
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive and critical review of the recent progress in electrocatalysts for the ethanol oxidation reaction
Lubna Yaqooba,
Tayyaba Noor *b and
Naseem Iqbalc
*b and
Naseem Iqbalc
aSchool of Natural Sciences (SNS), National University of Sciences and Technology (NUST), Islamabad, Pakistan
bSchool of Chemical and Materials Engineering (SCME), National University of Sciences and Technology (NUST), Islamabad, Pakistan. E-mail: tayyaba.noor@scme.nust.edu.pk; Tel: +92 51 9085 5121
cU.S.-Pakistan Center for Advanced Studies in Energy (USPCAS-E), National University of Sciences and Technology (NUST), H-12 Campus, Islamabad 44000, Pakistan
First published on 6th May 2021
Abstract
The human craving for energy is continually mounting and becoming progressively difficult to gratify. At present, the world's massive energy demands are chiefly encountered by nonrenewable and benign fossil fuels. However, the development of dynamic energy cradles for a gradually thriving world to lessen fossil fuel reserve depletion and environmental concerns is currently a persistent issue for society. The discovery of copious nonconventional resources to fill the gap between energy requirements and supply is the extreme obligation of the modern era. A new emergent, clean, and robust alternative to fossil fuels is the fuel cell. Among the different types of fuel cells, the direct ethanol fuel cell (DEFCs) is an outstanding option for light-duty vehicles and portable devices. A critical tactic for obtaining sustainable energy sources is the production of highly proficient, economical and green catalysts for energy storage and conversion devices. To date, a broad range of research is available for using Pt and modified Pt-based electrocatalysts to augment the C2H5OH oxidation process. Pt-based nanocubes, nanorods, nanoflowers, and the hybrids of Pt with metal oxides such as Fe2O3, TiO2, SnO2, MnO, Cu2O, and ZnO, and with conducting polymers are extensively utilized in both acidic and basic media. Moreover, Pd-based materials, transition metal-based materials, as well as transition metal-based materials are also points of interest for researchers nowadays. This review article delivers a broad vision of the current progress of the EOR process concerning noble metals and transition metals-based materials.
1. Introduction
Over the last few decades, fuel cells have attracted the attention of researchers as a renewable energy source and a potential alternative to fossil fuels (a non-renewable energy source) for the alleviation of the energy crisis and greenhouse gas emission, due to its high fuel conversion and electrical efficiency. Fuel cells are broadly employed in numerous fields such as transportation, and stationary and portable power generation. Among the different types of fuel cells, Solid Oxide Fuel Cells (SOFC), and Molten Carbonate Fuel Cells (MCFC) are examples of high-temperature fuel cells while Alkaline Fuel Cells (AFC), Proton Exchange Membrane Fuel Cells (PEMFC), Phosphoric Acid Fuel Cells (PAFC), and Direct Alcohol Fuel Cells (DAFC) are examples of low-temperature Fuel Cells.1–4There are two types of direct alcohol fuel cells, namely, DMFC (Direct Methanol Fuel Cell) and DEFC (Direct Ethanol Fuel Cell). As compared to the direct methanol fuel cell, the direct ethanol fuel cell shows remarkable advantages, such as greater safety in transportation, and storage, as well as lower costs for ethanol production and handling.5–7 For example, methanol is more toxic and tends to cross over the membranes more easily than ethanol. Health experts observed that inhaled methanol may seriously damage the central nervous system, and may cause blindness.8 Ethanol is more economical than methanol because of its large-scale production in the agriculture sector, continuous availability, and cheaper resources. Ethanol is preferred over methanol due to its high energy density as complete oxidation of ethanol produces 8.0 k W h−1 Kg−1 while complete oxidation of methanol produces 6.1 kW h−1 kg−1. Moreover, due to low operating temperature and integrated system design, DEFCs demonstrate promising applications in portable power devices for vehicles and electronics.9,10
The main objective of a DEFC is the conversion of ethanol into carbon dioxide through a direct, and complete oxidation process involving 12 electrons. Therefore, as compared to the methanol oxidation process where complete oxidation proceeds via a 6-electron process, greater energy is produced in ethanol oxidation. The reasons for the sluggish kinetics are the type of catalyst, the ethanol structure, fuel structure, membrane thickness, and physical factors such as temperature and concentration, as well as reaction intermediates formation, which poison the fuel cell catalyst.11,12
1.1. Different types of alcohol fuel cell
At present, different types of under-developed fuel cells include both high (>400–500 °C), and low (<100 °C) temperature systems: oxygen-ion proton-conducting electrolytes, solid/liquid electrolyte-based systems, and direct and indirect ethanol fuel cells. In the case of indirect alcohol fuel cells, alcohol is first converted to H2 and CO, which can be further utilized as a fuel in the solid oxide or molten carbonate fuel cells (high-temperature fuel cells), while in the proton exchange membrane fuel cell (low-temperature fuel cell), the CO is converted to H2 and CO2. Besides ethanol, other alcoholic fuels being utilized in fuel cells include methanol (CH3OH), and ethylene glycol (CH2OH)2. The number of electrons involved in the complete oxidation of the hydrogen, methanol, ethanol, and ethylene glycol are 2, 6, 12, and 10, respectively. The operating temperature conditions for polymer membrane-based electrolytes are 20–120 °C and 600–800 °C for ceramic-centered electrolyte fuel cells. Moreover, in the case of alcohol-based systems, the water formation occurs on the anode side in oxygen and hydroxyl ion-conducting electrolyte and on the cathode side in the proton-conducting electrolyte. Water also participates as a reactant in both of the above-mentioned fuel cells as illustrated in Fig. 1.4,13,14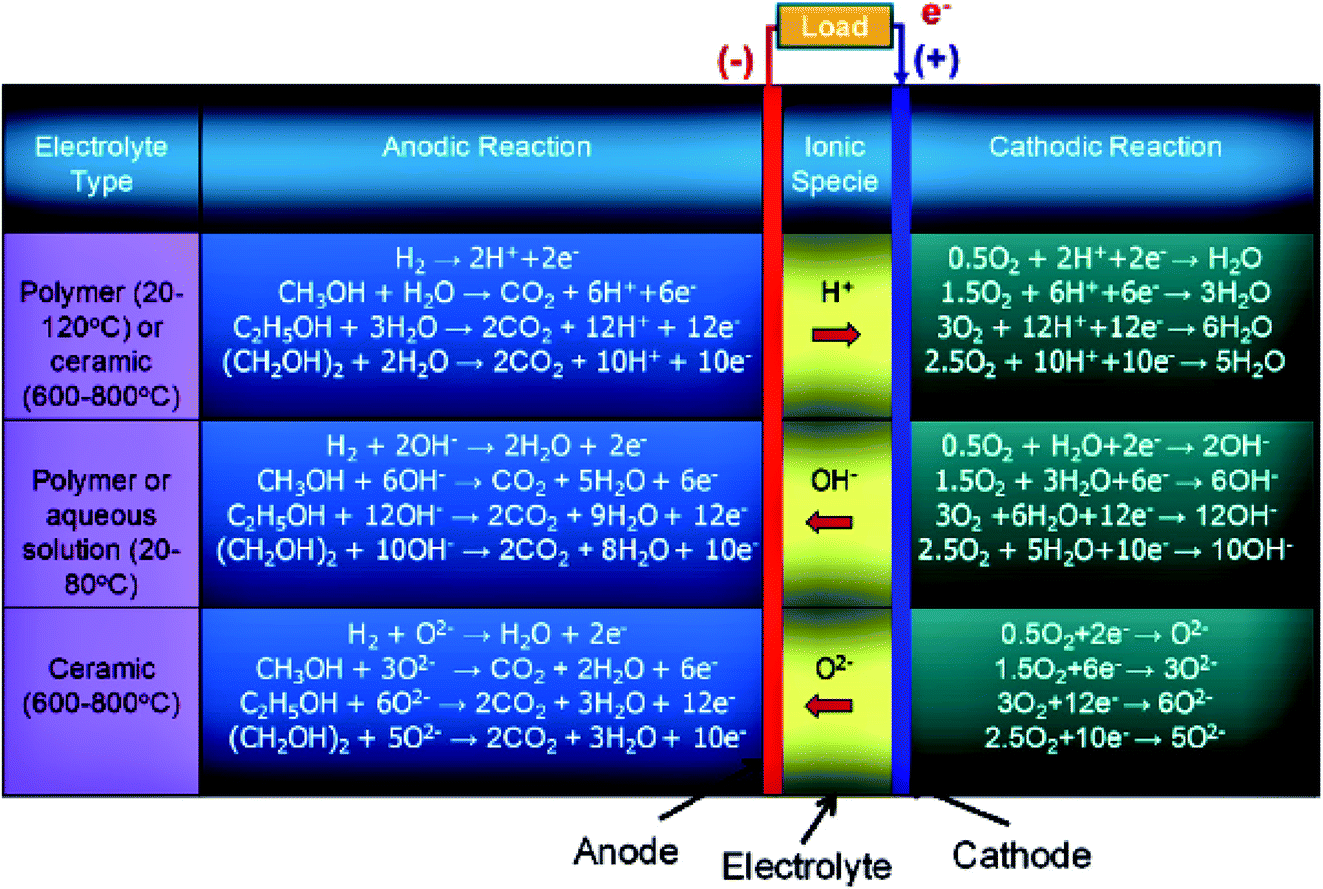 | ||
| Fig. 1 Schematic illustration of the electrochemical reactions of different types of alcohol fuel-based fuel cells. Reproduced from ref. 12, Copyright 2015, Elsevier. | ||
2. Ethanol oxidation
Ethanol is a renewable alcohol that is readily obtained from food crops (corn, sugarcane, and beetroot), unedible parts of food plants, or from other biomass resources such as poplar, grass, and forest residue. Ethanol is obtained by the fermentation of biomass, processing of biomass residues, and through the catalytic reaction of ethene with steam. Ethanol can be oxidized in two different types of environments: acidic and basic environments. The reaction mechanism is different in both environments, but the products are the same. The complete ethanol oxidation mechanism through the direct and indirect paths is represented here (Fig. 2).15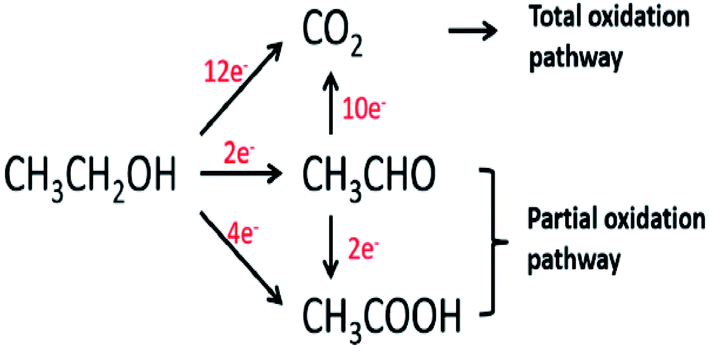 | ||
| Fig. 2 Schematic diagram of the parallel pathways during the ethanol oxidation reaction. Reproduced from ref. 13, Copyright 2005, Elsevier. | ||
2.1. Ethanol oxidation in an acidic medium
The ethanol oxidation reaction occurs at pH lower than 5 in an acidic type of fuel cell and the membrane used is called a proton exchange membrane (Nafion 115 or Nafion 117), which permits the movement of cations in the given electrolyte. Due to high H+ concentration, the cations of the membrane are easily exchanged in acidic media than in alkaline environments, and the Pt is effectively utilized as an electrocatalyst in an acidic medium. During the electrochemical reaction, the water consumption and then reformation occur at the anode and cathode, respectively. With time, the ethanol concentration tends to decrease in the anode compartment, which needs to be replenished, while in the cathode compartment, the concentration of water increases, which needs to be removed to prevent flooding and fuel cross-over problems. In acidic media, the Pt activity is maximum due to fewer blockages of active sites, and the dissolution of salts formed during the reaction. However, other noble metals such as Au, and Pd show less reactivity in an acidic medium owing to the corrosive nature of the co-catalyst, and the support, but gives a good response at higher pH.16–182.2. Ethanol oxidation in an alkaline medium
As compared to an acidic cell, ethanol oxidation occurs at higher pH (8–12) in an alkaline medium. Due to low H+ concentration, the proton exchange membrane (PEM) is not considered a good option because of slow kinetics, so the anion exchange membrane (AEM) is preferably employed, which allows anion exchange from the anode towards the cathode in the presence of an alkaline medium. During the electrochemical reaction, the water consumption and reformation occur at the cathode and anode surface, respectively (opposite to the PEM). Over time, the concentration of water in the cathode compartment tends to decrease, which may lead to the problem of fuel crossover.16,18There are a large number of differences in both types of cells. However, the following points indicate the alkaline cell superiority over the acidic cell. (i) In an alkaline cell at high pH, a large number of OH− ions provide oxygen for the oxidation process and result in improved reaction kinetics as compared to low pH. The catalyst is effectively utilized for the ethanol oxidation reaction instead of water dissociation to get more and more OH− ions. (ii) In an acidic cell, to promote the Pt catalytic activity, a support such as Ru, Ir, or Au is added since these supporting materials promote water dissociation to get more OH− ions, which further oxidizes the ethanol. (iii) In an alkaline medium, non-Pt catalysts show better activity as well as stability. For example, the literature shows that palladium performance is four times better as compared to Pt in basic media, so the expensive Pt can be easily replaced by the comparatively less costly Pd, Au, Ag, Fe, Co, Ni, etc. (iv) Catalysts show good stability and high current density response due to the less corrosive nature of an alkaline medium as compared to an acidic medium. (v) In an alkaline fuel cell, alcohol permeation can be restricted by reversing the ionic current direction due to OH− conduction against the traditional proton-conducting system. (vi) Low anodic overpotential.17,19–23 However, the problem associated with an alkaline medium is the formation of carbonate and bicarbonate ions by the reaction of OH− ions with the CO2, which then reacts with added base (KOH or NaOH) to form carbonated salts (e.g. Na2CO3 or K2CO3). These carbonated salts destroy the electroactive layer of the catalyst, block and restrict the diffusion of the ions in the medium, and lead to poor cell performance.18
2.3. Limitations of the direct ethanol fuel cells and their solutions
Direct ethanol fuel cells possess the prodigious potential for commercialization but the optimization of their performance encounters some challenges, particularly the sluggish kinetics of the ethanol oxidation process at the anode, incomplete oxidation, ethanol crossover, the high cost of cell materials, catalyst poisoning by the reaction intermediates, catalyst stability, water, and heat management. The catalyst in the direct ethanol oxidation process is a site that provides an active surface for the ethanol oxidation to proceed at a lower activation energy with a higher rate. The lower activation energy significantly affects the energy consumed during the breaking and the forming of the reactant and the product bonds, respectively, whereas, the higher rate reduces the time required for the reaction to proceed. The ethanol oxidation proceeds via different pathways, most of which result in partially oxidized products, e.g. acetaldehyde, and acetic acid; this partial oxidation, which is due to nearly unbreakable C–C bonds, leads to efficiency loss. An efficient catalyst is, therefore, required for the complete oxidation process.18,24,25 Ethanol crossover is comparatively lower in DEFC as compared to methanol crossover in DMFC. However, the permeation or ethanol crossover from the anode towards the cathode generates a mixed potential at the cathode and affects the cell performance. The fuel crossover is in turn greatly influenced by the temperature, ethanol concentration, and flow rate.26,27 Heysiattalab et al. recommended that there is a direct relationship between the cell operating temperature, and the electrocatalytic activity by (i) enhancing the electrochemical kinetics of both the anodic (EOR) and cathodic (ORR) processes, and by minimizing the activation loss; (ii) by improving the conductivity of the OH− ions, and by lowering the ohmic loss; (iii) elevating the transport of both the ethanol and oxygen with consequent inferior concentration loss.28Li et al. suggested that too-high ethanol concentrations mitigated the cell performance for two reasons: (i) coverage of electroactive sites with ethanol and less adsorption of OH− ions, and (ii) the excess ethanol reduces the level of OH− ionization and results in increased cell resistance due to the restricted movement of ions.29,30
Alzate et al. observed that the increased fuel solution flow rate improved the cell performance in the high current density region. This feature is due to the enhanced mass transfer of both the C2H5OH and the OH− ions from the flow-field towards the electrocatalyst layer.31 This problem can be resolved by the modification of the membrane (the core of the DEFC). Since the ethanol concentration entered at the anode catalyst layer, dilution of the ethanol solution can assist to prevent ethanol crossover, but this may cause specific energy losses in the direct-ethanol fuel system.26,32
Platinum has been recognized as an efficient electrocatalyst for a DEFC at the atomic level due to its distinctive, and tremendous electrocatalytic properties; however, (i) poisoning of Pt-catalyst by reaction intermediates (CO, CH3CHO, etc.) through an ethanol oxidation process results in a sharp decline in the current response (electrocatalytic activity); (ii) controlled platinum dosage due to high cost, as well as the incorporation of other metals into Pt, can reduce the cost.33,34
Heat management of the DEFC (Direct Ethanol Fuel Cell) is required since the products released during the electrochemical reaction include CO2, acetic acid, and acetaldehyde. Acetaldehyde selectivity increases with the increase of the operating temperature, and decreased selectivity of acetic acid occurs under constant current operation. The existing acetaldehyde and acetic acid could lower the direct ethanol fuel cell efficiency. Furthermore, high acetaldehyde concentration lowers the ethanol concentration and results in voltage decline. Therefore, by heat management, the products of the ethanol oxidation process can be controlled.35–37
Without H2O management, the fuel cell performance will be disturbed by the generated cell resistance. A marvelous amount of H2O crossover through the membrane can cause water loss in the anode compartment and flooding in the cathode compartment and can promote short-circuiting inside the device.38,39
To address these challenges, the research and development of an appropriate electrode catalyst play a vital role in fuel cell electrochemical reactions.40,41 These problems can be lessened or even removed by (a) the development of an economical non-Pt catalyst with high efficiency, and stability, controlled concentration, and the flow rate of the ethanol; (b) the establishment of a new membrane with mitigated ethanol crossover; (c) heat and water management; (d) the development of the catalyst crystal structure to ensure complete ethanol oxidation before reaching to the anode-membrane interface (Fig. 3 and 4).42
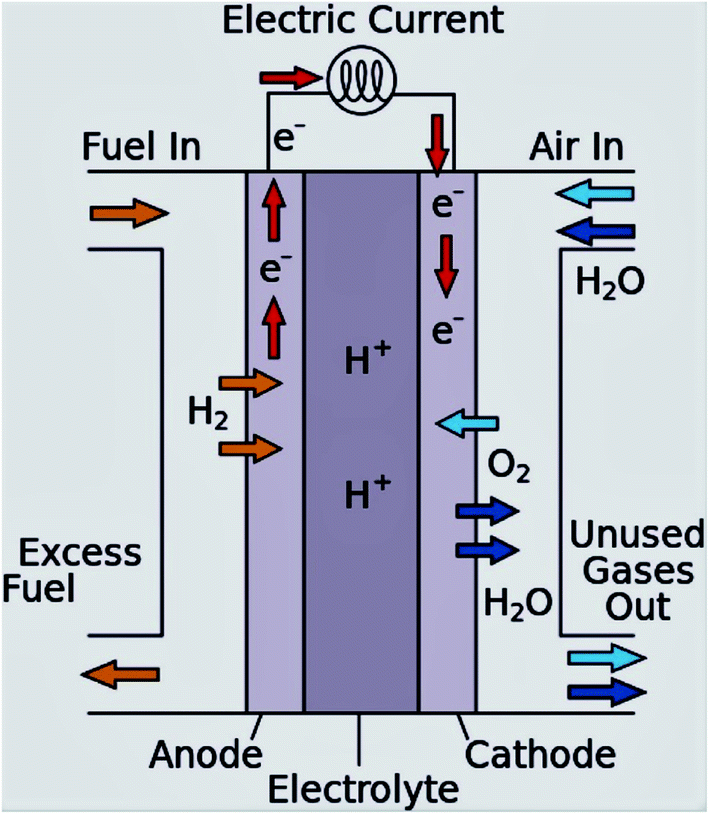 | ||
| Fig. 3 Schematic diagram of the proton exchange membrane fuel cell. Reproduced from ref. 1, Copyright 2016, Elsevier. | ||
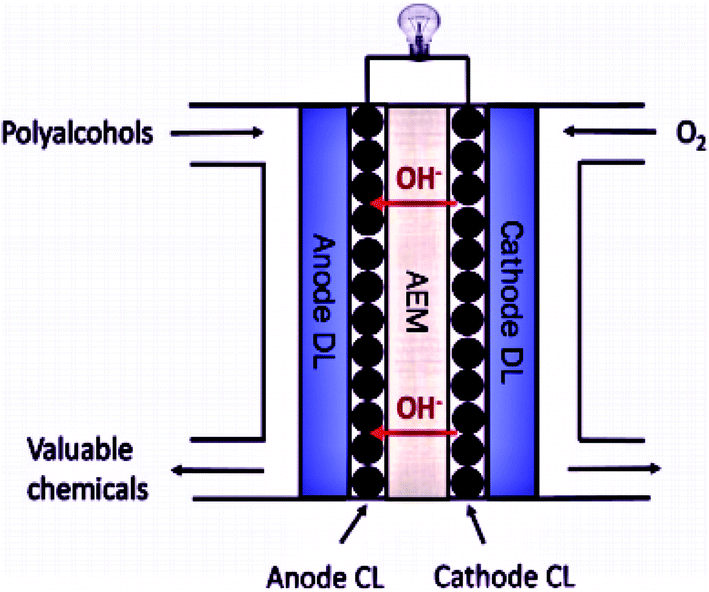 | ||
| Fig. 4 Schematic diagram of the Anion Exchange Membrane Fuel Cell. Reproduced from ref. 44, Copyright 2017, Elsevier [CL = catalyst layer, DL = diffusion layer, AEM = anion exchange membrane]. | ||
3. The direct ethanol fuel cell
The direct ethanol fuel cell is fundamentally a proton exchange membrane fuel cell with C2H5OH(aq) as a fuel, two electrodes where oxygen reduction and ethanol oxidation occur at the cathode and anode, respectively, and a state-of-the-art Nafion membrane separating the electrodes and allowing the proton diffusion from the anode towards the cathode. A membrane electrode assembly (MEA) is the collective term used for the combination of electrodes and the membrane. During the ethanol oxidation process (a) an aqueous solution of ethanol is fed from the anode side, and reaches the electrode surface, gets oxidized, and produces CO2 along with electrons and protons; (b) the diffusion of protons and electrons occurs through the membrane and external circuit, respectively; (c) oxygen is reduced at the cathode surface by accepting the external electrons, and the internal protons.11,43,44 The reaction is represented as follows:| CH3OH + 3O2 → 2CO2 + 3H2O |
3.1. Ethanol oxidation in acidic media
In acidic media, the ethanol reacts with water and produces carbon dioxide, protons, and electrons, while at the cathode, the reduction of oxygen generates the water. The complete reaction is given below:Reaction at the anode:
| C2H5OH + 3H2O → 2CO2 + 12H+ + 12e−, E0 = 0.09 V |
Reaction at the cathode:
| 3O2 + 12H+ +12e− → 6H2O, E0 = 1.23 V |
Overall reaction:24,45
| C2H5OH + 3O2 → 2CO2 + 3H2O, E0 = 1.14 V |
3.2. Ethanol oxidation in alkaline media
In alkaline media, ethanol reacts with hydroxyl ions and produces carbonate ions, water, and electrons, while at the cathode, the reduction of oxygen generates hydroxyl ions. The complete reaction is given below :24,46Reaction at the anode:
| C2H5OH + 12OH− → 2CO32− + 9H2O + 12e−, E0 = −0.743 V |
Reaction at the cathode:
| O2 + 2H2O + 4e− → 4OH−, E0 = 0.40 V |
Overall reaction:
| C2H5OH + 3O2 → 2CO2 + 3H2O, E0 = 1.143 V |
3.3. The mechanism of the ethanol oxidation process on the Pt surface
The ethanol oxidation process on the Pt surface follows sluggish kinetics due to the complex mechanism, and incomplete oxidation. Although a detailed reaction mechanism needs further research, good agreement about the reaction pathways validates that acetaldehyde and acetic acid are the main products of the electrooxidation of ethanol on the Pt electrode surface in acidic solution. At a potential of 0.6 V, even the C–C bond remains intact, and 2 or 4 electrons are produced; however, further oxidation results in the cleavage of the C–C bond after acetaldehyde readsorption and produces the CO intermediate. COads limits the adsorption of the reactants by blocking the active sites of the Pt; however, at potentials above 0.6 V, the water oxidation produces Pt–OH or Pt–O on the Pt surface, which then oxidizes the CO to CO2, so CO2 is produced at high potential only.47–49| Pt + CH3CH2OH → Pt(CHOH–CH3)ads + H+ + e− | (1) |
| Pt(CHOH–CH3)ads → Pt + CHO–CH3 + H+ + e | (2) |
| Pt + CHO–CH3 → Pt(CO–CH3)ads + H+ + e | (3) |
| Pt(CO–CH3)ads + Pt(OH−)ads → 2 Pt + CH3COOH | (4) |
| Pt(CO–CH3)ads + Pt → Pt(CO)ads + Pt(CH3)ads | (5) |
| Pt(CH3)ads + Pt(H)ads → 2Pt + CH4 | (6) |
| Pt(CO)ads + H2O → Pt + CO2 + 2H+ | (7) |
Dehydrogenation:
| Pt + R–OH → Pt + CO2 + nH2 | (8) |
Dehydration:
| Pt + R–OH → Pt–RO + H2O | (9) |
Hydration:
| Pt + H2O → Pt–OH + H+ + e− | (10) |
| Pt–RO + Pt–OH → 2Pt + CO2 + H+ + e− | (11) |
Dehydrogenation
| Pt + R–OH → Pt + CO2 + nH2 | (12) |
Dehydration:
| Pt + R–OH → Pt–RO + H2O | (13) |
Hydration:
| M + H2O → M–OH + H+ + e− | (14) |
| Pt–RO + M–OH → Pt + M + CO2 + H+ + e− | (15) |
3.4. The mechanism of the ethanol oxidation process on the Pd surface (alkaline medium)
The detailed ethanol oxidation mechanism on the Pd surface is shown below. In the 1st step, the Pd metal reacts with OH− ions provided by an alkaline medium. In the 2nd and 3rd steps, the Pt reaction with dissociated alcohol occurs in an alkaline medium, and gives Pd (CH3CO)ads at the potential of −0.7 V. The strongly adsorbed (CH3CO) ads on the active sites of the palladium metal surface was stripped off from the catalyst surface by the adsorbed OH− species, so the ethanol was continuously oxidized with an increase in the potential. The reaction in the fourth step is the rate-determining step. The adsorbed CH3CO on the Pd surface slows down the reaction process by blocking the active sites.50–53| Pd + OH− → Pd–OHads + e− | (16) |
| Pd + CH3CH2OH → Pd(CH3CH2OH)ads | (17) |
| Pd(CH3CH2OH)ads + 3OH− → Pd(CH3CO)ads + 3H2O + 3e− | (18) |
| Pd(CH3CO)ads + Pd–OHads + OH− → 2Pd + CH3COO− + H2O | (19) |
3.5. The mechanism of the ethanol oxidation process on the Pd–M surface
The first step is the adsorption of alcohol on the two metal surfaces followed by alcohol dehydrogenation in the 2nd step. In the 3rd step, metal hydroxylation gives M–OH ads, which combines with the ethoxy intermediate, and acetic acid is the final product in this reaction, which then reacts with hydroxyl anions to give acetate ions.| Pd–M + C2H5OH → Pd–M–(C2H5OH)ads | (20) |
| Pd–M–(C2H5OH)ads + 3OH− → Pd–M–(CH3CO)ads + 3H2O + 3e− | (21) |
| Pd–M + OH− → Pd–M–OHads + e− | (22) |
| Pd–M–(CH3CO)ads + Pd–M–OHads → 2Pd + 2M + CH3COOH | (23) |
| CH3COOH + OH− → CH3COO− + H2O | (24) |
3.6. The mechanism of the ethanol oxidation process on the Ni surface
The reaction on the Ni surface is mentioned below, and the reaction on the cobalt surface proceeded in the same way as that on the nickel surface. In the first step at the potential of 0.45 V vs. Hg/HgO, the Ni (OH)2 in an alkaline medium is oxidized to NiOOH, which then combines with alcohol and oxidizes it to CH3CHO, which is one of the possible products. However, another option is the reaction of the NiOOH with CH3CHO to give acetic acid as a final product.54,55| Ni(OH) 2 + OH− → NiOOH + H2O + e− | (25) |
| NiOOH + CH3CH2OH → CH3CHO + Ni(OH) 2 | (26) |
| NiOOH + CH3CHO → CH3COOH + Ni(OH) 2 | (27) |
| Ni (OH) 2 + OH− → (NiO)OH + H2O + e− | (28) |
| NiO(OH) + CH3CH2OH + 5OH− → Ni(OH) 2 + CH3COO + 4H2O + 4e− | (29) |
In the case of the OER process, the electrocatalyst microstructure plays an important role, so the controlled shape, size, and morphology are the main controlling factors. Nanostructure-engineered controlled morphology is normally employed to enhance the surface areas or to provide the special facets for promoting reaction kinetics of the metal catalysts, particularly Pt and Pd. At present, many novel schemes are utilized to synthesize Pd and Pt electrocatalysts with copious nanostructures including nanowires, nanosheets, nanoparticles, and nanopores. Moreover, different Pt-based alloys prepared by the mixing of Pt with oxyphilic atoms such as Bi, Sn, Zn, Cu, etc. also improve the catalytic activity. Specifically, the bimetallic Pt–Sn alloys display advanced electrocatalytic activity for the ethanol oxidation process in both basic and acidic media due to the bifunctional mechanism since Sn can provide OH− species by speeding up the water dissociation process at a lower overpotential that smoothens the intermediate oxidation process. The exceptional catalytic EOR performance in both the acidic and basic media also stems from the enormous exposed surface area of the (111) facet of the Pt–Sn nanosheets, which facilitates the C–C bond cleavage process, and removes poisonous reaction intermediates like CO.
Concerning the morphology, the nanowire array of the electrocatalysts generally has high aspect ratios, low-coordination sites, few grain boundaries, and privileged crystal growth favorable for the ethanol oxidation process. Pt nanowires show better performances than Pt nanoparticles due to the plentiful exposed active sites. In addition to the more developed nanowires, multiple nanostructures like nanoframes and nanocages were constructed to accommodate the huge surface to volume ratio and abundant active sites. The ethanol oxidation selectivity on the Pt surface markedly depends on the surface features. The ethanol oxidation process is comprised of (i) the initial ethanol dehydrogenation and (ii) the acetyl (CH3CO) oxidation. The open surface Pt facet is the best option to fully and efficiently oxidize ethanol at low potential.
As compared to Pt, Pd is more abundant, economical, and oxyphilic in nature to facilitate the oxidative desorption of reaction intermediates. The unique features of Pd-based electrocatalysts are also important for boosting electrocatalytic activity. Alloys of Pd with metals such as Ni, Ag, Sn, Au, Rh, Cu, Ru, metal oxides, and hydroxides develop multicomponent systems with operational durability and optimized catalytic performance. Moreover, the combination of the 2D configuration with the metal alloys (such as 2D PdAg nanodendrites) leads to remarkable electrocatalytic activity and stability due to abundant active sites, an enlarged surface area, as well as Ag-induced feeble binding between the adsorbate and Pd. The excellent performance of Pt–Pd composites with metal phosphide is credited to the hybridization between the metal phosphide and Pt/Pd during the synthesis and favors CO oxidation as a result of its dynamic role in the water activation process. Furthermore, metal oxides also arise as promising additives to enhance electrocatalytic activity. For example, the enhanced activity of the CeO2-modified Pt/Ni electrocatalyst is ascribed to the bifunctional mechanism, where CeO2 accelerates the CO oxidation on the Pt surface by providing chemisorbed oxygen. The substitution or partial replacement of the Pt/Pd with non-noble metals such as nickel not only reduces the cost, it also promotes surface oxidation by forming NiOOH, particularly in an alkaline solution. Other non-precious metal alloys based on Fe and Co have also attracted considerable attention from researchers. However, to improve the conductivity of these metals, the incorporation of other metals like Cd, and Cr to form co-catalysts also promotes the oxidation process since Cd is more prone to CdOOH formation.
Trimetallic catalysts showed more progressive catalytic performance towards the EOR than bimetallic catalysts, and monometallic electrocatalysts such as the hierarchical TiO2/ZnO nanostructures coupled with Pt showed very good responses towards the OER by delivering high current densities.56
4. Preparation methods for electrocatalytic materials
It is a well-known fact that the surface properties (shape and size) significantly affect the features of metal nanomaterials. Consequently, it is obligatory to enhance the efficient processes for the fabrication of nanoparticles with controlled morphology and size. One of the most promising approaches is the decoration of catalyst nanoparticles on the conductive supporting materials for the EOR. Innumerable synthetic approaches have been successfully established to prepare the metal nanoparticles on the supporting surfaces with valuable physical, and chemical characteristics as there is a strong relationship between the synthesis approaches and the surface coordination chemistry. However, in the synthetic process, even a single parameter variation influences the electrocatalytic activity. Recently, researchers have presented many synthetic schemes for loading the metal nanoparticles on the surface of the supporting materials, such as the solvothermal process, hydrothermal process, chemical reductions process, wet chemical method, microwave-assisted polyol process, and electrodeposition, etc.4.1. Hydrothermal synthesis
The hydrothermal process is a widely accepted method for the fabrication of materials for the MOR due to multiple advantages associated with it such as low-temperature synthesis, easy handling, precise control of the stoichiometry, and the one-step surfactant-free process. Different parameters such as precursor concentration, temperature, pressure, and reaction time all control the crystalline shape and particle size. This technique comprises a heterogeneous reaction in the presence of water under high temperature and pressure in a sealed system like Teflon-lined autoclaves inside a stainless-steel container. Ahmadi et al. in 2012 reported hydrothermally synthesized Fe3O4 nanoparticles and studied the effect of reaction temperature on the magnetic properties of the nanoparticles, while in 2019, Xu et al. introduced hydrothermally synthesized rod-like MnO nanoparticles and studied their use for dye removal and degradation purposes. Nanorods of cobalt-iron layered double hydroxides (CoFe-LDH) fixed with graphene oxides were directly fabricated by Yang et al. on nickel foam (NF) for use as an electrocatalyst for the methanol oxidation process.6,57–604.2. The solvothermal approach
The solvothermal technique is one of the most commonly used liquid phase techniques, where the reaction proceeds in a closed stainless steel vessel (autoclave) by developing an internal pressure above the solvent boiling point. A single solvent or mixture of solvents can be utilized for the synthesis depending on the reaction requirements. Most commonly used solvents include water, dimethylformamide (DMF), ethanol, methanol, and acetone, etc. Low-temperature reactions are carried out in a glass vial while for the high-temperature reactions, Teflon-lined autoclaves are utilized. In the solvothermal method, a solvent acts as a reacting medium that exists in the liquid and vapor phase. One end of the vessel is hot while the other end is cooler, so the temperature gradient is developed. By the process of convection of heat, the product moves from the hotter to the cooler part and is deposited at the low-temperature part with the size of the material being in nanometers. Yaqoob et al. in 2020 introduced the solvothermally synthesized hexagon-shaped Fe–NiNH2 BDC MOF/CNTs composites with high electrocatalytic activity. Noor et al. in 2019 reported the solvothermally synthesized NiO nanoparticles and their rGO composites and applied them as anode catalysts for the methanol oxidation reaction in an alkaline medium.61,624.3. Chemical reduction method
A chemical reduction approach was used for the synthesis of metal nanoparticles, as well as the conductive support. Materials required for the process include precursors salts, reducing agents and a stabilizer to fabricate the nanoparticles. In most cases, a catalyst along with heating is required. During the synthetic process, the metal precursors are reduced in the presence of reducing agents such as potassium borohydride, sodium borohydride, and ethylene glycol. The strong interaction between the metal nanoparticles and the supporting surface help in the growth of metal nanomaterials on the surface of the support. To prevent the agglomeration of the metal nanoparticles, polymer, ligands, and surfactants are added. The main features of the process are as follows: (1) production of the bulk quantities of the nano-alloys and nanoparticles, (2) easy scale-up process for mass-manufacturing needs, (3) particle size can be controlled to the size of 1 nm, (4) economical method as compared to other techniques. Alden and colleagues prepared PtPd nanoparticles through the chemical reduction method and studied their electrocatalytic activity for the methanol and formic acid oxidation process.634.4. Microwave-assisted polyol synthesis
The microwave-assisted polyol synthetic approach is a novel method for the development of metallic nanoparticles with well-controlled shape, size, and monodisperse precursors of metals in a liquid polyol (ethylene glycol). The polyol solvent is the most suitable option for the microwave method because of its high dipole moment and reducing power. The synthesis of nanostructures with novel morphologies and enhanced reaction kinetics are the salient features of this method. As compared to the polyol method, microwave synthesis requires less time but excess selective energy consumption. It is a no-solvent or very small amount of solvent reaction process through which a good quantity of material can be obtained. This process can be performed with various precursors, heat treatment, and dispersion agents to get the product of the desired morphology. The implementation of microwaves at different powers by providing different reaction energies leads to different kinetics during the ultrafast nucleation process and growth step. Hsieh and collaborators in 2015, tested the electrocatalytic activity of the rGO-supported Pd–Rh nanoparticles for the methanol oxidation process after their successful synthesis by the microwave-assisted polyol method.644.5. Wet chemical method
Wet chemical synthesis is one of the promising approaches for the precise control of the synthesis of complex nanoparticles involving various interfaces, i.e. metal–metal, and metal-support. The wet-chemical synthetic method is usually initiated from the formation of metal atoms by the reduction or thermal decomposition of noble metals, followed by nanoparticle-controlled aggregation. Wet-chemical synthesis has proven to be a very promising alternative, leading to the low cost, high-yield, and mass production of materials in the solution phase. Chen et al. prepared Au–Pt nanoparticles on the carbon-based support by an easy and facile wet-chemical strategy in the presence of caffeine, working as a capping agent and shape director without the precipitation of polymer or surfactant. Lv and co-workers processed a proficient, and facile single-step wet chemical approach for the synthesis of the porous Pt–Au nanodendrites homogeneously spread on the surface of rGO at room temperature by utilizing cytosine as a morphology director in the absence of any surfactant, seed, organic solvent, or complex instruments, and studied the electrocatalytic activity of the material towards the reduction of 4-nitrophenol.65–674.6. Electrodeposition method
The electrodeposition is a simple synthetic strategy for the well-defined control of the size, shape, and dispersion of noble-metal NPs without any intrusive additive. The other advantages associated with the electrodeposition include its being a clean, rapid, and facile process, with high particle purity, oversight control, distribution of trivial sized particles, improved control of distribution densities, and uniform dispersion of the metal nanoparticles on the support surface without any reductant or oxidant. Generally, a galvanostatic or potentiostatic technique, including a direct and pulse approach, is employed for the electrodeposition. This process involves both phase change and electron transfer and can be classified as cathodic electrodeposition or anodic electrodeposition based on the mechanism of the reaction. Different types of electrodeposition methods depending on the different applied current signals include square wave potential, square wave current, cyclic voltammetry, and constant current pulse electrodeposition. Surfactants such as polyethylene glycol and polymers with their nonionic nature have been added to electrolytic baths due to their selective adsorption to control the morphology and crystal growth. In electrocatalyst synthesis, the purpose of the addition of surfactants is to prevent catalyst agglomeration. Feng et al. in 2018 introduced PtRuIr nanoparticles synthesized by the electrodeposition method and analyzed their electrocatalytic behavior towards the oxygen evolution reaction (OER), and oxygen reduction reaction (ORR), while Thulasi Radhakrishnan utilized the same technique to fabricate Pt nanocrystals (100 plane) on the rGO surface to test it for the methanol oxidation process.66,68–715. Materials for the ethanol oxidation reaction
5.1. Platinum and platinum-based compounds
At present, the Pt-based electrocatalysts have been established as anode catalysts with the most potential in the DEFCs owing to their loftier activity towards C2H5OH electro-oxidation. However, the poisonous reaction intermediates (e.g. CO and CHx) engendered during the electrocatalytic process results in the surface poisoning of Pt-comprising catalysts and lead to the sluggish kinetics of the ethanol oxidation process along with pathetic service performance. Therefore, the extraordinary cost of Pt and the comparatively low stability are the main obstacles in the wide range of applications of the Pt electro-catalysts. Extensive efforts have been made by researchers to develop binary Pt-based catalysts to reduce the Pt loading or to carefully control the microstructures as well as to fabricate the Pd-based and other non-noble metal-based potential substitutes.72–75 Fabrication of the nanosized electrocatalyst with specified shape, geometry, and composition is also very fruitful for the oxidation process. As compared to bulk particles, the Pt-based materials, with narrow pore size, and completely ordered structure, possess a huge surface and show excellent electrocatalytic activity due to the smooth mass transfer of reactants, and the facile electron movement. The incorporation of a second metal can bring variations in the particle shape, size, surface structures, chemical selectivity, catalytic activity, as well as their alloy structures. Compared with single-metal systems, the rational design of the bimetallic nanostructures can provide more active sites through an enlarged surface area for the enhancement of the catalytic properties. A variety of dimetallic nanostructures have been fabricated such as nanotubes, nanowires, dendrites, and flowers.76–79 Moreover, the incorporation of the mesoporous conductive support further promotes electrocatalytic activity and stability. The supportive materials such as carbon-based CNTs, GO, rGO, graphite, and the non-carbon-based silica and metal oxides exhibit a promoting effect on the Pt-based materials.80,81Overall the key features of supportive materials for practical implementation are as follows:
(i) A strong contact between the catalyst and support
(ii) Huge surface area
(iii) Excellent water management
(iv) Facile catalyst recovery
(v) Mesoporous morphology
(vi) Good resistivity towards corrosion in acidic media
Pt possesses three types of morphologies, namely, Pt (100), Pt (110), and Pt (111), and it has been observed that Pt (111) produces greater current density than the other two forms. To study the influence of the morphology on alcohol oxidation, Dimos and colleagues in 2010 reported nanoporous and planar Pt and studied their catalytic activities for the methanol and ethanol oxidation reactions. Surface area analysis confirmed the greater surface area of the nanoporous Pt as compared to planar Pt, and the electrocatalytic response of the nanoporous Pt was also superior to the planar one. The reason was the presence of a greater fraction of Pt (111) than the other two phases, which was responsible for the high current density. For ethanol under basic conditions, the onset potential for nanoporous carbon was more positive than planar Pt accompanied by comparatively high stability. The greater anodic peak ratio of nanoporous Pt also reflects its tendency to oxidize the CO more efficiently. Nanoporous Pt also delivers greater current density in basic medium as compared to an acidic medium.82
Huang et al. in 2011 fabricated concave Pt nanocrystals containing high-index facets comprised of a high density of low-coordinated surface atoms, which therefore had enhanced electrocatalytic activity as compared to low-index faces. A solvothermal approach was employed to develop a concave nanocrystal in the presence of PVP and methylamine surfactant. The presence of high-index {411} exposed facets endowed the concave Pt nanocrystal with outstanding electrocatalytic activity for the ethanol oxidation reaction. The current density of 2 mA cm−2 was produced by Pt nanocrystals at the peak potential of 0.6 V vs. SCE and this current response was 4.2 and 6.0 times greater than the Pt black and Pt/C, and the reason was high-index {411} facets.83
Mesoporous carbon (MPC), utilized as an individual nanoscale reactor, and a new melt-diffusion approach was employed to load the precursor by Chen and colleagues in 2010 to fabricate the ethanol oxidation catalyst. Highly dispersed Pt-NPs (nanoparticles) confined in the small mesopores of the channel (Pt/MPC) and the pore wall were synthesized after the reduction of the precursors by hydrogen gas. The Pt/MPC (Pt mesoporous carbon) catalyst displayed a much enhanced catalytic activity and long-term stability for ethanol oxidation. The as-synthesized catalysts possess higher reactivity towards the C–C bond-breaking process and better selectivity for the complete conversion of ethanol to CO2. The improved electrocatalytic behavior was attributed to 3 aspects: (a) MPC possesses a large pore volume and specific surface area, as well as interpenetrated small-sized mesopores in the walls of the pore, which help in the uniform dispersion of the Pt NPs confined in MPC, and smooth mass transport of the reacting species. (b) The formation of Pt nanoparticles with small coordination sites is due to small mesopores and mesochannels, which limits the growth of the Pt NPs. (c) The unremitting carbon components in the mesoporous carbon channels enhance the electrical conductivity and improve the effective utilization of the Pt nanoparticles due to the melt-diffusion strategy employed for the precursor loading. In the CV experiment, it was observed that the current density of 675 mA cm−2 was delivered at the peak potential of 0.64 V vs. SCE, and stability was ensured through cyclic stability and chronoamperometric tests and the maximum amount of current density was retained until 1800 seconds and 4000 cycles.84
To study the effect of the morphology on the electrocatalytic activity of the material, in 2013, MourdiKoudis and coworkers described the synthesis of water-soluble Pt nanodendrites in dimethylformamide by utilizing a polyethyleneimine (PEI) stabilizer. The porosity of the dendrites (size = 20–25 nm) was tuned by varying the concentration of the metal precursors. The high surface area of the dendrites (up to 22 m2 g−1) was ensured by BET measurements, while the presence of high-index facets was revealed by X-ray diffraction in the face-centered-cubic (FCC) crystal structure of Pt. The as-prepared nanodendrites showed excellent electrocatalytic activity towards the oxidation of ethanol in alkaline solution as the current density of 197 mA cm−2 was delivered at the peak potential 0.77 V vs. RHE, while the forward and backward current density ratio, an indication of ethanol oxidation activity, was also greater in porous Pt3PEI as compared to the Pt1PEI and Pt2PEI having a nonporous structure. The nanodendrites' high porosity appears to have a robust impact on the electrocatalytic process.85
In 2015, Bu and colleagues reported the synthesis of hierarchical Pt–Co nanowires enclosed with high-index facets through a vigorous wet-chemical approach on the large scale as an electrocatalyst for fuel cell applications. Among all the synthesized Pt–Co NWs/C catalysts, the Pt3Co NWs/C electrocatalyst displayed the highest electrochemical active surface area (ECSA) of 52.1 m2 g−1 because of its high-density dendritic structure analogous to that of the commercial Pt/C (58.8 m2 g−1) catalyst. This feature leads to impressive electrocatalytic activity, and durability towards the ethanol oxidation reaction. Pt3Co NWs/C showed excellent activity by displaying an exceptional specific activity of 1.55 mA cm−2 and great mass activity of 0.81 A mg−1, which are 3.78 and 3.37 times higher as compared to commercial Pt and Pt/C, respectively. Moreover, the reported catalyst delivers a current density of 14.2 mA cm−2 at the peak potential 0f 1.0 V vs. RHE.86
In 2017, Suleiman et al. successfully fabricated the octahedral Pt2.3Ni/C nanocatalyst of average size ∼10 nm as an electrocatalyst for the ethanol oxidation process. The as-synthesized octahedral Pt2.3Ni/C showed higher electrocatalytic activity than the conventional Pt2Ni/C and commercial Pt/C (around 2.4 and 3.7 times, respectively) regarding forward peak current density. To monitor the reaction intermediates and products in situ, FTIR studies were conducted and it was observed that Pt/C showed higher selectivity towards CO2 as compared to acetic acid. However, CO poisoning may lead to both low stability and activity. The alleviated issue of CO poisoning was noticed on the octahedral Pt2.3Ni/C owing to its greater preference for C2 reaction pathways and resulted in faster kinetics on C2 pathways than on the C1 pathway. Moreover, the catalyst morphology and the synergetic effect of two metals are also activity-controlling factors. The peak current density response of the octahedral Pt2.3Ni/C (1.46 mA cm−2) was 2.4 and 3.7 times higher than the conventional Pt2Ni/C and Pt/C, respectively. The forward to backward peak current ratio, an indication of complete ethanol oxidation, was also higher on the octahedral Pt2.3Ni/C as compared to the conventional Pt2Ni/C and Pt/C along with long-term stability for 1800 seconds.87
Ultrathin nanowires of Pt–Mo–Ni (Pt–Mo–Ni NWs) with lengths of several micrometers and an average diameter of ∼2.5 nm were prepared by Mao et al. in 2017 by the HASR (H2-assisted solution route). The purpose of this catalyst design was as follows: (a) a large number of surface atoms on the ultrathin NWs increases the efficiency of Pt at the atomic level and lowers the catalyst cost; (b) Ni incorporation produces defects on the Pt surface leading to high catalytic activity by the adsorption of OH− species and the removal of CO intermediates; (c) Mo incorporation increases the stability of both Pt and Ni atoms, confirmed by density functional theory and experiment. As compared to Pt black and Pt/C, the high current density (2.5 mA cm−2 at 0.6 V vs. SCE), mass, and specific activity, as well as greater stability of Pt–Mo–Ni NWs, validate its potential application for the ethanol oxidation process. To study the effect of Mo incorporation on the stability of Pt–Ni alloy, Density Functional Theory (DFT) calculations were performed on the step edges of Pt–Mo–Ni NWs. The comparatively higher atomic cohesive energy (Ecoh) of Mo as compared to Pt and Ni increases the Ecoh of both of the metals and also allows Mo to be retained in Pt–Ni NWs by creating strong bonds with both Pt and Ni. As a result, the Mo atoms in nanowires stabilize the Ni and Pt towards the diffusion and dissolution process.88
In 2017, Liu et al. reported a one-pot scheme to formulate the monodisperse, uniform, narrow size distributed (4.6–5.1 nm) PtCu alloy polyhedrons. The specific activity and the mass activity on the Pt68Cu32 nanoalloy (19.3 mA cm−2, 2.33 A mg−1) were greater than on Pt85Cu15 (5.76 mA cm−2, 0.81 A mg−1) and Pt/C (1.63 mA cm−2, 1.10 A mg−1), respectively, while the peak current density on the Pt68Cu32 nanoalloy was 11.8 times greater than that on the commercial Pt/C (−0.1 V vs. Ag/AgCl). Moreover, the onset potential of the Pt68Cu32 nanoalloy was much lower than that on Pt/C, which suggests the faster reaction kinetics toward the C2H5OH oxidation. In a chronoamperometry experiment, the current density response of the Pt68Cu32 nanoalloy was much larger than others and was maintained for a longer time (3600 s). The calculated If/Ib ratio on the Pt68Cu32 nanoalloy was (1.89) much higher than that on Pt/C (1.21) and it indicated less CO adsorption, and an easier ethanol oxidation process. The enhancement of the catalytic performance for the as-synthesized Pt68Cu32 nanoalloy may be ascribed to the following: (i) the monodisperse, small-sized polyhedron structure that can meaningfully increase the number of edges, low coordination atoms, and corners prevailing in the PtCu nanoalloy surface; (ii) the synergistic effect between the Cu and Pt modify the chemical, and electronic properties of Pt metal via ligand and strain effects; (iii) a down-shift of the Pt d-band, weakened binding of Pt with the OER-active OH− species instead of poisonous intermediates.89
In 2015, Zhang et al. fabricated graphene nanoplates (GNPs)-supported Pt3Cu1 nanocrystals (Pt3Cu1 NCs) with different morphologies via a facile wet-chemistry synthetic approach by only varying the solution pH before Pt and Cu precursor reduction, where the in situ growth of metal nanocrystals on the GNPs effectively maximized the contact between the metal NCs and GNPs and thus attained the desired high electrocatalytic performance. Among the tested samples, the peak potential of Pt3Cu1 was lower as compared to the Pt catalyst, which suggests that the introduction of Cu can significantly decrease the activation barrier for the ethanol oxidation process. The mass and specific activities of tested samples follow the order of Pt/C-JM < Pt/GNPs < Pt3Cu1/GNPs < W-/GNPs Pt3Cu1 < D-Pt3Cu1/GNPs where, in particular, the current decay response of the Pt3Cu1/GNPs was significantly slower than other samples and it also followed the order of their e− transfer coefficients in the C2H5OH oxidation. Moreover, high current density (3 A mg−1) at peak potential (−0.35 V vs. SCE), and long-term stability (3600 s) are the salient features of D-Pt3Cu1/GNPs in alkaline medium. The thorough analysis proved that GNPs act as a morphology controlling factor by effecting the nucleation and growth processes of nanocrystals.90
Kung et al. in 2014 reported a novel hierarchically structured composite material of platinum ruthenium nanoparticles uniformly grown on the huge surface area of the 3D graphene foam (3D GF) through the CVD method. PtRu NPs produced through the reduction method were tested for the methanol and ethanol oxidation process, respectively. A PtRu/3D GF nanocatalyst exhibited greater tolerance towards CO poisoning and showed improved electrocatalytic activity for both the ethanol oxidation reaction (EOR) and methanol oxidation reaction (MOR). The particle size of the PtRu NPs on 3D graphene was reduced to 3.5 nm and correspondingly, the active surface area was improved to 186.2 m2 g−1. Consequently, the EOR and MOR rates were almost doubled on the PtRu/3D GF as compared to PtRu/C and PtRu. Cyclic voltammetry results showed that a current density of 78.6 mA cm−2 was exhibited at 0.91 V vs. SCE and after about 900 cycles, the current density response was 25.5 mA cm−2 in cycling stability tests. The If/Ib ratio, an indication of CO tolerance (1.0) in the case of PtRu/3D GF, was also greater than PtRu and PtRu/C, which proves its excellent performance for ethanol oxidation. The 3D multilayered structure of graphene foam with huge surface area and conducting pathways works as a free-standing electrode with minimum resistance and facile charge transfer. Moreover, the fine dispersion of metal nanoparticles on the GF surface prevents material agglomeration and leads to excellent electrocatalytic activity.91
In 2020, Hang et al. prepared mixed Ru–Sn oxides and deposited them on a high surface area carbon support through the thermal decomposition of Sn and Ru acetylacetonate complexes. The adsorption of preformed Pt nanoparticles generated an electrocatalyst with enhanced activity at a low potential for ethanol oxidation in aqueous sulfuric acid under ambient conditions (80 °C) in a Proton Exchange Membrane Fuel Cell (PEMFC). The change in the oxide composition (Ru0.38Sn0.62O2 to Ru0.67Sn0.33O2) did not significantly affect the catalyst's activity but elevated the stability in the acidic solution. However, higher stability was experienced in the PEM cell, where the anode catalyst Pt/Ru0.55Sn0.45O2/C provided comparatively higher currents at a lower potential than a commercial Pt/C catalyst for C2H5OH oxidation. A thermal processing technique was utilized for the fabrication of the anode by coating a carbon fiber paper support layer repeatedly with carbon black, Ru acetate, Sn acetate, and Pt nanoparticles. Pt/Ru0.67Sn0.33O2/C showed a current density response of 113.2 mA cm−2 at the peak potential of 1.65 V vs. RHE, while the maximum amount of current was maintained until 3500 seconds (stability response) with minor variations. The synergistic effect between Sn and Ru in the mixed oxide support was attributed to the high conductivity and strong ligand effect of SnO2 with the strong bifunctional effect of RuO2 leading to greater electrochemical activity.92
In 2014, Asgardi et al. presented the catalytic behavior of a 20 wt% Pt–Sn supported anode catalyst for the Direct Ethanol Fuel Cell (DEFC). Pt–Sn NPs with different Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn ratios (1:1 and 3:1) were mounted on the carbon nanofibers and carbon black. Physicochemical characterization techniques showed the fine dispersion of metal nanoparticles (4.0 nm) on the carbon supports. XRD analysis confirmed the presence of both SnO2 and Pt–Sn (1:1). The as-prepared catalysts were thoroughly studied in acidic media by carbon monoxide stripping voltammetry, cyclic voltammetry (CV), linear sweep voltammetry (LSV), and chronoamperometry (CA) in a 3 electrode assembly. It was found that Pt–Sn-supported electrocatalysts on the carbon black displayed higher electrocatalytic activity as compared to those deposited on carbon nanofibers (CNFs). The reason for the poor performance towards ethanol oxidation on carbon nanofibers is the low tendency for C2H5OH adsorption, deprotonation, and C–C bond cleavage. In acidic media, Pt1Sn1/CNF showed paramount current response as compared to its counterparts as the current density of 8 mA cm−2 was produced at the peak potential of 0.8 V vs. RHE accompanied by long-term stability until 800 seconds.93
:Sn ratios (1:1 and 3:1) were mounted on the carbon nanofibers and carbon black. Physicochemical characterization techniques showed the fine dispersion of metal nanoparticles (4.0 nm) on the carbon supports. XRD analysis confirmed the presence of both SnO2 and Pt–Sn (1:1). The as-prepared catalysts were thoroughly studied in acidic media by carbon monoxide stripping voltammetry, cyclic voltammetry (CV), linear sweep voltammetry (LSV), and chronoamperometry (CA) in a 3 electrode assembly. It was found that Pt–Sn-supported electrocatalysts on the carbon black displayed higher electrocatalytic activity as compared to those deposited on carbon nanofibers (CNFs). The reason for the poor performance towards ethanol oxidation on carbon nanofibers is the low tendency for C2H5OH adsorption, deprotonation, and C–C bond cleavage. In acidic media, Pt1Sn1/CNF showed paramount current response as compared to its counterparts as the current density of 8 mA cm−2 was produced at the peak potential of 0.8 V vs. RHE accompanied by long-term stability until 800 seconds.93
In 2016, Rizo et al. employed a formic acid reduction method (FAM) for the preparation of Pt–Sn electrocatalysts supported on different carbonaceous materials with different metal loadings (Pt–Sn 3:1, 1:1, 1:3), and with good dispersion to study their behavior toward the ethanol oxidation reaction and CO stripping. Fine particle dispersion on a carbon support, identical particle sizes (∼4–5 nm), and the presence of SnO were observed in all samples. Sn present in a high oxidation state helped in the oxidation of Pt. Ethanol oxidation was thoroughly studied in both acid and alkaline media by cyclic voltammetry and chronoamperometry. A higher anodic current was obtained with a gradual increase in the amount of tin in all the samples when supported on CNF as compared to carbon black support. The addition of Sn also shifted the onset potential for ethanol oxidation towards more negative values than Pt. It appeared that instead of an atomic ratio, the carbonaceous support strongly affected the CO tolerance. Cyclic voltammograms showed the inferior activity of Pt–Sn 1:3 as compared to the Pt–Sn 1:1 catalyst but identical stationary currents were established for both formulations in current transient curves, irrespective of the electrolyte and the carbon support. This important outcome proved the low noble metal (Pt) content in a sample with good activity. In all the cases, the electrocatalytic activity of the Pt–Sn 1:3 was found to be higher in alkaline media than in acidic media. Long-term stability for 700 seconds and a current density of 2 mA cm−2 are the salient features of Pt1Sn3/CNFs.94
In 2014, Chen and collaborators reported a green, one-pot, surfactant-free approach to develop PtPd NPs on the graphene nanosheets (PtPdNPs/GNs) by using an economical and readily accessible ethanol solvent as a reducing agent, and graphene nanosheets as the supporting material for metal nanoparticles. Metal nanoparticles of different morphologies such as spherical NPs (Pt1Pd1NPs), nanoflowers (Pd@PtNFs), and nanodendrites (Pt3Pd1NPs) were produced on the surface of GNs. The GNs not only increased the surface area, but also promoted the electron transfer process during the EOR. Among the tested samples, the PtPdNPs/GNs demonstrated better electrocatalytic activity in terms of high current density and negative onset potential. The negative shift of the anodic onset potential reflects the easy oxidation of the ethanol on the PtPdNPs/GNs. The delivered peak current density of the PtPdNPs/GNs (22.4 mA cm−2 at −0.1 V vs. Ag/AgCl), was approximately 2.5, 3.0 and 1.6 times greater than PtNFs/GNs (9.1 mA cm−2), PdNPs/GNs (7.5 mA cm−2), and PtPdNPs/C (14.2 mA cm−2), respectively. These results also coincide with electrochemically active surface area data since a greater surface area results in higher catalytic activity. The grander electrocatalytic performance of the tested material is attributed to 3 factors: (i) the functionalized GNs facilitate the CO removal from the surface of metal nanoparticles, (ii) the modified electronic structure of Pt with increased d-band vacancies due to Pd addition and favorable interatomic Pt–Pt distance, (iii) the oxophilic nature of Pd promotes CO removal from the Pt surface, (iv) graphene nanosheets with high surface area help in the fine dispersion of electrocatalyst and lead to the remarkable performance of the material.95
Zhang et al. in 2020 prepared the Pt1−xPdx/C electrocatalyst by varying the Pd concentration (x = 0, 27, 53, 77, and 100%) by the formic acidic reduction method for C2H5OH electro-oxidation in acidic and basic media. The stability and the catalytic performance of the composite for the ethanol oxidation were studied under both acid and alkaline conditions by cyclic voltammetry and chronoamperometry in terms of the effects of the ethanol and electrolyte concentrations, as well as the effect of the scan rate. In acidic media, an obvious double layer region appeared, which is not visible in alkaline medium since OH ion adsorption may even start in the H2 adsorption–desorption region and result in metal oxide formation. The reason for this better activity is the higher oxyphilic character of Pd/C and the comparatively inert nature of Pd/C towards C–C bond cleavage. The Pt/Pd ratio shows the significant impact on the alcohol oxidation in acidic and basic media as the Pt23Pd77/C electrocatalysts present the highest electrocatalytic performance with a good mass and specific peak current, high current density (2453.7 mA mg−1 at −0.2 V vs. SCE), and long term stability (3600 s) in acidic and alkaline media.96
Ren and colleagues in 2013 introduced a clean and facile method for the development of PtPd/rGO (reduced graphene oxide) catalysts with different Pt/Pd ratios without using any surfactant, stabilizer, and external energy in the form of temperature and pressure. The obtained catalysts were completely characterized by TEM, XRD, Raman spectroscopy, XPS, ICP-AES, and electrochemical measurements. The composition of all synthesized samples was controlled by the ratio of PtPd precursors and the particle size was in the range of 4–7 nm. Both cyclic voltammetry and chronoamperometry results validated that bimetallic (PtPd) catalysts possess superior ethanol oxidation activity as compared to the single metal Pt or Pd catalyst and the best performance was shown by the PtPd (1:3)/RGO catalyst. The current density of 105.9 mA cm−2 was delivered by PtPd (1:3)/rGO at a peak potential of −0.3 V vs. SCE in 1.0 M KOH/1.0 M C2H5OH solution, while the stable current response was maintained until 3600 seconds. The reason for the enhanced catalytic activity was due to (i) the synergetic effect of two metals and (ii) the ligand effect according to the Hammer Norskov d-band theory, (iii) large surface, high conductivity, and more active sites provided by rGO support.97
In 2014 Li et al. revealed a simple, one-pot water-soluble approach for the synthesis of porous dendritic Pt–Pd nanogarlands on the rGO surface at room temperature by using NP-40 (octylphenoxypolyethoxyethanol) as a soft template. The electrochemical testing showed that the as-prepared composites displayed significantly improved electrocatalytic activity and enhanced stability towards ethanol oxidation in an alkaline media. The electrochemically active surface area of PtPd/RGO (83.1 m2 g−1) is greater than that of Pt/RGO (16.3 m2 g−1), commercial Pd (19.7 m2 g−1), and Pd/RGO (12.5 m2 g−1 under identical circumstances). A negative shift of the onset potential, high mass and specific activity, high current density (210 mA cm−2 at −0.1 V vs. SCE), and long-term stability (4000 s/200 cycles) are other salient features of the dendritic PtPd/rGO composite. The improved electrocatalytic activity of the PtPd/RGO can be attributed to larger EASA with more active sites, the synergistic effects between Pd and Pt, the high catalyst loading and fine dispersion of the porous dendritic Pt–Pd garlands on the rGO surface, and the outstanding conductive nature of the graphene.98
Monodisperse nanoparticles of dimetallic and trimetallic alloys involving Ni, Pt, and Pd on the rGO support, synthesized through a surfactant-free solvothermal approach, were reported by Bhunia et al. in 2018. The HRTEM image of optimized Pt70Pd24Ni6 NPs shows {111} planes of multi-metals through the lattice spacing of 2.4 A0 after the incorporation of small-sized Ni atoms into the Pd and Pt lattice. The calculated ECSA of the tested multimetallic alloy NPs/rGO composites follow the order of Pt70Pd24Ni6/rGO (185 m2 g−1) > Pt73Pd27/rGO (160 m2 g−1) > Pt74Ni26/rGO (147 m2 g−1) > Pd68Ni32/rGO (142 m2 g−1) > Pt/C (86 m2 g−1). The assessed Rct data follow the sequence of Pt70Pd24Ni6/rGO (8.3 Ω) < Pt73Pd27/rGO (9.57 Ω) < Pt74Ni26/rGO (15.45 Ω) < Pd68Ni32/rGO (20.74 Ω) < Pt/C (29.75 Ω). The reasons for the enhanced catalytic activity of Pt70Pd24Ni6/rGO are the electronic structure, composition, fast reactants diffusion, reactants concentration, and scan rate. Upon Ni addition to the PtPd (bimetallic) NPs, the increase in the binding energy of Pd 3d and Pt 4f decreased the activation energy barrier for the oxidation reaction and poisoning effect due to carbonaceous species made on the surface of the electrocatalyst. Moreover, the smaller resistance value of the Pt70Pd24Ni6/rGO is also believed to be from the high electronic conductivity and the close contact of metal NPs with 2D rGO sheets. The oxygen-containing groups present on the GO surface (rGO precursor) act as strong anchoring sites for positively charge metal ions, which allows the firm attachment of metal NPs to the rGO surface with subsequent faster charge transfer and high electrocatalytic activity.99
In 2017, to improve the catalytic activity of materials for the alcohol oxidation process, Themsirimongkon et al. functionalized carbon nanotubes and decorated them with noble metal alloys (PtxMy, where M = Pd and Au; x and y = 1–3) via a reduction method. Material characterization was carried out by XRD, SEM, and TEM while electrochemical testing was done through CV, LSV, CA, and EIS. A uniform distribution of the spherical-shaped PtAu metal nanoparticles with diameters of 2–6 nm on the CNTs surface resulted in a large surface area (206.6 m2 g−1), low onset potential (0.28 V), high peak current density (22.5 mA cm−2 at 0.62 V vs. Ag/AgCl), small charge transfer resistances, high If/Ib ratio (0.88), durability (800 cycles), and better tolerance towards the CO poisoning during the ethanol oxidation reaction. The enhanced electrocatalytic activity of the as-prepared sample was attributed to (i) the high surface area and conductivity achieved through the uniformly dispersed small-sized metal NPs on the surface of the functionalized CNTs, (ii) improved electron transfer between the catalyst, CNTs channels, and electrolyte due to the highly conductive nature of the functionalized CNTs, and (iii) the incorporation of a second metal (synergistic effect), leading to easy CO oxidation.48
A facile and simple approach was used by Gnanaprakasam et al. in 2015 to synthesize a Pt thin film on the Au as Au@Pt core–shell nanoparticles and deposited on the reduced graphene oxide (rGO) surface via copper under potential deposition (UPD) tracked by a galvanic platinum replacement reaction. The driving force for the deposition of Pt on the Au surface by replacing copper is the reduction potential, and the rGO support helped in the fine dispersion of NPs. The characterization of the as-synthesized catalysts was performed by energy-dispersive X-ray spectroscopy (EDX), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Raman spectroscopy, X-ray diffraction (XRD), and electrochemical studies. rGO-Au@Pt NPs exhibited exceptional electrocatalytic activity during the ethanol oxidation reaction in alkaline medium due to enhanced specific and mass activities. Furthermore, the stability and the electrocatalytic activity of the as-synthesized electrocatalyst were compared with the commercially available Pt40/C and Pt20/C decorated electrodes. It was proved that the mass activity was 6.2 A mg−1, and the durability of the synthesized catalyst was significantly greater, even after 1000 seconds, as compared to the commercial catalysts; the basic reasons for this behavior include the following. (a)The strain effect leads to dissimilar bond lengths between the two metal atoms in the shell (core–shell nanoparticles) as compared to single-metal nanoparticles, (b) heterometallic interactions between the substrate and surface atoms (ligand effect) alter the chemical properties of the surface by altering the material electronic structure. (c) In heterometallic systems, the mixing of two metals with different Fermi levels produces lower energy levels, which alters the metal work and enhances the electrocatalytic activity through the strong binding of metals with the substrate, like ethanol, and increases its oxidation. (d) The rGO surface prevents metal NPs agglomeration and functional groups on the rGO surface attract the OH− of alcohol and enhance the analyst concentration close to the Pt surface with consequently enhanced activity. (e) Pt is the active center for alcohol oxidation and Au acts as a promoter for Pt.100
Paulo et al. in 2019 studied the ethanol oxidation activity of Pt nanoparticles (10–20 nm) in acidic media without ceria and with the CeO2-modified (1 and 5 wt%) electrode prepared by a novel Pechini method. During the experiments, it was observed that the incorporation of 1 wt% of CeO2 remarkably improved the activity of Pt NPs. This enhancement was ascribed to the lower microstrain of this catalyst as compared to the pure Pt, and strong interaction between CeO2 and the Pt. The incorporation of minute amounts of CeO2 (Ce4+) was responsible for a negative shift in the binding energy of Pt and a more oxidized CeO2 resulted in weaker interactions between the Pt and CO and the easy oxidation of CO to CO2. The current density of 1.5 mA cm−2 was delivered at a peak potential of 0.75 V vs. SCE and the maximum current was retained until 900 seconds.101
In 2018, Bai et al. reported the fabrication of the bimetallic PtRh nanodendritic alloy with tunable configuration by a simple complex-reduction synthetic approach under hydrothermal conditions. During the synthetic process, the difference in interaction strength between the metal precursors and polyallylamine altered the original reduction order. The formation mechanism, structural/morphological features, and the electrocatalytic activity of PtRh nanodendrites were thoroughly investigated by various physical techniques and electrochemical methods. The preformed nuclei of the Rh crystal effectively catalyzed the Pt2+ precursor reduction and gave the PtRh alloy due to the atomic inter diffusion method and catalytic growth, and generated the dendritic morphology. Pt1Rh1 ANDs demonstrated solution pH-dependent and chemical composition-dependent electrocatalytic activity for the OER process. At 0.88 V vs. RHE, the anodic current of Pt1Rh1 ANDs (9462.1 A g−1) was 8 times higher than the Pt nanocrystals electrocatalyst (57.5 A g−1), so the lower onset potential and larger oxidation current reflected the catalyst's best electrocatalytic performance. The PtRh ANDs also showed 2 times higher electrocatalytic activity in basic media than in the H2SO4 electrolyte. The higher EOR peak current and lower EOR peak potential of Pt1Rh1 ANDs indicated that in an alkaline solution there was faster reaction kinetics than in an acidic solution, and composition, as well as morphology, are responsible for this activity.102
In 2010, Shen and colleagues studied the electrocatalytic activity of carbon-supported PtRh electrocatalysts, synthesized by the microwave-assisted polyol method. The CV results confirmed that under alkaline conditions, among all the samples, the Pt2Rh/C catalyst possessed a greater electrocatalytic response in terms of peak current density, If/Ib ratio, and the onset potential for the EOR process as compared to Pt/C. The onset potential on the Pt2Rh/C catalyst (0.55 V) was 50 mV less than that of Pt/C, the peak potential (0.08 V vs. Hg/HgO) was 20 mV lower than that of Pt/C, while the peak current density (0.172 A cm−2) was higher than that of Pt/C. Most interestingly, the jf/jb ratio on Pt2Rh/C (1.9) was twice as large as compared to Pt/C. Moreover, the Tafel slope of Pt2Rh/C (112 mV dec−1) was less than that of Pt/C at a lower overpotential and the exchange current density calculated through the Tafel slope was 1.5 × 10−6 A cm−2. The reason for the enhanced electrocatalytic activity was an improvement in the C–C bond cleavage process in the presence of Rh and faster oxidation of CO to CO 2 due to the synergistic effect of two metals.103
To study the effect of morphology and composition, in 2019 Jin et al. introduced a facile and effectual self-templating technique to synthesize uniform and well-defined 1D ternary Pt–Rh–Te nanotubes with diverse compositions as a highly efficient electrocatalyst for C2H5OH oxidation. The compositions of these ternary Pt–Rh–Te nanotubes were precisely controlled by regulating the amount of H2PtCl6. The correlation of the structure, composition, and electrocatalytic performance was also examined. As compared to the Pt/C and PtTe nanotubes, the optimized sample of Pt5RhTe6 nanotubes exhibited 5.08 and 2.98 times greater mass activity for the ethanol oxidation process. Physicochemical characterizations and electrocatalytic measurements revealed that the porous walls, hollow interior, and unique tubular morphology helped to expose more surface electroactive sites. The incorporation of Rh atoms stimulated the C–C bond cleavage with the remarkable elimination of poisonous COads intermediates. The high current density (3500 mA mg−1 at −0.15 v vs. Ag/AgCl), low onset potential, high mass activity, high If/Ib ratio (2.30), high TOF (216 h−1), and maintenance of current density for a longer time (3600 s) as compared to PtTe nanotubes and Pt/C are other salient features of the tested sample.104
In 2013, Lee et al. prepared dendritic-shaped PtIr alloy nanoparticles (NPs) as a 3D structure for an enhanced C2H5OH oxidation reaction (EOR) by employing the thermal-decomposition technique in the presence of surfactant, cetyltrimethylammonium chloride (CTAC). The PtIr alloy dendritic form with 3D structure was analyzed by XPS, TEM, and XRD. Specifically, the PtIr alloy showed 2.74 times greater electrochemical active surface area (EASA) than the commercially available Pt/C due to the 3D dendritic structure. The observed lower onset potential of the d-PtIr/C (0.341 V) than the spherical PtIr/C (0.344 V) and commercial Pt/C (0.353 V), indicated the improved electrochemical activity of the dendritic PtIr/C nanocatalyst for the EOR. The maximum current density response of the dendritic PtIr/C was 13.76 mA cm−2 at 0.9 V vs. RHE. The If/Ib ratios of the reported d-PtIr/C, and s-PtIr/C were 1.55, and1.54, respectively, which were ∼1.2 times higher than Pt/C and this implies the complete ethanol oxidation reaction as compared to that on the pure Pt electrocatalyst, due to the facile C–C bond breakage after Ir addition. The improved electrocatalytic activity and durability of the PtIr alloy in EOR are credited to dendritic structures, huge surface, the electrocatalyst surface state, and the precise electronic structure due to the presence of Ir metal.105
In 2014, Ramesh and colleagues successfully fabricated the ordered ZrPt3 NPs with two different morphologies, the cubic ZrPt3 NPs and the hexagonal ZrPt3 NPs, by endorsing the transformation of the bulk-structure through controlled annealing (900 and 1000 °C). It was observed that the h-ZrPt3 NPs displayed better catalytic activity than the c-ZrPt3 NPs by showing enhanced oxidation activity toward ethanol and enhanced stability towards the repeated electrochemical cycles (200 cycles). The current density of 1.2 mA cm−2 was delivered by h-ZrPt3 NPs at a peak potential of 0.7 V vs. Ag/AgCl. The reason for the enhanced electrocatalytic performance of the h-ZrPt3 NPs was the transformation of the bulk structure from cubic to hexagonal, which increased the surface energy to compensate for the lowering of the bulk free energy. DFT calculations supported that the higher surface energy of h-ZrPt3 NPs, 1.47 J m−2 (most stable facet) as compared to the c-ZrPt3 NPs, 1.34 J m−2 resulted in high electrochemical performance since it promotes the adsorption and decomposition of ethanol and formic acid molecules on the surface and increases the catalyst stability by lowering the bulk free energy.106
Zhang et al. in 2016 reported a facile one-pot strategy to prepare highly ordered dendritic Pt/PtxPb core/shell nanowires (NWs). As compared to the reported NWs, the reported hierarchical core/shell NWs displayed the assimilated features of the 1D structure, core/shell features, high surface area, and alloy effect. The sequential incorporation of Pb and precursors was mainly responsible for the large surface area; however, the composition of the ordered Pt–Pb NWs could be tuned by varying the amount of Pt (acac)2. Among the six tested samples, these newly-developed Pt–Pb NWs showed the highest EOR mass and specific activity. The PtPb0.27 NWs reported mass activity was 2.1, 3.9, and 4.8 times greater than Pt NWs, PtPb0.21 NPs, and commercial Pt/C and after 1000 cycles; 74.9% of the initial mass activity was retained, which is an indicator of high stability. Moreover, no change in the composition and morphology occurred after the stability test. The current of 1.8 mA was delivered at a potential of 0.6 V vs. SCE and minor attenuation in current was recorded after 1000 cycles (Table 1).107
| Electrocatalyst | Electrolyte/methanol molarity | Electrode | Peak potential V (RHE) | Tafel slope mV dec−1/stability (s) | Current density mA cm−2 | Ref. |
|---|---|---|---|---|---|---|
| Nanoporous Pt | 1 M KOH/1 M C2H5OH | Silica/Au | 0.855 V | —/1000 s | 24.3 | 82 |
| Concave Pt | 0.1 M HClO4/0.1 M C2H5OH | — | 1.65 V | — | 2.0 | 83 |
| Pt/MPC | 0.1 M HClO4/0.1 M C2H5OH | GCE | 1.69 V | —/800 s, 4000 cycles | 674.0 | 84 |
| Pt3-PEI | 1.0 M NaOH/1.0 M C2H5OH | GCE | 0.77 V | — | 197.0 | 85 |
| Pt3Co/C NWs | 0.1 M HClO4/0.1 M C2H5OH | GCE | 1.00 V | — | 14.2 | 86 |
| Pt2.3Ni/C | 0.1 M HClO4/0.2 M C2H5OH | GCE | 0.92 V | —/1800 s | 1.5 | 87 |
| Pt–Mo–Ni NWs | 0.5 M H2SO4/2.0 M C2H5OH | GCE | 1.65 V | —/1500 s | 2.5 | 88 |
| Pt68Cu32 nanoalloy | 0.5 M KOH/0.5 M C2H5OH | GCE | 1.03 V | —/3600 s | 19.3 | 89 |
| D-Pt3Cu1/GNPs | 0.5 M KOH/0.5 M C2H5OH | GCE | 0.72 V | —/3600 s | 42.4 | 90 |
| PtRu/3D GF | 0.1 M H2SO4/1.0 M C2H5OH | GCE | 1.96 V | —/900 cycles | 78.6 | 91 |
| Pt/Ru0.67Sn0.33O2 | 0.5 M H2SO4/0.2 M C2H5OH | CFP | 1.65 V | —/3500 s | 113.2 | 92 |
| Pt1Sn1/CNF | 0.5 M H2SO4/2.0 M C2H5OH | GCD | 0.80 V | —/800 s | 8.0 | 93 |
| Pt1Sn3/CNF | 0.1 M NaOH/1.0 M C2H5OH | GCD | 1.00 V | —/700 s | 2.0 | 94 |
| PtPd NPs/GNs | 1.0 M NaOH/1.0 M C2H5OH | GCE | 0.82 V | —/1000 s | 22.4 | 95 |
| Pt23Pd77/C | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.85 V | —/3600 s | 173.3 | 96 |
| PtPd (1:3)/rGO |
1.0 M KOH/1.0 M C2H5OH | GCE | 0.77 V | —/3600 s | 105.9 | 97 |
| D-PtPd/rGO | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.97 V | —/4000 s | 210.0 | 98 |
| Pt70Pd24Ni6/rGO | 1.0 M KOH/0.5 M C2H5OH | GCE | 0.80 V | — | ∼79.0 | 99 |
| PtAu/CNTs | 0.5 MH2SO4/0.5 M C2H5OH | GCE | 1.68 V | —/800 cycles | 22.5 | 48 |
| rGO-Au@Pt NPs | 0.5 M NaOH/0.5 M C2H5OH | GCE | 0.79 V | —/1000 s | 173.6 | 100 |
| PtCeO2 | 0.5 M H2SO4/1.0 M C2H5OH | GCE | 1.75 V | —/900 s | 1.5 | 101 |
| Pt1Rh1 (dendritic) | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.88 V | —/2000 s, 1000 cycle | 32.6 | 102 |
| Pt2Rh/C | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.86 V | 102 mV dec−1/— | 172.0 | 103 |
| Pt5RhTe6 nanotubes | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.88 V | —/3500 s, 500 cycles | 247.2 | 104 |
| d-PtIr/C | 0.1 M HClO4/0.2 M C2H5OH | GCE | 1.68 V | —/7200 s | 13.8 | 105 |
| h-ZrPt3 NPs | 0.5 M H2SO4/1.0 M C2H5OH | GCE | 1.73 V | —/200 cycles | 1.2 | 106 |
| PtPb0.27 NWs | 0.1 M HClO4/0.15 M C2H5OH | GCE | 1.70 V | —/1000 cycles | 9.2 | 107 |
| PtPbPd NWs | 0.5 M NaOH/0.5 M C2H5OH | GCE | 1.24 V | —/10000 s |
105 | 108 |
To study the effect of composition, structure, and morphology, Duan et al. in 2019 reported interconnected ultrathin trimetallic nanowires of PtPbPd synthesized by the octylphenoxypolyethoxyethanol (NP-40)-facilitated one-step aqueous method using in situ-generated H2 bubbles as a dynamic template. It was observed that the precursor type and NP-40 concentration are critical in the synthesis process. The as-synthesized material showed remarkable upgrades in the catalytic properties for the EOR (ethanol oxidation reaction), and ORR (oxygen reduction reaction) as compared to PtPd NWs, PdPb NWs, PtPb NWs, and commercial Pt/C (20 wt%). Specifically, the greater mass/specific activity, higher current density, positive shift in on set potential and long-term stability are salient features of the reported catalyst in 0.5 M KOH solution. The reasons for the excellent catalytic performance include (i) electronic modulation and synergies between three metals, and (ii) abundant and exposed electroactive sites due to interconnected nanowires (Fig. 5).108
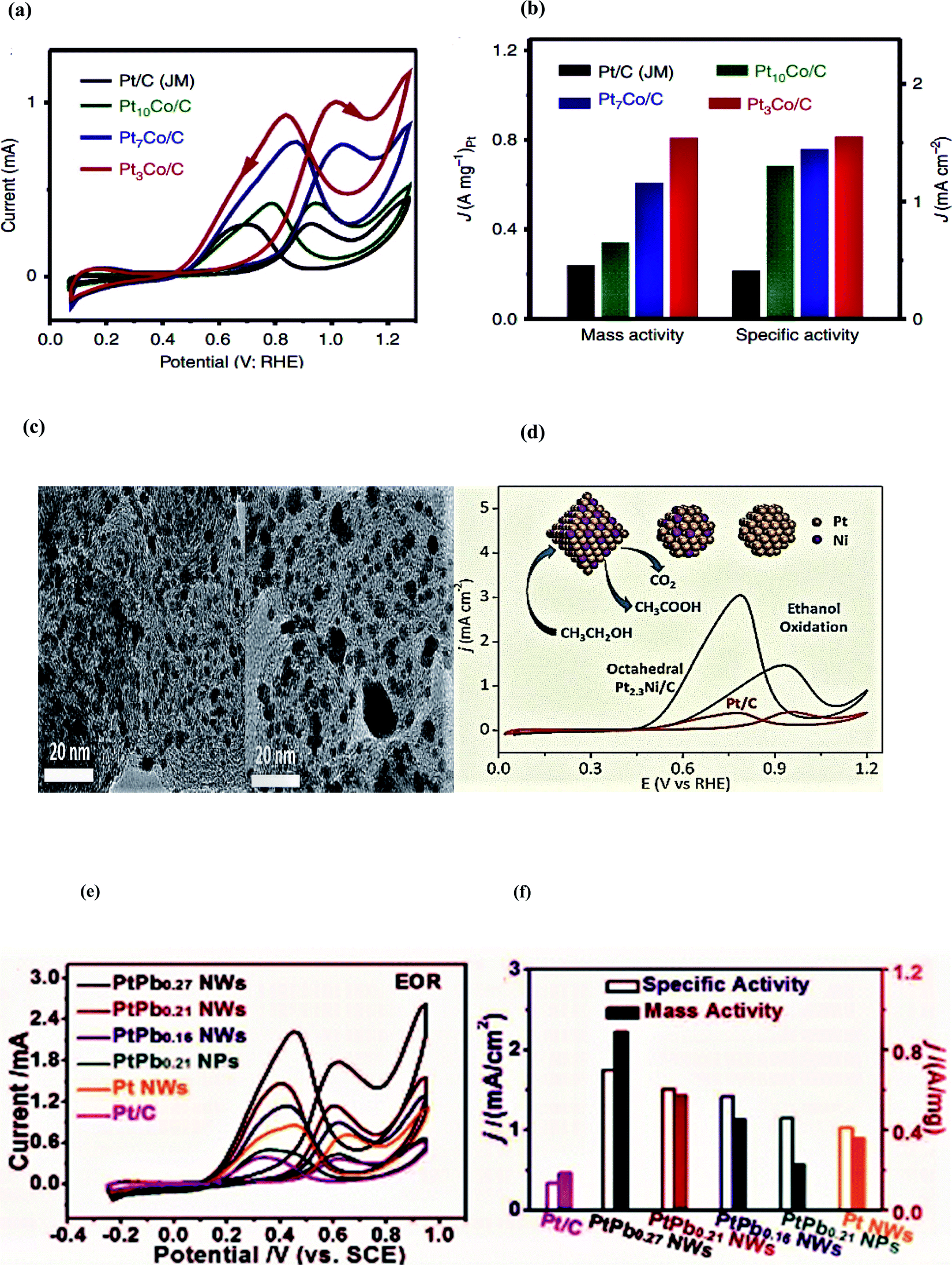 | ||
| Fig. 5 (a) CVs and (b) histogram of the mass activities and specific activities of different Pt-based catalysts for the EOR in 0.1 M HClO4 and 0.2 M ethanol solution. Reproduced from ref. 86, Copyright 2016, Nature. (c) SEM images and (d) CV polarization curves of Pt/C and Pt2.3Ni/C. Reproduced from ref. 87, Copyright 2017, ACS Catalysis. (e) CV polarization curves of Pt Pb nanowires and (f) comparison of the specific activity and mass activity of Pt Pb nanowires. Reproduced from ref. 107, Copyright 2017, ACS Catalysis. | ||
5.2. Palladium and palladium-based compounds
Among the numerous electrocatalysts, Pt seems to be the best electrocatalyst and is extensively utilized for the ethanol oxidation reaction. However, its high cost, limited resources, and deactivation/poisoning by the reaction intermediates, particularly CO, CH3COO−, etc., hinder its practical application. To resolve this issue, researchers have done a lot of work to prepare a catalyst with a low Pt dose to reduce the catalyst cost or to prepare a highly efficient non-Pt catalyst, and it was observed that Pd is the best alternative to Pt due to its comparatively lower cost, great abundance, and good catalytic activity at low onset and overpotential.109In 2016, Li et al. prepared a novel Pd@g-C3N4/GC composite with a width of ∼200 nm and thickness of ∼15 by an ethylene glycol microwave heating reduction scheme from the Na2PdCl4 precursor. In this scheme, the following features were reported: (i) the nanosheet-like structure of the graphite carbon support was prepared by an economical low-temperature (850 °C) Ni-catalytic route through a cheaper ion-exchange resin; (ii) the nano-islands of the ultra-thin g-C3N4 (5 nm) were successfully placed on the surface of the graphite C support, which facilitated the electron transfer process and diffusion of electrolyte; (iii) the electron coupling effect of the GC support and g-C3N4 can support the Pd nanoparticle fixation on the support surface; (iv) the pyridinic nitrogen-rich structural units of g-C3N4 induce a large number of electrochemically active points after interaction with the Pd metal. The analysis of the sample showed that the correct amount of g-C3N4 can enhance the electrocatalytic effect of Pd by lowering the activation energy required for ethanol oxidation, but an excessive amount would be counterproductive due to the tendency of graphitic nitrogen to combine with ethanol. The maximal peak current density of Pd@g-C3N4/GC (2156 A g−1 at 0.85 V vs. RHE) was 1.87 times greater than Pd/GC (1150 A g−1) and even after 200 CV cycles, the retained peak current density was 1904 A g−1, while for Pd/GC it was only 966 A g−1. In EIS, the curvature radius of Pd@g-C3N4/GC between Pd/AC and Pd/GC reflected the adequate electrical conductivity and desirable dynamics of Pd@g-C3N4/GC.110
In 2014, Jin et al. reported N-doped carbon-supported Pd nanoparticles and studied their electrocatalytic response towards the ethanol oxidation reaction. It has been confirmed that the fine dispersion of catalyst, the valence state of Pd, and the structural properties such as BET surface area (324 m2 g−1), as well as the porous structure of the support, may affect the performance of the catalysts. The reported electrochemical surface area ECSA of the Pd@CN electrocatalyst was is 1.4 and 1.9 times greater than the hydrothermal carbon-supported Pd (Pd@HC) and activated carbon-supported Pd (Pd@AC), confirmed by XPS. The current density of 2300 A g−1 was delivered at −0.22 V vs. SCE and the maximum current was retained even after 2000 seconds. Complete analysis proved that the incorporation of N into the carbon matrix upgraded the Pd0 percentage due to the positive inductive effect of nitrogen (proved by XPS), which offered more electroactive sites and amplified the Pd binding energy as a result of a down-shift of the d-band of Pd leading to the weak binding of the adsorbent on the Pd surface in Pd@CN and consequently enhanced the electrocatalytic activity. Moreover, the analogous particle size and uniform dispersion of Pd nanoparticles in the catalysts eliminated the strong CO adsorption and in turn, resulted in promoted activity.111
Hu et al. in 2012 synthesized self-assembled palladium nanocrystals on helical carbon nanofibers. First, the helical carbon was functionalized with benzyl mercaptan (Pd–S-HCNFs) and then Pd nanocrystals were fixed on the functionalized HCNF by Pd–S bond by a simple self-assembly method. The complete characterization of the as-prepared materials was done by X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction spectroscopy (XRD), cyclic voltammetry (CV), and fuel cell testing. TEM images of the synthesized materials displayed that surface nanofibers uniformly adhered to Pd nanocrystals (5 nm). Fuel cell testing and CV measurements demonstrated that both the electrocatalytic activity and the stability of Pd–S-HCNFs were significantly better than commercial electrocatalyst Pd/XC-72. This activity is attributed to the highly crystalline nature of the Pd nanoparticles, the good conductivity of HCNFs, their uniform structure, and the strong adherence of the carbon support to the nanocatalysts. The current density of 9 mA cm−2 was delivered at −0.35 V vs. SCE and a minor attenuation in current response was observed after 6000 seconds in stability testing.112
Zhang et al. in 2016, demonstrated a microwave-assisted ascorbic acid reduction method to fabricate NS co-doped graphene-braced PdOx-rich Pd catalysts. The NS co-doped graphene provided uniformly distributed active sites for the deposition of PdOx-rich Pd nanoparticles. The stronger interaction between the support and the PdOx-rich Pd particles effectively suppressed the agglomeration and exfoliation of the particles after a long-term stability test for 7000 seconds. The Pd/NS-rGO electrocatalyst also possessed a large electrochemically active surface area (35.7 m2 g−1), higher forward current density (1054 mA mg−1 at −0.17 v vs. SCE), forward to backward current density ratio (1.45), and tolerance towards the poisoning of reaction intermediates.51
Krittayavathananon et al. in 2019 studied the effect of PdO on the catalytic activity of Pd for the ethanol oxidation reaction. Experiments and a theoretical approach revealed that the oxide layer significantly enhanced the electrocatalytic performance of Pd in a basic solution as it altered the catalyst active sites, and modified the surface morphology, which led to the different reaction mechanisms and kinetics. Few nanometers thick nanoscale PdO {101} layers on the surface of Pd {111} strongly interacted with ethanol and reaction intermediate species (i.e. acetic acid and acetaldehyde), leading to an extraordinary current density response. The surface roughness and electroactive sites of the PdO {101} layer stabilized the adsorbed intermediates, accompanied by high autocatalytic decomposition and consequent overall productivity, even though the bulk PdO {101} was poor in terms of productivity and electrocatalytic activity. The modification of the metal surface by nanoscale metal oxide layers is, therefore, an uncertain process in terms of improving productivity and electrocatalytic activity. The binding energy between the catalyst and ethanol was determined by DFT calculation. The calculated binding energies of ethanol molecules on the surface of PdO and PdO/Pd were 6.51 kcal mol−1 and 12.53 kcal mol−1, respectively. Based on the Eads, it was observed that ethanol was preferably adsorbed on the surface of PdO/Pd, where oxygen is responsible for the stabilization of the ethanol layer with its subsequent decomposition and resulted in enhanced activity.113
To enhance the catalytic activity of the Pd, in 2014, Dong and collaborators reported copper-incorporated Pd nanoparticles braced on a graphene support. According to SEM, TEM, and EDX, the particle size, and the distribution of nanoparticles on the graphene surface can be tuned by varying the initial concentration of the reducing agent and Cu precursor. Electrochemical measurements showed that Pd/Cu/graphene-1 had the largest forward/backward current ratio (4.98) as compared to other materials. Pd/Cu/graphene-2 displayed the most optimal long-term stability and Pd/Cu/graphene-3 delivered the highest current density of 392.6 mA mg−1 at −0.26 V vs. SCE. The most probable reason for this excellent performance is an interaction between Cu and Pd, as well as the support of nanosheets of graphene. Graphene nanosheets not only provide support, they also control the growth of material. Moreover, oxygen-containing functional groups present on edges and surfaces strongly anchored the metal NPs and resulted in improved conductivity and reactivity.114
A series of PdCu-containing trimetallic nanocrystals (PdCuM NCs), including PdCu, PtCuCo, and PdCuNi, were fabricated on a large scale via a wet-chemical approach and were then readily converted into hierarchal intermetallic structures via simple annealing at 375 °C by Jiang et al. in 2014. The newly developed structurally ordered intermetallic PdCuM nanocrystals demonstrated an interesting metal composition-reliant electrocatalytic performance, where the Co-based sample showed the best ethanol oxidation activity with 12.9 and 17.5 times greater mass activity than commercial Pt/C and Pd/C electrocatalysts for the EOR. Moreover, PtCuCo also exhibited higher stability than the Pt/C, and Pd/C catalysts by retaining the maximum amount of the initial current density (0.72 A mg−1 at o.72 V vs. RHE) even after 10000 sweeping cycles. The DFT calculations disclosed that the improved ORR activity of the PdCuM NCs originated from the electrocatalytically active hollow sites arising from the ligand effect and the compressed strain on the surface of Pd due to the small size of Ni, Cu, and Co.115
Wang et al. in 2014 demonstrated an electrodeposition approach for the production of a novel and flexible PdCo nanotubes array on a carbon fiber cloth support and tested its ethanol oxidation activity. The calculated electrochemically active surface area (ESCA) of PdCo NTAs/CFC was 50.13 m2 g−1, remarkably greater than Pd NTAs/CFC (28.11 m2 g−1) and commercial Pd/C catalysts (23.82 m2 g−1). This enhanced ESCA of PdCo NTAs/CFC was due to the hollow nanotube structure, a high proportion of metallic Pd, the electronic effect of Co on Pd, and the hierarchical structure in the PdCo nanotube walls. The mass peak current density of PdCo NTAs/CFC was ∼1.7 times greater than Pd/CFC NTAs and ∼4 times higher than commercial Pd/C. The PdCo NTAs/CFC electrocatalysts displayed significantly enhanced catalytic activity and cycling stability during the ethanol oxidation process, indicating the auspicious potential of PdCo NTAs/CFC for the EOR. Most significantly, because of the invariant presentation under several distorted states, such as bending, normal, and twisting states, the PdCo NTAs/CFC provides a fundamental chance for the growth of flexible catalysts. The current density delivered by PdCo NTAs/CFC was about 1450 A mg−1 at a potential of 0.92 V vs. RHE and even after 600 s of stability testing, 90.6% of the original current was retained by the sample. The key reasons for the elevated electrocatalytic response are as follows: (i) due to their porosity, the nanotube arrays along with carbon fiber cloth provide a large surface area with easy electrolyte diffusion. (ii) The nanotube array and the carbon fiber cloth are both stable toward the dissolution and aggregation process. (iii) The CFC and PdCo NTAs are both flexible in nature. (iv) The optimized Pd Co ratio obtained through the electrodeposition technique resulted in improved catalytic activity.116
Zhang and colleagues, in 2017, employed a novel method to fabricate ultrafine PdCo bimetallic nanoparticles encapsulated in nitrogen-doped porous carbon nanocapsules (PdCo@NPCNs) through the one-pot Co and Pd precursor-mediated polymerization of dopamine on the surface of SiO2 nanospheres, accompanied by the carbonization and engraving of the SiO2 template. In this catalyst, the carbon shell of 5.6 nm thickness served as a barrier to thwart PdCo NPs aggregation during the annealing route to attain the 4.1 nm PdCo NPs. Moreover, the existence of cobalt in PdCo@NPCNs can meritoriously augment the content of N in NPCNs during the carbonization step, which results in the improved performance of the electrocatalysts. Consequently, the optimized PdCo@NPCNs comprising 3.68 wt% PdCo loading revealed a better electrocatalytic performance (current density of 1350 A mg−1 at 0.84 V vs. RHE), and stability (8000 s) toward EOR in an alkaline medium as compared to commercial Pd/C catalysts, and Pt/C respectively. According to DFT calculations, these nanocapsules exhibited the BET surface area pore volume of 730.2 m2 g−1 and 1.31 cm3 g−1, respectively. The pore size distribution data demonstrated that PdCo@NPCNs with mesopores and micropores centred at 4 and 0.6 nm, respectively possessed a hierarchical porous structure responsible for the growth and diffusion of PdCo NPs, the release of volatile material during the annealing step, and the carbonization of dopamine.117
Wang et al. in 2015 introduced a simplistic one-pot solution-based approach via the in situ thermal decomposition of lead acetate and iron pentacarbonyl in a refluxing dimethylformamide solution where the ultrafine Fe–Pd alloy was initially formed and mounted on the Fe2O3 NPs surface and then evenly dispersed on the surface of MWNTs. The Fe–Pd alloy formation was confirmed by the composition and crystal structures studied through XPS, XRD, and TEM, while electrochemical testing was done through CV, CA, and EIS, which depicted the enhanced electrocatalytic activity of the FePd–Fe2O3/MWNTs nanocatalyst towards the EOR as compared to Pd/MWNTs. The modified electronic structure of Pd by Fe incorporation prevented the formation of the poisonous intermediate, and the best combination of Fe2O3 (promoter) and MWNTs was mainly responsible for the greatly improved ethanol oxidation activity. The enhanced tolerance towards the poisoning species and depressed charge resistances obtained in CA and EIS correspond to the down-shifting of the d band center of the Fe–Pd alloy, easily generated oxygenated species on the surface of Fe2O3, and the stability provided by MWCNTs. High EASA (120.4 m2 g−1), greater current density (1.24 A mg−1 at −0.25 V vs. SCE), greater stability (1000 s), and minimum resistance are the prominent features of the FePd–Fe2O3/MWNTs nanocatalyst.118
In 2010, Maiyalagan and collaborators mounted Pd–Ni nanoparticles on the carbon nanofiber (CNF) support through a chemical reduction approach with NaBH4 as the reducing agent. The Pd–Ni/CNF anode catalysts were thoroughly characterized by X-ray diffraction, scanning electron microscopy, transmission electron microscopy, and electrochemical voltammetry analysis. TEM and SEM images showed that the 4.0 nm-sized Pd–Ni nanoparticles were quite homogeneously distributed on the surface of the carbon nanofibers, which provided a large surface area with a greater number of active sites. The onset potential was low at 0.2 V and the anodic peak current density (199.8 mA cm−2 at −0.15 V vs. Hg/HgO) was four times higher as compared to Pd/C for the ethanol oxidation reaction; the electrocatalytic response was further enhanced with an increase in the temperature from 20 to 60 °C. The reasons for the paramount activity were the uniform distribution of the metal nanoparticles on the CNF support, and the addition of Ni, which significantly decreased the overpotential, and the formation of NiOOH on the catalyst surface enhanced the electrocatalytic activity for C2H5OH oxidation.119
To design a carbon support and binder-free core–shell Ni@Pd–Ni nanowire array electrode for a superb ethanol electro-oxidation process, Guo et al. in 2018 reported facile template-assisted, self-supported nanowires of Ni@Pd–Ni with a solid Ni core (∼99.5 nm in diameter) and a shell of gauze-like Pd–Ni alloy (∼40 nm thick). The optimized sample Ni@Pd–Ni NAs possesses a huge electrochemically active surface area (ECSA) of 64.4 m2 g−1. Furthermore, the presence of Ni in the Pd–Ni alloy provides oxygen-based species that assist in the removal of C2H5OH molecules or intermediates. Consequently, the catalyst revealed an extraordinary peak current density of 1100 A g−1 at −0.15 V vs. SCE and retention of 96.7% current even after 50 cycles. The reported carbon and binder-free electrode of the Ni@Pd–Ni NAs microstructure with open and low-tortuosity circumvents the issues of active site blockage and ECSA degradation.120
The highly active Pd/Ni-NSC catalyst of size 200–300 nm was obtained by the annealing treatment of the metal–organic framework (M = Ni, L = vanillic thiosemicarbazone) and was tested for the ethanol oxidation reaction by Yang et al. in 2017. In the tested sample, the synergistic effect between Ni, N, S, and Pd supported the formation of OH radicals on the surface of a metal, N, and S at lower potential and reacted with species on the Pd surface to give the (CH3CO)ads intermediate, and provided free active sites. Besides, the e− donating effects of the S and N atoms improved the dispersion and prevented the agglomeration of the Pd NPs and consequently, Pd/Ni-NSC showed surprising stability (1600 s) and catalytic activity (current density = 110.3 mA cm−2 at −0.29 V vs. SCE) for the electrochemical ethanol oxidation reaction as compared to the commercial Pd/C catalyst. The good stability response of the Pd/Ni-NSC catalyst is ascribed to (a) well-dispersed Pd nanoparticles and (b) the stronger binding between the Pd NPs and co-doped S and N to inhibit particle agglomeration. Moreover, according to Density Functional Theory calculations (DFT), the improved catalytic activity and stability originated from the stimulated production of OH radicals on the Ni electroactive sites, which facilitated the removal of the carbonaceous intermediates through oxidation and interaction with CH3CO radicals on the contiguous Pd electroactive sites. The electron-accepting S and N species can divulge a comparatively high positive charge density on adjacent OH− in alkaline media, and the OHads is more easily generated on the surface of S and N than on the Pd surface at low voltage, leading to enhanced activity.121
To provide a detailed study of the effect of an increased number of active sites of oxyphilic metal and noble metal and the shortened distance between them on durability and electrocatalytic activity, in 2016, Chen et al. reported Pd–Ni–P ternary nanocatalysts prepared through a facile strategy. Electrochemical testing and DFT calculations highlighted that the integration of Ni/P nanoparticles and the shortening of the distance between the Ni and Pd active sites greatly expedited the formation of OH radicals and thus facilitated the interaction between CH3CO and OH radicals (rate-determining step). The CO tolerance tendency was also enhanced, and the catalyst had long-term stability and impressive EOR activity as compared to commercial Pd/C catalysts. With the upsurge in the Ni/Pd ratio from 4/6 to 5/5 and then 6/4, the NPs became more uniformly dispersed and prevented particle agglomeration, which is responsible for high activity. The peak current densities of Pd31Ni53P16 (2.74 A mg−1), Pd38Ni45P17 (4.42 A mg−1), ND Pd47Ni36P17 (4.9 A mg−1 at −0.9 V vs. RHE) NPs were respectively almost 3.81, 5.68, and 6.1 times higher than commercial Pd/C (0.72 A mg−1). Moreover, according to the TEM analysis, the nanocatalysts with no noticeable change in the particle size and shape after electrocatalysis accounted for the good stability of the material. A cluster model was also proposed to analyze the effect of nickel and phosphorus incorporation on OH formation and *OH desorption through DFT calculation. It was observed that Ni was preferentially adsorbed on the Ni surface due to attraction between the Ni (+ve charge) and the OH− (−ve charge). The Pd–Ni–P ternary nanoparticles showed the maximum adsorption energy for OH− (115.7 kcal mol−1) and the minimum desorption energy (44.2 kcal mol−1), which suggest that the Ni and P incorporation is beneficial for both OH− chemical absorption and *OH desorption, which thus expedites the CH3COOH formation. Therefore, the Ni and P incorporation in the ternary nanocatalysts pushes the EOR process favorably through the effectual reactive-intermediate pathway.122
Sarkar and collaborators, in 2015 fabricated Pd2Ge nanoparticles through a solvothermal super hydride reduction of Pd and Ge precursors (K2PdCl4 and GeCl4) without any surfactant, where particle size was controlled by varying the reaction time. The transmission electron microscopy (TEM) and powder X-ray diffraction (PXRD) data revealed that the Pd2Ge nanoparticles were designed as a well-organized intermetallic phase. In the crystal lattice, Ge and Pd atoms occupy the two dissimilar crystallographic positions with a vacant position at one of the Ge sites, which was proved by energy-dispersive X-ray (EDX) analysis, and PXRD. The catalyst is extremely efficient for the oxidation of the ethanol reaction and durable up to the 250th cycle in an alkaline medium. The reported electrochemical active surface area (EASA) and the current density values, 1.41 cm2 g−1 and 4.1 mA cm−2 at −0.074 V vs. Hg/HgO, respectively were superior to the commercial Pd/C values. This work also validates the fact that the p-block elements analogous to the first–row transition metals can also be utilized as active components along with the rare metals to form an intermetallic compound/alloy. Moreover, in the ordered Pd2Ge nanoparticles a perfect balance between the adsorption energies of OH− and CH3CO− on the electrocatalyst surface demonstrated its catalytic activity, and the occurrence of vacancies in the non-active sites affected the extent of reactivity. According to DFT calculation, the adsorption energy data of OH− and CH3CO− was compared and it was observed that the OH− strongly binds with the Pd2Ge surfaces as compared to the Pd {111} surface. After the adsorption of OH−, the Pd2Ge surface showed robust relaxation of ∼0.2 Å as compared to about 0.1 Å in the circumstance of the Pd surface. This robust relaxation of the surface of Pd2Ge indicated that the stout contact between the adsorbate and the surface and resulted in a strong interaction of OH− with the Pd {111} surface with consequent high electrochemical activity.123
In 2019, Rajamani and colleagues solvothermally synthesized Pd2−xNix Ge (x = 0, 0.2, 0.4, 0.5) with dissimilar Pd/Ni ratios by a solvothermal approach. Optimization of the Pd/Ni ratio and structural arrangement with nickel substitution at the Pd sites led to high ethanol electrooxidation activity and CO tolerance towards the reaction intermediate. The characterization of all compounds was carried out by powder X-ray diffraction, transmission electron microscopy (TEM), X-ray absorption fine-structure spectroscopy (XAFS), and ICP-OES. The electrochemical activities of the Pd2Ge and Ni-substituted Pd2−xNixGe were examined for the ethanol oxidation process in an alkaline medium through cyclic voltammetry (CV), and chronoamperometric (CA) techniques. The obtained results indicated that the Pd1.6Ni0.4Ge provides extraordinary electrochemical activity, exceptional stability, and a notable anti-poisoning tendency toward the ethanol oxidation reaction in terms of mass activity and current density. The Pd1.6Ni0.4Ge has the lowest onset potential (0.53 V) as compared to other compounds, which is 0.70 V less than pristine Pd2Ge, while the mass activity was 3.7 times greater than pristine Pd2Ge. The current density of 13 mA cm−2 was delivered at a potential of −0.05 V vs. Hg/HgO with a constant current response until 1000 seconds. To understand the Ni substitution in the as-synthesized sample, DFT calculations were performed. The Pd2Ge possesses four crystallographic sites: two Ge atoms occupy 2 Wyckoff sites, 2a and 1b, while 2 Pd atoms occupy the Wyckoff sites 3f and 3g. Ni replacement at the Pd site was found to be more stable than at Ge sites. Moreover, of the two (Wyckoff) sites of the Pd, Ni substitution at the 3g site was more favorable and was confirmed by the negative formation energy. Further analysis with different concentrations of Ni indicated that the Pd1.6Ni0.4Ge was the most fixed configuration. The main part of the oxyphilic nickel substituent in Pd2Ge was to increase the OH− adsorption energy on the catalyst as compared to CH3CO− adsorption to facilitate the CO oxidation and to improve the ethanol oxidation by lowering the d-band center of Pd Ge to the Fermi level.124
In 2020, Cermenek and colleagues reported Pd85Ni10Bi5/C and tested for the ethanol oxidation reaction in an alkaline medium. The as-synthesized catalyst displayed an exceptional current density of 2678 mA g−1 or 160 mA cm−2 (at 0.95 V vs. RHE) and it was found that the synthesis of the Pd85Ni10Bi5/C catalyst in an inert atmosphere, the use of HCl instead of NaCl, and the addition of solid NaBH4 reductant resulted in the highest EOR performance of this sample among all the prepared catalysts. They further declared that the addition of Bi and Ni to monometallic Pd increased the EOR stability and tolerance towards the by-products as compared to the commercial Pd/C electrocatalyst. However, the durability of the Pd85Ni10Bi5/C catalyst was lower than the PdxNiyBiz/C catalyst synthesized by the identical instant reduction approach, proposing that further optimization can be done in this process. Ni substitution in the ternary composite resulted in (a) the lowering of the binding energy of reaction intermediates, (b) a reduced poisoning effect of CO and CHx species, and (c) high affinity for OH−, which are crucial for the EOR process. Furthermore, Bi addition to Pd led to a high yield of additional oxide and hydroxide to enhance the OH− adsorption on the electrocatalyst surface to improve the activity for the EOR. Moreover, the carbon support helped in the fine dispersion of nanoparticles, increased the number of active sites and controlled the particle size and their distribution.125
In 2013, Shi et al. prepared an intermetallic Pd3Pb nanowires network (IM-Pd3Pb NNs) via a one-pot reduction approach by using an oil bath. The calculated electrochemically active surface area of IM-Pd3Pb NNs (44.8 m2 g−1) was greater than Pd black (13.3 m2 g−1). This enlarged surface area favors the accessibility of the mass to both the interior and the exterior active sites. Moreover, the kinetics, defects, steps, and boundaries, along with the ultrathin nanowire resulted in an increased number of electroactive sites. Additionally, the lower onset potential, high forward and backward current density ratio (1.5 times greater) and more negative onset potential reflect the greater CO oxidation and high electrocatalytic activity. During the durability test, slow current decay and maintenance of maximum current density (2 A mg−1 at 0.92 V vs. RHE) for 7200 seconds showed the maximum stability of the material. The enhanced activity was due to CO tolerance, enlarged active sites, geometric and electronic effects, accelerated electron transfer, reduced diffusion resistance, and stability, and all are interconnected with the porous network, ordered atomic structures, and the rationally designed 3D optimized intermetallic structure.126
In the same year, Luo and colleagues reported a series of Pd2Sn electrocatalysts prepared through the colloidal synthesis route in the presence of tri-n-octyl phosphine, methylamine hydrochloride, and oleylamine surfactants. During synthesis, the colloidal Pd2Sn nanorods were grown in the {010} direction. DFT calculations indicate that the {100} and {001} facets of orthorhombic Pd2Sn (optimized sample) are more suitable for EOR than {010}. Furthermore, DFT calculations displayed that OH− is favorably adsorbed on the Sn atoms at the {001}, and {100} facets, with adsorbed energy (Eads) values of −2.91, and-2.92, respectively. Such Pd2Sn NRs, braced on the graphitic carbon presents exceptional performance and firmness as an anode electrocatalyst for the ethanol oxidation reaction in an alkaline medium by delivering 10-fold and 3-fold higher anodic current density (at 0.0 V vs. Hg/HgO), respectively, as compared to Pd nanospheres and Pd2Sn. The reason for this performance was the development of favorable facets of nanorods during the colloidal synthesis.127
In 2020, Gao et al. reported well-dispersed Pd–Ag nanoparticles with identical size and abundant active sites prepared through an oil bath via the hydrothermal approach. The size of Pd–Ag NPs was first reduced with high Ag content and then it slightly increased. However, the lattice parameters gradually enlarged with the Ag content, and the parallel Pd–Pd bond length increased, which led to the lowering of the d-band center energy and limited adsorption of the toxic carbonaceous intermediates. The electrochemical measurements revealed that the anodic current density of PdAg-acac-4 Ag content could reach the highest value of 2246 mA mg−1 at 09.1 V vs. Hg/HgO, which can be attributed to the synergistic effect of the high EASA, Pd–Ag ratio, and small size of particles. In the stability test, it was observed that PdAg-acac-4 maintained its maximum current density even after 300 cycles. The calculated ECSAs of PdAg-acac-1 (112.4 cm2 mg−1), PdAg-acac-2 (259.6 cm2 mg−1), PdAg-acac-4 (486.3 cm2 mg−1), and PdAg-acac-5 (395.2 cm2 mg−1) also proved its stability response.128
The enhanced electrocatalytic activity and durability of the Pd–Ag bimetallic alloy network (Pd–Ag BANWs) towards the ethanol oxidation reaction in an alkaline media were studied by Fu et al. in 2015. The foam-like three-dimensional porous nanostructure was developed by a simple aqueous solution method. As compared to the traditional commercial Pd/C electrocatalyst, where Pd nanoparticles are supported on the carbon-based materials, the self-supported 3D BANWs offers many advantages to improve the stability and the electrocatalytic activity as 3D porous BANWs are not supported on the high surface area carbon materials, so carbon material degradation can be avoided. The 3D porous BANWs themselves are continuous sheet-like materials with huge surface areas, which prevents particle aggregation. According to the bi-functional mechanism theory, the Pd–Ag bimetallic alloy efficiently removes the carbonaceous intermediates and provides a large number of exposed active sites. Based on the d-band theory, the upward shift of the d-band center of Pd after Ag incorporation due to surface tensile strain caused by the altered lattice constant between Pd and Ag resulted in strong adsorption between the bimetallic alloy and OH−, and an improved catalytic response was obtained; however, more OH− adsorption suppressed the ethanol dissociative adsorption. Among the synthesized samples, the best response was shown by Pd33Ag67 BANWs as a current density of 2 A mg−1 was delivered at a potential of −0.2 V vs. Ag/AgCl, and in the stability test, the maximum current response was maintained until 2000 seconds. The enhanced electrocatalytic activity and stability are due to the distinctive 3D foam-like porous structure and the tendency to form the Pd and Ag alloy.129
In 2013, Zhang and colleagues reported the facile synthesis of nanodendrites of Pd/Ru by using an oil bath in the presence of potassium bromide, polyvinyl pyrrolidone, and ascorbic acid. The structure, morphology, and composition of the as-synthesized catalysts were thoroughly characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). Among the synthesized samples, the optimized Pd7/Ru1 nanocomposites showed the highest electrocatalytic activity and electrocatalytic durability for the ethanol oxidation reaction. The loftier electrocatalytic activity of Pd7/Ru1 can be ascribed to the following reasons: (i) Ru interaction with the OH− developed hydroxylated species (Ru–OH) at a comparatively lower potential; (ii) the reacting species serving as a proton-electron conductor assisted the oxidation of carbonaceous species (e.g. CH3CO, CO, CH3COOH); (iii) prevention of particle agglomeration by the addition of PVP. Therefore, the incorporation of Ru hastened the hydroxylated species formation and finally enhanced the catalytic response. However, the excessive amount of the Ru had a negative effect on the ethanol oxidation reaction. Active sites for ethanol oxidation were provided by the Pd, and the availability of Pd atoms for ethanol chemisorption decreased with the Ru excess. Pd7/Ru1 delivered a current density of 220.5 mA cm−2 at a peak potential of −0.2 V vs. SCE and showed a constant current response for 3600 seconds in an alkaline medium.130
To study the effect of Ru incorporation on the catalytic activity of the Pd, in 2017, Guo et al. prepared Pd2Ru/C through the reduction of metal precursors. Electrochemical measurements showed that as compared to Pd/C, the Pd2Ru/C favored the formation of acetaldehyde and prevented its oxidation. X-ray absorption data revealed that Ru promoted the electronic vacancy of the 4d band of the Pd and favored the ethoxy adsorption and its oxidation to acetaldehyde. However, an increase in the reactive species adsorption energy hindered the further oxidation of acetaldehyde. Furthermore, the degradation in the performance of the Pd2Ru/C occurred more rapidly than the Pd/C under identical conditions. The degradation is due to the accumulation of large amounts of acetaldehyde dimers formed by an aldol condensation reaction, which blocked the active sites and hindered further oxidation. A current of 0.024 A was produced at 0.0 V vs. Hg/HgO in 1 M NaOH solution along with minimum resistance and long-term stability for 3000 seconds.131
Sikeyi and collaborators in 2020 prepared a Pd–Ru nanocatalyst through the alcohol reduction method without changing the pH of the suspension and braced them on carbon nanofibers (CNFs) that were synthesized by a CVD method. The Pd–Ru/CNFs catalysts showed smaller particle size, high CO tolerance, and uniform dispersion of particles on the CNFs surface. The study revealed that the addition of ruthenium nanoparticles to Pd/CNFs greatly enhanced the electrocatalytic performance of the Pd–Ru/CNFs. The calculated (lf/lb) ratios for Pd/C (1:1), Pd/CNFs (2:0) and Pd–Ru/CNFs indicate that the Pd–Ru/CNFs have poisoning tolerance and according to the XPS, this behavior is Pd, Ru, and carbon concentration-dependent. The electrochemical data shows that the Pd–Ru/CNFs nanocatalyst displayed the best catalytic activity by delivering a high current (1500 μA at −0.2 V vs. Ag/AgCl) at low onset potential. In the long-term stability (2000 s) and impedance testing (11.20 Ohm), it was observed that during the ethanol oxidation test, the poisoning tolerance, current density, stability, and rate of charge transfer of the Pd–Ru/CNFs was much better than Pd/CNFs and commercial Pd/C nanocatalyst.132
Bimetallic Pd–W nanoparticles were successfully decorated on the fullerene-C60 thin film through a clean, simple, and proficient electrodeposition method without any surfactant, stabilizer, and capping agent addition as reported by Bhavani et al. in 2020. The proposed catalyst characterization was carried out in terms of structure, morphology, and elemental composition. Besides, the catalytic performance evaluation, electrochemical features, and long-term stability were explored by CV, LSV, CA, and EIS. The effects of the KOH and ethanol concentrations were examined to review the enhanced performance of the catalyst. Long-term stability, high If/Ib ratio, high EASA, and low resistance were demonstrated by Pd–W@Fullerene-C60 as compared to Pd@Fullerene-C60 and W@Fullerene-C60.These excellent outcomes were due to (a) the excellent thermal/mechanical properties and extended pi-conjugated system between the carbon atoms, (b) the additional accessible electroactive surface area (EASA = 96.56 m2 g−1) of fullerene-C60, (c) the uniform electrodeposition of bimetallic NPs for improved ethanol oxidation activity, (d) accelerated interfacial electron transfer between the solution and electrocatalyst interface by Pd–W@Fullerene-C60, and (e) the synergistic effect between the fullerene-C60 substrate and bimetallic nanoparticles.133
Bimetallic Pd/SnO2 electrocatalyst nanoparticles supported on the metal–organic framework (MOF)-derived carbon (MOFDC) were successfully produced by using a microwave-assisted scheme and were studied for the C2H5OH oxidation reaction (EOR) in alkaline solution by Adewale and colleagues in 2019. The complete characterization of materials was thoroughly carried out by XPS, XRD, TEM, and Raman spectroscopy. TEM analysis showed that Pd/SnO2/MOFDC produced nanoparticles of the average size of 5.5 nm, comparatively smaller than on carbon black. The Pd/SnO2/MOFDC displayed good catalytic activity in terms of the low onset and peak potential (at −0.1 V vs. Ag/AgCl), electrochemically active surface area (EASA) of 962 cm2 mg−1, resistance of 0.85 Ohm, Tafel slope of 216.2 mV dec−1, If/Ib ratio of 0.82, and long-term stability of 1200 seconds, and proved its potential application for the ethanol oxidation reaction.134
Mao et al. in 2014 reported a PdSn–SnO2/C composite fabricated by the impregnation reduction method. The characterization results showed that the Pd nanoparticles (4.25 nm) were uniformly distributed on the support and displayed higher catalytic activity towards the ethanol oxidation reaction. XRD analysis of the synthesized PdSn–SnO2/C exposed that the diffraction peaks of Pd shifted to a lower 2θ value as compared to the corresponding peaks of the Pd/C catalyst, demonstrating that Sn doping reduced the Pd crystalline lattice, and the XPS measurements proved the existence of SnO2 and Sn in the catalysts. The remarkable electrocatalytic activity of PdSn–SnO2/C catalysts as compared to the Pd–Sn/C and Pd/C catalysts was mainly due to the facile adsorption-dissociation of hydroxide ions on the surface of SnO2, which changed the electronic effect and augmented the ethanol adsorption on the Pd surface, and consequently resulted in improved catalytic activity by facile CO oxidation, while Sn addition modified the electric and geometric structure of Pd by varying the cell lattice. The main reason for this electrocatalytic response was the synergistic interaction between the PdSn and SnO2.135
Fahim and colleagues, in 2018, developed a core–shell designed M@Pd/SnO2-graphene [M = Cu, Ni, Co] catalyst using ethylene glycol reductant through a two-step microwave irradiation process. XRD, EDX, and TEM techniques physically characterized the as-prepared electrocatalysts and showed the particle size of 4.0, 6.0, and 6.5 nm at Cu@Pd/SnO2-Gr, Co@Pd/SnO2-Gr, and Ni@Pd/SnO2-Gr electrocatalysts, respectively. The Cu@Pd/SnO2-Gr electrocatalyst showed a negative shift of 133 mV in the onset potential as compared to Pd/Gr, and the ethanol oxidation mass activity was arranged in the following order: Cu@Pd/SnO2-Gr > Ni@Pd/SnO2-Gr Co@Pd/SnO2-Gr. The unique electrocatalytic behavior of Cu@Pd/SnO2-Gr can be attributed to the following: (i) the effective utilization of Pd NPs in the core–shell structure with its small size (4.0 nm) and augmented ECSA (467.49 m) values provided by graphene sheets. (ii) Its distinct core–shell structure enhanced the utilization of the Pd NPs. (iii) The electronic and geometric modifications due to the strong interaction between the Pd and Cu are responsible for the enhanced activity. In the case of the Pd–Cu composite, due to the more oxyphilic nature of Cu than Pd, the electrons transferred from Cu to Pd and, in turn, improved the CO oxidation tendency of Pd. (iv) The existence of SnO2 facilitated the adsorption of OH− ions, which are indispensable for CO oxidation. The current density of 105 mA mg−1 was produced at a peak potential of −0.1 V Ag/AgCl and the initial current density response was maintained until 1800 seconds.136
Chen et al., in 2013, reported bimetallic nanoporous PdAu catalysts with tunable Pd and Au ratios by electrochemically dealloying the Pd20−yAuyNi80 (y is 0, 5, 10, 15, 20) isomorphous alloys. The catalytic activities of the PdAu nanoparticles with uniform particle distribution were sturdily dependent on the Pd and Au atomic ratio, whereas the size and the geometric shape of the nanoporous alloys remained constant. In the nanoporous PdAu system, the Pd75Au25 nanoparticles possess superior electrocatalytic properties toward the ethanol oxidation reaction as compared to nanoparticles of the Pd and commercial Pt/C catalysts. The main reason for the excellent electrocatalytic activity is the optimal Pd and Au ratio, and the collective effect of the Pd surface dilution by Au. Besides, Au played a key role in the stability of the de-alloyed nanoparticles of PdAu bimetallic catalysts for the ethanol electrooxidation process.137
Zhong and associates developed a gold-decorated palladium bimetallic heterostructure by utilizing the Pd nanocubes as a structure-controlling parameter. The heterogeneous morphology and their elemental composition were verified by EDX, TEM, XRD, and XPS techniques. All lattice planes, as well as the Pd nanocubes direction, were observed parallel to the equivalent lattice planes or in the direction parallel to the Au layers, representing a static orientation relationship between the Au and Pd. The electrocatalytic behavior of the Au-decorated Pd heterostructure was demonstrated by the electrochemical method and it was noted that this decorated structure can improve the stability, catalytic activity, tolerance towards the poisonous species, and electrical conductivity. The CV results of the optimized Pd5Au1 catalyst expressed the synergistic effect between the Au and Pd surfaces. The onset potential on the Pd5Au1 was −0.61 V vs. SCE, which was 60 mV more negative than the monometallic Pd nanocubes catalysts, and indicated the easy oxidation of ethanol at low potential. The peak current density (1739.93 mA mg−1 at −0.1 V vs. SCE) was 2.2 times higher than on the monometallic Pd nanocubes catalyst. The long-term stability and low resistance are also responsible for the high activity of Pd5Au1 in an alkaline medium. The reasons for the enhanced catalytic activity are as follows: (i) the development of tensile strain and the uplifting of the Pd d-band center d-addition of Au resulted in elevated OH− adsorption and the elimination of the ethoxy intermediate from the Pd surface; (ii) enhanced EASA with more exposed electroactive sites.138
Shen et al. in 2018 successfully fabricated a series of PdxAuy/C catalysts with different Pd and Au atomic ratios by employing a co-reduction method and the optimal PdAu/C sample possessed good stability as well as strikingly improved electrocatalytic activity towards the C2H5OH oxidation reaction in alkaline media. Results displayed that the degradation percentage and the peak current density for PdAu/C were 12% and 84.16 mA cm−2 at −0.09 V vs. Hg/HgO, respectively. It was predicted that the striking electrocatalytic activity towards the EOR could be mainly attributed to the CO tolerance, electronically induced by the incorporation of the Au nanoparticles providing a large number of active sites, while the good stability was due to the development of the stable alloy phase of Pd1Au1. It was also remarkably revealed by the electrochemical and physicochemical characterizations that there was an unpredicted monotonically increasing trend to promote the effect of Au on the Pd, and the conciliation between the number of active sites, surface area and the specific activity led to the fact that the PdAu/C material with Au/Pd 1:1 atomic ratio showed the highest mass activity during the oxidation reaction in alkaline media (Table 2).139
| Electrocatalyst | Electrolyte/methanol molarity | Electrode | Peak potential mV (RHE) | Tafel slope mV dec−1/stability (s) | Current density mA cm−2 | Ref. |
|---|---|---|---|---|---|---|
| Pd@g-C3N4/GC | 1 M KOH/1 M C2H5OH | GCE | 0.85 V | —/200 cycles | 1692.0 | 110 |
| Pd@CN | 1 M KOH/1 M C2H5OH | GCE | 0.70 V | —/2000 s | ∼162.5 | 111 |
| Pd–S-HCNFs | 1 M KOH/0.5 M C2H5OH | GCE | 0.75 V | —/6000 s | 9.0 | 112 |
| Pd/NS-rGO | 0.5 MNaOH/1 M C2H5OH | GCE | 0.90 V | —/7000 s | 74.8 | 51 |
| Pd–PdOx | 1 M NaOH/1 M C2H5OH | GCE | 0.90 V | — | 14.0 | 113 |
| Pd/Cu/graphene | 1 M KOH/1 M C2H5OH | GCE | 0.80 V | —/3500 s | 26.6 | 114 |
| PdCuCo | 1 M NaOH/1 M C2H5OH | GCD | 0.75 V | —/10000 cycles |
1.54 | 115 |
| PdCo NTAs/CFC | 1 M KOH/1 M C2H5OH | CFC | 0.98 V | —/600 s, 500cycles | 11.7 | 116 |
| PdCo@ NPNCs | 1 M KOH/1 M C2H5OH | GCD | 0.84 V | —/8000 s | 176.4 | 117 |
| FePd–Fe3C/MWNTs | 1 M KOH/1 M C2H5OH | GCE | 0.90 V | —/1000 s | 84.8 | 118 |
| Pd–Ni/CNF | 1 M KOH/1 M C2H5OH | GCE | 0.77 V | 212/1800 s, 20 cycles | 199.8 | 119 |
| Ni@Pd–Ni NAs | 1 M KOH/0.5 M C2H5OH | GCE | 0.88 V | —/50 cycles | 50.0 | 120 |
| Pd (20 wt%)/Ni-NSC | 1 M KOH/1 M C2H5OH | GCE | 0.75 V | 190.24/1600 s | 110.0 | 121 |
| Pd47Ni36P17 | 1 M NaOH/1 M C2H5OH | GCE | 0.91 V | —/2000 s | 353.2 | 122 |
| Pd2Ge-36 NPS | 1 M KOH/1 M C2H5OH | GCE | ∼1.02 V | —/1000 s,2 50 cycles | 4.1 | 123 |
| Pd85Ni10Bi5/C | 1 M KOH/1 M C2H5OH | GC-RDE | 0.95 V | —/3600 s | 160.0 | 125 |
| Pd1.6Ni0.4Ge | 1 M KOH/1 M C2H5OH | GCE | 0.97 V | —/1000 s, 350 cycles | 13.0 | 124 |
| IM-Pd3Pb NNs | 1 M KOH/1 M C2H5OH | GCE | 0.91 V | —/7200 s | 230.0 | 126 |
| Pd2Sn NRs (9 × 26) | 0.5 M KOH/0.5 M C2H5OH | GCD | 0.92 V | —/7000 s | 45.6 | 127 |
| PdAg-acac-4 | 1 M KOH/1 M C2H5OH | GCE | 0.82 V | —/300 cycles | 159.7 | 128 |
| Pd33Ag67 BANWs | 1 M KOH/1 M C2H5OH | GCE | 0.83 V | —/2000 s | 142.0 | 129 |
| Pd7/Ru1 | 1 M KOH/1 M C2H5OH | GCD | 0.85 V | —/3600 s | 220.5 | 130 |
| Pd2Ru/C | 1 M NaOH/1 M C2H5OH | GCE | 0.97 V | —/3000 s | 169.9 | 131 |
| Pd–Ru/CNFs | 1 M KOH/1 M C2H5OH | GCE | 0.84 V | —/2000 s | 0.02 | 132 |
| Pd–W@Fullerene-C60 | 1 M KOH/1 M C2H5OH | GCE | — | —/2000 s, 350 cycles | 30.5 | 133 |
| Pd/SnO2/MOFDC | 0.25 M KOH/0.25 M C2H5OH | GCE | 0.88 V | 216.2/1200 s | 7.3 | 134 |
| PdSn–SnO2/C | 1 M KOH/1 M C2H5OH | Au | 0.85 V | —/3000 s | 68.7 | 135 |
| M@Pd/SnO2-Gr | 0.5 M NaOH/1 M C2H5H | GCE | 0.94 V | —/1800 s | 7.5 | 136 |
| Np–Pd75Au25 | 0.5 M NaOH/1 M C2H5H | GCE | 0.83 V | —/1800 s | 7.0 | 137 |
| Pd5Au1 | 1 M KOH/1 M C2H5OH | GCE | 0.95 V | —/1800 s | 123.5 | 138 |
| Pd1Au1 | 1 M KOH/1 M C2H5OH | GCE | 0.87 V | —/3000 cycles | 84.2 | 139 |
| PdRuCu NAs | 0.5 M KOH/0.5 M C2H5OH | GCE | 0.97 V | —/4000 s | 127 | 140 |
In 2019, a single-step aqueous approach was adopted by Zhang et al. for the construction of ordered multi-branched nanoassemblies of PdRuCu under the guidance of NaBr and octylphenoxypolyethoxyethanol (NP-40). The as-prepared PdRuCu NAs demonstrated enhanced electrocatalytic activity and upgraded stability for the ethanol, and ethylene glycol oxidation reactions in alkaline medium. The specific and mass activities of the as-synthesized catalyst for ethanol oxidation reaction, 3.78 mA cm−2 and 1.16 A mg−1, respectively, were greater as compared to commercial Pd black, 1.81 mA cm−2 and 0.17 A mg−1. Moreover, the current density of 127 mA cm−2 was delivered at a peak potential of 0.97 V (vs. RHE), and the maximum amount of the current density was retained till 4000 seconds in a stability test (Fig. 6).140
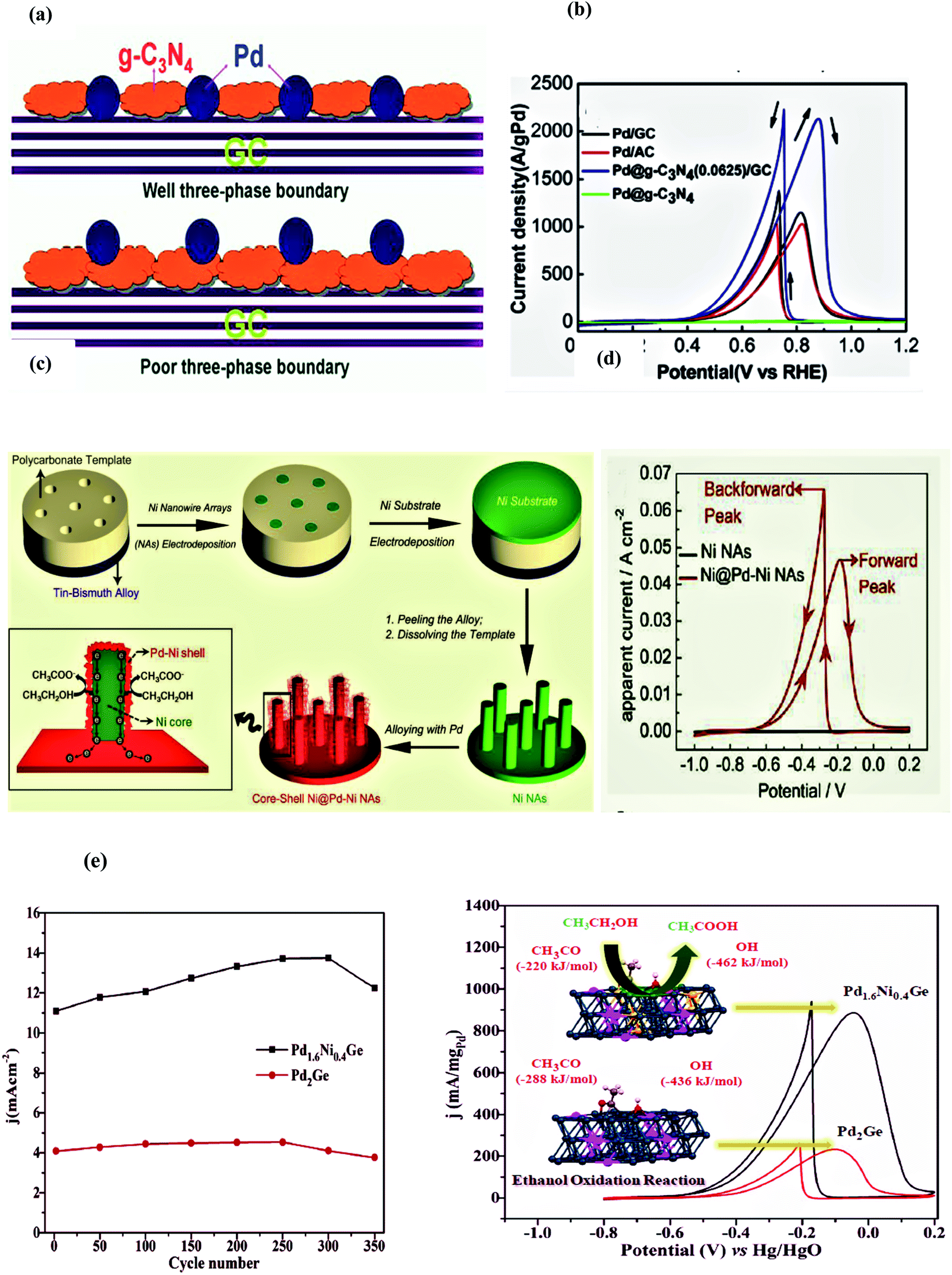 | ||
| Fig. 6 (a) The development of a three-phase boundary system and (b) CV polarization curves of Pd-based catalysts for the EO R in 1 KOH and 1 M ethanol solution. Reproduced from ref. 110, Copyright 2016, Elsevier. (c) Synthetic scheme and (b) CV polarization curves of Ni@Pd–Ni nanowire arrays for the EOR in 1 KOH and 0.5 M ethanol solution. Reproduced from ref. 120, Copyright 2018, ACS. (e) Stability response and CV polarization curves of Pd1.6Ni 0.4Ge for the EOR in 1 KOH and 1 M ethanol solution. Reproduced from ref. 124, Copyright 2019, ACS. | ||
5.3. Gold and silver-based compounds
Besides Pt and Pd, gold (Au) is also used as an anode catalyst in the ethanol oxidation reaction in both acidic and basic media. It was observed by Chen et al. that Au has characteristics similar to Pd such as being inert at low pH but highly active in high pH environments. It was also found that Au in alkaline media oxidizes the C2H5OH rather slowly as compared to the commercial Pt, as can be seen from its positive onset potential (greater than 0.7 V). However, incorporation of Au into Pt or Pd can improve their catalytic activity by facilitating the easy oxidation of the reaction intermediates.Silver, another noble metal, also plays a very important role in the improvement of the electrocatalytic response of Pt, and particularly Pt metal. Wang et al. observed that OH− adsorption on the surface of silver metal assists in removing the reaction intermediates on the surface of other metals (Pt, Pd) by the following equation:
M (CH3COO) + Ag OH− → Ag (CH3COOH) + M, and provides more active sites for enhanced oxidation.141,142
Hong et al. in 2014 introduced a facile and reproducible approach for the synthesis of Pd–Au nanowire networks in good yield. The whole synthetic process includes just mixing the precursors in an aqueous solution in the presence of polyvinylpyrrolidone surfactant, while sodium borohydride was used as a reductant. The complete synthetic process was carried out at room temperature and was completed in 30 minutes, which reduced the cost as well as time. Electrochemical measurements demonstrated that the synthesized catalysts exhibited intensely enhanced electrocatalytic activity towards the ethanol electrooxidation reaction in alkaline solution by delivering a current density of 781 mA cm−2 at the peak potential of 0.0 V vs. Ag/AgCl, along with the maximum retention of current density for 6000 seconds. The enhanced catalytic activity was ascribed to the following: (a) the 1D nanowire structure provides routes for facile electron transport, (b) the small-sized nanowires provide a large number of active sites, (c) C-support-free material avoids the chance of corrosion and oxidation. (d) The incorporation of Au into Pd increases the energy of the Pd d-band center, enhances the adsorption of OH−, and eliminates the ethoxy intermediates from the Pd metal surface with consequently enhanced activity.143
Chen and colleagues in 2017 reported a wet-chemical approach for the high-yield production of crystal-phase-heterostructured 4H/fcc gold nanorods in a one-pot process. Importantly, through the epitaxial growth of the palladium metal on the gold nanorods (Au NRs), bimetallic crystal-phase-heterostructured 4H/fcc Au@Pd core–shell nanorods were fabricated, which showed much higher stability and catalytic activity towards the EOR than Pt/C and commercial Pd black catalysts. The possible reasons for the enhanced catalytic activity are as follows. (i) A change in the coordination chemistry, crystal-phase-boundaries, and chemical bonding leads to an altered electron band structure with consequently enhanced activity. (ii) The atomic contact between different metals can modify their electronic structures and, therefore, result in improved activity. (iii) The epitaxial growth of the Pd shell on the Au NRs and full coverage of the metal surface promote the catalytic response. (iv) The Pd shell dendritic morphology in the 4H/fcc Au@Pd nanostructure comprises a large number of atomic steps/links (evidenced by TEM), which increases the activity. (v) Au incorporation elevates the stability of Pd metal. The current density of 3 A mg−1 was delivered at a peak potential of 0.78 V vs. RHE in an alkaline medium with a constant current response until 3600 seconds.144
Bi et al. in 2013 successfully fabricated a series of hexoctahedral (HOH), elongated tetrahexahedral (ETHH), and octahedral (OCT)-shaped Au@AumPdn nanoparticles with ultrathin facet-controlled bimetallic alloy-shells via the co-reduction of a minimal amount of Pd and Au precursors through ascorbic acid (AA) with the help of silver metal on their surfaces. The presence of elemental Ag on the surface of Au NPs enclosed with precise facets controls the synthesis of such types of nanoparticles. During the electrochemical measurements, it was found that the specific and mass activities of Au@AuPd NPs displayed “volcano-type” reliance concerning the Pd content on the surface of a thin AuPd alloy shell. Furthermore, their stability and activity were also highly reliant on the area ratio of the active and inactive facets on their surfaces, and these ratios depend on the selection of preformed Au NPs encircled by different facets. The Au@Au0.20Pd2.0 NPs (HOH-shaped) concurrently have excellent stability and the best activity due to the appropriate area ratio of the inactive and active facets on the surfaces of ultrathin bimetallic alloy-shells since they possess an ECSA value of 125.8 m2 g−1, a mass activity 11.9 A mg−1 and a specific activity 9.5 mA cm−2 for ethanol oxidation, which are greater as compared to commercial Pd/C catalysts.145
In 2015, the same author prepared unevenly shaped, concave, and cuboidal Au@AuPd nanoparticles (ISCC-Au@AuPd NPs) accompanied by high-index facets by growing Pd on the surface of pre-formed ISCC-Au nanoparticles with a 2% concentration of Pd. The alloy nature of the AuPd catalyst was ensured by X-ray photoelectron spectroscopy (XPS), cyclic voltammetry (CV), and energy-dispersive X-ray spectroscopy (EDX). Among the tested samples, the optimized ISCC-Au97.5@Au0.5 Pd2.0 NPs displayed the largest electrochemically active surface area (EASA up to 92.11 m2 g−1) due to the prevention of their close-packed agglomeration, and the unsurpassed long-term stability with respect to the oxidation process in an alkaline medium was due to efficiently removing the intermediates. Their mass and ECSA-normalized current density (4.15 A mg−1 at −0.2 V vs. Ag/AgCl, and 4.51 mA cm−2) were approximately 20.7 and 6.9 times greater than commercial Pd/C catalysts, respectively, along with a constant current response until 7000 seconds.146
Efficient electrocatalytic C–C bond splitting combined with 12-electron complete oxidation of the C2H5OH molecule to CO2 on the nanoscale catalysts involving a Pt monolayer (ML) and sub-monolayer (sML) mounted on the gold nanoparticles (Au@Pt ML/sML) was reported by Loukrakpam et al. in 2014. The Au@Pt electrocatalyst was synthesized by the surface-limited redox replacement (SLRR) of the Cu monolayer (under potentially deposited, UPD) in an electrochemical cell. Au@Pt ML exhibited an improved tendency for ethanol oxidation to CO2 at low potential, unlike the Pt sML counterparts, and Pt bulk. They explored the Pt–Au lattice mismatch and competing strain effects owing to Pt layer morphology/coverage, which controlled the surface chemisorption process and the overall activity. Controlled experiments were carried out by X-ray diffraction (XRD), in situ electrochemical FTIR spectroscopy (FTIR), pair-distribution function (PDF), and in situ scanning tunneling microscopy (STM). The vibrational impressions of the adsorbed CO authenticated the relationship between the layer strain, surface bond strength, morphology, and electrocatalytic activity. DFT calculations revealed that in Pt nanoclusters, the active strain showed strong radial dependence. In Pt nanoclusters, the average strain was found to be smaller than the strain developed by the epitaxial relationship between Au (111) and Pt owing to the finite size effect. The smaller cluster size of Pt in Au@Pt ML led to a greater compressive size-reliant strain, which worked opposite to the expansive epitaxial strain. Therefore, in Au@Pt sML, the sML surface covered by a larger Pt cluster size is anticipated to have extra tensile strain. As a result of this strain, the uplifting of the Pt d-band center leads to the facile adsorption of OH− with consequently enhanced activity.147
Kim et al. in 2013 introduced a facile process for the development of Au@Pd nanoparticles with controlled morphologies and sizes, from a flower-like shape to a cuboctahedral shape via seed-mediated growth, in which Au nanospheres were used as seeds, and cetylpyridinium chloride (CPC) was used as a surfactant. The most important parameter for the growth of flower-shaped Au@Pd nanostructures is the molar ratio of the surfactant with respect to the Pd salt, and its concentration-controlled size can be obtained by tuning the concentration of the Pd salt. Due to a larger number of electroactive sites and huge surface area, the flower-like Au@Pd catalyst showed 2.2 times boosted electrocatalytic activity as compared to cuboctahedral Au@Pd nanostructures. The current density of 198 mA mg−1 was produced at a peak potential of −0.1 V vs. Ag/AgCl with a stable current response for 1000 seconds in a stability test.148
In 2014, Qiu et al. prepared Au–Pd@Pd core–shell nanoflowers (Au–Pd@Pd CSNFs) through a polyallylamine-assisted water-based fabrication scheme. The morphology, composition, and structure analysis were carried out by XPS, XRD, EDX, TEM, and elemental mapping. During the synthesis, Pd (111) reduction gives the AuPd alloy core, while reduction of Pd (11) produces the Pd shell. The lower peak potential (0.8 V vs. RHE), the bigger specific and mass-activity (12 mA cm−2 and 2000 mA mg−1), and long term stability (3000 s) validated that Au–Pd@Pd CSNFs have admirable electrocatalytic activity for the EOR; the unique porous structure and synergetic effect between Pd and Au modified the geometric and electronic effect.149
To study the promotional effect of silver on the catalytic activity of other metals, in 2014, Feng et al. prepared dimetallic PdmAg alloy nanostructures via a facile co-reduction process and tested them for the ethanol oxidation process in an alkaline medium. The XPS results showed the modification of the Pd electronic structure after Ag incorporation in an appropriate amount. Electrochemical measurements revealed that the Pd catalytic activity strongly relied on the elemental composition of the PdmAg/C catalysts, as an optimized Pd0.5Ag/C composite showed 3.6 times higher mass activity and 2.4 times greater specific activity than commercial Pd/C, which might be attributed to the synergistic and electronic effects. During the testing, it was observed that the surface of Ag was fully covered with OH−, which is beneficial for limited adsorption of COads and CH3COads on the Pd surface, leading to remarkable performance during the EOR.20
In 2015, Peng and colleagues demonstrated a simple method to produce hollow raspberry-like nanospheres of PdAg alloy by using a galvanic displacement scheme between pre-synthesized palladium ions and the Ag nanoparticles. TEM and SEM showed that the fabricated PdAg alloy nanocrystals had an interesting porous and hollow structure. The CV and CA results revealed the superior electrocatalytic activity of PdAg alloy nanospheres as compared to commercial Pd/C for the ethanol electrooxidation reaction in an alkaline solution. Structural and electrocatalytic studies indicated that this material is advantageous in numerous ways: (a) excellent tolerance towards reaction intermediates, (b) effective usage of noble metals and a huge surface area due to the porous and hollow nanostructure, (c) the modification of the electronic structure due to interactions between Ag and Pd, and (d) the addition of silver promoted the oxidation reaction. During the electrochemical testing, the current density of 2.3 mA cm−2 was delivered at the potential of −0.2 V vs. Hg/HgO along with the If/Ib ratio of 2.28, and the constant current response for 1000 seconds in the chronoamperometric test reflected the material's activity, tendency to remove poisonous intermediates, and stability.150
Jo et al. in 2016 employed the same galvanic replacement reaction between Ag and lead salt to prepare a porous dendritic shaped PdAg alloy where the lead salt concentration controlled the composition of the material. The morphological and structural analysis revealed that silver-rich bimetallic dendrites possess a hollow and porous structure, while lead-rich dendrites tend to collapse into aggregates during the synthesis process. The electrochemical measurement showed that the dendritic Pd40Ag60 alloy exhibited better electrocatalytic activity than the simple PdAg catalyst due to the electronic effect and the morphology of porous dendrite. Porous Pd40Ag60 dendrites with high EASA of 41.75 m2 g−1 delivered a current density of 92.3 mA cm−2 at a peak potential of 0.0 V vs. Hg/HgO, and in the chronoamperometric test it showed minute current decay even after 50000 seconds. This stable response depicts the greater tolerance of the material towards the poisonous effect of reaction intermediates (CH3CO−) by the shifting of the d-band center of Pd towards the Fermi level and the increase in the surface area after Ag incorporation (Table 3).151
| Electrocatalyst | Electrolyte/methanol molarity | Electrode | Peak potential mV (RHE) | Tafel slope mV dec−1/stability (s per cycles)) | Current density mA cm−2 | Ref. |
|---|---|---|---|---|---|---|
| PdAu NNWs | 1 M KOH/1 M C2H5OH | GCE | 1.03 V | —/6000 s | 781.0 | 143 |
| 4H/fccAu@Pd NRs | 1 M KOH/1 M C2H5OH | GCE | 0.78 V | —/3200 s | 248.5 | 144 |
| Au@Au0.20Pd2.0NPs | 0.5 M KOH/0.5 M C2H5OH | GCE | 0.88 V | —/500 cycles | 852.0 | 145 |
| ISCC-Au97.5@Au0.5Pd2.0NPs | 0.3 M KOH/0.5 M C2H5OH | GCE | ∼0.85 V | —/7000 s | 294.6 | 146 |
| Au@Pt ML/ML | 0.5 M H2SO4/1 M C2H5OH | GCE | ∼0.98 V | — | 15.0 | 147 |
| Au@Pd nanostructure | 1 M KOH/0.5 M C2H5OH | GCE | 0.93 V | —/1000 s | 1.0 | 148 |
| Au–Pd@Pd CSNFs | 0.5 M KOH/0.5 M C2H5OH | GCE | 0.85 V | —/3000 s | 147.0 | 149 |
| Pd0.5Ag/C | 0.5 M KOH/2.0 M C2H5OH | GCE | 1.05 V | —/3500 s | 241.4 | 20 |
| PdAg-NS | 1 M KOH/1 M C2H5OH | GCE | 0.72 V | —/1000 s | 2.4 | 150 |
| Pd40Ag60 | 0.5 M NaOH/1 M C2H5OH | GCE | 0.92 V | —/5000 s | 92.3 | 151 |
| Pd–Ag/G | 1 M KOH/1 M C2H5OH | GCE | 0.82 V | —/7200 s | 355.0 | 152 |
In 2019, Douk and colleagues employed a simple, facile, green, low temperature, and surfactant-free approach for decorating the graphene surface with PdAg nanoparticles with the aid of the ultrasound technique. The uniformly dispersed nanoparticles (2–3 nm), verified by transmission electron microscopy showed exceptional stability (7200 s) with low activation energy, high If/Ib ratio, and high catalytic activity (J = 355 mA cm−2 at 0.82 V vs. RHE) towards the electro-oxidation of ethanol. According to the analysis results, it was established that (a) the amalgamation of Pd and Ag, and (b) the uniform distribution of ultra-small Pd–Ag nanoparticles led to the enhancement of the catalytic response of the Pd–Ag/G electrocatalyst. According to Hammer's theory, the uplifting of the Pd d-band center after Ag incorporation resulted in the elevation of the adsorbate binding energy and consequently promoted the Pd reactivity for CO oxidation and the graphene support prevented the particle agglomeration, which is responsible for the high surface area and excellent activity (Fig. 7).152
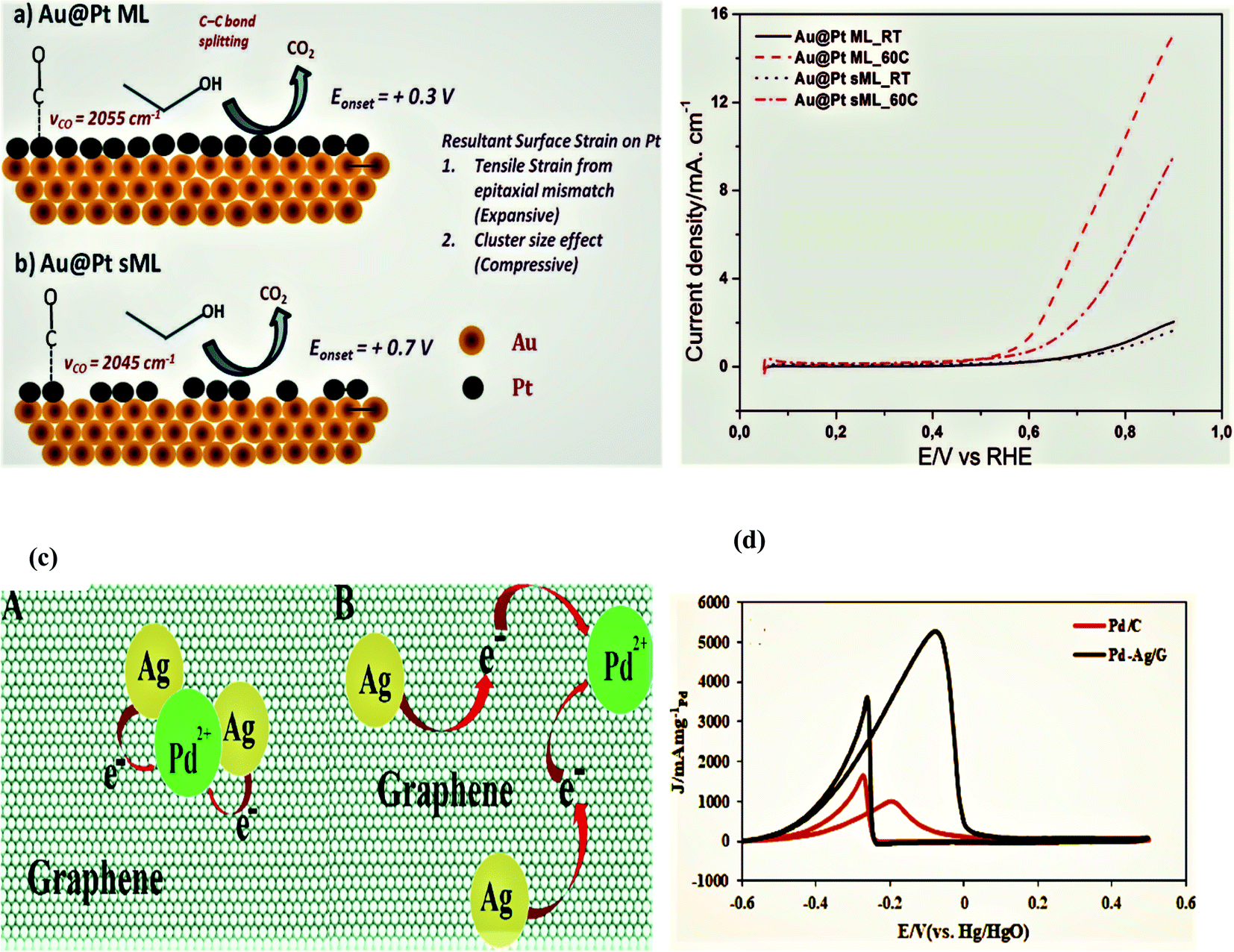 | ||
| Fig. 7 (a) Image of the Pt layer structure of Au@Pt ML and Au@Pt sML catalysts and their effects on the vibrational modes of the CO probe molecules, and the onset potential required for C–C bond splitting and CO2 formation from ethanol. (b) EOR comparison curves of Au@Pt ML at room temperature (RT), Au@Pt ML at 60 °C, Au@Pt sML at room temperature (RT), Au@Pt sML at 60 °C in 0.5 M H2SO4 + 1 M C2H5OH. Reproduced from ref. 147, Copyright 2014, Royal Society of Chemistry. (c) Galvanic displacement reaction by the (A) direct and (B) indirect paths, and (d) CV polarization curves of the Pd–Ag/G and Pd/C in 1.0 M KOH/1.0 M ethanol solution at 50 mV s−1. Reproduced from ref. 152, Copyright 2019, Elsevier. | ||
5.4. Transition metal-based compounds
To improve the ethanol oxidation reaction kinetics, Pt and Pd-based materials are potential candidates with excellent activity. However, high cost, less availability, poisoning by reaction intermediates, and slow kinetics are the main obstacles to their large-scale use. Keeping in mind the limitations of noble metals, and to gain the advantage of a basic electrolyte a large number of catalysts comprising mono, binary, ternary, and quaternary noble/non-noble metal NPs have been employed as anode electrocatalysts for the ethanol oxidation reaction (EOR). Materials synthesized by the fractional replacement of costly metals by non-noble metals (Fe, Ni, Co, etc.) or with semi-noble (Cu) metal NPs are the catalyst of interest to researchers to lessen the cost and improve the catalytic performance of DEFCs.Among the transition metals, nickel is the best option for various energy-associated applications such as supercapacitors, the hydrogen evolution reaction (HER), and alcohol oxidation (AOR), etc.
Researchers have further premeditated the effect of innumerable parameters like morphological modification, core–shell structure, temperature, and support. Moreover, Ni-containing compounds such as NiO, Ni (OH)2, nickel cobaltite, and Ni-based MOFs are also evidenced to be virtuous materials for energy-connected applications. Due to the benefits of nickel, such as renowned surface catalytic activity, good electric conductivity, easy availability, low price, and acceptable chemical stability, various hybrids or modified electrodes based on nickel and nickel-based alloys have been synthesized for use as electrocatalysts. Nickel in the form of Ni foam or foil as a substrate is also recognized as an eminent flexible support that researchers have used for solid-state supercapacitors. Oxides of nickel are also a very good option for the hydrogen evolution reaction.62,153–160
Kinetic and mechanistic details of the ethanol oxidation reaction (EOR) were thoroughly studied by Barbosa and colleagues in 2015 by using a nickel electrode in NaOH solution under controlled experimental conditions including the variation of the scan rate and solution temperature. The electrochemical analysis revealed that the slow reaction at −15 °C was due to a high activation energy barrier, and the ethanol oxidation reaction takes place through chemical interactions between the C2H5OH molecule and the β-NiOOH electrochemically formed on the electrode surface at potentials higher than 1.3 V. The kinetics of the reaction tends to be slow at low temperatures and results in the shift of the peak potential; however, the scan rate significantly affects the formation of β-NiOOH and reduction reactions. Moreover, in situ FTIR spectroscopic analysis showed that alcohol is selectively converted into the corresponding carboxylate under the testing conditions.161
Wang et al. in 2018 reported a carbon-coated Ni/NiO hybrid prepared by the calcination of nickel acetate along with glucose at 800 °C in an inert (N2) atmosphere, accompanied by calcination in air at multiple temperatures. The oxidative temperature of the carbon content and crystal structure control the morphology of hollow NiO. As compared to the Ni–C electrocatalyst, the Ni/NiO–C2 and Ni/NiO–C3 electrocatalysts parade superior electrocatalytic activity towards the ethanol oxidation reaction in alkaline media. The hollow-structured Ni/NiO–C3 showed the greatest catalytic performance in terms of the current density of 120 mA cm−2 at a potential of 0.7 V vs. SCE, along with a stable current response up to 3500 seconds due to the synergistic effects between carbon coating in moderate amount, NiO crystals, and the hollow structure, which shorten the diffusion paths between ethanol and the particles. This unique structure is responsible for the ethanol oxidation mechanism since the diffusion-controlled mechanism is followed in the case of decreased concentration and scan rate, but at high concentration and scan rate, the reaction occurs by the electron transfer process. Furthermore, the reason for the great stability of Ni/NiO–C3 in low concentration is the stable NiO crystal structure.162
A borohydride reduction hydrothermal method was used by Hasan and collaborators in 2012 to fabricate the Ni nanowire array (NiNWA)-braced 3D flower-like Pd nanocatalyst with high activity towards the ethanol oxidation reaction in alkaline media. This novel composite NiNWA/PdNF electrocatalyst possesses a huge electrochemically active surface area (45 m2 g−1), high CO tolerance, excellent electrocatalytic activity (1.7 mA cm−2 at 0.1 V vs. Hg/HgO), a high If/Ib ratio (1.2), Tafel slope of 96 mV dec−1, and 14 hours of constant current response to express the excellent electrocatalytic activity. The reasons for the high electrocatalytic activity are abundant grain boundaries and the 3D open nanostructure of the Pd nanoflowers. The open structure of the highly robust and conductive Ni metallic core guarantees substantial alcohol access to the electrocatalyst active sites for ion adsorption/desorption. The 3D flower-like Pd nanostructures with highly branched morphology and interspaces play an important role in the efficient electrocatalysis in DEFCs.163
A simple, economical, and benign method for the preparation of a metallic Ni-based electrocatalyst was investigated by Cuna and colleagues in 2016. In the first step of the synthesis, NiO NPs-supported hydrothermal carbon (NiO/HC) was obtained and in the second step, an activated hydrothermal C-supported Ni catalyst (Ni/aHC) was obtained as a result of NiO hydrothermal reduction and carbon activation by a hydrothermal process. The electrochemical testing of the Ni/aHC in the presence of ethanol in NaOH solution implies the contribution of the (NiO)OH species towards the EOR at 0.47 V vs. Hg/HgO electrode. The in situ ATR-FTIRS scrutiny demonstrated that the most significant product of the oxidation reaction on Ni/aHC in basic media was CH3COO−, appreciably formed at a potential of 0.5–0.6 V vs. Hg/HgO; the subsequent increase in current density is due to the oxygen evolution reaction. The current density of 28.5 mA cm−2 was shown at a potential of 0.7 V vs. Hg/HgO in an alkaline medium along with a stable current response for up to 35 minutes.55
Zhang et al. in 2016 employed a benign two-step strategy for the synthesis of the Ni@Pd/NG nanocatalyst and studied its electrocatalytic behavior towards the ethanol oxidation process. The as-synthesized material exhibited a large specific surface area, better kinetics, higher tolerance, and long-term stability as compared to the other electrocatalysts for the ethanol oxidation reaction in DEFCs. This response may be correlated with the core–shell architecture, synergistic effects between the Pd and Ni, and nitrogen doping in the graphene. The strong interactions of the metal nanoparticles with the N-doped graphene helped in the fine dispersion of the metal nanoparticles, as well as provided more active sites, stability, and a conductive network for efficient charge and mass transfer. The manufacturing of the Ni@Pd core/shell alloy nanostructure not only reduced the Pd consumption but also benefited from the ligand effect, a decline of the d-band energy, and is favorable for the promotion of the catalytic activity and stability of the Ni@Pd/NG nanostructure. The current density delivered by the material at the potential of −0.1 V vs. Hg/HgO was 82.5 mA cm−2 along with a constant current response with minor changes up to 3000 seconds.164
Hong et al. in 2019, successfully manufactured the Pt–Cu octahedra adorned by Ni (OH)2 in a one-pot fabrication scheme for EOR electrocatalysis in an acidic electrolyte. The capping agent (CO gas) was directly inoculated in a reaction process and Ni(OH)2 was naturally settled down on the alloy of Pt–Cu octahedra owing to the difference in the reduction potentials of Cu, Pt, and Ni. The occurrence of Ni (OH)2 on the Pt–Cu alloy octahedra surface was confirmed by XPS, EDX, TEM, and XRD. The improved CO tolerance of the Pt–Cu alloy was because of the lowering of the Pt d-band structure, and the consequent weakening of the CO binding with Pt, while the excellent electrocatalytic activity and stability of Ni (OH)2–PtCu/C were due to the co-existence of the surface Ni(OH)2 and Cu, which sufficiently endorsed the H2O dissociation and formation of OHads species for CO oxidation on the Pt surface. The lower onset potential of the Ni(OH)2–PtCu/C as compared to commercial Pt/C also indicated enhanced EOR kinetics due to the easy C–C bond-breaking process. The material possessed a current density of 7.76 mA cm−2 at the potential of 1.0 V vs. RHE, specific activity of 8.40 mA cm−2, mass activity of 1.97 mA mg−1, along with maintaining the current response for 3000 seconds under experimental conditions.165
Layered double hydroxide (LDH) based ethanol oxidation electrocatalyst was successfully synthesized by Xu et al. in 2017 through a simple and novel liquid-exfoliation method. The as-synthesized NiAl-LDH-NSs with a high concentration of Ni (+3) exhibits ultrathin 2D nanostructure (thickness = 0.75–0.88 nm), and shows excellent activity in terms of long durability and quick turnover frequency. This tremendous EOR performance, in terms of minimum resistance, a current density of 55 mA cm−2 at 0.58 V vs. Ag/AgCl, along with the maintenance of 81.8% of the original current response after 10000 seconds, is mainly attributed to the synergistic effect of two metal-exposed active sites, the production of active species, and improved conductivity after exfoliation as a large number of active species (NiOOH) were formed in the ultrathin nanosheets via the exfoliation process. These synergistic effects resulted in a 30-fold greater EOR activity of NiAl-LDH-NSs as compared to the bulk NiAl-LDHs. Furthermore, the product analysis by liquid chromatography revealed that acetaldehyde and acetic acid were the main oxidation products.166
In 2018, Hatamie and colleagues modified a flexible, and interconnected porous Ni foam with Gold NPs via an in situ electrochemical deposition method, and utilized the synthesized electrocatalyst for the ethanol oxidation reaction in an alkaline solution. For comparison, the planar gold NPs-modified Ni foil was also prepared by the same approach. During the electrochemical testing, it was observed that on the electrode surface, the NiOOH was the active species for the ethanol oxidation process, which reduced the NiOOH to NiO (forward scan), while in the reverse scan, the large peak was related to the carbonaceous species removal from the electrode surface. It was noted that the electrocatalytic performance of 3D NiF/AuNPs, in terms of current density, resistance and stability, was much better as compared to Ni foil/Au NPS in the presence of Au NPs and these gold nanoparticles were also responsible for low resistance, and enhanced activity. The oxidation and elimination of reaction intermediates was the rate-determining step, and the 3D porous structure of the electrode promoted the oxidation process by the facile removal of oxidation products. The effects of oxidation conditions, electrode structure, incorporation of gold nanoparticles, and resistance were also thoroughly studied.167
The in situ adornment of graphene by CoxNiy alloy nanoparticles was carried out by Barakat et al. in 2014. A modified chemical route was employed for the addition of metal precursors during the graphene preparation, followed by calcination at 850 °C. The physicochemical characteristics facilitated the formation of nanosheets of multi-layer graphene decorated by CoxNiy alloy nanoparticles. The influence of the NPs loading and chemical composition on the electrocatalytic performance was also investigated and it was concluded that 10 times greater activity was shown by the Co0.2Ni0.2/graphene nanosheets composite than pristine NPs, while the onset potential was shifted towards the negative side. The salient features of the studied material include a current density of 165 mA cm−2 at the potential of 0.75 V vs. Ag/AgCl, only 18% loss of current even after 40 cycles, and re-deposition of the catalyst after washing the electrode.168
In 2012, Wang and collaborators developed a novel, non-noble metal-based electrocatalyst for DEFCs by an economical, facile, and environmentally friendly scheme. The characterization of the as-prepared catalyst was carried out by TEM, EDX, XPS, XRD, and the electrochemical method. Electrochemical testing confirmed that the Ni–Co/graphene electrocatalyst showed better catalytic activity towards the ethanol oxidation reaction in an alkaline medium. The calculated electron-transfer coefficient (α) was 0.59 and 0.62, while the charge-transfer rate constants (k) were 0.072 and 0.157 s−1 for the Ni/graphene and Ni–Co/graphene electrocatalyst, respectively, along with the current density of 16.4 mA cm−2 at 0.7 V vs. SCE, and long term stability for 3500 seconds. The greater catalytic activity and better durability of the Ni–Co/graphene electrocatalyst for C2H5OH oxidation indicated that Co addition promoted the ethanol oxidation reaction at the surface of Ni/graphene and graphene nanosheets due to their large surface area not only leading to enhanced catalytic activity by preventing metal particle agglomeration, but also improving conductivity.169
In 2015, Kakati et al. synthesized and studied the electrochemical properties of N-doped reduced graphene oxide (NRGO), embraced Ni, Co, and Ni–Co composites in which the metal nanoparticles were inserted into a highly porous N-doped graphene medium by a simple one-pot method at mild temperature by using urea, urea choline chloride, and GO. The synthesized NiCo/NRGO displayed amazing electrocatalytic activity as seen by the low Tafel slope of (159.1 mV dec−1), current density of 80 mA cm−2 at 0.8 V vs. Ag/AgCl, and high stability (2000 s) for the ethanol electrooxidation reaction (EOR). The grander performance of the nanoalloy-based NRGO is credited to the huge surface area, homogeneous dispersion of high-density nitrogen, metal electroactive sites, and the synergistic effect.170
In 2011, Hasan et al. prepared a carbon electrode-supported nanocomposite of Ni–Cr2O3 by the electrodeposition method via Watts Ni bath in the presence of Cr2O3 NPs. Their electrochemical activities towards the ethanol oxidation were evaluated in a 1.0 M NaOH solution through cyclic voltammetry (CV), chronoamperometry (CA), and Tafel plots. The electrocatalytic activity of the synthesized electrodes increased with the increase in the Cr2O3 volume fraction percent up to 7 Vf%. The Ni–Cr2O3/C (7 Vf%) electrode exhibited significantly enhanced electrocatalytic activity and durability towards the ethanol oxidation reaction as compared to the Ni/C electrode. The deposition of nano-sized particles of Cr2O3 in the Ni deposit improved the nickel matrix surface morphology along with the reduction of the size of Ni grains. In contrast to the Ni/C electrode, the Ni–Cr2O3/C (7 Vf%) nanocomposite showed a huge surface area, high stability (14 minutes), elevated catalytic activity (current density of 327 mA cm−2 at 0.7 V vs. Hg/HgO), greater charge transfer rate constant, and charge transfer coefficient under testing conditions.171
A one-pot facile method (stirring and pyrolysis) was utilized by Shi et al. in 2016 to develop nitrogen-doped activated carbon accompanied by governable Ni nanorod (NiNC) electrocatalysts for the ethanol oxidation reaction in alkaline medium. The morphology, and size of the Ni nanorods were mainly controlled by the precursor concentration as it affected the electrocatalytic activity of the samples. The optimized NiNC-3 catalyst with the appropriate size and content of Ni exhibited upgraded catalytic activity during the ethanol oxidation by delivering a 5 times greater current density (47.5 mA cm−2 at 0.5 V vs. SCE), and 16 times greater rate constant than the NiNC-1 catalysts, along with a resistance of 4.5 ohm and long-term stability for 200 s. Furthermore, the retention of 80.7% of the current density after the 1500 cycle suggests excellent cyclic stability. The main reasons for this enhanced catalytic activity are (a) the relatively uniform size of the nanorods, (b) excellent electrical conductivity, and (c) the stability of the activated carbon support.172
Shamsuddin et al. in 2014 successfully prepared a series of G-supported NixPd100−x binary alloyed catalysts (BACs) with superior activity via a simple chemical method activity. Among the as-synthesized samples, the optimized Ni50Pd50/G catalyst revealed higher mass activity (614.6 mA mg−1), peak current density (16 mA cm−2 at −0.28 V vs. SCE), more negative onset potential (−0.8 V), lower Tafel slope (142 mV dec−1), and the greatest alloy content with facilitated charge transfer process. The CV and Tafel slope comparison of all samples indicated that the reaction proceeded through the formation of acetic acid. The main reasons for the enhanced catalytic activity are as follows: (i) the development of Ni (OH)2 increased the concentration of OH−, and facilitated the EOR process; (ii) the appropriate OH− amount provides active sites since excess Ni may block the active sites of Pd and lead to poor activity; (iii) excellent alloy formation occurs at Ni50Pd50/G and Ni25Pd75/G and less alloy formation at Ni75Pd25/G; (iv) limited particle agglomeration due to the high surface area graphene sheets.56
Muench et al. in 2012 employed an electroless plating bath scheme for the uniform deposition of spiky nickel films on the surface of complex-shaped substrates. Nanostructured Ni and Ni–B tubes were obtained by blending new and polymer templates of a borane-based plating reaction. The morphology, composition, and crystallinity of the reaction products were systematically investigated by scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), energy-dispersive X-ray spectroscopy (EDS), transmission electron microscopy (TEM), and X-ray diffraction (XRD). For ethanol oxidation reaction in an alkaline environment, the outstanding EOR performance of NiNT electrocatalysts is mainly attributed to (i) the easy oxidation of the surface, represented by the minor potential difference in the forward and backward peak, (ii) fast re-oxidation of the catalyst surface in the presence of ethanol, (iii) the formation of NiOOH at the low potential of ∼0.6 V. The current density of 1.6 mA cm−2 was delivered at the potential of 0.5 V vs. Ag/AgCl along with specific and mass activity of 2.18 mA cm−2 and 0.0098 mA mg−1, respectively.173
Among all the transition metals, cobalt (Co) has renowned catalytic activity in numerous chemical reactions; however, due to the comparatively low performance, cobalt cannot be used as a prime catalyst in alcohol fuel cells, though it can be used as a co-catalyst to reduce Pt poisoning since the Pt–Co alloy shows excellent CO–tolerance during the oxidation reaction.174
Subramani and colleagues successfully prepared a significantly active catalyst by the electroless deposition of Co NPS on the rGO surface through a sensitization-activation method that involves the incorporation of catalytic nuclei on the surface of a non-catalyst. These nuclei then facilitate the redox reaction on the activated rGO surface. In the two steps of the sensitization and activation process, the sensitization by the tin layer improved the Pd adsorption and its strong binding with the surface. The structure, morphology, elemental composition, and electrocatalytic activity of the Co–Pd/Sn/RGO nanocomposite were characterized by EDX, XPS, PXRD, Raman, FESEM, EIS, and CV studies, respectively. XPS results validated the equivalent binding energy and oxidation state of the palladium, carbon, cobalt, and tin. The HRTEM and FESEM images show the adornment of metal nanoparticles on the rGO surface. PXRD depicts the peaks of the metals and RGO, while Raman spectroscopy indicated the strong binding between the cobalt nanoparticles and RGO, and EDX reveals the elemental configuration of the coating on the rGO surface.175
Mohamed et al. in 2017 investigated the electrocatalytic activity of the Co/Cr nanocomposites synthesized by the electrospinning of the precursor mixture in the presence of polyvinyl alcohol (PVA) polymer, followed by the calcination in the inner atmosphere. The synthesized nanofibers possess good electrocatalytic activity towards the C2H5OH electrooxidation, as well as good stability in basic media owing to the alloy structure. However, the optimized Cr content of 10 wt% with respect to the Co content showed the maximum current density (105 mA cm−2 at 0.8 V vs. Ag/AgCl) in cyclic voltammetry along with long-term stability for 5000 seconds.176
Barakat and associates in 2012 synthesized novel Cd-doped cobalt NPs, encapsulated in a graphite shell, via a simple sol–gel technique. During synthesis, the electrocatalyst development is possible based on the polycondensation of Cd and Co acetates, and further grinding followed by calcination in an argon atmosphere led to Cd-doped Co NPs encapsulated in a thin layer of graphite. A high current density response (75 mA cm−2 at 0.8 V vs. Ag/AgCl), stability for 900 s, and significantly low onset potential of ∼585 mV vs. NHE were attained during the electrocatalytic application in the ethanol oxidation reaction in an alkaline medium. The main reason for the enhanced catalytic activity was the formation of CdOOH, an EOR active site by Cd dopant.177
In 2019, Chaitree et al. prepared a Ni, Co, and Mn-containing trimetallic anode catalyst (CoNiMo/CCS-15) through the electroless deposition scheme and evaluated it for the ethanol oxidation reaction in an alkaline medium. During testing, it was observed that the CoNiMo-catalyzed carbon substrate was highly active in alkaline solution, and the current density increased with an increase in temperature. Moreover, the CVs results indicated that the ethanol oxidation activity originated from electroactive Co, Mo, and Ni species and not from Pd. Tafel analysis indicated the enhancement in the exchange current density with the increase in temperature. Furthermore, due to the high rate constant and low activation energy, the catalytic activity of the trimetallic alloy electrode was better than the bimetallic and monometallic electrodes, and the linear relationship of the scan rate with peak potential suggested the irreversible nature of ethanol oxidation on the proposed electrocatalysts. The onset potential of −0.23 V, current density of 15 mA mg−1 at the potential of 0.1 V vs. Ag/AgCl, and If/Ib ratio of 0.33 are prominent features of the tested sample.178
Copper, another cheap and easily available material is an auspicious assistant for noble metal-based catalysts for electrocatalytic applications, and a few studies are available on alcohol oxidation reactions using copper-based catalysts with controlled morphologies in alkaline media.179,180
Flower-like Cu/CuO nanocomposites (F–Cu/CuO) were effectively synthesized by Bai et al. in 2018 through a nano-assembly scheme employing a template-free pyrolysis and corrosion method, and were further utilized as a support for Pt nanoparticles. The characterization techniques showed the uniform dispersion of narrow-sized Pt nanoparticles on the surface of the F–Cu/CuO composite. Electrochemical characterizations showed that the Pt/F–Cu/CuO electrocatalyst exhibited higher electrocatalytic activity (38 mA mg−1 at −0.2 V vs. SCE), better stability (7000 s), and stronger CO tolerance than Pt/XC-72 and Pt/N–Cu/CuO during the alcohol oxidation process, and the main reason is the high EASA provided by the flower-like Cu/CuO support.181
In 2014, Cai et al. reported a novel Cu@PdCu/C electrocatalyst prepared by the galvanic replacement reaction between Cu and Pd2+. The Cu@PdCu nanoparticles of 0.5 nm thickness comprised the Cu core along with the PdCu alloy shell. The Cu@PdCu/C electrocatalyst with core–shell structure increased the Pd utilization and increased the specific surface area, as well as provided more Pd electroactive sites for C2H5OH oxidation. Moreover, the synergistic effect between Cu and Pd improved the electrocatalytic performance of the Cu@PdCu/C anode catalyst. The peak current density of ethanol oxidation on the Cu@PdCu/C catalyst surface was very high and reached the value of 166.0 mA cm−2 at −0.1 V vs. Hg/HgO; its stability (1800 s), If/Ib ratio (1.36), and anti-poisoning tendency were especially promoted. Thus, the superior performance of Cu@PdCu/C marks it as a feasible anodic catalyst in ADEFC.182
The nanoparticles of the CuO/TiO2/Pd–NiO composite anode catalysts were produced by a dealloying technique in an acid medium by Niu et al. in 2017. The composite catalysts displayed a continuous three-dimensional nanoporous structure. Due to the NiO introduction, the electrocatalytic performances of the tested catalysts for ethanol and methanol oxidation in an alkaline medium were heightened enormously. Three suggested reasons for this activity are as follows: (a) the incorporation of Ni reduced the size of the Pd nanoparticles, and consequently increased the surface area; (b) the excess OH− ions produced as a result of synergistic effect between the Pd and NiO/TiO2 removed the carbonaceous intermediates; (c) the reduced charge transfer resistance due to electronic effects between the Pd and Ni. Based on the active surface area, stability, and electrocatalytic activity, the np-CuO/TiO2/Pd–NiO-3 electrocatalyst showed the best performance for the ethanol and methanol oxidation reactions. The highest peak current density was ∼1.72 and 4.40 and times greater than that of the commercial Pd/C electrocatalyst for ethanol and methanol oxidation, respectively. TiO2 and NiO are responsible for the maximum adsorption of OH− at low potential, and the presence of Ni promoted this process further and caused a negative shift in the onset potential. The maximum delivered current density by the sample at a potential of −0.3 V vs. RHE was 2.614 mA cm−2 (Table 4 and 5).183
| Electrocatalyst | Electrolyte/methanol molarity | Electrode/supporting substrate | Peak potential mV (RHE) | Tafel slope mV dec−1/stability (s per cycles)) | Current density mA cm−2 | Ref. |
|---|---|---|---|---|---|---|
| Polycrystalline Ni electrode | 1.0 M NaOH/0.5 M C2H5OH | Polycrystalline Ni foil | ∼1.55 V | — | 16.3 | 161 |
| Ni/NiO–C3 | 0.5 M NaOH/0.5 M C2H5OH | GCE | 1.50 V | —/3600 s | 120 | 162 |
| NiNWA/PdNF | 1.0 M KOH/0.5 M C2H5OH | Ni NWs | 0.99 V | 96/50400 s |
1.7 | 163 |
| Ni/aHC | 1.0 M NaOH/1.0 M C2H5OH | Graphite disc | 1.47 V | —/2100 s | 17.5 | 55 |
| Ni@Pd/NG | 1.0 M KOH/1.0 M C2H5OH | GCR | 0.74 V | —/3000 s | 82.5 | 164 |
| Ni(OH)2–PtCu/C | 0.5 M HClO4/1.0 M C2H5OH | GCD | 1.10 V | —/3000 s, 2000 cycles | 27.3 | 165 |
| NiAl-LDH-NSs | 1.0 M NaOH/1.0 M C2H5OH | GCE | 1.61 V | —/10000 s |
55 | 166 |
| NiF/AuNPs | 0.5 M NaOH/1.0 M C2H5OH | Ni foam | 1.23 V | —/1000 s | 6.5 | 167 |
| Ni0.2Co0.2/G | 1.0 M KOH/1.0 M C2H5OH | GCE | 1.73 V | —/1200 cycles | ∼165.0 | 168 |
| NiCo/graphene/GCE | 0.1 M KOH/0.1 M C2H5OH | GCE | 1.75 V | —/3500 s | 16.4 | 169 |
| Ni–Co/NRGO | 0.5 M KOH/0.1 M C2H5OH | GCE | 1.88 V | 159.1/2000 s | 80.0 | 170 |
| Ni–Cr2O3/C (7Vf%) | 1.0 M NaOH/2.0 M C2H5OH | GCE | 1.69 V | —/840 s | 327.0 | 171 |
| NiNC-3 | 0.1 M NaOH/0.5 M C2H5OH | GCE | 1.60 V | —/200 s, 1500 cycles | 47.5 | 172 |
| Ni50Pd50/G | 1.0 M KOH/0.5 M C2H5OH | GCE | 0.73 V | 142/10000, sec 500 cycles |
16.0 | 56 |
| Ni–B NTs | 0.5 M KOH/0.5 M C2H5OH | GCE | 1.53 V | — | 1.6 | 173 |
| Co–Pd/Sn/RGO | 0.5 M H2SO4/0.5 M C2H5OH | GCE | ∼1.62 V | — | 0.02 | 175 |
| Co/Cr | 1.0 M NaOH/1.0 M C2H5OH | GCE | 1.83 V | —/5000 s | 105.0 | 176 |
| Cd-doped Co/C | 1.0 M KOH/1.0 M C2H5OH | GCE | 1.83 V | —/900 s | 75.0 | 177 |
| CoNiMo/CCS-15 | 0.5 M NaOH/1.0 M C2H5OH | GCE | 1.33 V | 20.6/1800 s | 3.9 | 178 |
| Pt/F–Cu/CuO | 1.0 M KOH/1.0 M C2H5OH | GCE | 0.68 V | —/7000 s | 2.7 | 181 |
| Cu@PdCu | 1.0 M KOH/1.0 M C2H5OH | GCE | 1.02 V | —/1800 s, 1000 cycles | 182 | |
| np-CuO/TiO2/Pd–NiO-3 | 0.5 M NaOH/1.0 M C2H5OH | GCE | −0.32 V | 269/3000 s | 2.6 | 183 |
| PtPd3/Mn3O4-CNT | 0.5 M H2SO4/0.5 M C2H5OH | GCE | 1.58 V | —/3600 s | 16.7 | 184 |
| Metal | Contribution to electrocatalytic activity |
|---|---|
| Pt | • Pt shows good catalytic activity and stability in acidic and basic media |
| • Pt possess three types of morphologies; Pt (100), Pt (110), and Pt (111) and it has been observed that Pt (111) produces greater current density than the other two forms | |
| • Pt electrocatalysts with copious nanostructures including nanowires, nanosheets, nanoparticles, and nanopores show good activity due to a large number of active sites | |
| • The incorporation of a 2nd metal such as Pd, Ru, Ir, Bi, Co, Fe, Ni, etc. further improves the catalytic activity of Pt due to their oxyphilic nature | |
| • Pt composites with carbon-based material show remarkable performance due to high surface area, fine dispersion of material and high conductivity | |
| Pd | • Pd is comparatively abundant and economical |
| • The Pd oxyphilic nature favors the oxidative desorption of reaction intermediates | |
| • Alloys of Pd with metals such as Ni, Ag, Sn, Au, Rh, Cu, Ru, metal oxides, and hydroxides developed multi-component systems with operational durability and optimized catalytic performance | |
| • The combination of 2D configuration with the metal alloys (such as 2D PdAg nanodendrites) possess remarkable electrocatalytic activity and stability due to abundant active sites and enlarged surface area | |
| • Pt composites with carbon-based material show remarkable performance due to high surface area, fine dispersion of material and high conductivity | |
| Au | • Au is inert at low pH but highly active at high pH |
| • Au composites with different morphologies such as core–shell, layered double hydroxide, hollow, porous structure, etc. display superb activity | |
| • Au composites with GO, rGO, CNTs, graphene, carbon fiber, etc., show exceptional activity due to huge surface area, limited agglomeration of particles, and high conductivity | |
| • Au incorporation into Pt promotes OH− adsorption and the easy oxidation of poisonous intermediates | |
| • Au incorporation into Pt and Pd downshifts the d-band center and promotes the OH− adsorption | |
| Ag | • The role of Ag is identical to Au |
| Ni | • Ni is a good option for the EOR due to its renowned surface catalytic activity, good electric conductivity, easy availability, low price, and acceptable chemical stability |
| • Nickel and nickel-based alloys have been synthesized for use as electrocatalysts where the synergistic effect promotes the EOR activity | |
| • Multiple oxidation states of Ni, particularly the +3 state in the form of NiOOH is EOR electroactive | |
| • Nickel in the form of Ni foam or foil as a substrate is also recognized as an eminent flexible support for EOR catalysts | |
| • Ni composites with carbon-based materials possess remarkable catalytic activity | |
| • Different morphologies obtained through different synthetic schemes also play an important role in enhancing the catalytic activity | |
| Co | • Due to comparatively less reactivity, Cobalt cannot be used alone, but it can be used as a co-catalyst to reduce the cost and to enhance the catalytic activity |
| • The high oxidation states of cobalt such as +3 and +4 are EOR electroactive sites | |
| • Alloys of Co with other metals show better performance than pure Co due to the synergistic effect | |
| • Co composites with carbonaceous substances, as well as different morphologies with small particle size, high surface area, and easy diffusion of electrolyte lead to enhanced catalytic activity | |
| Cu and Mn and Cd | • Pure metals and their oxides are abundantly available at a low cost. They can be effectively used as co-catalysts to reduce the cost, shift the d-band energies of Pt and Pd, facilitate the adsorption of hydroxide ions as compared to reaction intermediates and promote the ethanol oxidation process |
| • Different morphologies and composites with carbon-based supporting material or polymers result in enhanced activity |
Themsirimongko et al. in 2016 deposited noble metals (Pt, Pd) on Mn3O4-modified carbon nanotubes (CNT) through an improved polyol method. The morphology, crystallinity, and composition of the as-synthesized electrocatalysts were completely characterized by transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and energy-dispersive X-ray spectroscopy (EDX). It was established that the Mn3O4 nanoparticles were uniformly dispersed on the CNTs surface, and covered the whole surface, and several atomic ratios of PtxPdy (10 wt%) nanoparticles (1–4 nm) were also uniformly distributed on the Mn3O4-CNTs surface. The electrocatalytic performance for ethanol oxidation was investigated by cyclic voltammetry (CV), and chronoamperometry (CA). The as-prepared catalyst unveiled enhanced electrocatalytic activity and stability as compared to the commercially available PtRu/Vulcan XC-72 electrocatalyst. It was proposed that a combination of noble metals and metal oxides could develop electroactive sites that improved their catalytic performances towards the ethanol oxidation process The current density of 540 mA mg−1 delivered at a potential of 0.55 V vs. Ag/AgCl, onset potential of 0.125, If/Ib ratio of 2.30, and stability for 3600 seconds are salient features of the reported material (Fig. 8).184
 | ||
| Fig. 8 (a) Synthetic scheme and the electrocatalytic response of NiAl-LDH-NSs and bulk NiAl-LDH under alkaline conditions. Reproduced from ref. 166, Copyright 2017, Elsevier. (b) SEM image and electrochemical behavior of Ni–B nanotubes and CVs of the three Ni materials recorded with a scan rate of 50 mV s−1 in 0.1 M NaOH solution. Reproduced from ref. 173, Copyright 2013, Elsevier. (C) Synthetic scheme and the electrocatalytic response of Pt/F–Cu/CuO under alkaline conditions. Reproduced from ref. 181, Copyright 2018, Elsevier. | ||
6. Conclusion and future perspectives
Clean and economical energy sources are the solution to numerous worldwide health, energy, and environmental problems. The utilization of low-cost, abundant and efficient materials in fuel cells is the utmost requirement of modern time to replace non-renewable fossil fuel sources. So far, noble metal (Pt and Pd)-based electrocatalysts are considered to be potential candidates for the EOR due to their firm and electroactive nature, but dearth and high cost have bound their commercialization. The potential alternatives to noble-metal-based materials are transition metal compounds, which can be effectively exploited in basic media with good stability, and further research is ongoing to develop transition metal-based compounds with excellent stability in acidic medium to cover the full pH range. Carbon-based conductive and stable supports can be employed to enhance the electrocatalytic response of the materials. Regardless of the substantial progress, the following challenges still exist and further research should be undertaken before the wide-range commercial application of these materials. (i) Complete ethanol oxidation requires 12 electrons per molecule, as well as breakage of the C–C bond. Research has disclosed that the ethanol is partially oxidized to acetic acid/acetaldehyde (2–4 electron in oxidation reactions) despite 12 electrons full oxidization. These reaction intermediates occupy catalyst active sites, limit further oxidation processes, and reduce the energy produced during the reaction. As such, a highly active EOR catalyst is required to replace the expensive noble metals for complete oxidation during the OER process. (ii) The size, shape, and architecture of the metal nanoparticles can prominently affect the electrocatalytic activity, therefore, metal nanoparticles phase engineering has become a promising idea for the future, and is gaining the attention of researchers. (iii) The utilization of single-metal atom NPs with assessed electrocatalytic activity and usual crystal phases through surfactant-free synthetic routes would be a good option. (iv) More research efforts are required to understand the fundamental correlation between the conductive support and the metal at the interface to achieve a high electrocatalytic activity. (v) A complete understanding of the reaction mechanism and the rate-controlling step through in situ characterization techniques still need to be addressed. (vi) It is required that different tools be utilized to completely analyze the reaction process to identify the side products obtained during the reaction via in situ FTIR spectroscopy, Raman spectroscopy, and Differential Electrochemical Mass Spectrometry (DEMS). (vii) More efforts are required to enhance the durability of the anode electrocatalyst in direct ethanol fuel cells. Although the hybrid structural design is an important strategy to homogeneously disperse the electrocatalyst on the support surface, the long-term cyclic stability test results in weak binding between the support material and catalyst. As such, the post-analysis of the composition, structure, and morphology is requisite to comprehend the stability of the catalyst during the EOR process. (viii) Investigation of the temperature-dependent performance of an electrocatalyst for the ethanol oxidation reaction, and determination of the optimized temperature conditions for a catalyst's highest conversion efficiency are also very important. (ix) There is a need to improve the electrolyte diffusibility between the electrodes and membrane for better contact with the electrode material. (x) Binary and ternary composites need to be further utilized for the EOR process since controlled composition and morphology result in different pathways and mechanisms and lead to remarkable performance. (xi) Membrane thickness, electrolyte concentration, management of water and heat, ethanol and oxidant flow rate also need to be controlled to obtain better efficiency. (xii) The performance of an electrocatalyst in alkaline media DEFC is much better than in an acidic electrolyte, but several problems such as a decline in ionic conductivity of membrane, and reduced hydrophobicity of the electrode still arise due to carbonate formation in the presence of a base, As such, there is a need to address these issues to attain the outstanding electrocatalytic performance of the catalyst. (xiii) Theoretical and simulation studies can lead to a better understanding of catalytic mechanisms, structural determination of the active sites, intrinsic reactivities, and identification of reaction intermediates, while modeling kinetics can do a great job in the design of an electrocatalyst with grander activity and stability. (xiv) Quantum mechanistic calculations-based Density Functional Theory (DFT), which simulates the catalytic surface and active centers, can be utilized for better study of the electrocatalytic activity of the material, the geometrical and electronic structure of the electric double layer, and the design of metal/non-metal-based electrocatalysts. However, the drawback of the DFT theory lies in its ability to simulate a limited number of atoms of a system instead of a large number of atoms, molecules and bonding interactions participating in a reaction in reality.Author contributions
Conceptualization, L. Y. and T. N; data curation and investigation, L. Y.; supervision T. N. and N. I.; original draft preparation L. Y.; writing-reviews and editing all authors.Conflicts of interest
There are no conflicts to declare.References
- M. Akhairi and S. Kamarudin, Int. J. Hydrogen Energy, 2016, 41, 4214–4228 CrossRef CAS.
- X. Li and A. Faghri, J. Power Sources, 2013, 226, 223–240 CrossRef CAS.
- L. Carrette, K. Friedrich and U. Stimming, Fuel cells, 2001, 1, 5–39 CrossRef CAS.
- S. Giddey, S. Badwal, A. Kulkarni and C. Munnings, Prog. Energy Combust. Sci., 2012, 38, 360–399 CrossRef CAS.
- J. Tayal, B. Rawat and S. Basu, Int. J. Hydrogen Energy, 2011, 36, 14884–14897 CrossRef CAS.
- H. Yang, T. Guo, D. Yin, Q. Liu and X. Zhang, J. Taiwan Inst. Chem. Engrs., 2020, 112, 212–221 CrossRef CAS.
- S. Kianfar, A. N. Golikand and B. ZareNezhad, J. Nanostruct. Chem., 2020, 4, 1–13 Search PubMed.
- Q. Wang, X. Lu, Q. Xin and G. Sun, Chin. J. Catal., 2014, 35, 1394–1401 CrossRef CAS.
- A. Azam, S. Lee, M. Masdar, A. Zainoodin and S. Kamarudin, Int. J. Hydrogen Energy, 2019, 44, 8566–8574 CrossRef CAS.
- H. R. Corti and E. R. Gonzalez, Direct Alcohol Fuel Cells, Springer, 2014, vol. 1, pp. 300–325 Search PubMed.
- B. Braunchweig, D. Hibbitts, M. Neurock and A. Wieckowski, Catal. Today, 2013, 202, 197–209 CrossRef CAS.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS.
- T. Zhao, Z. Liang, and J. Xu, Encyclopedia of electrochemical power sources, 2009, vol. 362 Search PubMed.
- S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- G. Camara and T. Iwasita, J. Electroanal. Chem., 2005, 578, 315–321 CrossRef CAS.
- E. Antolini and E. Gonzalez, J. Power Sources, 2010, 195, 3431–3450 CrossRef CAS.
- C. Yougui, L. Zhuang and L. Juntao, Chin. J. Catal., 2007, 28, 870–874 CrossRef.
- M. Kamarudin, S. Kamarudin, M. Masdar and W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438–9453 CrossRef CAS.
- Z. Zhang, L. Xin, K. Sun and W. Li, Int. J. Hydrogen Energy, 2011, 36, 12686–12697 CrossRef CAS.
- Y. Y. Feng, Z. H. Liu, W. Q. Kong, Q. Y. Yin and L. X. Du, Int. J. Hydrogen Energy, 2014, 39, 2497–2504 CrossRef CAS.
- J. Xu, T. Zhao, Y. Li and W. Yang, Int. J. Hydrogen Energy, 2010, 35, 9693–9700 CrossRef CAS.
- L. An, T. Zhao and J. Xu, Int. J. Hydrogen Energy, 2011, 36, 13089–13095 CrossRef CAS.
- S. Shen, T. Zhao and Q. Wu, Int. J. Hydrogen Energy, 2012, 37, 575–582 CrossRef CAS.
- L. An, T. Zhao and Y. Li, Renewable Sustainable Energy Rev., 2015, 50, 1462–1468 CrossRef CAS.
- M. C. Figueiredo, A. S. Aarnio, F. J. V. Iglesias, J. S. Gullón, J. M. Feliu and K. Kontturi, Appl. Catal., B, 2013, 140, 378–385 CrossRef.
- D. D. James and P. G. Pickup, Electrochim. Acta, 2010, 55, 3824–3829 CrossRef CAS.
- S. Song, W. Zhou, J. Tian, R. Cai, G. Sun and Q. Xin, J. Power Sources, 2005, 145, 266–271 CrossRef CAS.
- S. Heysiattalab, M. Shakeri, M. Safari and M. Keikha, J. Ind. Eng. Chem., 2011, 17, 727–729 CrossRef CAS.
- Y. Li, Y. He and W. Yang, Int. J. Hydrogen Energy, 2013, 38, 13427–13433 CrossRef CAS.
- Y. Li, T. Zhao and Z. Liang, J. Power Sources, 2009, 190, 223–229 CrossRef CAS.
- V. Alzate, K. Fatih and H. Wang, J. Power Sources, 2011, 196, 10625–10631 CrossRef CAS.
- G. Andreadis, S. Song and P. Tsiakaras, J. Power Sources, 2006, 157, 657–665 CrossRef CAS.
- A. Halder, S. Sharma, M. Hegde and N. Ravishankar, J. Phys. Chem. C, 2009, 113, 1466–1473 CrossRef CAS.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 CrossRef CAS PubMed.
- A. Ghumman, G. Li, D. V. Bennett and P. G. Pickup, J. Power Sources, 2009, 194, 286–290 CrossRef CAS.
- A. Jablonski, P. J. Kulesza and A. Lewera, J. Power Sources, 2011, 196, 4714–4718 CrossRef CAS.
- K. Taneda and Y. Yamazaki, Electrochim. Acta, 2006, 52, 1627–1631 CrossRef CAS.
- L. Jiang, G. Sun, S. Wang, G. Wang, Q. Xin and Z. Zhou, et al., Electrochem. Commun., 2005, 7, 663–668 CrossRef CAS.
- G. Lu, F. Liu and C.-Y. Wang, Electrochem. Solid-State Lett., 2004, 8, 400–420 CrossRef.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani and E. S. Smotkin, Science, 1998, 280, 1735–1737 CrossRef CAS PubMed.
- Y. Lin, X. Cui, C. H. Yen and C. M. Wai, Langmuir, 2005, 21, 11474–11479 CrossRef CAS PubMed.
- L. Gong, Z. Yang, K. Li, W. Xing, C. Liu and J. Ge, J. Energy Chem., 2018, 27, 1618–1628 CrossRef.
- A. Hacquard, Improving and understanding direct methanol fuel cell (DMFC) performance, 2005 Search PubMed.
- O. Zerbinati, J. Chem. Educ., 2002, 79, 829 CrossRef CAS.
- J. Sánchez-Monreal, M. Vera and P. A. García-Salaberri, Fundamentals of electrochemistry with application to direct alcohol fuel cell modeling, in Proton Exchange Membrane Fuel Cell, Intech, Rijeka, 2018, pp. 121–154 Search PubMed.
- Z. Pan, R. Chen, L. An and Y. Li, J. Power Sources, 2017, 365, 430–445 CrossRef CAS.
- C. Lamy, C. Coutanceau and J. M. Leger, The Direct Ethanol Fuel Cell: a Challenge to Convert Bioethanol Cleanly into Electric Energy, Wiley Online Library, 2009, pp. 1–42 Search PubMed.
- S. Themsirimongkon, T. Sarakonsri, S. Lapanantnoppakhun, J. Jakmunee and S. Saipanya, Int. J. Hydrogen Energy, 2019, 44, 30719–30731 CrossRef CAS.
- J. Cai, Y. Huang, B. Huang, S. Zheng and Y. Guo, Int. J. Hydrogen Energy, 2014, 39, 798–807 CrossRef CAS.
- R. Jiang, D. T. Tran, J. P. McClure and D. Chu, ACS Catal., 2014, 4, 2577–2586 CrossRef CAS.
- J. Zhang, X. Yang, H. Shao, C. Tseng, D. Wang and S. Tian, Fuel Cells, 2017, 17, 115–122 CrossRef CAS.
- M. F. R. Hanifah, J. Jaafar, M. Othman, A. Ismail, M. Rahman and N. Yusof, J. Alloys Compd., 2019, 793, 232–246 CrossRef CAS.
- K. I. Ozoemena, RSC Adv., 2016, 6, 89523–89550 RSC.
- S. C. Lai, S. E. Kleyn, V. Rosca and M. T. Koper, J. Phys. Chem. C, 2008, 112, 19080–19087 CrossRef CAS.
- A. Cuña, C. R. Plascencia, E. L. da Silva, J. Marcuzzo, S. Khan and N. Tancredi, Appl. Catal., B, 2017, 202, 95–103 CrossRef.
- M. S. Ahmed and S. Jeon, ACS Catal., 2014, 4, 1830–1837 CrossRef CAS.
- S. S. Turkyılmaz, N. Guy and M. O zacar, J. Photochem. Photobiol., A, 2017, 341, 39–50 CrossRef.
- G. R. Xu, J. J. Hui, T. Huang, Y. Chen and J. M. Lee, J. Power Sources, 2015, 285, 393–399 CrossRef CAS.
- S. Ahmadi, C. H. Chia, S. Zakaria, K. Saeedfar and N. Asim, J. Magn. Magn. Mater., 2012, 324, 4147–4150 CrossRef CAS.
- Y. Xu, B. Ren, R. Wang, L. Zhang, T. Jiao and Z. Liu, Nanomaterials, 2019, 9, 10–12 CrossRef PubMed.
- L. Yaqoob, T. Noor, N. Iqbal, H. Nasir, N. Zaman and K. Talha, J. Alloys Compd., 2021, 850, 156583–156590 CrossRef CAS.
- T. Noor, N. Zaman, H. Nasir, N. Iqbal and Z. Hussain, Electrochim. Acta, 2019, 307, 1–12 CrossRef CAS.
- L. R. Alden, C. Roychowdhury, F. Matsumoto, D. K. Han, V. B. Zeldovich and H. D. Abruña, Langmuir, 2006, 22, 10465–10471 CrossRef CAS PubMed.
- C. T. Hsieh, P. Y. Yu, D. Y. Tzou, J. P. Hsu and Y. R. Chiu, J. Electroanal. Chem., 2016, 761, 28–36 CrossRef CAS.
- J. Zhang, M. Chaker and D. Ma, J. Colloid Interface Sci., 2017, 489, 138–149 CrossRef CAS PubMed.
- J. J. Lv, A. J. Wang, X. Ma, R. Y. Xiang, J. R. Chen and J. J. Feng, J. Mater. Chem. A, 2015, 3, 290–296 RSC.
- D. J. Chen, Q. L. Zhang, J. X. Feng, K. J. Ju, A. J. Wang and J. Wei, J. Power Sources, 2015, 287, 363–369 CrossRef CAS.
- Z. Bo, D. Hu, J. Kong, J. Yan and K. Cen, J. Power Sources, 2015, 273, 530–537 CrossRef CAS.
- T. Radhakrishnan and N. Sandhyarani, Int. J. Hydrogen Energy, 2017, 42, 7014–7022 CrossRef CAS.
- F. Ye, C. Xu, G. Liu, M. Yuan, Z. Wang and X. Du, Energy Convers. Manage., 2018, 160, 85–92 CrossRef CAS.
- F. Ye, C. Xu, G. Liu, J. Li, X. Wang and X. Du, Energy Convers. Manage., 2018, 155, 182–187 CrossRef CAS.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang and A. Liu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- B. W. Zhang, H. L. Yang, Y. X. Wang, S. X. Dou and H. K. Liu, Adv. Energy Mater., 2018, 8, 1703597–1703600 CrossRef.
- A. N. Vyas, G. D. Saratale and S. D. Sartale, Int. J. Hydrogen Energy, 2020, 45, 5928–5947 CrossRef CAS.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- Z. Chen, A. Yu, D. Higgins, H. Li, H. Wang and Z. Chen, Nano Lett., 2012, 12, 1946–1952 CrossRef CAS PubMed.
- C. Koenigsmann, A. C. Santulli, K. Gong, M. B. Vukmirovic, W. p. Zhou and E. Sutter, J. Am. Chem. Soc., 2011, 133, 9783–9795 CrossRef CAS PubMed.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem.Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- J. Xu, A. R. Wilson, A. R. Rathmell, J. Howe, M. Chi and B. J. Wiley, ACS Nano, 2011, 5, 6119–6127 CrossRef CAS PubMed.
- X. Liu, L. Ning, M. Deng, J. Wu, A. Zhu and Q. Zhang, Nanoscale, 2019, 11, 3311–3317 RSC.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- M. M. Dimos and G. Blanchard, J. Phys. Chem. C, 2010, 114, 6019–6026 CrossRef CAS.
- X. Huang, Z. Zhao, J. Fan, Y. Tan and N. Zheng, J. Am. Chem. Soc., 2011, 133, 4718–4721 CrossRef CAS PubMed.
- M. H. Chen, Y. X. Jiang, S. R. Chen, R. Huang, J. L. Lin and S. P. Chen, J. Phys. Chem. C, 2010, 114, 19055–19061 CrossRef CAS.
- S. Mourdikoudis, M. Chirea, T. Altantzis, I. P. Santos, J. P. Juste and F. Silva, Nanoscale, 2013, 5, 4776–4784 RSC.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su and G. Lu, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- J. E. Sulaiman, S. Zhu, Z. Xing, Q. Chang and M. Shao, ACS Catal., 2017, 7, 5134–5141 CrossRef CAS.
- J. Mao, W. Chen, D. He, J. Wan, J. Pei and J. Dong, Sci. Adv., 2017, 3, 1603068–1603075 CrossRef PubMed.
- T. Liu, K. Wang, Q. Yuan, Z. Shen, Y. Wang and Q. Zhang, Nanoscale, 2017, 9, 2963–2968 RSC.
- G. Zhang, Z. Yang, W. Zhang, H. Hu, C. Wang and C. Huang, Nanoscale, 2016, 8, 3075–3084 RSC.
- C. C. Kung, P. Y. Lin, Y. Xue, R. Akolkar, L. Dai and X. Yu, J. Power Sources, 2014, 256, 329–335 CrossRef CAS.
- H. Hang, R. M. Altarawneh, T. M. Brueckner and P. G. Pickup, J. Electrochem. Soc., 2020, 167, 054518 CrossRef.
- J. Asgardi, J. C. Calderón, F. Alcaide, A. Querejeta, L. Calvillo and M. J. Lázaro, Appl. Catal., B, 2015, 168, 33–41 CrossRef.
- R. Rizo, D. Sebastián, M. J. Lázaro and E. Pastor, Appl. Catal., B, 2017, 200, 246–254 CrossRef CAS.
- X. Chen, Z. Cai, X. Chen and M. Oyama, J. Mater. Chem. A, 2014, 2, 315–320 RSC.
- Q. Zhang, T. Chen, R. Jiang and F. Jiang, RSC Adv., 2020, 10, 10134–10143 RSC.
- F. Ren, H. Wang, C. Zhai, M. Zhu, R. Yue and Y. Du, ACS Appl. Mater. Interfaces, 2014, 6, 3607–3614 CrossRef CAS PubMed.
- S. S. Li, J. N. Zheng, X. Ma, Y. Y. Hu, A. J. Wang and J. R. Chen, Nanoscale, 2014, 6, 5708–5713 RSC.
- K. Bhunia, S. Khilari and D. Pradhan, ACS Sustainable Chem. Eng., 2018, 6, 7769–7778 CrossRef CAS.
- P. Gnanaprakasam, S. Jeena and T. Selvaraju, J. Mater. Chem. A, 2015, 3, 18010–18018 RSC.
- M. J. Paulo, R. H. Venancio, R. G. Freitas, E. C. Pereira and A. C. Tavares, J. Electroanal. Chem., 2019, 840, 367–375 CrossRef CAS.
- J. Bai, X. Xiao, Y. Y. Xue, J. X. Jiang, J. H. Zeng and X. F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 19755–19763 CrossRef CAS PubMed.
- S. Shen, T. Zhao and J. Xu, Int. J. Hydrogen Energy, 2010, 35, 12911–12917 CrossRef CAS.
- L. Jin, H. Xu, C. Chen, H. Shang, Y. Wang and C. Wang, Inorg. Chem. Front., 2020, 7, 625–630 RSC.
- Y. W. Lee, E. T. Hwang, D. H. Kwak and K. W. Park, Catal. Sci. Technol., 2016, 6, 569–576 RSC.
- G. V. Ramesh, R. Kodiyath, T. Tanabe, M. Manikandan, T. Fujita and N. Umezawa, ACS Appl. Mater. Interfaces, 2014, 6, 16124–16130 CrossRef CAS PubMed.
- N. Zhang, S. Guo, X. Zhu, J. Guo and X. Huang, Chem. Mater., 2016, 28, 4447–4452 CrossRef CAS.
- J. J. Duan, J. J. Feng, L. Zhang, J. Yuan, Q. L. Zhang and A. J. Wang, Int. J. Hydrogen Energy, 2019, 44, 27455–27464 CrossRef CAS.
- H. Li, G. Chang, Y. Zhang, J. Tian, S. Liu and Y. Luo, Catal. Sci. Technol., 2012, 2, 1153–1156 RSC.
- Z. Li, R. Lin, Z. Liu, D. Li, H. Wang and Q. Li, Electrochim. Acta, 2016, 191, 606–615 CrossRef CAS.
- H. Jin, T. Xiong, Y. Li, X. Xu, M. Li and Y. Wang, Chem. Commun., 2014, 50, 12637–12640 RSC.
- G. Hu, F. Nitze, H. R. Barzegar, T. Sharifi, A. Mikołajczuk and C. W. Tai, J. Power Sources, 2012, 209, 236–242 CrossRef CAS.
- A. Krittayavathananon, S. Duangdangchote, P. Pannopard, N. Chanlek, S. Sathyamoorthi and J. Limtrakul, Sustainable Energy Fuels, 2020, 4, 1118–1125 RSC.
- Q. Dong, Y. Zhao, X. Han, Y. Wang, M. Liu and Y. Li, Int. J. Hydrogen Energy, 2014, 39, 14669–14679 CrossRef CAS.
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen and G. Lu, Angew. Chem., 2016, 128, 9176–9181 CrossRef.
- A. L. Wang, X. J. He, X. F. Lu, H. Xu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2015, 54, 3669–3673 CrossRef CAS PubMed.
- Z. Zhang, S. Liu, X. Tian, J. Wang, P. Xu and F. Xiao, J. Mater. Chem. A., 2017, 5, 10876–10884 RSC.
- Y. Wang, Q. He, J. Guo, J. Wang, Z. Luo and T. D. Shen, ACS Appl. Mater. Interfaces, 2015, 7, 23920–23931 CrossRef CAS PubMed.
- T. Maiyalagan and K. Scott, J. Power Sources, 2020, 195, 5246–5251 CrossRef.
- F. Guo, Y. Li, B. Fan, Y. Liu, L. Lu and Y. Lei, ACS Appl. Mater. Interfaces, 2018, 10, 4705–4714 CrossRef CAS PubMed.
- Z. R. Yang, S. Q. Wang, J. Wang, A. J. Zhou and C. W. Xu, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- L. Chen, L. Lu, H. Zhu, Y. Chen, Y. Huang and Y. Li, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- S. Sarkar, R. Jana, U. V. Waghmare, B. Kuppan, S. Sampath and S. C. Peter, Chem. Mater., 2015, 27, 7459–7467 CrossRef CAS.
- A. R. Rajamani, P. Ashly, L. Dheer, S. C. Sarma, S. Sarkar and D. Bagchi, ACS Appl. Energy Mater., 2019, 2, 7132–7141 CrossRef CAS.
- B. Cermenek, B. Genorio, T. Winter, S. Wolf, J. G. Connell and M. Roschger, Electrocatalysis, 2020, 11, 203–214 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, C. Bi, H. Xia, M. H. Engelhard and D. Du, J. Mater. Chem. A., 2017, 5, 23952–23959 RSC.
- Z. Luo, J. Lu, C. Flox, R. Nafria, A. Genç and J. Arbiol, J. Mater. Chem. A, 2016, 4, 16706–16713 RSC.
- F. Gao, Y. Yin, Z. Cao, H. Li and P. Guo, J. Chem., 2020, 2020 Search PubMed.
- S. Fu, C. Zhu, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 13842–13848 CrossRef CAS PubMed.
- K. Zhang, D. Bin, B. Yang, C. Wang, F. Ren and Y. Du, Nanoscale, 2015, 7, 12445–12451 RSC.
- J. Guo, R. Chen, F. C. Zhu, S. G. Sun and H. M. Villullas, Appl. Catal., B, 2018, 224, 602–611 CrossRef CAS.
- L. L. Sikeyi, T. Matthews, A. S. Adekunle and N. W. Maxakato, Electroanalysis, 2020, 32, 2681–2692 CrossRef CAS.
- K. S. Bhavani, T. Anusha, J. S. Kumar and P. K. Brahman, Electroanalysis, 2021, 33, 97–110 CrossRef CAS.
- A. K. Ipadeola, R. Barik, S. C. Ray and K. I. Ozoemena, Electrocatalysis, 2019, 10, 366–380 CrossRef CAS.
- H. Mao, L. Wang, P. Zhu, Q. Xu and Q. Li, Int. J. Hydrogen Energy, 2014, 39, 17583–17588 CrossRef CAS.
- A. E. Fahim, R. A. Hameed and N. K. Allam, New J. Chem., 2018, 42, 6144–6160 RSC.
- L. Chen, N. Chen, Y. Hou, Z. Wang, S. Lv and T. Fujita, et al., ACS Catal., 2013, 3, 1220–1230 CrossRef CAS.
- J. Zhong, D. Bin, Y. Feng, K. Zhang, J. Wang and C. Wang, Catal. Sci. Technol., 2016, 6, 5397–5404 RSC.
- S. Shen, Y. Guo, L. Luo, F. Li, L. Li and G. Wei, J. Phys. Chem. C, 2018, 122, 1604–1611 CrossRef CAS.
- R. L. Zhang, J. J. Duan, Z. Han, J. J. Feng, H. Huang and Q. L. Zhang, Appl. Surf. Sci., 2020, 506, 144791 CrossRef CAS.
- S. T. Nguyen, H. M. Law, H. T. Nguyen, N. Kristian, S. Wang and S. H. Chan, Appl. Catal., B, 2009, 91, 507–515 CrossRef CAS.
- S. T. Nguyen, Y. Yang and X. Wang, Appl. Catal., B, 2012, 113, 261–270 CrossRef.
- W. Hong, J. Wang and E. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 9481–9487 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, Z. Luo, X. Liu, Z. Lai and B. Li, Adv. Mater., 2017, 29, 1701331 CrossRef PubMed.
- C. Bi, Y. Song, H. He, C. Wu, W. Du and L. Huang, J. Mater. Chem. A, 2018, 6, 7675–7685 RSC.
- C. Bi, C. Feng, T. Miao, Y. Song, D. Wang and H. Xia, Nanoscale, 2015, 7, 20105–20116 RSC.
- R. Loukrakpam, Q. Yuan, V. Petkov, L. Gan, S. Rudi and R. Yang, Phys. Chem. Chem. Phys., 2014, 16, 18866–18876 RSC.
- S. W. Kang, K. W. Choi, S. W. Choi, S. W. Han, S. H. Im and O. O. Park, CrystEngComm, 2013, 15, 7113–7120 RSC.
- X. Qiu, Y. Dai, Y. Tang, T. Lu, S. Wei and Y. Chen, J. Power Sources, 2015, 278, 430–435 CrossRef CAS.
- C. Peng, Y. Hu, M. Liu and Y. Zheng, J. Power Sources, 2015, 278, 69–75 CrossRef CAS.
- Y.-G. Jo, S.-M. Kim, J.-W. Kim and S.-Y. Lee, J. Alloys Compd., 2016, 688, 447–453 CrossRef CAS.
- A. S. Douk, H. Saravani, M. Farsadrooh and M. Noroozifar, Ultrason. Sonochem., 2019, 58, 10461 CrossRef PubMed.
- D. P. Dubal, G. S. Gund, C. D. Lokhande and R. Holze, ACS Appl. Mater. Interfaces, 2013, 5, 2446–2454 CrossRef CAS PubMed.
- F. Wolfart, D. P. Dubal, M. Vidotti and P. Gómez-Romero, RSC Adv., 2016, 6, 15062–15070 RSC.
- S. Ghaderi and B. Habibi, Iran. J. Hydrogen Fuel Cell, 2015, 2, 109–119 Search PubMed.
- S. Jongsomjit, P. Prapainainar and K. Sombatmankhong, Solid State Ionics, 2016, 288, 147–153 CrossRef CAS.
- L. S. Parreira, J. C. M. da Silva, M. D'Villa-Silva, F. C. Simões, S. Garcia and I. Gaubeur, Electrochim. Acta, 2013, 96, 243–252 CrossRef CAS.
- L. Yaqoob, T. Noor, N. Iqbal, H. Nasir and N. Zaman, Catalysts, 2019, 9, 856 CrossRef CAS.
- L. Yaqoob, T. Noor and N. Iqbal, Int. J. Energy Res., 2020, 5, 45–50 Search PubMed.
- T. Noor, S. Pervaiz, N. Iqbal, H. Nasir, N. Zaman and M. Sharif, Nanomaterials, 2020, 10, 1601–1608 CrossRef CAS PubMed.
- A. Barbosa, V. Oliveira, J. Van Drunen and G. Tremiliosi-Filho, J. Electroanal. Chem., 2015, 746, 31–38 CrossRef CAS.
- H. Wang, Y. Cao, J. Li, J. Yu, H. Gao and Y. Zhao, Ionics, 2018, 24, 2745–2752 CrossRef CAS.
- M. Hasan, S. B. Newcomb, J. F. Rohan and K. M. Razeeb, J. Power Sources, 2012, 218, 148–156 CrossRef CAS.
- M. Zhang, Y. Li, D. Pan, Z. Yan, S. Meng and J. Xie, RSC Adv., 2016, 6, 33231–33239 RSC.
- Y. Hong, H. J. Kim, H. J. Lee, J. Kim and S. I. Choi, Front. Chem., 2019, 7, 608 CrossRef CAS PubMed.
- L. Xu, Z. Wang, X. Chen, Z. Qu, F. Li and W. Yang, Electrochim. Acta, 2018, 260, 898–904 CrossRef CAS PubMed.
- A. Hatamie, E. Rezvani, A. S. Rasouli and A. Simchi, Electroanalysis, 2019, 31, 504–511 CAS.
- N. A. Barakat, M. Motlak, B. H. Lim, M. H. El Newehy and S. S. Al Deyab, J. Electrochem. Soc., 2014, 161, F1194 CrossRef.
- Z. Wang, Y. Du, F. Zhang, Z. Zheng, Y. Zhang and C. Wang, J. Solid State Electrochem., 2013, 17, 99–107 CrossRef CAS.
- K. Kakaei and K. Marzang, J. Colloid Interface Sci., 2016, 462, 148–153 CrossRef CAS PubMed.
- H. Hassan and Z. A. Hamid, Int. J. Hydrogen Energy, 2011, 36, 5117–5127 CrossRef CAS.
- W. Shi, H. Gao, J. Yu, M. Jia, T. Dai and Y. Zhao, Electrochim. Acta, 2016, 220, 486–492 CrossRef CAS.
- F. Muench, M. Oezaslan, M. Rauber, S. Kaserer, A. Fuchs and E. Mankel, J. Power Sources, 2013, 222, 243–252 CrossRef CAS.
- U. Paulus, A. Wokaun, G. Scherer, T. Schmidt, V. Stamenkovic and V. Radmilovic, J. Phys. Chem. B, 2002, 106, 4181–4191 CrossRef CAS.
- S. M. Subramani and K. Gantigiah, Mater. Res. Express, 2019, 6, 124001 CrossRef CAS.
- I. Mohamed, M. Motlak, M. Obaid, M. S. Alsoufi, T. M. Bawazeer and A. F. Mohamed, J. Nanosci. Nanotechnol., 2017, 17, 1280–1286 CrossRef CAS PubMed.
- N. A. Barakat, M. A. Abdelkareem and H. Y. Kim, Appl. Catal. A. Gen., 2013, 455, 193–198 (J. Electrochem. Soc., 2019, 166, 392–400) CrossRef CAS.
- W. Chaitree and E. E. Kalu, J. Electrochem. Soc., 2019, 166, H392 CrossRef CAS.
- M. Gong, G. Fu, Y. Chen, Y. Tang and T. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 7301–7308 CrossRef CAS PubMed.
- M. Ammam and E. B. Easton, J. Power Sources, 2013, 222, 79–87 CrossRef CAS.
- Z. Bai, S. Li, Q. Zhang, M. Shi, J. Fu and L. Yang, Catal. Today, 2018, 318, 137–143 CrossRef CAS.
- J. Ledesma-García, J. Maya-Cornejo, N. Arjona, S. Rivas, L. Á. Contreras and M. G. Balcázar, J. Electrochem. Soc., 2015, 162, 1439–1450 CrossRef.
- M. Niu, W. Xu, S. Zhu, Y. Liang, Z. Cui and X. Yang, J. Power Sources, 2017, 362, 10–19 CrossRef CAS.
- S. Themsirimongko, N. Promsawan and S. Saipanya, Int. J. Electrochem. Sci., 2016, 11, 967–982 CAS.
| This journal is © The Royal Society of Chemistry 2021 |