
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissecting the activation of insulin degrading enzyme by inositol pyrophosphates and their bisphosphonate analogs†
Sarah
Hostachy
 a,
Tillmann
Utesch
a,
Katy
Franke
a,
Gillian Leigh
Dornan
a,
David
Furkert
ab,
Berke
Türkaydin
a,
Volker
Haucke
a,
Han
Sun
a and
Dorothea
Fiedler
*ab
a,
Tillmann
Utesch
a,
Katy
Franke
a,
Gillian Leigh
Dornan
a,
David
Furkert
ab,
Berke
Türkaydin
a,
Volker
Haucke
a,
Han
Sun
a and
Dorothea
Fiedler
*ab
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle Str. 10, 13125 Berlin, Germany. E-mail: fiedler@fmp-berlin.de
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
First published on 8th July 2021
Abstract
Inositol poly- and pyrophosphates (InsPs and PP-InsPs) are densely phosphorylated eukaryotic messengers, which are involved in numerous cellular processes. To elucidate their signaling functions at the molecular level, non-hydrolyzable bisphosphonate analogs of inositol pyrophosphates, PCP-InsPs, have been instrumental. Here, an efficient synthetic strategy to obtain these analogs in unprecedented quantities is described – relying on the use of combined phosphate ester-phosphoramidite reagents. The PCP-analogs, alongside their natural counterparts, were applied to investigate their regulatory effect on insulin-degrading enzyme (IDE), using a range of biochemical, biophysical and computational methods. A unique interplay between IDE, its substrates and the PP-InsPs was uncovered, in which the PP-InsPs differentially modulated the activity of the enzyme towards short peptide substrates. Aided by molecular docking and molecular dynamics simulations, a flexible binding mode for the InsPs/PP-InsPs was identified at the anion binding site of IDE. Targeting IDE for therapeutic purposes should thus take regulation by endogenous PP-InsP metabolites into account.
Introduction
Inositol-based signaling molecules are central eukaryotic messengers and include the highly phosphorylated, diffusible inositol polyphosphates (InsPs) and inositol pyrophosphates (PP-InsPs). The PP-InsPs are most densely phosphorylated, and one or two high-energy pyrophosphate groups are attached to the myo-inositol scaffold. Genetic studies have linked PP-InsPs to a wide range of biological processes in mammals, including insulin signaling, spermatogenesis, and blood coagulation.1–3 To mediate such varied cellular responses, distinct molecular mechanisms of action have been proposed for PP-InsPs. These mechanisms comprise allosteric regulation of enzyme activity, competition with phosphatidyl inositols for lipid binding domains, and protein pyrophosphorylation.4–7 Although allosteric regulation of protein function by PP-InsPs is readily conceivable, it is currently not well understood how subtle structural changes of the ligand are translated to notable alterations in protein activity.Insulin-degrading enzyme (IDE) is a conserved zinc metalloprotease, which is involved in the degradation of numerous bioactive peptides, including insulin, glucagon, amyloid beta peptides, bradykinin and angiotensin.8,9 Consequently, potent and selective modulation of IDE activity could provide a gateway to treat major diseases, such as type 2 diabetes and Alzheimer's disease. Despite the large therapeutic potential of targeting IDE, the regulatory mechanisms governing IDE substrate selectivity are not well characterized. Notably, IDE possesses an anion binding site remote from its active-site, and binding of polyanions, such as ATP, can promote the proteolytic cleavage of some IDE substrates, in particular short peptide substrates.10–14 It was recently demonstrated that also phosphatidylinositols and inositol polyphosphates enhanced proteolytic activity of IDE, but only limited data was presented on the effect of PP-InsPs.15
The investigation of the properties and functions of PP-InsPs is often hindered by the lability of the pyrophosphate moieties, which can undergo hydrolysis in physiological contexts or during synthesis. Therefore, non-hydrolyzable methylene bisphosphonate analogs of PP-InsPs have been developed (PCP-InsPs, Fig. 1). These stable mimics have been applied as PP-InsP surrogates in biochemical assays,16–19 structural studies,20 as mechanistic tools17,21,22 and as affinity reagents.23,24 Previous strategies to obtain the PCP-InsPs relied on phosphoryl chloride (PCP-Cl) reagents to append the PCP moiety in modest yield, while requiring high stoichiometry of the reagents and stringent anhydrous conditions (Scheme S1†).16,18,20 These limitations constricted the amounts of PCP-analogs available, imposing a bottleneck on subsequent biological, biochemical, and structural studies. We now employed phosphoramidite reagents to install the PCP-groups onto relatively hindered hydroxyl groups on the inositol scaffold, on a large scale. Using a convenient precipitation-based purification strategy, the PCP-analogs were obtained in excellent yields. The PCP-analogs, alongside their natural counterparts, were then used to investigate how PP-InsPs fine-tune the activity of insulin degrading enzyme (IDE). The PP-InsPs differentially modulated the activity of IDE towards distinct short peptide substrates, likely by targeting the anion binding site, as suggested by molecular dynamics simulations.
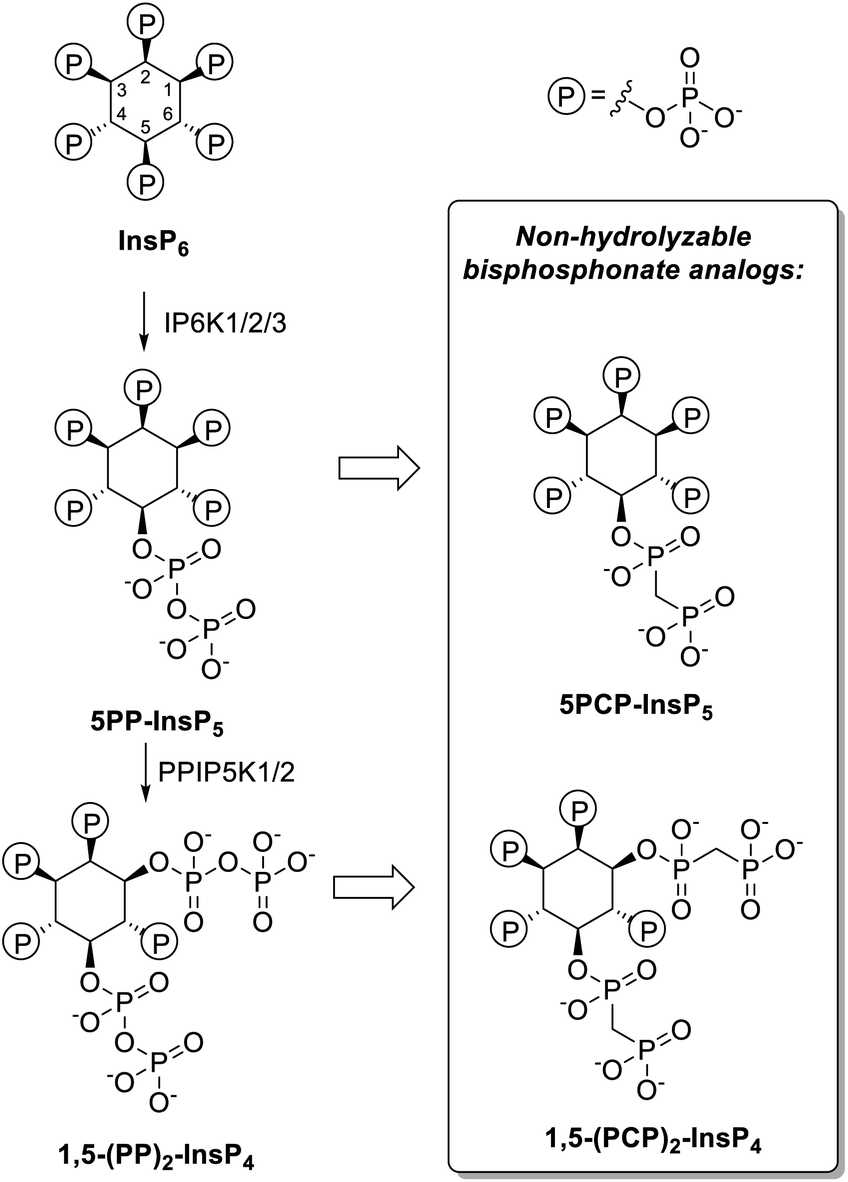 | ||
| Fig. 1 Left: Major biosynthetic pathway for the PP-InsPs in mammals. Right: non-hydrolyzable bisphosphonate analogs. | ||
Results and discussion
Efficient large-scale synthesis of PCP-InsPs
Phosphoramidite reagents display high reactivity towards O-nucleophiles, and exhibit lower steric hinderance at PIII centers, compared to PV reagents. We thus sought to generate and apply phosphoramidite-containing PCP-building blocks, containing both a phosphoester moiety (PV center) and a phosphoramidite functional group (PIII center), for the synthesis of PCP-InsP analogs. Related PCP-amidite reagents have recently been utilized to obtain non-hydrolyzable analogs of nucleotides and flavin adenine dinucleotide (FAD).25–27 Synthetic routes to obtain PCP-InsPs involve either acid-resistant, hydrogenation-labile benzyl protecting groups (Scheme S1,† route A) or acid-labile, hydrogenation-resistant ethyl protecting groups (Scheme S1,† route B) on the bisphosphonate moiety. Consequently, we synthesized benzyl- and ethyl-protected PCP-amidites 3a and 3b (Scheme 1). Briefly, methylphosphonate was deprotonated using lithium diisopropylamide (LDA), subsequently reacted with the corresponding chlorophosphoramidite, and purified by silica gel chromatography. Both 3a and 3b were obtained in good yields (60–66%) and could be stored for several months at −20 °C without significant degradation. This synthetic strategy was also applied to obtain a 13C-labeled version of 3b in similarly good yields (69%, see the ESI†).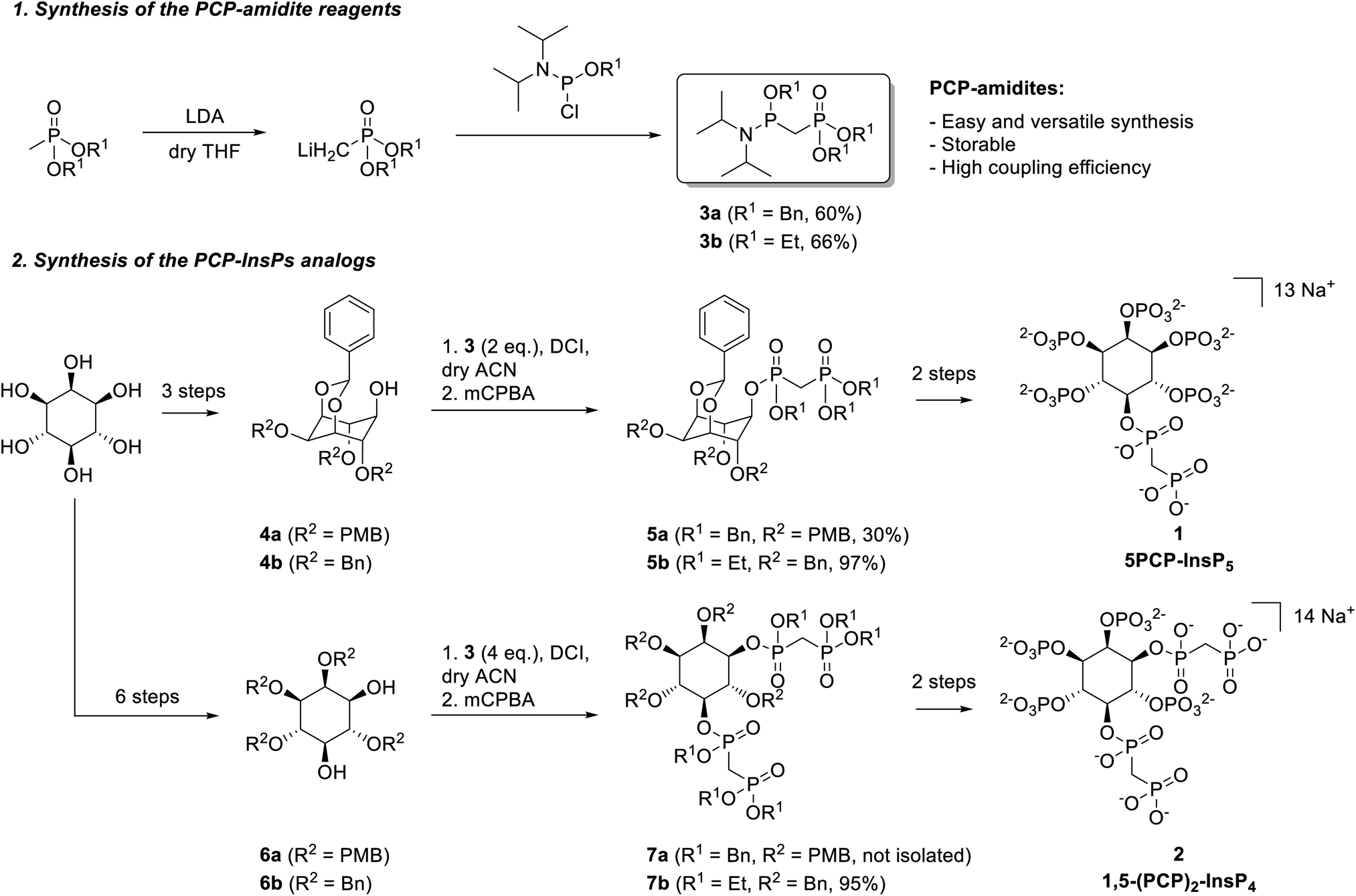 | ||
| Scheme 1 PCP-amidite reagents for the synthesis of PCP-InsPs. | ||
PCP-amidite reagents 3a and 3b were then employed for the synthesis of 5PCP-InsP5 (1) (Scheme S2†). Benzyl-protected PCP-phosphoramidite 3a (2 eq.) was reacted with 1 eq. of protected inositol 4a28,29 in the presence of 4,5-dicyanoimidazole (DCI), followed by oxidation with 3-chloroperbenzoic acid (mCPBA) (Scheme 1). While the secondary hydroxyl group at the 5-position is quite sterically hindered in 4a, this material can be obtained in only three steps from myo-inositol. Nevertheless, the reaction with phosphoramidite 3a provided the desired coupled product 5a in 30% yield. Although this yield was an improvement compared to previous phosphoryl chloride-based strategies, we also explored the less hindered, ethyl protected, PCP-amidite reagent 3b. Using the same reaction conditions as for 5a, the desired product 5b was obtained in excellent yield (up to 97%). Scaling up this key-coupling step enabled us to generate intermediate 5b on a gram-scale. Overall, product 5b was generated (i) with higher yields than previous literature reports for appending the PCP moiety, (ii) using a lower stoichiometry of the PCP reagent, and (iii) by derivatizing a sterically hindered hydroxyl group.
Hydrogenolysis of compound 5b afforded the pentaol, which was directly submitted to phosphitylation and oxidation, to yield compound S3 in high purity after HPLC purification. This compound was deprotected using TMSBr18 and the product was isolated via selective precipitation with magnesium ions, followed by resolubilization with IRC-748 Amberlite resin.30 Although TMSBr deprotection required extended reaction times to ensure complete removal of the ethyl protecting groups (3 days), the reaction worked robustly and in good yield (76%). Altogether, this synthesis was easily scalable, and 5PCP-InsP5 (1) could be isolated in batches of 150–200 mg at a time (Scheme 1).
Encouraged by these results, we next applied the PCP-amidite reagents to the synthesis of 1,5-(PCP)2-InsP4 (Scheme S3†). Appending two PCP-moieties had been painstaking in the past using the phosphoryl chloride reagents, which required reagent recycling, and provided only modest yields (20–30%).18,20 Likewise, reacting 4 eq. of PCP-amidite 3a with diol 6a resulted in a mixture of the desired product 7a with compounds bearing only one bisphosphonate moiety. The steric hindrance of the benzyl group presumably prevented the full conversion to the di-functionalized product. By contrast, when 4 eq. of PCP-amidite 3b were reacted with diol 6b, we obtained the doubly modified bisphosphonate 7b in excellent yield (95%). Once again, this strategy proved scalable, providing over one gram of compound 7b. The bisphosphonate analog 1,5-(PCP)2-InsP4 (2) was obtained from intermediate 7b following a synthetic route similar to the one used for 1, and batches of 50–100 mg of 2 were isolated at a time.
Differential modulation of IDE activity by PP-InsPs
We next wanted to investigate the influence of PCP-InsPs/PP-InsPs on the activity of insulin degrading enzyme (IDE) (Fig. 2A). Membrane-bound and diffusible inositol phosphates were recently shown to stimulate cleavage of IDE substrates, in particular the cleavage of small peptide substrates.15 However, only little data was provided for PP-InsPs. Given the wide substrate scope of IDE, and the increasing interest in PP-InsP signaling and metabolism, elucidating whether PP-InsPs govern IDE activation and substrate selectivity is important before further exploring IDE as a pharmacological target.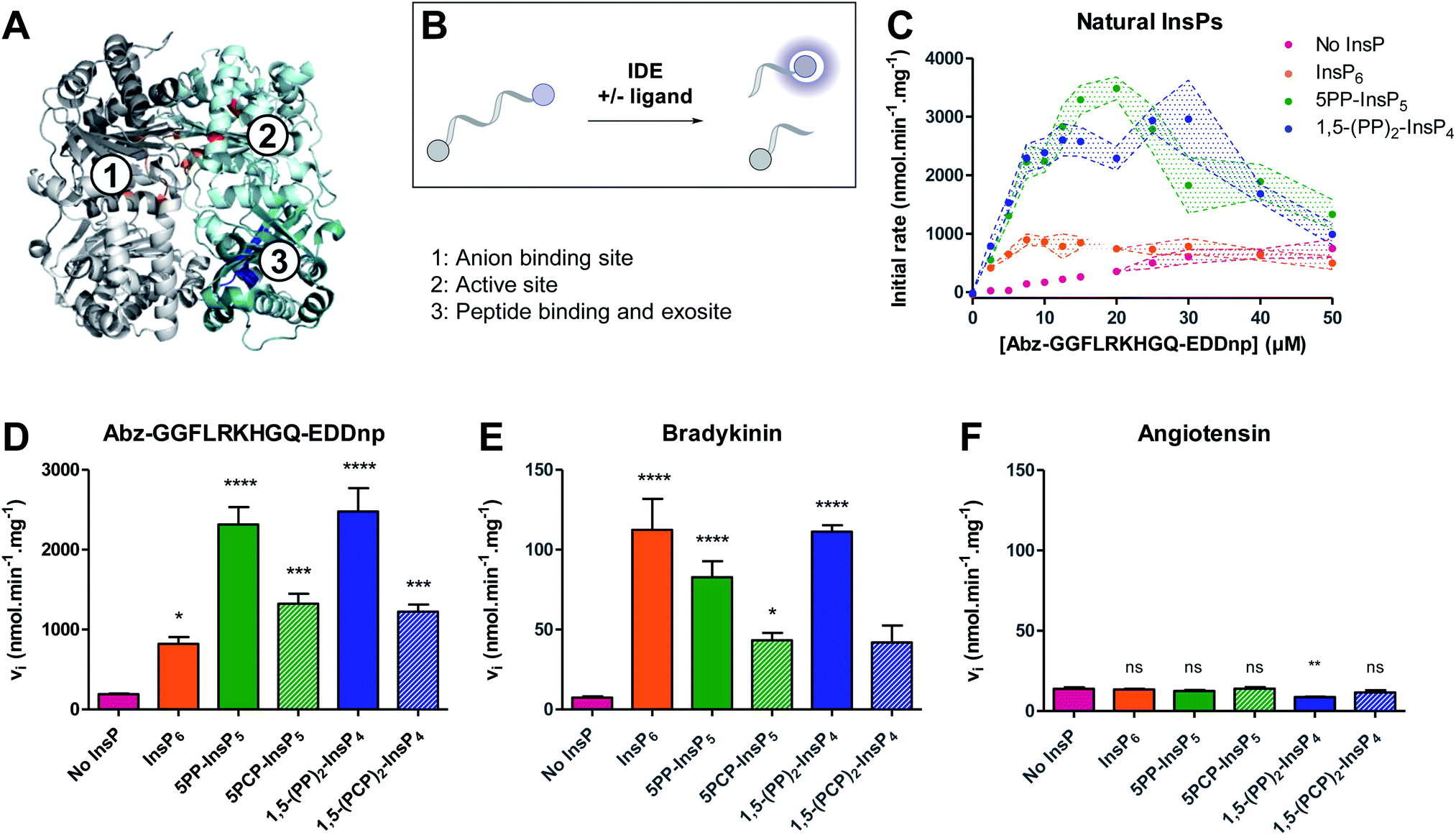 | ||
| Fig. 2 Inositol pyrophosphates modulate IDE activity distinctly, depending on the small peptide substrate. (A) Structure of rat IDE (pdb: 3tuv; identity to human IDE: 95%), highlighting relevant binding regions. IDE is composed of four domains, represented here in pale cyan (domain 1), teal (domain 2), light grey (domain 3) and dark grey (domain 4). Amino acids of relevant sites are highlighted in different colors: (1) anion binding site, orange; (2) active site, red; (3) peptide exosite, blue. (B) Principle of the fluorogenic activity assay. When intact, the fluorescence of the peptide is quenched. Upon cleavage of the peptide by IDE, fluorescence is recovered and can be measured as a read-out for enzyme activity. (C) Activity of IDE on a fluorogenic peptide in presence or absence of InsPs (5 μM). Experiments were performed at 37 °C in 50 mM Tris buffer, pH 7.4, 50 mM NaCl. All experiments were performed at least in triplicate, and standard error of the mean of these replicates are shown as intervals. (D–F) Activity of IDE for various peptides (peptide concentration: 10 μM, InsP concentration if present: 5 μM; IDE concentration: 0.05 μg mL−1). Error bars show standard error of the mean of at least three independent experiments. Statistical significance was assessed using one-way ANOVA. All samples containing InsP6/PP-InsPs were compared to the “No InsP” sample. The result of Dunnett's multiple comparison test is shown above each bar. (ns: non significant, p >0.1; no marker: p <0.1; *: p <0.05; **: p <0.01; ***: p <0.001, ****: p <0.0001). (F) is reproduced on a magnified scale in the ESI (Fig. S3†). | ||
The activation of IDE by PP-InsPs was assayed using a well-established model fluorogenic peptide (Fig. 2B).31,32 Cleavage of the model peptide by IDE results in a fluorescence recovery that can be followed over time. Initial rates were measured for various peptide concentrations in the absence or presence of InsP6, 5PP-InsP5, 1,5-(PP)2-InsP4, or their non-hydrolyzable counterparts. All ligands activated IDE, in particular at low peptide concentrations (<20 μM) (Fig. 2C and S1†). At higher peptide concentrations, all ligands displayed a substrate inhibition profile. Such behavior had already been observed in the case of the lower inositol phosphates, and could be due to competition with the peptide for a secondary substrate binding site.15 Interestingly, InsP6 was the least efficient activator, whereas 5PP-InsP5 and 1,5-(PP)2-InsP4 activated the cleavage of the peptide most strongly (Fig. 2C and S1†). The additional pyrophosphate on 1,5-(PP)2-InsP4 did not seem to promote further activation of the enzyme as compared to 5PP-InsP5. The PCP analogs mirrored this trend and induced the proteolytic cleavage by IDE more strongly than InsP6, although they exhibited lower potency than the natural molecules (Fig. 2C, D and S1†). Because the pKa values for the methylene bisphosphonate group are slightly elevated compared to the pyrophosphate group,33 the overall protonation states of the non-hydrolyzable analogs differ at physiological pH, potentially altering their interaction with the polyanion binding site of the protein.
We next determined the effect of PP-InsPs on IDE activation with respect to the natural substrates bradykinin and angiotensin. Using an HPLC assay, the initial rates of the enzyme were measured in the absence and presence of InsP6 and PP-InsP/PCP-InsP ligands. Cleavage of bradykinin was strongly activated by InsP6 and PP-InsPs (Fig. 2E). Contrary to the model peptide substrate, the strength of activation was very similar for InsP6, 5PP-InsP5 and 1,5-(PP)2-InsP4, and not more pronounced for the inositol pyrophosphates. The PCP-InsPs again displayed less potency as activators, compared to the natural molecules. Unexpectedly, InsP6 and PP-InsP/PCP-InsPs had no activating effect on the proteolytic cleavage of angiotensin. Neither InsP6, nor the PP-InsPs/PCP-InsPs accelerated the rate of angiotensin cleavage. In fact, angiotensin cleavage was even slightly inhibited by 1,5-(PP)2-InsP4 (Fig. 2F).
Overall, the InsPs/PP-InsPs influenced IDE activity in markedly different ways, depending on the peptide substrate. While cleavage of the model peptide substrate was most strongly promoted by the pyrophosphate-containing metabolites 5PP-InsP5 and 1,5-(PP)2-InsP4, IDE catalyzed cleavage of bradykinin was activated similarly by InsP6, 5PP-InsP5 and 1,5-(PP)2-InsP4. By contrast, no activating effect of InsPs/PP-InsPs was observed with angiotensin as the substrate, indicating a high degree of flexibility and adaptability in the system.
PP-InsPs target the anion binding site of IDE
Deciphering the interplay between IDE, InsPs/PP-InsPs and distinct peptide substrates at the molecular level could advance our understanding of the regulation of this central enzyme. Unfortunately, attempts to obtain co-crystal structures of IDE and the PCP-InsP analogs remained unsuccessful. Also, HDX-MS data could not pinpoint distinct conformational changes upon binding of PP-InsP ligands. While a decrease in deuterium exchange rate was evident in the peptide binding region (peptide 351–359) in samples containing both the bradykinin substrate and 5PCP-InsP5, a significant effect on the PCP-InsP–IDE-substrate complex could not be described (Fig. S4†). These results may indicate a more dynamic nature of the PP-InsP–IDE interactions, making it challenging to capture them experimentally.As an alternative approach, we employed computational methods to explore possible interactions between PP-InsPs and IDE. Potential binding modes of InsP6 and 5PCP-InsP5 to IDE were first investigated by molecular docking using GLIDE module implemented in the program Schrödinger.34,35 These two ligands were chosen, since DFT-optimized structures of InsP6 and 5PCP-InsP5 based on density functional theory (DFT) (Table S2†) had been described previously.20,36 Both ligands were docked to the ATP binding site of rat IDE, which exhibits a sequence identity of 95% with respect to human IDE (pdb: 3tuv).10 For both ligands, two binding poses were identified close to the ATP binding site, independent of their ionization state (Fig. 3A and B). In pose 1, similar side-chain interactions (R429, R892, K896, and K898) to those reported for ATP10 were involved in the recognition of InsP-phosphoryl groups. As InsP6 and 5PCP-InsP5 do not, or only slightly, overlap with ATP, further contacts of the ligands in pose 1 were established with R893, K906, and R920. In contrast, in pose 2, which was more remote from the ATP binding site, the ligands interacted mostly with K425, K571, and K632. Both poses were populated in the molecular docking analysis by various energetically comparable sub-poses with minor structural variations, including rotation and translation (Fig. S5†). These observations indicate a high degree of flexibility of InsP6 and 5PCP-InsP5 when binding to IDE. As a result, it was impossible to assign interactions of specific phosphoryl groups with individual amino acids.
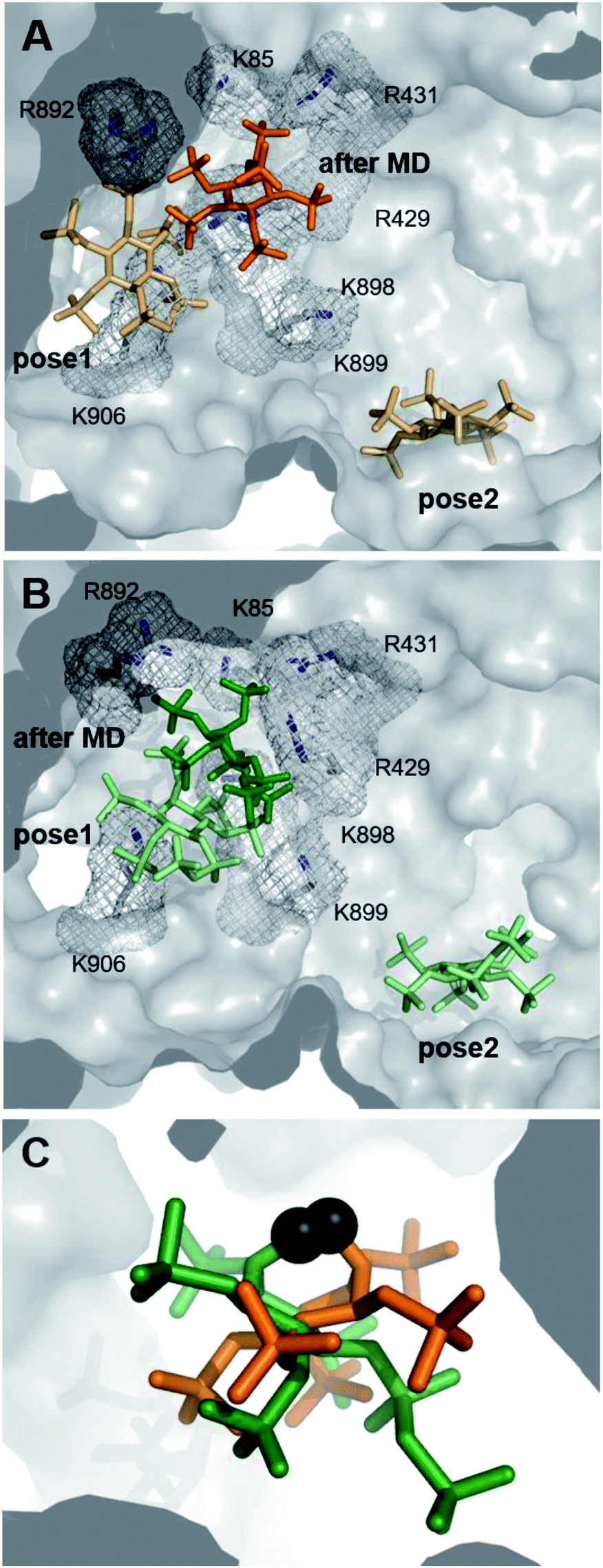 | ||
| Fig. 3 Computational investigation of the interactions of InsP6 and 5PCP-InsP5 with IDE. Two binding poses for InsP6 (A) and 5PCP-InsP5 (B) were obtained by docking experiments, which converged to one binding position after MD simulations. One exemplary pose after 100 ns MD simulation (pose after MD) is shown for both InsP. Basic amino acids (Lys and Arg) in pose after MD are highlighted as sticks and in the mesh representation. Protons are omitted for clarity. (C) Superimposition of MD binding sites of InsP6 (orange) and 5PCP-InsP5 (green) illustrate the variable orientations of InsPs when binding to IDE. Protons are omitted for clarity. Phosphate groups at the 2-position of the inositol ring are represented as dark grey spheres to facilitate comparison. | ||
To optimize the predicted docking poses in a flexible protein environment, and to validate our hypothesis of heterogeneous InsP–IDE complexes, both poses of InsP6 and 5PCP-InsP5 were subjected to all-atom molecular dynamics (MD). During the MD simulations, both InsP molecules underwent noticeable reorientation with respect to their starting poses, which reveals the limitations of the rigid docking approach. Interestingly, all models converged to only one principal binding mode, as illustrated in Fig. 3A and B. In this binding mode, however, the InsP/PP-InsP molecules adopted a wide range of orientations (Fig. 3C and S6†), varying in rotation and slightly in translation. Comparison of the binding poses of InsP6 and 5PCP-InsP5 revealed that both ligands share interactions with the same key amino acids (R429, R431, R892, R899) (Fig. S6†). The interaction with K85, K898, and K906 occurred sporadically but not specifically for either of the two InsP molecules.
Taken together, the computational approach identified one main binding mode for InsP6 and 5PCP-InsP5 at the anion binding site of IDE. The binding to this basic cavity, however, was highly flexible, with a heterogeneous population of ligand conformations, which might explain the experimental difficulty for characterizing these protein–ligand complexes by common structural biology techniques. The observed heterogeneity also provides a first clue as to how these structurally very similar molecules may be able to dictate substrate specificity.
Conclusion
In sum, we utilized combined phosphoester–phosphoramidite reagents to efficiently synthesize and isolate non-hydrolyzable analogs of PP-InsPs. Two PCP-amidite reagents, compatible with distinct protecting group schemes, were synthesized in good yields. These new building blocks consolidate existing PCP-amidite reagents and will enable alternative synthetic strategies to install methylene bisphosphonate groups on chemically and/or pharmaceutically relevant target structures. The PCP-amidite reagents were then applied to the synthesis of PCP-InsPs, yielding unprecedented amounts of these analogs, due to the efficient coupling of the PCP-amidites to sterically encumbered hydroxyl groups. Beyond the high coupling efficiency, this strategy also holds promise for further derivatization on the bisphosphonate moiety, as exemplified here by the introduction of a 13C-label into the PCP group. In light of our current computational and biochemical data, it appears that the protonation states of InsPs/PP-InsPs may be an important factor governing the interactions with IDE. In the future, incorporating a fluorinated bisphosphonate group could be of interest, since it better mimics the pyrophosphate group with regard to the pKa values.33We then utilized the PCP-InsP analogs, alongside their natural counterparts, to interrogate the mechanism of activation of IDE by PP-InsPs. Biochemical characterization revealed a strong dependence of protein activity on both the InsP/PP-InsP ligands and the peptide substrates. Subsequent MD simulations could pinpoint the interaction between IDE and InsPs/PP-InsPs to the anion binding site of IDE, and highlighted the high degree of flexibility in these interactions, as illustrated by the wide range of orientations of the ligands. To capture these dynamic interactions experimentally with high resolution will require the development of new molecular tools, such as PP-InsP–protein cross-linking reagents.
Because several IDE substrates have opposing functions (bradykinin versus angiotensin, insulin versus glucagon), it is interesting to speculate whether a fine-tuned balance of PP-InsP metabolites can influence complex organismal responses, such as vasodilation and carbohydrate metabolism, via IDE. As potent and selective pharmacological tools targeting PP-InsP metabolism are now becoming available,37,38 we should be able to address these questions in a more complex environment in the future. In return, understanding the modulation of IDE substrate multiplicity by PP-InsPs would also inform current drug development programs, targeting the inositol kinases.
Data availability
The data supporting this article have been included as part of the supplementary material.Author contributions
S. H. and Do. F. designed the experiments and wrote the manuscript. S. H., Da. F. and K. F. synthesized and characterized the compounds described in this paper. S. H. performed all biochemical assays and analyzed the data. G. L. D. performed the HDX experiments and analyzed the data, supported by V. H., H. S. and T. U. designed the computational studies, T. U. and B. T. performed them and T. U. and H. S. analyzed the data. All authors reviewed the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully thank the Tang lab for the generous gift of plasmid constructs for IDE expression, Dr Nicolás Veiga for kindly providing DFT-optimized structures of InsP6 and 5PCP-InsP5, and Dr Anja Schütz for crystallization experiments, and Dr Edgar Specker for HRMS analysis. G. L. D. was supported by a CIHR Banting Fellowship. Da. F. and S. H. were supported by funding from the Swiss National Foundation Sinergia Grant CRSII5_170925. S. H. also thank DAAD for a postdoctoral fellowship. The work was supported by the Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat (to T. U. and H. S.). We thank North-German Supercomputing Alliance (HLRN) for providing us computational time. We thank all members of the Fiedler group for careful reading of the manuscript.References
- R. Bhandari, K. R. Juluri, A. C. Resnick and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2349–2353 CrossRef CAS PubMed.
- A. Chakraborty, M. A. Koldobskiy, N. T. Bello, M. Maxwell, J. J. Potter, K. R. Juluri, D. Maag, S. Kim, A. S. Huang, M. J. Dailey, M. Saleh, A. M. Snowman, T. H. Moran, E. Mezey and S. H. Snyder, Cell, 2010, 143, 897–910 CrossRef CAS PubMed.
- S. Ghosh, D. Shukla, K. Suman, B. J. Lakshmi, R. Manorama, S. Kumar and R. Bhandari, Blood, 2013, 122, 1478–1486 CrossRef CAS PubMed.
- N. A. Gokhale, A. Zaremba, A. K. Janoshazi, J. D. Weaver and S. B. Shears, Biochem. J., 2013, 453, 413–426 CrossRef CAS PubMed.
- R. Bhandari, A. Saiardi, Y. Ahmadibeni, A. M. Snowman, A. C. Resnick, T. Z. Kristiansen, H. Molina, A. Pandey, J. K. Werner Jr, K. R. Juluri, Y. Xu, G. D. Prestwich, Parang K. and S. H Snyder, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15305–15310 CrossRef CAS PubMed.
- L. Lorenzo-Orts, D. Couto and M. Hothorn, New Phytol., 2020, 225, 637–652 CrossRef PubMed.
- S. B. Shears, J. Cell. Physiol., 2018, 233, 1897–1912 CrossRef CAS PubMed.
- E. Malito, L. A. Ralat, M. Manolopoulou, J. L. Tsay, N. L. Wadlington and W.-J. Tang, Biochemistry, 2008, 47, 12822–12834 CrossRef CAS PubMed.
- E.-S. Song, M. A. Juliano, L. Juliano and L. B. Hersh, J. Biol. Chem., 2003, 278, 49789–49794 CrossRef CAS PubMed.
- N. Noinaj, E. S. Song, S. Bhasin, B. J. Alper, W. K. Schmidt, L. B. Hersh and D. W. Rodgers, J. Biol. Chem., 2012, 287, 48–57 CrossRef CAS PubMed.
- E. S. Song, M. Ozbil, T. Zhang, M. Sheetz, D. Lee, D. Tran, S. Li, R. Prabhakar, L. B. Hersh and D. W. Rodgers, PLoS One, 2015, 10, e0133114 CrossRef PubMed.
- H. Im, M. Manolopoulou, E. Malito, Y. Shen, J. Zhao, M. Neant-Fery, C.-Y. Sun, S. C. Meredith, S. S. Sisodia, M. A. Leissring and W.-J. Tang, J. Biol. Chem., 2007, 282, 25453–25463 CrossRef CAS PubMed.
- Y. Shen, A. Joachimiak, M. R. Rosner and W.-J. Tang, Nature, 2006, 443, 870–874 CrossRef CAS PubMed.
- E. S. Song, M. A. Juliano, L. Juliano, M. G. Fried, S. L. Wagner and L. B. Hersh, J. Biol. Chem., 2004, 279, 54216–54220 CrossRef CAS PubMed.
- E. S. Song, H. Jang, H.-F. Guo, M. A. Juliano, L. Juliano, A. J. Morris, E. Galperin, D. W. Rodgers and L. B. Hersh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2826–E2835 CrossRef CAS PubMed.
- M. Wu, B. E. Dul, A. J. Trevisan and D. Fiedler, Chem. Sci., 2012, 4, 405–410 RSC.
- M. Wu, L. S. Chong, S. Capolicchio, H. J. Jessen, A. C. Resnick and D. Fiedler, Angew. Chem., 2014, 126, 7320–7325 CrossRef.
- A. M. Riley, H. Wang, S. B. Shears and B. V. L. Potter, Chem. Commun., 2015, 51, 12605–12608 RSC.
- H. Wang, H. Y. Godage, A. M. Riley, J. D. Weaver, S. B. Shears and B. V. L. Potter, Chem. Biol., 2014, 21, 689–699 CrossRef CAS PubMed.
- A. Hager, M. Wu, H. Wang, N. W. Brown, S. B. Shears, N. Veiga and D. Fiedler, Chem.–Eur. J., 2016, 22, 12406–12414 CrossRef CAS PubMed.
- X. Li, C. Gu, S. Hostachy, S. Sahu, C. Wittwer, H. J. Jessen, D. Fiedler, H. Wang and S. B. Shears, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3568–3574 CrossRef CAS PubMed.
- S. Sahu, Z. Wang, X. Jiao, C. Gu, N. Jork, C. Wittwer, X. Li, S. Hostachy, D. Fiedler, H. Wang, H. J. Jessen, M. Kiledjian and S. B. Shears, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19245–19253 CrossRef CAS PubMed.
- M. Wu, L. S. Chong, D. H. Perlman, A. C. Resnick and D. Fiedler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6757–E6765 CrossRef CAS PubMed.
- D. Furkert, S. Hostachy, M. Nadler-Holly and D. Fiedler, Cell Chem. Biol., 2020, 27, 1097–1108 CrossRef CAS PubMed.
- S. B. Engelsma, N. J. Meeuwenoord, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Angew. Chem., Int. Ed., 2017, 56, 2955–2959 CrossRef CAS PubMed.
- R. Q. Kim, M. Misra, A. Gonzalez, I. Tomašković, D. Shin, H. Schindelin, D. V. Filippov, H. Ovaa, I. Đikić and G. J. van der H. van Noort, Chem.–Eur. J., 2021, 27, 2506–2512 CrossRef CAS PubMed.
- Q. Liu, G. A. van der Marel and D. V. Filippov, Org. Biomol. Chem., 2019, 17, 5460–5474 RSC.
- S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912–6916 CrossRef CAS PubMed.
- S. Capolicchio, H. Wang, D. T. Thakor, S. B. Shears and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 9508–9511 CrossRef CAS PubMed.
- R. K. Harmel, R. Puschmann, M. N. Trung, A. Saiardi, P. Schmieder and D. Fiedler, Chem. Sci., 2019, 10, 5267–5274 RSC.
- E. Csuhai, M. A. Juliano, J. S. Pyrek, A. C. Harms, L. Juliano and L. B. Hersh, Anal. Biochem., 1999, 269, 149–154 CrossRef CAS PubMed.
- E.-S. Song, A. Mukherjee, M. A. Juliano, J. S. Pyrek, J. P. Goodman, L. Juliano and L. B. Hersh, J. Biol. Chem., 2001, 276, 1152–1155 CrossRef CAS PubMed.
- T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, MedChemComm, 2012, 3, 735–751 RSC.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- J. Torres, N. Veiga, J. S. Gancheff, S. Domínguez, A. Mederos, M. Sundberg, A. Sánchez, J. Castiglioni, A. Díaz and C. Kremer, J. Mol. Struct., 2008, 874, 77–88 CrossRef CAS.
- G. Liao, W. Ye, T. Heitmann, G. Ernst, M. DePasquale, L. Xu, M. Wormald, X. Hu, M. Ferrer, R. K. Harmel, D. Fiedler, J. Barrow and H. Wei, ACS Pharmacol. Transl. Sci., 2021, 4, 780–789 CrossRef CAS PubMed.
- Y. Terao, M. Takahashi, R. Hara, K. Hidaka, H. Furukawa, T. Yamasaki and S. T. Kasai, WO 2018/182051Al, 2018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02975d |
| This journal is © The Royal Society of Chemistry 2021 |