
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-labeling protein based on the small ultra-red fluorescent protein, smURFP†
John-Hanson
Machado
a,
Richard
Ting
bc,
John Y.
Lin
d and
Erik A.
Rodriguez
 *a
*a
aDepartment of Chemistry, The George Washington University, Washington, DC 20052, USA. E-mail: erik_rodriguez@gwu.edu
bDepartment of Radiology, Molecular Imaging Innovations Institute (MI3), Weill Cornell Medicine, New York, NY 10065, USA
cAntelope Surgical, Biolabs@NYULangone, New York, NY 10014, USA
dTasmanian School of Medicine, University of Tasmania, Hobart, Tasmania 7000, Australia
First published on 29th June 2021
Abstract
Self-labeling proteins have revolutionized super-resolution and sensor imaging. Tags recognize a bioorthogonal substrate for covalent attachment. We show the small Ultra-Red Fluorescent Protein (smURFP) is a self-labeling protein. The substrate is fluorogenic, fluoresces when attached, and quenches fluorescent cargo. The smURFP-tag has novel properties for tool development.
Fluorescent proteins and genetically encoded sensors have revolutionized fluorescence imaging in living organisms but are limited by the endogenous genetic code and available amino acids. Fluorescent proteins are typically less photostable than spectrally similar fluorescent small molecules commonly used for super-resolution microscopy.1–8 The endogenous amino acids allow for chemical modification9,10 but are not specific for an individual protein of interest. Unnatural nucleic acids and amino acids allow for the expansion of the genetic code and incorporation of L-unnatural amino acids tolerated by biological ribosomes, including fluorescent unnatural amino acids for high photostability and single-molecule imaging.11–20 Unnatural amino acid incorporation remains limited by the ribosome for L-enantiomers and the evolution of aminoacyl-tRNA synthetases that recognize unnatural amino acids.
Self-labeling proteins are genetically encoded and fused to proteins of interest to allow for the covalent attachment of a bioorthogonal chemical substrate that is linked to a cargo molecule. Self-labeling proteins go beyond these limitations by covalently attaching bioorthogonal modified substrates that bear cargo molecules of larger size than tolerated by the ribosome, any enantiomer, and limited only by the chemists’ imagination. There are currently three self-labeling proteins known as Halo-, SNAP-, and CLIP-tags. Halo-tag (33 kDa) was evolved from a bacterial haloalkane dehalogenase and covalently attaches an aliphatic hydrocarbon with a terminal chlorine with a cargo molecule.21,22 SNAP-tag (20 kDa) is a mutant of the human O6-alkylguanine-DNA-alkyltransferase, and the substrate molecule is O6-benzylguanine linked to a cargo molecule.23 The CLIP-tag (20 kDa) was also derived from the human O6-alkylguanine-DNA-alkyltransferase, and the substrate is O2-benzylcytosine with a cargo.24 These self-labeling proteins and substrates are commercially available from Promega and New England Biolabs.
The most common use for self-labeling proteins is the attachment of small molecule chemical dyes as the cargo for increased fluorescent intensity, greater photostability, temporal attachment of different color dyes, and fluorescence persists in living and fixed cells.25–28 These tags allow for protein localization beyond the diffraction limit of light, temporal labeling faster than fluorescent protein chromophore maturation, and protein–protein interactions. Biotin,29 halogenated fluorophores for correlative light and electron microscopy,30 and biorthogonal reactive molecules31 can serve as cargo molecules. Halo-tag was used to create sensitive voltage sensors of multiple colours for fluorescence imaging in living cells and entire organisms.32 All three self-labeling protein substrates only serve as recognition molecules for covalent attachment. Fluorescence tracking of a non-fluorescent small-molecule cargo would require the attachment of a second fluorescent dye or genetic attachment to a fluorescent protein.
The small Ultra-Red Fluorescent Protein (smURFP, 32 kDa dimer) was evolved from a light-harvesting phycobilisome protein α-allophycocyanin.7 The evolution involved the manual selection of >106E. coli colonies starting with autocatalytic covalent attachment of phycocyanobilin, changing the substrate to biliverdin IXα (BV), and evolving high expression and stability. During this evolution, smURFP evolved a highly unusual recognition of BV. From crystal structures, most BV binding proteins and fluorescent proteins (iRFPs and IFPs) recognize the carboxylates on BV for binding or covalent attachment.33–38 The crystal structure of a smURFP mutant showed no recognition of the BV carboxylates.39 We showed that methylation of the carboxylates created biliverdin dimethyl ester (BVMe2) and is covalently attached to smURFP in human embryonic kidney (HEK) 293A cells.7 smURFP lacks recognition of the BV carboxylates.
We hypothesized that smURFP could serve as a self-labeling protein and named the smURFP-tag. Biliverdin was modified with a polyethylene glycol (PEG) linker to attach a cargo molecule (Fig. 1). The PEG linker was chosen as a linear molecule to mimic biliverdin dimethyl ester. The BV recognition molecule is a substrate for covalent attachment and is a fluorogenic molecule. Upon covalent attachment of BV to smURFP, BV fluorescence is turned “on” by >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhancement of the quantum yield and emits far-red fluorescence.7 The smURFP-tag is unique in that the recognition molecule allows covalent attachment and far-red fluorescence tracking. To test this hypothesis, we targeted the smURFP-tag to the nucleus to test if the BV substrate can pass through the outer cell membrane and the nuclear envelope (Fig. 1B).
000-fold enhancement of the quantum yield and emits far-red fluorescence.7 The smURFP-tag is unique in that the recognition molecule allows covalent attachment and far-red fluorescence tracking. To test this hypothesis, we targeted the smURFP-tag to the nucleus to test if the BV substrate can pass through the outer cell membrane and the nuclear envelope (Fig. 1B).
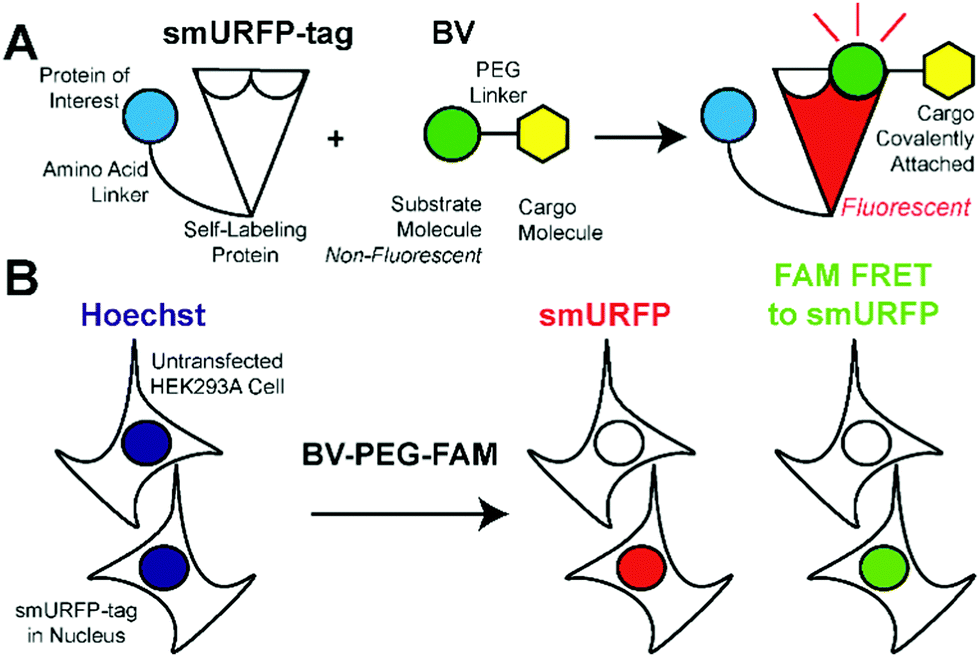 | ||
| Fig. 1 smURFP-tag recognition of biliverdin (BV) modified substrate and experimental design. (A) The self-labeling protein, smURFP-tag, is genetically fused to a protein of interest by an amino acid linker and is non-fluorescent. Modified BV substrate is added to the media and is covalently attached to the smURFP-tag with the cargo molecule and becomes fluorescent in the far-red. (B) Hoechst 3342 labels DNA in nuclei of all cells. The modified BV substrate (BV-PEG-FAM) is a polyethylene glycol (PEG) linker, and the cargo molecule is 5(6)-carboxyfluorescein (FAM). Untransfected HEK293A cells serve as a control and should be non-fluorescent. HEK293A cells are transfected with the smURFP-tag in the nucleus and become fluorescent. | ||
The BV substrate synthesis is relatively simple and uses amide bond formation for stability. For attachment of a single cargo, BV is reacted with H2N-PEG-R2 (Scheme 1). R2, in theory, can be any small molecule and is tracked by far-red fluorescence of BV when covalently attached to smURFP-tag. Steric hindrance limited the formation of BV with two cargo molecules. To attach two cargo molecules, BV was first reacted with H2N-PEG-NH2 in excess, followed by amide bond formation with cargo molecule (R2) to couple two cargo molecules. In this study, R2 is 5(6)-carboxyfluorescein (FAM) to allow for detection by fluorescence imaging to confirm that the cargo is not removed within the cell. The BV substrates are BV-PEG-FAM and BV-(PEG-FAM)2 for the single and double cargo molecules, respectively (Scheme 1).
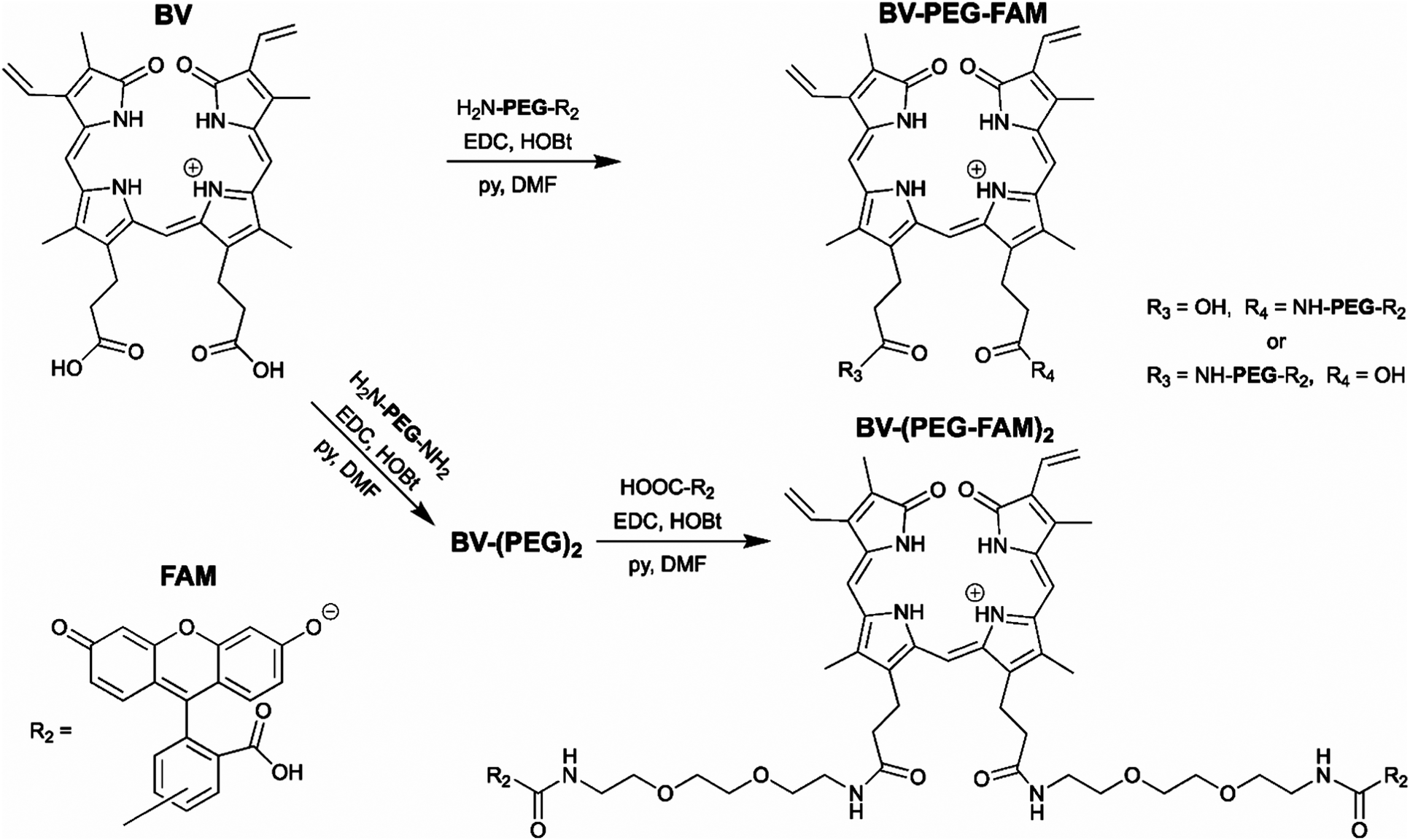 | ||
| Scheme 1 Synthesis of the biliverdin (BV) substrates. A polyethylene glycol (PEG) linker separates the cargo molecule, R2, from the recognition molecule, BV, to allow covalent attachment to the genetically encoded protein, smURFP-tag. R2 is 5(6)-carboxyfluorescein (FAM) in this study. (top) BV-PEG-FAM is synthesized by amide bond coupling of PEG and FAM (ESI† Scheme S1) followed by amide bond attachment to BV. Steric hindrance yields BV-PEG-FAM as the major product. (bottom) To avoid steric hindrance, BV-(PEG-FAM)2 is synthesized by amide coupling of PEG to BV followed by amide bond formation between BV-(PEG)2 and FAM. The synthesis allows for simple and stable synthesis of the BV substrates. | ||
BV-PEG-FAM was synthesized by amide bond formation between 5(6)-carboxyfluorescein (FAM) and 2,2′-(ethylenedioxy)bis(ethylamine) (PEG) to create PEG-FAM (ESI† Scheme S1). Analytical HPLC–MS of PEG-FAM showed the major product at 11 minutes with FAM absorbance at 492 nm, and MS confirmed the molecular weight (ESI† Fig. S1). PEG-FAM was coupled to the BV carboxylates to create BV-PEG-FAM (ESI† Scheme S2). Analytical HPLC–MS of BV-PEG-FAM showed a significant peak at 16 minutes with BV absorbance at 645 and 780 nm and correct mass (ESI† Fig. S2). Proton NMR of starting material, BV, and BV-PEG-FAM confirmed the structure (ESI† Fig. S3). The synthesis of BV-PEG-FAM showed no di-substituted BV-(PEG-FAM)2 due to steric hindrance.
The synthesis of BV-(PEG-FAM)2 used an alternative strategy to avoid steric hindrance (Scheme 1). BV was reacted with PEG to obtain amide bond attachment to both BV carboxylates (ESI† Scheme S3). Analytical HPLC–MS of BV-(PEG)2 confirmed synthesis (ESI† Fig. S4). BV-(PEG)2 was reacted with FAM, and the doubly substitute, BV-(PEG-FAM)2 was purified (ESI† Scheme S4). Analytical HPLC–MS of BV-(PEG-FAM)2 confirmed the molecule at 16 minutes by detecting the doubly charged species by MS (ESI† Fig. S5). Proton NMR of BV-(PEG-FAM)2 confirmed the structure of the molecule (ESI† Fig. S6). Attaching unmodified PEG to BV removed steric hindrance to allow the attachment of two cargo, FAM molecules. The synthesis of the mono- and di-substituted BV-PEG-R2 and BV-(PEG-R2)2 molecules are general strategies and should allow the attachment of any cargo molecules. The amide bond is exceptionally stable, surviving for ∼270 years in water at pH = 7,10 and should survive the cellular environment.
BV substrates were added to purified smURFP-tag without chromophore to demonstrate covalent attachment in vitro. Ten-fold excess of BV, BV-PEG-FAM, and BV-(PEG-FAM)2 were added to smURFP-tag overnight, and the free BV substrates were removed by nickel column purification. Absorbance showed smURFP-tag covalently attached BV by the characteristic Soret and Q band at 385 and 642 nm, respectively (ESI† Fig. S7). BV-PEG-FAM was covalently attached to the smURFP-tag and retained the absorbance of FAM at 492 nm in vitro. smURFP-tag did not covalently attach BV-(PEG-FAM)2in vitro by lack of absorbance from 340–700 nm.
BV substrates were initially added to HEK293A cells (ThermoFisher, R70507) that lacked any genetic modification to visualize background fluorescence. Cells were imaged without BV substrate removal. A substrate concentration of 25 μM for BV, BV-PEG-FAM, and BV-(PEG-FAM)2 was chosen because BV and BV dimethyl ester saturated smURFP-tag at 25 μM. The labeling time was 4 hours because fluorescence rise fit a first-order, exponential rate equation, and BV saturation is expected after ≤1.3 hours.7 Four hours ensures ample time to saturate the smURFP-tag with the substrates. Hoechst 3342 was used to visualize the DNA in the nucleus and showed all the cells within an image (Fig. 2). smURFP, FAM, and FAM Förster Resonance Energy Transfer (FRET) to smURFP fluorescence were imaged. HEK293A cells without transfection or untransfected showed minimal background fluorescence as expected (Fig. 1B). Addition of 25 μM BV or BV-PEG-FAM showed no significant increase in fluorescence signal. The lack of FAM fluorescence from BV-PEG-FAM is due to the quenching by the recognition molecule BV. The FAM fluorescence increases with the di-substituted BV-(PEG-FAM)2, and the single BV cannot quench both FAMs. The lack of significant fluorescence on untransfected cells is desired to only image the smURFP-tag fluorescence.
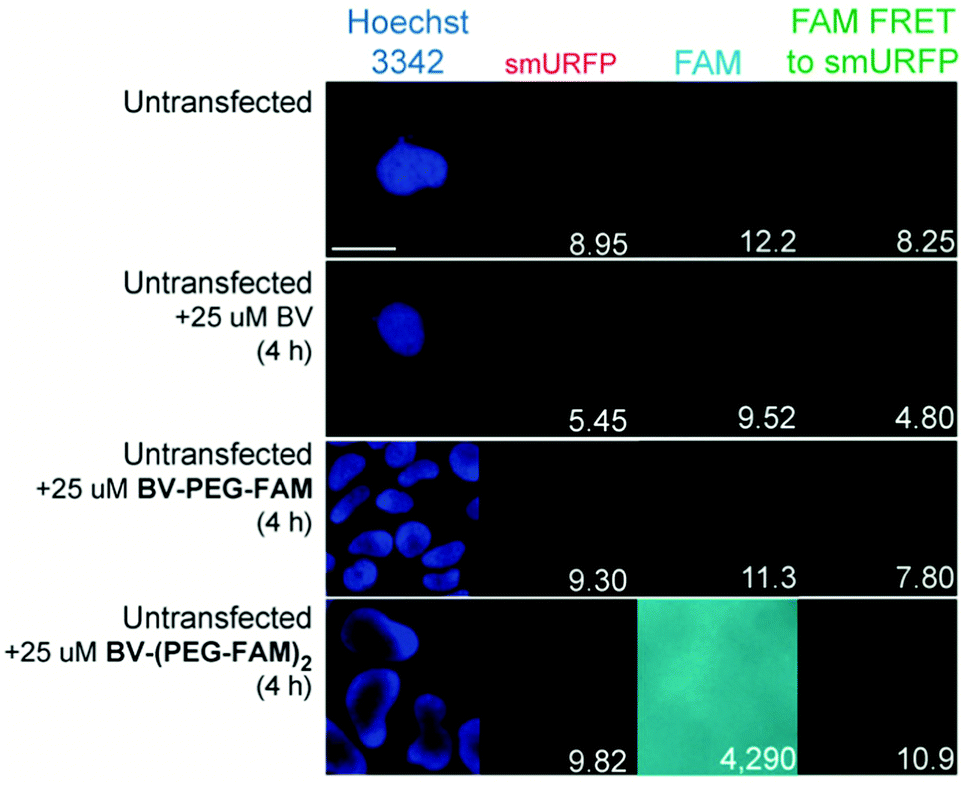 | ||
| Fig. 2 Representative images of BV substrate addition to untransfected cells. BV substrates were added for 4 h, and HEK293A cells were imaged without removal. Lack of fluorescence is expected (Fig. 1B). Scale bar is 10 μm and numbers are mean fluorescence intensity of 40 cells. | ||
HEK293A cells were transiently transfected with smURFP-tag localized within the nucleus by a nuclear localization sequence. We choose this location to test the ability of the BV substrates to pass the outer and nuclear envelope membranes (Fig. 1). Previously, we showed that the negatively charged BV carboxylates limited membrane permeability and utility in cell culture and mice.7 Modifying the carboxylates is predicted to enhance permeability to HEK293A cell membranes to increase the smURFP fluorescence relative to BV. Without BV addition, smURFP-tag is fluorescent due to BV present in the fetal bovine serum (FBS). There is slightly greater fluorescence in the FAM FRET to smURFP channel because BV covalently attached to smURFP-tag is somewhat excited by the blue excitation light (Fig. 3). The addition of BV and BV-PEG-FAM shows a 3.3- and 34-fold, respectively, increase in smURFP fluorescence. FAM fluorescence was slightly increased for BV-PEG-FAM, and FAM fluorescence was seen as FRET to smURFP. BV-(PEG-FAM)2 showed no increase in smURFP fluorescence, and the BV is not covalently attached in vivo, which agrees with the lack of in vitro attachment. The FAM fluorescence increases on the di-substituted BV-(PEG-FAM)2 because BV does not quench both FAMs. This experiment shows that BV-PEG-FAM can pass the cell outer and nuclear membranes to deliver the cargo (FAM) for covalent attachment to the smURFP-tag.
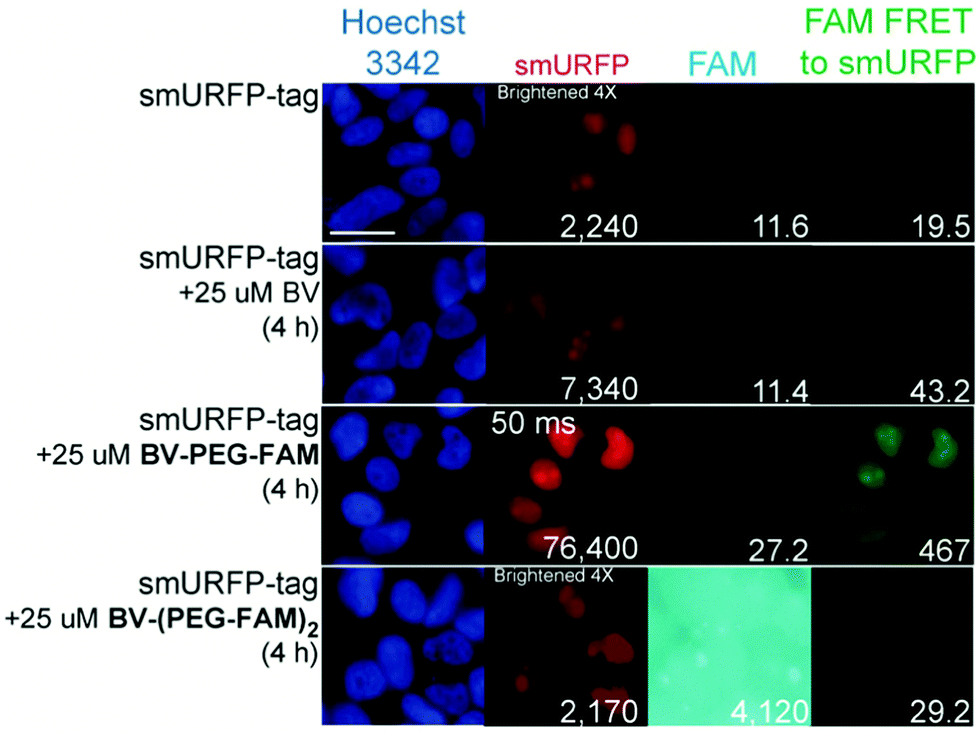 | ||
| Fig. 3 Representative images of BV substrates added to HEK293A cells transfected with nucleus localized smURFP-tag. BV substrates were added for 4 h, and cells were imaged without removal. Scale bar is 10 μm and numbers are mean fluorescence intensity of 40 cells. | ||
A region of interest was drawn on 40 cells for each sample, and the mean fluorescence intensity was calculated. The 40 measurements are plotted with statistical comparison in Fig. 4. Untransfected cells shown minimal smURFP fluorescence and are plotted on a Y-axis log scale to visualize points (Fig. 4A). HEK293A cells with smURFP-tag within the nucleus show a statistically significant increase in fluorescence. smURFP-tag without substrate and with BV-(PEG-FAM)2 show the same amount of fluorescence and confirms that BV-(PEG-FAM)2 is not attached in vivo. The addition of BV and BV-PEG-FAM show a statistically significant increase in fluorescence. BV membrane permeability is limited by the negatively charged carboxylates. BV-PEG-FAM masks a single carboxylate to create a molecule with an overall neutral charge (Scheme 1) that allows the molecule to pass the outer and nuclear membranes to increase the fluorescence of smURFP-tag by 34-fold.
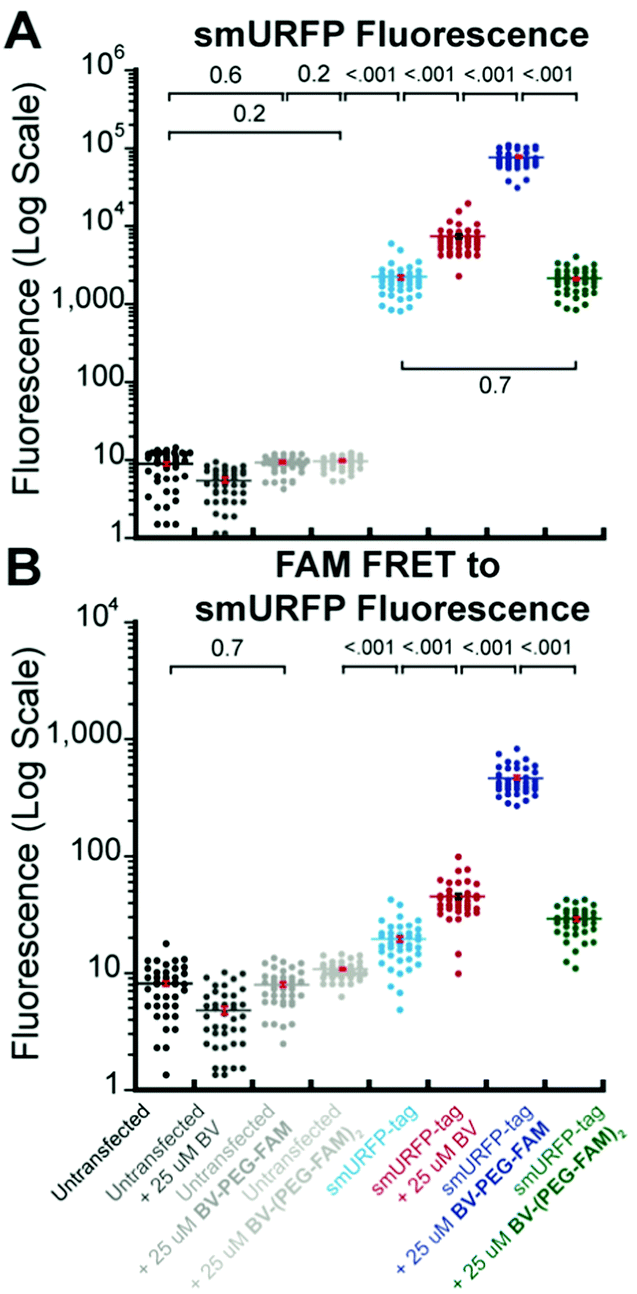 | ||
| Fig. 4 Comparison of data points and mean fluorescence intensity of (A) smURFP and (B) FAM FRET to smURFP from Fig. 2 and 3. The Y-axis is shown on a log scale to visualize untransfected HEK293A cell data points. Mean fluorescence intensity is written in Fig. 2 and 3 and indicated by a horizontal bar. A one-way ANOVA with α = 0.05 and a post hoc Tukey honestly significant difference (HSD) compared samples. All statistically insignificant comparisons are written, and all unwritten comparisons are p < 001. n = 40 cells and error bars are standard error of the mean (SEM). | ||
Unexpectedly, FAM fluorescence was seen almost entirely as FRET to smURFP. The FRET is due to the proximity of FAM to smURFP-tag and the substantial extinction coefficient of 180000 M−1 cm−1 for smURFP. Untransfected cells showed a minimal increase in FAM FRET to smURFP fluorescence, and BV-(PEG-FAM)2 showed the greatest increase due to the lack of quenching of both FAM molecules. smURFP-tag without substrate and BV show slightly increased signal relative to the untransfected cells due to slight excitation of smURFP by the blue light. The largest FAM FRET to smURFP signal is seen with the BV-PEG-FAM and confirms that the cargo molecule is delivered to the smURFP-tag and remains covalently attached inside the nucleus.
Conclusions
The small Ultra-Red Fluorescent Protein (smURFP) is also a self-labeling protein capable of recognizing and covalently attaching carboxylate modified BV substrates. The smURFP-tag is unique compared to Halo-, SNAP-, and CLIP-tags because BV serves as a recognition molecule for covalent attachment, is a fluorogenic dye for fluorescence tracking of covalently attached cargo, and quenches a fluorescent dye for imaging without substrate removal. The smURFP-tag within the nucleus could covalently attach the mono-substituted BV-PEG-FAM, which was cell and nuclear membrane permeant. BV-(PEG-FAM)2 did not covalently attach to the smURFP-tag in vitro and in vivo. Further evolution of the smURFP-tag binding pocket is necessary to tolerate di-substituted BV substrates. In the presence of ten molar equivalents of the chromophore, smURFP-tag covalently attached two biliverdin but could only covalently attach a single biliverdin dimethyl ester by mass spectrometry and biophysical properties.7BV-PEG-FAM increases steric hindrance, and the stoichiometry is expected to be a single BV-PEG-FAM per smURFP-tag, which is desired for increased brightness without diminishing the quantum yield. The unique properties of the smURFP-tag are useful for genetically targeting small molecules to proteins of interest with far-red fluorescence tracking without the need to add a chemical dye or genetically fuse to a fluorescent protein. Live-cell super-resolution microscopy will utilize the smURFP-tag for imaging small-molecule fluorophores without substrate washing due to biliverdin quenching to ensure saturation of newly translated proteins in the presence excess of substrate for the maximum signal with low background fluorescence. The smURFP-tag should find further use for the development of novel biosensors that utilize the smURFP-tag fluorescence with chemical and protein sensors3 that recognize small molecules, peptides, and other biomolecules. smURFP-tag exogenous labelling of proteins with sortase-mediated attachment,40 inclusion into virus particles,41 and smURFP-tag nanoparticles42 will allow for bioorthogonal, site-specific attachment of molecules without misfolding or optimization of reaction conditions for targeted or biosensor imaging in entire living animals.Author contributions
EAR conceptualized the methodology, designed experiments, and analysed fluorescence images. JHM, EAR, and RT synthesized and characterized molecules. All authors performed experiments, wrote the paper, created figures, and reviewed the final manuscript.Conflicts of interest
The described technology is patented (US20180201655A1).8Acknowledgements
This project was funded by startup funds provided by The George Washington University to EAR and US National Institutes of Health grant U01NS090590 to JYL.Notes and references
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS.
- F. Montecinos-Franjola, J. Y. Lin and E. A. Rodriguez, Biochem. Soc. Trans., 2020, 48, 2657–2667 CrossRef CAS.
- G. C. H. Mo, C. Posner, E. A. Rodriguez, T. Sun and J. Zhang, Nat. Commun., 2020, 11, 1848 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- E. A. Rodriguez, G. N. Tran, L. A. Gross, J. L. Crisp, X. Shu, J. Y. Lin and R. Y. Tsien, Nat. Methods, 2016, 13, 763–769 CrossRef CAS PubMed.
- E. A. Rodriguez, J. Y. Lin, R. Ting, G. N. Tran and R. Y. Tsien, US Pat., US20180201655A1, 2019 Search PubMed.
- O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- S. Mahesh, K. C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef PubMed.
- R. Pantoja, E. A. Rodriguez, M. I. Dibas, D. A. Dougherty and H. A. Lester, Biophys. J., 2009, 96, 226–237 CrossRef CAS PubMed.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8650–8655 CrossRef CAS.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, RNA, 2007, 13, 1703–1714 CrossRef CAS.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, RNA, 2007, 13, 1715–1722 CrossRef CAS.
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182–188 CrossRef CAS PubMed.
- D. R. Liu, T. J. Magliery, M. Pastrnak and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10092–10097 CrossRef CAS PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- D. L. Beene, D. A. Dougherty and H. A. Lester, Curr. Opin. Neurobiol., 2003, 13, 264–270 CrossRef CAS PubMed.
- D. D. Young and P. G. Schultz, ACS Chem. Biol., 2018, 13, 854–870 CrossRef CAS PubMed.
- S. Hoshika, N. A. Leal, M. J. Kim, M. S. Kim, N. B. Karalkar, H. J. Kim, A. M. Bates, N. E. Watkins, Jr., H. A. SantaLucia, A. J. Meyer, S. DasGupta, J. A. Piccirilli, A. D. Ellington, J. SantaLucia, Jr., M. M. Georgiadis and S. A. Benner, Science, 2019, 363, 884–887 CrossRef CAS PubMed.
- G. V. Los and K. Wood, Methods Mol. Biol., 2007, 356, 195–208 CAS.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa, Jr., M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- R. S. Erdmann, S. W. Baguley, J. H. Richens, R. F. Wissner, Z. Xi, E. S. Allgeyer, S. Zhong, A. D. Thompson, N. Lowe, R. Butler, J. Bewersdorf, J. E. Rothman, D. St Johnston, A. Schepartz and D. Toomre, Cell Chem. Biol., 2019, 26(584–592), e586 Search PubMed.
- B. Barlag, O. Beutel, D. Janning, F. Czarniak, C. P. Richter, C. Kommnick, V. Goser, R. Kurre, F. Fabiani, M. Erhardt, J. Piehler and M. Hensel, Sci. Rep., 2016, 6, 31601 CrossRef CAS PubMed.
- F. Stagge, G. Y. Mitronova, V. N. Belov, C. A. Wurm and S. Jakobs, PLoS One, 2013, 8, e78745 CrossRef CAS.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS.
- S. Svendsen, C. Zimprich, M. G. McDougall, D. H. Klaubert and G. V. Los, BMC Cell Biol., 2008, 9, 17 CrossRef.
- V. Liss, B. Barlag, M. Nietschke and M. Hensel, Sci. Rep., 2015, 5, 17740 CrossRef CAS.
- H. E. Murrey, J. C. Judkins, C. W. Am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS.
- A. S. Abdelfattah, T. Kawashima, A. Singh, O. Novak, H. Liu, Y. Shuai, Y. C. Huang, L. Campagnola, S. C. Seeman, J. Yu, J. Zheng, J. B. Grimm, R. Patel, J. Friedrich, B. D. Mensh, L. Paninski, J. J. Macklin, G. J. Murphy, K. Podgorski, B. J. Lin, T. W. Chen, G. C. Turner, Z. Liu, M. Koyama, K. Svoboda, M. B. Ahrens, L. D. Lavis and E. R. Schreiter, Science, 2019, 365, 699–704 CrossRef CAS PubMed.
- J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra and K. T. Forest, J. Biol. Chem., 2007, 282, 12298–12309 CrossRef CAS PubMed.
- S. Ghosh, C. L. Yu, D. J. Ferraro, S. Sudha, S. K. Pal, W. F. Schaefer, D. T. Gibson and S. Ramaswamy, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11513–11518 CrossRef CAS PubMed.
- D. Yu, W. C. Gustafson, C. Han, C. Lafaye, M. Noirclerc-Savoye, W. P. Ge, D. A. Thayer, H. Huang, T. B. Kornberg, A. Royant, L. Y. Jan, Y. N. Jan, W. A. Weiss and X. Shu, Nat. Commun., 2014, 5, 3626 CrossRef CAS PubMed.
- L. Lad, J. Friedman, H. Li, B. Bhaskar, P. R. Ortiz de Montellano and T. L. Poulos, Biochemistry, 2004, 43, 3793–3801 CrossRef CAS PubMed.
- O. Cunningham, A. Dunne, P. Sabido, D. Lightner and T. J. Mantle, J. Biol. Chem., 2000, 275, 19009–19017 CrossRef CAS PubMed.
- P. J. Pereira, S. Macedo-Ribeiro, A. Parraga, R. Perez-Luque, O. Cunningham, K. Darcy, T. J. Mantle and M. Coll, Nat. Struct. Biol., 2001, 8, 215–220 CrossRef CAS.
- J. P. Fuenzalida-Werner, R. Janowski, K. Mishra, I. Weidenfeld, D. Niessing, V. Ntziachristos and A. C. Stiel, J. Struct. Biol., 2018, 204, 519–522 CrossRef CAS PubMed.
- I. Deshpande, J. Liang, D. Hedeen, K. J. Roberts, Y. Zhang, B. Ha, N. R. Latorraca, B. Faust, R. O. Dror, P. A. Beachy, B. R. Myers and A. Manglik, Nature, 2019, 571, 284–288 CrossRef CAS.
- F. C. Herbert, O. R. Brohlin, T. Galbraith, C. Benjamin, C. A. Reyes, M. A. Luzuriaga, A. Shahrivarkevishahi and J. J. Gassensmith, Bioconjugate Chem., 2020, 31, 1529–1536 CrossRef CAS.
- F. An, N. Chen, W. J. Conlon, J. S. Hachey, J. Xin, O. Aras, E. A. Rodriguez and R. Ting, Int. J. Biol. Macromol., 2020, 153, 100–106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00127b |
| This journal is © The Royal Society of Chemistry 2021 |