
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpoc group: transformable protecting group with gold(III)-catalyzed fluorene formation†
Tomoya
Yamamoto
 a,
Tsung-Che
Chang
a and
Katsunori
Tanaka
*abc
a,
Tsung-Che
Chang
a and
Katsunori
Tanaka
*abc
aBiofunctional Synthetic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako-shi, Saitama, 351-0198, Japan. E-mail: kotzenori@riken.jp
bDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152-8552, Japan
cBiofunctional Chemistry Laboratory, Alexander Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Street, 420008, Kazan, Russian Federation
First published on 9th July 2021
Abstract
This study presents the novel concept of a transformable protecting group, which changes its properties through structural transformation. Based on this concept, we developed a 2-(2-ethynylphenyl)-2-(5-methylfuran-2-yl)-ethoxycarbonyl (Epoc) group. The Epoc group was transformed into an Fmoc-like structure with gold(III)-catalyzed fluorene formation and was removable under Fmoc-like mild basic conditions post-transformation even though it was originally stable under strongly basic conditions. As an application for organic synthesis, the Epoc group provides the novel orthogonality of gold(III)-labile protecting groups in solid-phase peptide synthesis. In addition, the high turnover number of fluorene formation in aqueous media is suggestive of the applicability of the Epoc group to biological systems.
Introduction
The mild deprotection of the Fmoc protecting group is advantageous to avoid side reactions and to obtain free functional groups efficiently.1 Thus, the Fmoc group has been applied to the synthesis of a wide variety of compounds including natural products, glycans, and peptides.2–5 This lability is also applicable to the controlled release of drugs in vivo; the modification of peptide drugs with an Fmoc structure prolongs their lifetime in circulation.6,7 In addition, some Fmoc-protected amino acids, peptides, and sugars have been used for low-molecular-weight gelators and applied as biomaterials.8–13 These compounds form fibrils with the π–π or CH–π stacking of fluorene rings, which results in the formation of hydrogels.Although this base lability is useful, the Fmoc group is not tolerant to reactions using strong basic reagents. A protecting group that achieves both Fmoc-like mild deprotection and high stability would solve this problem. However, it is difficult to develop such a protecting group because the stability of the protecting group is generally inseparable from its harsh deprotection conditions.
The base lability of the Fmoc group originates from the aromaticity of fluorenes, which stabilizes its anion and accelerates Ec1b elimination. The pKa value of fluorene is 10 less than that of diphenylmethane (Fig. 1a).14,15 Thus, the substitution of diarylmethane for fluorene in the Fmoc group increases stability. Based on this, we proposed the novel concept of a transformable protecting group, which changes its properties through structural transformation (Fig. 1b). For example, if the protecting group with diarylmethane is transformable into an Fmoc-like structure, its lability will be changed by the transformation, and this protecting group will achieve both high stability (before the transformation) and Fmoc-like lability (after the transformation). This system can also be applied to biological systems. Recently, metal catalysts have been used for prodrug activation and fluorescence labeling in biological systems.16–20 If these catalysts perform the transformation in vivo, this could create potential new application of the Fmoc group in biological systems such as triggering drug release and hydrogelations.
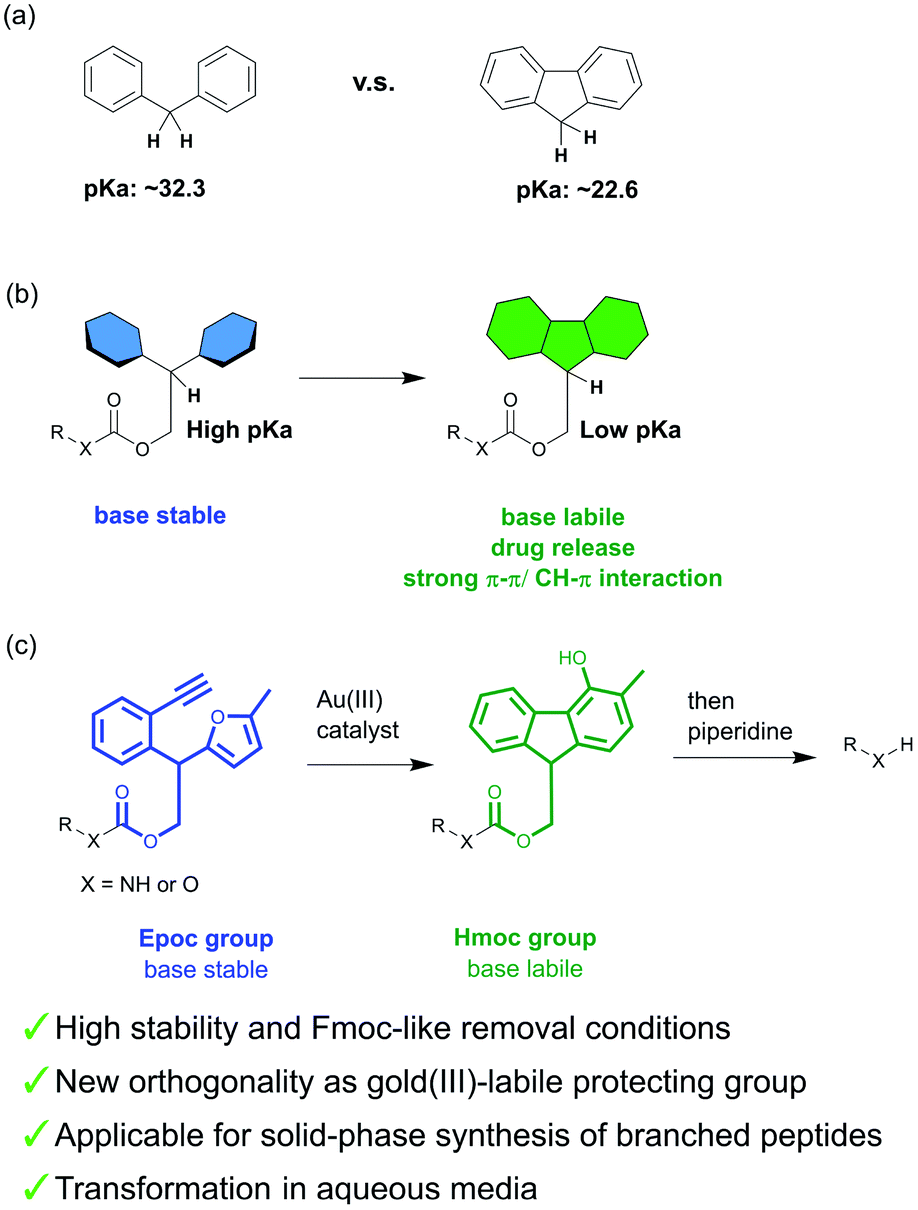 | ||
| Fig. 1 (a) The difference in pKa between diphenylmethane and fluorene. (b) Our strategy for the development of a transformable protecting group. (c) The novel protecting group developed in this work. The Epoc group is removed with a unique two-step procedure: gold(III)-catalyzed fluorene formation and base-induced Ec1b elimination. | ||
For the fluorene formation of this design, we focused on the gold(III)-catalyzed21,22 phenol synthesis reported by Hashmi et al.23–27 They have reported that the furan-yne system tethered with an aromatic linker is transformed into 4-OH-fluorenol with gold(III) catalysts within 10 minutes.28 Thus, we envisioned that the protecting group containing this sub-structure could meet our criteria for the design of a transformable protecting group.
In this study, we developed a novel protecting group containing the precursor structure of fluorene, 2-(2-ethynylphenyl)-2-(5-methylfuran-2-yl)-ethoxycarbonyl (Epoc) (Fig. 1c), which is the first example of a transformable protecting group. The Epoc group is stable under strongly basic conditions. On the other hand, a catalytic amount of gold(III) salt transforms the Epoc group into a (4-hydroxy-3-methyl-9H-fluoren-9-yl)-methoxycarbonyl (Hmoc) group, and shows Fmoc-like lability. The gold(III)-labile protecting group has not been reported to the best of our knowledge, and thus this group provided the novel orthogonality of the protecting group strategy. The Epoc group was successfully applied for the synthesis of branched peptides on resins. In addition, the fluorene formation proceeded even in aqueous media, which is suggestive of the application of the Epoc group for controlling the function of the Fmoc group in vivo.
Results and discussions
Synthesis of reagents for Epoc protecting groups
To develop the Epoc protecting group explained above, we first established the synthetic route of the reagent for introducing the Epoc group to various functional groups (Scheme 1). The TIPS-ethynyl group was introduced to 2-bromo-1-iodobenzene 1 with Sonogashira coupling, followed by lithiation and nucleophilic attack of 5-methyl-2-furfural to obtain alcohol 2. Ketone 3 was obtained via the oxidation of alcohol 2 with 2-iodoxybenzoic acid and the subsequent Wittig reaction gave enol ether 4. After hydrolysis of enol ether 4 under acidic conditions to obtain the aldehyde, this aldehyde was subsequently reduced with sodium borohydride to yield alcohol 5 as a racemic mixture. The TIPS group of alcohol 5 was removed with tetrabutylammonium fluoride to obtain EpOH 6.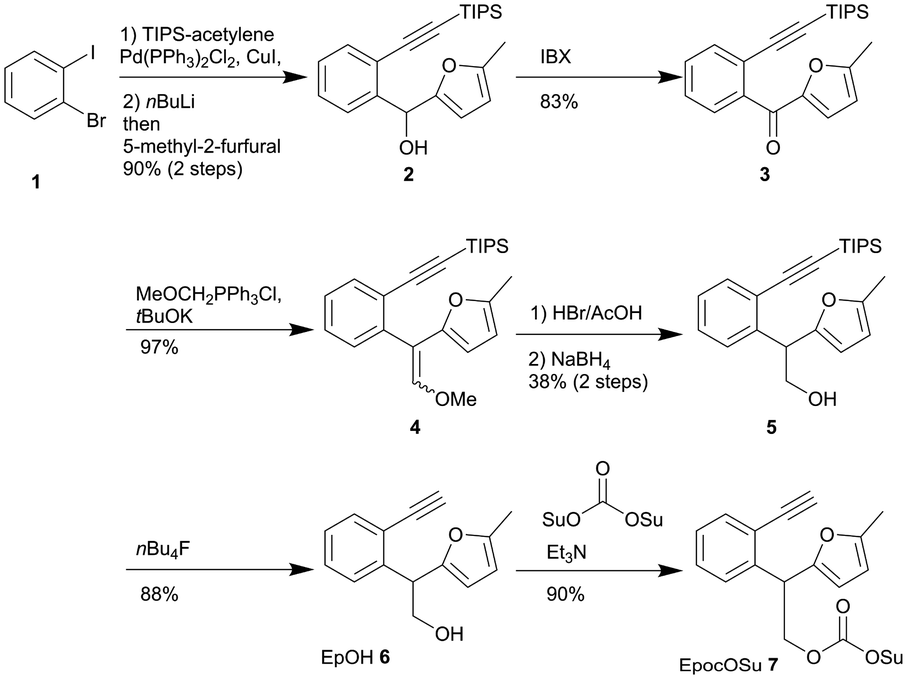 | ||
| Scheme 1 Synthesis of reagents for introducing Epoc group. | ||
EpOH 6 can be used for the introduction of the 2-(2-ethynylphenyl)-2-(5-methylfuranyl)-ethyl (Ep) protecting group to carboxylic acid, which is transformed to a structure similar to the fluorenylmethyl (Fm) group by gold catalysis. EpocOSu 7 was prepared by the reaction of EpOH 6 with di(N-succinimidyl) carbonate, which can be used to introduce the Epoc group to amines.
Introduction and deprotection conditions for protecting group
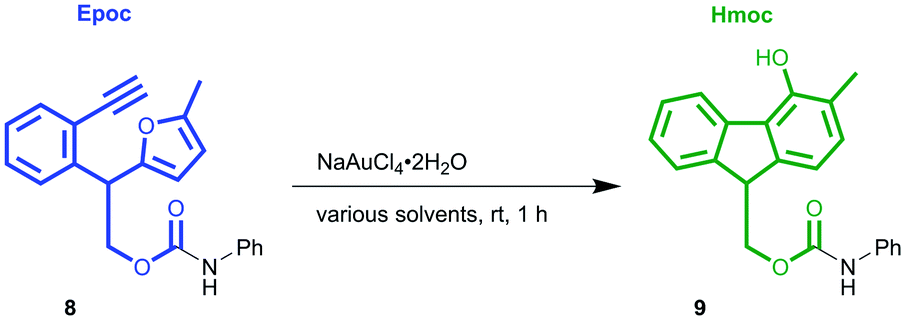
Before investigating the applicability of the Epoc group for protecting groups, we sought suitable conditions for gold-catalyzed fluorene formation. The Epoc-protected aniline 8 (Table 1) was obtained in the reaction with EpOH 6 and phenyl isocyanate and used as the substrate for screening reaction conditions. A catalytic amount of commercially available gold(III) salt, NaAuCl4·2H2O, transformed the Epoc group into the Hmoc group. This reaction proceeded the most efficiently in CH2Cl2, where 1 mol% of gold(III) catalyst smoothly transformed the Epoc group into the Hmoc group. Thus, we decided to use this solvent as the standard deprotection condition. Although the reaction proceeded in 1,4-dioxane, a larger amount of gold catalyst is needed for its completion. On the other hand, in other polar solvents such as THF, acetone, DMF, CH3CN, and DMSO, this reaction did not proceed at all.|
|
|||
|---|---|---|---|
| Entry | Solvent | NaAuCl4·2H2O (mol%) | Yielda (%) |
| a Yields determined by HPLC. The reactions were performed with 300 nmol of compound 8 in 200 μL of solvents (1.5 mM), at room temperature for 1 hour. The reactions were run in triplicate and the average yields were shown. | |||
| 1 | CH2Cl2 | 10 | >99 |
| 2 | CH2Cl2 | 1 | >99 |
| 3 | 1,4-Dioxane | 10 | 94 |
| 4 | 1,4-Dioxane | 1 | 15 |
| 5 | THF | 10 | 0 |
| 6 | Acetone | 10 | 0 |
| 7 | DMF | 10 | 0 |
| 8 | CH3CN | 10 | 0 |
| 9 | DMSO | 10 | 0 |
Next, we tested the reaction conditions for introducing the Epoc group (Scheme 2a). The introduction of the Epoc group into the amino group in compound 10 was successful with EpocOSu 7, which is a general condition for introducing carbamate protecting groups into amino groups. Carboxylic acids can also be protected with the Ep group. The Ep group was introduced to the carboxylic acid of compound 12 by esterification with EpOH 6. This is similar to the fluorenylmethyl (Fm) group, which is used for the protecting group of carboxylic acid. For the protection of alcohols with the Epoc group, the in situ formation of Epoc chloride with EpOH 6 and triphosgene and the subsequent addition of substrate 14 gave the best yield. The Epoc- and Ep- protected amino acids and sugars were obtained as the mixtures of diastereomers because the reagents for introducing the Epoc groups were obtained as racemic mixtures using the current technique.
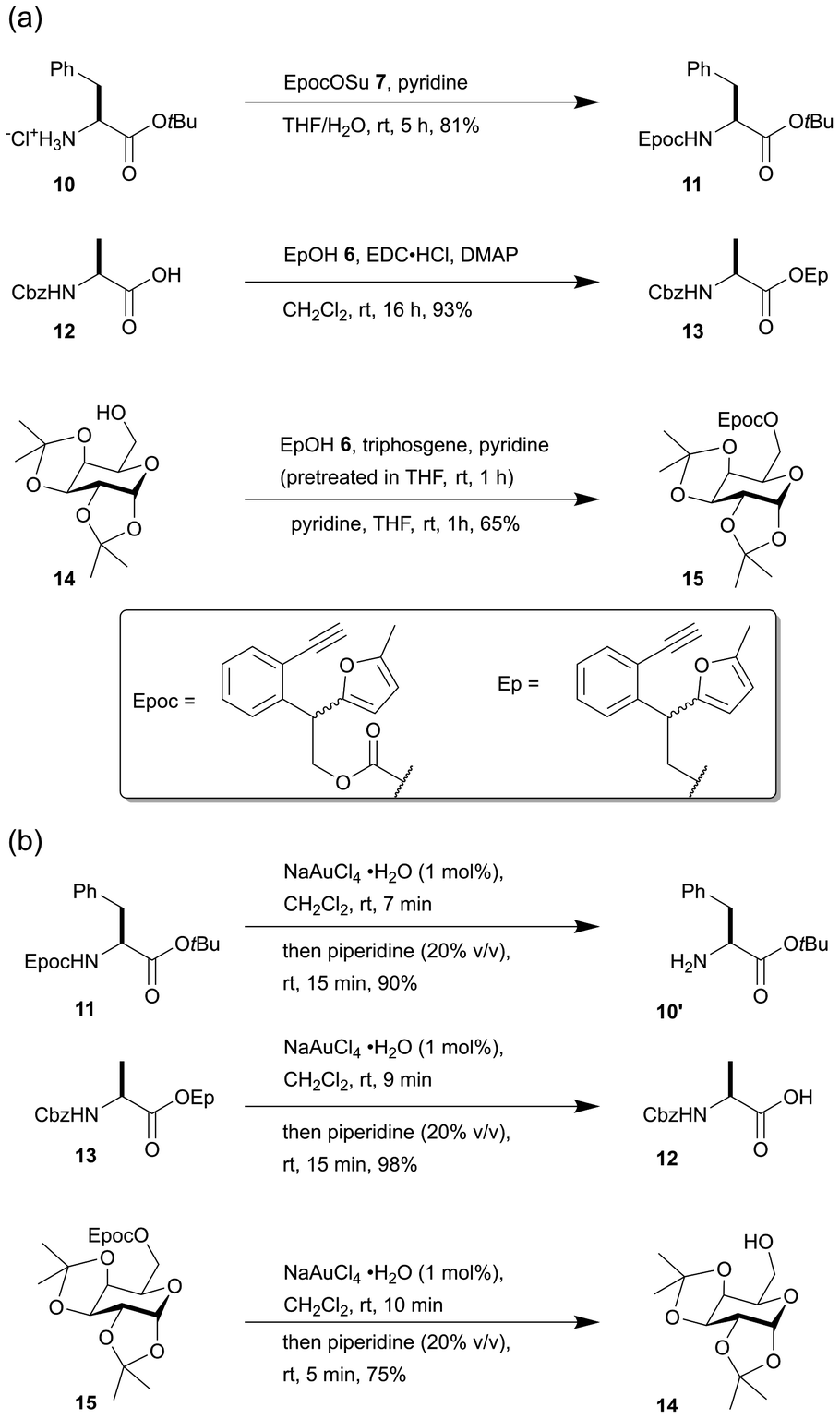 | ||
| Scheme 2 (a) Introduction and (b) removal of the Epoc and Ep groups. | ||
The fluorene formation of the Epoc and Ep group in substrates 11, 13, 15 proceeded smoothly (within 10 minutes) with 1 mol% of gold(III) catalyst in CH2Cl2 (Scheme 2b). The transformed protecting groups were easily removed with the subsequent addition of piperidine in a one-pot procedure after the completion of the fluorene formation. Although the yield for the deprotection of substrate 15 was relatively low (75%) compared to substrate 11 and 13, the fluorene formation was complete within 10 minutes. Production of a small amount of byproducts and loss of the product 14 during its purification decreased the overall yield. These results indicate that the Epoc and Ep groups are removed under mild conditions in almost the same manner as the Fmoc group.
Orthogonality of Epoc group
To demonstrate the stability and the mild removal conditions of the Epoc group, we tested the orthogonality of the Epoc group against other commonly used protecting groups. When compound 16, which is protected with both the Epoc and the Fmoc groups, was treated with piperidine, the Fmoc group was chemoselectively removed (Scheme 3a). This indicates that the Epoc group is tolerant to the conditions of Fmoc deprotection, and thus can be applied to the orthogonal protecting group for Fmoc solid-phase peptide synthesis (SPPS) and automated glycan assembly. The selective deprotection experiments for compound 18, possessing the Epoc, Boc, and ester groups, indicate that the Epoc group is also tolerant to acidic conditions for the Boc deprotection, and the conditions of the Epoc deprotection do not affect either the Boc or the ester groups (Scheme 3b). As expected, in the hydrolysis of methyl ester in compound 18 under strongly basic conditions, the Epoc group was stable and compound 20 was selectively obtained, where the Fmoc group is known to be labile. This indicates that the Epoc group achieves the criteria of our initial design for a transformable protecting group: high stability against bases and Fmoc-like lability post-transformation. Thus, the Epoc group will be useful for the protection of the amino group of compounds that are planned to undergo reactions under strongly basic conditions and require mild conditions for removal at the end of the synthetic route, where the Fmoc group is not applicable.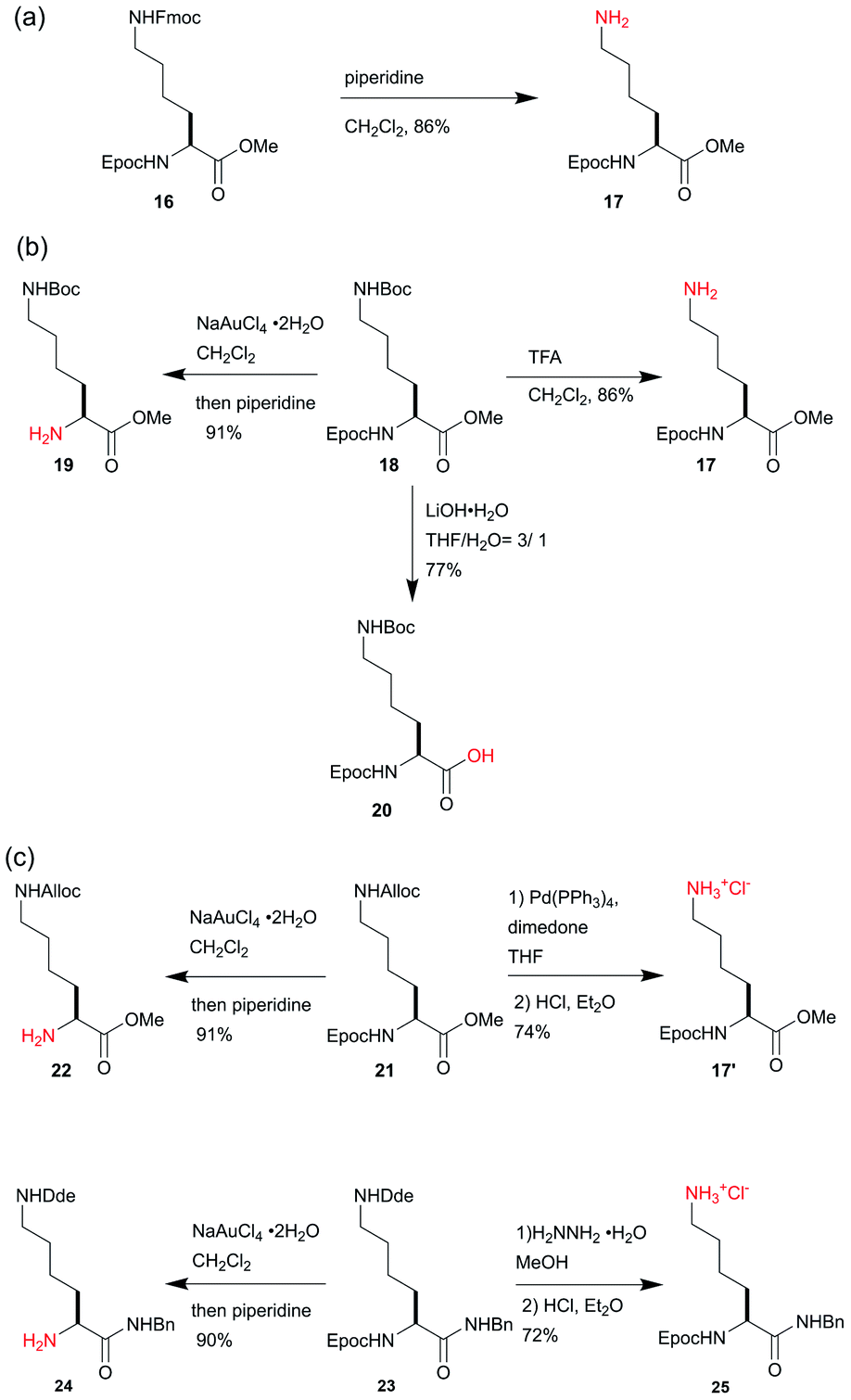 | ||
| Scheme 3 Orthogonality of the Epoc group. (a) Fmoc-selective deprotection. (b) Orthogonality of the Epoc group to acid- and base-labile protecting groups. (c) Orthogonality of Epoc group to Pd- and hydrazine-labile protecting groups. | ||
As well as the acid-labile and base-labile protecting groups, the Epoc group is orthogonal to the Pd-labile Alloc protecting group29 and the hydrazine-labile Dde protecting group (Scheme 3c),30 which are commonly used as the orthogonal protecting groups for amines. When PhSiH3 was used as a nucleophilic scavenger, the Alloc deprotection of compound 21 was unsuccessful because of the undesired reduction of alkyne in the Epoc group. On the other hand, the use of dimedone instead improved this problem. The Alloc group was chemoselectively removed with a palladium catalyst and compound 17′ was obtained. The treatment of an excess amount of hydrazine to compound 23 gave compound 25, where the Dde group was selectively removed. Thus, the Epoc group was also stable under the conditions required for Dde removal. The Epoc group was selectively removed from compounds 21 and 23 to obtain compounds 22 and 24 respectively, indicating that the removal of the Epoc group requires relatively mild conditions to keep other protecting groups unaffected. These results indicate the orthogonality of the Epoc group against the Alloc and Dde groups. The Alloc and Dde groups have been used for the solid-phase synthesis of cyclic and branched peptides. Thus, the addition of the Epoc group to a series of these orthogonal protecting groups enables the synthesis of more complex cyclic and branched peptides on resins.
Application to solid-phase peptide synthesis
We tested whether the transformation from Epoc to Hmoc and its subsequent deprotection proceeded on resins using N-Epoc-valylphenylalanine bound to Trt(2-Cl) resin 26 (Fig. 2). The transformation from the Epoc to the Hmoc group proceeded smoothly even on resins with the treatment of 10 mol% of NaAuCl4·2H2O for 10 minutes. The resulting peptide on resin 27 was then treated with piperidine to remove the Hmoc group. After the coupling with Fmoc-Gly-OH and cleavage, Fmoc-Gly-Val-Phe-OH 29 was successfully obtained. This indicates that the deprotection of the Epoc group proceeds properly even on resins and the Epoc group works as a protecting group for solid-phase synthesis.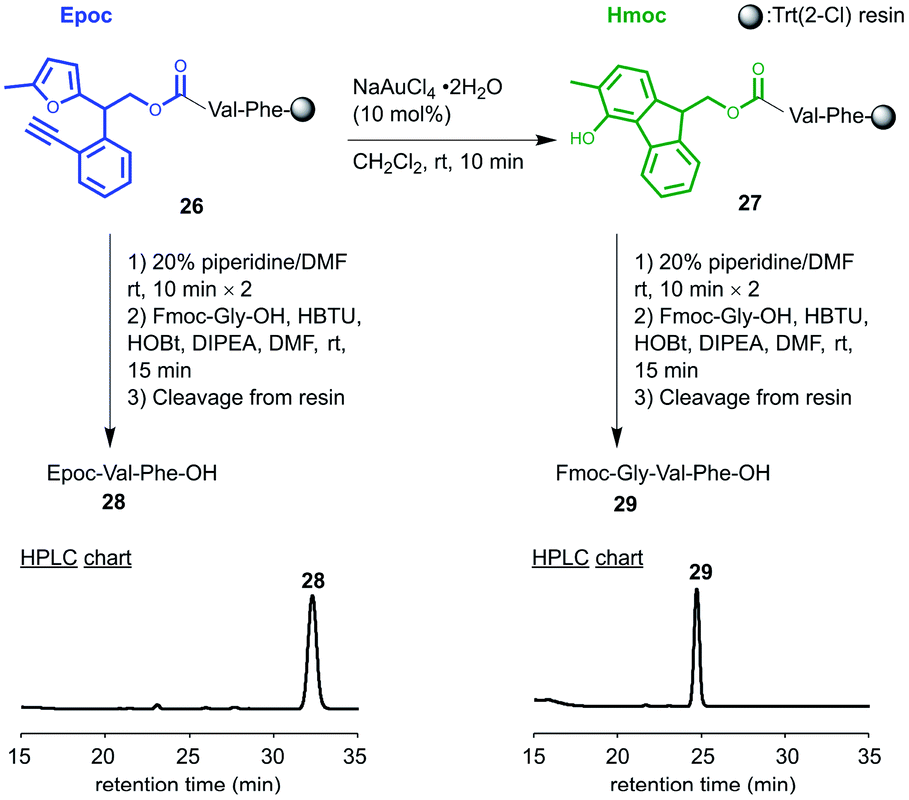 | ||
| Fig. 2 Transformation and removal of the Epoc group on resin. HPLC charts of the cleaved peptide are shown. HPLC methods: Table S2. | ||
As a negative control, we treated a 20% piperidine/DMF solution and the coupling reagents with the same resin 26 without the transformation from the Epoc to the Hmoc group. The Epoc group did not react with piperidine or the coupling reagents without transformation into the Hmoc group, and Epoc-Val-Phe-OH 28 was selectively obtained after cleavage. In this experiment, the peptides other than Epoc-Val-Phe-OH 28, such as the Fmoc-Gly-Val-Phe-OH 29 and other peptides derived from side reactions on the Epoc group were not obtained based on the HPLC chart (Fig. S5†). Therefore, the Epoc group is tolerant of the conditions for Fmoc removal and coupling on resins, and applicable to the orthogonal protecting group for the on-resin synthesis of peptides.
The use of the Epoc group for SPPS enabled the elongation of the branched peptides in two different directions with different amino acid sequences. In the first example, Epoc-Lys(Fmoc)-OH was incorporated into the elongation of the linear peptide with 7 amino acids on resin 30, whose N-terminus was protected with the Boc group (Scheme 4a). Although the Epoc group in the middle part of peptides on the resin was less reactive compared to the case of N-Epoc-valylphenylalanine, the transformation into the Hmoc group proceeded with a longer reaction time (5 hours total). After the removal of the Hmoc group with piperidine to obtain the resin-bound peptide 31, several amino acids were coupled from Nα at lysine. After the cleavage from resin and HPLC purification, the branched peptide 32 was obtained in a 34% yield. Fmoc-Lys(Epoc)-OH was applied in the second example, and peptides with 10 amino acids on resin 33 were synthesized (Scheme 4b). The transformation and removal of the Epoc group proceeded well also in this peptide. After the elongation from Nε at the lysine, peptides were cleaved and the branched peptide 35 was obtained at 26% yield after HPLC purification. In these syntheses, we confirmed that acid-labile protecting groups such as Trt, tBu, Boc, and Pbf, which are commonly used as permanent protecting groups in SPPS, were not affected by treatment with NaAuCl4·2H2O. These experiments demonstrate that the Epoc group is tolerant to multiple cycles of Fmoc deprotection and couplings. In addition, the transformation into the Hmoc group and its removal were shown to proceed even in the sterically hindered middle parts of peptides. Thus, the Epoc group is applicable for the synthesis of complex branched or cyclic peptides on resins. Similar to the Fmoc group, the absorbance of the Hmoc-piperidine adduct cleaved from resins is detectable using UV spectroscopy, which helps monitor the completion of the deprotections. At this point, the Epoc protecting group is more useful than other orthogonal protecting groups previously applied for the solid-phase synthesis of peptides and glycans.
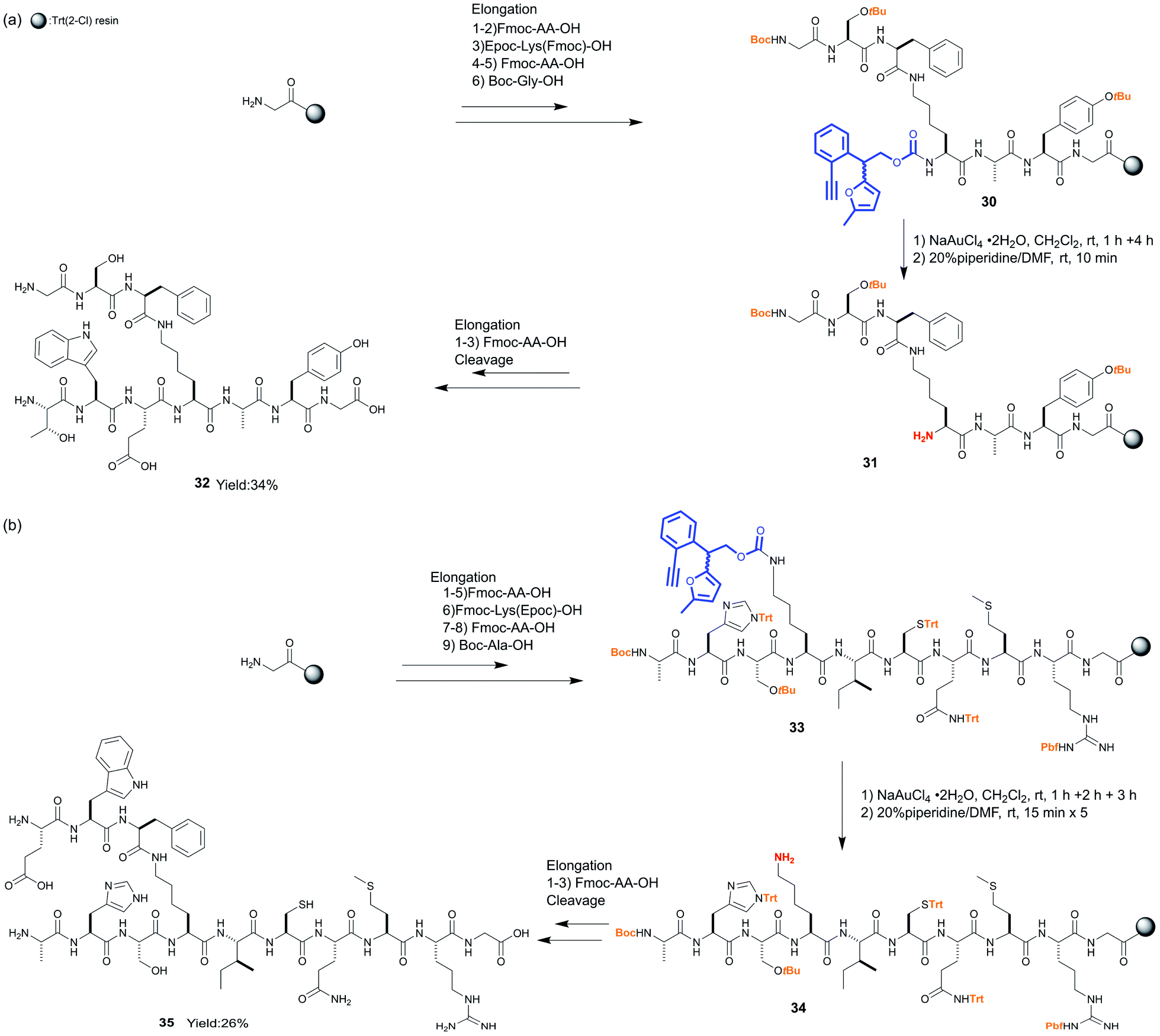 | ||
| Scheme 4 Application of the Epoc group as an orthogonal protecting group for branched peptide synthesis. | ||
Transformation of the Epoc group under aqueous conditions
To demonstrate the applicability of the Epoc group to biological systems, we performed the fluorene formation of the Epoc group under aqueous conditions (Table 2). The fluorene formation of compound 8 proceeded in PBS buffer at 37 °C for 24 hours and gave 50% yield with 1 mol% of NaAuCl4·2H2O. The reaction itself proceeded even with a very small amount of gold(III) catalyst (0.1 mol%) with a turnover number of approximately 340.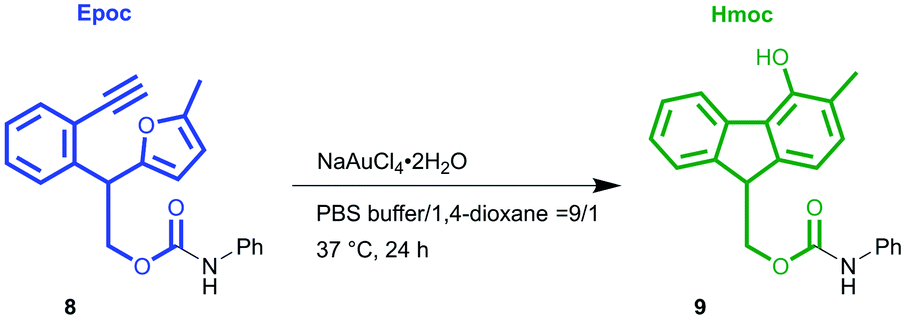
These results indicate that the fluorene formation of the Epoc group proceeds under conditions similar to biological settings, where a small concentration of gold catalysts exists in an aqueous solution. Although the gold catalysts, the substrate structure, pH of the solvent etc. have to be further optimized before its applications to biological systems, we were able to demonstrate that the transformation of the Epoc group could be applied to trigger drug release and hydrogelations.
Conclusions
In this study, we designed the novel concept of a transformable protecting group, which includes the transformation of the protecting group, changing its properties. To the best of our knowledge, a protecting group with this type of unique two-step deprotection procedure has not previously been developed. Based on this concept, we developed the Epoc and Ep groups, which transform from a stable to Fmoc-like labile structure with gold(III) catalysts. Because of the requirement for synthesizing complex molecules and constructing efficient synthetic strategies, the novel orthogonal protecting groups have still been developed recently such as reduction-labile, tetrazine-labile and Cu(I)-labile protecting groups.31–34 This work provided the novel orthogonality in the series of protecting groups as a gold(III)-labile protecting group with the extremely mild conditions for its removal. Its applicability to SPPS enables the efficient synthesis of branched and cyclic peptides including complex natural products, which require multiple orthogonal protecting groups.35 This study also demonstrated that the transformation of the Epoc group proceeds in aqueous media. Thus, the Epoc group is expected to be useful for future applications in biological systems for prodrugs and hydrogelators.Data availability
The experimental procedure, characterization data, and NMR spectra are provided in the ESI.Author contributions
Conceptualization: T. Y. and K. T.; funding acquisition: K. T.; investigation: T. Y. and T.-C. C.; writing-original draft preparation: T. Y.; writing-review and editing T. Y., T.-C. C., and K. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the AMED Grant JP15KM0908001, research grants from the Astellas Foundation, Mizutani Foundation and JSPS KAKENHI Grant Number, JP21H02065 (to K. T.) as well as additional support from a RIKEN Incentive Research Project grant (to T. Y.). This work was also funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (0671-2020-0063), with the support of the Kazan Federal University Strategic Academic Leadership Program.Notes and references
- L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404–3409 CrossRef CAS.
- C.-D. Chang and J. Meienhofer, Int. J. Pept. Protein Res., 1978, 11, 246–249 CrossRef CAS PubMed.
- E. Atherton, H. Fox, D. Harkiss, C. J. Logan, R. C. Sheppard and B. J. Williams, J. Chem. Soc., Chem. Commun., 1978, 537–539 RSC.
- L. Kröck, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschädler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS.
- E. Gershonov, I. Goldwaser, M. Fridkin and Y. Shechter, J. Med. Chem., 2000, 43, 2530–2537 CrossRef CAS PubMed.
- Y. Shechter, L. Preciado-Patt, G. Schreiber and M. Fridkin, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1212–1217 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- C. Diaferia, G. Morelli and A. Accardo, J. Mater. Chem. B, 2019, 7, 5142–5155 RSC.
- P. Chakraborty, Y. Tang, T. Yamamoto, Y. Yao, T. Guterman, S. Zilberzwige-Tal, N. Adadi, W. Ji, T. Dvir, A. Ramamoorthy, G. Wei and E. Gazit, Adv. Mater., 2020, 32, 1906043 CrossRef CAS.
- F. G. Bordwell, W. S. Matthews and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 442–443 CrossRef CAS.
- W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006–7014 CrossRef CAS.
- P. Destito, C. Vidal, F. López and J. L. Mascareñas, Chem.–Eur. J., 2021, 27, 4789–4816 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS PubMed.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- K. Vong, T. Tahara, S. Urano, I. Nasibullin, K. Tsubokura, Y. Nakao, A. Kurbangalieva, H. Onoe, Y. Watanabe and K. Tanaka, Sci. Adv., 2021, 7, eabg4038 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejović, Angew. Chem., Int. Ed., 2004, 43, 6545–6547 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Rudolph, J. P. Weyrauch, M. Wölfle, W. Frey and J. W. Bats, Angew. Chem., Int. Ed., 2005, 44, 2798–2801 CrossRef CAS PubMed.
- A. S. K. Hashmi, R. Salathé and W. Frey, Chem.–Eur. J., 2006, 12, 6991–6996 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Rudolph, H.-U. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem.–Eur. J., 2008, 14, 3703–3708 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. Hofmann, S. Shi, A. Schütz, M. Rudolph, C. Lothschütz, M. Wieteck, M. Bührle, M. Wölfle and F. Rominger, Chem.–Eur. J., 2013, 19, 382–389 CrossRef CAS PubMed.
- F. Guibé, Tetrahedron, 1998, 54, 2967–3042 CrossRef.
- B. W. Bycroft, W. C. Chan, S. R. Chhabra and N. D. Hone, J. Chem. Soc., Chem. Commun., 1993, 778–779 RSC.
- M. Staderini, A. Gambardella, A. Lilienkampf and M. Bradley, Org. Lett., 2018, 20, 3170–3173 CrossRef CAS PubMed.
- S. Chithanna, S. Vyasamudri and D.-Y. Yang, Org. Lett., 2020, 22, 2391–2395 CrossRef CAS.
- H. Liu, S.-Y. Zhou, G.-E. Wen, X.-X. Liu, D.-Y. Liu, Q.-J. Zhang, R. R. Schmidt and J.-S. Sun, Org. Lett., 2019, 21, 8049–8052 CrossRef CAS PubMed.
- A. Kumar, V. Gannedi, S. A. Rather, R. A. Vishwakarma and Q. N. Ahmed, J. Org. Chem., 2019, 84, 4131–4148 CrossRef CAS PubMed.
- H. Itoh and M. Inoue, Org. Biomol. Chem., 2019, 17, 6519–6527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, compound data, NMR spectra. See DOI: 10.1039/d1sc03125b |
| This journal is © The Royal Society of Chemistry 2021 |