
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomation isn't automatic
Melodie
Christensen
 ab,
Lars P. E.
Yunker
a,
Parisa
Shiri
a,
Tara
Zepel
a,
Paloma L.
Prieto
a,
Shad
Grunert
a,
Finn
Bork
a and
Jason E.
Hein
*a
ab,
Lars P. E.
Yunker
a,
Parisa
Shiri
a,
Tara
Zepel
a,
Paloma L.
Prieto
a,
Shad
Grunert
a,
Finn
Bork
a and
Jason E.
Hein
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: jhein@chem.ubc.ca
bDepartment of Process Research and Development, Merck & Co., Inc., Rahway, NJ 07065, USA
First published on 27th October 2021
Abstract
Automation has become an increasingly popular tool for synthetic chemists over the past decade. Recent advances in robotics and computer science have led to the emergence of automated systems that execute common laboratory procedures including parallel synthesis, reaction discovery, reaction optimization, time course studies, and crystallization development. While such systems offer many potential benefits, their implementation is rarely automatic due to the highly specialized nature of synthetic procedures. Each reaction category requires careful execution of a particular sequence of steps, the specifics of which change with different conditions and chemical systems. Careful assessment of these critical procedural requirements and identification of the tools suitable for effective experimental execution are key to developing effective automation workflows. Even then, it is often difficult to get all the components of an automated system integrated and operational. Data flows and specialized equipment present yet another level of challenge. Unfortunately, the pain points and process of implementing automated systems are often not shared or remain buried deep in the SI. This perspective provides an overview of the current state of automation of synthetic chemistry at the benchtop scale with a particular emphasis on core considerations and the ensuing challenges of deploying a system. Importantly, we aim to reframe automation as decidedly not automatic but rather an iterative process that involves a series of careful decisions (both human and computational) and constant adjustment.
Introduction
Synthesis is often characterized as much as an art as it is a science.1,2 Successful synthesis requires the careful execution of a highly variable series of complex reactions and steps, not to mention interaction with a wide range of equipment. Deciding what the next step in a particular workflow should be is often based on experience and chemists' intuition. However, the image of the creative synthesist at work is not all glamorous. Many of the steps involved include repetitive, time consuming tasks carried out on largely identical objects.Automation offers a way to reduce human intervention in such processes. In the 1960s, Merrifield and Stewart proposed the first automated system for solid-phase peptide synthesis, successfully reducing the amount of time needed for stepwise addition and purification as well as cutting material losses.3 Since then, automation has slowly crept into the synthetic chemistry laboratory, originally in the form of mechanized systems designed to perform largely identical tasks.4,5 These systems laid the groundwork for the development of specialized platforms capable of automating combinatorial chemistry6 and high-throughput experimentation,7 which are now industry standards in pharmaceutical research and development. More recently, there has been a shift towards flexible, modular systems with a focus on autonomous decision-making rather than simple automation.8–11
The appeal of automation often lies in the potential for increased efficiency (by offloading repetitive tasks and increasing throughput), reproducibility (given the high precision of robotic tools), and safety (where harmful chemicals or reactions can be performed with reduced human exposure). Importantly, automation opens up capabilities that are difficult to carry out by humans in a practical manner (for example, the automated sampling of 10 reactions in parallel). The problem is that the why is only a small part of the automation process. What remains far less discussed in the literature outside the implementation of specific systems is the question of how. How does one go about automating synthetic chemistry in general? This perspective article takes the premise that automating synthetic chemistry in a broadly applicable way is challenging as a starting point and systematically walks through key considerations and decisions that must be made. We draw both from exemplar systems and our own experience using robotics and computer algorithms to drive synthetic chemistry workflows.
Considerations for automating synthetic chemistry
Automation isn't automatic. Rather, it is an iterative process that involves careful interrogation and continuous adjustment on the part of both humans and machines. Different chemical systems and experimental parameters often fundamentally change what is required of an automated system. Therefore, even small considerations when getting started, such as whether a pump can accommodate different phase states, are nontrivial.A hypothetical experience in developing an automated platform
Imagine being a PhD student in organic chemistry and having interest in automating a reaction optimization workflow. Faced with the issue of lack of budget to purchase a standardized robotic platform, the quest to build one begins. How hard could it be?The first hurdle is identifying the required unit operation modules. At the very least, one would need liquid handling, stirring, and some form of temperature control. These modules could be supplemented by solid handling or filtration tools, or external peripherals such as a phase separation module or a camera. An even more advanced system could include a robotic arm to move vials to an analytical platform. These modules must be neither too expensive nor unwieldy, but at the same time need to fit in the allocated space and allow for ease of control. With these options in mind, the great search for the physical components begins.
Collecting a conglomeration of different devices unearths multitude of control softwares, ranging from binary input/output systems to full graphical user interfaces. Since the devices should ideally work in unison, studying the programming landscape to identify the best and, more importantly, easiest solution is commenced. Upon arriving at a decision around component control one would need to identify a nice space for the robot to live. After connecting everything together and a few leaking inlets, minor logic errors, volume calibrations, cabling problems, and some more leakages, the first successful run with water is completed. Therefore the system is ready for chemistry right?
Here the experimental problems begin. Pumping becomes inaccurate with organic solvents due to lower surface tension. Stock solutions decay so one of the components is added via solid handling, which was avoided earlier, because now the platform needs a larger habitat. And, oh look, a valve reacted with a starting material. Order an inert replacement and clean out all the tubing due to recurring obstructions. After the 20th iteration all of the components are out of sync and everything must be rebuilt. Also, the data format is inconsistent, plus no more stable USB-ports remain.
This experience in automation development is all too familiar for many. The goal of this perspective article is to critically examine the key factors and decisions required for implementing successful automation systems for synthetic chemistry. We divide these topics into three categories: (1) Equipment considerations, (2) Experimental considerations, and (3) Data and software. With the exception of automated flow systems,12 which are beyond the scope of this work, we consider examples from the recent literature and our lab's own experience in designing and deploying automated workflows at the benchtop scale.
Equipment considerations
 | ||
| Fig. 1 Flow chart for decision-making around whether to buy or build an automation module. | ||
 | ||
| Fig. 2 Types of synthetic chemistry automation modules with select examples. | ||
Solid handling. Solid dispensing is ubiquitous in synthetic chemistry, with scientists spending hours at the scale, performing manual weighing tasks. While desirable, automating this task is challenging due to the wide range of properties that solids can exhibit, from density to particle size distribution and flowability. Currently, there are two main types of solid dispensing modules utilized in chemistry automation and both rely on gravimetric dispensing: the first type involves hopper/feeder modules and second involves positive displacement modules.
Hopper/feeder modules are gravimetric solid dispense tools that consist of a hopper to which the solid is loaded with an opening at the bottom. Various types of feeders guide the solid flow through to the bottom port. Typically, the port opening is controlled through a rotary valve and the feeder action controlled through various mechanical means. In some instances, the solid flow can be additionally controlled through tapping or vibrational actions. The Mettler-Toledo Auto Chem Quantos utilizes a hopper-based module where the flow of solids is controlled through rotary tapping (Fig. 3a). Hopper/feeder modules are best suited for milligram to gram quantity solid dispensing.14,15 In addition, hopper/feeder modules are best suited to dispense free-flowing solids, as interruptions in solid flow may lead to a device timeout.
 | ||
| Fig. 3 Axelsemrau Chronect outfitted with a Mettler-Toledo Auto Chem Quantos solid dispense module (left). Chemspeed Technologies GDU-S SWILE with a positive-displacement module (right). Reprinted with permission from ref. 13. Copyright 2020 American Chemical Society. | ||
Positive displacement modules are also gravimetric, but rely on capillaries outfitted with pistons which move up and down to pick up and dispense solids through positive displacement. The Chemspeed Technologies GDU-S SWILE is an example of a positive-displacement module (Fig. 3b). The SWILE in particular has been shown to be highly effective in sub-milligram to low-milligram dispense quantities.14,15 Positive displacement modules are effective in the automated dispensing of a wider range of solids with varying physical properties, including sticky or oily solids.
Liquid handling. Liquid handling modules aspirate and dispense solvents, liquid reagents, and stock mixtures. The simplest liquid handling systems consist of a pump for liquid displacement, valves for guiding the flow, a variety of tubes, and a dispensing head outfitted with a pipette or needle. For example, in our work developing an automated platform for evaluating solubility, we used tubing to connect a syringe pump with the probe of a robotic arm.16 The arm was used to pick up needles on the probe to pierce through vial septa and transfer solutions.
The two most common pump types observed in chemistry automation systems are peristaltic pumps and syringe pumps (Fig. 4). A peristaltic pump displaces liquids by means of pressure waves generated through the compression of tubing by rotor-mounted rollers. Peristaltic pumps are relatively inexpensive and can be used for the continuous dispensing of larger volumes in the milliliter range. Syringe pumps contain a syringe and plunger (typically driven by a stepper motor) outfitted with a distributive or non-distributive valve for flexible flow path planning. These pumps are programmable for small, precise dispense volumes in microliter range. Valves are often used to enable different flow paths. Ports and positions are the main characteristics of valves. A port is the tubing connection point and a position is the directional state that the valve can achieve. Two-position rotary valves can have a large number of ports (typically six) where fluidic connections between adjacent ports are toggled by the position. Selector valves function in a similar manner, but in this case, a common port is connected to a variety of selectable ports through toggling the position. The tip of a liquid handling system plays an important role in the quality and accuracy of dispensing. Dispense heads can be outfitted with needles and pipettes. Needles have the advantage of being able to pierce through septa-capped vials.
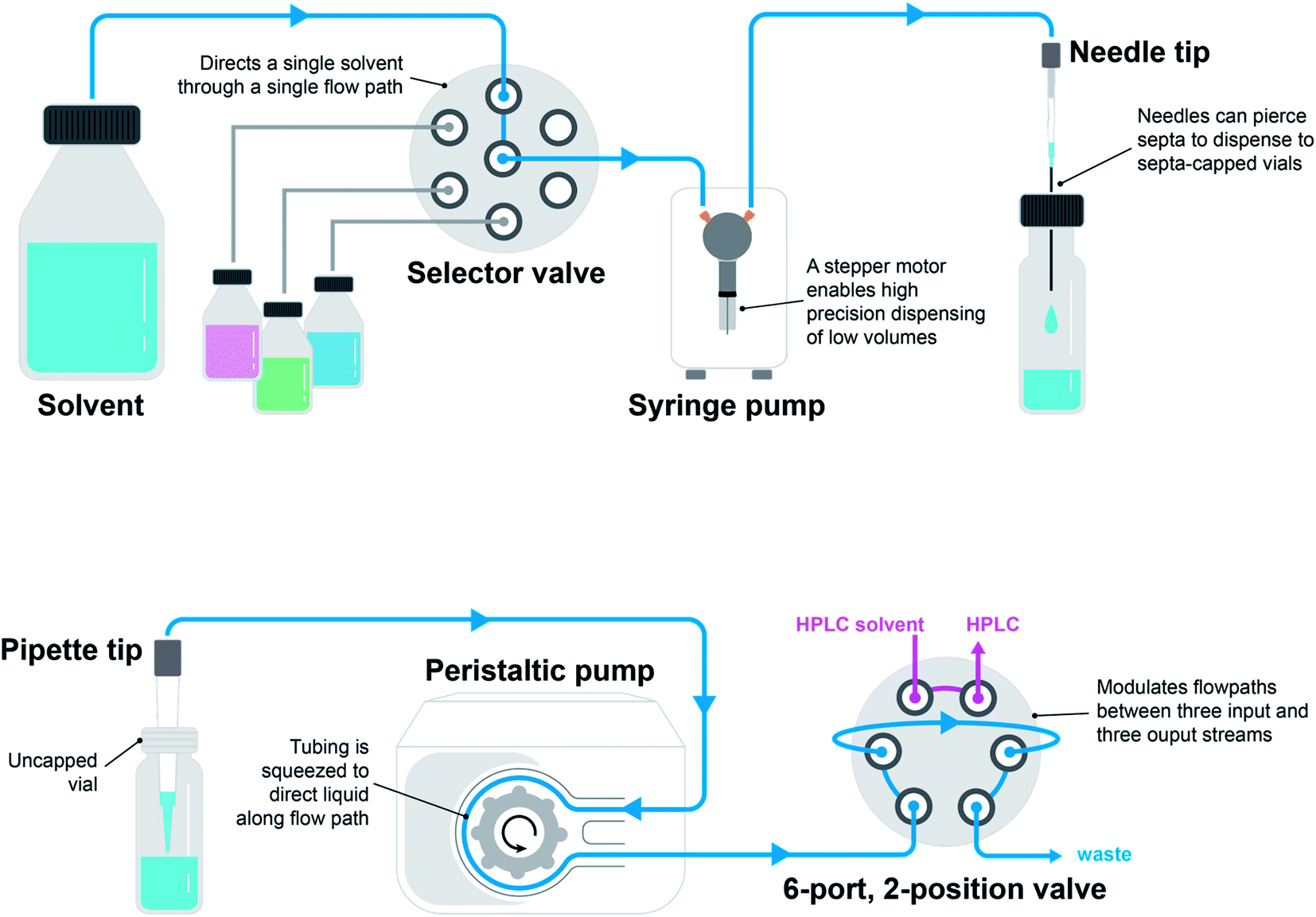 | ||
| Fig. 4 Liquid handling modules arranged in two configurations: a syringe pump, selector valve, and needle tip (top) and a peristaltic pump, 6-port, 2-position valve, and pipette tip (bottom). | ||
Liquid handling modules are relatively simple to put together, but achieving reliability and robustness can be a challenge. Calibrating liquid handling modules for small volume dispenses of various liquid types, such as low-viscosity organic solvents, may be necessary. High-viscosity liquids may pose unique challenges that are insurmountable using common liquid handling modules. These liquids are best handled through positive displacement pipettes, which have been incorporated in the GDU-V tool offered through Chempseed Technologies and the Dragonfly Discovery robot offered through SPT Labtech. Clogging and air bubbles are other common challenges of liquid handling.17 When aspirating and dispensing from capped vials, a vent is required for pressure equalization. Some solutions to this problem include the utilization of special needles with an extra groove for pressure equalization and the utilization of pre-slit septa caps.
Filtration. Filtration modules for separating solid and liquid phases can be utilized in workflows involving salt, crystal polymorph, co-crystal, and solubility screening. These workflows are typically designed to be high-throughput in order to maximize the diversity of parameters under evaluation. In addition, researchers are generally interested in both solid and liquid component analysis. Slurries are filtered through plate assemblies containing a cellulose- or membrane-based top filter plate outfitted with a bottom collection plate. Separation is promoted through the application of vacuum or centrifugal force to the filter plate assembly. Janey and coworkers describe a custom plate assembly with a XRPD-compatible (X-ray powder diffraction) filter plate for solid polymorph screening and analysis (Fig. 5).18 Qiu and Albrecht report the implementation of a commercial filter plate assembly with a similar configuration developed by Unchained Labs for solubility screening.19 The advantage of the Unchained Labs filter plate is the ability to carry out solid–liquid separations at various temperatures.
 | ||
| Fig. 5 Custom filtration apparatus developed for an Unchained Labs platform for polymorph screening. Reprinted with permission from ref. 17. Copyright 2016 American Chemical Society. | ||
Filtration modules can also be implemented in the automated sampling of solid–liquid mixtures for downstream analysis.20 Here, the goal is to analyze the liquid reaction components, thus, tubing outfitted with a small frit is typically sufficient for these purposes.
Stirring. The importance of stirring in chemical reactions is heavily influenced by the physical phases present in the reaction mixture. Homogeneous reactions consisting of a single liquid phase are less susceptible to mixing effects. Heterogeneous reactions consisting of multiple phases, including gas–liquid, liquid–liquid or solid–liquid phases necessitate even and robust mixing.
Automation for chemical discovery and optimization often involves parallelization of reactions in order to maximize the experimental throughput. The Society for Biomedical Sciences (SBS) has standardized reaction microplates for the automated execution of multiple reactions in individual wells. However, the rectangular shape and footprint of the SBS format plate requires the careful consideration of the mixing module. Key considerations include mixing efficiency and uniformity. Fig. 6 illustrates various stirring modules. The most ubiquitous stir plate designs for chemical applications utilize magnetic rotary stirring, but this technique is not uniform across rectangular SBS format plates, where stirring efficiency is reduced around the outer edges furthest from the magnet of the stirplate. Tumble stirring is instead more compatible with SBS format plates. V&P Scientific offers powerful tumble stirrers for this purpose, and these modules can be easily integrated into automation platforms. In addition to the stir bar based design, orbital shakers such as those offered by Glas-Col eliminate stir bars from the process, preventing undesired effects from stir bars such as grinding of solids.
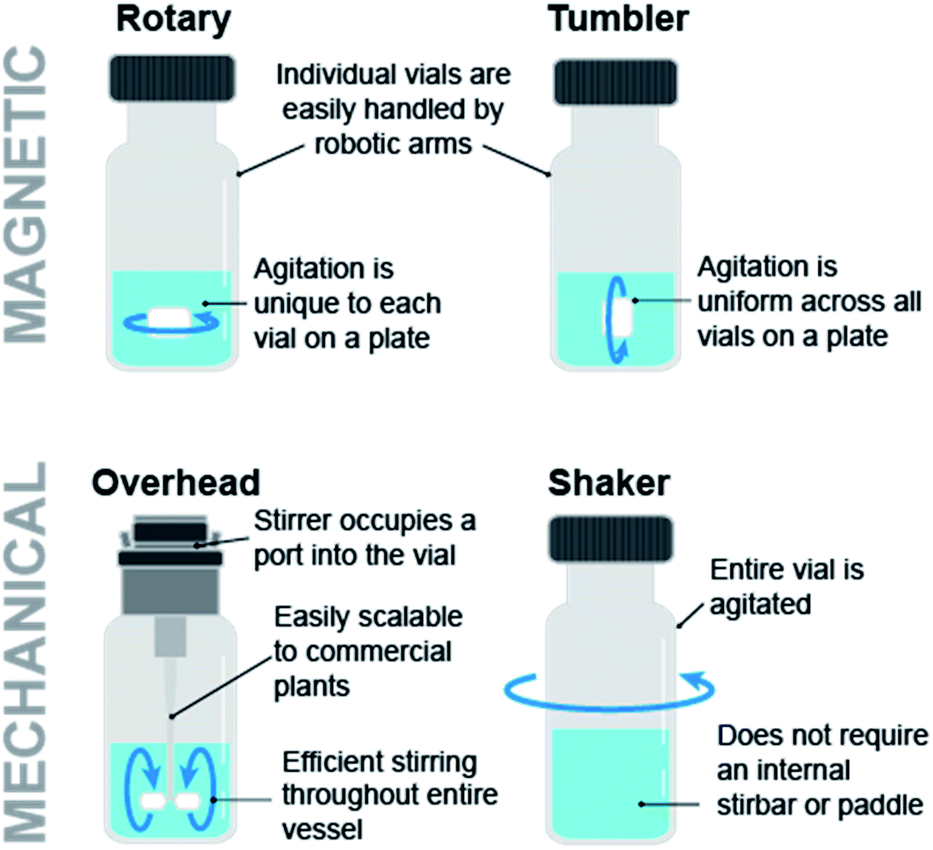 | ||
| Fig. 6 Magnetic and mechanical stirring modules impart different agitation homogeneity across a reaction plate, efficiency, and scalability. | ||
Automation for chemical development typically involves multi gram-scale automated lab reactors with overhead stirring capabilities to better simulate commercial plant-scale equipment. These reactors offer advantages to microplate reactors with respect to hydrodynamics, mixing and surface to volume ratio, which tend to be important factors in the execution of heterogeneous reactions. Mettler-Toledo Auto Chem automated lab reactor systems provide both rotary and overhead stirring capabilities.
Temperature control. Temperature control is key to the effective execution of synthetic reactions. Again, homogeneity of temperature is critical to experimentation, and this is generally achieved through circulator block/chiller modules such as the circulator blocks offered by MeCour and Analytical Sales. High-efficiency chillers can be obtained from companies such as Huber. On the other hand, Peltier-based heating/cooling capabilities can be utilized in automated reactors, such as those utilized in Integrity-10 systems. The advantage with the Peltier-based technology is that a larger number of reactor zones with individual temperature control can be accessed. A circulating chiller with sufficient cooling power is still required to remove heat from the back of the Peltier elements.
Analysis. Perhaps among the most challenging modules to integrate, analytical instruments provide the most useful data regarding the chemistry that has been automated. The complications arising from integration of analytics into a system derive mainly from the fact that most instruments on the market are designed and sold as walk-up systems, in many cases incorporating their own automation technology (such as an autosampler). When broken down to the core actions of taking a sample, performing an action on it (quenching, dilution, filtration), and moving the modified sample into the analysis environment, there is not much that differs from the basic actions in place for reaction automation.
For spectroscopic methods, the sample need only be put in the beam path or electromagnetic field (Fig. 7, top). This is as simple as using a pump to aspirate and move a sample plug into a flow cell, or even using a probe such as Mettler-Toledo Auto Chem ReactIR (infrared spectroscopy) placed directly in the reaction mixture. Similarly, NMR (nuclear magnetic resonance spectroscopy) requires either moving a sample into a flow cell or loading an NMR tube and moving it into the reading frame. Low-field continuous-flow NMR systems are particularly attractive for integration with automated systems due to their low cost and small footprint. However, gains in practicality come at the price of lower data quality stemming from lower external magnetic fields. High-field NMR systems offer high resolution but are typically not integrated with automation systems due to size, cost and maintenance considerations.21
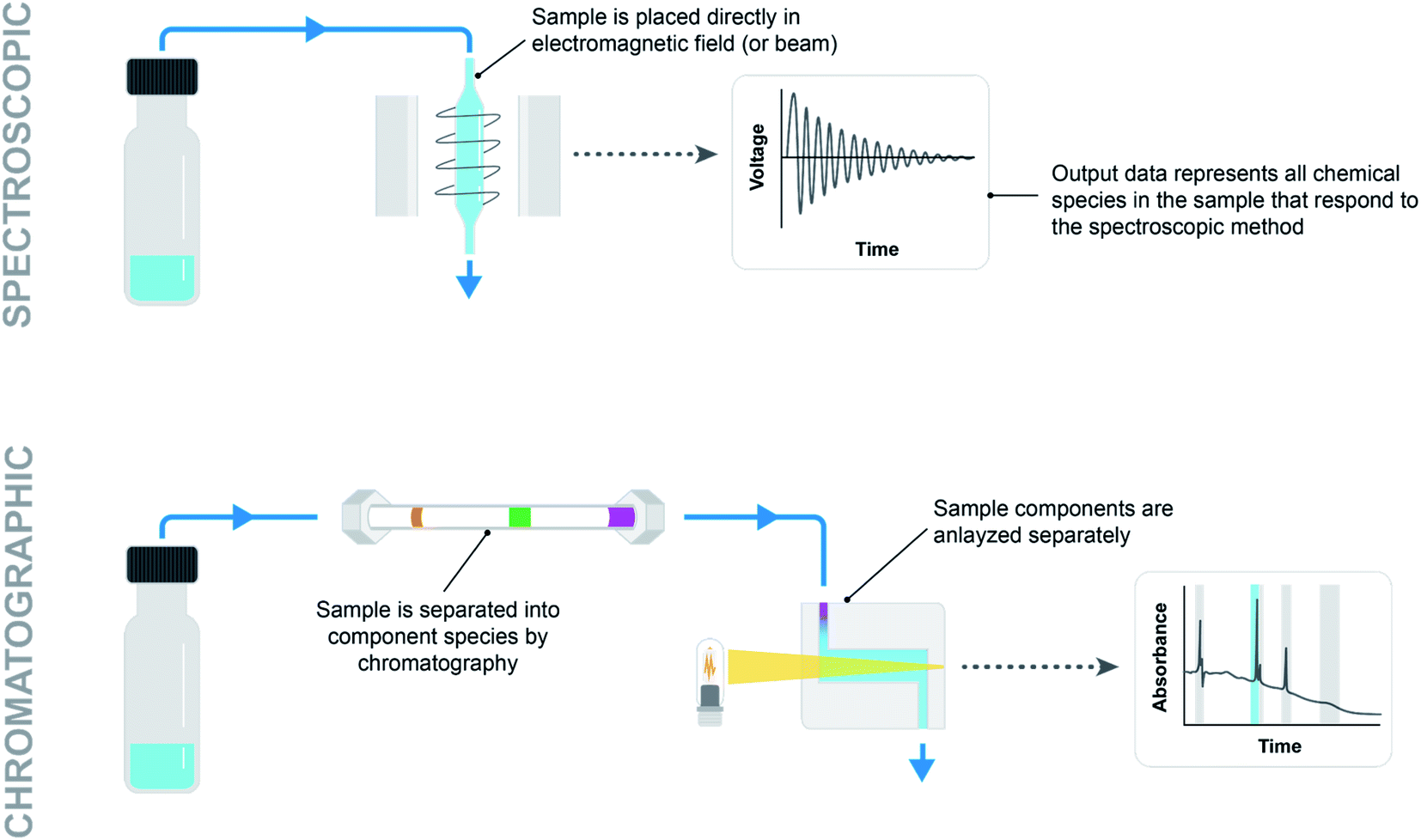 | ||
| Fig. 7 Comparison of simple spectroscopic (top) and chromatographic (bottom) analytical methods. | ||
For chromatographic methods, a pump is used to move the sample into a loop on a high pressure injection valve plumbed into the flowpath ahead of a column (Fig. 7, bottom). Solid, liquid, and gas handling modules provide an endless combination of parts to creatively assemble into an analytical sampling system. Difficulties arise in being able to move sample volumes accurately and reproducibly to ensure that the recorded data is useful. Rigorous testing under a variety of reaction conditions (pressurized vs. unpressurized, homogeneous vs. heterogeneous, heated vs. room temperature) must go into demonstrating the validity of a sampling system before any inferences can be drawn from collected reaction data.
In addition to spectroscopic and chromatographic methods, computer vision modules have gained more traction in the synthetic chemistry automation realm in recent years. Pipetting robots such as the Andrew Alliance liquid handling robot rely on machine vision to assess pipet tip positioning and dispense volumes. Our lab has reported the use of computer vision in automated solubility screening, where turbidity (the measure of the cloudiness or haziness of a liquid) can be monitored through the incorporation of a webcam (Fig. 8).16
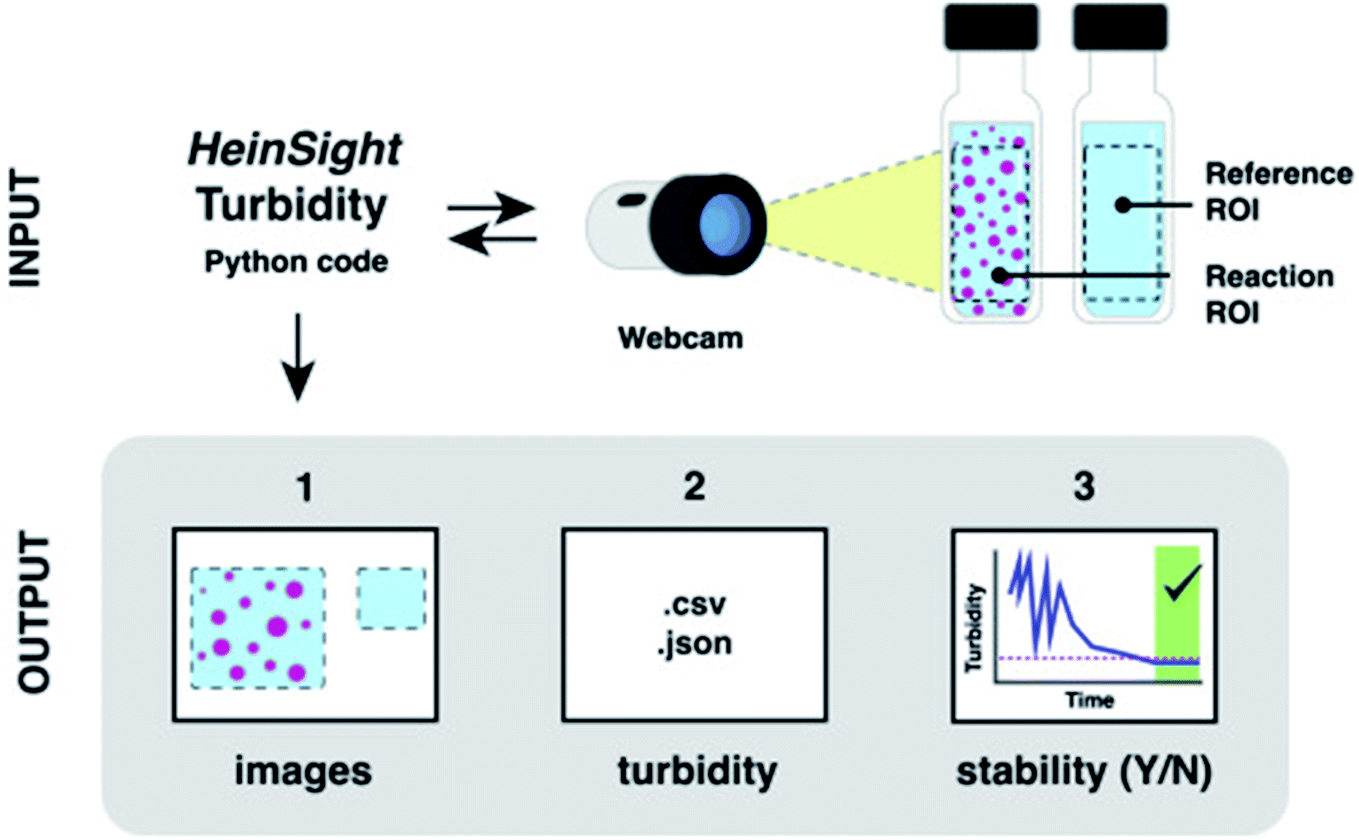 | ||
| Fig. 8 The use of a webcam for computer-vision based turbidity measurement. Reproduced with permission from ref. 15. Copyright 2021 Elsevier. | ||
Translocation. The three separate classes of translocation technology discussed here can be packaged under the umbrella ‘robotics’ (Fig. 9), the general purpose technology underpinning more and more complex laboratory automation solutions. Complex laboratory automation solutions typically consist of a collection of modules, thus, translocation between individual modules is an important consideration.
 | ||
| Fig. 9 Robotic modules for translocation. | ||
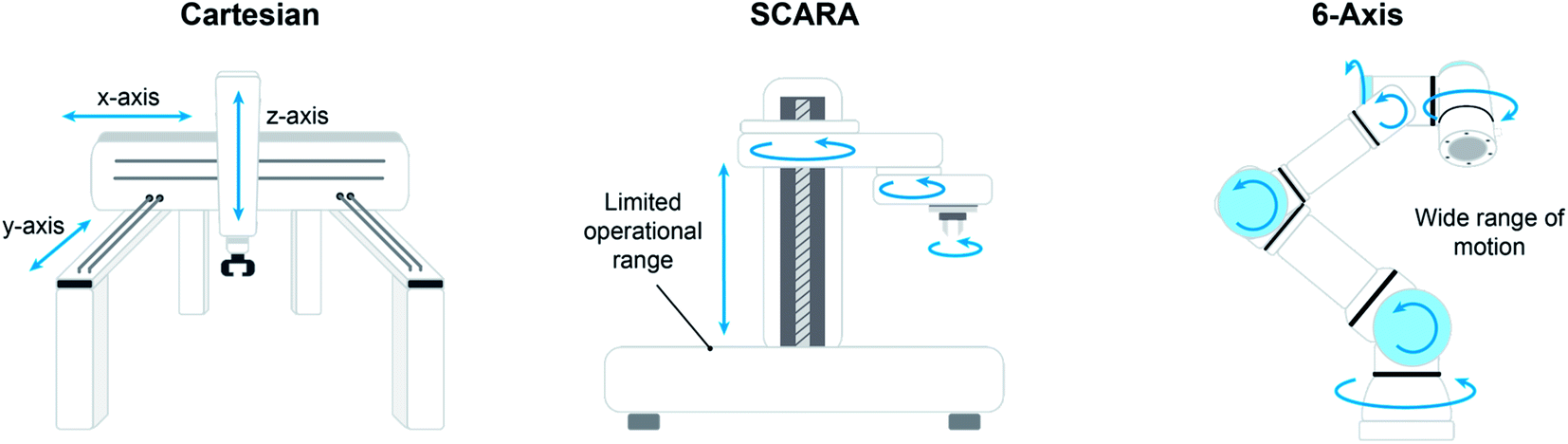
The most accessible class of robotics is the Cartesian system, where an Z axis-actuated end effector (typically a needle probe) moves along rigid rails in the XY plane. Platforms geared toward HTE (high-throughput experimentation) are often equipped with Cartesian robotics, offering the benefit of a turnkey solution but coming at a relatively high space penalty. For example, the Chemspeed Technologies SWING benchtop system is an enclosed box with a footprint of 1.5 m by 0.9 m, while the Chemspeed Technologies SWING XL measures 2.4 m by 0.9 m and comes on a custom mobile bench. In the latter case especially, it is evident that significant accommodations must be afforded which may require costly laboratory modifications such as removal of benches and cabinets. One outstanding advantage of this constrained box design is that it facilitates environment management, in that the entire enclosure can be a swept/inerted atmosphere or simply connected to ventilation systems for removal of hazardous fumes.
While Cartesian platforms excel at automated experimentation within their capabilities, the researcher must often adapt their experimental workflow to the actions the platform can execute. More articulated robotic systems, or robotic arms, can more closely mimic the actions of the researcher at the bench. Although the range of motions offered by robotic arms enables more human-like interaction with the work environment, enabling access to a multitude of custom-designed modules or other commercial laboratory instrumentation, the tradeoff comes in that this is very much not a turnkey solution; significant effort goes into programming positions, timing, module actions, and the like to execute a workflow. SCARA (selective compliance assembly robot arm) platforms offer excellent locational precision and reproducibility within a limited operational range. Similar to a Cartesian system, the arm on a SCARA platform operates in any number of z-planes, but it differs in that the end effector is supported from a main pillar instead of overhead supports. This provides access to a greater range of motions, most notably the ability to move material laterally into environments inaccessible to an overhead system, such as loading a sample onto an enclosed analytical balance. Despite this advantage, movement locations are still somewhat limited.
Multi-axis robotic arms such as the Universal Robots UR3 allow for the widest range of motion. The layout of the workspace is limited only by the imagination of the researcher, providing access to human-like interactions with any number of analytical or processing modules. The movement of the robotics is not constrained to an enclosed box, rather only by the reach of the arm. Finally, the mobile robotic chemist of Burger et al. is the natural extension of a stationary articulated robotic arm to a mobile human analogue.11 Rather than using the arm to pass samples between modules/stations, they took the approach of giving the arm wheels to transport samples around the laboratory for processing and analysis at conventional benchtop instruments.
The full package. The discussed modules can be combined in various configurations to develop a variety of integrated platforms, including micromole scale, high throughput systems and millimole scale, medium throughput systems up to mole scale, single reactor systems.
In the high-throughput space, vendors such as Unchained Labs, Chempseed Technologies and Tecan dominate. Unchained Labs and Chemspeed Technologies systems can be purchased with insertable enclosures, whereas Tecan systems are typically open to the atmosphere. These systems consist of a solid handler, liquid handler, stirring and temperature modules, a translocation module and a large robotic deck capable of holding multiple microplate reactors. Filtration and online analytical capabilities are also possible. These systems are best suited for parallel synthesis and wide-net reaction parameter screening.
In the medium-throughput space, vendors such as AmigoChem offer 10-reactor systems with individual stirring and temperature control modules, along with a liquid handling and translocation module for automated sampling of reactions over time. These systems are best suited for kinetic studies to gain mechanistic insights into the chemistry under evaluation.
In the low-throughput space, vendors such as Mettler-Toledo Auto Chem and H.E.L. Group dominate with offerings of single or double automated reactor systems outfitted with various stirring options, temperature control, and reactor ports for reagent addition, automated sampling and online analytics. Mettler Toledo Autochem offers the EasySampler liquid handling module for automated sampling of reactions over time, as well as the ReactIR and ReactRaman probes for in situ reaction analysis. These systems are best suited for reaction parameter optimization, reaction parameter range-finding, and process characterization experiments to define the parameters ranges to deliver consistent product quality.
The path to preserving long-term robustness is an effective maintenance strategy. We caution against relying on one subject matter expert for system maintenance in the absence of written documentation and suggest well-documented procedures and schedules. Aside from maintenance, user training should also be taken into consideration, as proper usage will ensure the longevity of the equipment. For user training, we also suggest recorded documentation (images and videos can be helpful here). Finally, if budget allows, commercial vendors can be approached for yearly preventative maintenance plans.
Experimental considerations
Due to the specialized nature of synthetic organic reaction procedures, unique operational considerations dictate the process of automating experimental workflows. These include, but are not limited to: throughput, scale, special chemical requirements (i.e., timing, rate of addition, mixing, temperature, water or oxygen sensitivity), as well as sampling and analysis. Below we outline how these considerations impact the automation systems under development.When estimating throughput to evaluate the potential of a new automated system, a 4-digit reaction count per day is impressive, but may not necessarily be the best solution to every synthetic challenge. This section focuses on identifying optimal throughput based on the experimental goal, the importance of timing, and the required analytical methods.
HTE is especially useful in reaction discovery. In reaction discovery studies, a diverse array of categorical parameters are typically examined to determine conditions that afford the desired bond disconnection. In an example published by our group, both Suzuki–Miyaura cross-coupling conditions (Fig. 10a) and asymmetric hydrogenation conditions (Fig. 10b) for the enantioselective synthesis of α-methyl-β-cyclopropyldihydrocinnamates were discovered through HTE.27 Although these studies were carried out in semi-automated fashion, the automated dispensing of phosphine ligands significantly shortened cycle times in both cases. If the number of experiments exceeds limits imposed by material, equipment or timing limitations, the use of statistical tools such as Design of Experiments (DoE) and Principal Component Analysis (PCA) can reduce the necessary number of reactions. Although DoE approaches are typically utilized in continuous parameter optimizations, in an impressive example by Moseley and coworkers, categorical ligand and solvent space were explored efficiently through a combined PCA and DoE approach.28
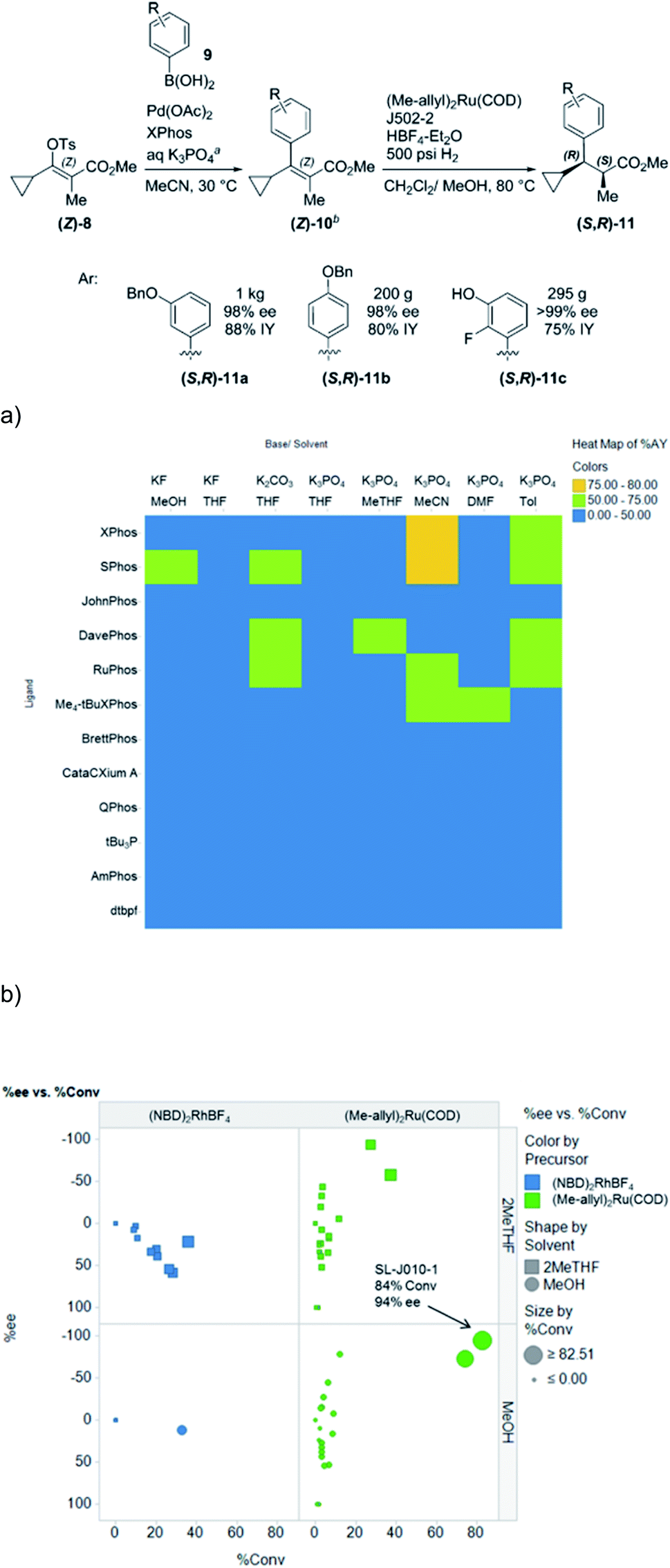 | ||
| Fig. 10 (a) HTE to discover optimal conditions for a Suzuki cross-coupling reaction (b) HTE to discover optimal conditions for an asymmetric hydrogenation reaction. Reprinted with permission from ref. 25. Copyright 2016 American Chemical Society. | ||
When considering throughput, timing is a key consideration. If a reaction is especially sensitive to timing, fewer reactions can be carried out in parallel because the system must be ready for use at specific times to execute specific steps (reagent addition, quench, sampling and analysis). This limitation can be addressed by using faster robots. The upper speed limit was reported by Dreher and coworkers at the nano-scale with 6144 reagents in 30 min (which is equivalent to an impressive 3.4 pipetting steps per second).25 While 30 minutes is not a limiting time period for multi-hour reactions, some reaction mixtures are highly active and can be converted in a few seconds or decompose within minutes. Dispense, sampling, and quenching times can therefore pose significant challenges for batch setups. If the throughput does not allow for the timing requirements to be met, parallel experiments can be broken down into smaller blocks and executed sequentially. The run may take longer to execute with sequential blocks, but would not require additional human intervention.
The sampling strategy is also an important factor in determining the optimal throughput. High-throughput experiments are typically sampled once at the end of each reaction. However, reaction profiling studies, where each reaction is sampled at multiple time points over time, can provide valuable mechanistic insights into the system under evaluation. Here, the sampling resolution is determined by the minimum sampling time, which is heavily influenced by the throughput and the sampling module. The sampling resolution can be especially critical when the influence of factors on the system change over time, as demonstrated in an in automated DoE reaction profiling study by Jurica and McMullen on the optimization of a pyridone synthesis (Fig. 11).29 Here, the use of DoE reduced the experimental throughput, allowing for experimental execution via a Mettler-Toledo Auto Chem EasyMax reactor outfitted with an EasySampler module.
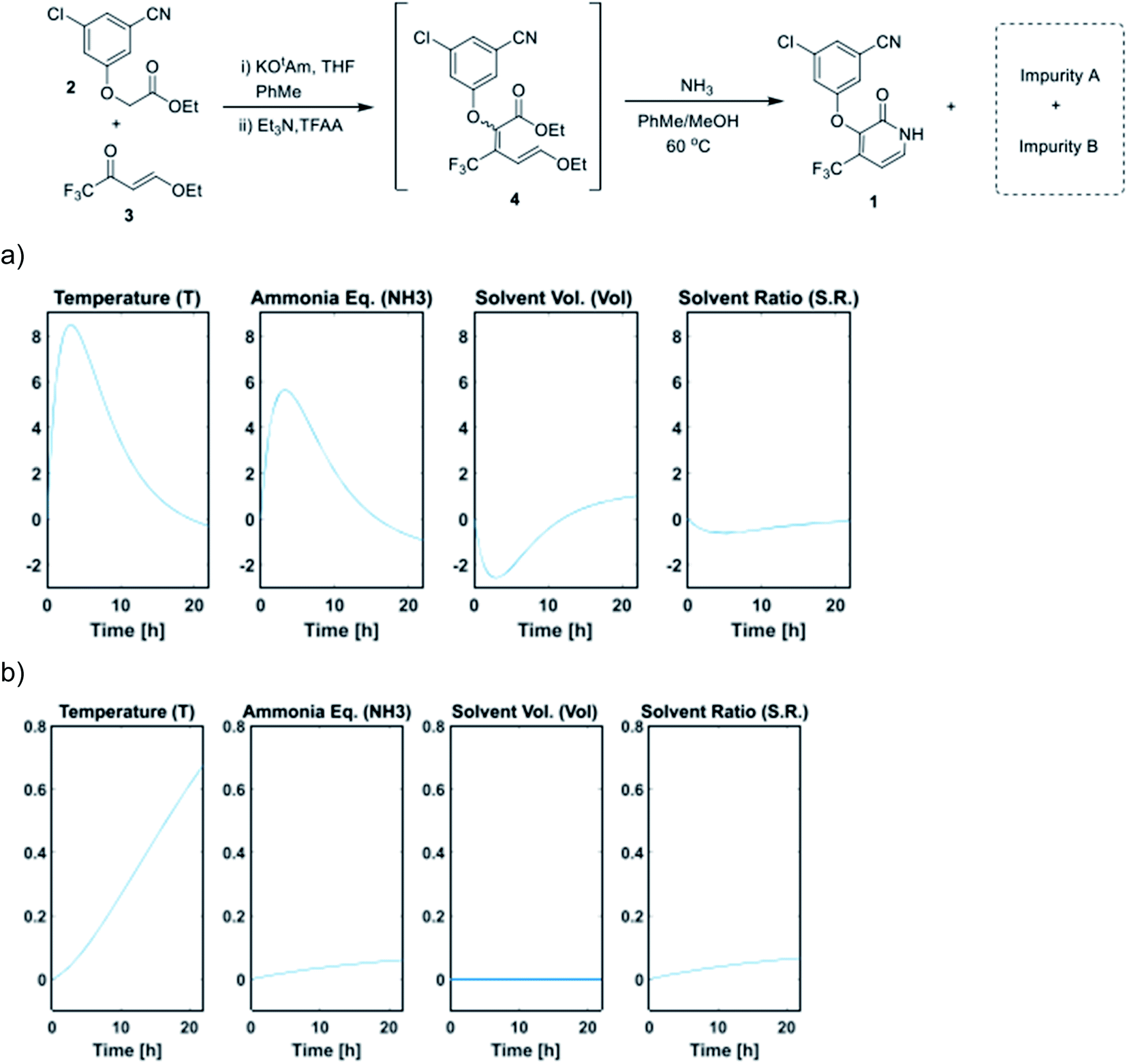 | ||
| Fig. 11 (a) Time profile for the main effects on pyridone formation. (b) Time profile for the main effects on impurity B formation. Adapted with permission from ref. 27. Copyright 2021 American Chemical Society. | ||
The final consideration is analysis of experimental outcomes. In high-throughput studies, the time spent analyzing the outcome far exceeds the time spent setting up reactions. In the above-mentioned study by Dreher and coworkers it took 30 minutes to dose all reagents, 1 hour for sampling and 52 hours to analyze all samples via UPLC.25 A general approach for faster analytics is to run all samples sequentially via flow injection analysis (FIA) or multiple injections in a single experimental run (MISER). In exchange for detailed tracking of all species, one species can be tracked much faster through selected ion monitoring (SIM). This MISER variant is the most common workflow improvement for mass spectrometry coupled to liquid, gas, and supercritical fluid chromatography, but other techniques have been automated as well in an attempt to increase analytical throughput (i.e. MALDI, DESI, AE-MS (acoustic ejection) and even NMR). Fig. 12 shows a comparison of the suitability of analytical techniques for high-throughput systems, and an in-depth discussion of automated sampling and analytics follows in Section 4.22
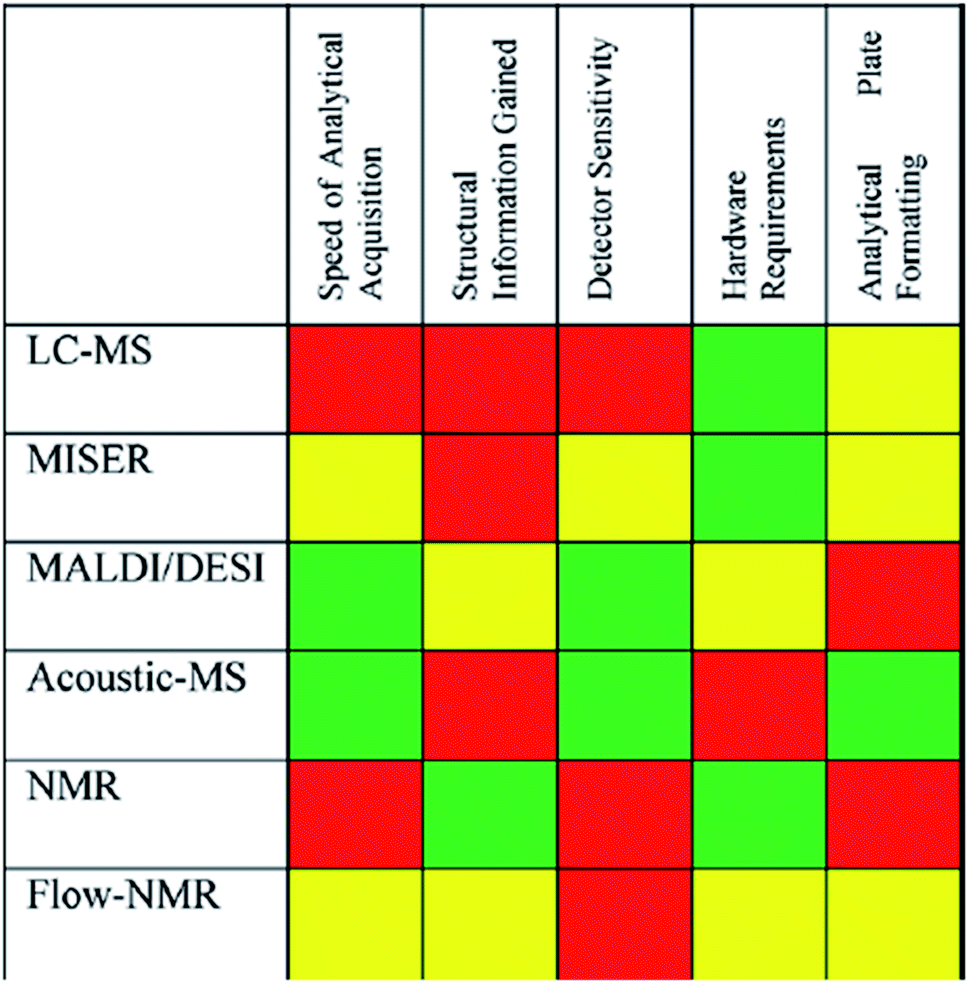 | ||
| Fig. 12 Qualitative factors that contribute to the selection of high-throughput analysis techniques. Red = least favorable; yellow = moderately favorable, green = most favorable. Reproduced with permission from ref. 20. Copyright 2021 American Chemical Society. | ||
Overall, the determination of appropriate throughput depends on the study goals, the material cost and availability, the scope of parameters under consideration, the sampling strategy, and the appropriate analytical technique. Several statistical methods such as DoE and PCA exist to narrow the search space if needed, and this can especially prove useful in high-throughput time course studies where short sampling interval times are required.
The cost and accessibility of starting materials can dictate the reaction scale: if the study requires complex fragments of a total synthesis, a chiral ligand or catalyst, or other rare starting materials, a large number of reactions per day can become prohibitively expensive. A thrifty solution is to reduce the required throughput through statistical methods (see throughput section on DoE). If that is not possible the scale may need to be decreased to save material. Screenings in nanomole scale are possible but require a solubilizing, low-volatility, plastic-compatible reaction medium (DMSO or NMP) and aging at room temperature.25 Very small scales also require highly specialized equipment that can be too expensive for most research groups. An example is acoustic ejection pipetting, a technique capable of multiple single digit nanoliter volume dispensing steps per second.22 Due to those restrictions most automated experiments are carried out on the micromole7,13,23,24,26 or millimole scale.8,29
One interesting concept would be to add “scale” as a factor to the multi-categorical screening, but in practice most automated experimentation equipment cannot handle both nanomole and millimole scale reactions. This is due to the different reactor volumes, as well as liquid and solid handling modules, that become inaccurate for small scales (1 μL ± 5 μL) or take too much time for large scales (a 1 mL syringe dispensing 2 L solvent is simply unreasonable). However, smaller variations in scale, for example from 250 μL to 8 mL can still provide insight on how a reaction responds to upscaling (Fig. 13).22,30,31
 | ||
| Fig. 13 Suzuki–Miyaura reaction screening across different scales. Figure reproduced with permission form the authors of ref. 29. | ||
In process chemistry applications, chemists sometimes prefer to carry out automated experiments in larger scale automated reactors such as Mettler-Toledo Auto Chem EasyMax reactors in order to mimic pilot plant reactor geometries. These millimole-scale automated reactors allow for overhead stirring, which becomes especially important in reactions sensitive to mixing efficiency, such as heterogeneous reactions.29
Some automated workflows mirror the set of operations familiar to bench chemists: dispensing of solids, followed by liquids, followed by specialty gas delivery, if applicable.29,32,33 However, parallelized micromole-scale workflows typically involve the automated dispensing of stock mixtures. Boga and Christensen at Merck implemented the latter method in their automated strong base screening workflow (Fig. 14).26 The key to successful implementation was to determine which stock mixtures would be stable over the course of the experimental set-up. In this case, robust outcomes were achieved by preparing separate substrate, strong base, and electrophile stock mixtures in compatible solvents with stringent humidity level control to prevent water-mediated decomposition. Equally important was the order and timing of operations, where the substrate was first treated with base solution, aged for a set time period, and then treated with electrophile. This ensured deprotonation could occur prior to functionalization. Finally, temperature control was paramount to reaction control. High-capacity chillers coupled with tumble-stirred cooling blocks are highly effective in achieving temperature ranges between −50 and 150 °C. Using this set-up, the strong base screening workflow was reported to reach block temperatures as low as −50 °C. Uniform mixing across all positions of a 96-well plate was also important. This was achieved through incorporation of a tumble stirring module.
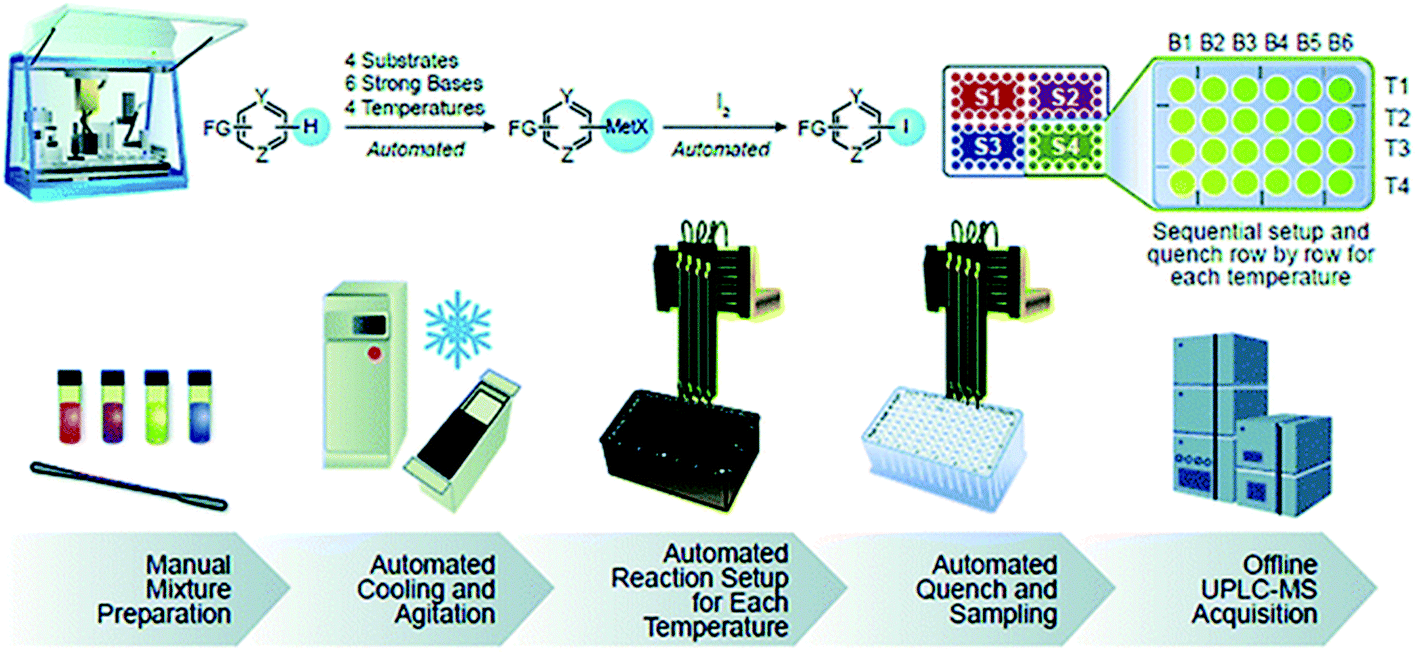 | ||
| Fig. 14 Strong base screening workflow involving liquid stock mixture dispensing. Reproduced from ref. 24 with permission from the Royal Society of Chemistry. | ||
Another demonstration of parallelized micromole-scale reactions involving the automated dispensing of stock mixtures was reported by Bristol Myers Squibb.34 In this study, humidity level control was only required during certain steps of the automated workflow. They observed that acetoin dimer 2 (Fig. 15) exhibited hygroscopicity in a highly automated annulation reaction. A lack of humidity control during the weighing of the acetoin dimer led to a very high coefficient of variation (CV) of 60% in automated runs. The issue was corrected through weighing the acetoin dimer and internal standard in a glovebox. This simple update to the workflow decreased the CV to 19%.
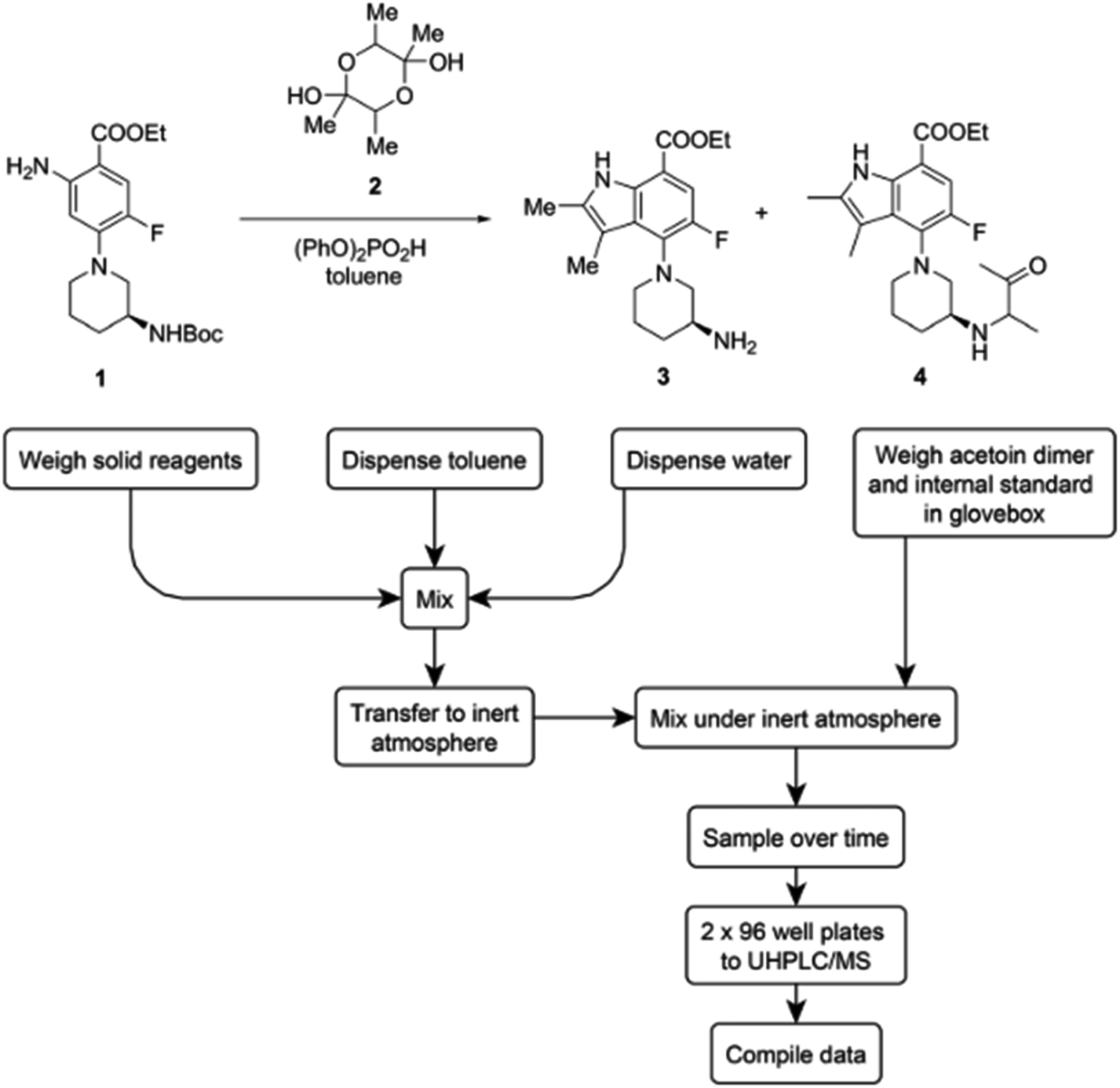 | ||
| Fig. 15 The weighing step of the acetoin dimer is moved to a glove box to account for hygroscopicity. Reproduced from ref. 32 with permission from the Royal Society of Chemistry. | ||
The physical properties of stock mixtures can also significantly impact dispense accuracy and precision.35 For example, it is very difficult to accurately dispense mixtures in volatile solvents such as diethyl ether, or dense solvents such as dichloromethane. Shevlin highlights the use of dichloroethane and dimethoxyethane as viable alternatives to these solvents in the screening of a tandem Heck–Suzuki reaction (Fig. 16).36
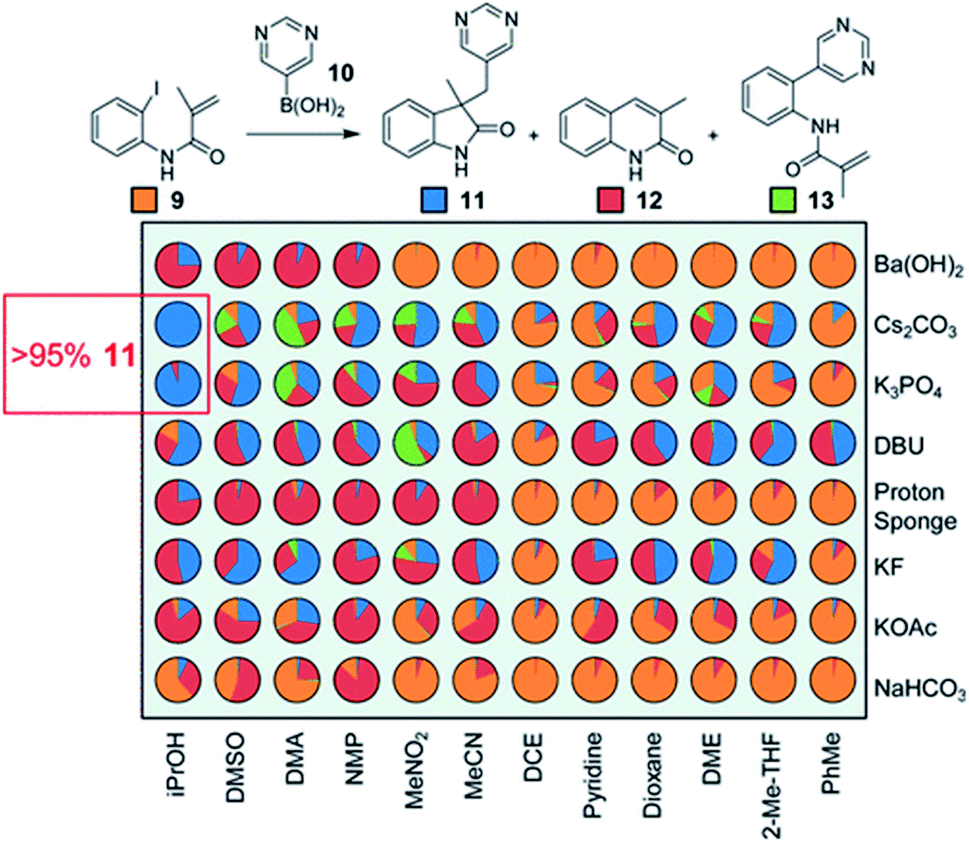 | ||
| Fig. 16 HTE in the optimization of a tandem Heck–Suzuki reaction. Reproduced with permission from ref. 34. Copyright 2017 American Chemical Society. | ||
Finally, the compatibility of the dispensed chemicals with each robotic platform component requires careful thought. This is why it is best to invest in robots developed for chemistry applications. Even with chemistry-compatible robotic systems, we have encountered issues with stainless steel needle compatibility with highly acidic media. Surprisingly, most robotic platforms in the chemistry space are outfitted with stainless steel needles, but several companies offer various inert coating options.
The key to successful offline LC analysis is to effectively quench the reaction aliquot. A recent report from Merck indicates that a streamlined acidic quench protocol enabled DoE studies in combination with reaction profiling to fine-tune the conditions of the Vilsmeier Haack bromination of γ-cyclodextrin (Fig. 17).33
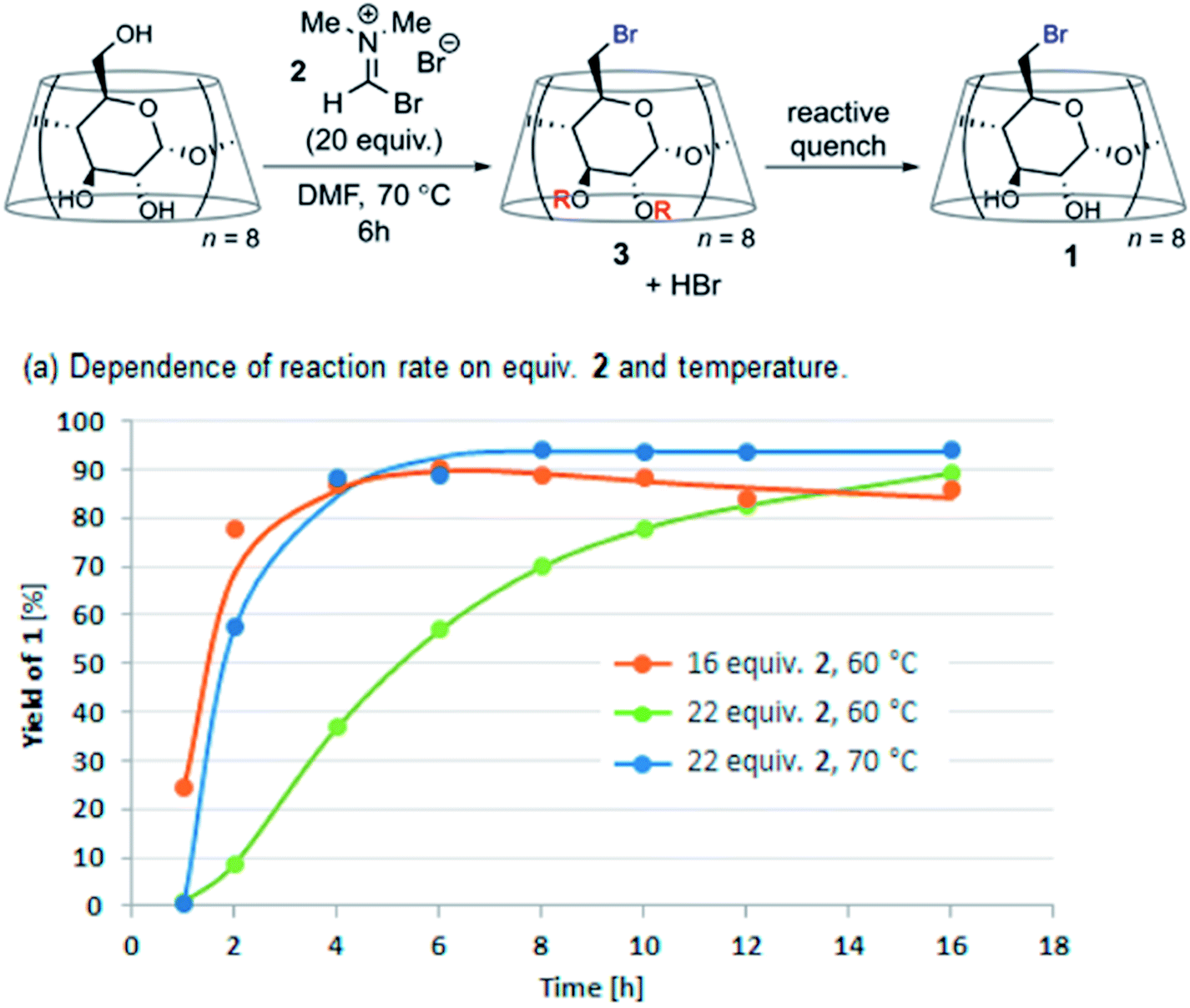 | ||
| Fig. 17 DoE time course studies enabled by an acidic quench in the Vilsmeier Haack bromination of γ-cyclodextrin. Adapted with permission from ref. 31. Copyright 2021 American Chemical Society. | ||
In instances where an effective quench cannot be developed or certain analytes are not stable, online systems that enable near real-time analysis have become increasingly prevalent. We have reported numerous systems of this nature, including a Chemspeed Technologies SWING system with online HPLC (high performance LC) analysis,38 an automated reactor system with online HPLC-MS analysis,39 and an automated reactor system capable of acquiring accurate kinetic profiles from heterogeneous reactions.20 Welch and coworkers have developed a similar online sampling capability for the Mettler-Toledo Auto Chem EasySampler, allowing for real-time MISER analysis of reaction mixtures.40 A key example highlighting the utility of online HPLC systems is in the Suzuki coupling of an E-vinyl tosylate and aryl boronic acid under palladium catalysis (Fig. 18).38 In this example, online HPLC analysis allows for accurate quantitation of reactant and product concentrations without requiring reactant stability over time. The comparison plots between online and offline HPLC analysis strikingly highlight the disparity between the two techniques. The offline HPLC analysis reveals that the absence of an effective quench leads to decomposition of the reactants in the presence of the palladium catalyst and oxygen upon sample aging. Thus, online analysis is essential to the accurate representation of the reactant concentrations over time.
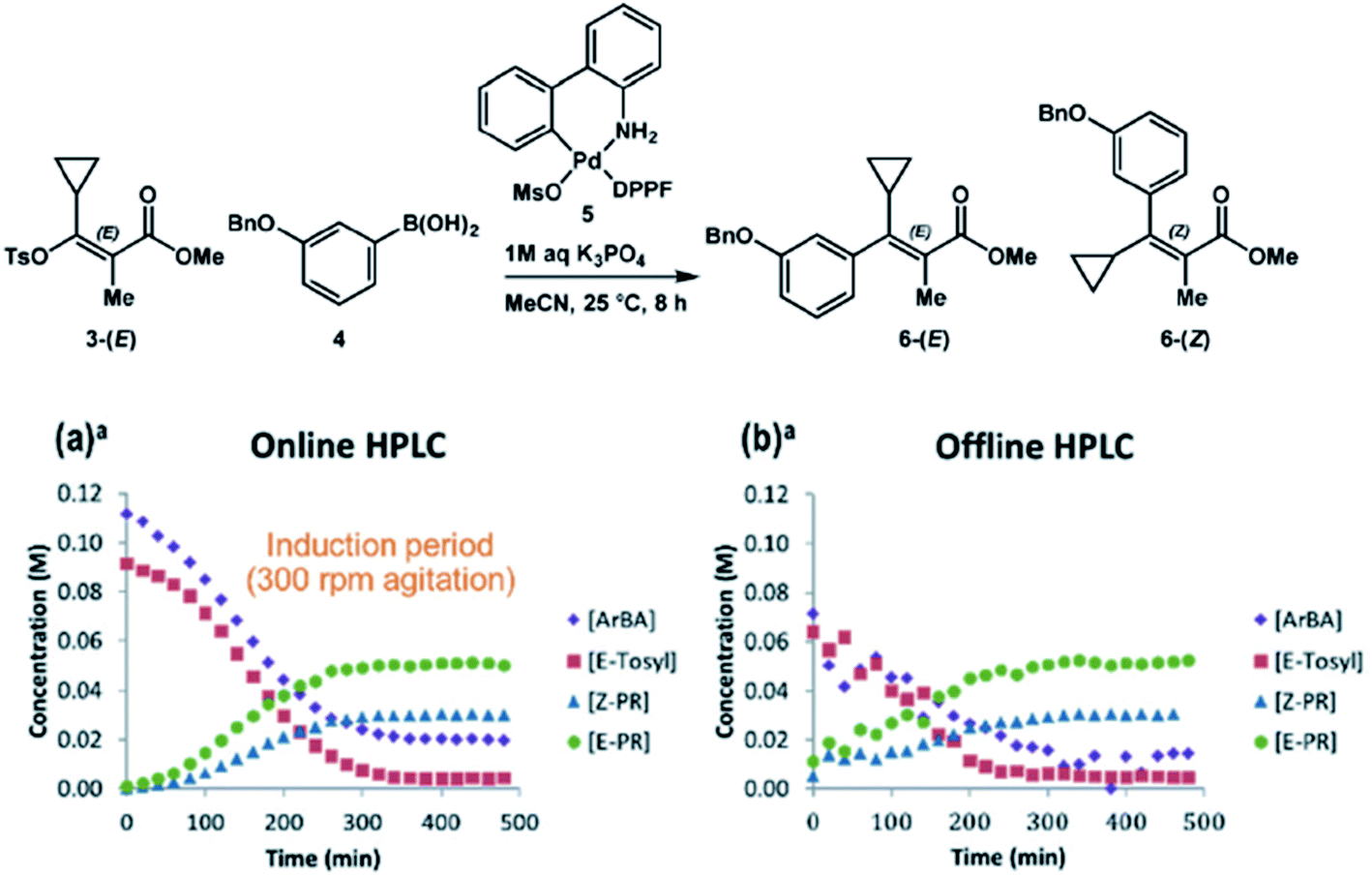 | ||
| Fig. 18 Reaction monitoring with online HPLC analysis enables a more accurate reaction snapshot. Reproduced from ref. 36 with permission from the Royal Society of Chemistry. | ||
Aside from developing an effective quench and consideration of online versus offline sampling, aspiration of a representative aliquot from a reaction mixture brings about an additional set of challenges. For example, it is difficult to aspirate multiple samples from capped vial reactions at elevated temperature or pressure without the loss of solvent or gas reactant. Unchained Labs OSR platforms have overcome this hurdle through the use of a novel sampling mechanism to allow for aspiration under elevated temperature and pressure. Nunn and coworkers have demonstrated this platform in the optimization of a diastereoselective oxazolidine synthesis, where sampling a DoE campaign over time at elevated temperature provided key mechanistic insights into the formation of a diastereomeric impurity (Fig. 19).32
 | ||
| Fig. 19 DoE with time course sampling to optimize a diastereoselective oxazolidine synthesis through Unchained Labs OSR sampling at elevated temperature. Reprinted with permission from ref. 30. Copyright 2018 American Chemical Society. | ||
Analysis of crystallization experiments typically involve in situ focused beam reflectance measurement (FBRM) and powder X-ray powder diffraction (XRPD) of isolated solids.18 High throughput instrumentation for XRPD is currently an offering on the market, but plates utilized in such measurements are expensive and cumbersome to assemble. XRPD-compatible filter plates would fill a current gap in these types of analyses.
One potential application of feedback control is autonomous reaction optimization. Many examples have been focused on flow reactor systems, which are outside of the scope of this review.8 However, a recent demonstration of feedback control integrates high-throughput batch reactions performed by commercial robotic systems with online HPLC analysis and Bayesian optimization techniques (Fig. 20).41 Over a four-day optimization campaign that was free of human intervention, this system developed an optimized stereoselective Suzuki cross-coupling protocol with a 76% yield. While there is still much to learn about the interpretation of data stemming from experimental planning algorithms, it is evident that the application of autonomous technologies is the next frontier in chemistry automation.
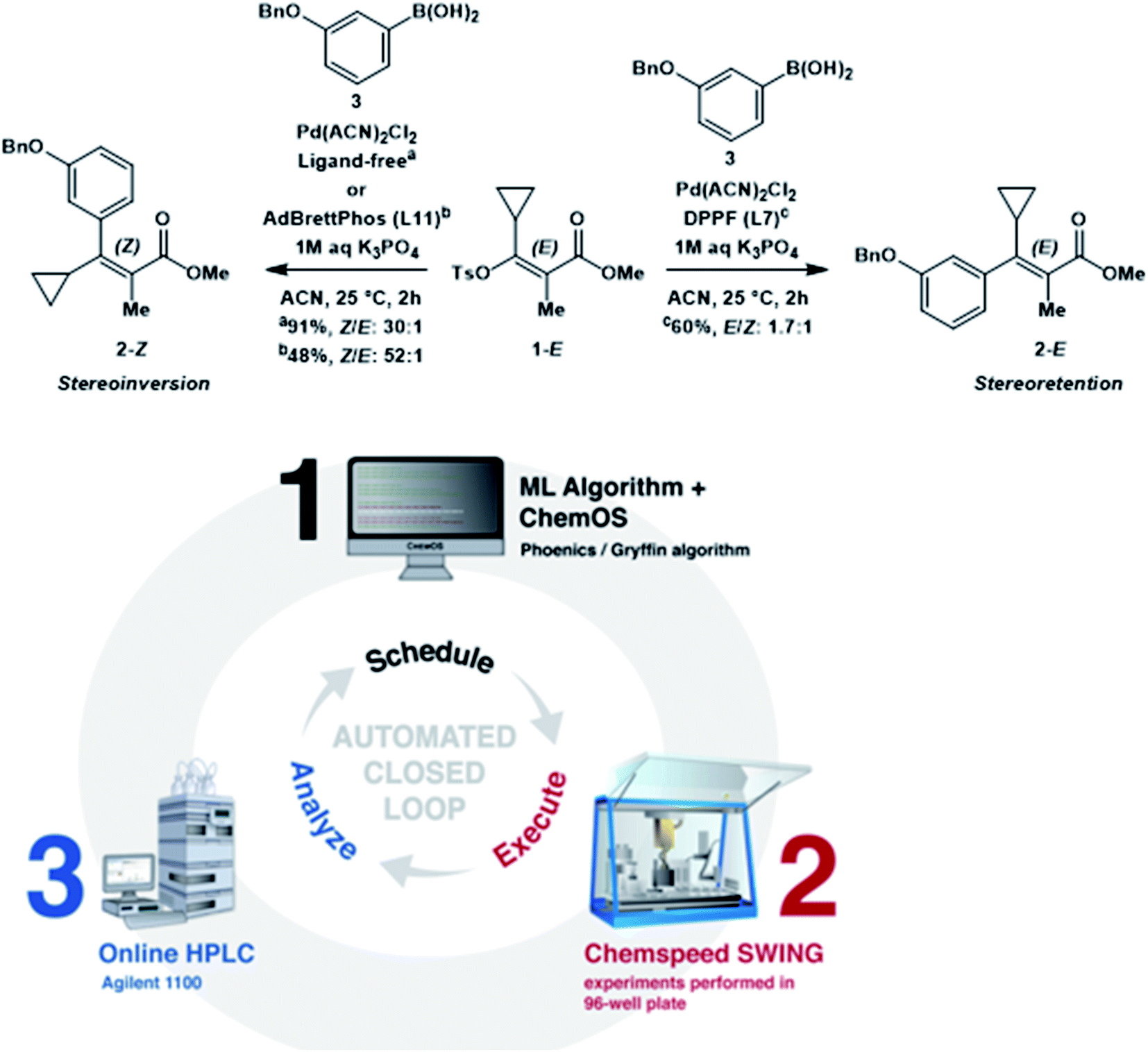 | ||
| Fig. 20 Automated closed-loop for reaction optimization. Reproduced with permission from ref. 39. | ||
Data/software considerations
A significant component in any automation workflow is the software integration. While the selected hardware may be physically capable of performing the desired tasks, this capability is irrelevant if the instrument is not controlled by accessible software or firmware. In most cases, each instrument or hardware component has its own dedicated software, and it falls to the experimentalist to connect these disparate softwares. It is worth noting that in commercial integrated robots, all components are typically packaged under one software ecosystem, unless integration of a custom component not offered by the vendor is desired (Chemspeed Technologies, Unchained Labs, Tecan, Mettler-Toledo Auto Chem etc.). Beyond component control and automation software, error handling and data management must also be considered. These non-trivial tasks are rarely or only superficially discussed in automation publications, even though they can make or break an automation workflow.6Software APIs vary greatly in complexity, ranging from simple one-way serial strings to bi-directional protocols with checksums for error catching across a wide variety of communication methods (TCP/IP, serial etc.). Generally, the more complicated the hardware, the more complicated the interface unless the manufacturer has gone to great lengths to simplify their API. This simplification is referred to as “abstraction” and refers to the process of simplifying the interface by hiding the working details of the hardware (“low level” processes), providing a “higher level” interface. In the context of automation hardware, higher-level interfaces tend to group together commonly conducted sequences or steps into single processes. In many cases, hardware integration benefits from abstraction as those are likely to be sensible groupings of actions that will need to be done anyway, but in some cases APIs do not provide a sufficiently low-level interface to do what is required by a workflow. The abstraction level of a hardware's API should be considered before integration into an automation workflow.
If no API exists for the hardware, then a hardware workaround will be required. If the hardware has a method of being remotely triggered, leveraging that method is straightforward. The hardware can be preconfigured to execute whatever actions are required of it, and an automation workflow can simply trigger that sequence of actions by, for example, a simple contact closure. In effect, the hardware's entire workflow will be abstracted into a single signal from the perspective of the automation workflow. Note that if this is the selected integration method for a piece of hardware, careful consideration of the execution duration of the hardware will be required to ensure that the hardware's portion is completed before the next requisite step.
There is also the option to leverage an existing automation software package (e.g. LabVIEW, Cycle Composer), which can communicate with, control, and orchestrate many components. Although convenient, not every possible component or instrument is controllable by these softwares, and it may become necessary to develop custom-built scripts in a programming language. If this becomes necessary, there is significant overhead involved in the development and implementation of these scripts.
It would seem that a simple solution to any error state would be the “catch and continue” method, where if an error occurs, the error is ignored and the workflow continues as it normally would. We would strongly caution against the implementation of this method, as it turns the automation workflow into a complete black box, rendering any data generated by that workflow untrustworthy (if it is not known whether there was an error, how can one know that each step completed successfully). While it requires a great deal more effort to selectively handle each error as it arises, this approach will result in a much more robust workflow whose produced data may be relied upon.
The future of automation in chemistry
The future of chemistry automation is intelligent, flexible automation, where the integration of computer science, machine vision, rapid analytics, and robotics will enable the execution of if-then loops. For example, the utilization of machine vision in automation systems will allow for a certain set of steps to be executed once certain requirements are met. The same scenario can be envisioned with ML algorithms, where the interpretation of analytical outcomes will determine the next set of actions. We have highlighted two examples that demonstrate the utility of such systems here,16,41 but are looking forward to a new era in which intelligent automation becomes the norm, and not the exception. Only with this feedback control will automation platforms be able to master the art of synthesis.Data availability
We do not have experimental or computational data associated with this perspective article.Author contributions
J. E. H. and M. C. conceived and supervised the project. M. C. and F. B. led the development of the experimental considerations sections. L. P. E. Y. directed the sections on data and software considerations. P. S. and S. G. contributed to equipment module descriptions and commentary. T. Z. and P. L. P. structured the manuscript and created the original artwork. All authors participated in the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Shane T. Grosser for his insightful comments around this manuscript.References
- K. C. Nicolaou, Proc. R. Soc. A, 2014, 470, 20130690 CrossRef CAS PubMed.
- S. J. Lippard, Nature, 2002, 416, 587 CrossRef CAS PubMed.
- R. B. Merrifield and J. M. Stewart, Nature, 1965, 207, 522–523 CrossRef CAS PubMed.
- E. Farrant, ACS Med. Chem. Lett., 2020, 11, 1506–1513 CrossRef CAS PubMed.
- R. L. Sharp, R. G. Whitfield and L. E. Fox, Anal. Chem., 1988, 60, 1056A–1062A CrossRef.
- J. H. Hardin and F. R. Smietana, Mol. Diversity, 1996, 1, 270–274 CrossRef CAS PubMed.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef PubMed.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Science, 2019, 363(6423) DOI:10.1126/science.aav2211.
- B. P. MacLeod, F. G. L. Parlane, T. D. Morrissey, F. Häse, L. M. Roch, K. E. Dettelbach, R. Moreira, L. P. E. Yunker, M. B. Rooney, J. R. Deeth, V. Lai, G. J. Ng, H. Situ, R. H. Zhang, M. S. Elliott, T. H. Haley, D. J. Dvorak, A. Aspuru-Guzik, J. E. Hein and C. P. Berlinguette, Sci. Adv., 2020, 6, eaaz8867 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- J. A. Selekman, J. Qui, K. Tran, J. Stevens, V. Rosso, E. Simmons, Y. Xiao and J. Janey, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 525–547 CrossRef PubMed.
- M. N. Bahr, M. A. Morris, N. P. Tu and A. Nandkeolyar, Org. Process Res. Dev., 2020, 24, 2752–2761 CrossRef CAS.
- M. N. Bahr, D. B. Damon, S. D. Yates, A. S. Chin, J. D. Christopher, S. Cromer, N. Perrotto, J. Quiroz and V. Rosso, Org. Process Res. Dev., 2018, 22, 1500–1508 CrossRef CAS.
- P. Shiri, V. Lai, T. Zepel, D. Griffin, J. Reifman, S. Clark, S. Grunert, L. P. E. Yunker, S. Steiner, H. Situ, F. Yang, P. L. Prieto and J. E. Hein, iScience, 2021, 24, 102176 CrossRef CAS PubMed.
- F. Kong, L. Yuan, Y. F. Zheng and W. Chen, J. Lab. Autom., 2012, 17, 169–185 CrossRef CAS.
- J. A. Selekman, D. Roberts, V. Rosso, J. Qiu, J. Nolfo, Q. Gao and J. Janey, Org. Process Res. Dev., 2016, 20, 70–75 CrossRef CAS.
- J. Qiu and J. Albrecht, Org. Process Res. Dev., 2018, 22, 829–835 CrossRef CAS.
- C. Rougeot, H. Situ, B. H. Cao, V. Vlachos and J. E. Hein, React. Chem. Eng., 2017, 2, 226–231 RSC.
- T. Maschmeyer, P. L. Prieto, S. Grunert and J. E. Hein, Magn. Reson. Chem., 2020, 58, 1234–1248 CAS.
- R. Grainger and S. Whibley, Org. Process Res. Dev., 2021, 25, 354–364 CrossRef CAS.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- A. Buitrago Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49–53 CrossRef CAS.
- S. B. Boga, M. Christensen, N. Perrotto, S. W. Krska, S. Dreher, M. T. Tudge, E. R. Ashley, M. Poirier, M. Reibarkh, Y. Liu, E. Streckfuss, L.-C. Campeau, R. T. Ruck, I. W. Davies and P. Vachal, React. Chem. Eng., 2017, 2, 446–450 RSC.
- M. Christensen, A. Nolting, M. Shevlin, M. Weisel, P. E. Maligres, J. Lee, R. K. Orr, C. W. Plummer, M. T. Tudge, L.-C. Campeau and R. T. Ruck, J. Org. Chem., 2016, 81, 824–830 CrossRef CAS PubMed.
- J. D. Moseley and P. M. Murray, J. Chem. Technol. Biotechnol., 2014, 89, 623–632 CrossRef CAS.
- J. A. Jurica and J. P. McMullen, Org. Process Res. Dev., 2021, 25, 282–291 CrossRef CAS.
- J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71–80 Search PubMed.
- S. Berritt, S. D. Dreher, S. D. Goble, M. Tudge and D. Conway, Presented in part at the 240th National Meeting of the American Chemical Society, Poster ORGN 1054, Boston, MA, 2010 Search PubMed.
- C. Nunn, A. DiPietro, N. Hodnett, P. Sun and K. M. Wells, Org. Process Res. Dev., 2018, 22, 54–61 CrossRef CAS.
- S. L. Zultanski, N. Kuhl, W. Zhong, R. D. Cohen, M. Reibarkh, J. Jurica, J. Kim, L. Weisel, A. R. Ekkati, A. Klapars, D. R. Gauthier Jr and J. M. McCabe Dunn, Org. Process Res. Dev., 2021, 25, 597–607 CrossRef CAS.
- V. Rosso, J. Albrecht, F. Roberts and J. M. Janey, React. Chem. Eng., 2019, 4, 1646–1657 RSC.
- S. C. Chai, A. N. Goktug, J. Cui, J. Low and T. Chen, in Drug Discovery, ed. H. A. El-Shemy, IntechOpen, Rijeka, 2013 Search PubMed.
- M. Shevlin, ACS Med. Chem. Lett., 2017, 8, 601–607 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- M. Christensen, F. Adedeji, S. Grosser, K. Zawatzky, Y. Ji, J. Liu, J. A. Jurica, J. R. Naber and J. E. Hein, React. Chem. Eng., 2019, 4, 1555–1558 RSC.
- T. C. Malig, J. D. B. Koenig, H. Situ, N. K. Chehal, P. G. Hultin and J. E. Hein, React. Chem. Eng., 2017, 2, 309–314 RSC.
- K. Zawatzky, S. Grosser and C. J. Welch, Tetrahedron, 2017, 73, 5048–5053 CrossRef CAS.
- M. Christensen, L. Yunker, F. Adedeji, F. Häse, L. Roch, T. Gensch, G. dos Passos Gomes, T. Zepel, M. Sigman, A. Aspuru-Guzik and J. Hein, Commun. Chem., 2021, 4, 112 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |