-Slater, M. Vedadi, C. H. Arrowsmith, A. N. Plotnikov and M. Schapira, PLoS One, 2010, 5, e8570 CrossRef PubMed.
- N. Liu, Z. Zhang, H. Wu, Y. Jiang, L. Meng, J. Xiong, Z. Zhao, X. Zhou, J. Li, H. Li, Y. Zheng, S. Chen, T. Cai, S. Gao and B. Zhu, Genes Dev., 2015, 29, 379–393 CrossRef CAS PubMed.
- M. Tachibana, K. Sugimoto, M. Nozaki, J. Ueda, T. Ohta, M. Ohki, M. Fukuda, N. Takeda, H. Niida, H. Kato and Y. Shinkai, Genes Dev., 2002, 16, 1779–1791 CrossRef CAS PubMed.
- T. H. W. Dobson, R. J. Hatcher, J. Swaminathan, C. M. Das, S. Shaik, R.-H. Tao, C. Milite, S. Castellano, P. H. Taylor, G. Sbardella and V. Gopalakrishnan, Mol. Cancer Res., 2017, 15, 1073–1084 CrossRef CAS PubMed.
- T. Huang, P. Zhang, W. Li, T. Zhao, Z. Zhang, S. Chen, Y. Yang, Y. Feng, F. Li, X. Shirley Liu, L. Zhang, G. Jiang and F. Zhang, Cell Death Dis., 2017, 8, e2726 CrossRef CAS PubMed.
- T. Kleefstra, M. Smidt, M. J. Banning, A. R. Oudakker, H. Van Esch, A. P. de Brouwer, W. Nillesen, E. A. Sistermans, B. C. Hamel, D. de Bruijn, J. P. Fryns, H. G. Yntema, H. G. Brunner, B. B. de Vries and H. van Bokhoven, J. Med. Genet., 2005, 42, 299–306 CrossRef CAS PubMed.
- F. Antignano, K. Burrows, M. R. Hughes, J. M. Han, K. J. Kron, N. M. Penrod, M. J. Oudhoff, S. K. Wang, P. H. Min, M. J. Gold, A. L. Chenery, M. J. Braam, T. C. Fung, F. M. Rossi, K. M. McNagny, C. H. Arrowsmith, M. Lupien, M. K. Levings and C. Zaph, J. Clin. Invest., 2014, 124, 1945–1955 CrossRef CAS PubMed.
- Q. Liu, X. Cai, D. Yang, Y. Chen, Y. Wang, L. Shao and M. W. Wang, Bioorg. Med. Chem., 2017, 25, 4579–4594 CrossRef CAS PubMed.
- K. Devkota, B. Lohse, Q. Liu, M. W. Wang, D. Stærk, J. Berthelsen and R. P. Clausen, ACS Med. Chem. Lett., 2014, 5, 293–297 CrossRef CAS PubMed.
- S. Kubicek, R. J. O'Sullivan, E. M. August, E. R. Hickey, Q. Zhang, M. L. Teodoro, S. Rea, K. Mechtler, J. A. Kowalski, C. A. Homon, T. A. Kelly and T. Jenuwein, Mol. Cell, 2007, 25, 473–481 CrossRef CAS PubMed.
- Y. Yuan, Q. Wang, J. Paulk, S. Kubicek, M. M. Kemp, D. J. Adams, A. F. Shamji, B. K. Wagner and S. L. Schreiber, ACS Chem. Biol., 2012, 7, 1152–1157 CrossRef CAS PubMed.
- Y. Chang, X. Zhang, J. R. Horton, A. K. Upadhyay, A. Spannhoff, J. Liu, J. P. Snyder, M. T. Bedford and X. Cheng, Nat. Struct. Mol. Biol., 2009, 16, 312–317 CrossRef CAS PubMed.
- I. A. Ciechomska, P. Przanowski, J. Jackl, B. Wojtas and B. Kaminska, Sci. Rep., 2016, 6, 38723 CrossRef CAS PubMed.
- Z. Rahman, M. R. Bazaz, G. Devabattula, M. A. Khan and C. Godugu, J. Biochem. Mol. Toxicol., 2020, e22674, DOI:10.1002/jbt.22674.
- Y. Chang, T. Ganesh, J. R. Horton, A. Spannhoff, J. Liu, A. Sun, X. Zhang, M. T. Bedford, Y. Shinkai, J. P. Snyder and X. Cheng, J. Mol. Biol., 2010, 400, 1–7 CrossRef CAS PubMed.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, G. A. Wasney, A. Dong, D. Barsyte, I. Kozieradzki, G. Senisterra, I. Chau, A. Siarheyeva, D. B. Kireev, A. Jadhav, J. M. Herold, S. V. Frye, C. H. Arrowsmith, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2009, 52, 7950–7953 CrossRef CAS PubMed.
- F. Liu, X. Chen, A. Allali-Hassani, A. M. Quinn, T. J. Wigle, G. A. Wasney, A. Dong, G. Senisterra, I. Chau, A. Siarheyeva, J. L. Norris, D. B. Kireev, A. Jadhav, J. M. Herold, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, P. J. Brown, A. Simeonov, M. Vedadi and J. Jin, J. Med. Chem., 2010, 53, 5844–5857 CrossRef CAS PubMed.
- M. Vedadi, D. Barsyte-Lovejoy, F. Liu, S. Rival-Gervier, A. Allali-Hassani, V. Labrie, T. J. Wigle, P. A. Dimaggio, G. A. Wasney, A. Siarheyeva, A. Dong, W. Tempel, S. C. Wang, X. Chen, I. Chau, T. J. Mangano, X. P. Huang, C. D. Simpson, S. G. Pattenden, J. L. Norris, D. B. Kireev, A. Tripathy, A. Edwards, B. L. Roth, W. P. Janzen, B. A. Garcia, A. Petronis, J. Ellis, P. J. Brown, S. V. Frye, C. H. Arrowsmith and J. Jin, Nat. Chem. Biol., 2011, 7, 566–574 CrossRef CAS PubMed.
- F. Liu, D. Barsyte-Lovejoy, F. Li, Y. Xiong, V. Korboukh, X. P. Huang, A. Allali-Hassani, W. P. Janzen, B. L. Roth, S. V. Frye, C. H. Arrowsmith, P. J. Brown, M. Vedadi and J. Jin, J. Med. Chem., 2013, 56, 8931–8942 CrossRef CAS PubMed.
- F. Liu, D. Barsyte-Lovejoy, A. Allali-Hassani, Y. He, J. M. Herold, X. Chen, C. M. Yates, S. V. Frye, P. J. Brown, J. Huang, M. Vedadi, C. H. Arrowsmith and J. Jin, J. Med. Chem., 2011, 54, 6139–6150 CrossRef CAS PubMed.
- C. Milite, A. Feoli, J. R. Horton, D. Rescigno, A. Cipriano, V. Pisapia, M. Viviano, G. Pepe, G. Amendola, E. Novellino, S. Cosconati, X. Cheng, S. Castellano and G. Sbardella, J. Med. Chem., 2019, 62, 2666–2689 CrossRef CAS PubMed.
- E. San Jose-Eneriz, X. Agirre, O. Rabal, A. Vilas-Zornoza, J. A. Sanchez-Arias, E. Miranda, A. Ugarte, S. Roa, B. Paiva, A. Estella-Hermoso de Mendoza, R. M. Alvarez, N. Casares, V. Segura, J. I. Martin-Subero, F. X. Ogi, P. Soule, C. M. Santiveri, R. Campos-Olivas, G. Castellano, M. G. F. de Barrena, J. R. Rodriguez-Madoz, M. J. Garcia-Barchino, J. J. Lasarte, M. A. Avila, J. A. Martinez-Climent, J. Oyarzabal and F. Prosper, Nat. Commun., 2017, 8, 15424 CrossRef CAS PubMed.
- R. F. Sweis, M. Pliushchev, P. J. Brown, J. Guo, F. Li, D. Maag, A. M. Petros, N. B. Soni, C. Tse, M. Vedadi, M. R. Michaelides, G. G. Chiang and W. N. Pappano, ACS Med. Chem. Lett., 2014, 5, 205–209 CrossRef CAS PubMed.
- T. Wagner, H. Greschik, T. Burgahn, K. Schmidtkunz, A. K. Schott, J. McMillan, L. Baranauskiene, Y. Xiong, O. Fedorov, J. Jin, U. Oppermann, D. Matulis, R. Schule and M. Jung, Nucleic Acids Res., 2016, 44, e88 CrossRef PubMed.
- V. Fagan, C. Johansson, C. Gileadi, O. Monteiro, J. E. Dunford, R. Nibhani, M. Philpott, J. Malzahn, G. Wells, R. Faram, A. P. Cribbs, N. Halidi, F. Li, I. Chau, H. Greschik, S. Velupillai, A. Allali-Hassani, J. Bennett, T. Christott, C. Giroud, A. M. Lewis, K. V. M. Huber, N. Athanasou, C. Bountra, M. Jung, R. Schule, M. Vedadi, C. Arrowsmith, Y. Xiong, J. Jin, O. Fedorov, G. Farnie, P. E. Brennan and U. Oppermann, J. Med. Chem., 2019, 62, 9008–9025 CrossRef CAS PubMed.
- K. Katayama, K. Ishii, H. Terashima, E. Tsuda, M. Suzuki, K. Yotsumoto, K. Hiramoto, I. Yasumatsu, M. Torihata, T. Ishiyama, T. Muto and T. Katagiri, ACS Med. Chem. Lett., 2021, 12, 121–128 CrossRef CAS PubMed.
- K. Katayama, K. Ishii, E. Tsuda, K. Yotsumoto, K. Hiramoto, M. Suzuki, I. Yasumatsu, W. Igarashi, M. Torihata, T. Ishiyama and T. Katagiri, Bioorg. Med. Chem. Lett., 2020, 30, 127475 CrossRef CAS PubMed.
- H. G. Chin, D. Patnaik, P. O. Esteve, S. E. Jacobsen and S. Pradhan, Biochemistry, 2006, 45, 3272–3284 CrossRef CAS PubMed.
- T. Wang, C. Xu, Y. Liu, K. Fan, Z. Li, X. Sun, H. Ouyang, X. Zhang, J. Zhang, Y. Li, F. Mackenzie, J. Min and X. Tu, PLoS One, 2012, 7, e52977 CrossRef CAS PubMed.
- D. O'Carroll, H. Scherthan, A. H. Peters, S. Opravil, A. R. Haynes, G. Laible, S. Rea, M. Schmid, A. Lebersorger, M. Jerratsch, L. Sattler, M. G. Mattei, P. Denny, S. D. Brown, D. Schweizer and T. Jenuwein, Mol. Cell. Biol., 2000, 20, 9423–9433 CrossRef PubMed.
- M. K. Schuhmacher, S. Kudithipudi, D. Kusevic, S. Weirich and A. Jeltsch, Biochim. Biophys. Acta, 2015, 1849, 55–63 CrossRef CAS PubMed.
- A. Allali-Hassani, G. A. Wasney, A. Siarheyeva, T. Hajian, C. H. Arrowsmith and M. Vedadi, J. Biomol. Screen, 2012, 17, 71–84 CrossRef CAS PubMed.
- K. Sone, L. Piao, M. Nakakido, K. Ueda, T. Jenuwein, Y. Nakamura and R. Hamamoto, Nat. Commun., 2014, 5, 5691 CrossRef CAS PubMed.
- M. Puschendorf, R. Terranova, E. Boutsma, X. Mao, K. Isono, U. Brykczynska, C. Kolb, A. P. Otte, H. Koseki, S. H. Orkin, M. van Lohuizen and A. H. Peters, Nat. Genet., 2008, 40, 411–420 CrossRef CAS PubMed.
- M. García-Cao, R. O'Sullivan, A. H. Peters, T. Jenuwein and M. A. Blasco, Nat. Genet., 2004, 36, 94–99 CrossRef PubMed.
- A. H. Peters, D. O'Carroll, H. Scherthan, K. Mechtler, S. Sauer, C. Schöfer, K. Weipoltshammer, M. Pagani, M. Lachner, A. Kohlmaier, S. Opravil, M. Doyle, M. Sibilia and T. Jenuwein, Cell, 2001, 107, 323–337 CrossRef CAS PubMed.
- C. R. Keenan, N. Iannarella, G. Naselli, N. G. Bediaga, T. M. Johanson, L. C. Harrison and R. S. Allan, Blood, 2020, 135, 2049–2058 CrossRef PubMed.
- Y. Zheng, B. Li, J. Wang, Y. Xiong, K. Wang, Y. Qi, H. Sun, L. Wu and L. Yang, Clin. Epigenet., 2018, 10, 129 CrossRef CAS PubMed.
- W. Shuai, J. Wu, S. Chen, R. Liu, Z. Ye, C. Kuang, X. Fu, G. Wang, Y. Li, Q. Peng, W. Shi, Y. Li, Q. Zhou and W. Huang, Cancer Lett., 2018, 422, 56–69 CrossRef CAS PubMed.
- V. Jagannathan, J. Bannoehr, P. Plattet, R. Hauswirth, C. Drögemüller, M. Drögemüller, D. J. Wiener, M. Doherr, M. Owczarek-Lipska, A. Galichet, M. M. Welle, K. Tengvall, K. Bergvall, H. Lohi, S. Rüfenacht, M. Linek, M. Paradis, E. J. Müller, P. Roosje and T. Leeb, PLoS Genet., 2013, 9, e1003848 CrossRef PubMed.
- X. Y. Liu and H. Li, J. Diabetes Res., 2017, 2017, 8365762 Search PubMed.
- Z. Fan, L. Li, M. Li, X. Zhang, C. Hao, L. Yu, S. Zeng, H. Xu, M. Fang, A. Shen, T. Jenuwein and Y. Xu, Hepatology, 2017, 65, 1904–1919 CrossRef CAS PubMed.
- D. Greiner, T. Bonaldi, R. Eskeland, E. Roemer and A. Imhof, Nat. Chem. Biol., 2005, 1, 143–145 CrossRef CAS PubMed.
- A. V. Paschall, D. Yang, C. Lu, J.-H. Choi, X. Li, F. Liu, M. Figueroa, N. H. Oberlies, C. Pearce, W. B. Bollag, A. Nayak-Kapoor and K. Liu, J. Immunology, 2015, 195, 1868–1882 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. M. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Muller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, R. Ross, B. Roth, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundstrom, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- C. Lu, D. Yang, J. D. Klement, I. K. Oh, N. M. Savage, J. L. Waller, A. H. Colby, M. W. Grinstaff, N. H. Oberlies, C. J. Pearce, Z. Xie, S. K. Kulp, C. C. Coss, M. A. Phelps, T. Albers, I. O. Lebedyeva and K. Liu, Cancer Immunol. Res., 2019, 7, 414–427 CrossRef CAS PubMed.
- C. Lu, J. D. Klement, D. Yang, T. Albers, I. O. Lebedyeva, J. L. Waller and K. Liu, Cancer Lett., 2020, 476, 87–96 CrossRef CAS PubMed.
- T. Vougiouklakis, V. Saloura, J. H. Park, N. Takamatsu, T. Miyamoto, Y. Nakamura and Y. Matsuo, Oncotarget, 2018, 9, 31820–31831 CrossRef PubMed.
- J. Müller, C. M. Hart, N. J. Francis, M. L. Vargas, A. Sengupta, B. Wild, E. L. Miller, M. B. O'Connor, R. E. Kingston and J. A. Simon, Cell, 2002, 111, 197–208 CrossRef.
- R. Margueron, G. Li, K. Sarma, A. Blais, J. Zavadil, C. L. Woodcock, B. D. Dynlacht and D. Reinberg, Mol. Cell, 2008, 32, 503–518 CrossRef CAS PubMed.
- A. Kuzmichev, K. Nishioka, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Genes Dev., 2002, 16, 2893–2905 CrossRef CAS PubMed.
- R. Cao, L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage, P. Tempst, R. S. Jones and Y. Zhang, Science, 2002, 298, 1039–1043 CrossRef CAS PubMed.
- D. Pasini, A. P. Bracken, M. R. Jensen, E. Lazzerini Denchi and K. Helin, EMBO J., 2004, 23, 4061–4071 CrossRef CAS PubMed.
- R. Margueron, N. Justin, K. Ohno, M. L. Sharpe, J. Son, W. J. Drury, 3rd, P. Voigt, S. R. Martin, W. R. Taylor, V. De Marco, V. Pirrotta, D. Reinberg and S. J. Gamblin, Nature, 2009, 461, 762–767 CrossRef CAS PubMed.
- B. Czermin, R. Melfi, D. McCabe, V. Seitz, A. Imhof and V. Pirrotta, Cell, 2002, 111, 185–196 CrossRef CAS PubMed.
- H. Wang, L. Wang, H. Erdjument-Bromage, M. Vidal, P. Tempst, R. S. Jones and Y. Zhang, Nature, 2004, 431, 873–878 CrossRef CAS PubMed.
- S. Varambally, S. M. Dhanasekaran, M. Zhou, T. R. Barrette, C. Kumar-Sinha, M. G. Sanda, D. Ghosh, K. J. Pienta, R. G. Sewalt, A. P. Otte, M. A. Rubin and A. M. Chinnaiyan, Nature, 2002, 419, 624–629 CrossRef CAS PubMed.
- C. G. Kleer, Q. Cao, S. Varambally, R. Shen, I. Ota, S. A. Tomlins, D. Ghosh, R. G. Sewalt, A. P. Otte, D. F. Hayes, M. S. Sabel, D. Livant, S. J. Weiss, M. A. Rubin and A. M. Chinnaiyan, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11606–11611 CrossRef CAS PubMed.
- B. D. Karanikolas, M. L. Figueiredo and L. Wu, Mol. Cancer Res., 2009, 7, 1456–1465 CrossRef CAS PubMed.
- M. Yamagishi, M. Hori, D. Fujikawa, T. Ohsugi, D. Honma, N. Adachi, H. Katano, T. Hishima, S. Kobayashi, K. Nakano, M. Nakashima, M. Iwanaga, A. Utsunomiya, Y. Tanaka, S. Okada, K. Tsukasaki, K. Tobinai, K. Araki, T. Watanabe and K. Uchimaru, Cell Rep., 2019, 29, 2321–2337 e2327 CrossRef CAS PubMed.
- S. Fujita, D. Honma, N. Adachi, K. Araki, E. Takamatsu, T. Katsumoto, K. Yamagata, K. Akashi, K. Aoyama, A. Iwama and I. Kitabayashi, Leukemia, 2018, 32, 855–864 CrossRef CAS PubMed.
- S. K. Knutson, T. J. Wigle, N. M. Warholic, C. J. Sneeringer, C. J. Allain, C. R. Klaus, J. D. Sacks, A. Raimondi, C. R. Majer, J. Song, M. P. Scott, L. Jin, J. J. Smith, E. J. Olhava, R. Chesworth, M. P. Moyer, V. M. Richon, R. A. Copeland, H. Keilhack, R. M. Pollock and K. W. Kuntz, Nat. Chem. Biol., 2012, 8, 890–896 CrossRef CAS PubMed.
- S. K. Knutson, N. M. Warholic, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, M. Porter Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, R. M. Pollock, K. W. Kuntz and H. Keilhack, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7922–7927 CrossRef CAS PubMed.
- S. K. Knutson, S. Kawano, Y. Minoshima, N. M. Warholic, K. C. Huang, Y. Xiao, T. Kadowaki, M. Uesugi, G. Kuznetsov, N. Kumar, T. J. Wigle, C. R. Klaus, C. J. Allain, A. Raimondi, N. J. Waters, J. J. Smith, M. Porter-Scott, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon, T. Uenaka, R. M. Pollock, K. W. Kuntz, A. Yokoi and H. Keilhack, Mol. Cancer Ther., 2014, 13, 842–854 CrossRef CAS PubMed.
- I. Epizyme, Study of Tazemetostat With Enzalutamide or Abiraterone/Prednisone in Subjects With Castration Resistant Prostate Cancer Who Have Not Received Chemotherapy, https://ClinicalTrials.gov/show/NCT04179864, 2019.
- I. Epizyme, Open-Label, Multicenter, Phase 1/2 Study of Tazemetostat (EZH2 Histone Methyl Transferase HMT Inhibitor) as a Single Agent in Subjects With Adv. Solid Tumors or With B-cell Lymphomas and Tazemetostat in Combination With Prednisolone in Subjects With DLBCL, https://ClinicalTrials.gov/show/NCT01897571, 2013.
- N. C. Institute, Tazemetostat and Pembrolizumab in Treating Patients With Locally Advanced or Metastatic Urothelial Carcinoma, https://ClinicalTrials.gov/show/NCT03854474, 2019.
- J. E. Campbell, K. W. Kuntz, S. K. Knutson, N. M. Warholic, H. Keilhack, T. J. Wigle, A. Raimondi, C. R. Klaus, N. Rioux, A. Yokoi, S. Kawano, Y. Minoshima, H. W. Choi, M. Porter Scott, N. J. Waters, J. J. Smith, R. Chesworth, M. P. Moyer and R. A. Copeland, ACS Med. Chem. Lett., 2015, 6, 491–495 CrossRef CAS PubMed.
- B. Lu, X. Shen, L. Zhang, D. Liu, C. Zhang, J. Cao, R. Shen, J. Zhang, D. Wang, H. Wan, Z. Xu, M. H. Ho, M. Zhang, L. Zhang, F. He and W. Tao, ACS Med. Chem. Lett., 2018, 9, 98–102 CrossRef CAS PubMed.
- M. T. McCabe, H. M. Ott, G. Ganji, S. Korenchuk, C. Thompson, G. S. Van Aller, Y. Liu, A. P. Graves, A. Della Pietra, 3rd, E. Diaz, L. V. LaFrance, M. Mellinger, C. Duquenne, X. Tian, R. G. Kruger, C. F. McHugh, M. Brandt, W. H. Miller, D. Dhanak, S. K. Verma, P. J. Tummino and C. L. Creasy, Nature, 2012, 492, 108–112 CrossRef CAS PubMed.
- D. Zeng, M. Liu and J. Pan, Oncotarget, 2017, 8, 3396–3411 CrossRef PubMed.
- GlaxoSmithKline, A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2816126 in Subjects With Relapsed/Refractory Diffuse Large B Cell Lymphoma, Transformed Follicular Lymphoma, Other Non-Hodgkin's Lymphomas, Solid Tumors and Multiple Myeloma, https://ClinicalTrials.gov/show/NCT02082977, 2014.
- S. Huang, Z. Wang, J. Zhou, J. Huang, L. Zhou, J. Luo, Y. Y. Wan, H. Long and B. Zhu, Cancer Res., 2019, 79, 2009–2020 CrossRef CAS PubMed.
- M. Bratkowski, X. Yang and X. Liu, Sci. Rep., 2018, 8, 9092 CrossRef PubMed.
- S. K. Verma, X. Tian, L. V. LaFrance, C. Duquenne, D. P. Suarez, K. A. Newlander, S. P. Romeril, J. L. Burgess, S. W. Grant, J. A. Brackley, A. P. Graves, D. A. Scherzer, A. Shu, C. Thompson, H. M. Ott, G. S. Aller, C. A. Machutta, E. Diaz, Y. Jiang, N. W. Johnson, S. D. Knight, R. G. Kruger, M. T. McCabe, D. Dhanak, P. J. Tummino, C. L. Creasy and W. H. Miller, ACS Med. Chem. Lett., 2012, 3, 1091–1096 CrossRef CAS PubMed.
- W. Qi, H. Chan, L. Teng, L. Li, S. Chuai, R. Zhang, J. Zeng, M. Li, H. Fan, Y. Lin, J. Gu, O. Ardayfio, J. H. Zhang, X. Yan, J. Fang, Y. Mi, M. Zhang, T. Zhou, G. Feng, Z. Chen, G. Li, T. Yang, K. Zhao, X. Liu, Z. Yu, C. X. Lu, P. Atadja and E. Li, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21360–21365 CrossRef CAS PubMed.
- R. G. Vaswani, V. S. Gehling, L. A. Dakin, A. S. Cook, C. G. Nasveschuk, M. Duplessis, P. Iyer, S. Balasubramanian, F. Zhao, A. C. Good, R. Campbell, C. Lee, N. Cantone, R. T. Cummings, E. Normant, S. F. Bellon, B. K. Albrecht, J. C. Harmange, P. Trojer, J. E. Audia, Y. Zhang, N. Justin, S. Chen, J. R. Wilson and S. J. Gamblin, J. Med. Chem., 2016, 59, 9928–9941 CrossRef CAS PubMed.
- C. Pharmaceuticals, A Study Evaluating CPI-1205 in Patients With B-Cell Lymphomas, https://ClinicalTrials.gov/show/NCT02395601, 2015.
- C. Pharmaceuticals, ProSTAR: A Study Evaluating CPI-1205 in Patients With Metastatic Castration Resistant Prostate Cancer, https://ClinicalTrials.gov/show/NCT03480646, 2017.
- S. Goswami, I. Apostolou, J. Zhang, J. Skepner, S. Anandhan, X. Zhang, L. Xiong, P. Trojer, A. Aparicio, S. K. Subudhi, J. P. Allison, H. Zhao and P. Sharma, J. Clin. Invest., 2018, 128, 3813–3818 CrossRef PubMed.
- X. Song, T. Gao, N. Wang, Q. Feng, X. You, T. Ye, Q. Lei, Y. Zhu, M. Xiong, Y. Xia, F. Yang, Y. Shi, Y. Wei, L. Zhang and L. Yu, Sci. Rep., 2016, 6, 20864 CrossRef CAS PubMed.
- P. P. Kung, P. Bingham, A. Brooun, M. Collins, Y. L. Deng, D. Dinh, C. Fan, K. S. Gajiwala, R. Grantner, H. J. Gukasyan, W. Hu, B. Huang, R. Kania, S. E. Kephart, C. Krivacic, R. A. Kumpf, P. Khamphavong, M. Kraus, W. Liu, K. A. Maegley, L. Nguyen, S. Ren, D. Richter, R. A. Rollins, N. Sach, S. Sharma, J. Sherrill, J. Spangler, A. E. Stewart, S. Sutton, S. Uryu, D. Verhelle, H. Wang, S. Wang, M. Wythes, S. Xin, S. Yamazaki, H. Zhu, J. Zhu, L. Zehnder and M. Edwards, J. Med. Chem., 2018, 61, 650–665 CrossRef CAS PubMed.
- Pfizer, PF-06821497 Treatment Of Relapsed/Refractory SCLC, Castration Resistant Prostate Cancer, and Follicular Lymphoma, https://ClinicalTrials.gov/show/NCT03460977, 2018.
- K. D. Konze, A. Ma, F. Li, D. Barsyte-Lovejoy, T. Parton, C. J. Macnevin, F. Liu, C. Gao, X. P. Huang, E. Kuznetsova, M. Rougie, A. Jiang, S. G. Pattenden, J. L. Norris, L. I. James, B. L. Roth, P. J. Brown, S. V. Frye, C. H. Arrowsmith, K. M. Hahn, G. G. Wang, M. Vedadi and J. Jin, ACS Chem. Biol., 2013, 8, 1324–1334 CrossRef CAS PubMed.
- B. Xu, D. M. On, A. Ma, T. Parton, K. D. Konze, S. G. Pattenden, D. F. Allison, L. Cai, S. Rockowitz, S. Liu, Y. Liu, F. Li, M. Vedadi, S. V. Frye, B. A. Garcia, D. Zheng, J. Jin and G. G. Wang, Blood, 2015, 125, 346–357 CrossRef CAS PubMed.
- D. Honma, O. Kanno, J. Watanabe, J. Kinoshita, M. Hirasawa, E. Nosaka, M. Shiroishi, T. Takizawa, I. Yasumatsu, T. Horiuchi, A. Nakao, K. Suzuki, T. Yamasaki, K. Nakajima, M. Hayakawa, T. Yamazaki, A. S. Yadav and N. Adachi, Cancer Sci., 2017, 108, 2069–2078 CrossRef CAS PubMed.
- W. Qi, K. Zhao, J. Gu, Y. Huang, Y. Wang, H. Zhang, M. Zhang, J. Zhang, Z. Yu, L. Li, L. Teng, S. Chuai, C. Zhang, M. Zhao, H. Chan, Z. Chen, D. Fang, Q. Fei, L. Feng, L. Feng, Y. Gao, H. Ge, X. Ge, G. Li, A. Lingel, Y. Lin, Y. Liu, F. Luo, M. Shi, L. Wang, Z. Wang, Y. Yu, J. Zeng, C. Zeng, L. Zhang, Q. Zhang, S. Zhou, C. Oyang, P. Atadja and E. Li, Nat. Chem. Biol., 2017, 13, 381–388 CrossRef CAS PubMed.
- Y. He, S. Selvaraju, M. L. Curtin, C. G. Jakob, H. Zhu, K. M. Comess, B. Shaw, J. The, E. Lima-Fernandes, M. M. Szewczyk, D. Cheng, K. L. Klinge, H. Q. Li, M. Pliushchev, M. A. Algire, D. Maag, J. Guo, J. Dietrich, S. C. Panchal, A. M. Petros, R. F. Sweis, M. Torrent, L. J. Bigelow, G. Senisterra, F. Li, S. Kennedy, Q. Wu, D. J. Osterling, D. J. Lindley, W. Gao, S. Galasinski, D. Barsyte-Lovejoy, M. Vedadi, F. G. Buchanan, C. H. Arrowsmith, G. G. Chiang, C. Sun and W. N. Pappano, Nat. Chem. Biol., 2017, 13, 389–395 CrossRef CAS PubMed.
- H. Dong, S. Liu, X. Zhang, S. Chen, L. Kang, Y. Chen, S. Ma, X. Fu, Y. Liu, H. Zhang and B. Zou, Cancer Res., 2019, 79, 5587–5596 CrossRef CAS PubMed.
- N. Pharmaceuticals and Novartis, Safety and Efficacy of MAK683 in Adult Patients With Advanced Malignancies, https://ClinicalTrials.gov/show/NCT02900651, 2016.
- A. Ma, E. Stratikopoulos, K. S. Park, J. Wei, T. C. Martin, X. Yang, M. Schwarz, V. Leshchenko, A. Rialdi, B. Dale, A. Lagana, E. Guccione, S. Parekh, R. Parsons and J. Jin, Nat. Chem. Biol., 2020, 16, 214–222 CrossRef CAS PubMed.
- Z. Liu, X. Hu, Q. Wang, X. Wu, Q. Zhang, W. Wei, X. Su, H. He, S. Zhou, R. Hu, T. Ye, Y. Zhu, N. Wang and L. Yu, J. Med. Chem., 2021, 64, 2829–2848 CrossRef CAS PubMed.
- X. Wang, W. Cao, J. Zhang, M. Yan, Q. Xu, X. Wu, L. Wan, Z. Zhang, C. Zhang, X. Qin, M. Xiao, D. Ye, Y. Liu, Z. Han, S. Wang, L. Mao, W. Wei and W. Chen, EMBO J., 2017, 36, 1243–1260 CrossRef CAS PubMed.
- J. H. Hsu, T. Rasmusson, J. Robinson, F. Pachl, J. Read, S. Kawatkar, O. D. Dh, S. Bagal, E. Code, P. Rawlins, A. Argyrou, R. Tomlinson, N. Gao, X. Zhu, E. Chiarparin, K. Jacques, M. Shen, H. Woods, E. Bednarski, D. M. Wilson, L. Drew, M. P. Castaldi, S. Fawell and A. Bloecher, Cell Chem. Biol., 2020, 27, 41–46 e17 CrossRef CAS PubMed.
- F. Potjewyd, A. W. Turner, J. Beri, J. M. Rectenwald, J. L. Norris-Drouin, S. H. Cholensky, D. M. Margolis, K. H. Pearce, L. E. Herring and L. I. James, Cell Chem. Biol., 2020, 27, 47–56 e15 CrossRef CAS PubMed.
- K. Nishioka, S. Chuikov, K. Sarma, H. Erdjument-Bromage, C. D. Allis, P. Tempst and D. Reinberg, Genes Dev., 2002, 16, 479–489 CrossRef CAS PubMed.
- H. Wang, R. Cao, L. Xia, H. Erdjument-Bromage, C. Borchers, P. Tempst and Y. Zhang, Mol. Cell, 2001, 8, 1207–1217 CrossRef CAS PubMed.
- T. Kwon, J. H. Chang, E. Kwak, C. W. Lee, A. Joachimiak, Y. C. Kim, J. Lee and Y. Cho, EMBO J., 2003, 22, 292–303 CrossRef CAS PubMed.
- J. R. Wilson, C. Jing, P. A. Walker, S. R. Martin, S. A. Howell, G. M. Blackburn, S. J. Gamblin and B. Xiao, Cell, 2002, 111, 105–115 CrossRef CAS PubMed.
- P. Hu, S. Wang and Y. Zhang, J. Am. Chem. Soc., 2008, 130, 3806–3813 CrossRef CAS PubMed.
- S. Chuikov, J. K. Kurash, J. R. Wilson, B. Xiao, N. Justin, G. S. Ivanov, K. McKinney, P. Tempst, C. Prives, S. J. Gamblin, N. A. Barlev and D. Reinberg, Nature, 2004, 432, 353–360 CrossRef CAS PubMed.
- J. Wang, S. Hevi, J. K. Kurash, H. Lei, F. Gay, J. Bajko, H. Su, W. Sun, H. Chang, G. Xu, F. Gaudet, E. Li and T. Chen, Nat. Genet., 2009, 41, 125–129 CrossRef CAS PubMed.
- P.-O. Estève, H. G. Chin, J. Benner, G. R. Feehery, M. Samaranayake, G. A. Horwitz, S. E. Jacobsen and S. Pradhan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5076–5081 CrossRef PubMed.
- Y. Li, M. A. Reddy, F. Miao, N. Shanmugam, J. K. Yee, D. Hawkins, B. Ren and R. Natarajan, J. Biol. Chem., 2008, 283, 26771–26781 CrossRef CAS PubMed.
- B. Duan, J. Bai, J. Qiu, J. Wang, C. Tong, X. Wang, J. Miao, Z. Li, W. Li, J. Yang and C. Huang, EBioMedicine, 2018, 37, 134–143 CrossRef PubMed.
- Y. Gu, X. Wang, H. Liu, G. Li, W. Yu and Q. Ma, Oncol. Rep., 2018, 40, 1863–1874 CAS.
- S. Mori, K. Iwase, N. Iwanami, Y. Tanaka, H. Kagechika and T. Hirano, Bioorg. Med. Chem., 2010, 18, 8158–8166 CrossRef CAS PubMed.
- H. Niwa, N. Handa, Y. Tomabechi, K. Honda, M. Toyama, N. Ohsawa, M. Shirouzu, H. Kagechika, T. Hirano, T. Umehara and S. Yokoyama, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 595–602 CrossRef CAS PubMed.
- Y. Takemoto, A. Ito, H. Niwa, M. Okamura, T. Fujiwara, T. Hirano, N. Handa, T. Umehara, T. Sonoda, K. Ogawa, M. Tariq, N. Nishino, S. Dan, H. Kagechika, T. Yamori, S. Yokoyama and M. Yoshida, J. Med. Chem., 2016, 59, 3650–3660 CrossRef CAS PubMed.
- T. Hirano, T. Fujiwara, H. Niwa, M. Hirano, K. Ohira, Y. Okazaki, S. Sato, T. Umehara, Y. Maemoto, A. Ito, M. Yoshida and H. Kagechika, ChemMedChem, 2018, 13, 1530–1540 CrossRef CAS PubMed.
- F. Meng, S. Cheng, H. Ding, S. Liu, Y. Liu, K. Zhu, S. Chen, J. Lu, Y. Xie, L. Li, R. Liu, Z. Shi, Y. Zhou, Y. C. Liu, M. Zheng, H. Jiang, W. Lu, H. Liu and C. Luo, J. Med. Chem., 2015, 58, 8166–8181 CrossRef CAS PubMed.
- D. Barsyte-Lovejoy, F. Li, M. J. Oudhoff, J. H. Tatlock, A. Dong, H. Zeng, H. Wu, S. A. Freeman, M. Schapira, G. A. Senisterra, E. Kuznetsova, R. Marcellus, A. Allali-Hassani, S. Kennedy, J. P. Lambert, A. L. Couzens, A. Aman, A. C. Gingras, R. Al-Awar, P. V. Fish, B. S. Gerstenberger, L. Roberts, C. L. Benn, R. L. Grimley, M. J. Braam, F. M. Rossi, M. Sudol, P. J. Brown, M. E. Bunnage, D. R. Owen, C. Zaph, M. Vedadi and C. H. Arrowsmith, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12853–12858 CrossRef CAS PubMed.
- B. Tang, B. Li, B. Li, J. Qin, J. Zhao, J. Xu, Y. Qiu, Z. Wu and M. Fang, RSC Adv., 2019, 9, 9218–9227 RSC.
- M. A. Brown, R. J. Sims, 3rd, P. D. Gottlieb and P. W. Tucker, Mol. Cancer, 2006, 5, 26 CrossRef PubMed.
- S. Weirich, M. K. Schuhmacher, S. Kudithipudi, C. Lungu, A. D. Ferguson and A. Jeltsch, ChemBioChem, 2020, 21, 256–264 CrossRef CAS PubMed.
- J. Huang, L. Perez-Burgos, B. J. Placek, R. Sengupta, M. Richter, J. A. Dorsey, S. Kubicek, S. Opravil, T. Jenuwein and S. L. Berger, Nature, 2006, 444, 629–632 CrossRef CAS PubMed.
- B. Sesé, M. J. Barrero, M. C. Fabregat, V. Sander and J. C. Izpisua Belmonte, Int. J. Dev. Biol., 2013, 57, 357–364 CrossRef PubMed.
- F. Diehl, M. A. Brown, M. J. van Amerongen, T. Novoyatleva, A. Wietelmann, J. Harriss, F. Ferrazzi, T. Böttger, R. P. Harvey, P. W. Tucker and F. B. Engel, PLoS One, 2010, 5, e9748 CrossRef PubMed.
- S. Komatsu, D. Ichikawa, S. Hirajima, H. Nagata, Y. Nishimura, T. Kawaguchi, M. Miyamae, W. Okajima, T. Ohashi, H. Konishi, A. Shiozaki, H. Fujiwara, K. Okamoto, H. Tsuda, I. Imoto, J. Inazawa and E. Otsuji, Br. J. Cancer, 2015, 112, 357–364 CrossRef CAS PubMed.
- N. Reynoird, P. K. Mazur, T. Stellfeld, N. M. Flores, S. M. Lofgren, S. M. Carlson, E. Brambilla, P. Hainaut, E. B. Kaznowska, C. H. Arrowsmith, P. Khatri, C. Stresemann, O. Gozani and J. Sage, Genes Dev., 2016, 30, 772–785 CrossRef CAS PubMed.
- S. Komatsu, I. Imoto, H. Tsuda, K. I. Kozaki, T. Muramatsu, Y. Shimada, S. Aiko, Y. Yoshizumi, D. Ichikawa, E. Otsuji and J. Inazawa, Carcinogenesis, 2009, 30, 1139–1146 CrossRef CAS PubMed.
- L. Liu, S. Kimball, H. Liu, A. Holowatyj and Z. Q. Yang, Oncotarget, 2015, 6, 2466–2482 CrossRef PubMed.
- A. D. Ferguson, N. A. Larsen, T. Howard, H. Pollard, I. Green, C. Grande, T. Cheung, R. Garcia-Arenas, S. Cowen, J. Wu, R. Godin, H. Chen and N. Keen, Structure, 2011, 19, 1262–1273 CrossRef CAS PubMed.
- R. F. Sweis, Z. Wang, M. Algire, C. H. Arrowsmith, P. J. Brown, G. G. Chiang, J. Guo, C. G. Jakob, S. Kennedy, F. Li, D. Maag, B. Shaw, N. B. Soni, M. Vedadi and W. N. Pappano, ACS Med. Chem. Lett., 2015, 6, 695–700 CrossRef CAS PubMed.
- S. Cowen, EpiCongress 2013, Boston, MA, July 23–24, 2013, 2013.
- S. Throner, S. Cowen, D. Russell, L. Dakin, H. W. Chen, N. Larsen, R. Godin, X. L. Zheng, A. Molina, J. Q. Wu, T. Cheung, T. Howard, R. Garcia-Arenas, N. Keen and A. Ferguson, Abstr. Pap. Am. Chem. Soc., 2015, 250, 1 Search PubMed.
- S. D. Cowen, D. Russell, L. A. Dakin, H. Chen, N. A. Larsen, R. Godin, S. Throner, X. Zheng, A. Molina, J. Wu, T. Cheung, T. Howard, R. Garcia-Arenas, N. Keen, C. S. Pendleton, J. A. Pietenpol and A. D. Ferguson, J. Med. Chem., 2016, 59, 11079–11097 CrossRef CAS PubMed.
- H. Nguyen, A. Allali-Hassani, S. Antonysamy, S. Chang, L. H. Chen, C. Curtis, S. Emtage, L. Fan, T. Gheyi, F. Li, S. Liu, J. R. Martin, D. Mendel, J. B. Olsen, L. Pelletier, T. Shatseva, S. Wu, F. F. Zhang, C. H. Arrowsmith, P. J. Brown, R. M. Campbell, B. A. Garcia, D. Barsyte-Lovejoy, M. Mader and M. Vedadi, J. Biol. Chem., 2015, 290, 13641–13653 CrossRef CAS PubMed.
- E. Eggert, R. C. Hillig, S. Koehr, D. Stockigt, J. Weiske, N. Barak, J. Mowat, T. Brumby, C. D. Christ, A. Ter Laak, T. Lang, A. E. Fernandez-Montalvan, V. Badock, H. Weinmann, I. V. Hartung, D. Barsyte-Lovejoy, M. Szewczyk, S. Kennedy, F. Li, M. Vedadi, P. J. Brown, V. Santhakumar, C. H. Arrowsmith, T. Stellfeld and C. Stresemann, J. Med. Chem., 2016, 59, 4578–4600 CrossRef CAS PubMed.
- M. J. Thomenius, J. Totman, D. Harvey, L. H. Mitchell, T. V. Riera, K. Cosmopoulos, A. R. Grassian, C. Klaus, M. Foley, E. A. Admirand, H. Jahic, C. Majer, T. Wigle, S. L. Jacques, J. Gureasko, D. Brach, T. Lingaraj, K. West, S. Smith, N. Rioux, N. J. Waters, C. Tang, A. Raimondi, M. Munchhof, J. E. Mills, S. Ribich, M. Porter Scott, K. W. Kuntz, W. P. Janzen, M. Moyer, J. J. Smith, R. Chesworth, R. A. Copeland and P. A. Boriack-Sjodin, PLoS One, 2018, 13, e0197372 CrossRef PubMed.
- A. P. Taylor, M. Swewczyk, S. Kennedy, V. V. Trush, H. Wu, H. Zeng, A. Dong, R. Ferreira de Freitas, J. Tatlock, R. A. Kumpf, M. Wythes, A. Casimiro-Garcia, R. A. Denny, M. D. Parikh, F. Li, D. Barsyte-Lovejoy, M. Schapira, M. Vedadi, P. J. Brown, C. H. Arrowsmith and D. R. Owen, J. Med. Chem., 2019, 62, 7669–7683 CrossRef CAS PubMed.
- G. S. Van Aller, N. Reynoird, O. Barbash, M. Huddleston, S. Liu, A. F. Zmoos, P. McDevitt, R. Sinnamon, B. Le, G. Mas, R. Annan, J. Sage, B. A. Garcia, P. J. Tummino, O. Gozani and R. G. Kruger, Epigenetics, 2012, 7, 340–343 CrossRef CAS PubMed.
- S. Xu, J. Wu, B. Sun, C. Zhong and J. Ding, Nucleic Acids Res., 2011, 39, 4438–4449 CrossRef CAS PubMed.
- M. A. Brown, K. Foreman, J. Harriss, C. Das, L. Zhu, M. Edwards, S. Shaaban and H. Tucker, Oncotarget, 2015, 6, 4005–4019 CrossRef PubMed.
- M. Kunizaki, R. Hamamoto, F. P. Silva, K. Yamaguchi, T. Nagayasu, M. Shibuya, Y. Nakamura and Y. Furukawa, Cancer Res., 2007, 67, 10759–10765 CrossRef CAS PubMed.
- P. K. Mazur, N. Reynoird, P. Khatri, P. W. Jansen, A. W. Wilkinson, S. Liu, O. Barbash, G. S. Van Aller, M. Huddleston, D. Dhanak, P. J. Tummino, R. G. Kruger, B. A. Garcia, A. J. Butte, M. Vermeulen, J. Sage and O. Gozani, Nature, 2014, 510, 283–287 CrossRef CAS PubMed.
- S. Suzuki, Y. Nozawa, S. Tsukamoto, T. Kaneko, H. Imai and N. Minami, Reproduction, 2015, 150, 21–30 CAS.
- X. G. Luo, T. Xi, S. Guo, Z. P. Liu, N. Wang, Y. Jiang and T. C. Zhang, IUBMB Life, 2009, 61, 679–684 CrossRef CAS PubMed.
- A. M. Cock-Rada, S. Medjkane, N. Janski, N. Yousfi, M. Perichon, M. Chaussepied, J. Chluba, G. Langsley and J. B. Weitzman, Cancer Res., 2012, 72, 810–820 CrossRef CAS PubMed.
- R. Hamamoto, Y. Furukawa, M. Morita, Y. Iimura, F. P. Silva, M. Li, R. Yagyu and Y. Nakamura, Nat. Cell Biol., 2004, 6, 731–740 CrossRef CAS PubMed.
- A. Peserico, A. Germani, P. Sanese, A. J. Barbosa, V. Di Virgilio, R. Fittipaldi, E. Fabini, C. Bertucci, G. Varchi, M. P. Moyer, G. Caretti, A. Del Rio and C. Simone, J. Cell. Physiol., 2015, 230, 2447–2460 CrossRef CAS PubMed.
- G. S. Van Aller, A. P. Graves, P. A. Elkins, W. G. Bonnette, P. J. McDevitt, F. Zappacosta, R. S. Annan, T. W. Dean, D. S. Su, C. L. Carpenter, H. P. Mohammad and R. G. Kruger, Structure, 2016, 24, 774–781 CrossRef CAS PubMed.
- L. H. Mitchell, P. A. Boriack-Sjodin, S. Smith, M. Thomenius, N. Rioux, M. Munchhof, J. E. Mills, C. Klaus, J. Totman, T. V. Riera, A. Raimondi, S. L. Jacques, K. West, M. Foley, N. J. Waters, K. W. Kuntz, T. J. Wigle, M. P. Scott, R. A. Copeland, J. J. Smith and R. Chesworth, ACS Med. Chem. Lett., 2016, 7, 134–138 CrossRef CAS PubMed.
- C. Huang, S. S. Liew, G. R. Lin, A. Poulsen, M. J. Y. Ang, B. C. S. Chia, S. Y. Chew, Z. P. Kwek, J. L. K. Wee, E. H. Ong, P. Retna, N. Baburajendran, R. Li, W. Yu, X. Koh-Stenta, A. Ngo, S. Manesh, J. Fulwood, Z. Ke, H. H. Chung, S. Sepramaniam, X. H. Chew, N. Dinie, M. A. Lee, Y. S. Chew, C. B. Low, V. Pendharkar, V. Manoharan, S. Vuddagiri, K. Sangthongpitag, J. Joy, A. Matter, J. Hill, T. H. Keller and K. Foo, ACS Med. Chem. Lett., 2019, 10, 978–984 CrossRef CAS PubMed.
- D. S. Su, J. Qu, M. Schulz, C. W. Blackledge, H. Yu, J. Zeng, J. Burgess, A. Reif, M. Stern, R. Nagarajan, M. B. Pappalardi, K. Wong, A. P. Graves, W. Bonnette, L. Wang, P. Elkins, B. Knapp-Reed, J. D. Carson, C. McHugh, H. Mohammad, R. Kruger, J. Luengo, D. A. Heerding and C. L. Creasy, ACS Med. Chem. Lett., 2020, 11, 133–140 CrossRef CAS PubMed.
- V. O. Talibov, E. Fabini, E. FitzGerald, D. Tedesco, D. Eriksson, M. J. Talu, M. M. Rachman, F. Mihalic, E. Manoni, M. Naldi, P. Sanese, G. Forte, M. L. Signorile, X. Barril, C. Simone, M. Bartolini, D. Dobritzsch, A. Del Rio and U. H. Danielson, ChemBioChem, 2021, 22, 1597–1608 CrossRef CAS PubMed.
- J. W. Edmunds, L. C. Mahadevan and A. L. Clayton, EMBO J., 2008, 27, 406–420 CrossRef CAS PubMed.
- B. D. Strahl, P. A. Grant, S. D. Briggs, Z. W. Sun, J. R. Bone, J. A. Caldwell, S. Mollah, R. G. Cook, J. Shabanowitz, D. F. Hunt and C. D. Allis, Mol. Cell. Biol., 2002, 22, 1298–1306 CrossRef CAS PubMed.
- I. Y. Park, R. T. Powell, D. N. Tripathi, R. Dere, T. H. Ho, T. L. Blasius, Y. C. Chiang, I. J. Davis, C. C. Fahey, K. E. Hacker, K. J. Verhey, M. T. Bedford, E. Jonasch, W. K. Rathmell and C. L. Walker, Cell, 2016, 166, 950–962 CrossRef CAS PubMed.
- K. Chen, J. Liu, S. Liu, M. Xia, X. Zhang, D. Han, Y. Jiang, C. Wang and X. Cao, Cell, 2017, 170, 492–506 e414 CrossRef CAS PubMed.
- M. K. Schuhmacher, S. Beldar, M. S. Khella, A. Brohm, J. Ludwig, W. Tempel, S. Weirich, J. Min and A. Jeltsch, Commun. Biol., 2020, 3, 511 CrossRef CAS PubMed.
- M. Li, H. P. Phatnani, Z. Guan, H. Sage, A. L. Greenleaf and P. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17636–17641 CrossRef CAS PubMed.
- X. J. Sun, J. Wei, X. Y. Wu, M. Hu, L. Wang, H. H. Wang, Q. H. Zhang, S. J. Chen, Q. H. Huang and Z. Chen, J. Biol. Chem., 2005, 280, 35261–35271 CrossRef CAS PubMed.
- I. Sanidas, C. Polytarchou, M. Hatziapostolou, S. A. Ezell, F. Kottakis, L. Hu, A. Guo, J. Xie, M. J. Comb, D. Iliopoulos and P. N. Tsichlis, Mol. Cell, 2014, 53, 577–590 CrossRef CAS PubMed.
- N. Cancer Genome Atlas Research, W. M. Linehan, P. T. Spellman, C. J. Ricketts, C. J. Creighton, S. S. Fei, C. Davis, D. A. Wheeler, B. A. Murray, L. Schmidt, C. D. Vocke, M. Peto, A. A. Al Mamun, E. Shinbrot, A. Sethi, S. Brooks, W. K. Rathmell, A. N. Brooks, K. A. Hoadley, A. G. Robertson, D. Brooks, R. Bowlby, S. Sadeghi, H. Shen, D. J. Weisenberger, M. Bootwalla, S. B. Baylin, P. W. Laird, A. D. Cherniack, G. Saksena, S. Haake, J. Li, H. Liang, Y. Lu, G. B. Mills, R. Akbani, M. D. Leiserson, B. J. Raphael, P. Anur, D. Bottaro, L. Albiges, N. Barnabas, T. K. Choueiri, B. Czerniak, A. K. Godwin, A. A. Hakimi, T. H. Ho, J. Hsieh, M. Ittmann, W. Y. Kim, B. Krishnan, M. J. Merino, K. R. Mills Shaw, V. E. Reuter, E. Reznik, C. S. Shelley, B. Shuch, S. Signoretti, R. Srinivasan, P. Tamboli, G. Thomas, S. Tickoo, K. Burnett, D. Crain, J. Gardner, K. Lau, D. Mallery, S. Morris, J. D. Paulauskis, R. J. Penny, C. Shelton, W. T. Shelton, M. Sherman, E. Thompson, P. Yena, M. T. Avedon, J. Bowen, J. M. Gastier-Foster, M. Gerken, K. M. Leraas, T. M. Lichtenberg, N. C. Ramirez, T. Santos, L. Wise, E. Zmuda, J. A. Demchok, I. Felau, C. M. Hutter, M. Sheth, H. J. Sofia, R. Tarnuzzer, Z. Wang, L. Yang, J. C. Zenklusen, J. Zhang, B. Ayala, J. Baboud, S. Chudamani, J. Liu, L. Lolla, R. Naresh, T. Pihl, Q. Sun, Y. Wan, Y. Wu, A. Ally, M. Balasundaram, S. Balu, R. Beroukhim, T. Bodenheimer, C. Buhay, Y. S. Butterfield, R. Carlsen, S. L. Carter, H. Chao, E. Chuah, A. Clarke, K. R. Covington, M. Dahdouli, N. Dewal, N. Dhalla, H. V. Doddapaneni, J. A. Drummond, S. B. Gabriel, R. A. Gibbs, R. Guin, W. Hale, A. Hawes, D. N. Hayes, R. A. Holt, A. P. Hoyle, S. R. Jefferys, S. J. Jones, C. D. Jones, D. Kalra, C. Kovar, L. Lewis, J. Li, Y. Ma, M. A. Marra, M. Mayo, S. Meng, M. Meyerson, P. A. Mieczkowski, R. A. Moore, D. Morton, L. E. Mose, A. J. Mungall, D. Muzny, J. S. Parker, C. M. Perou, J. Roach, J. E. Schein, S. E. Schumacher, Y. Shi, J. V. Simons, P. Sipahimalani, T. Skelly, M. G. Soloway, C. Sougnez, A. Tam, D. Tan, N. Thiessen, U. Veluvolu, M. Wang, M. D. Wilkerson, T. Wong, J. Wu, L. Xi, J. Zhou, J. Bedford, F. Chen, Y. Fu, M. Gerstein, D. Haussler, K. Kasaian, P. Lai, S. Ling, A. Radenbaugh, D. Van Den Berg, J. N. Weinstein, J. Zhu, M. Albert, I. Alexopoulou, J. J. Andersen, J. T. Auman, J. Bartlett, S. Bastacky, J. Bergsten, M. L. Blute, L. Boice, R. J. Bollag, J. Boyd, E. Castle, Y. B. Chen, J. C. Cheville, E. Curley, B. Davies, A. DeVolk, R. Dhir, L. Dike, J. Eckman, J. Engel, J. Harr, R. Hrebinko, M. Huang, L. Huelsenbeck-Dill, M. Iacocca, B. Jacobs, M. Lobis, J. K. Maranchie, S. McMeekin, J. Myers, J. Nelson, J. Parfitt, A. Parwani, N. Petrelli, B. Rabeno, S. Roy, A. L. Salner, J. Slaton, M. Stanton, R. H. Thompson, L. Thorne, K. Tucker, P. M. Weinberger, C. Winemiller, L. A. Zach and R. Zuna, N. Engl. J. Med., 2016, 374, 135–145 CrossRef PubMed.
- A. M. Fontebasso, J. Schwartzentruber, D. A. Khuong-Quang, X. Y. Liu, D. Sturm, A. Korshunov, D. T. Jones, H. Witt, M. Kool, S. Albrecht, A. Fleming, D. Hadjadj, S. Busche, P. Lepage, A. Montpetit, A. Staffa, N. Gerges, M. Zakrzewska, K. Zakrzewski, P. P. Liberski, P. Hauser, M. Garami, A. Klekner, L. Bognar, G. Zadeh, D. Faury, S. M. Pfister, N. Jabado and J. Majewski, Acta Neuropathol., 2013, 125, 659–669 CrossRef CAS PubMed.
- X. Zhu, F. He, H. Zeng, S. Ling, A. Chen, Y. Wang, X. Yan, W. Wei, Y. Pang, H. Cheng, C. Hua, Y. Zhang, X. Yang, X. Lu, L. Cao, L. Hao, L. Dong, W. Zou, J. Wu, X. Li, S. Zheng, J. Yan, J. Zhou, L. Zhang, S. Mi, X. Wang, L. Zhang, Y. Zou, Y. Chen, Z. Geng, J. Wang, J. Zhou, X. Liu, J. Wang, W. Yuan, G. Huang, T. Cheng and Q. F. Wang, Nat. Genet., 2014, 46, 287–293 CrossRef CAS PubMed.
- C. Kandoth, M. D. McLellan, F. Vandin, K. Ye, B. Niu, C. Lu, M. Xie, Q. Zhang, J. F. McMichael, M. A. Wyczalkowski, M. D. M. Leiserson, C. A. Miller, J. S. Welch, M. J. Walter, M. C. Wendl, T. J. Ley, R. K. Wilson, B. J. Raphael and L. Ding, Nature, 2013, 502, 333–339 CrossRef CAS PubMed.
- A. A. Hakimi, I. Ostrovnaya, B. Reva, N. Schultz, Y. B. Chen, M. Gonen, H. Liu, S. Takeda, M. H. Voss, S. K. Tickoo, V. E. Reuter, P. Russo, E. H. Cheng, C. Sander, R. J. Motzer, J. J. Hsieh and R. C. C. C. G. A. R. N. I. cc, Clin. Cancer Res., 2013, 19, 3259–3267 CrossRef CAS PubMed.
- N. Kanu, E. Gronroos, P. Martinez, R. A. Burrell, X. Yi Goh, J. Bartkova, A. Maya-Mendoza, M. Mistrik, A. J. Rowan, H. Patel, A. Rabinowitz, P. East, G. Wilson, C. R. Santos, N. McGranahan, S. Gulati, M. Gerlinger, N. J. Birkbak, T. Joshi, L. B. Alexandrov, M. R. Stratton, T. Powles, N. Matthews, P. A. Bates, A. Stewart, Z. Szallasi, J. Larkin, J. Bartek and C. Swanton, Oncogene, 2015, 34, 5699–5708 CrossRef CAS PubMed.
- W. Zheng, G. Ibanez, H. Wu, G. Blum, H. Zeng, A. Dong, F. Li, T. Hajian, A. Allali-Hassani, M. F. Amaya, A. Siarheyeva, W. Yu, P. J. Brown, M. Schapira, M. Vedadi, J. Min and M. Luo, J. Am. Chem. Soc., 2012, 134, 18004–18014 CrossRef CAS PubMed.
- K. Nishioka, J. C. Rice, K. Sarma, H. Erdjument-Bromage, J. Werner, Y. Wang, S. Chuikov, P. Valenzuela, P. Tempst, R. Steward, J. T. Lis, C. D. Allis and D. Reinberg, Mol. Cell, 2002, 9, 1201–1213 CrossRef CAS PubMed.
- T. S. Girish, R. K. McGinty and S. Tan, J. Mol. Biol., 2016, 428, 1531–1543 CrossRef CAS PubMed.
- X. Shi, I. Kachirskaia, H. Yamaguchi, L. E. West, H. Wen, E. W. Wang, S. Dutta, E. Appella and O. Gozani, Mol. Cell, 2007, 27, 636–646 CrossRef CAS PubMed.
- G. K. Dhami, H. Liu, M. Galka, C. Voss, R. Wei, K. Muranko, T. Kaneko, S. P. Cregan, L. Li and S. S. Li, Mol. Cell, 2013, 50, 565–576 CrossRef CAS PubMed.
- M. Takawa, H. S. Cho, S. Hayami, G. Toyokawa, M. Kogure, Y. Yamane, Y. Iwai, K. Maejima, K. Ueda, A. Masuda, N. Dohmae, H. I. Field, T. Tsunoda, T. Kobayashi, T. Akasu, M. Sugiyama, S. Ohnuma, Y. Atomi, B. A. Ponder, Y. Nakamura and R. Hamamoto, Cancer Res., 2012, 72, 3217–3227 CrossRef CAS PubMed.
- S. Jorgensen, I. Elvers, M. B. Trelle, T. Menzel, M. Eskildsen, O. N. Jensen, T. Helleday, K. Helin and C. S. Sorensen, J. Cell Biol., 2007, 179, 1337–1345 CrossRef PubMed.
- G. Schotta, R. Sengupta, S. Kubicek, S. Malin, M. Kauer, E. Callen, A. Celeste, M. Pagani, S. Opravil, I. A. De La Rosa-Velazquez, A. Espejo, M. T. Bedford, A. Nussenzweig, M. Busslinger and T. Jenuwein, Genes Dev., 2008, 22, 2048–2061 CrossRef CAS PubMed.
- H. Oda, M. R. Hubner, D. B. Beck, M. Vermeulen, J. Hurwitz, D. L. Spector and D. Reinberg, Mol. Cell, 2010, 40, 364–376 CrossRef CAS PubMed.
- F. Yang, L. Sun, Q. Li, X. Han, L. Lei, H. Zhang and Y. Shang, EMBO J., 2012, 31, 110–123 CrossRef CAS PubMed.
- R. Huang, Y. Yu, X. Zong, X. Li, L. Ma and Q. Zheng, Cancer Lett., 2017, 390, 1–10 CrossRef CAS PubMed.
- D. E. Williams, F. Izard, S. Arnould, D. S. Dalisay, C. Tantapakul, W. Maneerat, T. Matainaho, E. Julien and R. J. Andersen, J. Org. Chem., 2016, 81, 1324–1332 CrossRef CAS PubMed.
- D. E. Williams, D. S. Dalisay, F. Li, J. Amphlett, W. Maneerat, M. A. Chavez, Y. A. Wang, T. Matainaho, W. Yu, P. J. Brown, C. H. Arrowsmith, M. Vedadi and R. J. Andersen, Org. Lett., 2013, 15, 414–417 CrossRef CAS PubMed.
- G. Blum, G. Ibanez, X. Rao, D. Shum, C. Radu, H. Djaballah, J. C. Rice and M. Luo, ACS Chem. Biol., 2014, 9, 2471–2478 CrossRef CAS PubMed.
- A. Ma, W. Yu, F. Li, R. M. Bleich, J. M. Herold, K. V. Butler, J. L. Norris, V. Korboukh, A. Tripathy, W. P. Janzen, C. H. Arrowsmith, S. V. Frye, M. Vedadi, P. J. Brown and J. Jin, J. Med. Chem., 2014, 57, 6822–6833 CrossRef CAS PubMed.
- L. Herviou, F. Izard, O. Karmous-Gadacha, C. Gourzones, C. Bellanger, E. Desmedt, A. Ma, L. Vincent, G. Cartron, K. Vanderkerken, J. Jin, E. De Bruyne, C. Grimaud, E. Julien and J. Moreaux, bioRxiv, 2019, 776930, DOI:10.1101/776930.
- K. V. Butler, A. Ma, W. Yu, F. Li, W. Tempel, N. Babault, F. Pittella-Silva, J. Shao, J. Wang, M. Luo, M. Vedadi, P. J. Brown, C. H. Arrowsmith and J. Jin, J. Med. Chem., 2016, 59, 9881–9889 CrossRef CAS PubMed.
- H. Yang, J. J. Pesavento, T. W. Starnes, D. E. Cryderman, L. L. Wallrath, N. L. Kelleher and C. A. Mizzen, J. Biol. Chem., 2008, 283, 12085–12092 CrossRef CAS PubMed.
- H. Wu, A. Siarheyeva, H. Zeng, R. Lam, A. Dong, X. H. Wu, Y. Li, M. Schapira, M. Vedadi and J. Min, FEBS Lett., 2013, 587, 3859–3868 CrossRef CAS PubMed.
- X. Lu, M. D. Simon, J. V. Chodaparambil, J. C. Hansen, K. M. Shokat and K. Luger, Nat. Struct. Mol. Biol., 2008, 15, 1122–1124 CrossRef CAS PubMed.
- G. Schotta, M. Lachner, K. Sarma, A. Ebert, R. Sengupta, G. Reuter, D. Reinberg and T. Jenuwein, Genes Dev., 2004, 18, 1251–1262 CrossRef CAS PubMed.
- R. Benetti, S. Gonzalo, I. Jaco, G. Schotta, P. Klatt, T. Jenuwein and M. A. Blasco, J. Cell Biol., 2007, 178, 925–936 CrossRef CAS PubMed.
- R. M. Marion, G. Schotta, S. Ortega and M. A. Blasco, PLoS One, 2011, 6, e25680 CrossRef CAS PubMed.
- L. Piao, X. Yuan, L. Wang, X. Xu, M. Zhuang, J. Li, R. Kong and Z. Liu, Oncol. Lett., 2020, 20, 26 CAS.
- M. Viotti, C. Wilson, M. McCleland, H. Koeppen, B. Haley, S. Jhunjhunwala, C. Klijn, Z. Modrusan, D. Arnott, M. Classon, J. P. Stephan and I. Mellman, J. Cell Biol., 2018, 217, 763–777 CrossRef CAS PubMed.
- A. Brohm, H. Elsawy, P. Rathert, S. Kudithipudi, T. Schoch, M. K. Schuhmacher, S. Weirich and A. Jeltsch, J. Mol. Biol., 2019, 431, 3068–3080 CrossRef PubMed.
- K. D. Bromberg, T. R. Mitchell, A. K. Upadhyay, C. G. Jakob, M. A. Jhala, K. M. Comess, L. M. Lasko, C. Li, C. T. Tuzon, Y. Dai, F. Li, M. S. Eram, A. Nuber, N. B. Soni, V. Manaves, M. A. Algire, R. F. Sweis, M. Torrent, G. Schotta, C. Sun, M. R. Michaelides, A. R. Shoemaker, C. H. Arrowsmith, P. J. Brown, V. Santhakumar, A. Martin, J. C. Rice, G. G. Chiang, M. Vedadi, D. Barsyte-Lovejoy and W. N. Pappano, Nat. Chem. Biol., 2017, 13, 317–324 CrossRef CAS PubMed.
- R. Zheng, D. Liang, N. Jiang, J. Zhou, X. Long, X. Wang, M. Wu, C. Wu and J. Bao, J. Biomol. Struct. Dyn., 2021, 39, 526–537 CrossRef CAS PubMed.
- W. Li, W. Tian, G. Yuan, P. Deng, D. Sengupta, Z. Cheng, Y. Cao, J. Ren, Y. Qin, Y. Zhou, Y. Jia, O. Gozani, D. J. Patel and Z. Wang, Nature, 2021, 590, 498–503 CrossRef CAS PubMed.
- M. Morishita, D. Mevius and E. di Luccio, BMC Struct. Biol., 2014, 14, 25 Search PubMed.
- J. Marango, M. Shimoyama, H. Nishio, J. A. Meyer, D. J. Min, A. Sirulnik, Y. Martinez-Martinez, M. Chesi, P. L. Bergsagel, M. M. Zhou, S. Waxman, B. A. Leibovitch, M. J. Walsh and J. D. Licht, Blood, 2008, 111, 3145–3154 CrossRef CAS PubMed.
- J. Y. Kim, H. J. Kee, N. W. Choe, S. M. Kim, G. H. Eom, H. J. Baek, H. Kook, H. Kook and S. B. Seo, Mol. Cell. Biol., 2008, 28, 2023–2034 CrossRef CAS PubMed.
- G. V. Rayasam, O. Wendling, P. O. Angrand, M. Mark, K. Niederreither, L. Song, T. Lerouge, G. L. Hager, P. Chambon and R. Losson, EMBO J., 2003, 22, 3153–3163 CrossRef CAS PubMed.
- J. Douglas, S. Hanks, I. K. Temple, S. Davies, A. Murray, M. Upadhyaya, S. Tomkins, H. E. Hughes, T. R. Cole and N. Rahman, Am. J. Hum. Genet., 2003, 72, 132–143 CrossRef CAS PubMed.
- N. Kurotaki, K. Imaizumi, N. Harada, M. Masuno, T. Kondoh, T. Nagai, H. Ohashi, K. Naritomi, M. Tsukahara, Y. Makita, T. Sugimoto, T. Sonoda, T. Hasegawa, Y. Chinen, H. A. Tomita Ha, A. Kinoshita, T. Mizuguchi, K. Yoshiura Ki, T. Ohta, T. Kishino, Y. Fukushima, N. Niikawa and N. Matsumoto, Nat. Genet., 2002, 30, 365–366 CrossRef CAS PubMed.
- K. Nimura, K. Ura, H. Shiratori, M. Ikawa, M. Okabe, R. J. Schwartz and Y. Kaneda, Nature, 2009, 460, 287–291 CrossRef CAS PubMed.
- N. L. Shannon, E. L. Maltby, A. S. Rigby and O. W. Quarrell, J. Med. Genet., 2001, 38, 674–679 CrossRef CAS PubMed.
- P. O. Angrand, F. Apiou, A. F. Stewart, B. Dutrillaux, R. Losson and P. Chambon, Genomics, 2001, 74, 79–88 CrossRef CAS PubMed.
- M. Chesi, E. Nardini, R. S. C. Lim, K. D. Smith, W. M. Kuehl and P. L. Bergsagel, Blood, 1998, 92, 3025–3034 CrossRef CAS PubMed.
- J. L. Brito, B. Walker, M. Jenner, N. J. Dickens, N. J. Brown, F. M. Ross, A. Avramidou, J. A. Irving, D. Gonzalez, F. E. Davies and G. J. Morgan, Haematologica, 2009, 94, 78–86 CrossRef CAS PubMed.
- M. Morishita, D. E. H. F. Mevius, Y. Shen, S. Zhao and E. di Luccio, Med. Chem. Res., 2017, 26, 2038–2047 CrossRef CAS.
- H. Huang, C. A. Howard, S. Zari, H. J. Cho, S. Shukla, H. Li, J. Ndoj, P. Gonzalez-Alonso, C. Nikolaidis, J. Abbott, D. S. Rogawski, M. A. Potopnyk, K. Kempinska, H. Miao, T. Purohit, A. Henderson, A. Mapp, M. L. Sulis, A. Ferrando, J. Grembecka and T. Cierpicki, Nat. Chem. Biol., 2020, 16, 1403–1410 CrossRef CAS PubMed.
- D. Tisi, E. Chiarparin, E. Tamanini, P. Pathuri, J. E. Coyle, A. Hold, F. P. Holding, N. Amin, A. C. Martin, S. J. Rich, V. Berdini, J. Yon, P. Acklam, R. Burke, L. Drouin, J. E. Harmer, F. Jeganathan, R. L. van Montfort, Y. Newbatt, M. Tortorici, M. Westlake, A. Wood, S. Hoelder and T. D. Heightman, ACS Chem. Biol., 2016, 11, 3093–3105 CrossRef CAS PubMed.
- N. P. Coussens, S. C. Kales, M. J. Henderson, O. W. Lee, K. Y. Horiuchi, Y. Wang, Q. Chen, E. Kuznetsova, J. Wu, S. Chakka, D. M. Cheff, K. C. Cheng, P. Shinn, K. R. Brimacombe, M. Shen, A. Simeonov, M. Lal-Nag, H. Ma, A. Jadhav and M. D. Hall, J. Biol. Chem., 2018, 293, 13750–13765 CrossRef CAS PubMed.
- R. Ferreira de Freitas, Y. Liu, M. M. Szewczyk, N. Mehta, F. Li, D. McLeod, C. Zepeda-Velazquez, D. Dilworth, R. P. Hanley, E. Gibson, P. J. Brown, R. Al-Awar, L. I. James, C. H. Arrowsmith, D. Barsyte-Lovejoy, J. Min, M. Vedadi, M. Schapira and A. Allali-Hassani, J. Med. Chem., 2021, 64, 1584–1592 CrossRef CAS PubMed.
- D. Dilworth, R. P. Hanley, R. Ferreira de Freitas, A. Allali-Hassani, M. Zhou, N. Mehta, M. R. Marunde, S. Ackloo, R. A. Carvalho Machado, A. Khalili Yazdi, D. D. G. Owens, V. Vu, D. Y. Nie, M. Alqazzaz, E. Marcon, F. Li, I. Chau, A. Bolotokova, S. Qin, M. Lei, Y. Liu, M. M. Szewczyk, A. Dong, S. Kazemzadeh, T. Abramyan, I. K. Popova, N. W. Hall, M. J. Meiners, M. A. Cheek, E. Gibson, D. Kireev, J. F. Greenblatt, M.-C. Keogh, J. Min, P. J. Brown, M. Vedadi, C. H. Arrowsmith, D. Barsyte-Lovejoy, L. I. James and M. Schapira, Nat. Chem. Biol., 2022, 18, 56–63 CrossRef CAS PubMed.
- J. Bottcher, D. Dilworth, U. Reiser, R. A. Neumuller, M. Schleicher, M. Petronczki, M. Zeeb, N. Mischerikow, A. Allali-Hassani, M. M. Szewczyk, F. Li, S. Kennedy, M. Vedadi, D. Barsyte-Lovejoy, P. J. Brown, K. V. M. Huber, C. M. Rogers, C. I. Wells, O. Fedorov, K. Rumpel, A. Zoephel, M. Mayer, T. Wunberg, D. Bose, S. Zahn, H. Arnhof, H. Berger, C. Reiser, A. Hormann, T. Krammer, M. Corcokovic, B. Sharps, S. Winkler, D. Haring, X. L. Cockcroft, J. E. Fuchs, B. Mullauer, A. Weiss-Puxbaum, T. Gerstberger, G. Boehmelt, C. R. Vakoc, C. H. Arrowsmith, M. Pearson and D. B. McConnell, Nat. Chem. Biol., 2019, 15, 822–829 CrossRef PubMed.
- T. A. Milne, S. D. Briggs, H. W. Brock, M. E. Martin, D. Gibbs, C. D. Allis and J. L. Hess, Mol. Cell, 2002, 10, 1107–1117 CrossRef CAS PubMed.
- S. A. Shinsky, K. E. Monteith, S. Viggiano and M. S. Cosgrove, J. Biol. Chem., 2015, 290, 6361–6375 CrossRef CAS PubMed.
- Y. H. Takahashi, G. H. Westfield, A. N. Oleskie, R. C. Trievel, A. Shilatifard and G. Skiniotis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20526–20531 CrossRef CAS PubMed.
- N. T. Crump and T. A. Milne, Cell. Mol. Life Sci., 2019, 76, 2885–2898 CrossRef CAS PubMed.
- P. M. Ayton, E. H. Chen and M. L. Cleary, Mol. Cell. Biol., 2004, 24, 10470–10478 CrossRef CAS PubMed.
- L. Wu, S. Y. Lee, B. Zhou, U. T. Nguyen, T. W. Muir, S. Tan and Y. Dou, Mol. Cell, 2013, 49, 1108–1120 CrossRef CAS PubMed.
- D. Hu, X. Gao, M. A. Morgan, H. M. Herz, E. R. Smith and A. Shilatifard, Mol. Cell. Biol., 2013, 33, 4745–4754 CrossRef CAS PubMed.
- C. Caslini, Z. Yang, M. El-Osta, T. A. Milne, R. K. Slany and J. L. Hess, Cancer Res., 2007, 67, 7275–7283 CrossRef CAS PubMed.
- S. Yin, J. Yang, B. Lin, W. Deng, Y. Zhang, X. Yi, Y. Shi, Y. Tao, J. Cai, C. I. Wu, G. Zhao, L. D. Hurst, J. Zhang, L. Hu and X. Kong, Sci. Rep., 2014, 4, 6036 CrossRef CAS PubMed.
- M. Le Gallo, M. L. Rudd, M. E. Urick, N. F. Hansen, S. Zhang, N. C. S. Program, F. Lozy, D. C. Sgroi, A. Vidal Bel, X. Matias-Guiu, R. R. Broaddus, K. H. Lu, D. A. Levine, D. G. Mutch, P. J. Goodfellow, H. B. Salvesen, J. C. Mullikin and D. W. Bell, Cancer, 2017, 123, 3261–3268 CrossRef CAS PubMed.
- T. Sjoblom, S. Jones, L. D. Wood, D. W. Parsons, J. Lin, T. D. Barber, D. Mandelker, R. J. Leary, J. Ptak, N. Silliman, S. Szabo, P. Buckhaults, C. Farrell, P. Meeh, S. D. Markowitz, J. Willis, D. Dawson, J. K. Willson, A. F. Gazdar, J. Hartigan, L. Wu, C. Liu, G. Parmigiani, B. H. Park, K. E. Bachman, N. Papadopoulos, B. Vogelstein, K. W. Kinzler and V. E. Velculescu, Science, 2006, 314, 268–274 CrossRef PubMed.
- T. R. Chern, L. Liu, E. Petrunak, J. A. Stuckey, M. Wang, D. Bernard, H. Zhou, S. Lee, Y. Dou and S. Wang, ACS Med. Chem. Lett., 2020, 11, 1348–1352 CrossRef CAS PubMed.
- A. Patel, V. E. Vought, V. Dharmarajan and M. S. Cosgrove, J. Biol. Chem., 2008, 283, 32162–32175 CrossRef CAS PubMed.
- H. Karatas, E. C. Townsend, D. Bernard, Y. Dou and S. Wang, J. Med. Chem., 2010, 53, 5179–5185 CrossRef CAS PubMed.
- H. Karatas, E. C. Townsend, F. Cao, Y. Chen, D. Bernard, L. Liu, M. Lei, Y. Dou and S. Wang, J. Am. Chem. Soc., 2013, 135, 669–682 CrossRef CAS PubMed.
- F. Cao, E. C. Townsend, H. Karatas, J. Xu, L. Li, S. Lee, L. Liu, Y. Chen, P. Ouillette, J. Zhu, J. L. Hess, P. Atadja, M. Lei, Z. S. Qin, S. Malek, S. Wang and Y. Dou, Mol. Cell, 2014, 53, 247–261 CrossRef CAS PubMed.
- H. Karatas, Y. Li, L. Liu, J. Ji, S. Lee, Y. Chen, J. Yang, L. Huang, D. Bernard, J. Xu, E. C. Townsend, F. Cao, X. Ran, X. Li, B. Wen, D. Sun, J. A. Stuckey, M. Lei, Y. Dou and S. Wang, J. Med. Chem., 2017, 60, 4818–4839 CrossRef CAS PubMed.
- G. Senisterra, H. Wu, A. Allali-Hassani, G. A. Wasney, D. Barsyte-Lovejoy, L. Dombrovski, A. Dong, K. T. Nguyen, D. Smil, Y. Bolshan, T. Hajian, H. He, A. Seitova, I. Chau, F. Li, G. Poda, J. F. Couture, P. J. Brown, R. Al-Awar, M. Schapira, C. H. Arrowsmith and M. Vedadi, Biochem. J., 2013, 449, 151–159 CrossRef CAS PubMed.
- Y. Bolshan, M. Getlik, E. Kuznetsova, G. A. Wasney, T. Hajian, G. Poda, K. T. Nguyen, H. Wu, L. Dombrovski, A. Dong, G. Senisterra, M. Schapira, C. H. Arrowsmith, P. J. Brown, R. Al-Awar, M. Vedadi and D. Smil, ACS Med. Chem. Lett., 2013, 4, 353–357 CrossRef CAS PubMed.
- D. D. Li, W. L. Chen, X. L. Xu, F. Jiang, L. Wang, Y. Y. Xie, X. J. Zhang, X. K. Guo, Q. D. You and H. P. Sun, Eur. J. Med. Chem., 2016, 118, 1–8 CrossRef CAS PubMed.
- D. D. Li, Z. H. Wang, W. L. Chen, Y. Y. Xie, Q. D. You and X. K. Guo, Bioorg. Med. Chem., 2016, 24, 6109–6118 CrossRef CAS PubMed.
- D. D. Li, W. L. Chen, Z. H. Wang, Y. Y. Xie, X. L. Xu, Z. Y. Jiang, X. J. Zhang, Q. D. You and X. K. Guo, Eur. J. Med. Chem., 2016, 124, 480–489 CrossRef CAS PubMed.
- M. Getlik, D. Smil, C. Zepeda-Velazquez, Y. Bolshan, G. Poda, H. Wu, A. Dong, E. Kuznetsova, R. Marcellus, G. Senisterra, L. Dombrovski, T. Hajian, T. Kiyota, M. Schapira, C. H. Arrowsmith, P. J. Brown, M. Vedadi and R. Al-Awar, J. Med. Chem., 2016, 59, 2478–2496 CrossRef CAS PubMed.
- F. Grebien, M. Vedadi, M. Getlik, R. Giambruno, A. Grover, R. Avellino, A. Skucha, S. Vittori, E. Kuznetsova, D. Smil, D. Barsyte-Lovejoy, F. Li, G. Poda, M. Schapira, H. Wu, A. Dong, G. Senisterra, A. Stukalov, K. V. M. Huber, A. Schonegger, R. Marcellus, M. Bilban, C. Bock, P. J. Brown, J. Zuber, K. L. Bennett, R. Al-Awar, R. Delwel, C. Nerlov, C. H. Arrowsmith and G. Superti-Furga, Nat. Chem. Biol., 2015, 11, 571–578 CrossRef CAS PubMed.
- F. Wang, K. O. Jeon, J. M. Salovich, J. D. Macdonald, J. Alvarado, R. D. Gogliotti, J. Phan, E. T. Olejniczak, Q. Sun, S. Wang, D. Camper, J. P. Yuh, J. G. Shaw, J. Sai, O. W. Rossanese, W. P. Tansey, S. R. Stauffer and S. W. Fesik, J. Med. Chem., 2018, 61, 5623–5642 CrossRef CAS PubMed.
- A. Yokoyama, T. C. Somervaille, K. S. Smith, O. Rozenblatt-Rosen, M. Meyerson and M. L. Cleary, Cell, 2005, 123, 207–218 CrossRef CAS PubMed.
- J. Grembecka, S. He, A. Shi, T. Purohit, A. G. Muntean, R. J. Sorenson, H. D. Showalter, M. J. Murai, A. M. Belcher, T. Hartley, J. L. Hess and T. Cierpicki, Nat. Chem. Biol., 2012, 8, 277–284 CrossRef CAS PubMed.
- A. Shi, M. J. Murai, S. He, G. Lund, T. Hartley, T. Purohit, G. Reddy, M. Chruszcz, J. Grembecka and T. Cierpicki, Blood, 2012, 120, 4461–4469 CrossRef CAS PubMed.
- D. Borkin, S. He, H. Miao, K. Kempinska, J. Pollock, J. Chase, T. Purohit, B. Malik, T. Zhao, J. Wang, B. Wen, H. Zong, M. Jones, G. Danet-Desnoyers, M. L. Guzman, M. Talpaz, D. L. Bixby, D. Sun, J. L. Hess, A. G. Muntean, I. Maillard, T. Cierpicki and J. Grembecka, Cancer Cell, 2015, 27, 589–602 CrossRef CAS PubMed.
- D. Borkin, J. Pollock, K. Kempinska, T. Purohit, X. Li, B. Wen, T. Zhao, H. Miao, S. Shukla, M. He, D. Sun, T. Cierpicki and J. Grembecka, J. Med. Chem., 2016, 59, 892–913 CrossRef CAS PubMed.
- D. Borkin, S. Klossowski, J. Pollock, H. Miao, B. M. Linhares, K. Kempinska, Z. Jin, T. Purohit, B. Wen, M. He, D. Sun, T. Cierpicki and J. Grembecka, J. Med. Chem., 2018, 61, 4832–4850 CrossRef CAS PubMed.
- S. Klossowski, H. Miao, K. Kempinska, T. Wu, T. Purohit, E. Kim, B. M. Linhares, D. Chen, G. Jih, E. Perkey, H. Huang, M. He, B. Wen, Y. Wang, K. Yu, S. C. Lee, G. Danet-Desnoyers, W. Trotman, M. Kandarpa, A. Cotton, O. Abdel-Wahab, H. Lei, Y. Dou, M. Guzman, L. Peterson, T. Gruber, S. Choi, D. Sun, P. Ren, L. S. Li, Y. Liu, F. Burrows, I. Maillard, T. Cierpicki and J. Grembecka, J. Clin. Invest., 2020, 130, 981–997 CrossRef CAS PubMed.
- A. V. Krivtsov, K. Evans, J. Y. Gadrey, B. K. Eschle, C. Hatton, H. J. Uckelmann, K. N. Ross, F. Perner, S. N. Olsen, T. Pritchard, L. McDermott, C. D. Jones, D. Jing, A. Braytee, D. Chacon, E. Earley, B. M. McKeever, D. Claremon, A. J. Gifford, H. J. Lee, B. A. Teicher, J. E. Pimanda, D. Beck, J. A. Perry, M. A. Smith, G. M. McGeehan, R. B. Lock and S. A. Armstrong, Cancer Cell, 2019, 36, 660–673 e611 CrossRef CAS PubMed.
- S. He, T. J. Senter, J. Pollock, C. Han, S. K. Upadhyay, T. Purohit, R. D. Gogliotti, C. W. Lindsley, T. Cierpicki, S. R. Stauffer and J. Grembecka, J. Med. Chem., 2014, 57, 1543–1556 CrossRef CAS PubMed.
- S. Xu, A. Aguilar, T. Xu, K. Zheng, L. Huang, J. Stuckey, K. Chinnaswamy, D. Bernard, E. Fernandez-Salas, L. Liu, M. Wang, D. McEachern, S. Przybranowski, C. Foster and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 1601–1605 CrossRef CAS PubMed.
- S. Xu, A. Aguilar, L. Huang, T. Xu, K. Zheng, D. McEachern, S. Przybranowski, C. Foster, K. Zawacki, Z. Liu, K. Chinnaswamy, J. Stuckey and S. Wang, J. Med. Chem., 2020, 63, 4997–5010 CrossRef CAS PubMed.
- K. Hayashi, K. Yoshida and Y. Matsui, Nature, 2005, 438, 374–378 CrossRef CAS PubMed.
- H. Wu, N. Mathioudakis, B. Diagouraga, A. Dong, L. Dombrovski, F. Baudat, S. Cusack, B. de Massy and J. Kadlec, Cell Rep., 2013, 5, 13–20 CrossRef CAS PubMed.
- I. Fumasoni, N. Meani, D. Rambaldi, G. Scafetta, M. Alcalay and F. D. Ciccarelli, BMC Evol. Biol., 2007, 7, 187 CrossRef PubMed.
- S. Myers, R. Bowden, A. Tumian, R. E. Bontrop, C. Freeman, T. S. MacFie, G. McVean and P. Donnelly, Science, 2010, 327, 876–879 CrossRef CAS PubMed.
- A. Sorrentino, A. Federico, M. Rienzo, P. Gazzerro, M. Bifulco, A. Ciccodicola, A. Casamassimi and C. Abbondanza, Int. J. Mol. Sci., 2018, 19, 3250 CrossRef PubMed.
- A. E. Zou, H. Zheng, M. A. Saad, M. Rahimy, J. Ku, S. Z. Kuo, T. K. Honda, J. Wang-Rodriguez, Y. Xuan, A. Korrapati, V. Yu, P. Singh, J. R. Grandis, C. C. King, S. M. Lippman, X. Q. Wang, A. Hinton and W. M. Ongkeko, Oncotarget, 2016, 7, 51211–51222 CrossRef PubMed.
- J. Hussin, D. Sinnett, F. Casals, Y. Idaghdour, V. Bruat, V. Saillour, J. Healy, J. C. Grenier, T. de Malliard, S. Busche, J. F. Spinella, M. Lariviere, G. Gibson, A. Andersson, L. Holmfeldt, J. Ma, L. Wei, J. Zhang, G. Andelfinger, J. R. Downing, C. G. Mullighan and P. Awadalla, Genome Res., 2013, 23, 419–430 CrossRef CAS PubMed.
- A. A. Houle, H. Gibling, F. C. Lamaze, H. A. Edgington, D. Soave, M. J. Fave, M. Agbessi, V. Bruat, L. D. Stein and P. Awadalla, Genome Res., 2018, 28, 1611–1620 CrossRef CAS PubMed.
- A. Allali-Hassani, M. M. Szewczyk, D. Ivanochko, S. L. Organ, J. Bok, J. S. Y. Ho, F. P. H. Gay, F. Li, L. Blazer, M. S. Eram, L. Halabelian, D. Dilworth, G. M. Luciani, E. Lima-Fernandes, Q. Wu, P. Loppnau, N. Palmer, S. Z. A. Talib, P. J. Brown, M. Schapira, P. Kaldis, R. C. O'Hagan, E. Guccione, D. Barsyte-Lovejoy, C. H. Arrowsmith, J. M. Sanders, S. D. Kattar, D. J. Bennett, B. Nicholson and M. Vedadi, Nat. Commun., 2019, 10, 5759 CrossRef CAS PubMed.
- N. Tripoulas, D. LaJeunesse, J. Gildea and A. Shearn, Genetics, 1996, 143, 913–928 CrossRef CAS PubMed.
- H. Miyazaki, K. Higashimoto, Y. Yada, T. A. Endo, J. Sharif, T. Komori, M. Matsuda, Y. Koseki, M. Nakayama, H. Soejima, H. Handa, H. Koseki, S. Hirose and K. Nishioka, PLoS Genet., 2013, 9, e1003897 CrossRef PubMed.
- S. An, K. J. Yeo, Y. H. Jeon and J. J. Song, J. Biol. Chem., 2011, 286, 8369–8374 CrossRef CAS PubMed.
- C. Huang, F. Yang, Z. Zhang, J. Zhang, G. Cai, L. Li, Y. Zheng, S. Chen, R. Xi and B. Zhu, Nat. Commun., 2017, 8, 1649 CrossRef PubMed.
- W. Shen, P. Krautscheid, A. M. Rutz, P. Bayrak-Toydemir and S. L. Dugan, Eur. J. Med. Genet., 2019, 62, 55–60 CrossRef PubMed.
- B. Skawran, D. Steinemann, A. Weigmann, P. Flemming, T. Becker, J. Flik, H. Kreipe, B. Schlegelberger and L. Wilkens, Mod. Pathol., 2008, 21, 505–516 CrossRef CAS PubMed.
- B. Xu, T. Qin, J. Yu, T. J. Giordano, M. A. Sartor and R. J. Koenig, J. Biol. Chem., 2020, 295, 8834–8845 CrossRef CAS PubMed.
- D. S. Rogawski, J. Deng, H. Li, H. Miao, D. Borkin, T. Purohit, J. Song, J. Chase, S. Li, J. Ndoj, S. Klossowski, E. Kim, F. Mao, B. Zhou, J. Ropa, M. Z. Krotoska, Z. Jin, P. Ernst, X. Feng, G. Huang, K. Nishioka, S. Kelly, M. He, B. Wen, D. Sun, A. Muntean, Y. Dou, I. Maillard, T. Cierpicki and J. Grembecka, Nat. Commun., 2021, 12, 2792 CrossRef CAS PubMed.
Footnote |
| † A. F. and M. V. equally contributed to this work and are listed in alphabetical order. |
| This journal is © The Royal Society of Chemistry 2022 |