
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted disruption of PKC from AKAP signaling complexes†
Ameya J.
Limaye
 *,
George N.
Bendzunas
and
Eileen J.
Kennedy
*
*,
George N.
Bendzunas
and
Eileen J.
Kennedy
*
Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA. E-mail: ekennedy@uga.edu
First published on 19th July 2021
Abstract
Protein Kinase C (PKC) is a member of the AGC subfamily of kinases and regulates a wide array of signaling pathways and physiological processes. Protein–protein interactions involving PKC and its scaffolding partners dictate the spatiotemporal dynamics of PKC activity, including its access to activating second messenger molecules and potential substrates. While the A Kinase Anchoring Protein (AKAP) family of scaffold proteins universally bind PKA, several were also found to scaffold PKC, thereby serving to tune its catalytic output. Targeting these scaffolding interactions can further shed light on the effect of subcellular compartmentalization on PKC signaling. Here we report the development of two hydrocarbon stapled peptides, CSTAD5 and CSTAD6, that are cell permeable and bind PKC to disrupt PKC–gravin complex formation in cells. Both constrained peptides downregulate PMA-induced cytoskeletal remodeling that is mediated by the PKC–gravin complex as measured by cell rounding. Further, these peptides downregulate PKC substrate phosphorylation and cell motility. To the best of our knowledge, no PKC-selective AKAP disruptors have previously been reported and thus CSTAD5 and CSTAD6 are novel disruptors of PKC scaffolding by AKAPs and may serve as powerful tools for dissecting AKAP-localized PKC signaling.
Protein Kinase C (PKC) is a serine/threonine kinase that is involved in regulation of a multitude of fundamental cellular processes.1 It plays a critical role in several signal transduction pathways that modulate cell growth, differentiation, proliferation, migration, gene expression and apoptosis.2 PKC is activated by generation of lipid second messengers and/or an increase in cytoplasmic calcium levels in response to stimuli such as hormones, growth factors and cytokines.3 Given its ubiquitous expression across virtually all cell types and overlapping substrate specificities within the family, PKC activity is tightly regulated.4,5 Protein–protein interactions (PPIs) involving PKC play an important role in determining localization, activity and amplitude of PKC signaling as well as its accessibility to various substrates.6–8
A Kinase Anchoring Proteins (AKAPs) serve as a class of scaffolding proteins that partly regulate PKC. AKAPs are a family of over 40 proteins that are diverse but share the commonality of binding to the regulatory (R) subunits of Protein Kinase A (PKA) through a conserved helical binding interface called the A Kinase Binding (AKB) domain.9,10 This anchoring interaction allows AKAPs to spatially confine PKA to specific subcellular compartments. This spatial confinement dictates the accessibility of PKA to a pool of activating second messenger molecules as well as potential substrates.10 AKAPs form multivalent protein assemblies that involve several different kinases, phosphatases, and their substrates in order to integrate multiple second messenger signaling cascades as well as their feedback loops to create localized signalosomes for PKA.11–13
However, PKA scaffolding only denotes one aspect of AKAP function. While all AKAPs can bind and anchor PKA, PKC can also be scaffolded by some AKAPs. Various members of the AKAP family have been reported to interact with PKC through binding regions that are distinct from that of PKA anchoring and with PKC isozyme specificities including AKAP79 (AKAP5), gravin (AKAP12, AKAP250), AKAP9 (AKAP350, yotiao), AKAP-Lbc (AKAP13), AKAP149 (AKAP1, D-AKAP-1), AKAP7 (AKAP15, AKAP18), ezrin and pericentrin.14–23 PKC scaffolding by AKAPs can have a profound effect on PKC activity via altered substrate engagement and modulation of downstream signaling cascades.23–26
Disruption of PPI-mediated scaffolding and subcellular localization of PKC presents a unique avenue for tuning PKC output while avoiding outright inhibition of its catalytic activity. Given the importance of PKC localization and protein–protein interactions in a myriad of cellular functions, we sought to develop conformationally constrained peptide disruptors that target PKC scaffolding by AKAPs (Fig. 1A). We focused on the PKC binding site on AKAP79 which is also one of the only well-characterized AKAP–PKC interactions which occurs through an N-terminal polybasic domain on AKAP79 (AKAP79 residues 31–52).27 Two arginine (R) residues at positions 39 and 40 were reported to be indispensable for binding.27 This predicted helical binding site also contains a potential helix-disrupting proline (P) residue at position 49.27–29 A helical wheel projection of residues 31–48 reveals the amphipathic nature of the binding site with positively charged lysine (K) residues clustered onto the hydrophilic face while the two arginine (R) residues critical for binding are located on the opposite face (Fig. 1B). Due to the lack of any structural information pertaining to this binding interaction, we proceeded to develop a small library of peptides corresponding to residues 31–48 of AKAP79 that were synthetically constrained with all-hydrocarbon staples to chemically reinforce the secondary structure of the peptide. The native sequence also contains a methionine residue at position 34. It was replaced with an isosteric norleucine in the library design so as to avoid possible oxidation of the methionine side chain. Peptides were synthesized using standard Fmoc chemistry on solid support and synthetic olefinic amino acid residues ((S)-2-(4′-pentenyl)alanine, S5) were placed at i, i + 4 positions to allow for stabilization across a predicted single helical turn (Fig. 1C). Ring closing metathesis was performed using Grubb's 1st generation catalyst (Fig. 1D).
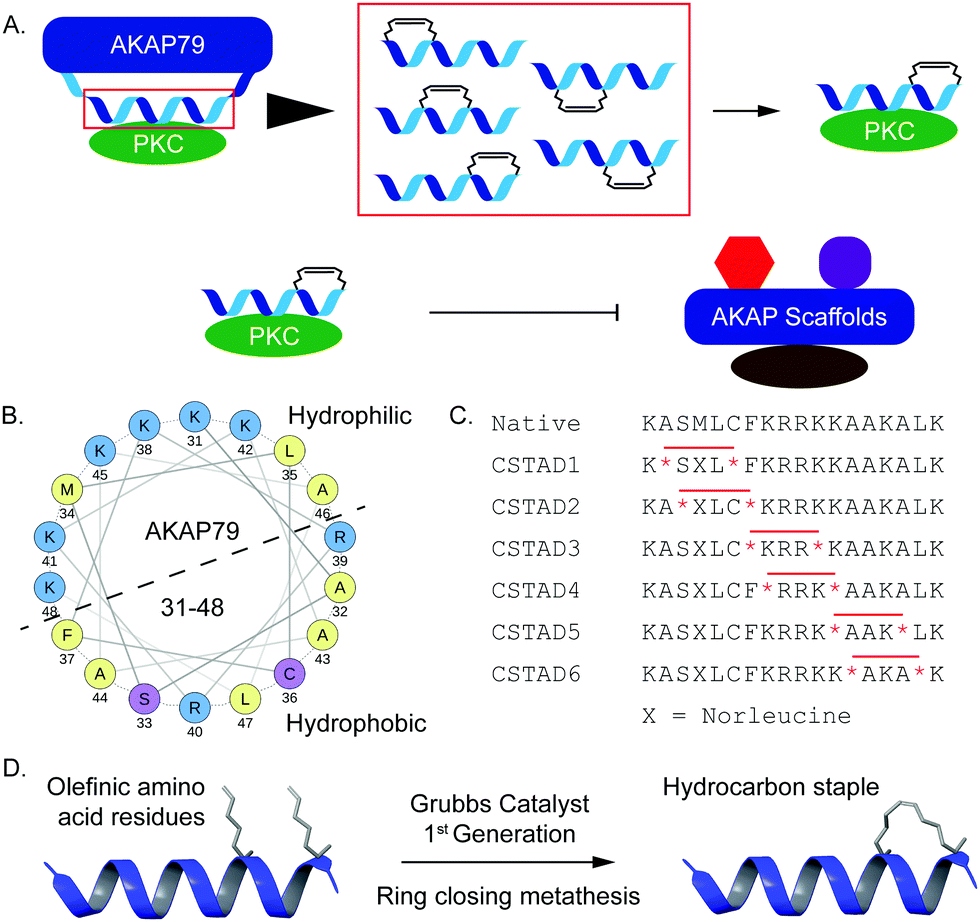 | ||
| Fig. 1 Design and synthesis of all-hydrocarbon stapled peptides to target PKC scaffolding by AKAPs. (A) Schematic representation of the rationale behind the design of all-hydrocarbon stapled peptides derived from AKAP79 and designed to disrupt PKC scaffolding by AKAPs. (B) Helical wheel projection of the PKC binding site present on AKAP79 (residues 31–48) with basic residues in blue, polar residues in lilac and nonpolar residues in yellow. Helical wheel was generated using NetWheels. (C) The native AKAP79 PKC binding site sequence was used to design a library of stapled peptides where olefinic amino acids (*) were placed in various positions to place the staple on the non-binding peptide interface. The hydrocarbon staple is represented by a red line drawn across two olefinic amino acids (*). CSTAD stands for PKC-Stapled Anchoring Disruptor. (D) Ring closing metathesis of olefinic amino acid residues placed at i, i + 4 positions was performed using Grubb's 1st generation catalyst to result in a hydrocarbon staple across individual helical turns. | ||
To first screen which peptides could disrupt PKC–AKAP docking, we used a cell morphology assay that was previously described.30 It was shown that phorbol ester-induced remodeling of actin cytoskeleton and cell rounding in epithelial prostate cancer cells is dependent on a direct scaffolding interaction between PKC and the AKAP gravin. We used prostate derived PC-3 cells for cell-based experiments due to their native high expression of gravin and tested for Phorbol 12-myristate 13-acetate (PMA)-induced cytoskeletal remodeling and cell rounding as a function of PKC scaffolding by gravin in the presence and absence of our peptides. Following an initial screen of PKC-stapled anchoring disruptors (CSTADs), CSTAD5 and CSTAD6 were chosen as the lead candidates due to their ability to downregulate cytoskeletal remodeling in PC-3 cells (Fig. S1, ESI†).
Next, the two lead compounds were evaluated for cellular uptake and target binding. Cells were treated with 5 μM of 5(6)-carboxyfluorescein (FAM)-labeled CSTAD5 or CSTAD6 for four hours prior to imaging. Both CSTAD5 and CSTAD6 showed considerable permeation at the four hours time point with diffuse distribution throughout the cytoplasm as well as some degree of nuclear localization (Fig. 2A and Fig. S2, ESI†). Notably, the non-modified native sequence demonstrated poor cell permeability and was thus not carried forward for cell-based experiments (Fig. S2–S4, ESI†). We then analyzed whether the lead CSTAD peptides could bind PKC using pulldown assays. Cell lysates were treated with 5 μM biotin-labeled CSTAD5 and CSTAD6 peptides along with their corresponding scrambled controls for four hours. Pulldowns were then performed using streptavidin–agarose beads followed by immunoblotting with a pan-PKC antibody (Fig. 2B). Whole cell lysates were used as input controls and lysate treated with vehicle (DMSO) was used as a negative control. Both lead peptides were able to successfully pull-down PKC while their scrambled controls did not (Fig. 2C). Elutes resulting from streptavidin–biotin pulldown assays, using biotin labelled CSTAD5 and CSTAD6 along with their scrambled controls, were also analyzed using mass spectroscopy. This analysis showed that PKC was present in CSTAD5 and CSTAD6 pulldowns but not in their scrambled controls (Table S1, ESI†).
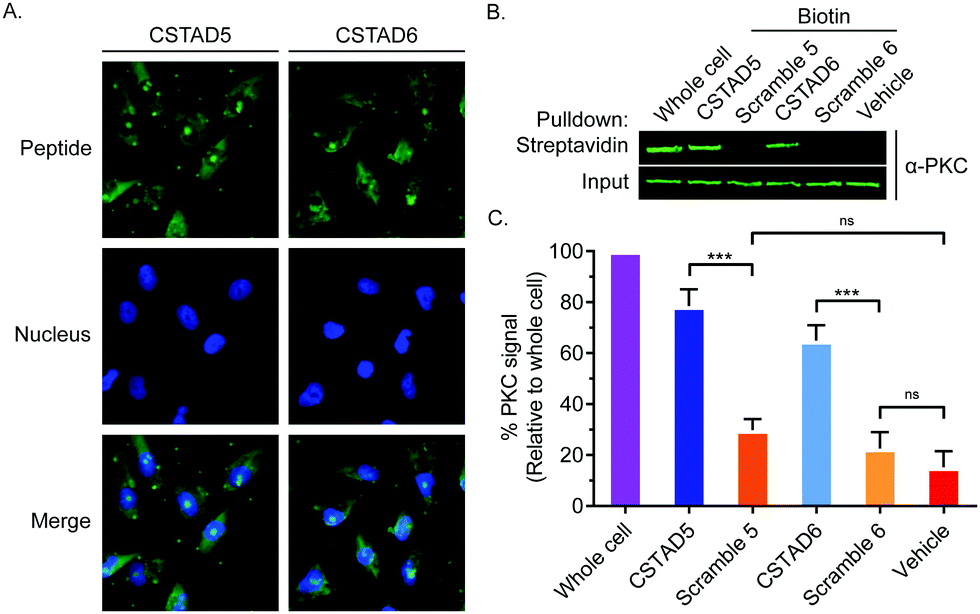 | ||
| Fig. 2 CSTAD5 and CSTAD6 permeate cells and bind PKC. (A) Fluorescence microscopy of PC-3 cells following treatment with CSTAD5 or CSTAD6 (5 μM) for four hours. Both peptides show diffuse fluorescence throughout the cytoplasm along with detectable puncta in the nucleus. (B) Representative western blot (n = 6) demonstrating that the CSTAD peptides, but not their scrambled controls, bind their intended target of PKC. PC-3 cell lysates were treated with biotin-labeled peptides for four hours. Pulldowns were performed using streptavidin–agarose resin. PKC was detected by immunoblotting. (C) Densitometric quantification of six independent streptavidin–biotin pulldown assays using LI-COR Image Studio shows that CSTAD5 and CSTAD6 bound to PKC but their scramble controls did not. *** p < 0.01; ns, not significant as assessed by one-way ANOVA and Bonferroni's multiple comparisons test. Error bars represent standard deviation. | ||
After confirming cellular uptake and PKC binding, we next investigated whether these candidate peptides could successfully disrupt PKC binding to an AKAP scaffold. To evaluate this, we first performed pulldown assays using biotin labeled CSTAD5 and CSTAD6 peptides and immunoblotted for PKC and the AKAP gravin. Although PKC was present in both CSTAD5- and CSTAD6-treated pulldowns, no detectable levels of gravin were observed (Fig. 3A). Next, we performed the reciprocal experiment where gravin was immunoprecipitated from cell lysates that were pretreated with either of the lead peptides, their respective scrambled controls or vehicle (DMSO) and immunoblotted for PKC. PKC levels observed with CSTAD5 and CSTAD6 treatment were nearly undetectable as compared to the scrambled controls or vehicle thus demonstrating the successful disruption of binding between gravin and PKC (Fig. 3B and Fig. S5, ESI†). We then utilized a previously established PMA-induced cytoskeletal remodeling assay which showed that a direct scaffolding interaction between gravin and PKC was required for PMA-induced remodeling of the actin cytoskeleton and cell morphology to assess cell rounding.30 PC-3 cells were serum starved for 24 hours, followed by treatment with 5 μM of CSTAD5 or CSTAD6 for four hours. Cells were then stimulated with 500 nM PMA for 45 minutes followed by imaging to analyze altered morphology as defined by cell rounding (Fig. 3C and Fig. S6, ESI†). Cells pre-treated with CSTAD5 or CSTAD6 predominantly maintained a flat, elongated morphology even after PMA stimulation, whereas cells pre-treated with scrambled controls or vehicle displayed radically altered morphology with collapse of flat cytoplasmic surfaces towards perinuclear regions (Fig. S7, ESI†). The average percentage of rounded cells following PMA stimulation in CSTAD5 and CSTAD6 treated cells was ∼30% and ∼27%, respectively, as compared to over 60% rounding for vehicle treated cells (Fig. 3D). Although these data do not directly measure the disruption of the scaffolding interaction, they support the hypothesis that CSTAD5 and CSTAD6 disrupt PKC scaffolding by gravin.
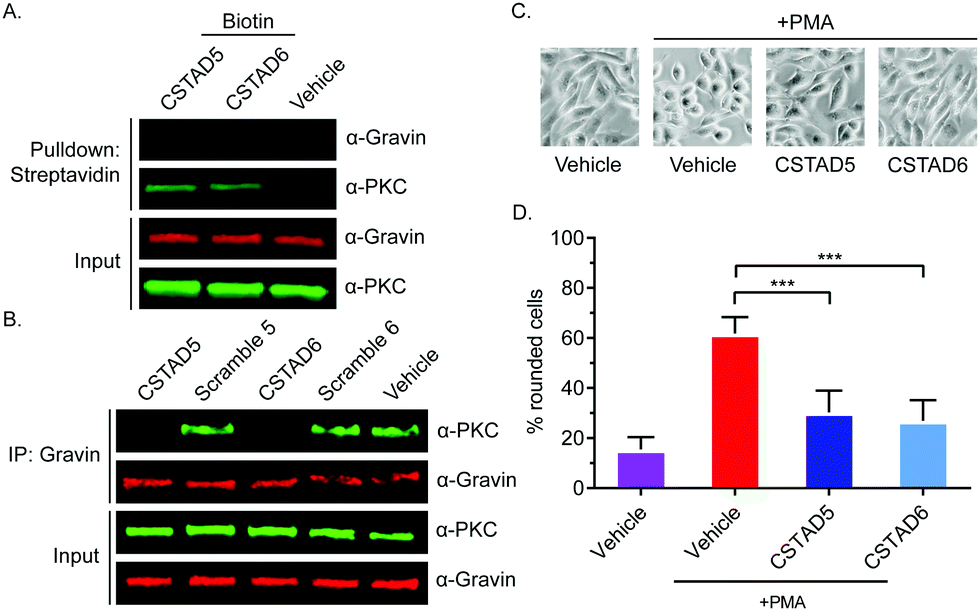 | ||
| Fig. 3 CSTAD5 and CSTAD6 disrupt the interaction between PKC and gravin and downregulate PMA-induced cytoskeletal remodeling in PC-3 cells. (A) Representative western blot showing disruption of the PKC-gravin interaction by CSTAD5 and CSTAD6 peptides (n = 3). A549 cell lysates were treated with biotin-labeled peptides for four hours. Pulldowns were performed using streptavidin–agarose resin. Gravin and PKC were probed by immunoblotting. CSTAD5 and CSTAD6 bound to PKC, but gravin was not detected in complex with PKC. (B) To probe complex formation, gravin was immunoprecipitated and PKC was probed by western blotting using A549 cell lysates (n = 4). In the presence of CSTAD5 or CSTAD6, PKC levels were significantly reduced. The gravin–PKC complex was not affected by vehicle or scrambled control treatments. (C) Representative phase contrast images showing morphology of PC-3 cells treated with 5 μM CSTAD peptides or vehicle for four hours and stimulated with 500 nM PMA for 45 minutes. CSTAD5 and CSTAD6 downregulated PMA-induced cell rounding. (D) Quantification of PMA induced cytoskeletal remodeling in PC-3 cells treated with CSTAD5 or CSTAD6 peptides versus vehicle (n = 4). Both peptides significantly inhibited rounding as compared to vehicle-treated cells. *** p < 0.01 by one-way ANOVA and Bonferroni's multiple comparisons test. Error bars represent standard deviation. | ||
Additionally, we tested the effect of CSTAD5 and CSTAD6 peptides on PKC substrate phosphorylation since its localization by AKAPs is known to influence substrate access. Serum starved PC-3 cells were treated with increasing concentrations of CSTAD5 and CSTAD6, sotrastaurin (selective catalytic inhibitor of PKC) or vehicle (DMSO) for four hours. Cells were then stimulated with PMA and harvested. Western blots probed with an anti-PKC p-Serine substrate antibody revealed a dose dependent reduction in phosphorylated PKC substrates in cells treated with either CSTAD5 or CSTAD6. Intriguingly, cells treated with the highest concentrations (5 μM) of CSTAD5 and CSTAD6 showed lower levels of PKC p-Serine substrates as compared to the serum starved, untreated unstimulated control (Fig. 4A).
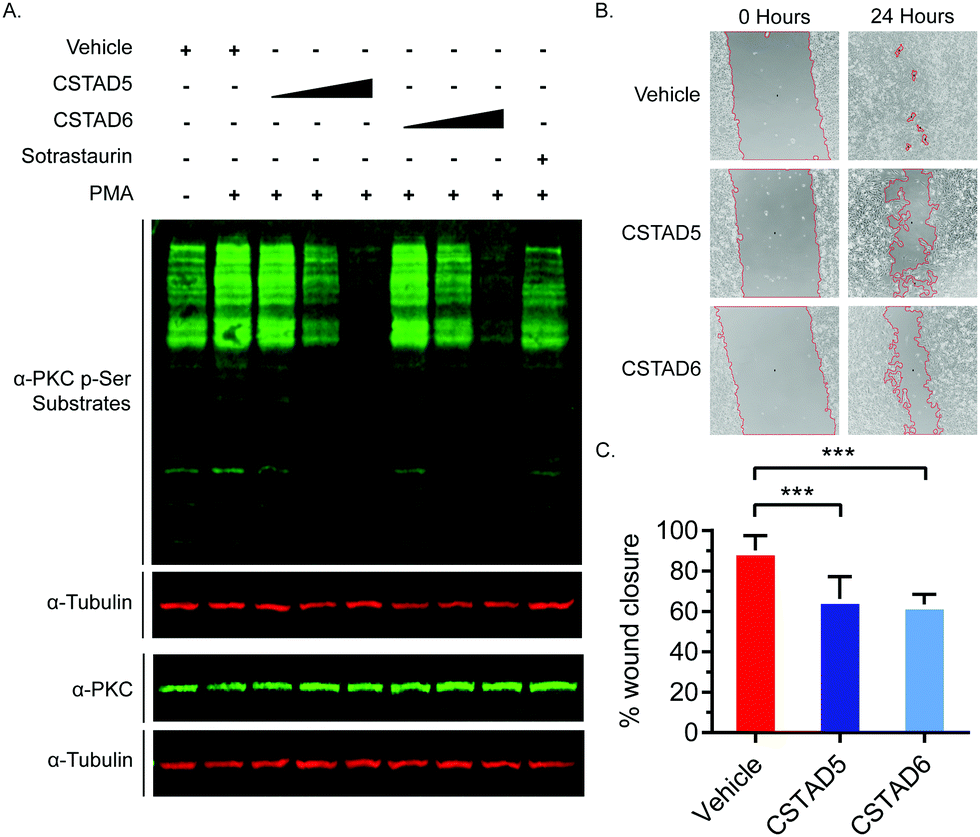 | ||
| Fig. 4 CSTAD5 and CSTAD6 downregulate PKC substrate phosphorylation and cell migration. (A) Representative western blot from serum starved PC-3 cells treated with CSTAD5, CSTAD6 (either 1, 2.5, 5 μM, four hours) or vehicle (DMSO) along with a PKC specific catalytic inhibitor (sotrastaurin, 500 nM, 90 minutes) followed by PMA stimulation (500 nM) for five minutes (n = 3). Both peptides caused a significant reduction in PKC signaling at 5 μM treatments. (B) Wound healing assays were performed using PC-3 cells treated with 5 μM CSTAD5, CSTAD6 or vehicle (DMSO) for 24 hours. The wound area is marked by red boundary lines generated using MRI Wound Healing Tool for ImageJ. Representative images (n = 3) show CSTAD5 and CSTAD6 slowed wound healing over this time course. (C) Quantification of PC-3 cell motility as assessed by measuring percentage wound closure from 0 to 24 hours across three independent wound healing assays. Wound area was calculated using MRI Wound Healing Tool for ImageJ. *** p < 0.01 by one-way ANOVA and Bonferroni's multiple comparisons test. Error bars represent standard deviation. | ||
Since gravin has been implicated in controlling cell migration and motility through its ability regulate PKC and PKA signaling,31–33 we assessed whether the lead CSTAD peptides would have an effect on cell motility. To measure this, wound healing assays were performed in the presence of 5 μM CSTAD5, CSTAD6 or vehicle (DMSO) in media supplemented with 5% fetal bovine serum. After 24 hours, both CSTAD5 and CSTAD6 showed a statistically significant reduction in wound healing with only approximately 62% closure in the presence of either peptide as compared to vehicle-treated cells which achieved over 90% wound healing (Fig. 4B and C).
In conclusion, here we describe the development of AKAP-derived stapled peptides that were designed to disrupt PKC scaffolding by AKAPs. Two peptides, CSTAD5 and CSTAD6, could permeate cells and bind PKC to disrupt the PKC-gravin complex. Notably, both peptides were able to downregulate cytoskeletal remodeling mediated by AKAP-scaffolded PKC as measured by cell rounding and could also downregulate PKC-mediated signaling and cell motility. To the best of our knowledge, no PKC-selective AKAP disruptors have previously been reported and thus this work establishes CSTAD5 and CSTAD6 as the first disruptors of PKC scaffolding by AKAPs.
Indeed, such an approach has been applied to develop disruptors for other PPIs involving PKC. Most notable are the peptide disruptors aimed at a scaffolding interaction between PKC and Receptor for Activated C Kinase (RACK).34,35 With several AKAPs being identified as scaffolding partners that regulate the spatiotemporal dynamics of PKC, these AKAP–PKC scaffolds provide targets for the development of new disruptors that can aid in elucidating the intricacies of localized PKC signaling.
Multiple iterations of peptide disruptors for AKAP-PKA complexes have been developed and reported, however, no such disruptors have been designed to target AKAP–PKC complexes.36–40 The overall structural and functional conservation in the AKB domain across the AKAP family provides for a relatively streamlined development of peptide disruptors to target PKA anchoring by AKAPs.38,40–42 Conversely, no such conserved binding interface has been identified for AKAP–PKC binding and selectivity of certain AKAPs for some PKC isoforms has further confounded the complexity of these interactions. Taken together, these results provide tantalizing hints at a complex role for scaffolding and subcellular localization in determining the PKC signaling output. It also demonstrates that extraneous disruption of spatiotemporal regulation of PKC can directly result in alteration of various cellular functions. This work provides two original stapled peptides that can disrupt AKAP scaffolding of PKC, thereby providing a powerful new tool for further exploration of the intricacies of intracellular PKC activity. Combining these peptides with previously established PKA anchoring disruptors can allow for systematic dissection of AKAP signalosomes to gain a deeper understanding of how they orchestrate specific cellular activities.
Author contributions
The project was conceptualized by EJK. Peptides were designed, synthesized and purified by AJL. All experiments were performed by AJL and GNB. Data analysis was performed by all authors. The original draft of the manuscript was written by AJL and all authors reviewed and edited the final draft of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (GM134168 to EJK).Notes and references
- A. C. Newton, C. E. Antal and S. F. Steinberg, Clin. Sci., 2016, 130, 1499–1510 CrossRef CAS PubMed.
- I. Merida, J. Arranz-Nicolas, C. Rodriguez-Rodriguez and A. Avila-Flores, Biochem. J., 2019, 476, 1205–1219 CrossRef CAS PubMed.
- N. Isakov, Semin. Cancer Biol., 2018, 48, 36–52 CrossRef CAS PubMed.
- A. Mukherjee, S. Roy, B. Saha and D. Mukherjee, Front. Immunol., 2016, 7, 45 Search PubMed.
- A. Tarafdar and A. M. Michie, Biochem. Soc. Trans., 2014, 42, 1556–1562 CrossRef CAS PubMed.
- C. E. Antal and A. C. Newton, Biochem. Soc. Trans., 2014, 42, 1477–1483 CrossRef CAS PubMed.
- K. Mackay and D. Mochly-Rosen, J. Mol. Cell. Cardiol., 2001, 33, 1301–1307 CrossRef CAS PubMed.
- M. A. Lanuza, M. M. Santafe, N. Garcia, N. Besalduch, M. Tomas, T. Obis, M. Priego, P. G. Nelson and J. Tomas, J. Anat., 2014, 224, 61–73 CrossRef CAS PubMed.
- P. P. Burgers, M. A. van der Heyden, B. Kok, A. J. Heck and A. Scholten, Biochemistry, 2015, 54, 11–21 CrossRef CAS PubMed.
- E. J. Kennedy and J. D. Scott, Methods Mol. Biol., 2015, 1294, 137–150 CrossRef CAS PubMed.
- L. G. Helton, A. J. Limaye, G. N. Bendzunas and E. J. Kennedy, in Next Generation Kinase Inhibitors: Moving Beyond the ATP Binding/Catalytic Sites, ed. P. Shapiro, Springer International Publishing, Cham, 2020, pp. 141–167 DOI:10.1007/978-3-030-48283-1_7.
- A. J. L. Leah, G. Helton, G. N. Bendzunas and E. J. Kennedy, in Next Generation Kinase Inhibitors, ed. P. Shapiro, Springer, Cham, 2020, ch. 7, pp. 141–167 DOI:10.1007/978-3-030-48283-1_7.
- A. J. Limaye, L. G. Helton, G. N. Bendzunas and E. J. Kennedy, in Next Generation Kinase Inhibitors, ed. P. Shapiro, Springer, Cham, 2020, ch. 7, pp. 141–167 DOI:10.1007/978-3-030-48283-1_7.
- G. Pidoux and K. Tasken, J. Mol. Endocrinol., 2010, 44, 271–284 CAS.
- W. Wong and J. D. Scott, Nat. Rev. Mol. Cell Biol., 2004, 5, 959–970 CrossRef CAS PubMed.
- T. M. Klauck, M. C. Faux, K. Labudda, L. K. Langeberg, S. Jaken and J. D. Scott, Science, 1996, 271, 1589–1592 CrossRef CAS PubMed.
- J. B. Nauert, T. M. Klauck, L. K. Langeberg and J. D. Scott, Curr. Biol., 1997, 7, 52–62 CrossRef CAS PubMed.
- M. Takahashi, H. Mukai, K. Oishi, T. Isagawa and Y. Ono, J. Biol. Chem., 2000, 275, 34592–34596 CrossRef CAS PubMed.
- T. Ng, M. Parsons, W. E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P. J. Verveer, P. I. Bastiaens and P. J. Parker, EMBO J., 2001, 20, 2723–2741 CrossRef CAS PubMed.
- G. K. Carnegie, F. D. Smith, G. McConnachie, L. K. Langeberg and J. D. Scott, Mol. Cell, 2004, 15, 889–899 CrossRef CAS PubMed.
- T. Kuntziger, M. Rogne, R. L. Folstad and P. Collas, Biochemistry, 2006, 45, 5868–5877 CrossRef PubMed.
- A. Bengrine, J. Li and M. S. Awayda, FASEB J., 2007, 21, 1189–1201 CrossRef CAS PubMed.
- D. Chen, A. Purohit, E. Halilovic, S. J. Doxsey and A. C. Newton, J. Biol. Chem., 2004, 279, 4829–4839 CrossRef CAS PubMed.
- E. C. Dempsey, A. C. Newton, D. Mochly-Rosen, A. P. Fields, M. E. Reyland, P. A. Insel and R. O. Messing, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 279, L429–438 CrossRef CAS PubMed.
- G. A. Perkins, L. Wang, L. J. Huang, K. Humphries, V. J. Yao, M. Martone, T. J. Deerinck, D. M. Barraclough, J. D. Violin, D. Smith, A. Newton, J. D. Scott, S. S. Taylor and M. H. Ellisman, BMC Neurosci., 2001, 2, 17 CrossRef CAS PubMed.
- N. Hoshi, L. K. Langeberg, C. M. Gould, A. C. Newton and J. D. Scott, Mol. Cell, 2010, 37, 541–550 CrossRef CAS PubMed.
- M. C. Faux, E. N. Rollins, A. S. Edwards, L. K. Langeberg, A. C. Newton and J. D. Scott, Biochem. J., 1999, 343(2), 443–452 CrossRef CAS.
- N. Patel, F. Stengel, R. Aebersold and M. G. Gold, Nat. Commun., 2017, 8, 1681 CrossRef PubMed.
- P. J. Nygren, S. Mehta, D. K. Schweppe, L. K. Langeberg, J. L. Whiting, C. R. Weisbrod, J. E. Bruce, J. Zhang, D. Veesler and J. D. Scott, eLife, 2017, 6 Search PubMed.
- L. W. Guo, L. Gao, J. Rothschild, B. Su and I. H. Gelman, J. Biol. Chem., 2011, 286, 38356–38366 CrossRef CAS PubMed.
- S. Akakura and I. H. Gelman, J. Signal Transduction, 2012, 2012, 529179 Search PubMed.
- Q. Fan, X. Yin, A. Rababa’h, A. Diaz Diaz, C. S. Wijaya, S. Singh, S. V. Suryavanshi, H. H. Vo, M. Saeed, Y. Zhang and B. K. McConnell, Am. J. Physiol., 2019, 317, H793–H810 Search PubMed.
- I. H. Gelman, E. Tombler and J. Vargas, Jr., Histochem. J., 2000, 32, 13–26 CrossRef CAS PubMed.
- T. Liron, L. E. Chen, H. Khaner, A. Vallentin and D. Mochly-Rosen, J. Mol. Cell. Cardiol., 2007, 42, 835–841 CrossRef CAS PubMed.
- R. Brandman, M. H. Disatnik, E. Churchill and D. Mochly-Rosen, J. Biol. Chem., 2007, 282, 4113–4123 CrossRef CAS PubMed.
- J. Troger, M. C. Moutty, P. Skroblin and E. Klussmann, Br. J. Pharmacol., 2012, 166, 420–433 CrossRef PubMed.
- D. W. Carr, Z. E. Hausken, I. D. Fraser, R. E. Stofko-Hahn and J. D. Scott, J. Biol. Chem., 1992, 267, 13376–13382 CrossRef CAS.
- Y. Wang, T. G. Ho, D. Bertinetti, M. Neddermann, E. Franz, G. C. Mo, L. P. Schendowich, A. Sukhu, R. C. Spelts, J. Zhang, F. W. Herberg and E. J. Kennedy, ACS Chem. Biol., 2014, 9, 635–642 CrossRef CAS PubMed.
- C. R. Carlson, B. Lygren, T. Berge, N. Hoshi, W. Wong, K. Tasken and J. D. Scott, J. Biol. Chem., 2006, 281, 21535–21545 CrossRef CAS PubMed.
- Y. Wang, T. G. Ho, E. Franz, J. S. Hermann, F. D. Smith, H. Hehnly, J. L. Esseltine, L. E. Hanold, M. M. Murph, D. Bertinetti, J. D. Scott, F. W. Herberg and E. J. Kennedy, ACS Chem. Biol., 2015, 10, 1502–1510 CrossRef CAS PubMed.
- K. Autenrieth, N. G. Bendzunas, D. D. Bertinetti, F. W. P. D. Herberg and E. J. P. D. Kennedy, ChemBioChem, 2016, 17, 693–697 CrossRef CAS PubMed.
- N. G. Bendzunas, S. Dorfler, K. Autenrieth, D. Bertinetti, E. M. F. Machal, E. J. Kennedy and F. W. Herberg, Bioorg. Med. Chem., 2018, 26, 1174–1178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00106j |
| This journal is © The Royal Society of Chemistry 2021 |