
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe 100 facets of the Passerini reaction
Luca
Banfi†
 *,
Andrea
Basso†
,
Chiara
Lambruschini†
,
Lisa
Moni†
and
Renata
Riva†
*,
Andrea
Basso†
,
Chiara
Lambruschini†
,
Lisa
Moni†
and
Renata
Riva†
Department of Chemistry and Industrial Chemistry, University of Genova, Via Dodecaneso 31, 16146 Genova, Italy. E-mail: luca.banfi@unige.it
First published on 30th September 2021
Abstract
This perspective aims at celebrating the 100th anniversary of the discovery of the Passerini three component reaction. After being nearly neglected for many years, now this reaction has become quite popular, thanks to the achievements of the last 30 years, which have revealed several chances of exploitation in organic synthesis. Though not being comprehensive, this review means to show the various ways that have been used in order to expand the utility of the Passerini reaction. Post-MCR transformations to give heterocycles or peptidomimetics, variants through single component replacement, stereochemical issues, and applications in total syntheses will be especially covered.
1. Introduction
The reaction of isocyanides, aldehydes/ketones and carboxylic acids to give α-acyloxyamides (Scheme 1) is the oldest isocyanide-based multicomponent reaction (IMCR). It was discovered in Florence by Mario Passerini (Fig. 1) and first reported in 1921.1 Therefore, 2021 represents an important anniversary, not only for the Passerini reaction, but also for all IMCRs.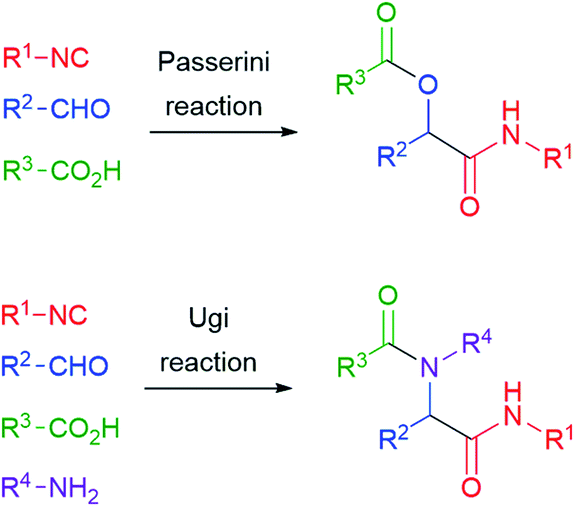 | ||
| Scheme 1 The Passerini and Ugi reactions. | ||
 | ||
| Fig. 1 Mario Passerini. This picture, was taken in 1921 and was kindly given us in 2003 by Passerini's son, Pietro. We thank his granddaughter Sara Passerini for authorizing the publication of this photography. © reserved; photo courtesy of the Passerini family. | ||
This valuable reaction was nearly neglected for 40 years after its discovery. In 1959, Ivar Ugi added a fourth component, a primary amine, obtaining α-acylaminoamides (Scheme 1). Most importantly, he was able to recognize the great potential of multicomponent reactions in the fast generation of large collections of similar molecules. His pioneering work provided the basis for the upsurge of combinatorial chemistry and diversity-oriented synthesis that took place in the 90's. In those years, the Ugi and Passerini reactions became well known to most organic chemists, and the publications related to them and to other IMCRs have continued to steadily increase in the new century till nowadays.
Nevertheless, in the 90's, the Passerini reaction was still much less exploited than its daughter, the Ugi reaction, probably because esters are less attractive than amides in medicinal chemistry. The renewal of interest in this reaction was thus achieved by extending the scope from simple α-acyloxyamides (depsipeptides) to a variety of different products, including heterocycles and peptidomimetics. Italy, which has been the cradle of the Passerini reaction, had an important role in this renaissance too, especially thanks to Stefano Marcaccini‡ (Florence University), but from 1998 also to our own group.
During the last 30 years, several new variants and/or applications of the Passerini reaction were reported, eventually granting to it a well deserved place in the Olympus of organic named reactions.2
1.1 Main features of the Passerini reaction
The generally accepted mechanism of the Passerini reaction is shown in Scheme 2.3 This is a concerted mechanism, where the H-bonded cluster formed by the interaction of the carboxylic acid with the carbonyl compound reacts with the isocyanide in a single step. Overall, this is an α-addition of the carbonyl component (as an electrophile) and of the carboxylate (as a nucleophile) to the isocyanide to give the intermediate 2. The intermediacy of a relatively non-polar cyclic transition state 1 agrees with the fact that the reaction is faster in apolar solvents. Then the α-addition adduct 2 rearranges to the final product 3. A still unresolved question is whether the rate limiting step is the α-addition or the rearrangement, which may have important consequences on the stereochemical course. A recent theoretical study4 suggests that the rate-limiting step may be the last one and that an additional molecule of the carboxylic acid is involved in the transition state. However, an old study,5 carried out using a ketone as the carbonyl component, reported that the Passerini reaction is first order in each of the three reactants. We think that both the rate limiting step and the intervention of an additional carboxylic molecule in the T.S. may strongly depend on the nature of substrates. This may explain the apparent contradiction between the theoretical study (done on formaldehyde) and the experimental one. | ||
| Scheme 2 The mechanism of the Passerini reaction. | ||
Being faster in a low polarity medium, the Passerini reaction is typically carried out in solvents such as dichloromethane, ethyl acetate, diethyl ether and tetrahydrofuran. In contrary to the related Ugi reaction, alcohols are not well suited as reaction solvents. Actually, when the Passerini reaction occurs as an unwanted side reaction in an Ugi process, it may be suppressed by using high polar alcohols, such as trifluoroethanol. The classical Passerini reaction is typically carried out at rt, with the reaction times varying from a few hours to several days (in the case of ketones). With bulky ketones, the reaction may be sluggish. Thus, in general yields are much better with aldehydes. The reaction takes place under mild conditions in a slightly acidic medium and thus tolerates a variety of different functional groups, which is very useful for the introduction of additional functionalities described in chapter 2. Only electron-poor phenols or primary and secondary amines are not accepted, giving the Passerini–Smile (see chapter 3) or the Ugi reaction instead. With aldehydes or unbulky ketones the scope is quite broad, although we found that some heteroaromatic aldehydes may be unreactive.6 Also α,β-unsaturated aldehydes/ketones are poor substrates.
As for the carboxylic acids or the isocyanide components, the scope is very broad. Aromatic isocyanides tend to be somehow unstable. In these cases, their in situ preparation may be advantageous.7
1.2 Organization of this perspective
In this perspective, we would like to review these advancements, demonstrating the many facets of this reaction. The Passerini reaction and its variants have been already reviewed in a comprehensive manner in 2005 and 2014 by us.3,8 This perspective, on the other hand, was created with the aim of rationally guiding the reader through the four main lines of research that have characterized this multicomponent reaction, in which our research group has often played an important role: (1) the introduction of post-condensation transformations; (2) the use of non-classical components; (3) the control of diastereoselectivity and enantioselectivity; (4) the synthesis of natural products and APIs.Each chapter will describe the more interesting, original, and recent applications; in addition, a “future perspective” paragraph will describe what, in our opinion, are the strategies and improvements, not yet explored, that could characterize the Passerini reaction of the second centennial.
2. The power of post-Passerini transformations: from cyclization to heterocycles to the PADAM strategy
2.1 Post-Passerini cyclizations to give heterocycles9,10
The typical Passerini products are acyclic depsipeptides, which are not much interesting, from a medicinal chemistry point of view, because of the lability of ester groups under physiological conditions. Cyclization to heterocycles can solve this problem. For this purpose, the presence of one or two additional functionalities in the three components is typically needed, although some cyclization may take advantage of the isocyanide-derived secondary amide, which can act either as a nucleophile or an electrophile. The oldest example of this approach was reported in 1983 by Sebti and Foucaud (Scheme 3)11 and involved a halogen as a single additional group. β-Lactams 4 were obtained through an SN2 reaction, where the secondary amide nitrogen acts as a nucleophile. A few years later, in 1991, Marcaccini reported a Paal–Knorr cyclization that used carboxylic derived C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and an additional oxo group installed in the aldehyde component. In this way, arylglyoxals 5 have been converted into oxazoles 6.12
O and an additional oxo group installed in the aldehyde component. In this way, arylglyoxals 5 have been converted into oxazoles 6.12
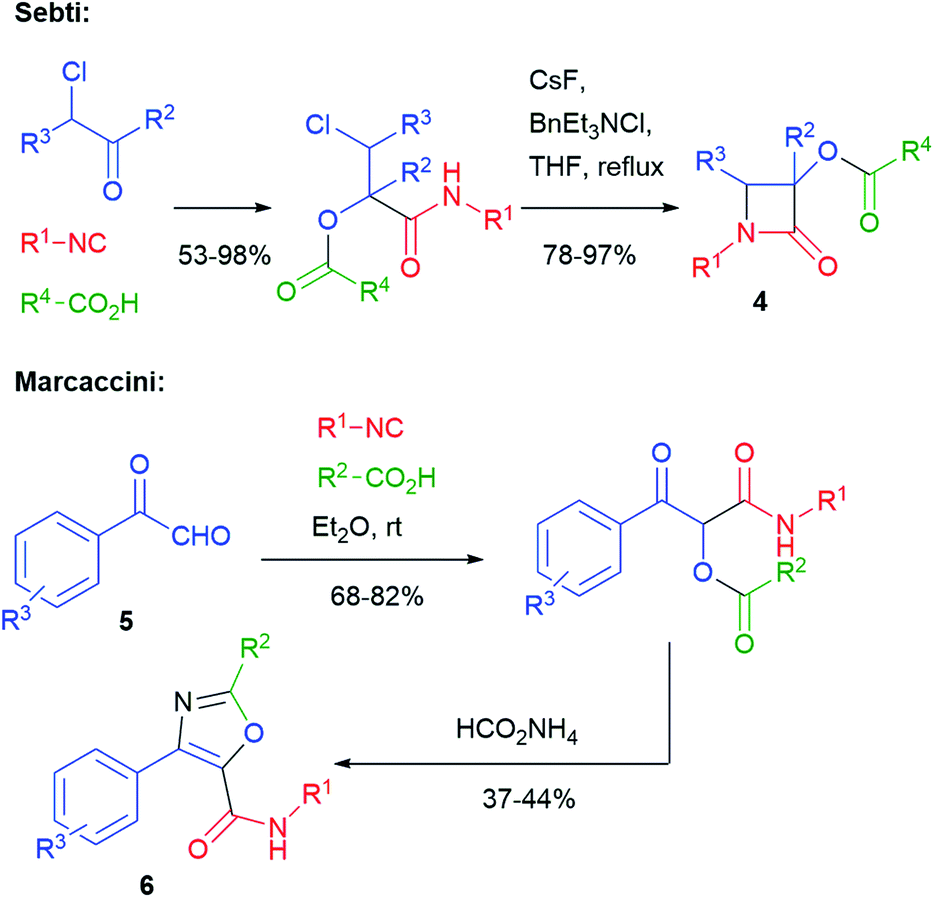 | ||
| Scheme 3 The oldest syntheses of heterocycles through post-Passerini cyclizations. | ||
In 2010 we have obtained oxazolines 9, this time exploiting, as an additional functional group, an azide placed on the aldehydic component (Scheme 4).13 Since α-azidoaldehydes 8 are rather unstable, we used an in situ oxidation protocol14 of α-azidoalcohols 7 with IBX. The Passerini adducts were then cyclized using supported triphenylphosphine in a Staudinger–aza-Wittig reaction. The use of a supported reagent makes this protocol well suited for combinatorial synthesis of these entities. Diastereomeric mixtures of oxazolines 9 were obtained, with poor stereoselectivity. The same products have been later obtained by Ding from chloroacetaldehyde derived Passerini products 10, by introducing the azide just before the Staudinger–aza-Wittig step.15 Using α,β-unsaturated azidoenals, oxazoles (instead of oxazolines) were obtained by Wang.16 Finally, benzoxazines 11 were prepared by Ding applying a similar protocol with ortho-azidobenzaldehyde.17
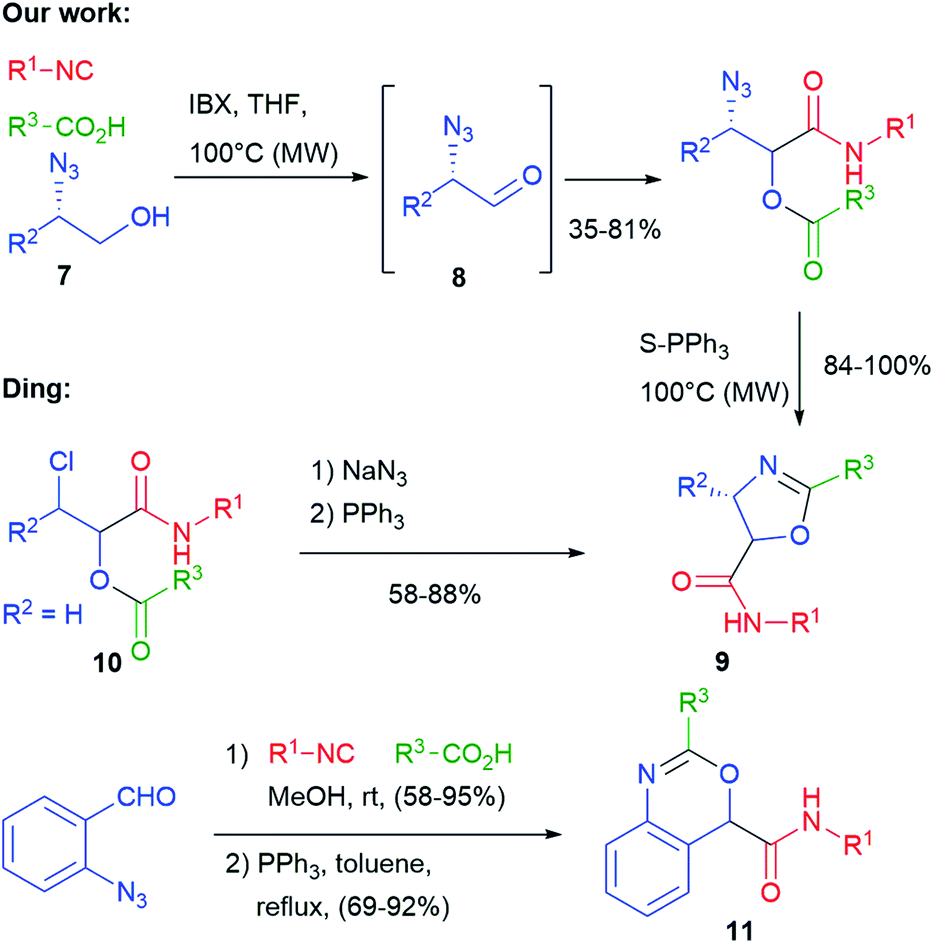 | ||
| Scheme 4 Post-Passerini Staudinger–aza-Wittig protocols involving the ester carbonyl. | ||
A wider exploration of chemical space can be achieved starting from two additional functional groups, such as an azide and a ketone. For example, the Staudinger–aza-Wittig protocol was applied by Wang to azidoketones 15, in turn obtained by a Passerini reaction of azidoacid 13 and aryl glyoxals 14 (Scheme 5).16 Interestingly, the authors employed a one-pot procedure, which also involved an in situ generation of the isocyanide from the corresponding formamide 12. Triphenylphosphine was used both for formamide dehydration (together with hexachloroethane) and for azide reduction.
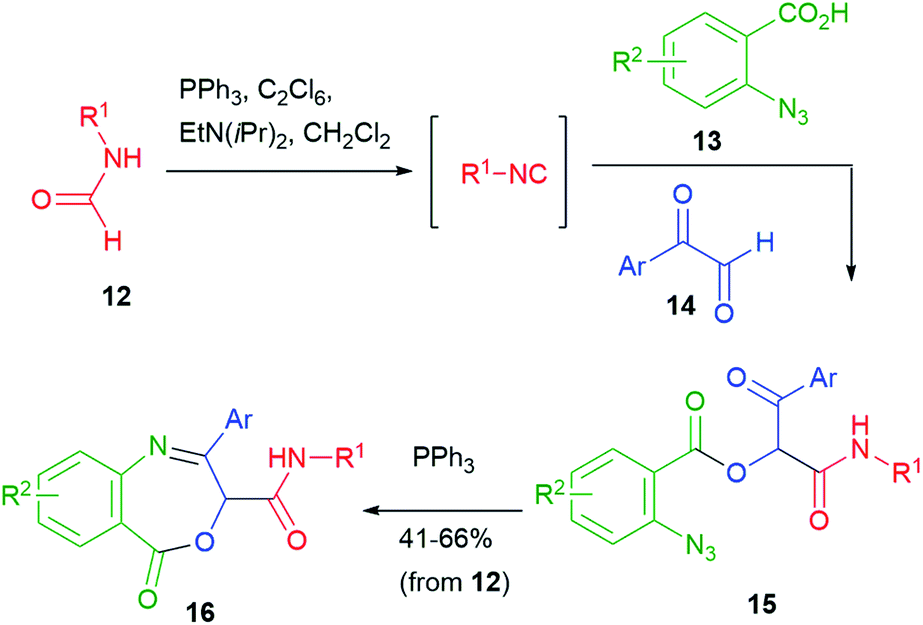 | ||
| Scheme 5 Synthesis of heterocycles via post-Passerini Staudinger–aza-Wittig involving an additional ketone. | ||
Dihydrobenzazepines 16 were obtained and thus this work represents one of the rare examples of use of the Passerini reaction for the synthesis of seven-membered heterocycles.18
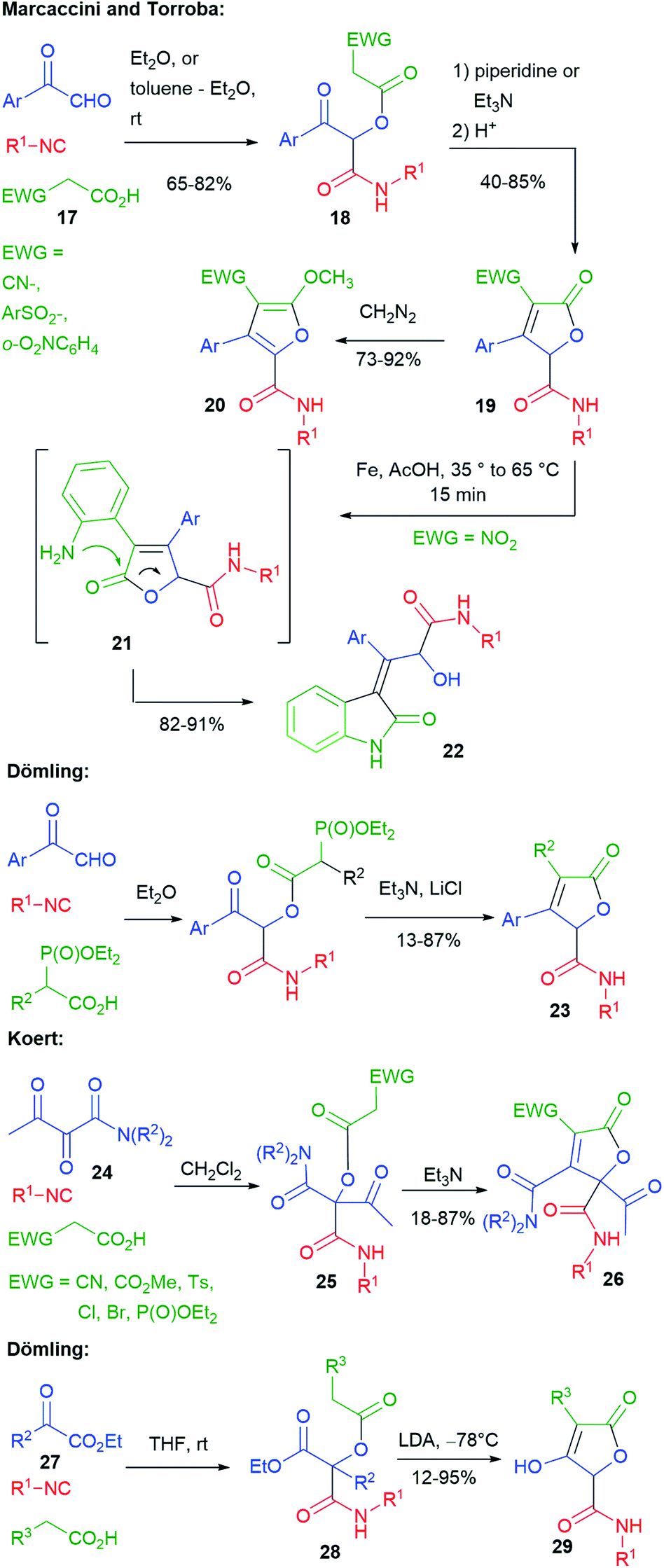
Several researchers have studied post-Passerini CC bond formation through Knoevenagel-type or Wittig-type reactions to give furanones (butenolides) (Scheme 6). The first ones have been Marcaccini and Torroba. In 1993 (ref. 19), they reported that, by using carboxylic acid 17, containing an electron-withdrawing group (EWG), the Passerini products 18 were acidic enough to undergo an intramolecular Knoevenagel-type reaction in the presence of an amine, affording furanones 19. The electron-withdrawing group can be either CN,19 a sulphonyl group,20 or an o-nitroaryl.21 Treatment with diazomethane gave methoxyfuranes 20.
 | ||
| Scheme 6 Synthesis of butenolides via tandem Passerini–Knoevenagel or Passerini–HWE or Passerini–Dieckmann. | ||
Moreover, furanones 19 with EWG = o-nitrophenyl have been submitted to a second post-MCR transformation, leading to indoles 22, through a domino process involving the reduction of the nitro group to 21 and acyl migration.21
Similar butenolides 23 have been prepared by Dömling in 2001 through a tandem (one pot) Passerini–HWE (Horner–Wadsworth–Emmons) strategy.22 Butenolides 26 were prepared in 2014 by Koert,23 through a Passerini–Knoevenagel sequence, using this time α,β-dioxoamides 24, instead of aryl glyoxals. The Passerini reactions were highly regioselective, involving the keto group α to the amide, thus providing 25. In this work, a broader variety of electron-withdrawing groups was exploited. Interestingly, when EWG = P(O)OEt2, the phosphonyl group was retained (only 6% of the HWE product was detected).
By using α-oxoesters 27 as the carbonylic component, Dömling has combined the Passerini reaction with a subsequent Dieckmann cyclization, affording hydroxybutenolides 29. The procedure can be carried out in a one pot manner, without the need to isolate intermediate diesters 28. In this case, the presence of an electron-withdrawing R3 substituent was not needed, since a stronger base (LDA) was employed.24
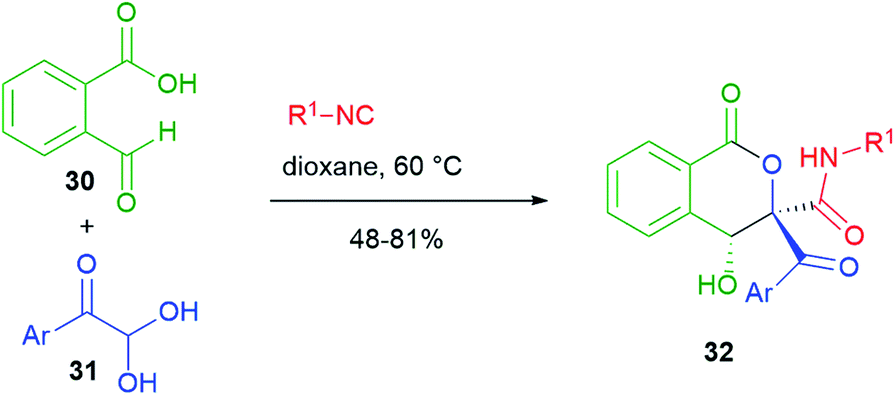
In the cyclizations reported in Scheme 6, the nucleophilic part is always played by the α-carbon of the carboxylic component of the Passerini reaction. On the other hand, Scheme 7 shows an interesting post-Passerini aldol-type reaction, where it is the carbon α to the secondary amide that plays the nucleophilic role. For this strategy, Jiang and Tu, in 2014,25 employed a system containing two aldehydes, 2-formylbenzoic acid 30 and an arylglyoxal 31. Substrate selectivity depends upon the solvent employed. In dioxane, the isocyanide selectively attacks aldehyde 31 and the resulting adduct, easily deprotonated, spontaneously cyclizes onto the aromatic aldehyde to give isocoumarins 32. This aldol reaction is highly diastereoselective.
 | ||
| Scheme 7 Synthesis of isocoumarins via the domino Passerini–Aldol reaction. | ||
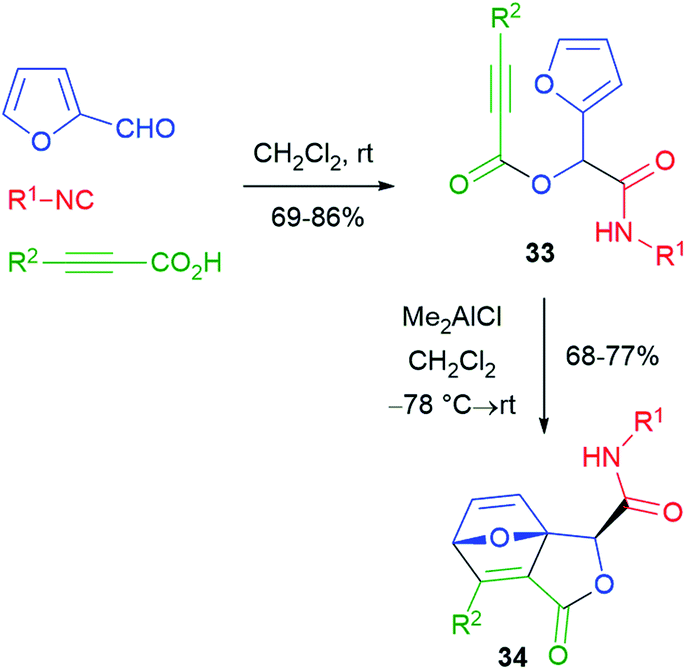
Various post-Passerini cycloaddition reactions have been implemented. The first example was reported by Wright26 in 2002 (Scheme 8), and furfural and a propiolic acid were used in order to install the needed reactive groups for an intramolecular Diels–Alder reaction. The thermal cycloaddition of Passerini adducts 33 did not work, but it could be promoted under Lewis acid catalysis, affording labile tricyclic lactones 34.
 | ||
| Scheme 8 Tandem Passerini–Diels–Alder sequence. | ||
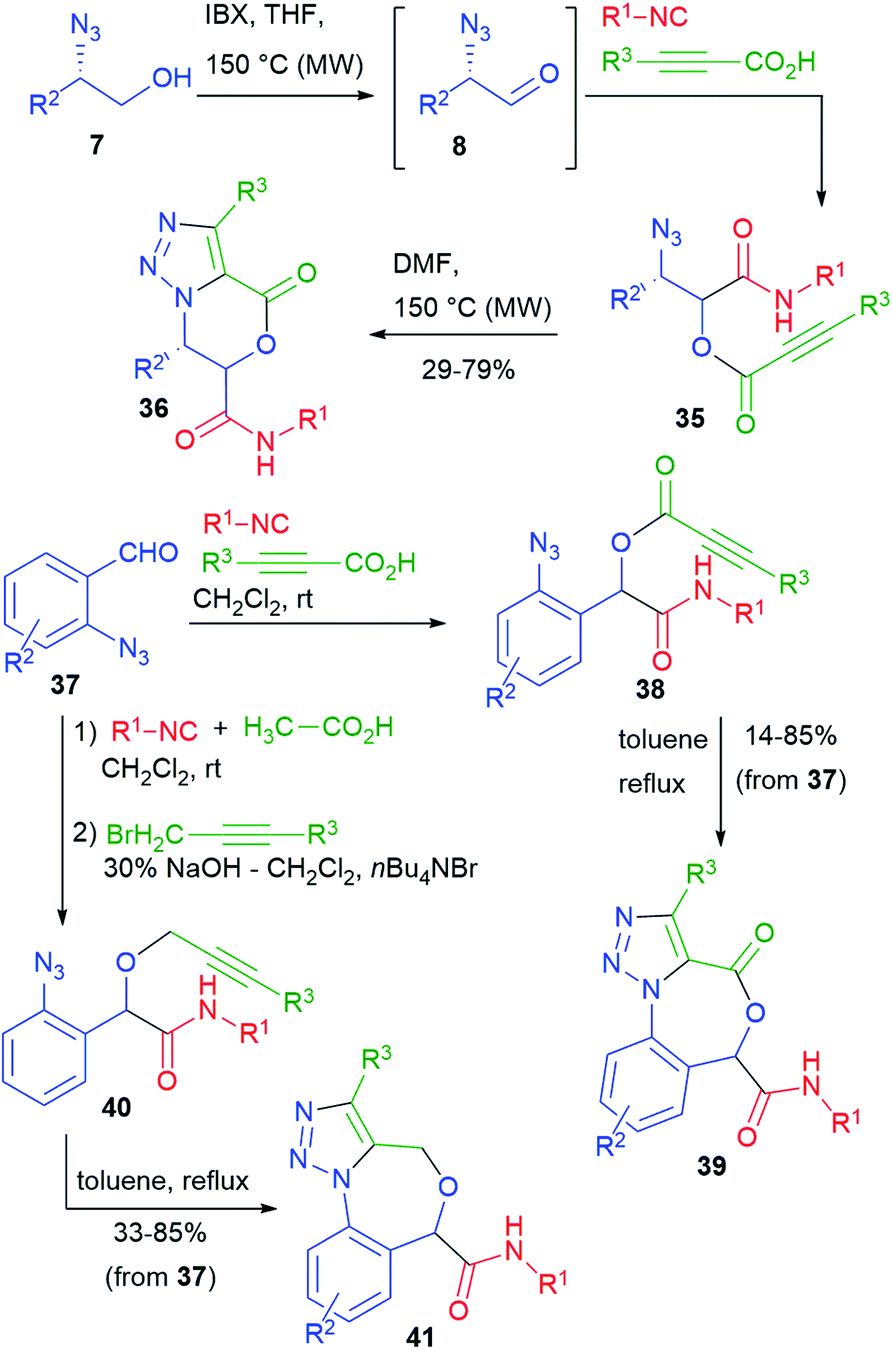
Our group has studied post-Passerini alkyne–azide 1,3-dipolar cycloadditions (Huisgen reaction).27 Towards that goal, we performed a series of Passerini reactions on α-azidoaldehydes 8, obtained in situ from azidoalcohols 7 (cf.Scheme 4), using as the carboxylic component a propiolic acid (Scheme 9). In some cases (e.g. with trimethylsilylpropiolic acid), we could not add all the reagents from the beginning, because of the instability of the propiolic acids under oxidative conditions. It is well known that the intramolecular variant of the Huisgen reaction must be carried out thermally, since the needed regiochemistry is opposite to the one strongly favoured by copper catalysis. Nevertheless, the cyclization of azidoalkynes 35 proceeded efficiently by warming at 150 °C, affording triazolo-fused dihydrooxazinones 36.28
 | ||
| Scheme 9 Tandem Passerini – azide–alkyne cycloaddition. | ||
By using different azidoaldehydes, namely 37, the same strategy leads to Passerini product 38 first, and then to seven-membered triazolo-fused benzoxazepinones 39.29
On the other hand, benzoxazepines 41 could be obtained by the thermal cyclization of azidoalkynes 40. The synthesis of 40 will be described in paragraph 3.3. The overall yields were in most cases good.
We have recently reported (2016)7 a double cyclization of a Passerini adduct by performing, in sequence, two metal catalyzed processes (Scheme 10). Towards this goal, we have designed a family of unsaturated isocyanides 42 and a family of aldehydes 43, containing an allyl carbonate. The Passerini adducts 44, obtained from various carboxylic acids, could be cyclized in a very high yield, and with moderate diastereoselectivity, through a Pd(0) catalyzed Tsuji–Trost process. Then, the resulting dienes 45 have been further cyclized by ring closing metathesis (RCM).
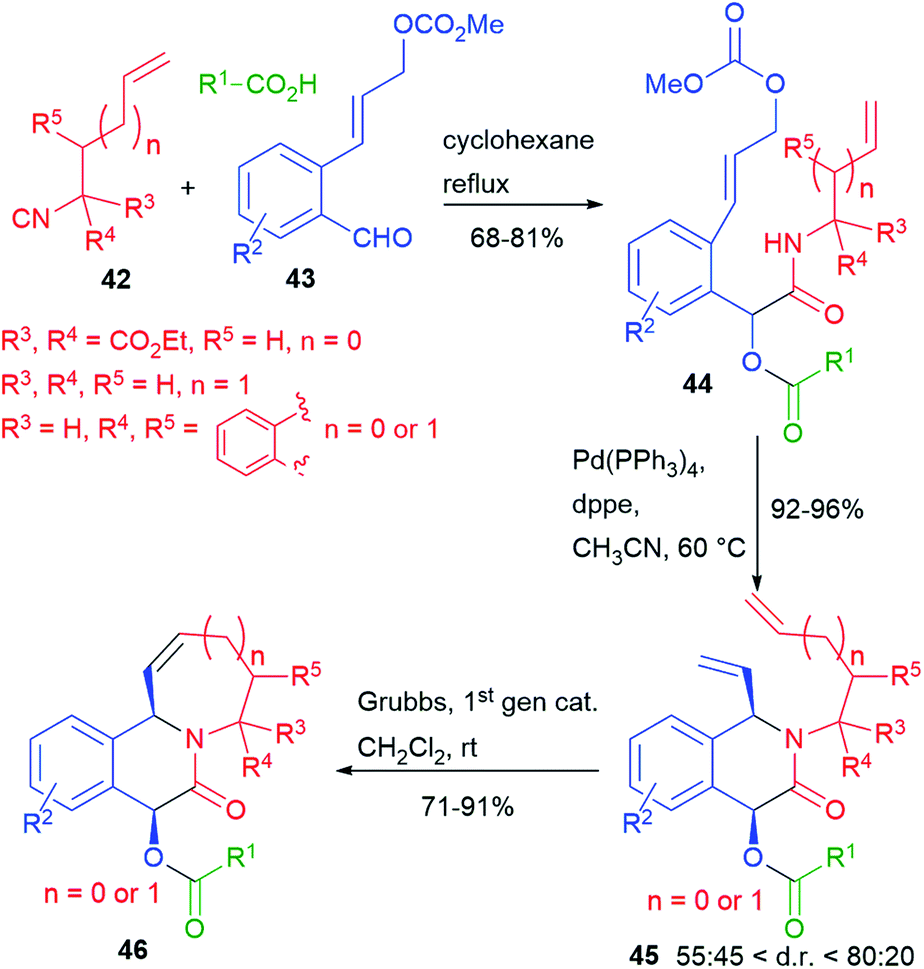 | ||
| Scheme 10 Tandem Passerini–Tsuji–Trost – RCM. | ||
Complex tricyclic adducts 46, resembling some natural alkaloids, were obtained, displaying either 6- or 7-membered rings.
Passerini reactions followed by RCM cyclizations were also reported by Dömling30 and Martens.31 In the first case, various natural-like macrocycles were obtained using unsaturated isocyanides and unsaturated carboxylic acids as MCR inputs. In the second case, six-membered lactones were synthesized starting from allyl ketones and β,γ-unsaturated carboxylic acids.
In chapter 4, we will describe the synthesis of other heterocycles by cyclizations after a diastereoselective Passerini reaction of erythritol derivatives.
2.2 The PADAM approach to peptidomimetics
In 1998, in order to overcome the problem of the physiologically labile ester functionality in the typical Passerini adducts, we designed a rapid and efficient way to convert the Passerini adducts into peptides, starting from protected α-aminoaldehydes 47 (Scheme 11).32 In this design, we have also been inspired by the work of Hulme on the UDC strategy (Ugi/De-Boc/Cyclize).33 After the Passerini reaction to afford 48, a simple deprotection of the amine promotes O → N acyl migration, to form α-hydroxy-β-acylaminoamides 49. With this protocol, which we called PADAM (Passerini-Amine Deprotection-Acyl Migration),34 we could produce, in just two easy steps, peptides 49 containing α-hydroxy-β-amino acid units (norstatine) or, after oxidation, ketones 50, characterized by an α-oxo-β-amino acid unit. These classes of compounds are widely explored for their potential as inhibitors of aspartic proteases (49) or of serine proteases (50). Prior to our work, they were typically synthesized through longer sequences of 6–7 steps. α-Aminoaldehydes can be obtained in a wide variety and in enantiomerically pure from α-amino acids, and thus the final peptidomimetics may be obtained in an optically pure form.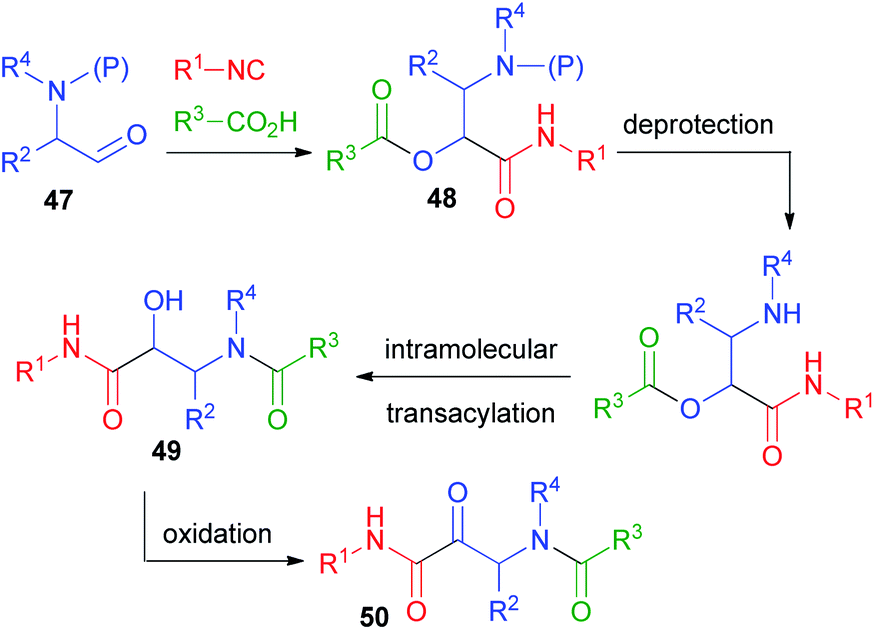 | ||
| Scheme 11 The PADAM protocol. | ||
Initially, we used the Boc protecting group.32 A library of Passerini adducts 48 was produced in good yields (59–95%) and with moderate stereoselectivity (52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48 < d.r. < 71:29). Then, adducts 48 were transformed into 49 using first CF3CO2H, and afterwards triethylamine in a one-pot procedure (yields ranging from 75 to 99%). Recently, the major stereoisomers have been recognized as syn.35 The moderate stereoselection is obviously not a problem if the desired final products are α-ketoamides 50, derived by the oxidation of 49.
:48 < d.r. < 71:29). Then, adducts 48 were transformed into 49 using first CF3CO2H, and afterwards triethylamine in a one-pot procedure (yields ranging from 75 to 99%). Recently, the major stereoisomers have been recognized as syn.35 The moderate stereoselection is obviously not a problem if the desired final products are α-ketoamides 50, derived by the oxidation of 49.
Additionally, we developed the same method on the solid phase, using a photocleavable linker34 (maintaining the Boc group on aldehyde 47) or employing an acid-labile linker (this time shifting to the Fmoc protection).36
The PADAM approach can be even more potent if a protected amino acid is used as the carboxylic component. This concept is exemplified in Scheme 12 by the synthesis of a known inhibitor of cytomegalovirus protease, a serine protease.37 During the deprotection/acyl migration step, the protecting group of the amino acid (in this example, the asparagine derivative 51) is concurrently removed, affording 52, leaving a free amino group available for the introduction of a fourth component by standard peptide coupling. Finally, the oxidation of 53 gives α-oxoamide 54. It should be noted that 52 was not isolated and thence the synthesis of 54 involves just three steps with the introduction of 4 diversity inputs.
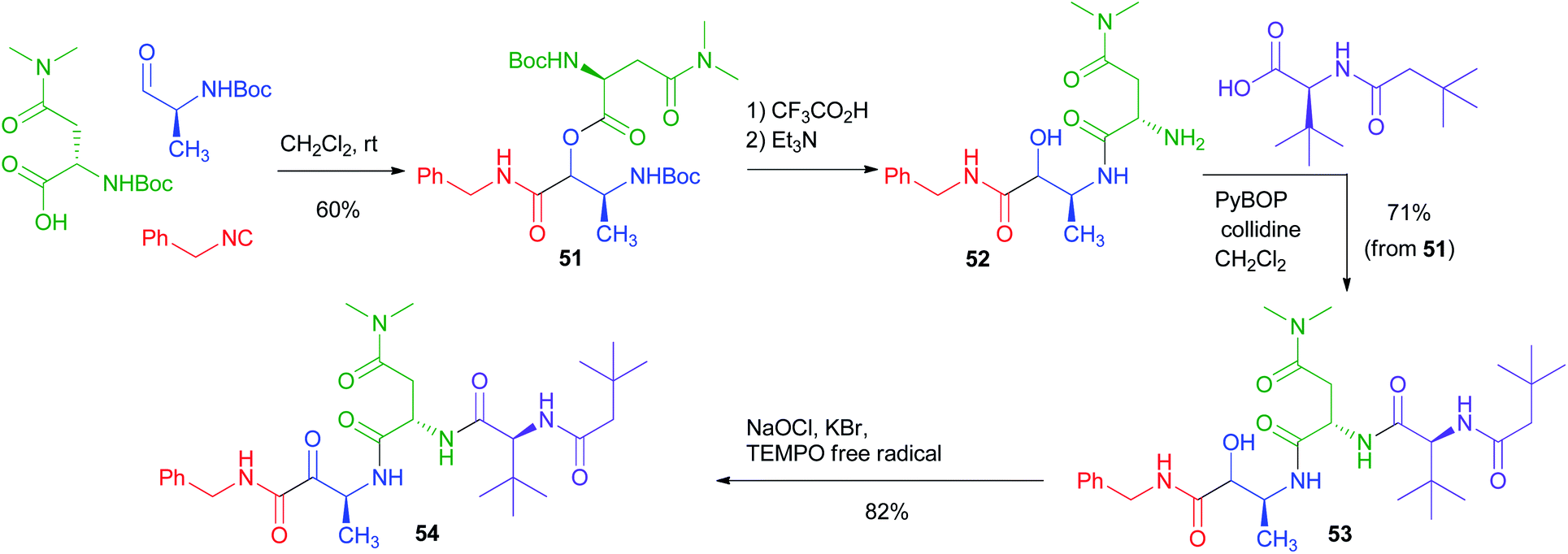 | ||
| Scheme 12 Application of the PADAM strategy to the synthesis of an inhibitor of Cytomegalovirus protease. | ||
In 2001, Semple used the protected amino acid 57 as the carboxylic component for a total synthesis of Eurystatin A (Scheme 13).38 This time, however, orthogonal protections were used for the acid and the α-aminoaldehyde 55. The third component, isocyanide 56, was also derived from a α-amino acid. The chosen aldehyde protecting group was Fmoc, and thus the deprotection–acyl migration stage was accomplished with Et2NH. The resulting peptidomimetic 58 already contains all the fragments of the final target. Simultaneous deblocking of benzyl ester and of Cbz afforded an amino acid that was cyclized to 59. Finally, a few steps afforded Eurystatin A, an α-oxoamide. Thus, also in this case, the poor diastereoselectivity of the Passerini reaction was not problematic, because the stereogenic centre was lost in the final oxidation.
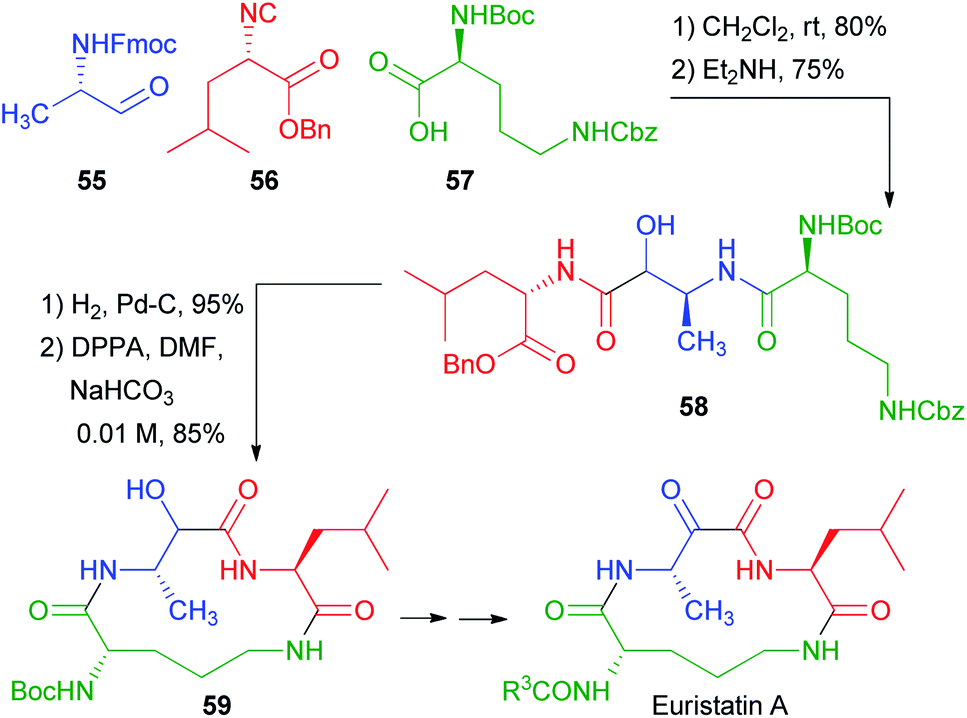 | ||
| Scheme 13 Synthesis of Eurystatin A. | ||
These examples point out PADAM as an ideal protocol for the combinatorial synthesis of libraries of peptidomimetics, in drug discovery.
For example, Bode in 2012 used this approach for the synthesis of α-hydroxy-β-acylaminoamides as HIV protease inhibitors, employing branched isocyanides.39 Other total syntheses of peptidomimetics will be described in chapter 5.
In 2012, Hulme coupled the PADAM protocol with a post-MCR cyclization. Using the special isocyanide 60, he obtained a series of benzimidazoles 63, isosteres of norstatines (Scheme 14).40 After the Passerini reaction, the depsipeptide 61 was converted as usual to 62. Further treatment under acidic conditions at higher temperatures brought about condensation to give the imidazole ring.
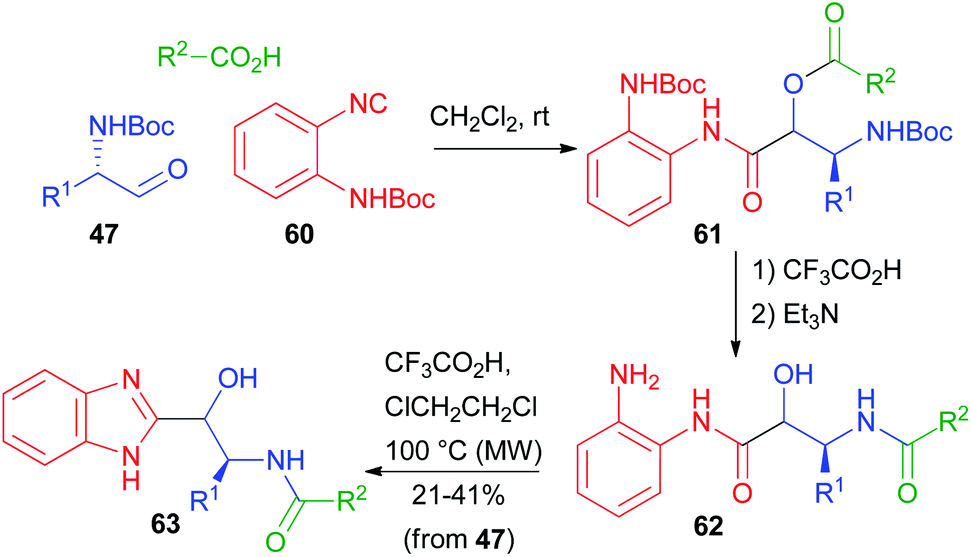 | ||
| Scheme 14 PADAM followed by cyclization as a route to benzimidazole isosteres of norstatines. | ||
Our group studied post-PADAM cyclization processes as well. In 2011 (ref. 41) we exploited the use of an additional amine placed in the carboxylic component (Scheme 15). In contrast to the above-described Hulme's work, we planned to perform the condensation of the amine onto the ketone resulting from post-PADAM oxidation. Since a free amine was expected to interfere with oxidation, we used orthogonal protections for the α-aminoaldehyde 64 (Fmoc) and the α-amino acid 65 (Boc) to obtain 66. The PADAM protocol was thus implemented using diethylamine for the deprotection/acyl migration step. After oxidation to the α-ketoamide 67, treatment with CF3CO2H was expected to afford dihydropyrazinones 68, but an unexpected aromatization took place, giving pyrazinones 69.
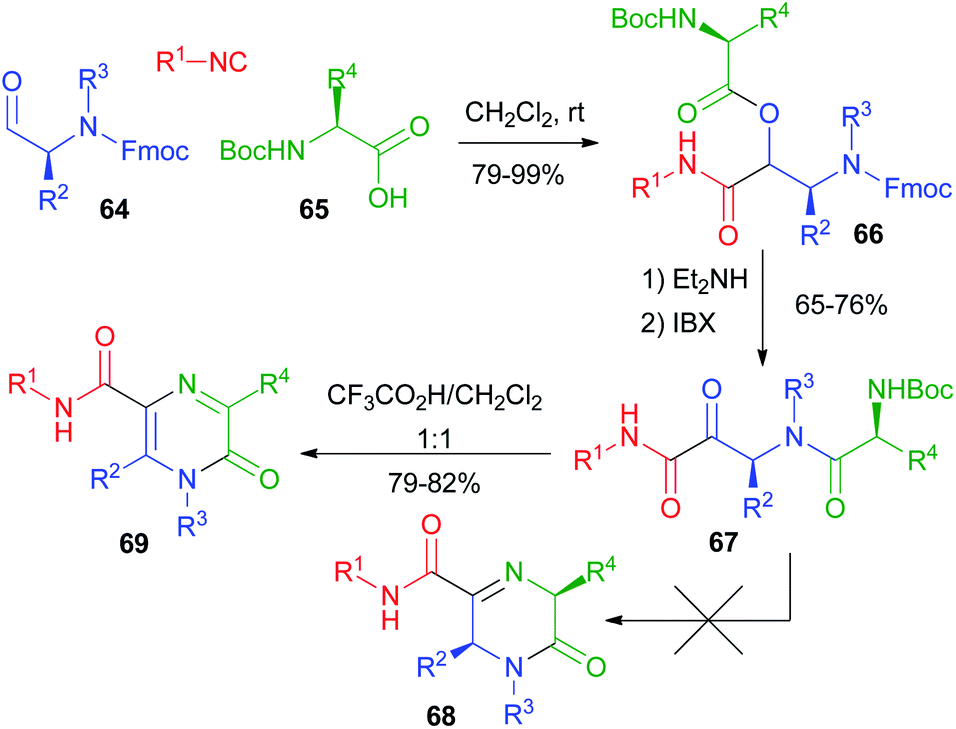 | ||
| Scheme 15 PADAM followed by cyclization as a route to pyrazinones. | ||
2.3 The homo-PADAM and bis-homo-PADAM protocols
Starting from β-aminoaldehydes, a homo-PADAM protocol could be applied, giving access to a new class of elongated peptidomimetics. However, β-aminoaldehydes are much less easily accessible than α-aminoaldehydes. Moreover, when possessing a stereogenic centre in the α position, they are known to be rather prone to racemisation.In 2013 (ref. 42), we reported a very efficient and stereoselective diversity-oriented route towards α-oxo-γ-acylaminoamides 74 and α-hydroxy-γ-acylaminoamides 73, taking advantage of the asymmetric organocatalytic synthesis of Boc protected β-aminoaldehydes 71 developed by List.43 Towards that goal, we prepared a series of aldehydes 71 from carbamoylsulphones 70 through a proline catalysed Mannich reaction (Scheme 16). These aldehydes are stereochemically labile and for this reason, in previous studies, they were promptly reduced or oxidized. Nevertheless, we found out that the mildness of Passerini conditions allowed the obtainment of products 72 with no epimerization at all. Deprotection–acyl migration to give α-hydroxyamides 73 takes place in high yields. Compared to the standard PADAM, treatment with Et3N for a longer time is necessary to promote the acyl migration. Finally, oxidation gave α-ketoamides 74. Thanks to the efficiency of the various steps, the overall sequence from 70 to 74 can be carried out without any intermediate purification, in good overall yields (typically >50% with a few exceptions).
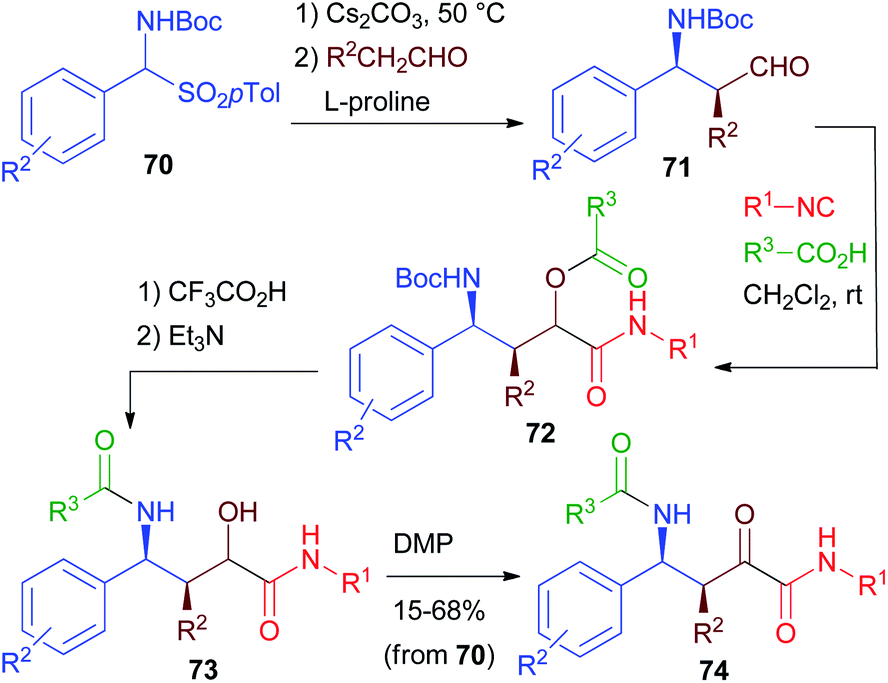 | ||
| Scheme 16 The homo-PADAM protocol. | ||
Unfortunately, as expected, the diastereoselection of the Passerini reaction was poor, giving compounds 73 as 1:1 diastereomeric mixtures. The back reduction of 74 to 73, however, can be highly diastereoselective, allowing the obtainment of 73 with high (>9:1) d.r. In some cases, both stereoisomers could be selectively obtained by simply changing the reducing agent.
For a bis-homo-PADAM approach, protected γ-aminoaldehydes are needed. Instead of the usual Boc or Fmoc protections, in this case we chose to use an azide, as amine synthetic equivalent. Once again, the main problem is the obtainment of γ-azidoaldehydes in an enantioselective form, but we solved it by applying a biocatalytic approach.44 Monoacetate 75 (Scheme 17) was obtained by the desymmetrization of the corresponding meso diol and was then converted, in a few high yielding steps, into azidoaldehyde 76. After the Passerini reaction to give 77, treatment with PPh3 brought about azide reduction and acyl transfer, to afford α-hydroxy-δ-acylaminoamide 78, demonstrating the feasibility of the method. On this product, we were able to carry out a cyclization through SN2 substitution to give pyrrolidine 79. In this case, no additional functional group was needed. Interestingly, cyclization was found to be stereoconvergent, probably because of the in situ epimerization of the stereogenic centre generated during the Passerini reaction. As a result, a 1:1 mixture of 78 was nearly completely converted into a single diastereomer of pyrrolidine 79. An application of this strategy to the total synthesis of an important active pharmaceutical ingredient (API) will be described in chapter 5.
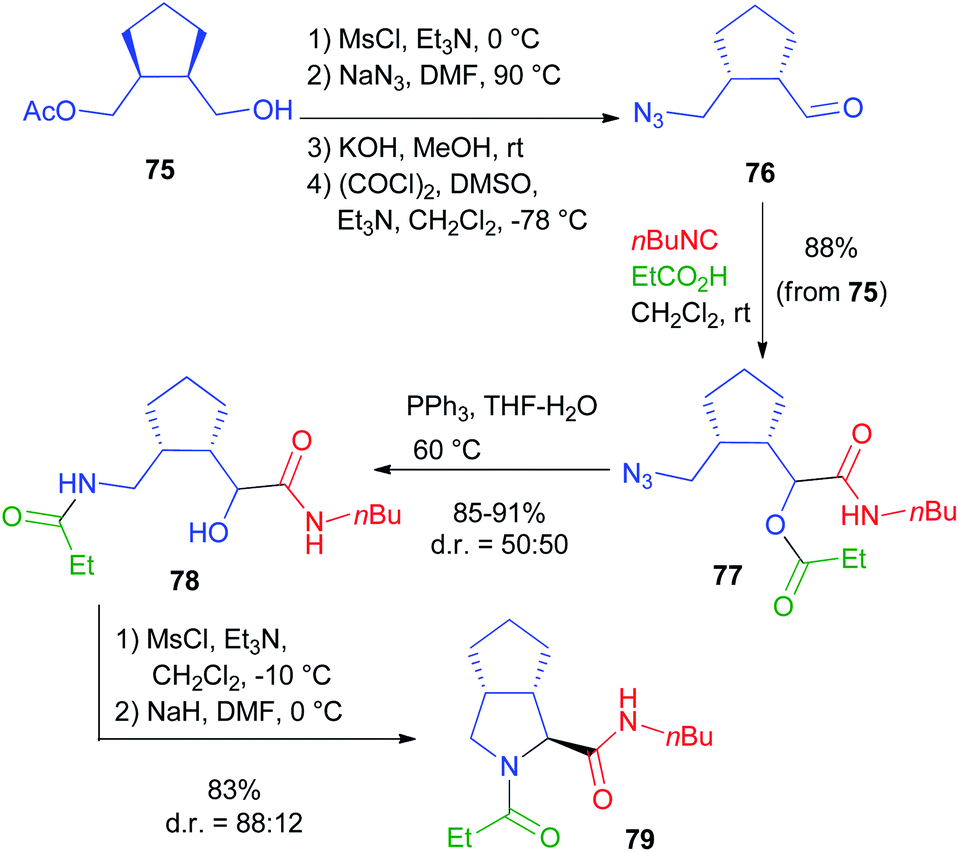 | ||
| Scheme 17 The bis-homo-PADAM protocol. | ||
2.4 Future prospects
While a huge variety of heterocyclic systems have been prepared through Ugi reactions followed by post-MCR cyclizations, a similar strategy was less frequently utilized for the Passerini reaction. There are indeed limitations due to the ester nature of the Passerini products and the fewer chances of including additional functional groups (Passerini is a 3-CR, while Ugi is a 4-CR). The PADAM protocol and its daughters (homo-PADAM and bis-homo-PADAM) offer a way to overcome these restraints. We think that coupling these strategies with subsequent cyclizations may offer many possibilities of diversity-oriented heterocyclic synthesis. Nevertheless, these applications are still rather infrequent and thus there is ample room for future achievements. We have already demonstrated that the problem of diastereoselectivity can be solved by the oxidation–reduction of the secondary alcohol derived from the PADAM protocol, and thus this fact can facilitate the stereocontrolled synthesis of chiral non-aromatic heterocycles.3. New opportunities for the Passerini reaction by the replacement of one or two components
The Single Reactant Replacement (SRR) approach was first coined by Ganem and has been extensively used to improve known MCRs and design new synthetic routes.45 The first SRR approach of the Passerini reaction was probably the Ugi reaction, and in the last two decades the scope of the Passerini reaction has experienced further expansion by replacing one or two components, leading to new interesting pathways and different final products. These new products can undergo in some cases post-Passerini transformations, further enlarging the accessible chemical diversity. The isocyanide component usually cannot be changed, although recently Cui reported the Passerini reaction employing tosylynamides 80 in place of isonitriles,46 affording β-acyloxyamides 81 with broad scope and functional group tolerance (Scheme 18). | ||
| Scheme 18 Passerini reaction with tosylynamides. | ||
Most studies, however, have focussed on the substitution of the carboxylic acid or, less often, of the carbonyl component. The subject has already been reviewed by us,3 while some recent examples will be reported in this chapter.
3.1 The ketene 3-component Passerini reaction
In 2008, Danishefsky reported on the two component reaction of isocyanides with carboxylic acids to yield formylamides 82.47,48In the same period, we made a serendipitous discovery, finding that, when an arylacetic acid was employed, a new three component variant of the Passerini reaction took place instead, involving two equivalents of the acid and leading to interesting captodative olefins 83 (Scheme 19).6 By studying the mechanism, we understood that the aryl group indeed played a crucial role. The initial α-adduct between an arylacetic acid and isocyanide, in fact, being an activated ester, preferentially underwent a nucleophilic acylic substitution (SNAc) with an additional molecule of isocyanide, leading to 84. The O → N acyl migration, leading to 82 and reported by Danishefsky, was not observed in this case.
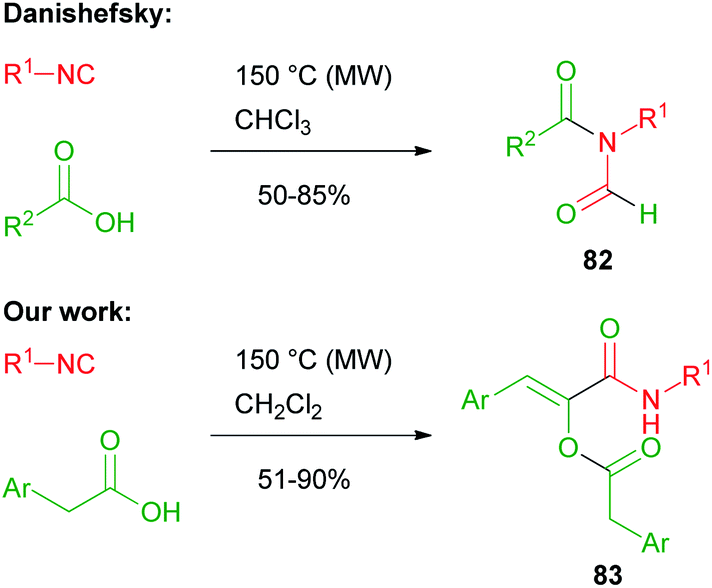 | ||
| Scheme 19 Different outcomes of the reaction between isocyanides and carboxylic acids. | ||
The formation of adduct 84 was favoured by the stabilization ensured by the aromatic ring, able to generate an extended conjugation in the enolic form. The addition of a second molecule of arylacetic acid onto the nitrilium functionality of 84, followed by Mumm's rearrangement of the resulting adduct, afforded captodative olefin 83. Adduct 84 can be considered as formally derived from the addition of an isocyanide to a protonated arylketene, which replaces the protonated carbonyl compound, while the subsequent steps occur similarly to a normal Passerini reaction (Scheme 20).
 | ||
| Scheme 20 Postulated mechanism for the reaction between isocyanides and arylacetic acids. | ||
This serendipitous finding prompted us to investigate if the reaction between ketenes, isocyanides and carboxylic acids could be generalized in a three component reaction involving three real diversity inputs. Due to the intrinsic instability of most ketenes, we devised the possibility to generate them in situ from diazoketones 85. The photoinduced ketene 3-component reaction (K-3CR) was therefore developed, taking advantage of the light-induced Wolff rearrangement of diazoketones, while thermal and silver-mediated procedures were unsuccessful (Scheme 21).49 This was one of the first examples of photoinduced multicomponent reactions50 and has been recently included in the ReactionFlash® App for named reactions.51
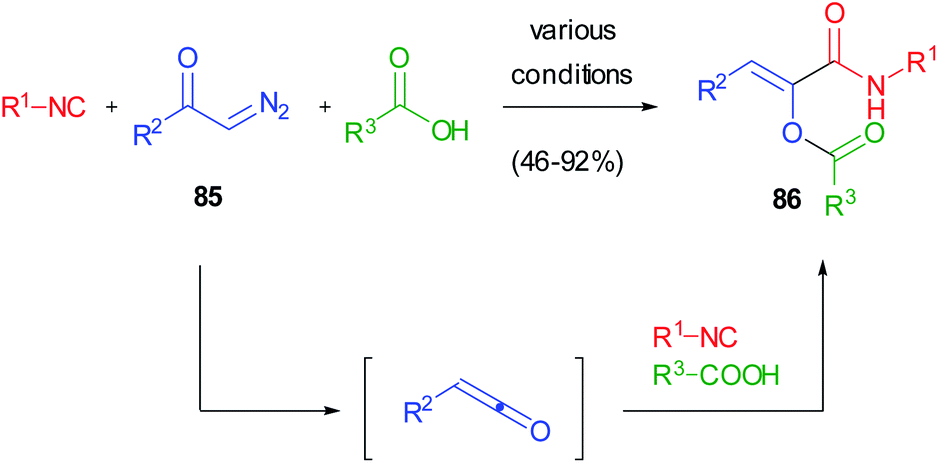 | ||
| Scheme 21 The ketene 3-component reaction (K-3CR). | ||
The reaction had a very broad scope, as both aromatic and aliphatic diazoketones could be used indifferently, together with any acid and isocyanide. However, it was poorly selective regarding the stereochemistry of the double bond, since E/Z mixtures of the captodative olefins were often isolated. This was a consequence of the UV-mediated isomerization of Z olefines 86, the sole products of the K-3CR, under the reaction conditions. This problem was solved in different ways: (i) by using, as additives, triplet quenchers (piperylene and trans-stilbene) able to inhibit the alkene isomerization but not the Wolff rearrangement;52 (ii) by performing the reaction under continuous flow conditions, in order to remove the olefin from the irradiation area immediately after its formation;53 (iii) very recently, by using visible light to induce Wolff rearrangement: although diazoketones usually display an absorption maximum around 250–300 nm, we have demonstrated that the molar extinction coefficient at 450 nm is not neglectable, and reactions performed under blue LED irradiation show very high quantum yields and better selectivities.54 The captodative olefins originating from this approach have been converted into pyrrolidine-2,5-diones or pyrrolones in a divergent manner, depending on the basic conditions employed.55
3.2 The modification of the ketene–Passerini reaction using silanols
The silylative Passerini reaction, a modified version where a silanol replaces a carboxylic acid, has been reported by Soeta and Inomata in 2010.56 Silanols can effectively act as surrogates of carboxylic acids because they display both a nucleophilic site, necessary to attack the nitrilium ion, and an electrophilic site, essential for the final Mumm's rearrangement. Due to the lower reactivity of silanols compared to carboxylic acids, the reaction had to be performed with excess reagents, at high temperatures and for prolonged times, affording α-silyloxy amides 87 in moderate to good yields. Triphenylsilanol performed better than other derivatives, due to the EWG character of the aromatic rings (Scheme 22).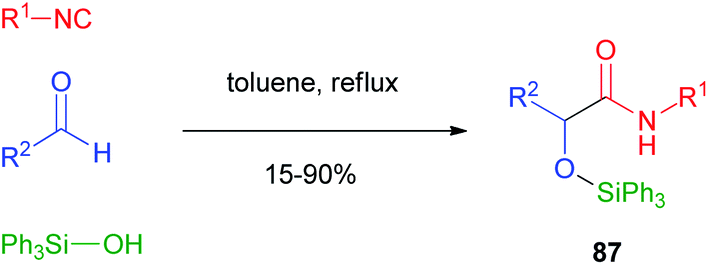 | ||
| Scheme 22 Silylative Passerini reaction. | ||
Also, the K-3CR could be effectively performed in the presence of silanols replacing the carboxylic acids, but in this case there was no need for excess reagents or higher temperatures. The same solutions used for the K-3CR also made it possible to stereoselectively obtain Z-silyloxy acrylamides 88 in this case. The scope of the reaction was quite broad, and also in this case triphenyl silanol performed better than others (Scheme 23).57
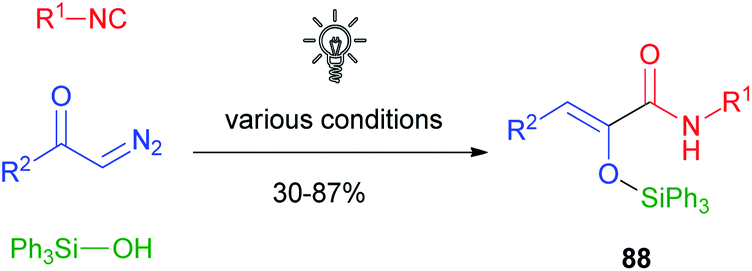 | ||
| Scheme 23 Silylative ketene 3-component reaction (SK-3CR). | ||
Compared to α-acyloxyacrylamides 86, silyl enolethers 88 displayed a lower stability, especially during chromatographic purification: the disadvantage in terms of isolated yields turned into an added value in terms of reactivity, as these compounds could be efficiently subjected to the Mannich reaction, Saegusa oxidation, decarboxylative cyclization with α-amino acids, photoredox acetonylation58 and interestingly to a tandem Michael/aldol dimerization reaction able to afford natural product-like compounds 89 in a very straightforward manner.59,60 Six new bonds and three stereogenic centres, including two quaternary carbons, could be formed stereoselectively in just two steps, starting from diazoketones, silanols and isocyanides (Scheme 24).
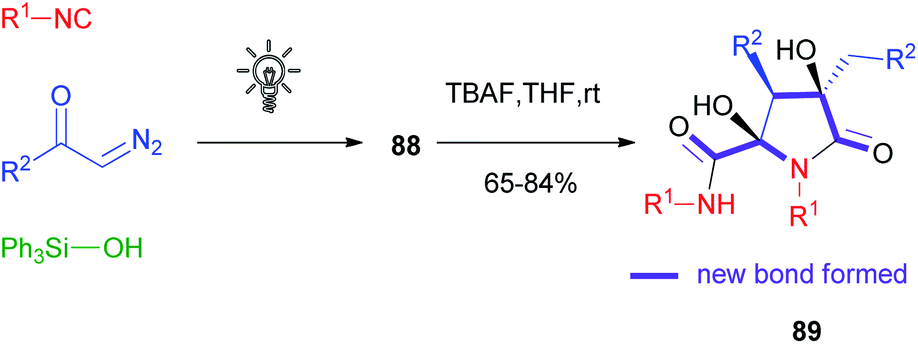 | ||
| Scheme 24 Two-step synthesis of the common skeleton of natural products eusynstyelamides and anchinopeptolides. | ||
3.3 The Passerini reaction using alcohols
According to what has been discussed in the previous paragraph, alcohols cannot replace carboxylic acids in a Passerini reaction, generating α-alkoxy amides, as they lack an electrophilic site to allow the Mumm rearrangement. However, when electron poor phenols 90 are used, a Smiles rearrangement through intermediate 91 can occur instead, and α-aryloxy amides 92 can be obtained, as reported in 2006 by El Kaïm and Grimaud.61 Yields, however, were lower than those of the parent Ugi–Smiles reaction, as aldehydes display a reduced electrophilicity compared to imines. About ten years later, the same authors62 reported improved conditions for the Passerini–Smiles reaction, with the use of a tertiary nucleophilic amine as an additive together with solvent free conditions (Scheme 25).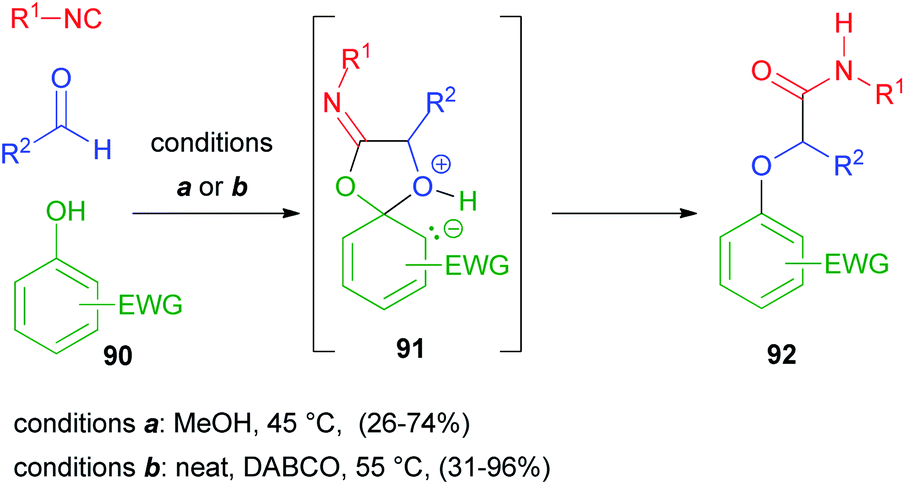 | ||
| Scheme 25 Passerini–Smiles reaction with activated phenols. | ||
The enol-Passerini reaction recently reported by Neo and Marcos,63 employing enolic pirrolidin-2,5-diones as substrates 93, is based on the same Smiles rearrangement. Through the formation of intermediate 94, it affords enolamides 95; however, if a polar protic solvent is used, a competitive enol-Ugi reaction is observed, due to the solvent promoted imine formation between the isocyanide and the aldehyde (Scheme 26).
 | ||
| Scheme 26 Enol–Passerini reaction. | ||
Attempts to achieve a general alkylative Passerini reaction to products 96 were made by Taguchi.64 The reaction was performed with the alcohol used as the solvent in the presence of Lewis acids such as lanthanoid(III) triflates. The method, however, was limited in scope as only aromatic and unsaturated aldehydes could be efficiently used, and the alcohol was also the solvent of the reaction. Therefore, its choice was limited to simple, low-boiling point compounds (Scheme 27).
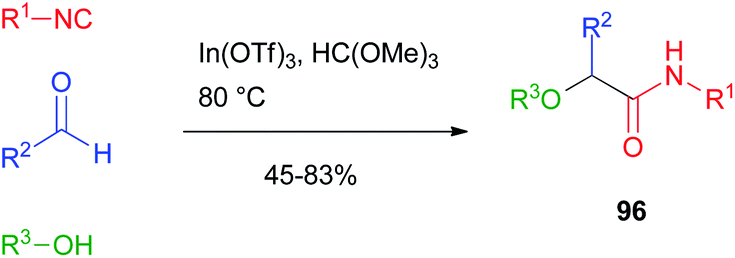 | ||
| Scheme 27 Alkylative Passerini reaction. | ||
This limitation proved insurmountable when a few years ago we needed to straightforwardly synthesise α-propargyloxy amides of general structure 40 (cf.Scheme 9).29 The solution was found applying a four-step one-pot reaction involving an Oxidative Passerini reaction, a Hydrolysis, and an Alkylation (OPHA).65 Starting from o-azido benzyl alcohols and using sacrificial acetic acid, all steps could be performed without isolation of the intermediates. This strategy paralleled a real multicomponent reaction in terms of operational simplicity and atom economy (Scheme 28). In Section 2.1, a Huisgen cyclization of compounds 40 has been described.
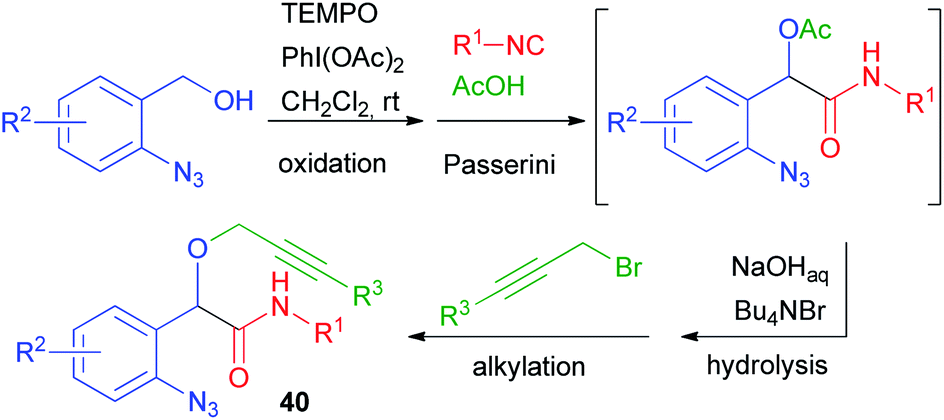 | ||
| Scheme 28 OPHA strategy as alternative to an alkylative Passerini reaction. | ||
In 2018, Ruijter66 reported the Passerini reaction with hexafluoroisopropanol as an acid surrogate: in this case, the intermediate imidate 97, unable to induce the Mumm rearrangement and to proceed to an alkylated Passerini adduct, was sufficiently stable and could be reduced under mild conditions to β-aminoalcohol 98 (Scheme 29).
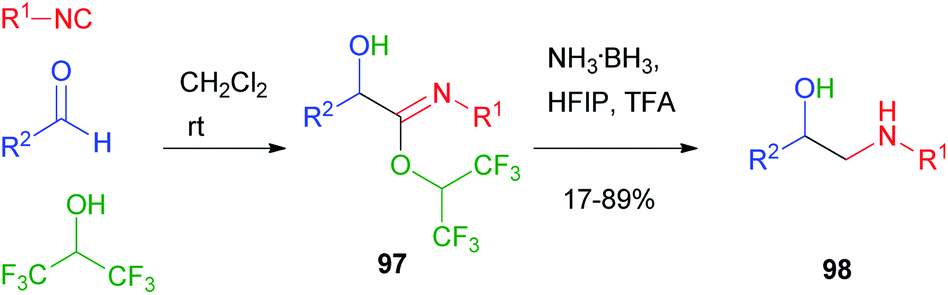 | ||
| Scheme 29 Passerini reaction with hexafluoroisopropanol (HFIP). | ||
3.4 Other recent Passerini variants using different components
Cramail and Meier67 have reported the Passerini four-component reaction between aldehydes, isocyanides, alcohols and carbon dioxide. The reaction proceeds through the formation of a carbonate ion, initiated by a base catalyst, and its attack on the nitrilium ion, followed by Mumm's rearrangement of intermediate 99 and formation of an α-alkoxycarbonyl amide 100 (Scheme 30). Final products were isolated in moderate yields and carbonate hydrolysis was observed, together with the formation of classic Passerini adducts, derived from the oxidation of the aldehyde to carboxylic acid.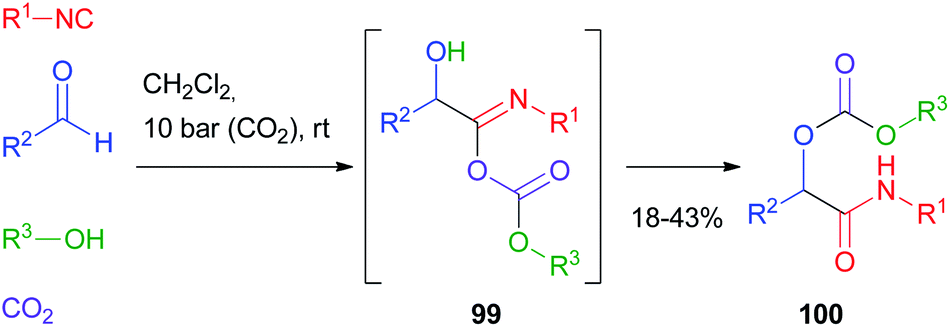 | ||
| Scheme 30 Passerini reaction with carbon dioxide. | ||
The replacement of the carboxylic acid with hydrazoic acid was first reported by Ugi in 1961,68 but only in 2002 Nixey and Hulme reported the use of less toxic/explosive and commercially available TMS-N3 (ref. 69) as the azide source. During the last few years, the methodology has been used independently by Ding70 and Xiong71 to assemble intermediates 101 for the synthesis of 4H-3,1-benzoxazine derivatives 102 (Scheme 31).
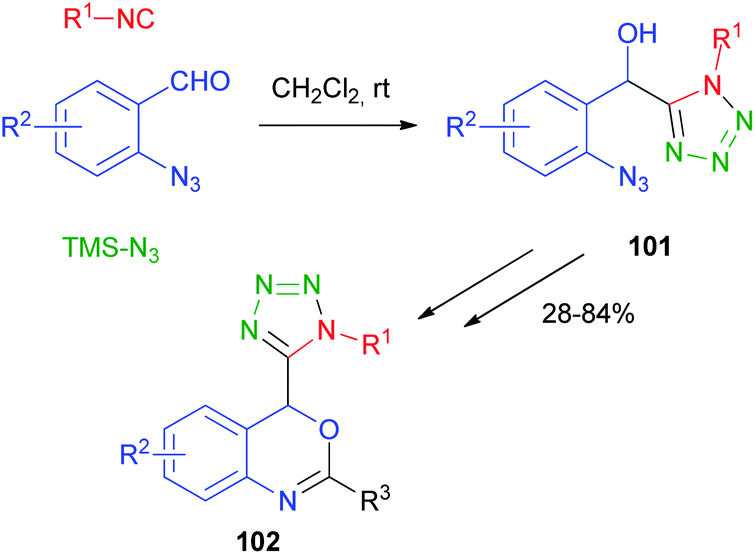 | ||
| Scheme 31 Passerini-azide reaction for the synthesis of tetrazole derivatives. | ||
A new MCR using arynes, carboxylic derivatives and isocyanides has been reported by Stoltz (Scheme 32).72 In this reaction, the aryne, generated in situ from o-silyl aryltriflate 103, acts as a carbonyl derivative and is subjected to nucleophilic attack by the isocyanide. The resulting zwitterion 104, in the original design, should have been intercepted by a carboxylate ion, following the typical Passerini mechanism, affording the α-adduct and finally 105 upon the migration of the acyl group onto the carbanionic site. However, the desired product was never isolated, as 104 was not reactive towards carboxylate ions. In contrast, when a carboxylate ester was employed instead, an alternative mechanism occurred, in which the carboxylate was first attacked by the carbanionic site, and then the resulting alkoxide 106 intramolecularly attacked the nitrilium ion, affording the final iminoisobenzofuran derivative 107. Compounds 105 could be eventually obtained by acid hydrolysis of 107.
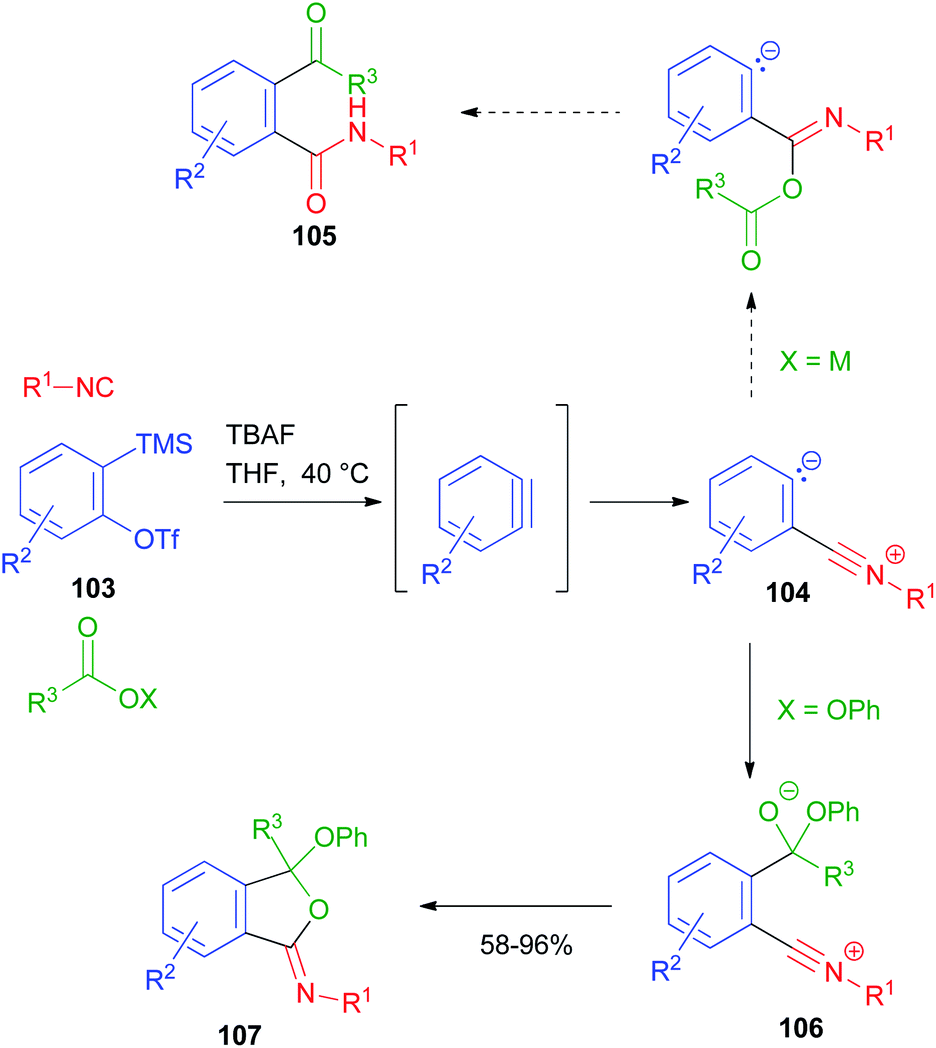 | ||
| Scheme 32 Three-component reaction between arynes, carboxylate esters and isocyanides. | ||
3.5 Future prospects
The substitution of one or two components has attracted high interest over the past two decades, since it has allowed overcoming the inherent limitations of the Passerini reaction. Carboxylic acids have been replaced by various species, ranging from phenols to azides to silanols and alcohols. Surprisingly, no reports about boron derivatives have been found in the recent literature. Boronic acids, in fact, possess both a nucleophilic hydroxyl group to attack the nitrilium ion and an electrophilic boron atom capable of enabling the Mumm rearrangement. Hopefully, the borono-Passerini reaction will be soon disclosed. If many carboxylic acid surrogates are available, the same does not apply for the electrophilic component, for which the only viable alternatives to aldehydes and ketones are, so far, only ketenes and arynes. Ketenes, in particular, have shown high versatility and could be in principle employed in other Passerini-like reactions, apart from the K-3CR and the SK-3CR. On the other hand, it would be interesting to see whether other electrophilic species, not necessarily carbon-based, could operate by a similar mechanism. Emerging fields such as photocatalysis and electrocatalysis could be used to generate in situ such species and widen the range of possible reactions.4. The still unresolved problem of stereoselectivity in the Passerini reaction
The Passerini reaction always generates a new stereogenic centre when an unsymmetrical carbonyl compound is employed. Therefore, stereoselectivity, either enantio- or diastereoselectivity, represents a major challenge. As it will be illustrated in this chapter, this issue is still in part unresolved.4.1 The state of the art in the enantioselective catalytic Passerini reaction
Although the catalytic enantioselective addition of nucleophiles to CO and CN double bonds is a well-established procedure, the extension of this protocol to the Passerini (and Ugi) reaction, where the isocyanide is the nucleophile, is an extremely challenging task.73 There are several reasons that hampered the discovery of very efficient enantioselective approaches such as for example: (a) the competition of the not enantioselective MCR reaction, as the Passerini reaction is a spontaneous process and the turnover of the catalyst is usually not efficient; (b) the fact that a Lewis acid, the most logical activating moiety for the aldehyde component, can also interact with other basic sites, first of all with the isocyanide.
The first enantioselective approach to the Passerini reaction was reported by Denmark in 2003 (Scheme 33),74 where an efficient enantioselective organocatalytic “truncated” two component Passerini reaction was described. The employment of silicon tetrachloride, a weak Lewis acid, instead of the carboxylic acid, enables interaction with the chiral bisphosphoroamide 110, resulting in the formation of a silicon cation which activates the aldehyde before the selective nucleophilic attack of the isocyanide on one of the enantiotopic faces of the carbonyl.
 | ||
| Scheme 33 The enantioselective “truncated” Passerini reaction. | ||
The intermediate nitrilium ion is trapped by the only available nucleophile, namely chloride, to afford imidoyl chloride 108, which decomposes under mild basic conditions giving α-hydroxyamide 109.
In the same year, in a pioneering study, Dömling reported the first enantioselective not truncated Passerini reaction where a screening of chiral catalysts allowed identifying Ti(IV)–TADDOL complex 112 as the best performing one. However, 111 was obtained only with a moderate e.e. and, due to the low turnover number, an equimolar amount of 112 was required (Scheme 34).75
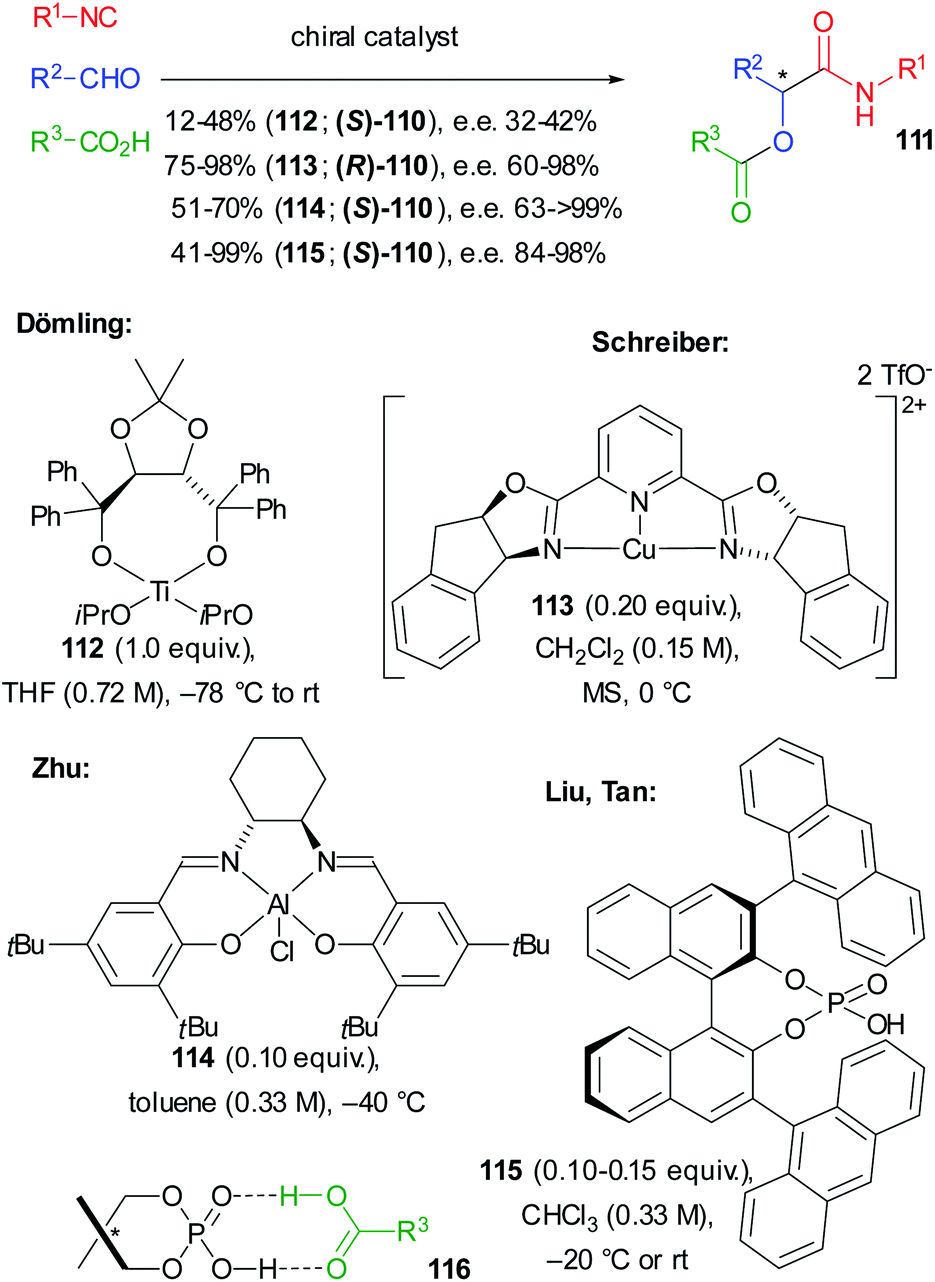 | ||
| Scheme 34 The enantioselective Passerini reaction. | ||
Shortly after, an efficient ligand accelerated approach, using 0.20 equivalents of tridentate Cu(II)–PyBOX complex 113, which usually allows high enantioselectivity, was reported by Schreiber, in particular using chloroacetic acid. High enantioselectivities were obtained, provided that dry conditions were used and that chelating aldehydes were employed. Water, in fact, could deactivate the catalyst, making the underground reaction competitive.76
Later Zhu employed (salen)Al(III)Cl complex 114 which allowed the transformation of a variety of nonchelating aldehydes into 111 with good to excellent enantiomeric excess, the best results being achieved with the less reactive aromatic isocyanides.73,77
The first organocatalytic enantioselective Passerini reaction was described by Liu and Tan, who obtained excellent results using chiral phosphoric acid 115 (CPA), which represents the best protocol available to date. With a few exceptions, the yields are high and the e.e.s are almost always >90%. The authors justified the good results as a consequence of the formation of heterodimer 116, responsible for the double activation of 115 (more acidic CPA) and of the carboxylic acid (enhanced nucleophilicity).78
As a matter of fact, the enantioselective Passerini reaction was made possible taking advantage of previous studies on the development of the enantioselective approach to 5-aminooxazoles 119 through a 2-component reaction involving a particular class of isocyanides, namely α-isocyanoacetamides 117 and aldehydes. In this reaction, with the carboxylate missing, the intermediate nitrilium ion 118 is intramolecularly trapped by the amide oxygen (Scheme 35). Good to excellent results were achieved using a Sn(II)–PyBOX complex,79 catalyst 114,80 and CPAs either as ligands for Al,81 or as Brønsted acid organocatalysts.82 In this regard the most efficient ligand, developed by Matsunaka and Shibasaki, is 120 which was transformed into several heterobimetallic Schiff base complexes such as, for example, 121.83 Despite the complex synthesis of the chiral catalyst, the results are excellent, with e.e.s in the range 95–98%, being lower (88%) only in one case, where 3-phenylpropanal was used as the aldehyde. The concurrent presence of two Lewis acid species allows most likely the concomitant activation of the aldehyde and the coordination of the amide oxygen, bringing the two reacting molecules close together with a controlled orientation during the enantiodiscriminating step (see 122). The very efficient catalyst 120 has a very peculiar structure, which enables only the coordination of α-isocyanoamides, making the application to the classic Passerini reaction unlikely, as these isocyanides gave 5-aminooxazoles even in the presence of a carboxylic acid.
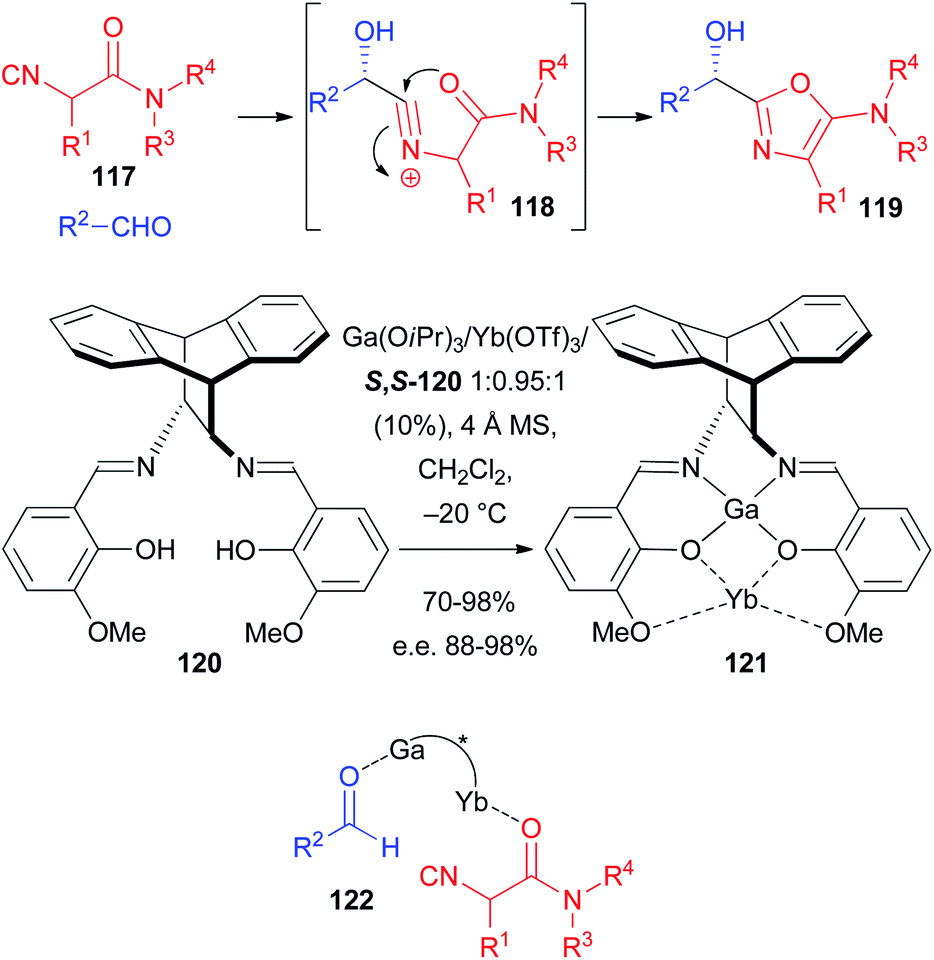 | ||
| Scheme 35 The enantioselective synthesis of 5-aminooxazoles. | ||
Finally, another approach to 5-membered nitrogen heterocycles based on the reactivity of isocyanide 117 was followed by Wang and Zhu to synthesize 5-(1-hydroxyalkyl)tetrazoles in moderate to good enantiomeric excess. This time a three component Passerini-like reaction involved an aldehyde and isocyanide 117, while the third component, which replaces the carboxylic acid, was hydrazoic acid. Once again a (salen)Al(III) complex, similar to 114 (Me instead of Cl), was used as the catalyst.84
4.2 Diastereoselective Passerini reactions
Although the diastereoselectivity control seems in principle much easier, this challenge is still far from being solved.85,86 Chiral isocyanides are usually unable to control the diastereoselectivity, one of the very few exceptions being a camphor-derived one described by Ugi with a single example.87 The main reason is that they have a linear, not sterically demanding, structure. The main chance to control the stereoselectivity relies on the use of chiral carbonyl compounds, which have been used both in intramolecular and, most of all, intermolecular processes. Alternatively, chiral carboxylic acids have also been employed, although less frequently.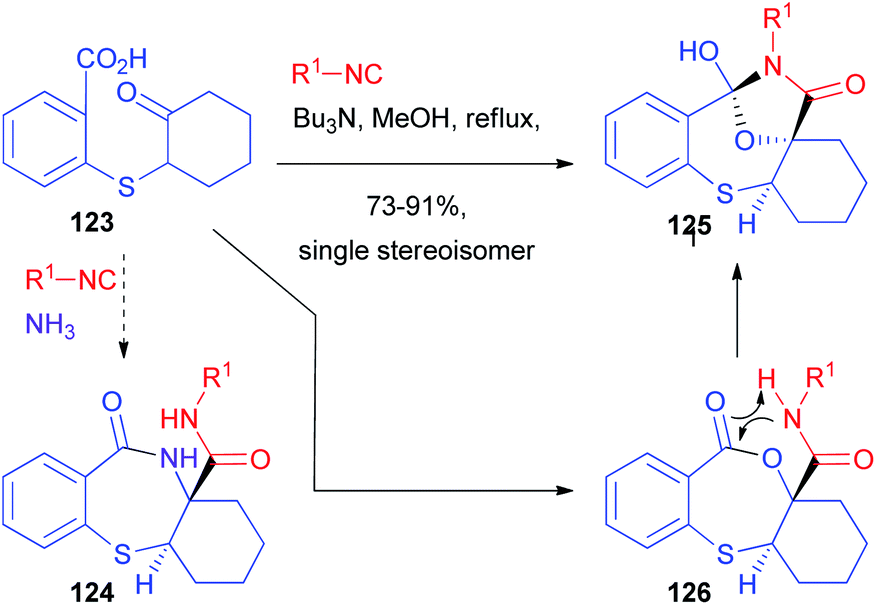 | ||
| Scheme 36 The diastereoselective synthesis of orthoamides. | ||
Another report, using racemic as well as enantioenriched ketoacids, was more recently published by Ruijter and Orru, who obtained the functionalized bicyclic scaffold 129 by the Passerini-3-centre-2-component reaction (Scheme 37).89 Starting from γ-ketoacid 127, bicyclic lactone 128 was obtained with moderate stereoselectivity (85:15, R1 = tBu) using CH2Cl2 as solvent. The d.r. could be slightly enhanced performing the reaction in dimethyl carbonate in the presence of catalytic zinc triflate.
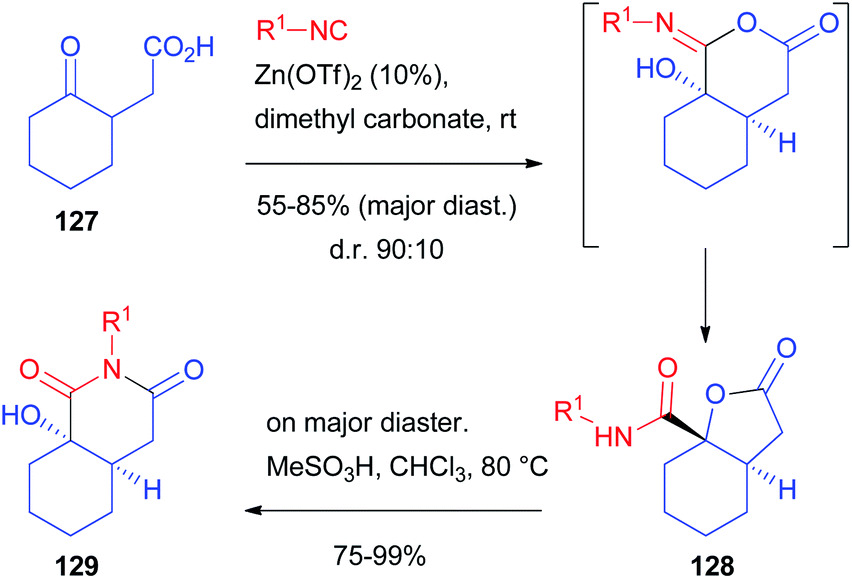 | ||
| Scheme 37 The diastereoselective synthesis of bicyclic derivatives through the Passerini-3 centre-2-component reaction. | ||
The prevailing formation of trans-fused isomer 128 is the result of the preferred axial attack of the not encumbered isocyanide to afford the cis-fused α-adduct, which, upon Mumm rearrangement, affords trans-128. The reaction works well regardless of the employed isocyanide, while the protocol is less tolerant with respect to other cyclic ketones. On the other hand, the diastereomeric ratio is always around 9:1. Moreover, by the treatment of the major diastereoisomer with a Brønsted acid, a complete rearrangement occurs, affording the less strained cis-hexahydroisoquinolinedione scaffold 129.
Among the first reports on the Passerini reaction with chiral aldehydes, 2,3-epoxyaldehydes 130, obtained from 132 by Sharpless epoxidation by Krishna and Lopinti90 as well as by an organocatalytic procedure by Corrêa and Paixão,91,92 have been used (Scheme 38).
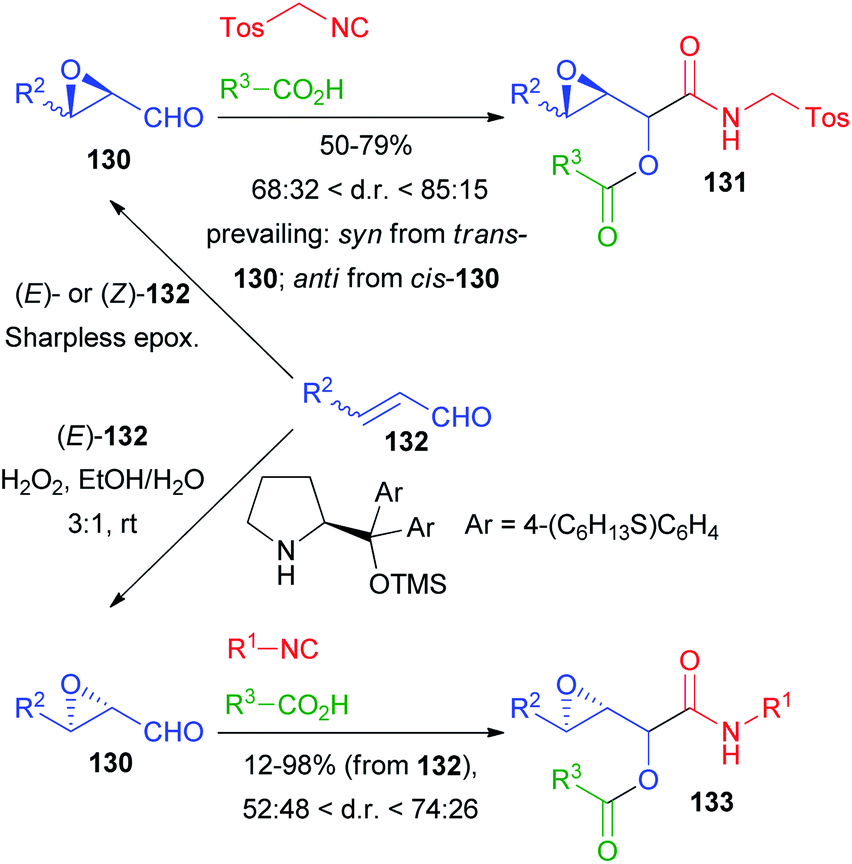 | ||
| Scheme 38 Diastereoselective Passerini reaction on 2,3-epoxy aldehydes. | ||
In the first case only TosMIC was used as an isocyanide and the d.r.s are moderate to good; the prevailing diastereoisomer is decided by the relative configuration of the starting 2,3-epoxyaldehyde. Interestingly, the Lewis acid-mediated reaction did not afford 131, most likely due to the instability of 130 under the reaction conditions. In the second approach, a one pot procedure for the direct epoxidation, followed by the addition of Passerini reagents, afforded 133 with poor to moderate stereoselectivity, and the relative configuration of compounds 133 was not established.
More recently, Jerić used sugar-derived aldehydes for the synthesis of very complex glycomimetics.93 Initially fructose derived aldehyde 134 was involved in Passerini reactions with either achiral or chiral sugar isocyanides, and achiral or chiral acids (N-protected amino acids or sugar acids) (Scheme 39). However, the control is always exerted by the chiral aldehydes. The yields are moderate to very good, while the d.r.s are up to 94:6. The very popular Felkin–Anh model cannot be invoked to explain the stereoselectivity, because of the presence of two oxygens bound to the stereogenic α carbon of 134. For this reason, the authors carried out DFT calculations, following the mechanistic pathway proposed by Ramozzi and Morokuma, which involves the presence of two molecules of carboxylic acid in the step where the new stereogenic centre is created, which is the one affording the nitrilium ion.94
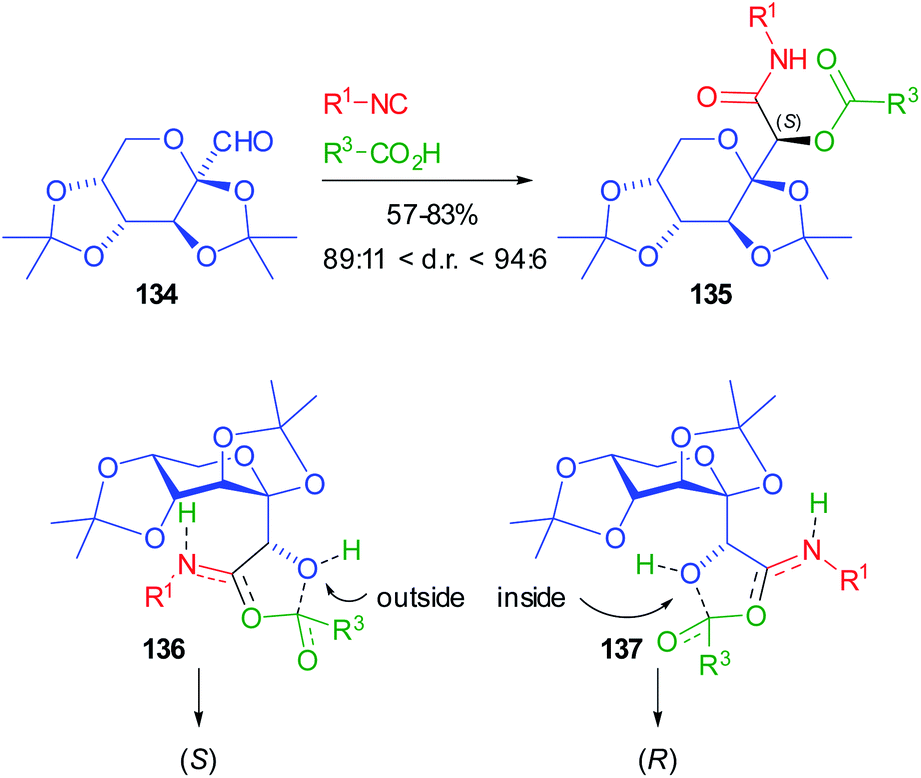 | ||
| Scheme 39 Diastereoselective Passerini reaction with sugar aldehydes. | ||
From these calculations, TS 136 with the aldehyde oxygen “outside” seems more stable than 137 with the same oxygen “inside”, which explains the highly preferred formation of the (S) configuration for 135. The same protocol was later applied to the reaction involving other sugar aldehydes (from sorbose, galactose, and allose), and sugar isocyanides and carboxylic acids: the robustness of the methodology was demonstrated and in some cases d.r.s are >99%, while the yields depend on the nature of the reagents.95
For more than 35 years, our group has developed biocatalytic methods for the synthesis of small enantioenriched chiral building blocks, through enzyme-catalysed desymmetrizations as well as kinetic resolutions, applying them in asymmetric synthesis and natural product synthesis.96–98
Moreover, since the exploitation of renewable biomass-based feedstocks has become a hot topic, we applied biocatalysis to the valorisation of natural meso-diols and the corresponding chiral aldehydes. The enzymatic desymmetrization of protected erythritol 138 allowed the preparation of the chiral building blocks 139 that were transformed into the Passerini products by a one-pot procedure (Scheme 40). After oxidation with TEMPO/PhI(OAc)2, only the isocyanide was added, because the carboxylic acid is the side product of the oxidation, which represents a nice example of waste recycling in a tandem reaction.99 The traditional Passerini reaction in dichloromethane without any additive turned out to be unexpectedly stereoselective, affording anti-140 in a 4:1 ratio, while the same reaction on the trans isomers of 139 was not stereoselective at all. In order to improve the stereoselectivity, we investigated the effect of different Lewis acids as additives, finding that catalytic ZnBr2 is able to improve the d.r. up to 96:4, depending on R2 and on the isocyanide. The reaction is reliable and different isocyanides can be efficiently incorporated.
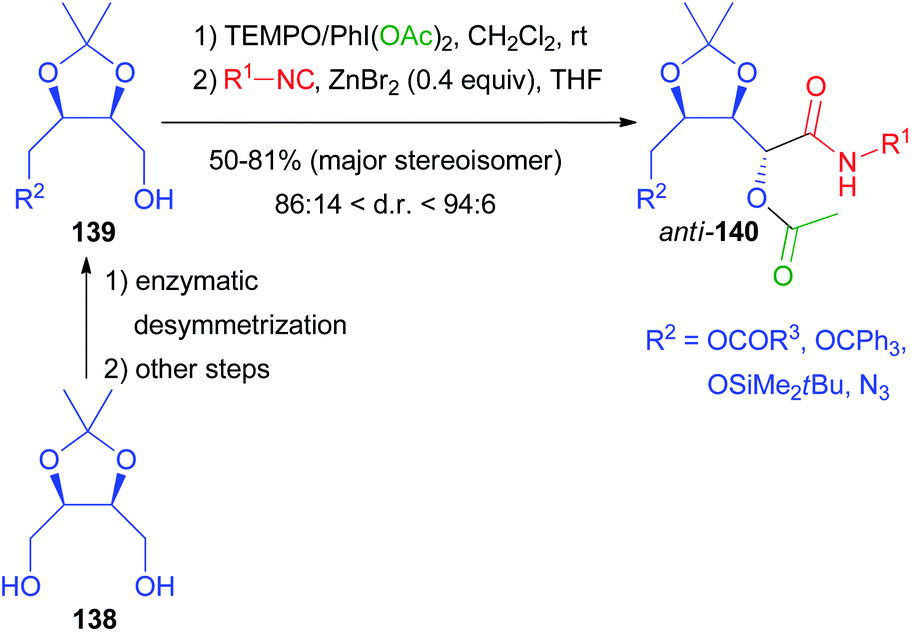 | ||
| Scheme 40 Diastereoselective Passerini reaction catalysed by ZnBr2. | ||
Compounds 140 were demonstrated to be pluripotent100 intermediates, as we were able to convert them into six O- or N-heterocyclic scaffolds (141–146) (Scheme 41).
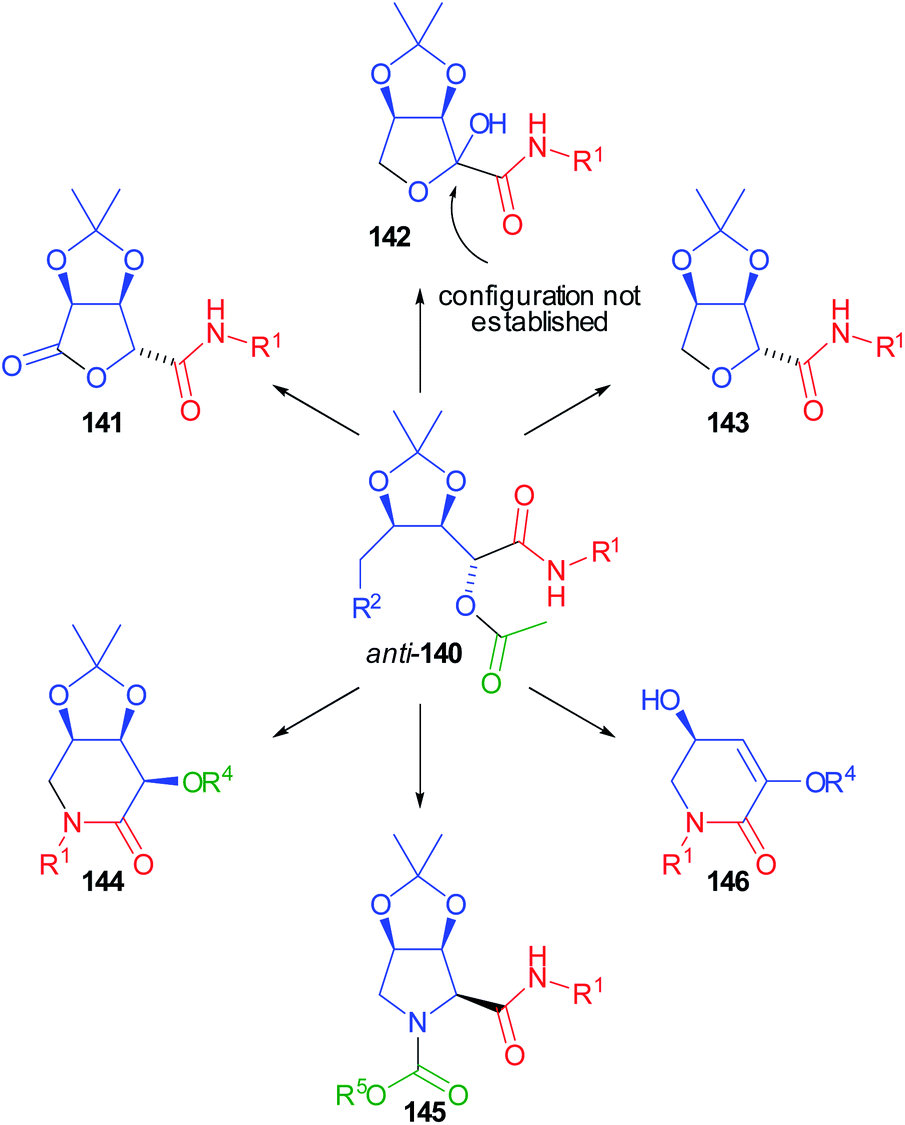 | ||
| Scheme 41 Transformation of 140 into different heterocycles (actually, the enantiomers of compounds 144–146 were obtained, as they were prepared from the enantiomer of 139, with R2 = N3 but, for sake of clarity, we reported here only one enantiomeric structure). | ||
Another application of erythritol-related chiral building blocks is summarized in Scheme 42. Monoacetate 147, resulting from the desymmetrization of 138, was homologated to α,β-unsaturated ester 148, with the aim of converting the acetoxymethyl moiety into the aldehyde.101 However, regardless of the reaction conditions (chemical or enzymatic), as soon as alcohol 149 was released, it immediately underwent a moderately stereoselective intramolecular Michael addition, affording 150 with the cis stereoisomer slightly prevailing. To obtain the chiral aldehyde needed for the Passerini reaction, we performed a protecting group exchange on 147 (tert-butyldimethylsilyl ether introduction, followed by acetate hydrolysis). The silyl ether was efficiently removed with pyridine·(HF)x after the introduction of the α,β-unsaturated ester moiety, avoiding the Michael cyclization completely. After oxidation we obtained 151.
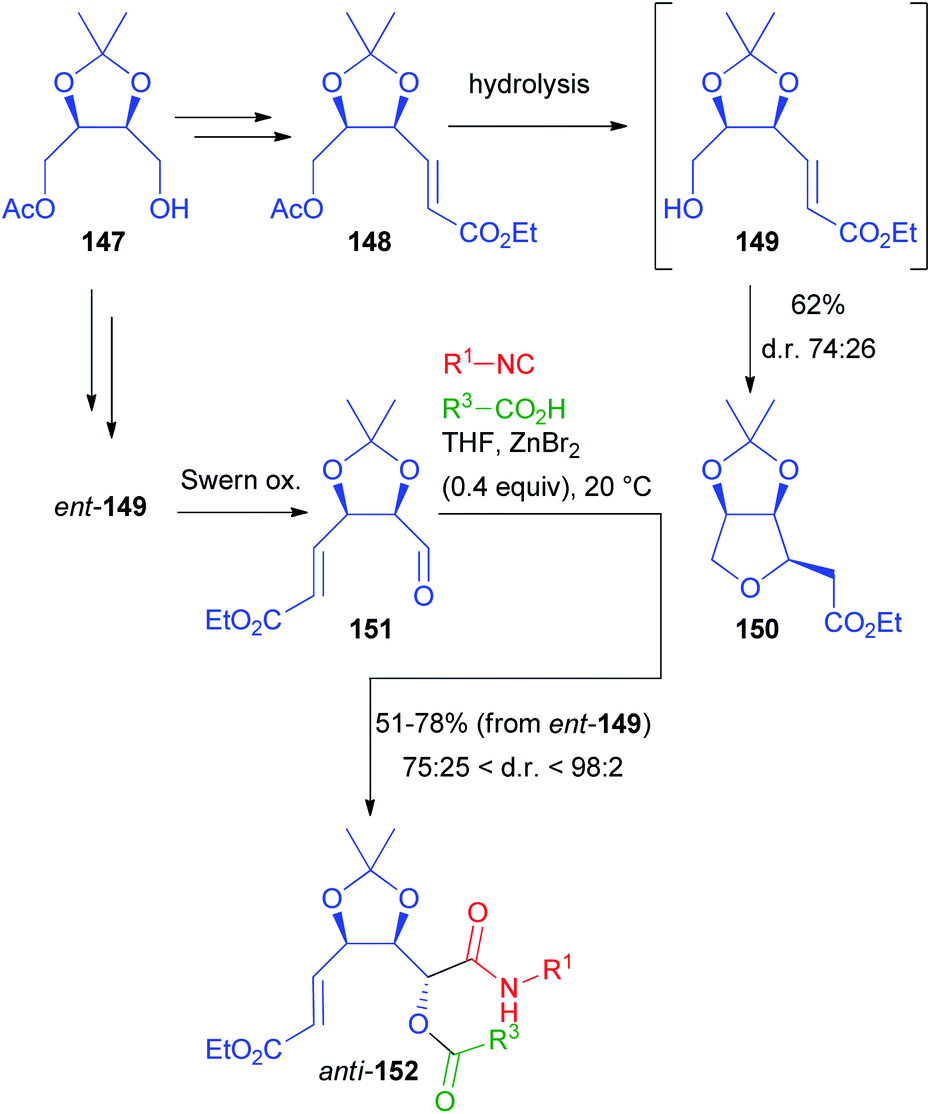 | ||
| Scheme 42 Another application of erythritol-derived monoacetate 147. | ||
Nevertheless, it is noteworthy that, while 147 can be prepared by enzymatic monoacylation of 138, also its enantiomer is easily accessible by the enzymatic monohydrolysis of the diacetate of 138, thus allowing access to both enantiomers of a given target.
As expected, the classic Passerini reaction was poorly diastereoselective (about 6:4). Applying the same one-pot protocol (oxidation/ZnBr2-catalysed Passerini) described for 139, a partial over-oxidation of aldehyde 151 was observed. Therefore, we preferred to switch to Swern oxidation, also because this allowed us to explore the carboxylic acid diversity. Under the catalysis of ZnBr2, we were able to obtain 152 in a moderate to excellent diastereomeric ratio (in most cases >4:1 and up to 98:2), with the anti-stereoisomer always prevailing. Again, a variety of structurally different isocyanides and carboxylic acids could be successfully employed.
Both diastereoisomers of 152, with R3 either Me or Ph, were separately hydrolysed and the corresponding secondary alcohols underwent spontaneous intramolecular Michael addition to afford preferentially tetrahydrofurans 153 (from anti-152) and 155 (from syn-152), highly prevailing over 154 and 156 respectively (Scheme 43). Moreover, submitting 153 to acidic conditions the densely functionalized tetrahydro[3,2-b]furan-5-one 157 was isolated, which is an analogue of naturally occurring (+)-goniofufurone.101
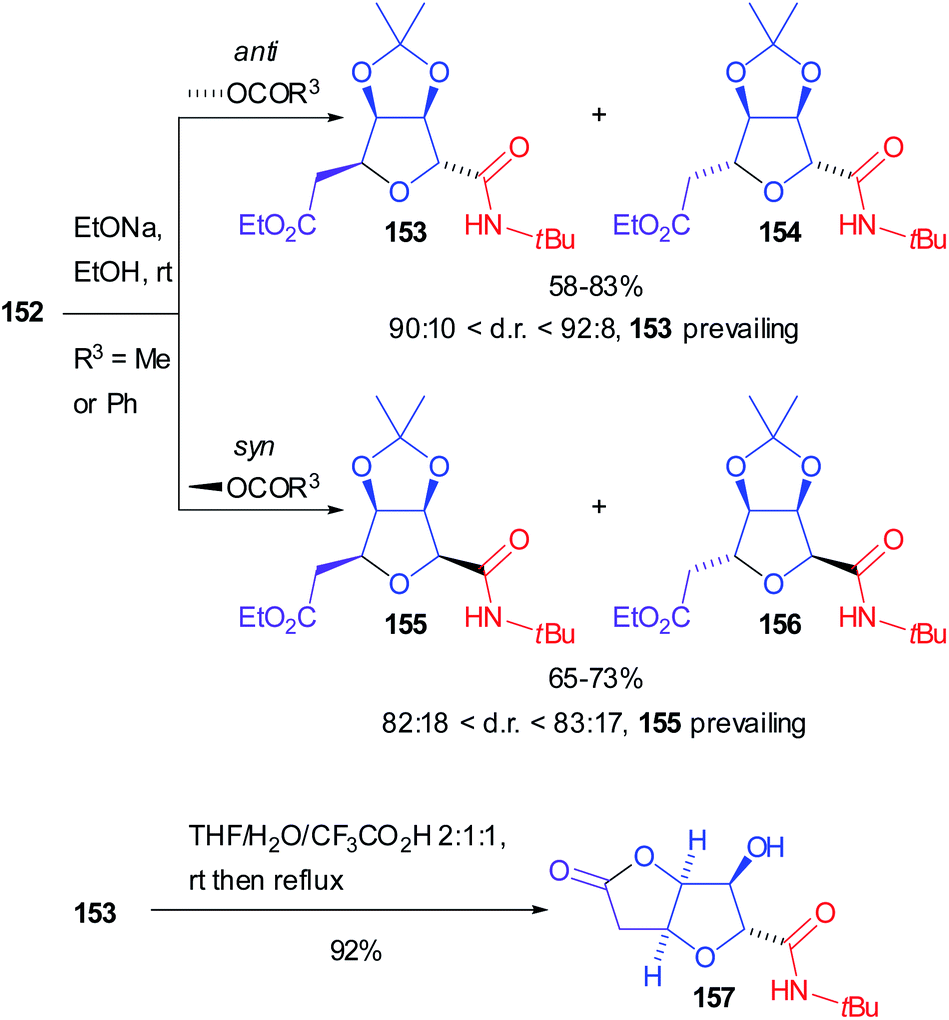 | ||
| Scheme 43 Further synthetic elaboration of 152. | ||
More recently, a stereoselective Passerini reaction has been applied on a more functionalized aldehyde, obtained from protected erythritol 138.102 Chiral intermediate 158 was stereoselectively homologated and the other hydroxymethyl group was oxidized, after deprotection, to the key aldehyde 159 (Scheme 44), which underwent a Passerini reaction with enantiopure isocyanide 160 in a double asymmetric induction reaction. The reaction afforded 161 with a moderate preference for the anti-stereoisomer, after the hydrolysis of the acetate.
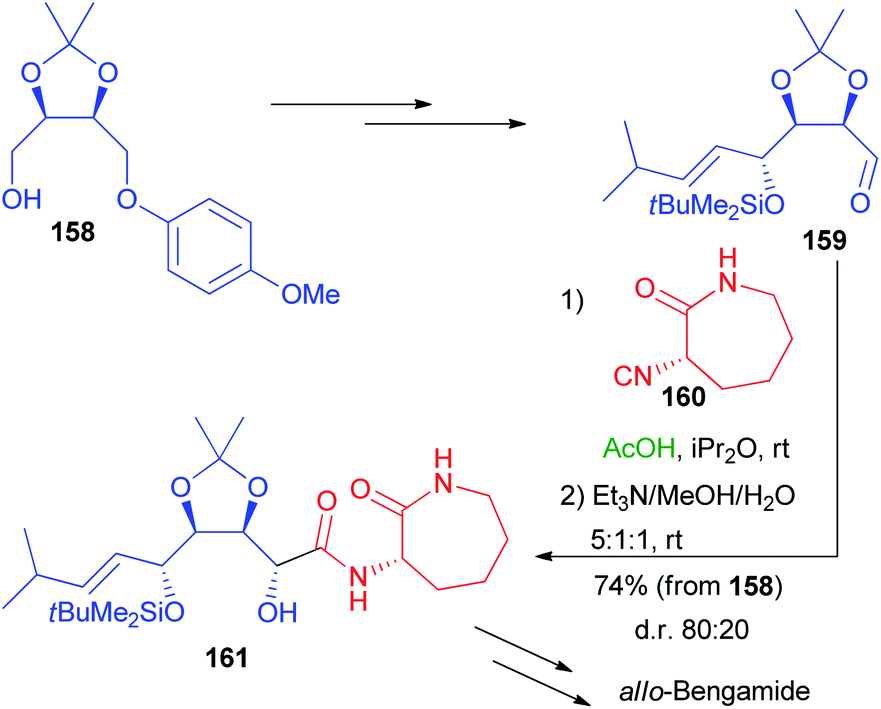 | ||
| Scheme 44 Synthesis of allo-Bengamide. | ||
Nevertheless, we were unable to further improve the stereoselectivity taking advantage of ZnBr2, because 160 is unstable in the presence of Lewis acids. Intermediate 161 was then converted into allo-Bengamide.
Another chiral α-alkoxyaldehyde considered by us is 164, obtained by lipase catalysed desymmetrization of 2,5-bis(hydroxymethyl)tetrahydrofuran 163, a reduced derivative of 5-(hydroxymethyl)furfural 162, an important building block available from lignocellulosic biomass (Scheme 45). The Passerini reaction, performed under standard conditions on a model compound with R1 = tBu and R2 = Ac, was, unlike 139, poorly diastereoselective, affording 166 as a 59:41 diastereomeric mixture.103 Then we applied the previously described protocol with ZnBr2 as the catalyst and we successfully increased the diastereomeric ratio to 81:19 (82% yield), provided that a carefully dried aldehyde was used to prevent the formation of the stabilized hydrated form 165, responsible for not reproducible results. The best results were obtained if a pre-mixed solution of 164 and the carboxylic acid (AcOH) in iPr2O was added to the mixture of isocyanide and ZnBr2. However, the scope of the reaction using the optimized conditions with other carboxylic acids and isocyanides was demonstrated to be troublesome because of the formation of variable amounts of the “truncated” products, as well as due to the insolubility of many carboxylic acids in iPr2O.
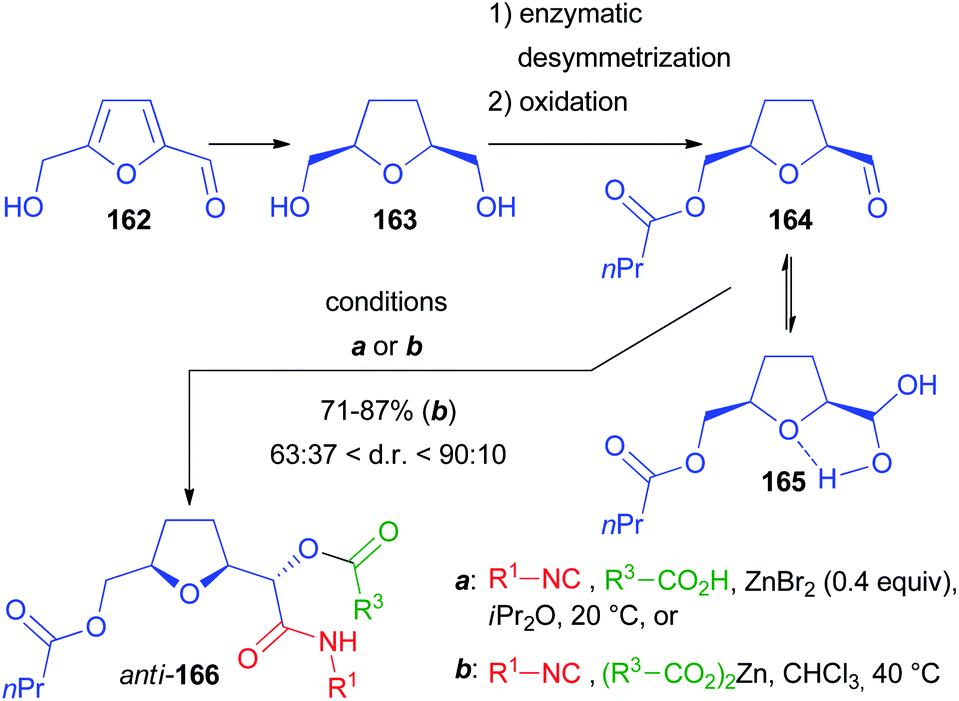 | ||
| Scheme 45 Diastereoselective Passerini reaction with Zn carboxylates. | ||
For this reason, we developed a more robust and unprecedented methodology using for the first time stoichiometric zinc(II) dicarboxylates. Under these conditions, the d.r. was improved up to 9:1 with the anti-stereoisomer prevailing, and most reactions affording at least a 4:1 ratio.
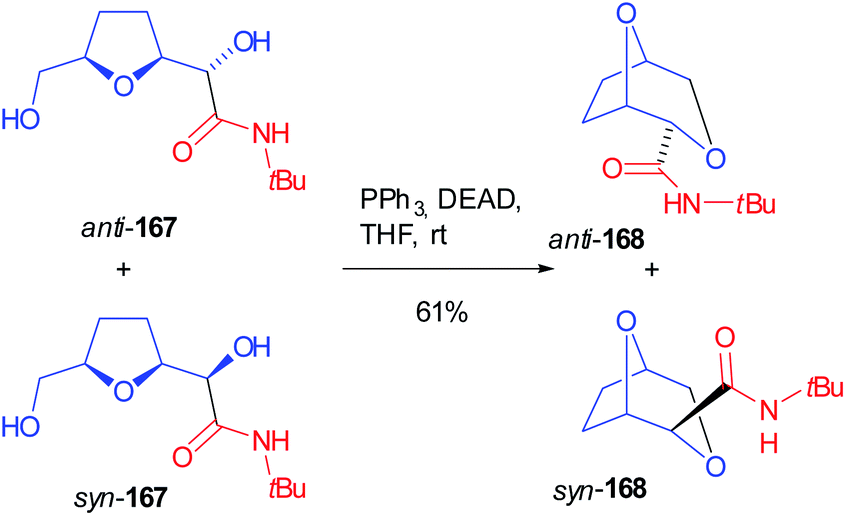
A mixture of both Passerini products 166 (R1 = tBu) was submitted to the contemporary hydrolysis of both ester functions and diols 167 were submitted to an intramolecular Mitsunobu reaction, where the primary alcohol cyclized onto the secondary one (168) in a stereospecific manner, without losing the initial diastereomeric ratio (Scheme 46).
 | ||
| Scheme 46 Synthetic elaboration of 166. | ||
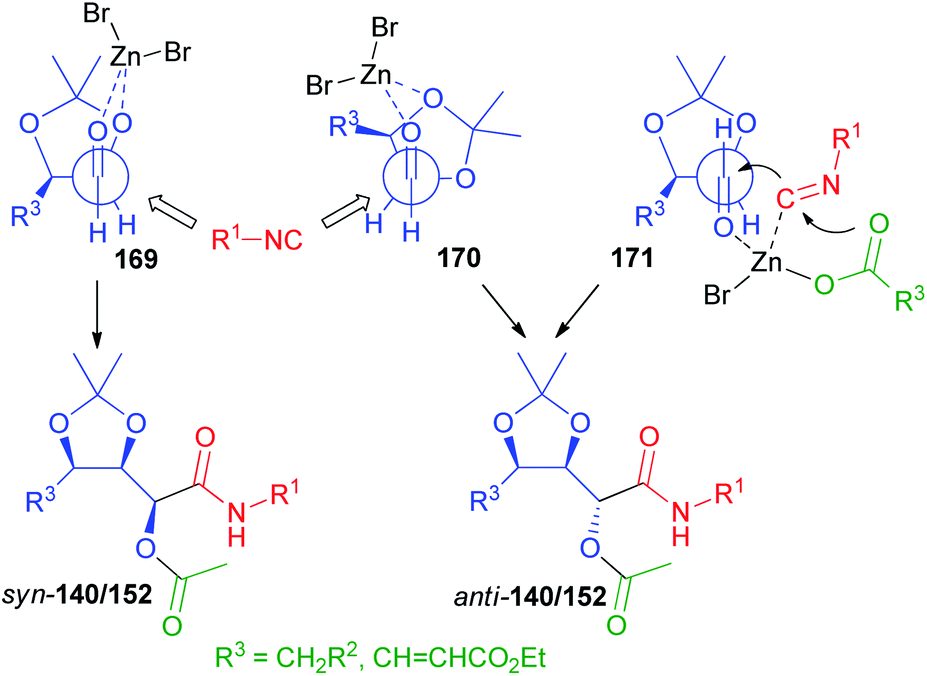
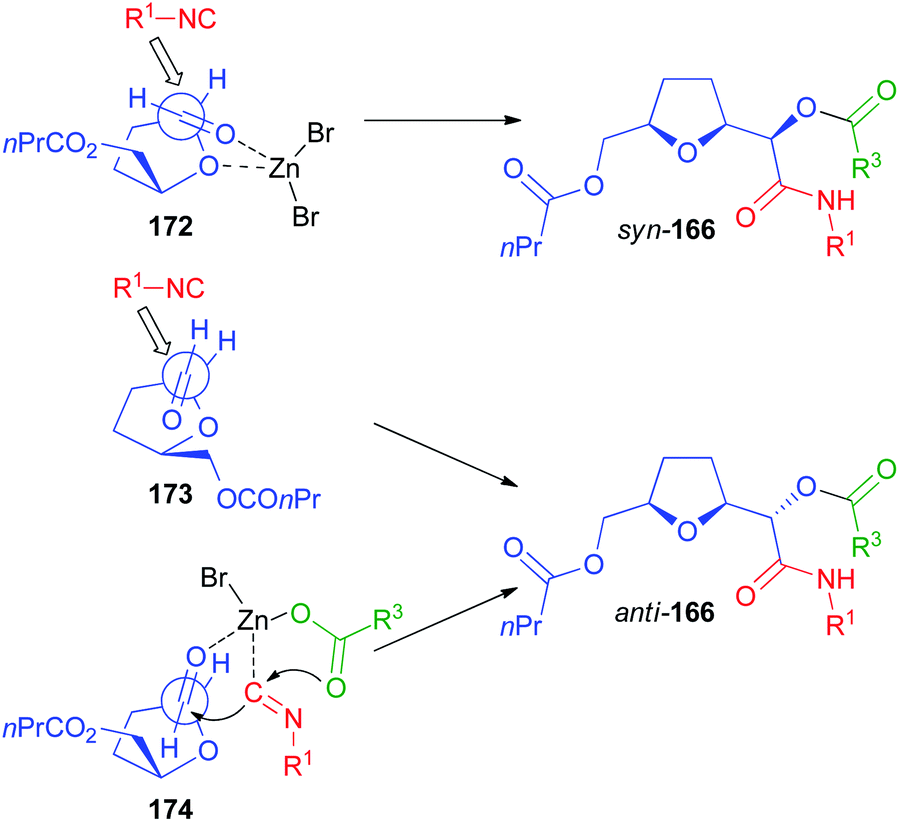
The rationalization of the stereochemical outcome of these reactions with α-alkoxyaldehydes is not as simple as it might seem at first sight. The increase of stereoselectivity on erythritol-derived aldehydes and on 164 in the presence of ZnBr2 could be explained if a chelated TS, in which the carbonyl and the α-alkoxy oxygen are both coordinated by the metal (TS 169, Scheme 47 and 172, Scheme 48 respectively), is operating. However, this would in all cases preferentially afford syn stereoisomers (140, 152, and 166). For the erythritol-derived aldehydes, another possibility is represented by the coordination of the β-alkoxy oxygen (TS 170), but this is not possible of course for 164. Nevertheless, the formation of the anti-stereoisomer (140, 152, and 166) is preferred also in the absence of Zn(II). The Felkin–Anh model (TS 170 or TS 173, Scheme 48) can explain the prevailing formation of anti-products both for aldehydes from erythritol and for 164.
 | ||
| Scheme 47 Stereochemical outcome rationalization for Passerini reactions with erythritol aldehydes. | ||
 | ||
| Scheme 48 Stereochemical outcome rationalization for Passerini reactions with aldehyde 164. | ||
However, the well-known Felkin–Ahn and chelation models are not always able to explain the stereochemical outcome in the nucleophilic additions to carbonyls,104 especially those promoted by weak nucleophiles, such as isocyanides. In this case the addition is most likely endoergonic and the TS is probably more product-like.
Also on the basis of DFT calculations, we think that in the isocyanide addition the position of the carbonyl with respect to the ring plays a crucial role. The “inside” position of the carbonyl (TS 170 and 173) would be disfavoured by steric interactions with the side arm substituent, and therefore an “outside” arrangement (TS 171 and 174) is preferred. Moreover, we believe that a concerted mechanism, where the metal, bound to the carboxylate, is coordinated by both the isocyanide and the carbonyl oxygen is working. In this way, a bulkier environment is created around the reaction centre, making one of the two diastereotopic faces more accessible.
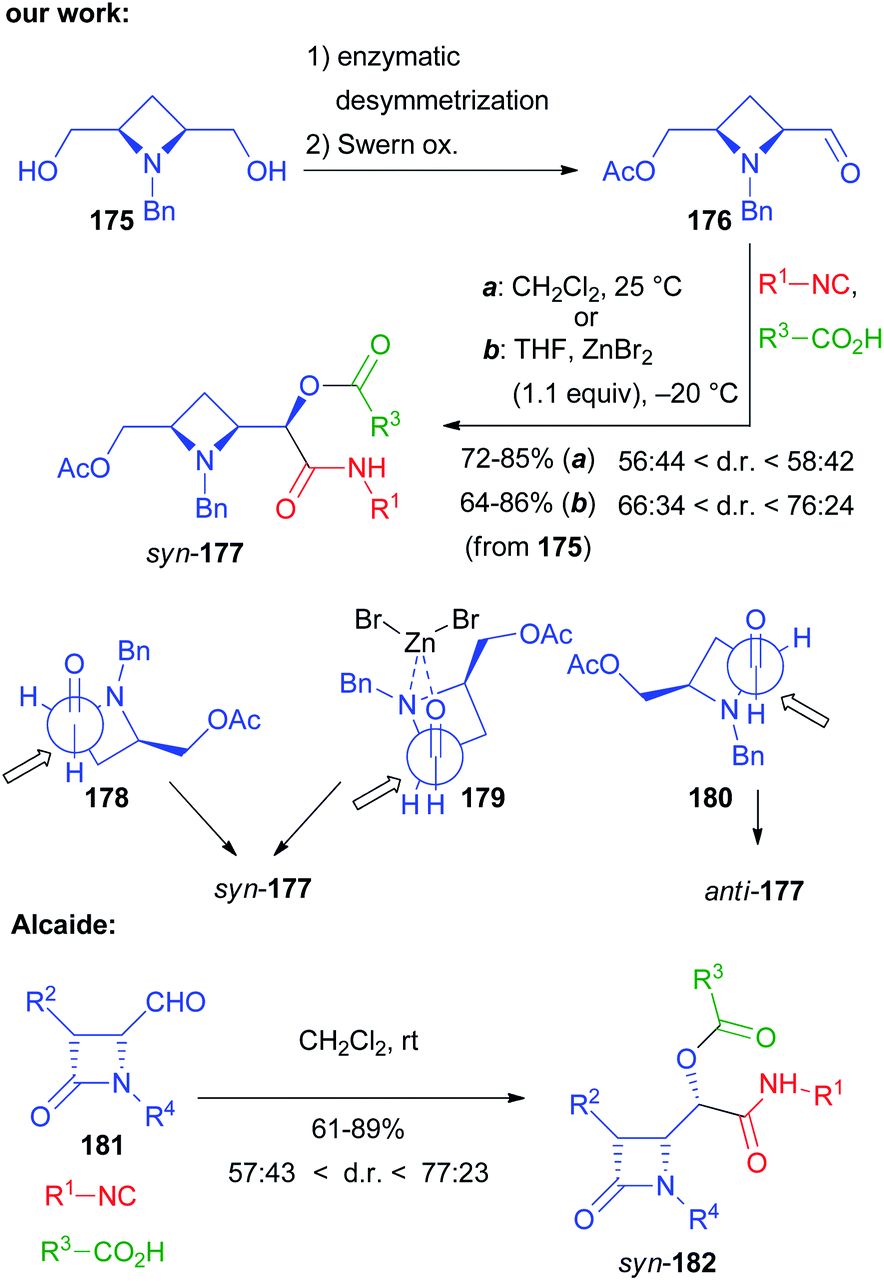
Not only chiral α-alkoxyaldehydes but also α-aminoaldehydes were explored as chiral carbonyl inputs. In particular we used biocatalytically derived azetidine aldehyde 176 (Scheme 49), obtained by a chemoenzymatic procedure from meso-diol 175.105
 | ||
| Scheme 49 Passerini reaction on chiral α-aminoaldehydes. | ||
In contrast to what we experienced with α-alkoxyaldehydes, this time the syn stereoisomer 177 prevailed but, using the classic Passerini conditions, the ratio was rather poor. The catalysis by ZnBr2 still was able to improve significantly the diastereoselectivity, though the best results were only around a 3:1 ratio. The explanation of the diastereoselectivity cannot be based on Felkin–Anh or Cram models, with the azetidine nitrogen behaving as a large group, because this would predict the prevailing formation of the anti-stereoisomer. For the uncatalyzed Passerini reaction the preference for syn-177 can be explained by the Cram model (TS 178). The poor stereoselectivity can be justified by the two possible TS 178 and 180, where the approach of the small nucleophile, the isocyanide, is not able to efficiently discriminate between the two diastereotopic faces of the carbonyl. In the presence of Zn(II) a chelated TS (179), where the metal is able to coordinate both the oxygen and the nitrogen, is most likely operating, which explains the observed enhanced stereoselectivity.
The prevailing formation of the syn product is in agreement with a previous report by Alcaide, where the classic Passerini reaction was performed using β-lactam aldehydes 181 from isopropylidene-D-glyceraldehyde to afford syn-182.106
As described in chapter 2, also acyclic α-NBoc-aminoaldehydes 47 (Scheme 11) afford preferentially syn derivatives, though with a moderate d.r.32,37 In contrast, α-alkyl-β-aminoaldehydes 71 (Scheme 16) gave, as expected, no diastereoselectivity at all, furnishing the homo-PADAM products 73 as a 1:1 mixture.42
Very recently Dömling optimized a diastereoselective Passerini reaction on a well-defined combination of 3 chiral reagents, which was aimed at the total synthesis of N14-desacetoxytubulysin H (Scheme 50) reported in detail in chapter 5.107
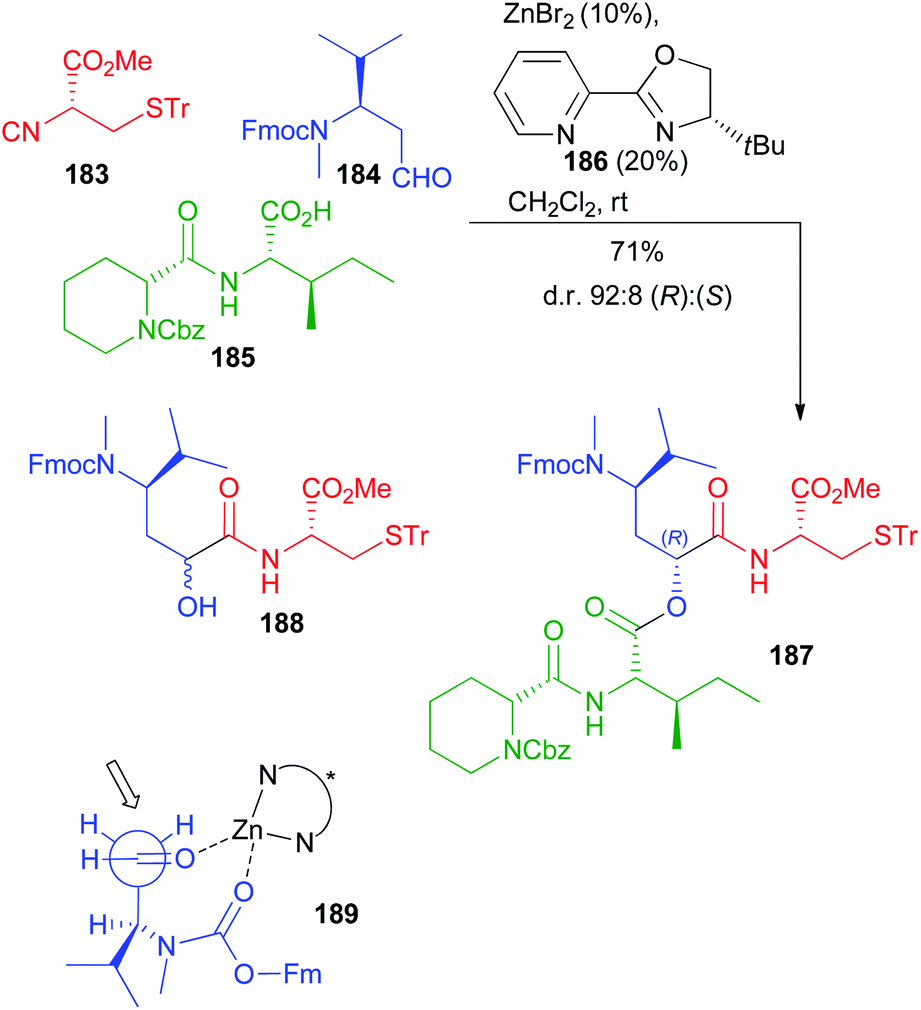 | ||
| Scheme 50 Stereoselective synthesis of tubulysin fragment 187 through the diastereoselective Passerini reaction (Fm = fluorenylmethyl). | ||
This time β-N-Fmoc-aminoaldehyde 184 undergoes a stereoselective attack by chiral isocyanide 183, and the intermediate is trapped by acid 185. As expected, the Passerini reaction under conventional conditions (CH2Cl2, 1 M) was not stereoselective at all, while the yield was excellent (90%). Higher dilution (up to 0.01 M) enhanced the stereoselectivity (up to 70:30), but the yield dramatically decreased to 15%, as the prevailing product became “truncated” 188.
In order to improve the stereoselectivity, favouring the formation of 187 with the needed (R) configuration at the new stereogenic centre, the authors used chiral CPAs, which recently allowed the first enantioselective Ugi reaction.108 Despite the high d.r. (95:5), the yields were poor, in part because of the unwanted formation of 188, which was almost the only product when the CPA was used in the presence of pyridine to mask its acidity. Switching to the activation by an achiral Lewis acid (ZnBr2), a moderate 80:20 diastereoselectivity was obtained with an appreciable 70% yield. Finally, the combination of ZnBr2 with a chiral ligand, with 186 being the best performing one, allowed obtaining 187 with a remarkable 92:8 diastereomeric ratio in 71% overall yield. The proposed TS 189 agrees with a Cram chelated model, where a chelation between the aldehyde and the urethane carbonyls is most likely working. The chiral ligand reinforces the stereoselectivity, favouring the attack of 183 in a matched approach from the less hindered Si face of the carbonyl.
A long range highly diastereoselective Passerini reaction is rarely reported, an exception being the reaction on desmycosin 190, a natural macrolide antibiotic, characterized by a CH2CHO group, reported by Wessjohann (Scheme 51).109 Only two reactions were reported affording 191 but, using propanoic acid, the d.r. was very good considering the distance between the functional group and the closest stereogenic centre.
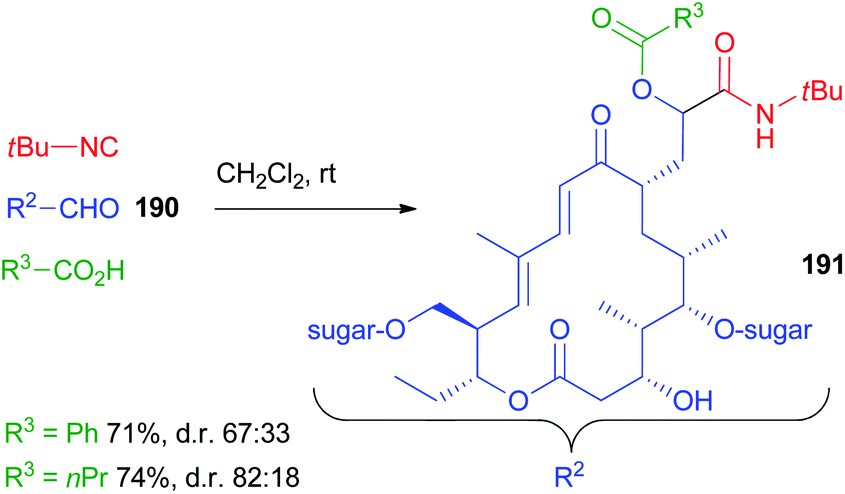 | ||
| Scheme 51 Long range diastereoselection in the Passerini reaction on macrolide aldehyde 190. | ||
In another paper by Ostaszewski, aimed at the synthesis of depsipeptides 194, L-phenylalanine methyl ester derived isocyanide 192 was reacted with differently N-protected valines 193 using cyclohexanone as the carbonyl component as well as solvent (Scheme 52).112 The reaction itself cannot be diastereoselective as a symmetric ketone is employed, which means that only one product should be obtained in principle. The formation of two diastereoisomers is therefore a consequence of the more or less extended racemisation of the isocyanide, which is heavily dependent on the protecting group of 193. Actually, the extent of the racemisation is negligible using trityl, phthalyl or Boc (d.r. > 99:1 and yield up to 99%) as protecting groups, which suggests that the irreversible Passerini reaction is faster than the racemisation (k1 > krac), while the epimerization after the MCR, as well as the possible dynamic kinetic resolution occurring after the racemisation of (S)-192 have been ruled out on the basis of appropriate experiments.
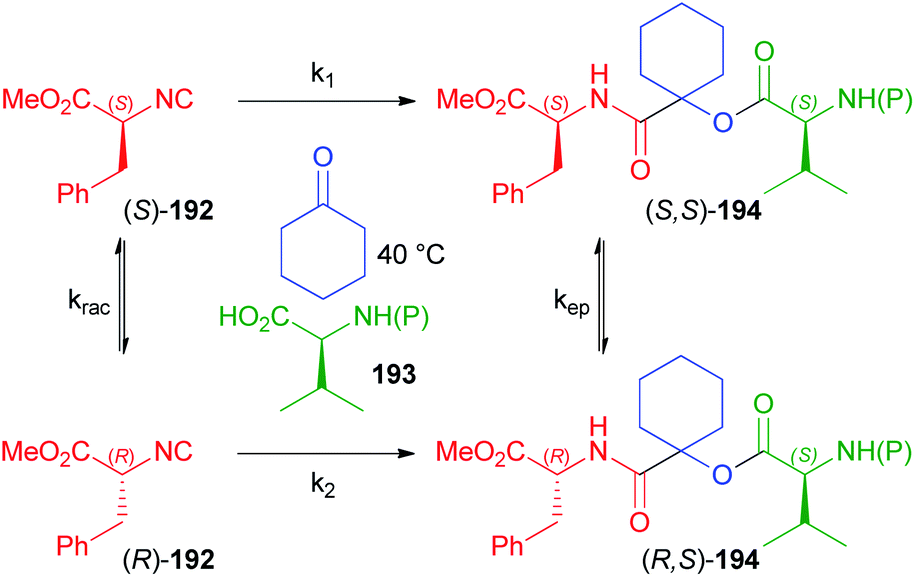 | ||
| Scheme 52 Effect of the racemisation of 192, depending on the protecting group (P) of 193. | ||
4.3 Future prospects
As witnessed by the relatively small number of papers on this topic, the stereochemistry issues of the Passerini reaction have not been fully solved yet. There is plenty of space to study new conditions and new combinations of catalysts/additives, and to focus on a double asymmetric induction approach. For these reasons, we think that in the future we will be able to better understand the factors affecting the stereochemistry control in the Passerini reaction.5. The use of the Passerini reaction in the total syntheses of API and natural substances
While initially the Passerini reaction, as other isocyanide-based MCRs, was considered useful only in diversity-oriented synthesis, in the last few decades several applications in target-oriented synthesis have appeared. In particular, we can find successful uses of the Passerini reaction and its modifications in the total synthesis of natural compounds and in the preparation of APIs. In this chapter, we report the most significant examples.5.1 Use of an intramolecular Passerini reaction
In 1981 Falck reported one of the oldest example, preparing the tetrahydroisoquinoline-based alkaloid hydrastine 199 (Scheme 53).113 The total synthesis of this compound was already published and involved a Bischler–Napieralski cyclization of 198, followed by catalytic hydrogenation with PtO2 of the resulting dihydroisoquinoline and a final methylation by reductive amination. The synthesis of the lactonic amide 198 was troublesome and Falck envisaged the construction of this key intermediate by an intramolecular Passerini reaction between the isocyanide 196 (achievable in one step from piperonyl bromide 195) and opianic acid 197. The reaction proceeds well under classical conditions and this example well highlights the power of multicomponent reactions.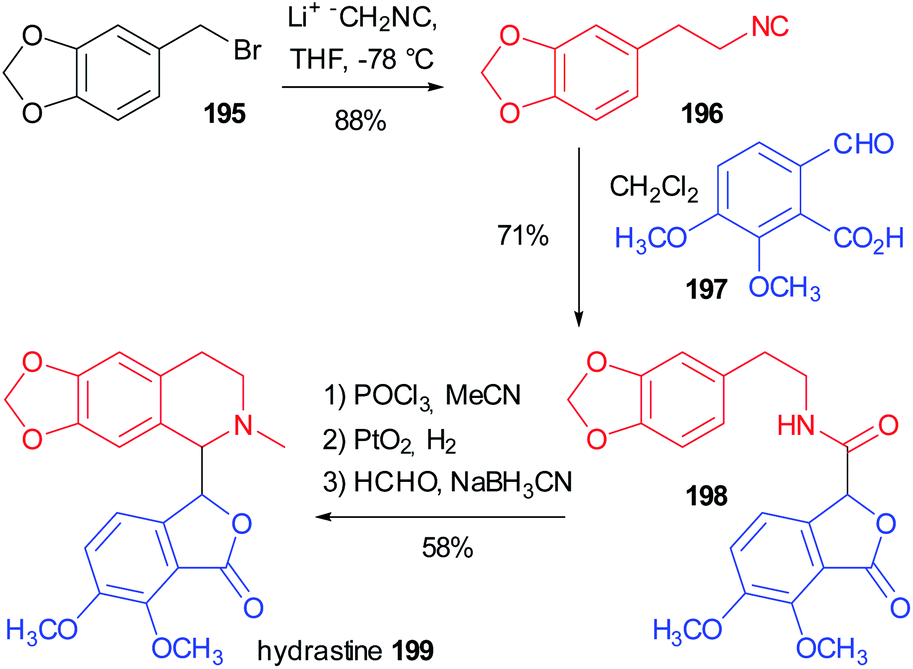 | ||
| Scheme 53 Synthesis of hydrastine 199 by an intramolecular Passerini reaction. | ||
5.2 Use of a modified Passerini reaction
The α-hydroxy amide framework is a recurrent motif in biologically active molecules and this important moiety can be obtained in a convergent fashion employing a “truncated” Passerini reaction.This strategy has been applied to the synthesis of bicalutamide (Casodex®) 203, an antiandrogen used to treat prostate cancer, which can be conveniently obtained in two steps, starting from commercially available reagents, employing a Passerini reaction assisted by TiCl4, as a key step (Scheme 54).114 The thioether group of the commercially available ketone 200 is oxidized to the sulfone 201, and, together with the isocyanide 202, was subjected to this modified version of the Passerini reaction, leading directly to α-hydroxy amides, without the need for an additional hydrolysis step. Bicalutamide was obtained with a yield of 66% and a purity >99% after recrystallization.
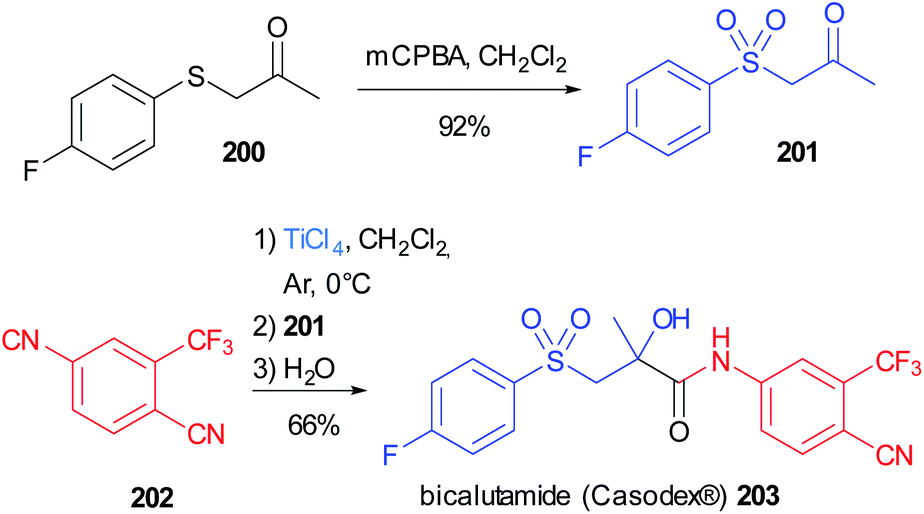 | ||
| Scheme 54 Synthesis of bicalutamide 203 through a modified Passerini reaction. | ||
The use of TiCl4 as a Lewis acid in the Passerini reaction was first reported by Seebach in 1983 (ref. 115 and 116) and ten years later Floriani definitively clarified the mechanism, thanks to X-ray diffraction studies.117 Although the original hypothesis involved the insertion of the isocyanide into the Ti–Cl bond, nowadays the accepted mechanism proceeds through the initial coordination of the ketone, thanks to the oxophilic nature of titanium(IV). Then, the nucleophilic attack of the isocyanide, to give adduct 204, is followed by the transfer of a chlorine atom from titanium to carbon (205) and the free coordination site is then filled by the nitrogen atom (206) (Scheme 55). This reactive imidoyl chloride intermediate 206 is prone to hydrolyse leading to α-hydroxyamide. Therefore, the role of TiCl4 is to enhance the electrophilicity of the carbonyl group and to bring together the two components of the reaction.
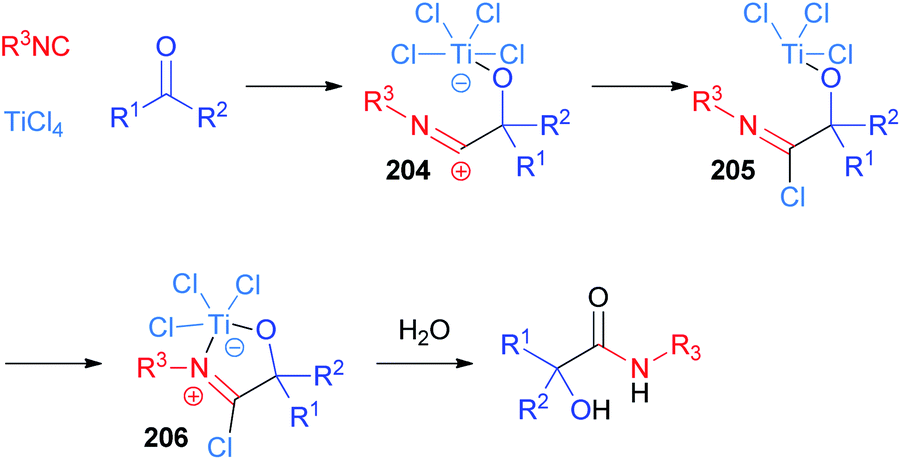 | ||
| Scheme 55 Mechanism of the TiCl4 mediated Passerini reaction. | ||
Seebach's Passerini variation was also applied in the synthesis of mandipropamid (Pergado®) 210, a potent fungicide used to protect crops from downy mildew. The isocyanide precursor 208 could be easily obtained in 4 steps from vanillin 207 through a Henry reaction followed by reduction, formylation and propargylation. It is noteworthy that in this case the isocyanide was not isolated, but the formamide was directly subjected to the one-pot isocyanide formation/modified Passerini reaction, leading to α-hydroxyamide 209 in good yield. The final propargylation of the mandelic acid moiety afforded the marketed agrochemical mandipropamid (Scheme 56).118
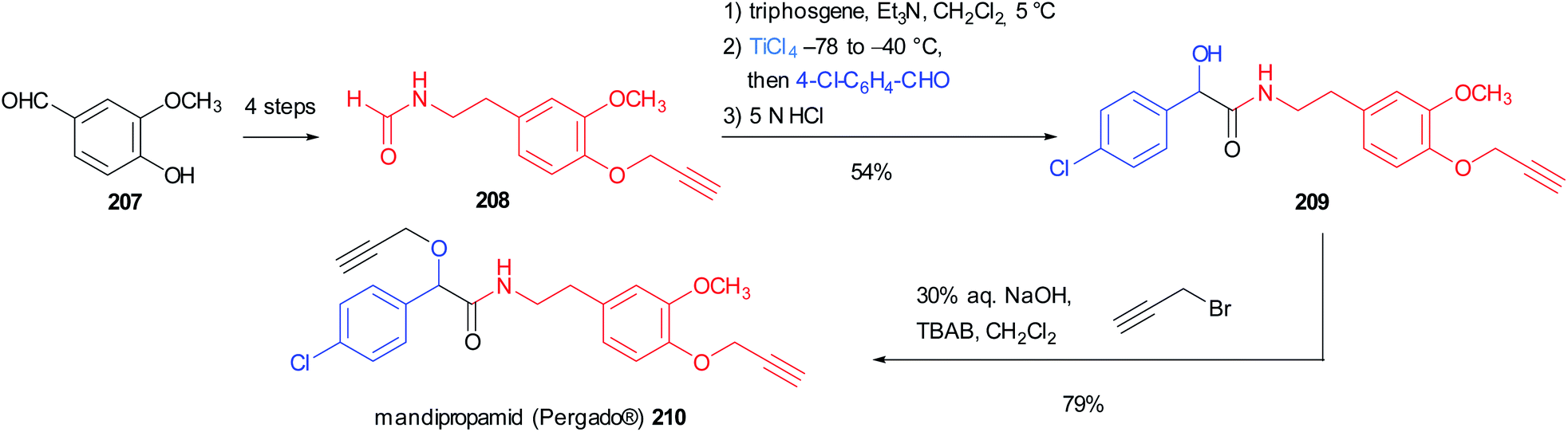 | ||
| Scheme 56 Synthesis of mandipropamid 210 through a modified Passerini reaction. | ||
Salbutamol (Ventolin®) 214, also known as albuterol, is one of the most notorious β2 adrenergic receptor agonists and it is used to treat asthma. In 2020 Virieux, Ayad reported the asymmetric synthesis of the biologically active (R) enantiomer of this β-amino alcohol on a gram-scale (Scheme 57)119 exploiting the work of Orru on the two-step one-pot synthesis of N-substituted-β-amino alcohol.120 This reductive Passerini-type process involves the variation of the asymmetric “truncated” Passerini reaction developed by Denmark,74 in which the α-trichlorosilyloxy imidoyl chloride intermediate 108 (see Scheme 33, Section 4.1) is reduced in situ by an ammonia borane complex.
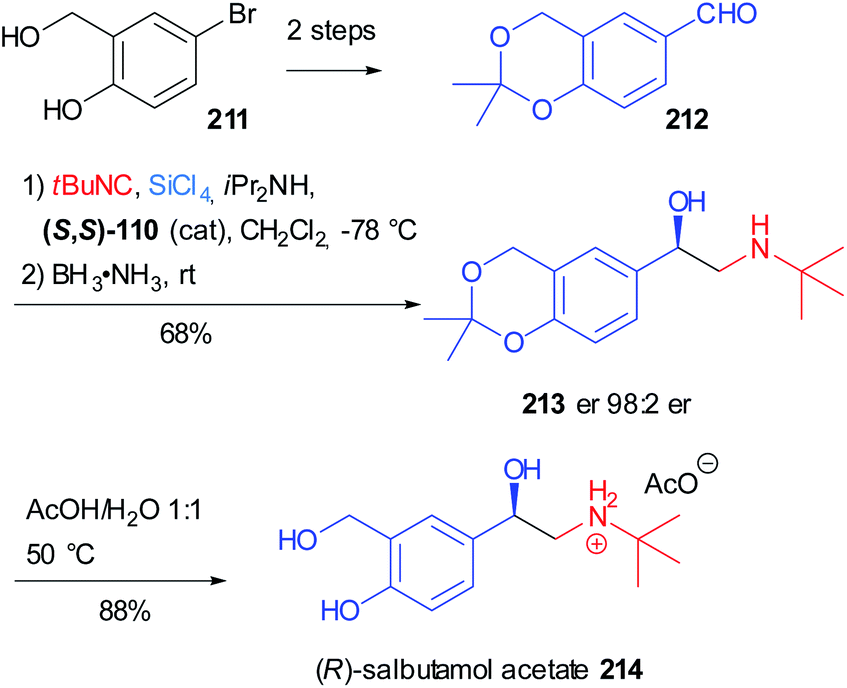 | ||
| Scheme 57 Synthesis of (R)-salbutamol acetate 214 through a two step one-pot reductive Passerini-type reaction. | ||
The aldehyde 212 could be easily obtained from commercially available 5-bromo-2-hydroxybenzyl alcohol 211 in two steps: isopropylidene acetal protection and formylation via bromo/lithium-exchange. The key β-amino alcohol 213 was obtained in good yield and the excellent enantiomeric ratio is due to the attack of the isocyanide on the Si face of the aldehyde, thanks to the induction given by the (S,S) enantiomer of the chiral bisphosphoramide Lewis base. The final acetal removal furnished salbutamol 214 as an acetate salt.
Applying the synthesis of β-amino alcohols trough the modified Passerini reaction discussed on chapter 3 (Scheme 29), Ruijter reported the preparation of propranolol 217 and rivaroxaban 223 (Scheme 58).66 Starting from (1-naphthyloxy)acetaldehyde 215 and isopropylisocyanide 216, propranolol can be obtained in one step with excellent yield. The preparation of Rivaroxaban started from the commercially available aniline 218, which was efficiently transformed into the isocyanide 219 in 2 steps. The reductive Passerini-type reaction of 219 and the Cbz protected aminoacetaldehyde 220 afforded the desired amino alcohol 221 in good yield.
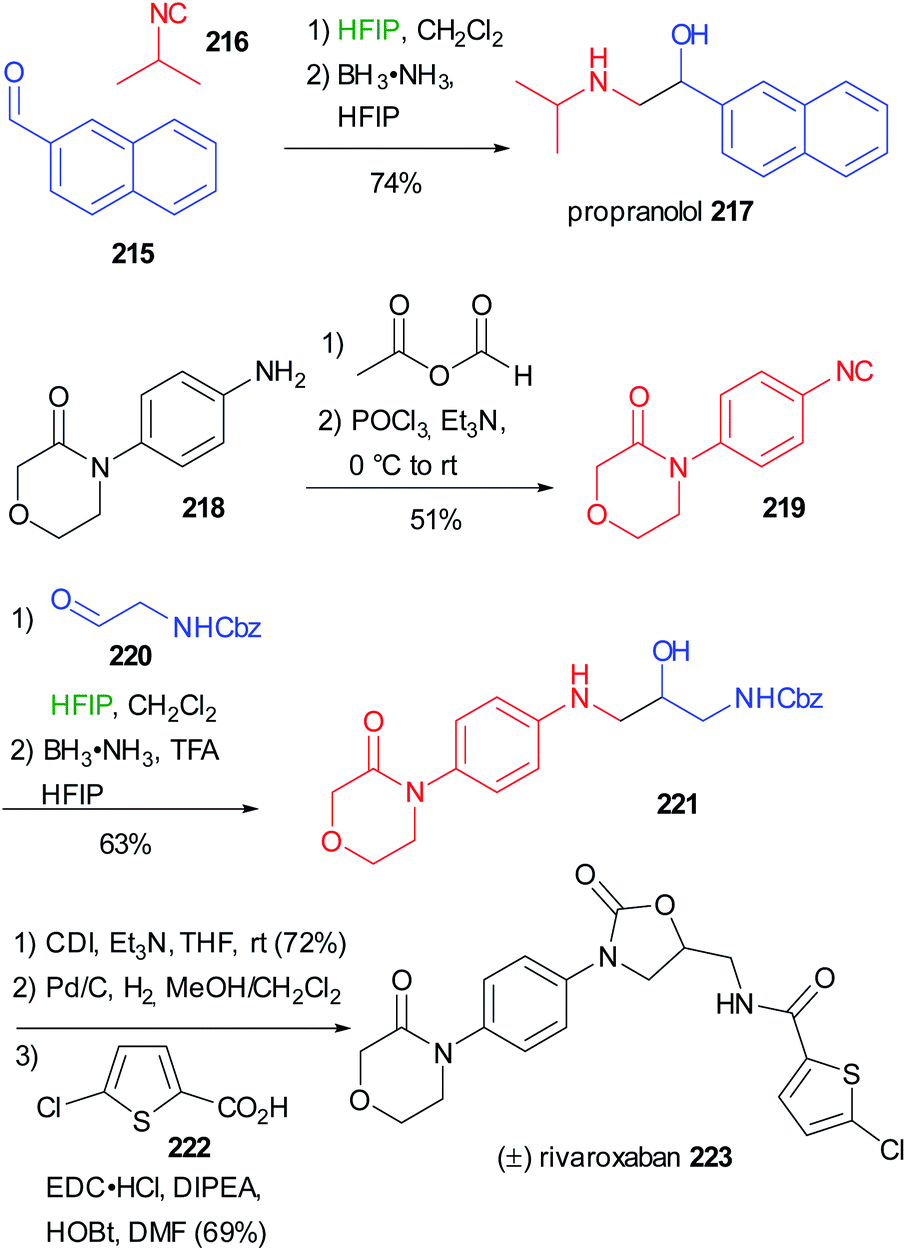 | ||
| Scheme 58 Synthesis of propranolol 217 and (±)rivaroxaban 223 through a two step one-pot reductive Passerini-type reaction. | ||
In this case, an excess of trifluoroacetic acid was necessary to activate the imidate toward the reduction, given its lower basicity derived from the use of an aromatic isocyanide. Finally, 221 could be converted into rivaroxaban in three steps involving the formation of a cyclic carbamate with 1,1′-carbonyldiimidazole (CDI), the deprotection of Cbz and the ultimate coupling with 5-chlorothiophene-2-carboxylic acid 222.
Recently, Echavarren reported the racemic and stereoselective total synthesis of seven members of the lapidilectine and grandilodine family (224–230) (Scheme 59).121 These natural molecules are indole-based alkaloids and preliminary studies on their biological activity showed their ability to reverse multidrug resistance in vincristine-resistant cancer cells.122 These alkaloids have a common pyrroloazocine indole core in which the central 8-membered ring is embedded in a rigid [4.2.2]azabicyclic structure.
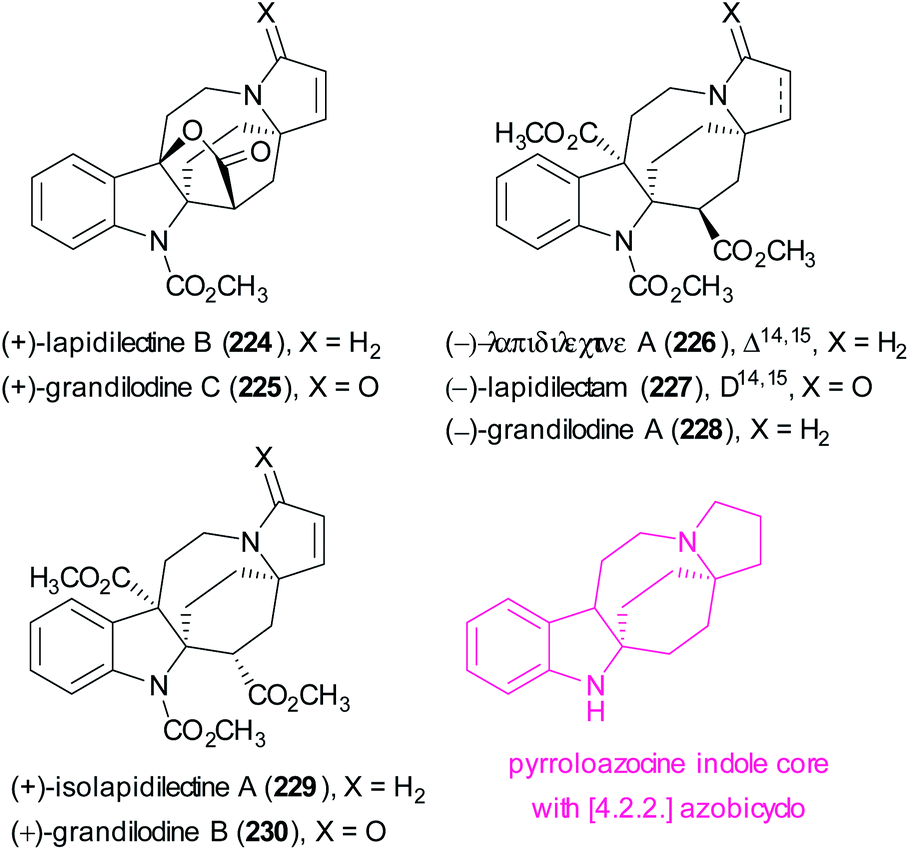 | ||
| Scheme 59 Structure of lapidilectine and grandilodine alkaloids. | ||
The pyrroloazicine indole skeleton was built via a gold-catalyzed 8-endo-dig hydroarylation, while the azabicyclo was created by a radical 6-exo-trig photoredox cyclization, where the catalyst was again an Au complex ([(dppm)AuCl2], dppm = bis(diphenylphosphanyl)methane) (Scheme 60).
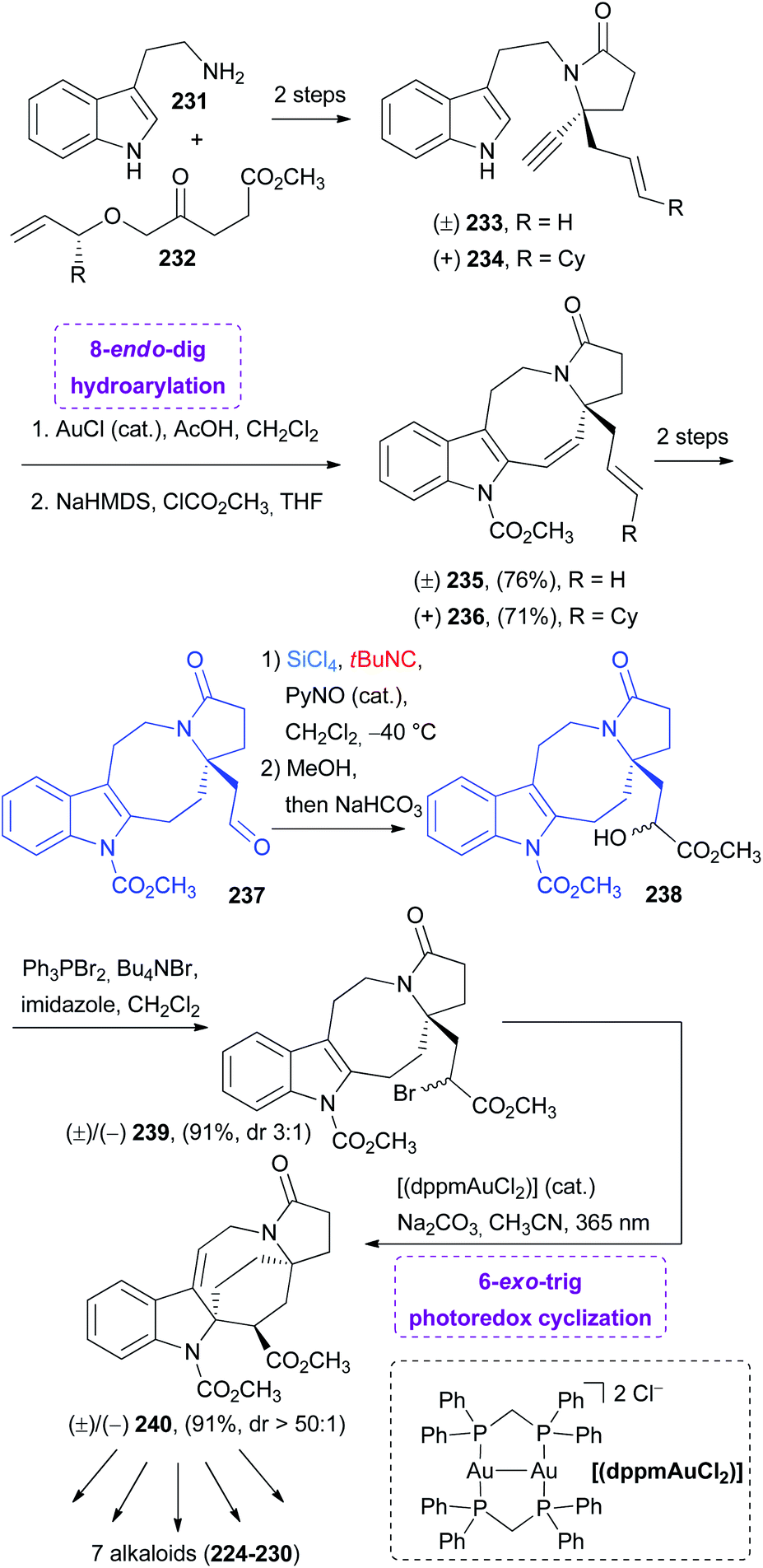 | ||
| Scheme 60 Synthesis of the key intermediate 240via hydroarylation “truncated” Passerini and photoredox cyclization. | ||
The alkynes 233 and 234 were prepared in two steps starting from tryptamine 231 and the oxoester 232, with an excellent transfer of chirality in the case of 234. These alkynes were subjected to gold catalysed hydroarylation, which proceeds with excellent 8-endo selectivity (>50:1 endo/exo) and high yield, and then the methylcarbamate moiety was installed. Compounds 235/236 were converted in two steps into the aldehyde 237 by the oxidative cleavage of the exocyclic double bond by OsO4/NaIO4 and hydrogenation of the endocyclic double bond. The substrate necessary for this second key transformation is the α-bromo ester 239, which in turn could be easily prepared by an Appel reaction from alcohol 238.
To install the key α-hydroxy methyl ester group, the authors seeked a transformation with excellent functional group tolerance, having the labile methyl carbamate moiety, and the Passerini-type reaction reported by Denmark74 was again selected. Here, the α-trichlorosilyloxy imidoyl chloride intermediate was treated with MeOH and the subsequent basic hydrolysis of the imino ether afforded the α-hydroxy ester. Thus, the nitrogen substituent of the isocyanide was not retained. In this case, the stereocontrol was not a goal because the photoredox cyclization proceeds through a radical intermediate; therefore an achiral Lewis base, such as pyridine N-oxide, was employed. Compound 240 is a common intermediate in the preparation of the natural alkaloids 224–230.
Neoxaline 241, oxaline 242 and melagrin A 243 are indole alkaloids containing a unique indoline spiroaminal framework and possess anti-cancer activity (Scheme 61). Their asymmetric total synthesis was reported by Ōmura and Sunazuka123,124 starting from tryptophol 244. The enantiopure aldehyde 245 was prepared in seven steps and then employed in a “truncated” Passerini reaction with methyl isocyanoacetate in the presence of boric acid affording the α-hydroxyamide 246, which bears suitable functional groups for the construction of the indoline spiroaminal skeleton. The authors screened many conditions and eventually they found that boric acid was the best reagent to mediate the α-addition of methyl isocyanoacetate to the aldehyde 245.125 The induction given by the chiral aldehyde was poor; in fact the diastereomeric ratio was only 1:2 in favour of desired isomer (9S)-246. Unfortunately, the (9S)-isomer was proved unable to give the spiroaminal framework; therefore, the authors tried to reverse the d.r., confident that the stereocentre C9 could be epimerized later in the synthesis. Through oxidation and reduction by IBX and NaBH4 at low temperature, the d.r. was reversed in favour of the other diastereoisomer (9R)-246. The two diastereoisomers could be separated by chromatography after the protection of the alcohol and deprotection of the carbamate.
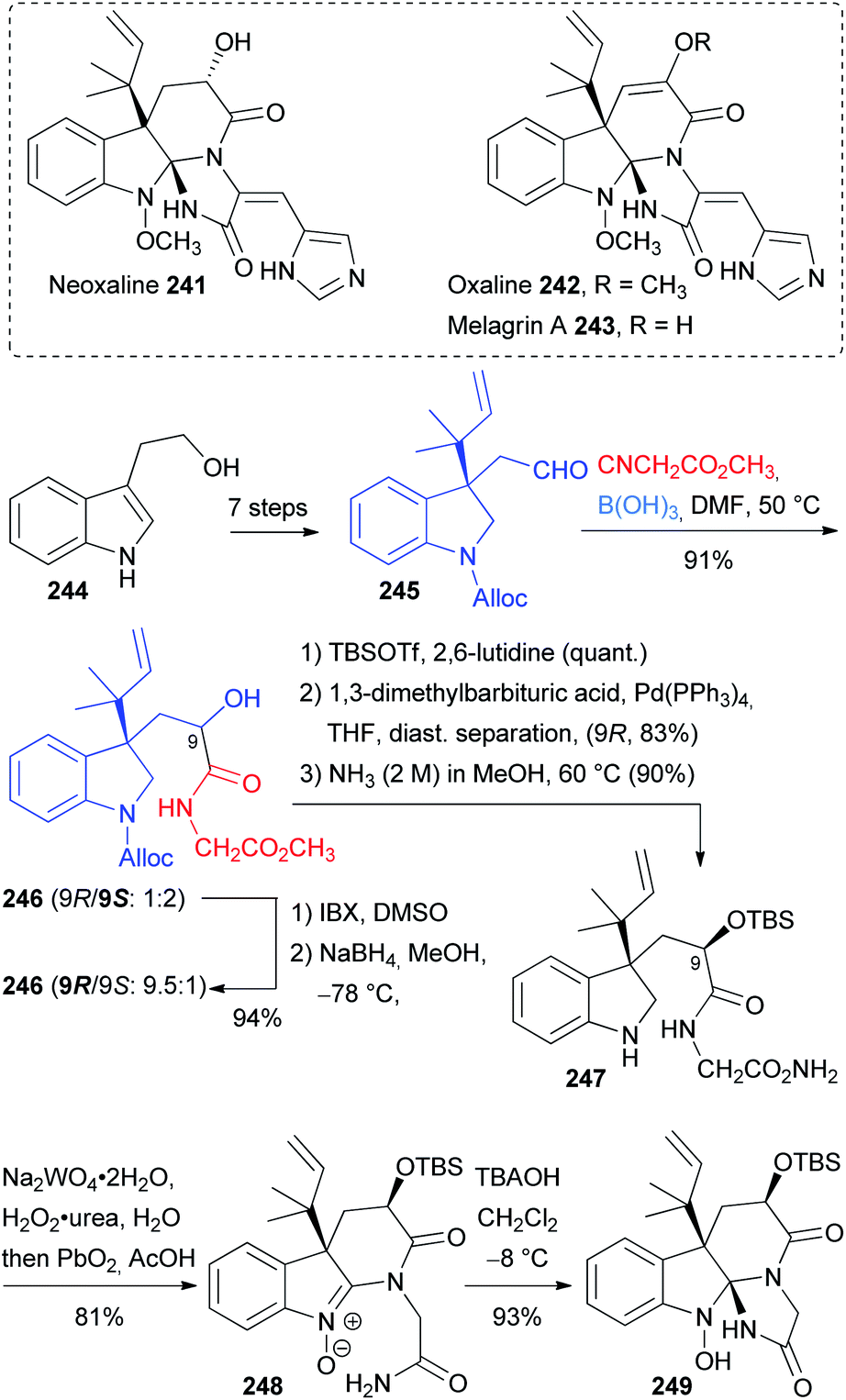 | ||
| Scheme 61 Synthesis of the key intermediate 249via “truncated” Passerini and oxidations/cyclizations. | ||
The final conversion of the ester in the primary amide furnished 247, which is the substrate for the oxidation/cyclization strategy that produces the spiroaminal framework in two steps. This transformation proceeds through the initial oxidation of the indoline 247 to the nitrone thanks to sodium tungstate, and then PbO2 and AcOH mediated the cyclization to the diaminal intermediate and its further oxidation to nitrone 248. Finally, treatment with tetrabutylammonium hydroxide (TBAOH) provided the desired indoline spiroaminal 249, which is a common intermediate for the preparation of natural alkaloids 241–243.
5.3 Use of the PADAM approach
The β-acylamino-α-ketoamide scaffold is present in many natural and synthetic protease inhibitors and can be obtained through the PADAM strategy followed by oxidation (as described in Section 2.2). This approach has been applied to the total synthesis of eurystatin A38 (cf.Scheme 13) and cyclotheonamide C,126 two potent inhibitors of serine proteases.Cyclotheonamide C 256 is a 19-membered pentapeptide containing the unusual amino acids vinylogous L-dehydrotyrosine (L-V-ΔTr), D-phenylalanine, L-α-keto-β-homoarginine (L-K-hArg) and L-diamminopropanoic acid (L-Dpr) that links L-proline and V-ΔTyr residues (Scheme 62). The retrosynthetic approach envisaged by Aitken is highly convergent; in fact cyclotheonamide C is obtained in only four steps after the PADAM protocol. The preparation of the aldehyde 251 and the carboxylic acid 252 was already accomplished by the same authors during previous efforts on the total synthesis of 256.127 The isocyanide 250 was obtained without racemisation by classical N-formylation/dehydration with POCl3, starting from the parent primary amine, the synthesis of which was already reported.127 These three components were subjected to the Passerini reaction and the desired product 253 was obtained with respectable yield. It is noteworthy that the reaction does not proceed when the Fmoc protecting group is installed on the acid and the Boc is on the aldehyde. Selective Fmoc deprotection and O- to N-acyl migration, induced by the addition of Et3N, provided the key linear pentapeptide 254 in 88% yield. Simultaneous N/C termini deprotection under acidic conditions is followed by macrocyclization mediated by a coupling agent. The late-stage oxidation of 255 with Dess-Martin Periodinane (DMP) and the complete deprotection of the phenol and guanidine moieties with HF·pyridine in the presence of anisole furnished cyclotheonamide C as a single stereoisomer.
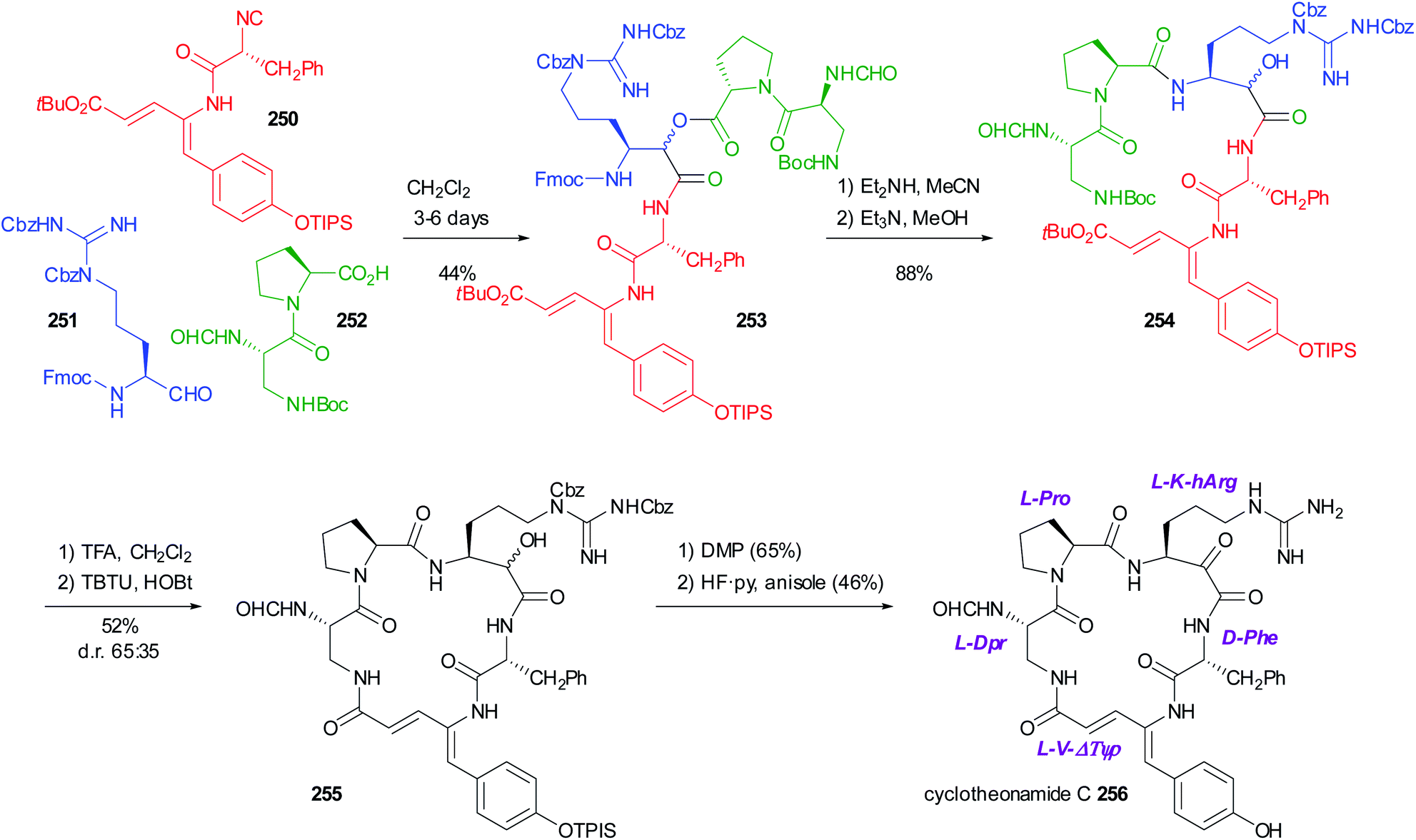 | ||
| Scheme 62 Total synthesis of cyclotheonamide C 256via the PADAM/oxidation strategy. | ||
Tubulysins are unusual peptides mainly composed of isoleucine (Ile) and non-proteinogenic amino acids, such as D-N-methyl pipecolic acid (Mep), tubuvaline (Tuv) and tubuphenylalanine (Tup). Recently, Dömling reported the highly convergent total synthesis of N14-desacetoxytubulysin H 263 employing a diastereoselective Passerini reaction applied in the PADAM protocol (Scheme 63).107 The three components needed in the MCR could be prepared on a multi-gram scale from commercially available amino acids. The isocyanide 257 was prepared in three steps from L-cysteine methyl ester by trityl protection and the classical formamide/dehydration protocol; the aldehyde 258 derived from Fmoc L-valine by the Arndt–Eistert homologation reaction and oxidation/reduction steps and the acid 259 was obtained by coupling Cbz D-pipecolic acid and L-isoleucine methyl ester and final hydrolysis of the ester.
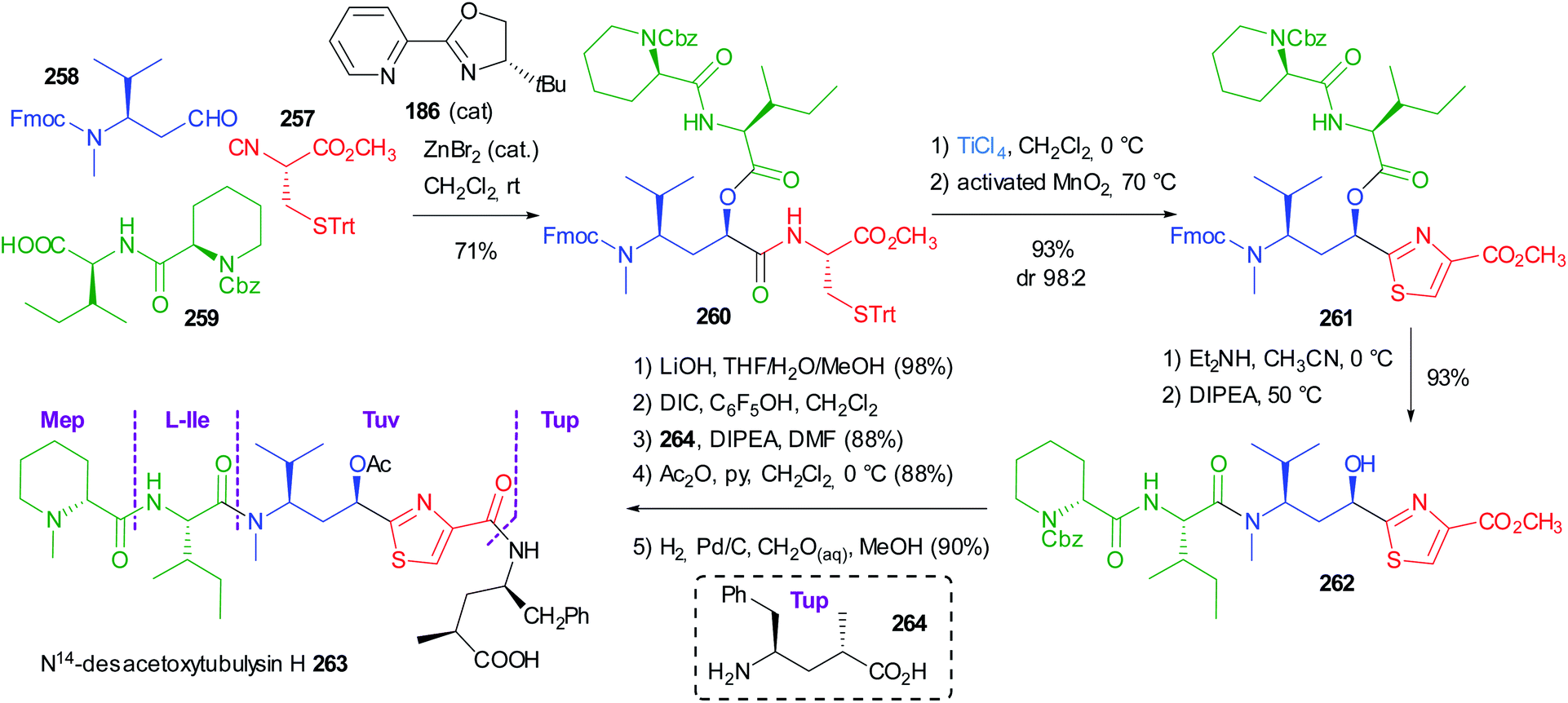 | ||
| Scheme 63 Total synthesis of N14-desacetoxytubulysin H 263via the diastereoselective PADAM strategy. | ||
The Passerini reaction proceeded in a highly diastereoselective fashion thanks to the bulky chiral ligand 186 together with ZnBr2 (cf.Scheme 49). Then the isocyanide residue of product 260 was converted in the peculiar 1,3-thiazole moiety of tubuvaline applying a one-pot two step method using TiCl4-mediated deprotection–cyclodehydration, followed by MnO2 oxidation of the resulting thiazoline to afford 261 in excellent yield without racemisation.128 Basic conditions promoted Fmoc deprotection and O- to N-acyl migration ending the PADAM process. The α-hydroxy peptide 262 was converted into the desired product in five trivial steps. The methyl ester of the tubuvaline residue was hydrolysed and coupled with tubphenylalanine (Tup) 264via the activation of the acid as pentafluorophenyl ester. Finally, the acetylation of the secondary alcohol and a late-stage N-methylation, accomplished by catalytic hydrogenation in the presence of formaldehyde, afford 263 in good yield.
To date, the total synthesis of cyclotheonamide C 256 and N14-desacetoxytubulysin H 263 has been the most elaborate application of the PADAM strategy. In 2015, we reported a new efficient synthesis of telaprevir 273, a specific viral serine protease inhibitor, approved by the FDA in 2011 for the treatment of hepatitis C.44
Since in this case, the bis-homo-PADAM approach using γ-azidoaldehyde 76 failed (see Scheme 17), we synthesized the key intermediate 269 exploring an alternative route (Scheme 64). The oxidation of enantiopure azidoalcohol 265 with stoichiometric iodosobenzene diacetate and catalytic TEMPO radical (2,2,6,6-tetramethyl-1-piperidinyloxy) generated the aldehyde 76 and acetic acid as a by-product, which reacted in a Passerini reaction after the addition of chiral isocyanide 266. The basic treatment of the crude gave alcohol 267 as a 1:1 mixture of diastereoisomers. Compound 268 was obtained with remarkable yield after azide reduction and coupling with L-Boc-tert-glycine. The lack of diastereoselection was compensated for by the stereoconvergent cyclization performed directly on the mixture, obtaining pyrrolidine 269 as a single diastereoisomer. This product was finally converted into intermediate 270 and, finally, to alcohol 271, by sequential deprotection and acylation. Finally, a second Passerini reaction was employed to convert 271 into α-acetoxyamide 272 and telaprevir 273 in further 2 steps.
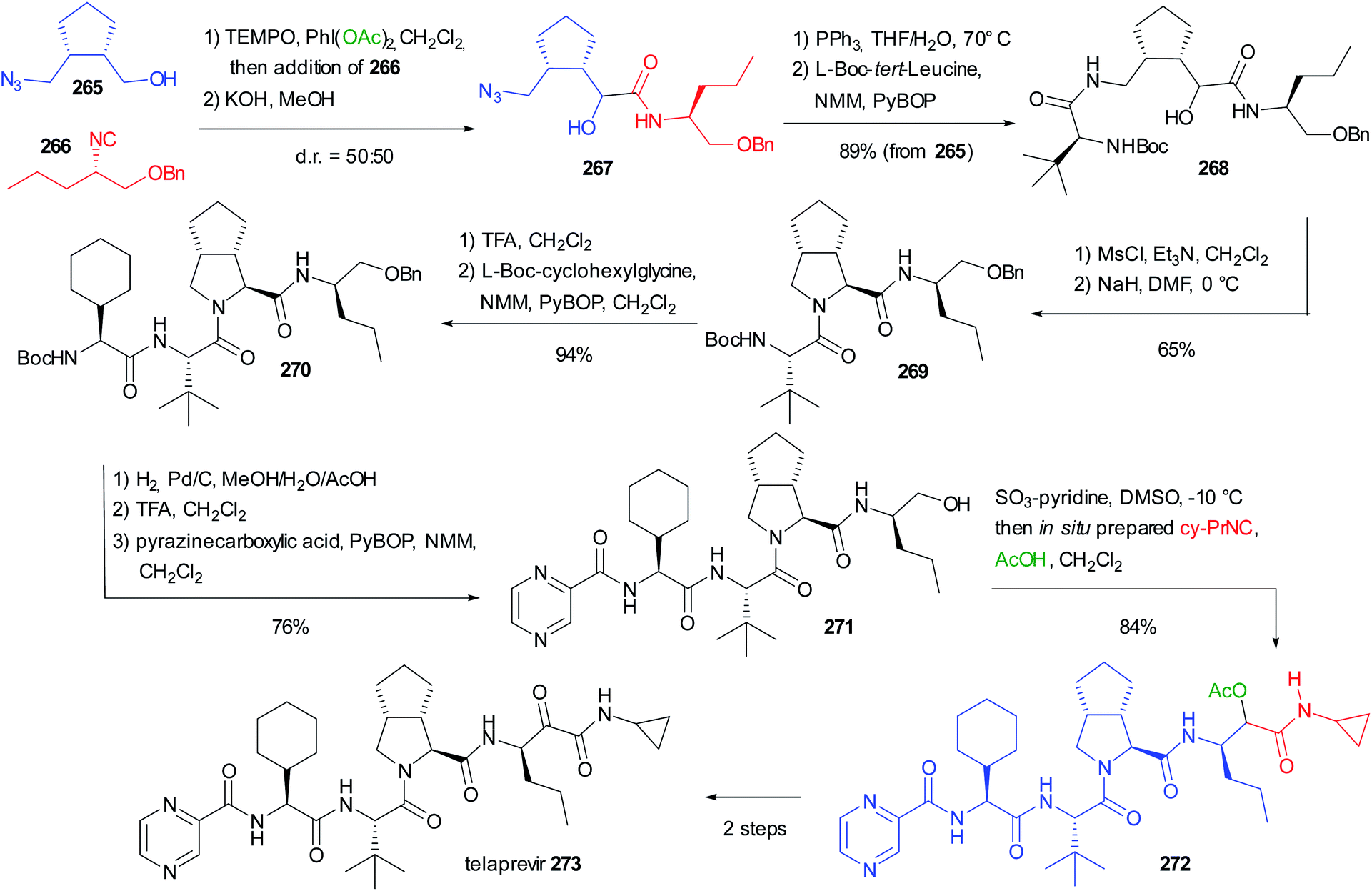 | ||
| Scheme 64 Total synthesis of telaprevir 273via the Passerini reaction and stereoconvergent cyclization. | ||
Furthermore, compound 272 represents an advanced intermediate in another total synthesis of telaprevir, reported by Ruijter.129 This efficient and convergent approach exploits an Ugi-Joullié three component reaction between chiral cyclic imine 277, obtained by enzymatic desymmetrization of the commercially available octahydrocyclopenta[c]pyrrole, carboxylic acid 278 and the chiral isocyanide 276, prepared trough a Passerini reaction (Scheme 65). N-Formylation of commercially available (S)-2-amino-1-pentanol furnished alcohol 274, which was subjected to the one-pot oxidation/Passerini reaction procedure to give formamide 275 in acceptable yield. This protocol avoids the difficult isolation of the aldehyde, which easily dimerized producing a low yield in the Passerini reaction. Dehydration under standard conditions afforded isocyanide 276 without any racemisation.
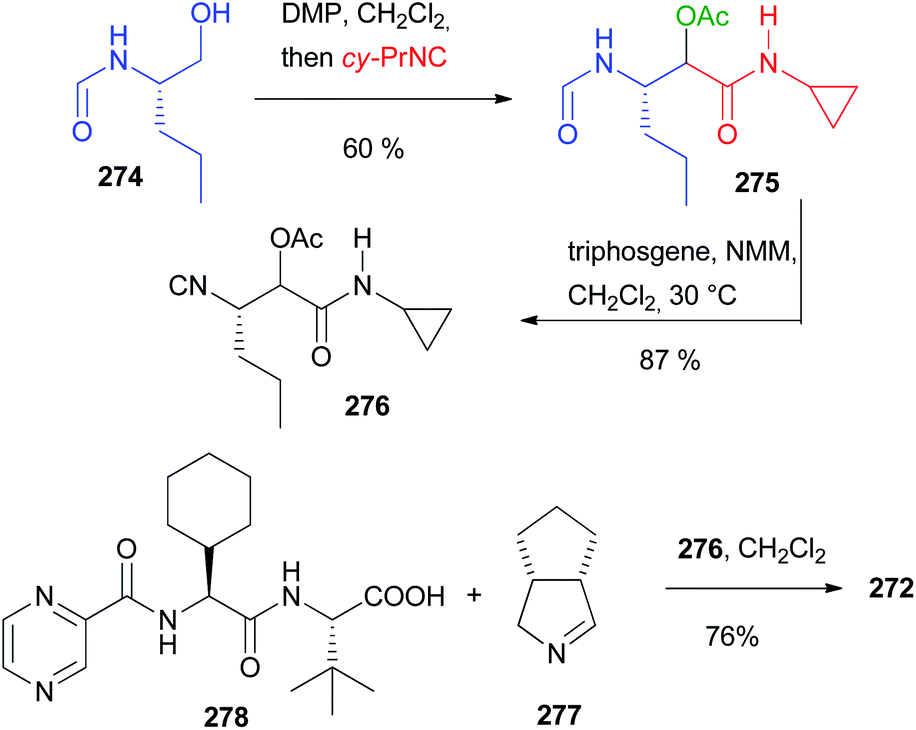 | ||
| Scheme 65 Synthesis of isocyanide 276via the Passerini reaction and its use for the synthesis of 272via the Ugi-Joullié three component reaction. | ||
5.4 Future prospects
As highlighted in the Introduction, the esters are rarely found in natural compounds and active pharmaceutical ingredients. Therefore, the “classical” Passerini reaction is seldom employed in this field. However, its modifications have been successfully applied in the synthesis of important bioactive compounds. Up to now, the most fruitful examples involved the well-studied “truncated” Passerini and the PADAM strategy; however we believe that the more recent modifications developed by the Single Reactant Replacement approach can find future applications in the area of total synthesis.6. Conclusions
In this review, we have tried to highlight the several facets revealed by this centennial reaction in recent years. Thanks to the brilliant work of many researchers around the world, a reaction that was nearly neglected for many years has now eventually entered the Olympus of the most useful methodologies in organic synthesis. It is a great pity that Mario Passerini has not been able to witness all these advancements and to celebrate with us this anniversary or maybe he is.Author contributions
Luca Banfi: conceptualization, visualization, writing – original draft, and writing – review & editing. Andrea Basso: conceptualization, visualization, writing – original draft, and writing – review & editing. Chiara Lambruschini: visualization, writing – original draft, and writing – review & editing. Lisa Moni: visualization, writing – original draft, and writing – review & editing. Renata Riva: conceptualization, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This perspective is dedicated to Prof. Giuseppe Guanti, who was the first to prompt us to engage in research concerning the Passerini reaction.Notes and references
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Academic Press, 2005 Search PubMed.
- R. Riva, L. Banfi and A. Basso, in Science of Synthesis: Multicomponent Reactions, ed. T. J. J. Müller, Thieme, 2014, vol. 1, pp. 327–414 Search PubMed.
- S. Maeda, S. Komagawa, M. Uchiyama and K. Morokuma, Angew. Chem., Int. Ed., 2011, 50, 644–649 CrossRef CAS PubMed.
- R. H. Baker and D. Stanonis, J. Am. Chem. Soc., 1951, 73, 699–702 CrossRef CAS.
- A. Basso, L. Banfi, A. Galatini, G. Guanti, F. Rastrelli and R. Riva, Org. Lett., 2009, 11, 4068–4071 CrossRef CAS PubMed.
- M. Spallarossa, L. Banfi, A. Basso, L. Moni and R. Riva, Adv. Synth. Catal., 2016, 358, 2940–2948 CrossRef CAS.
- L. Banfi and R. Riva, in Organic Reactions, 2005, pp. 1–140 Search PubMed.
- L. Banfi, A. Basso and R. Riva, in Synthesis of Heterocycles Via Multicomponent Reactions I, ed. R. V. A. Orru and E. Ruijter, 2010, vol. 23, pp. 1–39 Search PubMed.
- S. Marcaccini and T. Torroba, in Multicomponent Reactions, 2005, pp. 33–75 Search PubMed.
- S. Sebti and A. Foucaud, Synthesis, 1983, 1983, 546–549 CrossRef.
- R. Bossio, S. Marcaccini and R. Pepino, Liebigs Ann. Chem., 1991, 1107–1108 CrossRef CAS.
- F. De Moliner, S. Crosignani, L. Banfi, R. Riva and A. Basso, J. Comb. Chem., 2010, 12, 613–616 CrossRef CAS PubMed.
- L. Moni, L. Banfi, R. Riva and A. Basso, Synthesis, 2016, 48, 4050–4059 CrossRef CAS.
- J. Wu, J.-C. Liu, L. Wang and M.-W. Ding, Synlett, 2011, 2880–2882 CAS.
- N. Liu, F. Chao, M. G. Liu, N. Y. Huang, K. Zou and L. Wang, J. Org. Chem., 2019, 84, 2366–2371 CrossRef CAS PubMed.
- P. He, J. Wu, Y. B. Nie and M. W. Ding, Tetrahedron, 2009, 65, 8563–8570 CrossRef CAS.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Heterocycl. Compd., 2017, 53, 382–408 CrossRef CAS.
- R. Bossio, S. Marcaccini, R. Pepino and T. Torroba, Synthesis, 1993, 783–785 CrossRef CAS.
- R. Bossio, S. Marcaccini and R. Pepino, Liebigs Ann. Chem., 1994, 527–528 CrossRef CAS.
- S. Marcaccini, R. Pepino, C. F. Marcos, C. Polo and T. Torroba, J. Heterocycl. Chem., 2000, 37, 1501–1503 CrossRef CAS.
- B. Beck, M. Magnin-Lachaux, E. Herdtweck and A. Dömling, Org. Lett., 2001, 3, 2875–2878 CrossRef CAS PubMed.
- J. Rossbach, K. Harms and U. Koert, Eur. J. Org. Chem., 2014, 2014, 993–1006 CrossRef CAS.
- N. A. M. Yehia, W. Antuch, B. Beck, S. Hess, V. Schauer Vukasinovic, M. Almstetter, P. Furer, E. Herdtweck and A. Dömling, Bioorg. Med. Chem. Lett., 2004, 14, 3121–3125 CrossRef CAS PubMed.
- G.-H. Ma, B. Jiang, X.-J. Tu, Y. Ning, S.-J. Tu and G. Li, Org. Lett., 2014, 16, 4504–4507 CrossRef CAS PubMed.
- D. L. Wright, C. V. Robotham and K. Aboud, Tetrahedron Lett., 2002, 43, 943–946 CrossRef CAS.
- R. Huisgen, Proc. Chem. Soc., 1961, 357–396 Search PubMed.
- F. De Moliner, S. Crosignani, A. Galatini, R. Riva and A. Basso, ACS Comb. Sci., 2011, 13, 453–457 CrossRef CAS PubMed.
- F. De Moliner, M. Bigatti, C. De Rosa, L. Banfi, R. Riva and A. Basso, Mol. Divers., 2014, 18, 473–482 CrossRef CAS PubMed.
- B. Beck, G. Larbig, B. Mejat, M. Magnin Lachaux, A. Picard, E. Herdtweck and A. Dömling, Org. Lett., 2003, 5, 1047–1050 CrossRef CAS PubMed.
- A. Schwaeblein and J. Martens, Eur. J. Org. Chem., 2011, 2011, 4335–4344 CrossRef CAS.
- L. Banfi, G. Guanti and R. Riva, Chem. Commun., 2000, 985–986 RSC.
- C. Hulme, M. M. Morrissette, F. A. Volz and C. J. Burns, Tetrahedron Lett., 1998, 39, 1113–1116 CrossRef CAS.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Mol. Divers., 2003, 6, 227–235 CrossRef CAS PubMed.
- M. Zimuwandeyi, F. Kola, A. Lemmerer, D. Brady, A. L. Rousseau and M. L. Bode, Tetrahedron, 2018, 74, 2925–2941 CrossRef CAS.
- A. Basso, L. Banfi, R. Riva, P. Piaggio and G. Guanti, Tetrahedron Lett., 2003, 44, 2367–2370 CrossRef CAS.
- L. Banfi, G. Guanti, R. Riva, A. Basso and E. Calcagno, Tetrahedron Lett., 2002, 43, 4067–4069 CrossRef CAS.
- T. D. Owens, G.-A. Araldi, R. F. Nutt and J. E. Semple, Tetrahedron Lett., 2001, 42, 6271–6274 CrossRef CAS.
- D. Gravestock, A. L. Rousseau, A. C. U. Lourens, H. C. Hoppe, L. A. Nkabinde and M. L. Bode, Tetrahedron Lett., 2012, 53, 3225–3229 CrossRef CAS.
- A. Y. Shaw, F. Medda and C. Hulme, Tetrahedron Lett., 2012, 53, 1313–1315 CrossRef CAS PubMed.
- A. Basso, L. Banfi, G. Guanti, R. Riva and P. Tosatti, Synlett, 2011, 2009–2012 CrossRef CAS.
- F. Morana, A. Basso, R. Riva, V. Rocca and L. Banfi, Chem.–Eur. J., 2013, 19, 4563–4569 CrossRef CAS PubMed.
- J. W. Yang, M. Stadler and B. List, Angew. Chem., Int. Ed., 2007, 46, 609–611 CrossRef CAS PubMed.
- L. Moni, L. Banfi, A. Basso, L. Carcone, M. Rasparini and R. Riva, J. Org. Chem., 2015, 80, 3411–3428 CrossRef CAS PubMed.
- B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed.
- Y. Y. Shen, B. Huang, L. W. Zeng and S. L. Cui, Org. Lett., 2017, 19, 4616–4619 CrossRef CAS PubMed.
- X. Li and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 5446–5448 CrossRef CAS PubMed.
- G. O. Jones, X. Li, A. E. Hayden, K. N. Houk and S. J. Danishefsky, Org. Lett., 2008, 10, 4093–4096 CrossRef CAS PubMed.
- A. Basso, L. Banfi, S. Garbarino and R. Riva, Angew. Chem., Int. Ed., 2013, 52, 2096–2099 CrossRef CAS PubMed.
- S. Garbarino, D. Ravelli, S. Protti and A. Basso, Angew. Chem., Int. Ed., 2016, 55, 15476–15484 CrossRef PubMed.
- https://www.elsevier.com/solutions/reaxys/who-we-serve/reactionflash .
- S. Garbarino, L. Banfi, R. Riva and A. Basso, J. Org. Chem., 2014, 79, 3615–3622 CrossRef CAS PubMed.
- S. Garbarino, S. Protti and A. Basso, Synthesis, 2015, 47, 2385–2390 CrossRef CAS.
- P. Capurro, C. Lambruschini, P. Lova, L. Moni and A. Basso, J. Org. Chem., 2021, 86, 5845–5851 CrossRef CAS PubMed.
- A. Basso, L. Banfi and R. Riva, Molecules, 2011, 16, 8775–8787 CrossRef CAS.
- T. Soeta, Y. Kojima, Y. Ukaji and K. Inomata, Org. Lett., 2010, 12, 4341–4343 CrossRef CAS PubMed.
- F. Ibba, P. Capurro, S. Garbarino, M. Anselmo, L. Moni and A. Basso, Org. Lett., 2018, 20, 1098–1101 CrossRef CAS PubMed.
- M. Anselmo, A. Basso, S. Protti and D. Ravelli, ACS Catal., 2019, 9, 2493–2500 CrossRef CAS.
- E. Bergamaschi, P. Capurro, C. Lambruschini, R. Riva and A. Basso, Eur. J. Org. Chem., 2019, 2019, 5992–5997 CrossRef CAS.
- P. Capurro, E. Bergamaschi, A. Basso and L. Moni, Chem. Heterocycl. Compd., 2020, 56, 467–472 CrossRef CAS.
- L. El Kaim, M. Gizolme and L. Grimaud, Org. Lett., 2006, 8, 5021–5023 CrossRef CAS PubMed.
- E. Martinand-Lurin, A. Dos Santos, E. Robineau, P. Retailleau, P. Dauban, L. Grimaud and L. El Kaim, Molecules, 2016, 21, 14 CrossRef PubMed.
- A. G. Neo and C. F. Marcos, Org. Lett., 2018, 20, 3875–3878 CrossRef CAS PubMed.
- H. Yanai, T. Oguchi and T. Taguchi, J. Org. Chem., 2009, 74, 3927–3929 CrossRef CAS PubMed.
- F. De Moliner, M. Bigatti, L. Banfi, R. Riva and A. Basso, Org. Lett., 2014, 16, 2280–2283 CrossRef CAS PubMed.
- J. M. Saya, R. Berabez, P. Broersen, I. Schuringa, A. Kruithof, R. V. A. Orru and E. Ruijter, Org. Lett., 2018, 20, 3988–3991 CrossRef CAS PubMed.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, RSC Adv., 2018, 8, 31490–31495 RSC.
- I. Ugi and R. Meyr, Chem. Ber., 1961, 94, 2229–2233 CrossRef CAS.
- T. Nixey and C. Hulme, Tetrahedron Lett., 2002, 43, 6833–6835 CrossRef CAS.
- Z. L. Ren, J. C. Liu and M. W. Ding, Synthesis, 2017, 49, 745–754 CAS.
- Q. X. Feng, Z. Y. Mu, G. Yao, J. A. Zhang, H. T. He, Y. L. Pang and J. Xiong, Synlett, 2020, 31, 1003–1006 CrossRef CAS.
- K. M. Allan, C. D. Gilmore and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 4488–4491 CrossRef CAS PubMed.
- Q. Wang, D. X. Wang, M. X. Wang and J. P. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS PubMed.
- S. E. Denmark and Y. Fan, J. Am. Chem. Soc., 2003, 125, 7825–7827 CrossRef CAS PubMed.
- U. Kusebauch, B. Beck, K. Messer, E. Herdtweck and A. Dömling, Org. Lett., 2003, 5, 4021–4024 CrossRef CAS PubMed.
- P. R. Andreana, C. C. Liu and S. L. Schreiber, Org. Lett., 2004, 6, 4231–4233 CrossRef CAS PubMed.
- S. X. Wang, M. X. Wang, D. X. Wang and J. P. Zhu, Angew. Chem., Int. Ed., 2008, 47, 388–391 CrossRef CAS PubMed.
- J. Zhang, S.-X. Lin, D.-J. Cheng, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 14039–14042 CrossRef CAS PubMed.
- S. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Eur. J. Org. Chem., 2007, 2007, 4076–4080 CrossRef.
- S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Org. Lett., 2007, 9, 3615–3618 CrossRef CAS PubMed.
- T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, J. Org. Chem., 2009, 74, 8396–8399 CrossRef CAS PubMed.
- X. Zeng, K. Ye, M. Lu, P. J. Chua, B. Tan and G. Zhong, Org. Lett., 2010, 12, 2414–2417 CrossRef CAS PubMed.
- H. Mihara, Y. Xu, N. E. Shepherd, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 8384–8385 CrossRef CAS PubMed.
- T. Yue, M. X. Wang, D. X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2008, 47, 9454–9457 CrossRef CAS PubMed.
- L. Banfi, A. Basso, L. Moni and R. Riva, Eur. J. Org. Chem., 2014, 2014, 2005–2015 CrossRef CAS.
- C. Lambruschini, L. Moni and L. Banfi, Eur. J. Org. Chem., 2020, 2020, 3766–3778 CrossRef CAS.
- H. Bock and I. Ugi, J. Prakt. Chem., 1997, 339, 385–389 CrossRef CAS.
- S. Marcaccini, D. Miguel, T. Torroba and M. Garcia Valverde, J. Org. Chem., 2003, 68, 3315–3318 CrossRef CAS PubMed.
- R. C. Cioc, V. Estevez, D. J. Van der Niet, C. M. L. Vande Velde, N. G. Turrini, M. Hall, K. Faber, E. Ruijter and R. V. A. Orru, Eur. J. Org. Chem., 2017, 2017, 1262–1271 CrossRef CAS PubMed.
- P. Radha Krishna and K. Lopinti, Synlett, 2007, 2007, 0083–0086 CrossRef.
- D. A. Dos Santos, A. M. Deobald, V. E. Cornelio, R. M. D. Ávila, R. C. Cornea, G. C. R. Bernasconi, M. W. Paixão, P. C. Vieira and A. G. Corrêa, Bioorg. Med. Chem. Lett., 2017, 25, 4620–4627 CrossRef CAS PubMed.
- A. M. Deobald, A. G. Corrêa, D. G. Rivera and M. W. Paixão, Org. Biomol. Chem., 2012, 10, 7681–7684 RSC.
- K. Vlahovicek-Kahlina, M. Vazdar, A. Jakas, V. Smrecki and I. Jeric, J. Org. Chem., 2018, 83, 13146–13156 CrossRef CAS PubMed.
- R. Ramozzi and K. Morokuma, J. Org. Chem., 2015, 80, 5652–5657 CrossRef CAS PubMed.
- A. Jakas, A. Visnjevac and I. Jeric, J. Org. Chem., 2020, 85, 3766–3787 CrossRef CAS PubMed.
- G. Guanti, L. Banfi, E. Narisano, R. Riva and S. Thea, Tetrahedron Lett., 1986, 27, 4639–4642 CrossRef CAS.
- L. Banfi, G. Guanti and R. Riva, Tetrahedron: Asymmetry, 1995, 6, 1345–1356 CrossRef CAS.
- L. Banfi and G. Guanti, Eur. J. Org. Chem., 1998, 1998, 745–757 CrossRef.
- L. Moni, L. Banfi, A. Basso, E. Martino and R. Riva, Org. Lett., 2016, 18, 1638–1641 CrossRef CAS PubMed.
- W. R. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
- G. Vitali Forconesi, L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Molecules, 2020, 25, 3227 CrossRef PubMed.
- L. Moni, 2021, manuscript in preparation.
- L. Moni, L. Banfi, D. Cartagenova, A. Cavalli, C. Lambruschini, E. Martino, R. V. A. Orru, E. Ruijter, J. M. Saya, J. Sgrignani and R. Riva, Org. Chem. Front., 2020, 7, 380–398 RSC.
- N. D. Bartolo, J. A. Read, E. M. Valentin and K. A. Woerpel, Chem. Rev., 2020, 120, 1513–1619 CrossRef CAS PubMed.
- L. Moni, L. Banfi, A. Basso, A. Bozzano, M. Spallarossa, L. Wessjohann and R. Riva, Molecules, 2016, 21, 1153 CrossRef PubMed.
- B. Alcaide, P. Almendros, C. Aragoncillo, R. Callejo, M. P. Ruiz and M. R. Torres, Org. Biomol. Chem., 2015, 13, 1387–1394 RSC.
- T. M. Vishwanatha, B. Giepmans, S. K. Goda and A. Domling, Org. Lett., 2020, 22, 5396–5400 CrossRef CAS PubMed.
- J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, eaas8707 CrossRef PubMed.
- T. Budragchaa, B. Westermann and L. A. Wessjohann, Bioorg. Med. Chem., 2019, 27, 3237–3247 CrossRef CAS PubMed.
- R. Frey, S. G. Galbraith, S. Guelfi, C. Lamberth and M. Zeller, Synlett, 2003, 1536–1538 CAS.
- P. Radha Krishna, G. Dayaker and P. V. Narasimha Reddy, Tetrahedron Lett., 2006, 47, 5977–5980 CrossRef.
- S. Berlozecki, W. Szymanski and R. Ostaszewski, Tetrahedron, 2008, 64, 9780–9783 CrossRef CAS.
- J. R. Falck and S. Manna, Tetrahedron Lett., 1981, 22, 619–620 CrossRef CAS.
- C. Kalinski, H. Lemoine, J. Schmidt, C. Burdack, J. Kolb, M. Umkehrer and G. Ross, Synthesis, 2008, 2008, 4007–4011 CrossRef.
- M. Schiess and D. Seebach, Helv. Chim. Acta, 1983, 66, 1618–1623 CrossRef CAS.
- D. Seebach, G. Adam, T. Gees, M. Schiess and W. Weigand, Chem. Ber., 1988, 121, 507–517 CrossRef CAS.
- T. Carofiglio, P. G. Cozzi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 2002, 12, 2726–2736 CrossRef.
- C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H. J. Kempf and R. Zeun, Bioorg. Med. Chem., 2008, 16, 1531–1545 CrossRef CAS PubMed.
- A. Gernet, V. Ratovelomanana-Vidal, J. L. Pirat, D. Virieux and T. Ayad, Eur. J. Org. Chem., 2020, 2020, 6497–6500 CrossRef CAS.
- R. C. Cioc, D. J. van der Niet, E. Janssen, E. Ruijter and R. V. Orru, Chem.–Eur. J., 2015, 21, 7808–7813 CrossRef CAS PubMed.
- F. M. Miloserdov, M. S. Kirillova, M. E. Muratore and A. M. Echavarren, J. Am. Chem. Soc., 2018, 140, 5393–5400 CrossRef CAS PubMed.
- W. S. Yap, C. Y. Gan, Y. Y. Low, Y. M. Choo, T. Etoh, M. Hayashi, K. Komiyama and T. S. Kam, J. Nat. Prod., 2011, 74, 1309–1312 CrossRef CAS PubMed.
- T. Ideguchi, T. Yamada, T. Shirahata, T. Hirose, A. Sugawara, Y. Kobayashi, S. Omura and T. Sunazuka, J. Am. Chem. Soc., 2013, 135, 12568–12571 CrossRef CAS PubMed.
- T. Yamada, T. Ideguchi-Matsushita, T. Hirose, T. Shirahata, R. Hokari, A. Ishiyama, M. Iwatsuki, A. Sugawara, Y. Kobayashi, K. Otoguro, S. Omura and T. Sunazuka, Chem.–Eur. J., 2015, 21, 11855–11864 CrossRef CAS PubMed.
- J. S. Kumar, S. C. Jonnalagadda and V. R. Mereddy, Tetrahedron Lett., 2010, 51, 779–782 CrossRef PubMed.
- S. Faure, T. Hjelmgaard, S. P. Roche and D. J. Aitken, Org. Lett., 2009, 11, 1167–1170 CrossRef CAS PubMed.
- S. P. Roche, S. Faure, L. El Blidi and D. J. Aitken, Eur. J. Org. Chem., 2008, 2008, 5067–5078 CrossRef.
- P. Raman, H. Razavi and J. W. Kelly, Org. Lett., 2000, 2, 3289–3292 CrossRef CAS PubMed.
- A. Znabet, M. M. Polak, E. Janssen, F. J. J. de Kanter, N. J. Turner, R. V. A. Orru and E. Ruijter, Chem. Commun., 2010, 46, 7918–7920 RSC.
Footnotes |
| † All the authors equally contributed to this paper. |
| ‡ Unfortunately, Stefano Marcaccini passed away in 2012 and thus he cannot celebrate with us this anniversary, as he would have certainly liked. |
| This journal is © The Royal Society of Chemistry 2021 |