
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu(II)/BODIPY core co-encapsulated ratiometric nanotools for intracellular O2 sensing in live cancer cells†
Karmel Sofia
Gkika
a,
Anna
Kargaard
b,
Christopher S.
Burke
ab,
Ciaran
Dolan
a,
Andreas
Heise
 bcd and
Tia E.
Keyes
*a
bcd and
Tia E.
Keyes
*a
aSchool of Chemical Sciences, National Centre for Sensor Research, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: tia.keyes@dcu.ie
bDepartment of Chemistry, RCSI, Dublin, Ireland
cCÚRAM, SFI Research Centre for Medical Devices, RCSI, Dublin D02, Ireland
dAMBER, The SFI Advanced Materials and Bioengineering Research Centre, RCSI, Dublin D02, Ireland
First published on 28th July 2021
Abstract
Oxygen is a crucial reagent in many biochemical processes within living cells and its concentration can be an effective marker in disease, particularly in cancer where tissue hypoxia has been shown to indicate tumour growth. Probes that can reflect the oxygen concentration and distribution using ratiometric signals can be applied to a range of conventional methods without the need for specialised equipment and are particularly useful. The preparation and in cellulo study of luminescent ratiometric core–shell nanoparticles are presented. Here, a new lipophilic and oxygen-responsive Ru(II) tris-heteroleptic polypyridyl complex is co-encapsulated with a reference BODIPY dye into the core of poly-L-lysine-coated polystyrene particles. The co-core encapsulation ensures oxygen response but reduces the impact of the environment on both probes. Single wavelength excitation of the particles, suspended in aqueous buffer, at 480 nm, triggers well-resolved dual emission from both dyes with peak maxima at 515 nm and 618 nm. A robust ratiometric oxygen response is observed from water, with a linear dynamic range of 3.6–262 μM which matches well with typical biological ranges. The uptake of RuBDP NPs was found to be cell-line dependent, but in cancerous cell lines, the particles were strongly permeable with late endosomal and partial lysosomal co-staining observed within 3 to 4 hours, eventually leading to extensive staining of the cytoplasm. The co-localisation of the ruthenium and BODIPY emission confirms that the particles remain intact in cellulo with no indication of dye leaching. The ratiometric O2 sensing response of the particles in cellulo was demonstrated using a plate-based assay and by confocal xyλ scanning of cells exposed to hypoxic conditions.
Introduction
Molecular oxygen (O2) plays a central role in the biochemistry of mammalian cells including in oxidative phosphorylation, production of reactive oxygen species and hypoxia response.1–3 Hypoxia, a reduction in the tissue oxygen concentration below normal levels, is associated with a number of disease states including tissue injury and metastasis in cancer. It has also been identified as a marker of radiotherapeutic resistance in cancer disease.4 Indeed, recently, the Nobel Prize in Physiology and Medicine was awarded for the discoveries made on the molecular response and adaptation of cells to oxygen availability.5 Reliable and quantitative oxygen sensors with real-time responsivity that can be deployed within the cellular environment are of significant biomedical value, including for prognosis and therapy.6–8 Although there are a number of examples of very effective oxygen sensing luminescent probes reported for in-cell sensing, they primarily focus on phosphorescence lifetime imaging which is a rather specialist technique,9–11 whereas for wide applications, probes that are amenable to instrumentation such as fluorescence microscopy and plate reader assays that are widely available within biology laboratories are ideally required. Such applications require that sensing and mapping of intracellular O2 be based on intensity rather than lifetime measurements.12,13Luminescent metal complexes are attractive probes for sensing of O2 in live cells due to the following characteristics: (1) facile photophysical tuning by metal centers, i.e., Ru(II) or Ir(III) or ligand substitution,14–18 (2) red emission that coincides well with the photobiological optical window (650–950 nm) and avoids interfering autofluorescence from biological media, (3) good quantum yield, (4) long-lived, triplet-based excited states (i.e. hundreds of ns to ∼3 μs) that provide oxygen sensitivity and can be exploited in time-gating to eliminate background,19 and (5) the ability to incorporate ligand functionalities for post-modification such as the attachment of targeting vectors.20–30 The latter has been instrumental in overcoming a key limitation in the application of metal complex oxygen probes in cellular imaging. Metal complexes, because of their mass, charge and relative hydrophilicity, are typically not cell membrane permeable, but coupling to cargo-carrying moieties can promote permeation, enabling their application in the monitoring of oxygen levels even at the level of specific cellular organelles as reported in the case of the nuclear-targeting Ru(II) bis-bpy (bpy = 2,2′-bipyridine) complex.31
A significant drawback of intensity-based measurements for sensing is that even for probes with excellent responsivity, selectivity and large linear range, the intensity of emission is influenced by many parameters beyond the target analyte. These include technical issues around the stability of the excitation source, detector drift, stray light, and physiochemical issues such as photodamage, leaching of probes or interactions with other species within the cellular environment, for example, proteins or membranes. Also, if a probe is inhomogeneously distributed within the sensing environment, this will affect the intensity which is a key issue in cells.
Ratiometric sensing where a probe signal is referenced to a stable emission from a species that does not respond to the environment, but will be equally subject to fluctuations in the light source intensity or detector sensitivity etc., is a useful solution to this issue.32,33 Such ratiometric responses to O2 have been demonstrated both in molecular probes34–39 and in particle-based sensors.40–49
In practice, in ratiometric sensing, it is important to ensure that both the probe and reference signal can be generated with a single excitation source. One approach taken has been exciting the reference indirectly through energy transfer from the O2 indicator which is directly excited. Usually achieved by Förster resonance energy transfer (FRET), the extent of energy transfer is itself influenced by quenching, so the ratiometric signal is modulated by molecular oxygen.50 Such FRET-based ratiometric probes can be challenging to build, as it can be difficult to ensure that competing photophysical or photochemical processes that can be influenced by the environment, such as photoinduced electron transfer quenching, do not occur.51 Although this FRET-based approach has primarily been demonstrated in molecular species, it has also been demonstrated in conjugated polymer nanoparticles.32,52
An alternative, and a more direct approach, is to build structures in which both signal and reference luminophores are excited at the same wavelength, but emit at different and distinguishable wavelengths. This is a challenge for purely organic fluorophores but is feasible where a probe or reference is Stokes shifted; thus, metal complex luminophores are attractive for use in such systems. We recently demonstrated this approach in a ruthenium–BODIPY probe–reference dyad used as a ratiometric probe for oxygen sensing in non-aqueous media. Negligible electronic cross-talk between the photoactive moieties is an essential criterion for this approach.53
While molecular species have been widely explored in ratiometric sensing, there are particular advantages to encapsulating sensing species in particles, particularly for sensing within heterogeneous environments, such as in living cells. In relation to gaseous species such as oxygen, particles can isolate the sensor and reference from any confounding environmental impact the cellular interior may have on the photophysics of the probe and reference. Particles also can be very stable, may promote uptake through endocytosis and can, depending on materials, show low cytotoxicity relative to molecular species.54 Ratiometric particle-based approaches frequently involve the incorporation of both the O2 sensitive component and the reference probe in a single system for built-in correction of the O2 response.49,55–61 Examples include ratiometric dual-wavelength emission at 800 nm and 670 nm reported for the single excitation of polystyrene nanoparticles doped with Pd meso-tetraphenylporphyrin and the resulting DY-635 reference dye.58 Kopelman and co-workers reported ratiometric nanosensors by incorporating an O2-insensitive reference dye such as Alexa 647 with the O2-sensitive and NIR emitting Pd-tetra-(4-carboxyphenyl)tetrabenzoporphyrin dendrimer.62 Papkovsky et al. incorporated a phosphorescent O2 sensor dye, PtTFPP, and poly(9,9-dioctylfluorene) (PFO) reference dye also acting as a FRET donor in a single nanoparticle system for multimodal O2 sensing.60
However, there are relatively few examples of ratiometric nanoparticle sensors that have been applied to produce quantitative intensity-based ratiometric signals for oxygen sensitivity in live cells and none to our knowledge used direct co-excitation of the probe and reference rather than FRET.
We recently exploited the ratiometric particle-based approach using a core–shell particle design, where the Ru(II) component was conjugated to the poly-lysine shell as the oxygen sensor and the BODIPY reference was spatially isolated from the polystyrene particle core.63 These polymer-based particles showed excellent photostability and good ratiometric response to oxygen in aqueous media. However, without pre-treatment with a cationic surfactant, the particles were impermeable to the cell membrane. Furthermore, with the metal complex luminophore located at the exterior of the complex, it is difficult to distinguish the environmental impact, on the complex photophysics, e.g., from membranes and proteins, from that of oxygen.
Conversely, nanoparticles, polymer-based carriers, and liposomes have been used to facilitate the uptake and accumulation of ruthenium probes within cells, particularly for photodynamic therapy applications.64–67 The incorporation of ruthenium complexes into nanoparticles can also improve their photophysical properties by increasing their luminescence quantum yield for example.68,69
Here, using a simplified approach, we describe the fabrication of a core–shell ratiometric sensor in which both the oxygen sensor and reference dye are encapsulated within the particle core, and demonstrate that by isolating the sensor complex to the particle core, the poly-L-lysine shell promotes efficient live cell uptake of the nanoparticles. This approach eliminates the need for a membrane permeabilizing reagent and simultaneously permits the protection of the probe as well as the reference, resulting in a self-referenced oxygen-responsive signal that can be observed within live cells.
We demonstrate for the first time, using confocal microscopy and lambda (λ) scanning, the emission spectra ratiometric oxygen response to normoxic and oxygen deprived (hypoxia) conditions in A549 lung carcinoma and HeLa cells. We also demonstrate that this probe is suitable for use in assays with a conventional plate reader.
Experimental
Materials
All chemicals and reagents, cell culture media and corresponding components were purchased from Sigma Aldrich (Ireland) and were used as received. Co-localising dyes were purchased from Bio-Sciences and the resazurin agent from PromoKine.Synthesis
The BODIPY reference probe was synthesised as described previously.70 The Ru(II) parent complex was synthesized by modifying the reported synthetic route for the preparation of (tris)heteroleptic Ru(II) compounds via an oxalate route.71 The cis-Ru(DMSO)4Cl271,72 precursor (1) and ligands phen-NH273 and bpybenzCOOEt53 were synthesised according to procedures in the literature. The synthesis of the intermediate ruthenium compounds (2, 3) is described in the ESI.†Preparation of the Ru(II) parent complex, [Ru(dpp)(phen-NH2)(bpybenzCOOEt)][PF6]2 (4)
[Ru(dpp)(phen-NH2)(ox)] (22.1 mg, 0.031 mmol) was dissolved in acetonitrile (2 mL) prior to the addition of 1 M perchloric acid (2 mL). The mixture was heated to reflux overnight. The red-brown solution was cooled on an ice bath and 3 mL of stirring water was added to it. The Ru-intermediate precipitate was collected and transferred to a hot mixture of bpybenzCOOEt (0.031 mmol) in ethylene glycol (3 mL). Following overnight reflux, the mixture was cooled to RT and was added to stirring aqueous ammonium hexafluorophosphate. The deep red solids were collected via vacuum filtration, and washed with water and diethyl ether. Purification was carried out by performing column chromatography on silica gel using 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:1 (MeCN/H2O/20% w/v KNO3). The re-precipitation of product fractions yielded a mixture of geometric isomers of the Ru(II) complex as vivid red solids. Yield isomer mixture: 27.6 mg (72%). 1H NMR (600 MHz, MeCN-d3) δ (ppm): 8.88–8.78 (dd, 2 H), 8.35–8.30 (d, 1 H), 8.29–8.14 (m, 7 H), 8.13–8.07 (m,1 H), 8.04–7.94 (m, 3 H) 7.93–7.83 (m, 1 H), 7.81–7.69 (m, 3 H), 7.68–7.49 (m, 13 H), 7.48–7.31 (m, 2 H), 7.24–7.18 (d, 1 H), 5.58 (s, 1 H), 4.40–4.34 (q, 2 H), 1.41–1.35 (t, 3 H).
:20:1 (MeCN/H2O/20% w/v KNO3). The re-precipitation of product fractions yielded a mixture of geometric isomers of the Ru(II) complex as vivid red solids. Yield isomer mixture: 27.6 mg (72%). 1H NMR (600 MHz, MeCN-d3) δ (ppm): 8.88–8.78 (dd, 2 H), 8.35–8.30 (d, 1 H), 8.29–8.14 (m, 7 H), 8.13–8.07 (m,1 H), 8.04–7.94 (m, 3 H) 7.93–7.83 (m, 1 H), 7.81–7.69 (m, 3 H), 7.68–7.49 (m, 13 H), 7.48–7.31 (m, 2 H), 7.24–7.18 (d, 1 H), 5.58 (s, 1 H), 4.40–4.34 (q, 2 H), 1.41–1.35 (t, 3 H).
13C NMR (600 MHz, MeCN-d3) δ (ppm): 165.05, 151.69, 151.56, 147.69, 143.98, 135.13, 133.14, 129.59, 129.26, 129.21, 129.09, 128.54, 128.31, 127.15, 127.10, 126.97, 125.44, 125.16, 124.27, 123.46, 102.75. HR-MS (ESI-TOF) m/z: calculated for C55H41N7O2Ru [M−2PF6−]+: 466.6177; found: 466.6657.
Preparation of poly(styrene) nano-structures and characterization
A triblock copolymer poly(styrene-b-ε-benzyloxycarbonyl-L-Lysine-b-Fmoc-L-lysine) PS38-PZLL64-PFLL5 was prepared as previously described.63 For the first deprotection step of the Fmoc group, the triblock copolymer (0.8 g) was dissolved in 8 mL of DMF. To this, 2 mL of piperidine was added and the solution was stirred for 2 h at room temperature. The resulting polymer was recovered via precipitation in diethyl ether. The copolymer was then re-dissolved in chloroform and precipitated in diethyl ether three times and isolated as a white powder (yield: 0.70 g).To 80 mg of this copolymer, the ruthenium complex 4 (10 mg, 0.0082 mmol) was added and the mixture was exposed to a Z protecting group deprotection procedure by slowly adding a solution of HBr (33 wt% in acetic acid) (0.3 mL) at 0 °C to a solution of the copolymer and ligand in trifluoroacetic acid (6 mL). After 4 h, the diblock copolymer/ligand mixture was precipitated in diethyl ether. The precipitate was washed several times with diethyl ether and after drying, it was dissolved in DDI water and dialyzed against DDI water using Spectra/Por dialysis membranes (MWCO, 3.5 kDa) for 72 h at room temperature. The product was lyophilized and isolated as an orange powder (yield: 57 mg).
The mini-emulsion polymerization of a 95/5 (v/v) solution of styrene/divinyl benzene (DVB) and BODIPY was carried out in a 10 mL two-neck reactor equipped with a reflux condenser, nitrogen inlet and magnetic stirrer. In a typical reaction, the diblock copolymer PS38-PLys68/Ru(II)(4)2 mixture (40 mg) was added to the reactor under an inert atmosphere and dissolved in 4.5 mL of degassed distilled water. A styrene/DVB monomer solution (0.40 g) was deoxygenated separately for 20 min by bubbling nitrogen through it. The BODIPY dye (0.15 mg) was dissolved in this solution and injected into the reactor. The reaction mixture was left with maximum stirring (1400 rpm) for 5 min, while being kept on an ice bath. The reaction flask was transferred to a heated oil bath (70 °C) and a deoxygenated initiator solution (5 mg of potassium persulfate in 0.5 mL of water) was injected to start the polymerization. The reaction was left to proceed for 4 hours after which the resulting latex was dialyzed against DDI for 48 hours using Spectra/Por dialysis membranes (MWCO, 3.5 kDa).
Instrumentation
1H NMR, 13C NMR and COSY spectra were recorded using a 600 MHz Bruker spectrophotometer (unless stated otherwise) and processed and calibrated against solvent peaks using Bruker Topspin (v2.1) software. High-resolution mass spectrometry (HR-MS) with electrospray ionization in positive mode on a Waters Micromass LCT system was carried out at the Mass Spectrometry Facility in University College Dublin. Thin layer chromatography (TLC) was performed on glass silica gel (Merck, 250 μm thickness) or C18 plates (Sorbent Technologies, 250 μm thickness).Electronic absorption spectra were acquired with a Jasco V670 UV/vis NIR spectrophotometer using a quartz cuvette with a pathlength of 1 cm. Fluorescence spectra were collected using a Varian Cary Eclipse fluorescence spectrofluorometer with background correction. Luminescence lifetime data were acquired up to 10000 counts using a time correlated single photon counting (TCSPC) system by PicoQuant with a laser excitation source of 450 nm. PicoQuant NanoHarp and TimeHarp software were used for data analysis and fitting. The emission spectra and lifetimes were also collected under deaerated conditions. All photophysical measurements were performed at room temperature (293 K) and in triplicate (n = 3).
Delsa nano C submicron particle size and a zeta potential particle analyzer with standard size cell accessories were used for dynamic light scattering (DLS) and zeta potential measurements. Scanning electron microscopy was carried out using a Hitachi S3400 Variable Pressure SEM. RuBDP NPs were sonicated for 20 minutes at room temperature prior to characterization or cell culture studies.
Oxygen calibration studies
RuBDP NPs were dissolved in PBS (pH 7.4) at 0.01% w/v. Following purging of the solution with nitrogen for 20 minutes at room temperature, the solution was allowed to re-aerate while the emission spectra were recorded at various oxygen concentrations measured in μmol L−1 using a PreSens oxygen probe. The emission spectra were collected with an excitation and emission slit width of 10 nm, and the oxygen calibration curves were constructed (n = 3).Plate reader-based ratiometric O2 response assay
For the assessment of oxygen quenching in a plate reader-based assay, samples of RuBDP NPs (4.5 μg mL−1) were dispensed in aliquots of 100 μL into three wells of a 96-well plate and treated with sodium sulfite (Na2SO3; 5 mg mL−1). The emission spectra of RuBDP NPs were recorded using a CLARIOstar (plus) (v 5.70) plate reader with excitation at 480 nm and an emission range of 505–840 nm. The concentration of oxygen was monitored and measured in μmol L−1 using a PreSens oxygen probe and an oxygen calibration curve was constructed.A549 cells were seeded at 104 cells per well in 100 μL media for 24 h at 37 °C under 5% v/v CO2. RuBDP NPs were added under uptake conditions, 4.5 μg mL−1/4 h. The cells were then washed with PBS (×2) prior to exposure to sodium sulfite (5 mg mL−1). The intracellular NP ratiometric response to changing oxygen concentration following Na2SO3 treatment was monitored by recording the emission spectra using a CLARIOstar (plus) (v 5.70) plate reader with excitation at 480 nm and an emission range of 505–840 nm.
Cell culture
Eagle's minimum essential medium (MEM) supplemented with 10% foetal bovine serum (FBS), 1% penicillin/streptomycin and L-glutamine (2 mM) was used as HeLa cell culture media. The MEM supplemented with 10% FBS, 2 mM L-glutamine and 1% non-essential amino acids was used to subculture MCF 7 cells. Ham's F12K supplemented with 10% FBS and L-glutamine (2 mM) was used to subculture A549 cells. CHO cell culture media were composed of F-12 Ham/DMEM media (1:1) supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were grown at 37 °C with 5% CO2 and harvested or split at 90% confluency using 1× Trypsin. PBS was supplemented with 1.1 mM MgCl2 and 0.9 mM CaCl2.
RuBDP NP uptake
CHO, HeLa and A549 cells were seeded at 1.5 × 105 cells in a 35-mm glass-bottom culture dish (Ibidi, Germany) of 1.5 mL total volume and cultured for 2 days at 37 °C with 5% CO2. The growth medium was removed and RuBDP NPs were added at 4.5 μg mL−1. Following 4 h of incubation, the cells were washed with supplemented PBS three times and imaged directly using a Leica TSP DMi8 confocal microscope (100 × oil immersion objective lens unless stated otherwise) with a heated stage at 37 °C and a O2, CO2 chamber was used for the oxygen mapping studies. The RuBDP NPs were excited using a 480 nm white light laser and the emission range was set to 505–550 nm for the BODIPY component and 569–850 nm for the Ru(II) component. DRAQ7 was added (3 μM) to distinguish live cells from damaged/permeabilized cells. The 633 nm laser was used to excite DRAQ7 and the emission was collected between 635 and 900 nm. The time-lapse series was carried out using a Nikon Ti2 fluorescence microscope (100 × oil immersion objective lens) with a heated stage at 37 °C and 5% v/v CO2.Cytotoxicity assay
HeLa, CHO, MCF 7, and A549 cells were seeded separately in a 96-well plate in 100 μL media at 104 cells per well for 24 h at 37 °C under 5% CO2. RuBDP NPs were added at concentrations between 0.9 and 100 μg mL−1 in triplicate wells. Control samples were prepared with 1% PBS. Following 24 h of incubation of the nanoparticles, the resazurin reagent was added to each well (10 μL per well) and incubated for 6 h in the dark at 37 °C. The Alamar Blue assay was used to estimate viable cells based on the absorbance measured at 570 nm with a background measurement at 600 nm using a Tecan plate reader. The assay was performed at n = 3. Comparisons between the data were made using the two-way ANOVA post hoc Tukey analysis at a 95% confidence level. Differences between the tested groups were considered statistically significant if P ≤ 0.05.Co-localisation studies
Commercially available dyes DAPI, LysoTracker Deep Red (Invitrogen), Rab7a-GFP (CellLight BacMam 2.0, Invitrogen) and MitoTracker Deep Red were used in co-localisation studies for the determination of the localisation of RuBDP NPs following cell uptake. The nuclear staining DAPI dye was added at 3 μM concentration and incubated for 20 min prior to imaging. DAPI was excited using a 405 nm laser and the emission was collected between 425 and 580 nm. LysoTracker Deep Red, used for staining of lysosomes, was added at 50 nM and incubated for 75 min prior to imaging (λexc 647 nm, λem range: 650–800 nm). Rab7a-GFP, used to stain late endosomes, was added to the cell dish and incubated overnight at 37 °C prior to the addition of RuBDP NPs. Rab7a-GFP was excited using a 488 nm white light laser and the emission was collected between 490 and 540 nm. MitoTracker Deep Red (25 nM) was incubated for 40 min prior to imaging and was excited at 644 nm with emissions collected between 650 and 800 nm. Following incubation, the dye/growth medium was removed, and the cells were washed with supplemented PBS prior to imaging. The fluorescence intensity profiles were obtained using ImageJ.Results and discussion
Synthesis
The synthetic route to the metal complex, reference probe and oxygen sensor core–shell nanoparticles is summarized in Scheme 1. The oxygen insensitive BODIPY reference compound, 2,6 diethyl-1,3,5,7-tetramethyl-8-(2-fluorophenyl)–6 methoxy-1,5-naphthyridine-4,4′-difluoroboradiazaindacene, was prepared as reported previously.70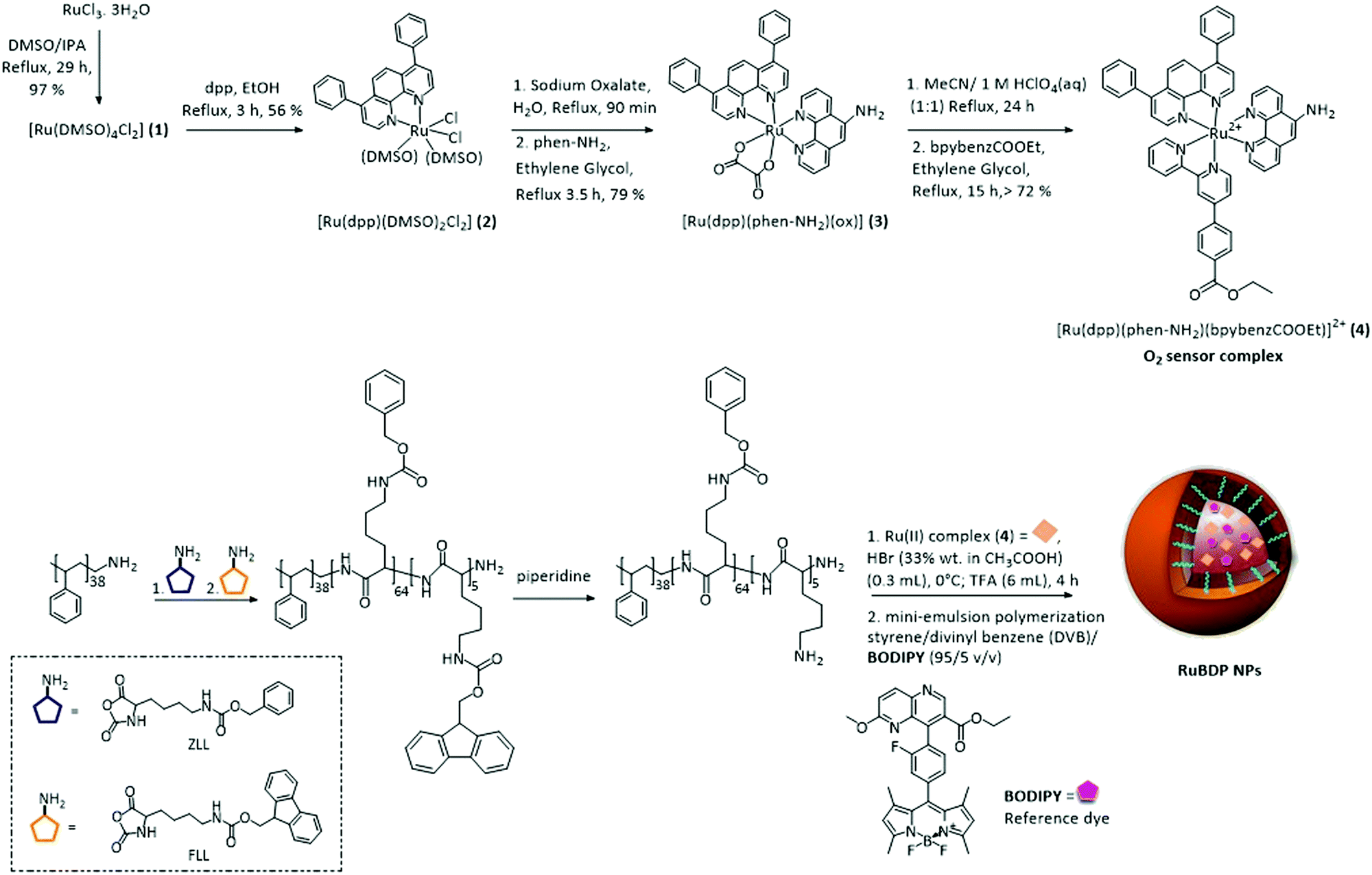 | ||
| Scheme 1 Route to synthesis of the Ru(II) tris-heteroleptic polypyridyl complex (4; O2 sensor) and preparation of self-referenced nanoparticles. | ||
In an effort to improve the photophysical response of the Ru(II) oxygen sensor, we prepared a novel tris-heteroleptic complex, [Ru(dpp)(phen-NH2)(bpybenzCOOEt)]2+. Ru(II) complexes comprising 4,7-diphenyl-1,10-phenanthroline (dpp) ligands have been shown extensively to promote sensitivity to O2.74–80 The bpybenzCOOEt was selected as a counter-ligand along with diphenyl phenanthroline to increase lipophilicity to facilitate the PS core encapsulation.
This tris-heteroleptic Ru(II) polypyridyl sensor was synthesised in high yield via an oxalate intermediate (3) to form the tris-heteroleptic Ru(II) complex (4), through a method adapted from the one previously reported by us.71
1H NMR, 13C NMR, COSY and mass spectrometry were used to confirm the structure and purity of the resulting complex. The 1H NMR showed the expected inequivalence of dpp and phen-NH2, arising from the cis-configuration of the bidentate complex, from the signals in the aromatic region of the 1H NMR spectrum. The presence of the ester moiety of bpybenzCOOEt was confirmed from the characteristic quartet and triplet signals in the aliphatic region of the 1H NMR spectrum integrating to 2 H and 3 H at 4.36 ppm and 1.34 ppm, respectively. The coupling interaction between these protons was confirmed by COSY analysis. The HRMS confirmed a mass cluster with the Ru isotope pattern that corresponded to [M–2PF6−]+ (m/z calculated: 466.6177; found: 466.6657).
The Ru(II) polypyridyl complex was co-encapsulated with the O2 insensitive BODIPY reference probe into the nanoparticle core for self-referenced ratiometric luminescence response to oxygen. The rationale for this approach was that with co-encapsulation into a core–shell structure, both the probe and reference are protected from any environmental effects which may interfere with the ratiometric response signal. The shell offering spatial separation between the probes and environment, and the high oxygen permeability of polystyrene along with the enhanced O2 sensitivity of dpp-coordinated Ru(II), permitting stable and selective O2 access to the sensor. Importantly, the exterior of the particle is free for surface modifications such as conjugation through the poly-L-lysine.
By adopting a modified synthesis, the particles were prepared by the miniemulsion polymerisation of styrene/divinyl benzene (DVB) using an amphiphilic diblock copolymer poly(styrene-b-L-lysine) (PS38-PLL69) as a surfactant.63,81,82 The lipophilicity of both the BODIPY and Ru(II) complex allowed for their co-encapsulation into the non-polar cross-linked polystyrene core at the miniemulsion step, yielding RuBDP NPs. The physical anchoring of the amphiphilic surfactant rendered the NP surface hydrophilic.
Dynamic light scattering (DLS) was carried out to determine the size and zeta potential of the RuBDP NPs in PBS (pH 7.4). The diameter of the particles was measured as 98.2 ± 1.09 nm and the average zeta potential was measured as +25 ± 1.37 mV. The positive charge on the particles can be attributed to the polylysine polymer. The spherical shape and size uniformity of the nanoparticles were confirmed by SEM imaging under 9.00 kV × 37.0k (Fig. S9, ESI†).
The magnitude of the zeta potential indicates that the particles form a stable dispersion in aqueous buffered solution and indeed it was confirmed that there was no decomposition or precipitate formation over seven months of the particles in suspension. In addition, the absolute emission intensity and the ratio of Ru(II) to BDP emission remained unchanged over this time window (Fig. S10, ESI†), indicating that there was no leaching of the probes from the particles.
In a separate study, to confirm the location of the probes within the particles, we soaked the particles in THF as a swelling agent and evaluated the fluorescence spectra of the supernatant followed by the centrifugation of the particles (Fig. S11, ESI†). We then compared the supernatant from a similar treatment of our previously reported core–shell RuBODIPY particles, in which the Ru is bound to the particle surface and the BODIPY to the core.63 From the latter, we observed the release of ruthenium from the particle outer shell under these conditions, whereas with the co-core encapsulated particles we did not observe any release of ruthenium to the supernatant. This confirms that Ru is confined to the PS core, where the PS cross-linkage prevents the leaching of the sensor from the core due to size exclusion. However, in both cases, the BODIPY, which is smaller, was observed to release on particle swelling.
Photophysical characterisation of RuBDP nanoparticles
The photophysical properties of the parent ruthenium complex were first characterised in acetonitrile (Table 1). [Ru(dpp)(phen-NH2)(bpybenzCOOEt)]2+ exhibits a metal-to-ligand charge-transfer (1MLCT) transition centred at λmax 460 nm (Fig. S12, ESI†). And, when excited at λmax, it exhibits a broad intense emission with a maximum at 622 nm. [Ru(dpp)(phen-NH2)(bpybenzCOOEt)]2+ emission shows strong oxygen sensitivity in acetonitrile. The quantum yield of 4 was averaged from triplicate measurements in aerated and deaerated acetonitrile as 0.0173 ± 0.0003 and 0.0291 ± 0.0003, respectively, using [Ru(bpy)3]2+ as the reference standard.83 The parent complex exhibits monoexponential luminescent decay in acetonitrile with a lifetime of τ ≈ 372 ns, within error, under air saturated conditions (Fig. S13, ESI†). The BODIPY derivative was selected as the reference in this study because control studies in solution confirmed the lack of quenching or other interactions that might predict the cross-talk in the particles. Similar to previously reported BODIPY–Ru(II) pairs in a molecular dyad and nanoparticle system, photophysical studies showed no evidence of cross-communication between the two dyes either in solution or when combined in the particle core.53,63 Indeed, as shown below, the photophysical properties of the particles confirm that the probe and reference do not cross-talk to any significant extent. Furthermore, both ruthenium and BODIPY derivatives can be simultaneously excited at 480 nm. The iteration of the ratios of probe and reference under this excitation wavelength was carried out in solution to ensure the appropriate relative scale of signals and a ratio of 1:2 (BODIPY/Ru(II)) for particle preparation was then used in all particle preparation processes.
| Compound | Solvent | λ abs/nm | λ em/nm | τ aerated /ns | τ deaerated /ns | φ lum aerated deaerated |
|---|---|---|---|---|---|---|
| a All measurements were performed at room temperature. b Percentage relative amplitudes are given in parentheses. c [Ru(bpy)3]2+ was used as a reference standard. Errors included as ±SD (n = 3). | ||||||
| [Ru(dpp)(phen-NH2)(bpy-benz-COOEt)]2+ (4) | MeCN | 460 | 626 | 371.9 ± 5.3 | 599.4 ± 7.3 | 0.0173 ± 0.0003, 0.0291 ± 0.0004 |
| BODIPY dye | MeCN | 498 | 512 | 3.85 ± 0.03 | — | — |
| RuBDP NPs BODIPY-core | PBS (pH 7.4) | 480 | 515 | 3.87 ± 0.01 | — | — |
| Ru(II)-core | 618 | 523.0 ± 12.1 | 708.9 ± 10.2 | |||
The photophysics of the RuBDP NPs was examined in aqueous PBS solution, pH 7.4, as this was the medium used for biological studies. The photophysical characteristics of the particles correlate well with the solution phase behaviour of the constituent luminophores. The RuBDP particles exhibited dual, well-resolved emission signals with maxima of 618 nm and 515 nm attributed to the ruthenium probe and BODIPY reference, respectively. While the relative emission intensity of each (Ru and BODIPY) luminophore varies with the excitation wavelength, the emission of both the probe and reference was retained with no evidence for cross-talk. An excitation wavelength of 480 nm was used throughout the subsequent measurements as this gave dual emission with the appropriate relative intensity of the probe and reference Fig. 1A.
 | ||
| Fig. 1 (A) RuBDP response to change in oxygen concentration measured in μmol L−1. Emission spectra of RuBDP NPs in PBS (pH 7.4) when excited at 480 nm; both excitation and emission slit widths set at 10 nm. (B) Stern–Volmer plot: the luminescence originating from the ruthenium component decreases with the increase in oxygen concentration while the BODIPY reference probe is moderately affected (n = 3). | ||
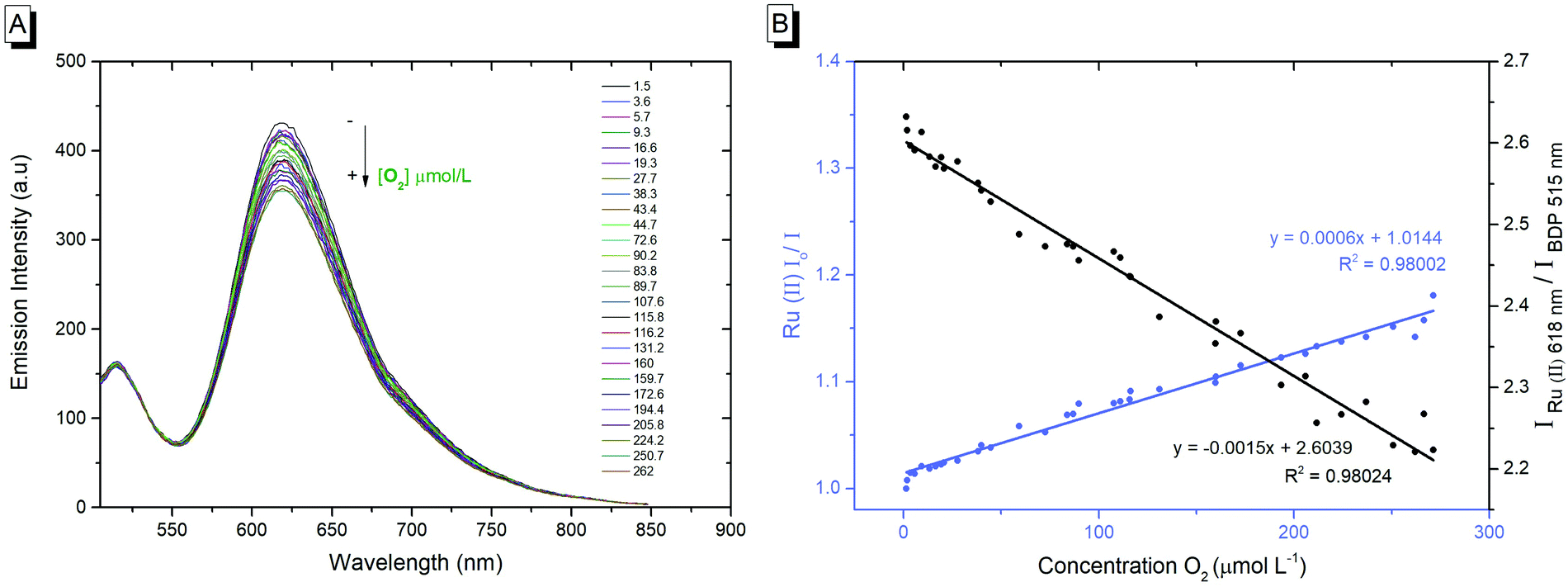
As shown in Fig. 1B, the emission intensity of the NP-encapsulated Ru(II) varies linearly with [O2], whereas, as expected, the emission intensity of the BODIPY reference probe remains constant within experimental errors. By calibrating the oxygen concentration in the contacting solution using a PreSens oxygen probe, the emission intensity data were fit to the Stern–Volmer equation (eqn (1))26 (Fig. 1B) where the ratiometric intensity data show good linearity (R2 = 0.9802).
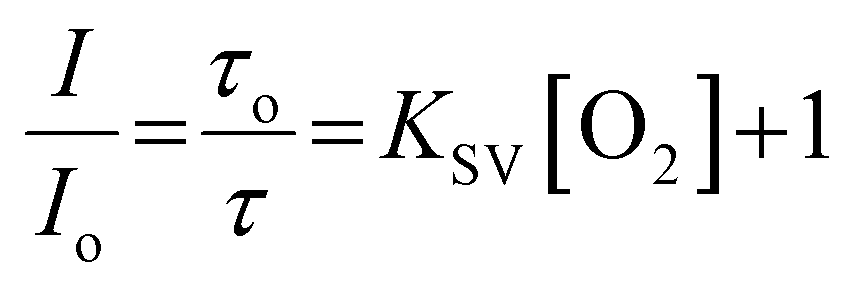 | (1) |
| KSV = kqτo | (2) |
The Stern–Volmer quenching constant (KSV) was obtained from the slope of the Stern–Volmer fit (--) and the rate of quenching (kq) was found to be 4.25 × 108 M−1 s−1 in PBS (pH 7.4), according to eqn (2) (where τ0 is the lifetime in the absence of oxygen). While the O2 permeability of polystyrene is high,84 the kq is lower than that of the metal complex reported in solution which is likely attributed to some impedance of the diffusion of oxygen in the PS core or the inaccessibility of some metal complexes to O2 on encapsulation.
The linear dynamic range for ratiometric signals from the RuBDP particles was measured as 3.6–262 μM O2, which coincides well with the O2 range anticipated in in vitro studies (0–250 μM),13 spanning oxygen concentration ranges indicative of hypoxia. Although the magnitude of signal change in response to oxygen is not as large as the one reported for molecular systems such as porphyrin or iridium coordination compounds, such species typically show high singlet oxygen yields and/or cytotoxicity, compared to the nanoparticle-encapsulated complexes reported here that show limited toxicity even under extended photoirradiation, as discussed vide infra.85–89
The emission decay of the RuBDP particles was collected under aerated and deaerated conditions. The emission decay from the particles was found to fit to a dual exponential kinetics. In air saturated PBS ([O2] 262 μmol L−1), lifetimes of 523 ± 12 ns and 3.87 ± 0.01 ns were recorded corresponding to the amplitude averaged lifetime of the Ru(II) and BODIPY components, respectively. Upon deaeration ([O2] 3.6 μmol L−1), the emission lifetime of the Ru(II) component of the decay increased to 708.9 ± 10.2 ns while the BODIPY lifetime remained unchanged (Fig. S14 and 15, ESI†). The photophysical data for the Ru(II)–parent compound, BODIPY core and RuBDP NPs are summarized in Table 1.
Cell studies
Having confirmed the RuBDP NP stability in PBS, ratiometric signals and oxygen responsiveness, we next investigated the permeability of the NPs to live cells.Uptake studies in live HeLa, CHO and A549 cell lines
The cell uptake of RuBDP nanoparticles was assessed across several concentrations in live HeLa cells over time by incubating the nanoparticles in the absence of light at 37 °C with 5% v/v CO2. Uptake of the nanoparticles at 4.5 μg mL−1 into the cytoplasm is observed to have commenced after 3 h. By 4 h, the particles are dispersed throughout the cytoplasm. Critically, in contrast to our previously reported nanoparticle system, the uptake was spontaneous and did not require facilitation by a solvent or surfactant.63 The cationic poly-lysine shell of the particles likely promotes the interaction with the cell membrane. This is in contrast to the previously reported particles, where it would seem that when the ruthenium complex was appended to the particle exterior, the uptake was inhibited.63Consistent with solution studies, 480 nm was used as the excitation wavelength in cell imaging to excite both the probe and reference. Fig. S16 (ESI†) shows the images collected from two channels within the ranges of 505–550 nm and 569–850 nm, coincident with the BODIPY reference and Ru(II) probe, respectively. The spatial coincidence of the two signals confirms that the particles are present and that dual emission is observed from each luminophore under this excitation wavelength. The spatial coincidence of each emission signal also confirms that the particle cores are intact.
Co-staining with the nuclear staining probe, DAPI, revealed that the RuBDP NPs are excluded from the nucleus which accumulate in the nuclear peripheral environment (Fig. 2). Crossing of the nuclear membrane in live cells typically requires the interaction with the nuclear pore complex via a small nuclear localization signal (NLS) or via a significantly smaller particle size.90–92
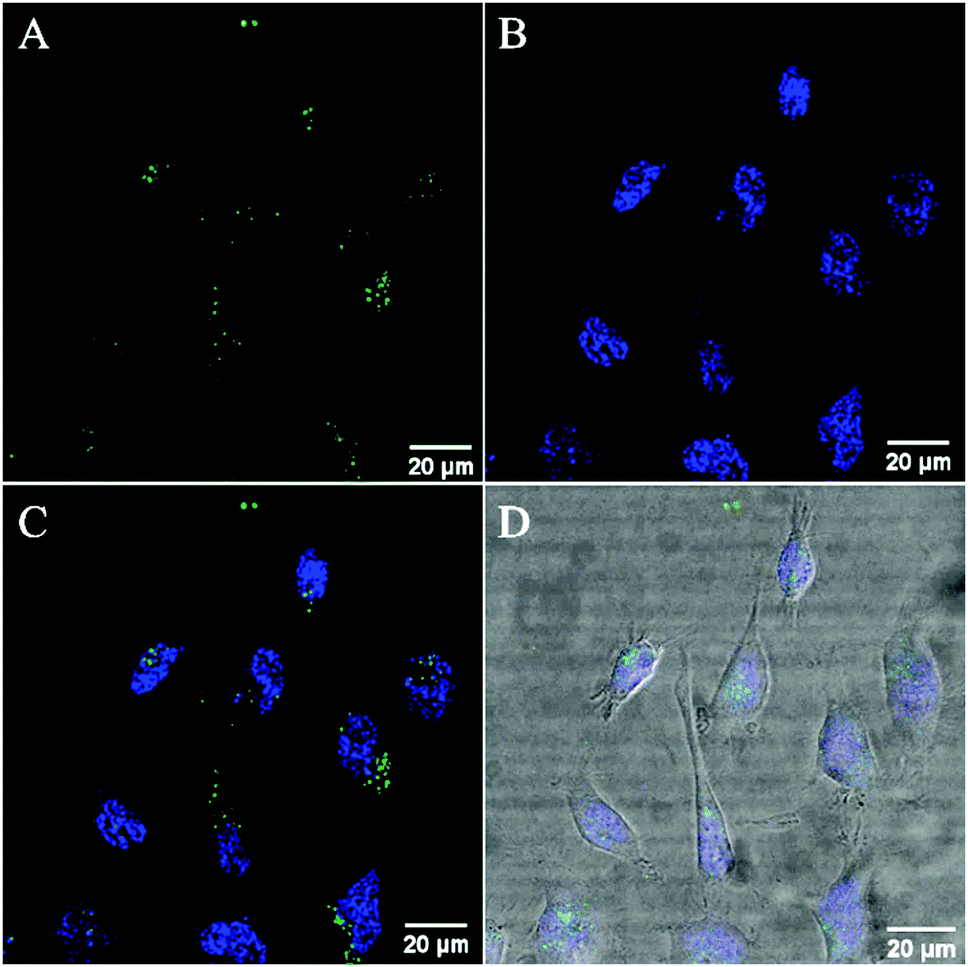 | ||
| Fig. 2 Confocal luminescence images of RuBDP NP uptake at 4.5 μg mL−1 in live HeLa cells. The cells were incubated in the absence of light for 4 h. (A) The RuBDP NPs were excited using a 480 nm white light laser and the emission for the Ru(II) component was collected between 569 nm and 850 nm. (B) The cells were co-stained with a nuclear staining probe, DAPI. (C) Overlay of the Ru(II) channel with DAPI. (D) Overlay with brightfield. | ||
While nuclear targeting is often desirable in drug delivery/therapeutic applications, the nuclear exclusion is preferable for the monitoring of oxygen levels in other cellular compartments involved in ATP production and cellular metabolism.93
Confocal imaging indicated that HeLa cells remain viable following post-incubation with RuBDP NPs for 24 h at 4.5 μg mL−1 with no evidence for cytotoxicity despite wide cytoplasmic distribution over extended windows (Fig. S17, ESI†).
A time-lapse study was carried out to investigate the fate of RuBDP NPs in live HeLa cells following uptake. HeLa cells were treated with RuBDP NPs under imaging conditions (4.5 μg mL−1/4 h) for uptake (Fig. 3A) and were monitored over time using a widefield fluorescence microscope. The emission from the Ru(II) channel was acquired every 10 minutes overnight on a heated stage (37 °C) under an atmosphere of 5% v/v CO2. As shown in Fig. 3, the emission intensity from the particles increased sharply following 4 h of incubation (Fig. 3B). This was also observed by confocal microscopy for HeLa cells pre-treated under the same conditions and imaged post 4 h of initial incubation. The precise origin of this increase in emission intensity signal from the Ru(II) component is unknown so far but we tentatively attribute it to the endosomal escape of the particles from late stage endosomes, where the acidity of the environment may impact the Ru intensity.
 | ||
| Fig. 3 Widefield fluorescence images of HeLa cells pre-treated with RuBDP NPs (4.5 μg mL−1) and monitored over time. Emission collected from the Ru(II) channel (A) following uptake of the particles and (B) at 4 h of incubation after NP uptake. The Ru(II) component of the NPs was excited at 470 nm using the GFP excitation filter. | ||
Contributions from proteolysis of the shell over extended windows in the lysosome are unlikely given the relatively low partitioning into the lysosome and also, the continuous co-localisation of the BODIPY and Ru emission signals over the extended range of time scales indicates that irrespective of origin, the particle core remains intact.
Furthermore, comparing the behaviour of the related nanoparticles where Ru was immobilised at the protein surface, the persistence of co-localisation indicated that the particles are not proteolyzed in cellulo under imaging conditions. Future studies will focus on assessing the promotion of particle release from late endosomal structures into other cellular compartments and organelles such as the mitochondria in surface-modified structures.94,95
RuBDP uptake in a non-cancerous cell line was also studied. Here, CHO cells were incubated with RuBDP NPs under the same conditions (4.5 μg mL −1 for 4 h); however, interestingly, at this concentration, in contrast to HeLa and the other cancer cell lines studied vide infra, there was no evidence of nanoparticle uptake. Rather, the particles adhered to the cell membrane exterior (Fig. S18, ESI†). The incubation of CHO cells with the increased concentration of 12 μg mL−1 NPs for 24 h did lead to particle uptake and non-specific distribution within the cytoplasm (Fig. S18C, ESI†).
However, in addition, at this concentration, cell debris was observed and by confocal imaging on DRAQ7 staining, damaged CHO cells were evident.
The contrasting uptake between CHO and HeLa is interesting and we speculate that this may be due to the differences widely noted between the cancerous and non-cancer cell lines.96–99 This includes the differences in pathways for endocytosis100 as well as the composition of the cell membrane which can differ significantly in terms of lipid composition, membrane fluidity and lipid rafts.101,102 Cancer cells are characterized by a highly negative surface charge due to the exposure of phosphatidylserine (PS) at the outer membrane.103,104
In order to investigate further, uptake studies were also carried out for a second cancerous cell line, the human lung carcinoma A549 cell line. A549 cells were treated with RuBDP NPs at 4.5 μg mL−1 and incubated at 37 °C under 5% CO2. Similar to HeLa cells, the RuBDP nanoparticles were internalized within 3–4 h of incubation and the same punctuate staining was observed throughout the cytoplasm (Fig. S19, ESI†).
Cytotoxicity
The Alamar Blue (resazurin) viability assay was used to evaluate the cytotoxicity of RuBDP NPs between 0.9 and 100 μg mL−1 in HeLa, A549 and CHO cells over 24 h in the absence of light at 37 °C. It was found that HeLa and A549 cell lines were remarkably tolerant to the particles up to 9 μg mL−1 (Fig. 4) which is twice the working concentration of 4.5 μg mL−1 used in our confocal imaging studies. Above 9 μg mL−1, the viability varied depending on the cell line. This may be attributed to the particle uptake mechanism and localization.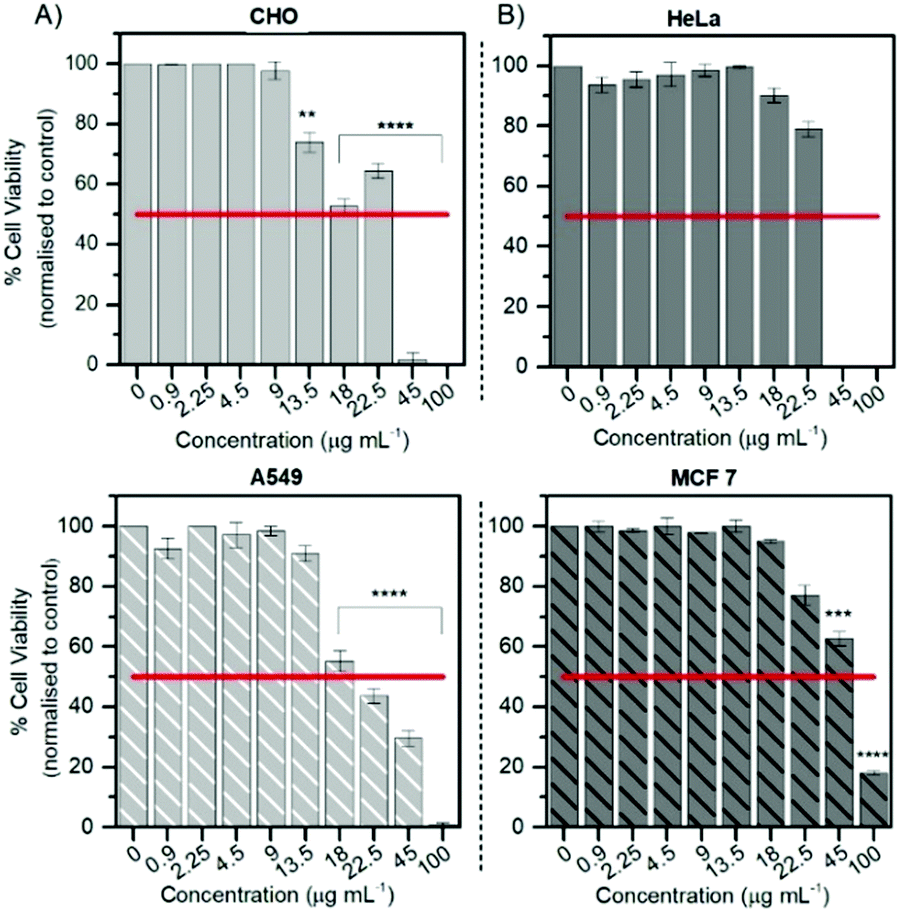 | ||
| Fig. 4 Cell viability of HeLa, MCF 7 and CHO cells after 24 h of exposure to RuBDP NPs over a range of concentrations. Live cells were treated with the nanoparticles followed by the addition of resazurin for 6 h. Absorbance readings at 570 nm with a background at 600 nm were performed. Data were expressed as averaged percentages and compared to non-treated cells. P ≤ 0.05; ***P ≤ 0.01, ****P ≤ 0.0001. | ||
The results for the cancer lines correspond well with the cell imaging where no cell death was observed at 4.5 μg mL−1 and a decrease in cell viability is observed for the CHO cell line above 9 μg mL−1. As mentioned previously, no particle uptake was observed for CHO cells at the working concentration and non-specific distribution within the cytoplasm could be observed post incubation at high particle concentration (12 μg mL−1). In addition, HeLa cells show good tolerance with 73% of cells still viable up to 22.5 μg mL−1. RuBDP NPs were found to be more toxic towards A549 cells at these concentrations with an IC50 between 18 and 22.5 μg mL−1. A decrease in the viability of A549 cells in comparison to HeLa cells may be attributed to the increased rate of nanoparticle uptake as suggested by confocal imaging.
Similarly, platinum(II)–porphyrin nanoparticles were shown to be non-toxic between 5 and 20 μg mL−1 and concentrations above 40 μg mL−1 resulted in cluster formation and cell morphological changes.105 The viability of particle-treated MCF 7 cells showed that these cells had superior tolerance, with an IC50 above 45 μg mL−1, ten times the working NP concentration for confocal imaging in A549 and HeLa cells. Overall, from these results, the uptake and toxicity of RuBDP NPs are both concentration and cell-line dependent.
Uptake mechanism and co-localization studies
Particle uptake studies in HeLa and A549 cells were also carried out at 4 °C. At low temperatures, uptake was inhibited with the accumulation of RuBDP NPs at the cell surface observed for both cell lines (Fig. S20 and S21, ESI†). And, notably, no particle permeation to the cell interior was observed, indicating that uptake occurs through an energy-dependent mechanism, likely, endocytosis.Nanomaterials are widely reported to be transported across the cell membrane of mammalian cells via endocytosis and specifically pinocytosis when the particle size, as is the case here, is <200 nm.106 Internalization of nanoparticles via an endocytic pathway typically involves multi-vesicular bodies known as endosomes.107 To evaluate particle localization to endosomes, co-localization studies in HeLa and A549 cells were carried out using the Late Endosome Rab7a-GFP.
Emission intensity profiling showed strong co-localization of RuBDP NPs with Rab7a-GFP following uptake at 4.5 μg mL−1 after 4 h of incubation (Fig. 5D). The Pearson's coefficient, which quantifies the degree of co-localization between the NPs and Rab7a-GFP, was determined to be 0.88. This observation strongly supports the notion that uptake in these cancer cell lines is through endocytosis where following this mechanism of uptake, the RuBDP NPs will be transported by endosomes which mature into late endosomes (LE).
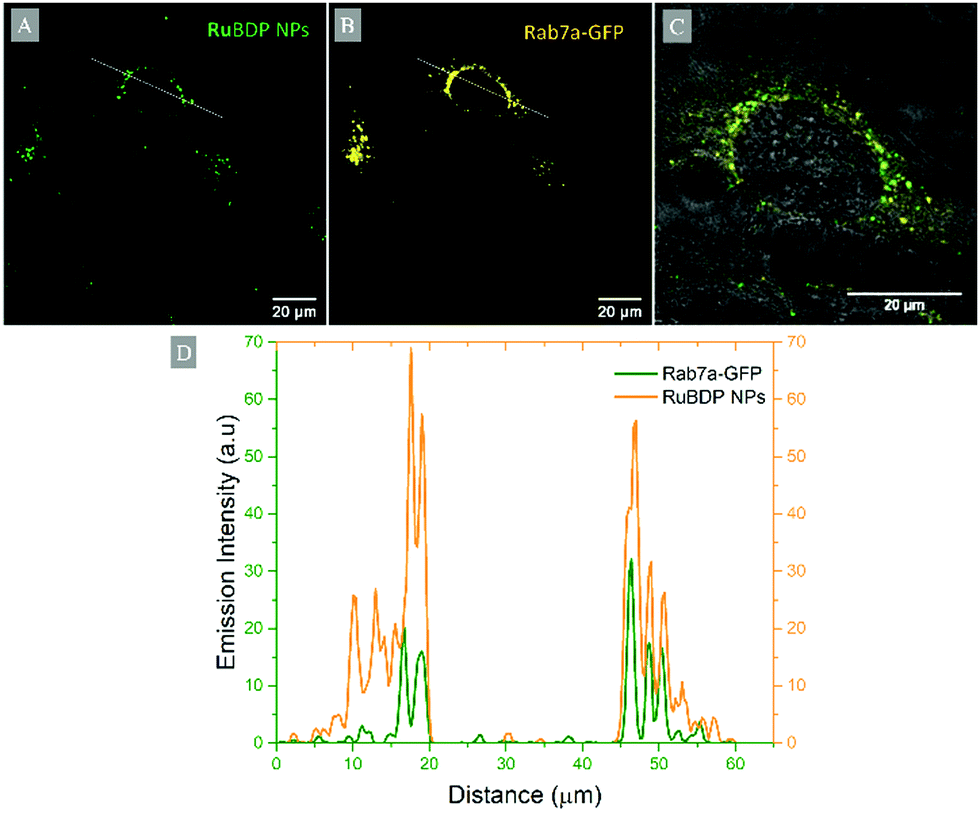 | ||
| Fig. 5 Co-localization of RuBDP NPs with late endosomal staining probe in live HeLa cells: confocal imaging of (A) RuBDP NPs at 4.5 μg mL−1/4 h (green), (B) Rab7a-GFP (yellow), and (C) overlay of RuBDP/Rab7a-GFP channels with the brightfield background. (D) The fluorescence intensity profile of RuBDP NPs and Rab7a-GFP obtained from the line profile across the cell is also shown (ImageJ). | ||
Co-localization studies with Lysotracker Deep Red and MitoTracker Deep Red were carried out to determine the fate of the particles at 4.5 μg mL−1 following 4 h of incubation and transportation in late endosomes.
Late endosomes and therefore potentially the NPs can undergo lysosomal fusion, exocytosis or can be released in the cytoplasm and/or then enter cellular compartments and organelles.108 Co-staining studies with Lysotracker Deep Red revealed a Pearson's coefficient value of 0.32 and thus a low degree of co-localization, suggesting only the partial lysosomal distribution of RuBDP NPs in HeLa cells (Fig. S23, ESI†). However, co-staining with MitoTracker Deep Red (Fig. S24, ESI†) showed that under these conditions, the RuBDP NPs do not localize to the mitochondria. More homogeneous emission, without the punctuate staining characteristic of endosomal entrapment, following 24 h of incubation, suggesting that the particles are released to the cytoplasm without targeting specific organelles, making them potentially useful oxygen probes. However, another consideration is that quenching of the triplet-excited state of Ru(II) complexes leads to singlet oxygen formation that can in turn lead to the damage of cellular components.109–111
Therefore, to assess the photo-toxicity of RuBDP NPs following uptake in HeLa cells, a ROI of cells was selected for continuous irradiation at 480 nm. The viability of the cells was monitored by the presence or absence of the nuclear staining DRAQ7 dye. A control sample was also irradiated under the same conditions in the absence of RuBDP NPs and the viability was monitored with DRAQ7. As shown in Fig. S25 (ESI†), toxicity was observed, but only after two hours of continuous irradiation which would be outside the time interval used in conventional imaging/sensing studies, where irradiation would rarely be continuous over such windows.
The absence of photo-toxic effects over shorter term may be attributed to the particle stability and enclosure within the LE. This is a key advantage to RuBDP NPs for continuous real-time oxygen sensing in live cells.
Ratiometric response under induced hypoxic conditions
To evaluate if a ratiometric signal that reflects O2 variation can be measured intracellularly, the emission spectrum of intracellular RuBDP NPs was collected using the xyλ scanning mode of a confocal microscope. The spectra were collected under normoxic (20% O2) and hypoxic conditions (5% O2). A549 cells were treated with the particles under normoxic conditions, washed with PBS and immediately imaged (Fig. 6A and B). The emission spectrum was collected at this point between 495 and 795 nm at 5 nm intervals using xyλ. The chamber conditions were adjusted from normoxic to hypoxic by gradually switching to 5% v/v O2. The images were normalised to BODIPY emission intensity (Fig. 6E), which was observed not to change with oxygen concentration but a clear increase in luminescence intensity for the Ru(II) channel was observed during the confocal imaging of the same region of cells (Fig. 6C) moving from normoxic to hypoxic conditions. An analogous response was also observed in HeLa cells under the same experimental conditions (Fig. S26, ESI†). Emission spectra were acquired in this manner for three different cell regions and averaged as shown in Fig. 6E. The figure shows an approximately two-fold increase in the intensity of the Ru(II) component while the reference BODIPY probe intensity remains unchanged. Using the ratio of IRu(II)616/IBDP515 under hypoxic conditions, the intracellular [O2] was estimated to be <2.6 μmol L−1.
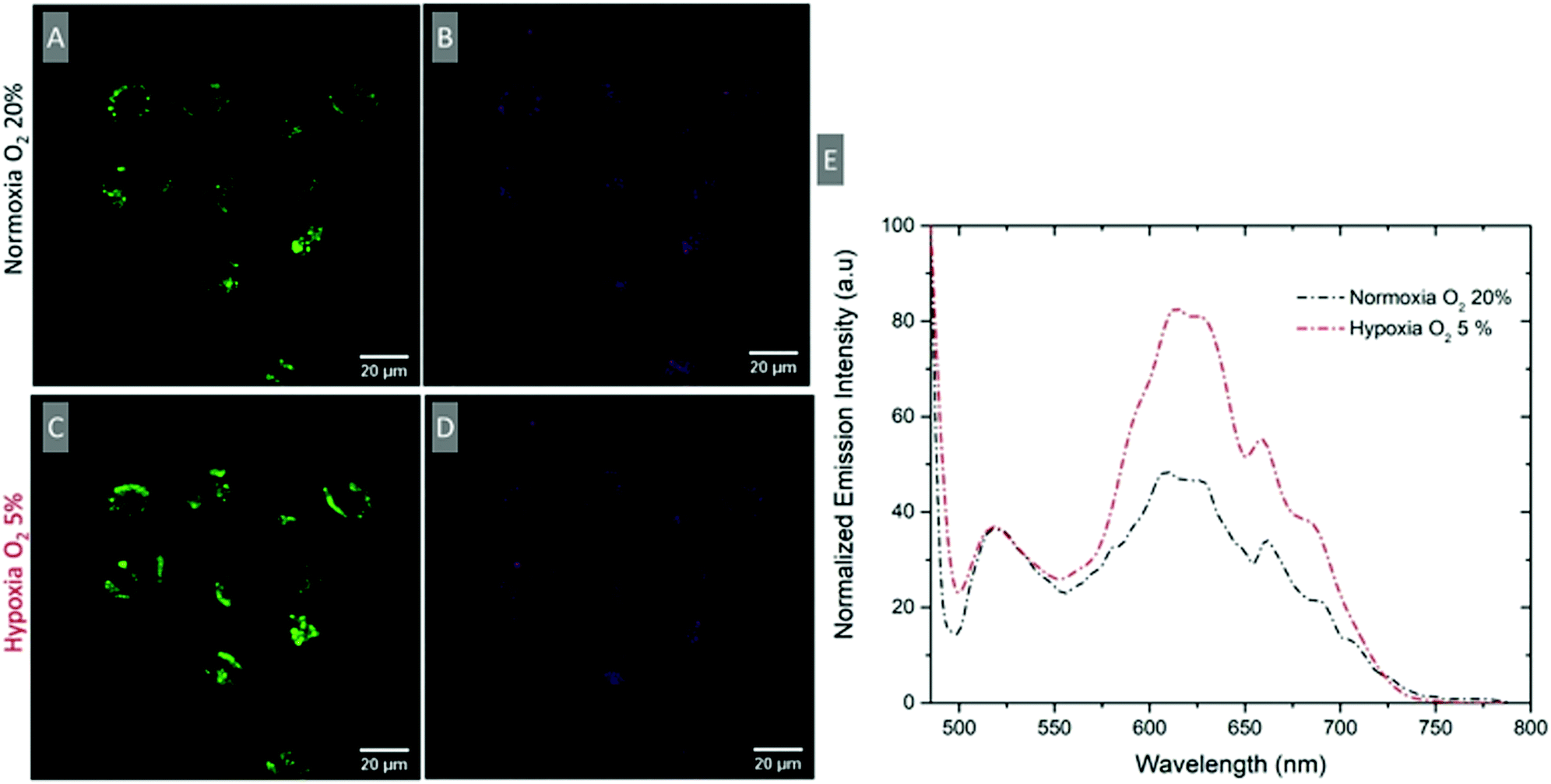 | ||
| Fig. 6 Confocal imaging of A549 cells treated with RuBDP NPs at 4.5 μg mL−1 for 4 h at 37 °C. (A and C) Ru (II) channel (569–850 nm) and (B and D) BODIPY channel (505–550 nm) under normoxic and hypoxic conditions. (E) Confocal lambda (λ) scan: emission spectra collected between 495 nm and 795 nm following single excitation at 480 nm at normoxic and hypoxic conditions where both the BODIPY and the Ru(II) component emission maximum is observed (n = 3). | ||
The particles show good uptake to the cytoplasm, low dark toxicity and phototoxicity, and good self-referenced response to oxygen in solution and in cells.
Finally, a key motivation in creating ratiometric oxygen sensing particles is that they are suitable for intensity-based studies with conventional lab instruments rather than specialised techniques such as lifetime imaging. Therefore, the ratiometric oxygen response of the particles in cells at oxygen levels between normoxic and hypoxic conditions was assessed using a plate reader-based assay. To establish instrument response (notably the detector sensitivity was greater towards the red region of the spectrum), a plate-based calibration plot was obtained by recording the particle spectra at various oxygen concentrations using a CLARIOStar (plus) reader and a PreSens oxygen probe. The emission intensity data were fit to the Stern–Volmer equation as shown in Fig. S27 (ESI†).
A quantitative cell-based study was then carried out where A549 cells were pre-treated with RuBDP NPs (4.5 μg mL−1) and using a plate reader, the emission spectra of the particles were collected prior to and sequentially in time, following exposure to oxygen scavenger, sodium sulfite112 (Fig. S28, ESI†). The absolute emission intensity was naturally lower for the cell-encapsulated nanoparticles but the RuBDP NPs showed a quantitative ratiometric response to gradually changing oxygen concentration on incubation of the cells with Na2SO3 (5 mg mL−1). The [O2] could be readily quantified as 156.6, 98.9, 72.6 and 32.8 μmol L−1 at several timepoints by the intracellular probe following treatment with Na2SO3 using the ratiometric signal with the calibration plot.
Conclusions
The preparation of highly stable and lipophilic ratiometric nanoparticles is described. Here, the probe and reference – an O2 sensitive novel Ru(II) complex and a BODIPY fluorophore – are co-encapsulated in the particle polystyrene (PS) core that is decorated with a poly-L-lysine exterior. This approach promotes the cell permeability and isolates the core bound components from environmental effects except for oxygen which is PS permeable. Solution studies of RuBDP NPs confirm the sensitive ratiometric response to oxygen with a dynamic range that is expected to be suitable for biological studies. The particles in cells were studied. Uptake of RuBDP NPs in cancer cell lines – HeLa and A549 cells – was observed at 4.5 μg mL−1 within 4 h of incubation at 37 °C but was inhibited at 4 °C. In contrast, the particles were impermeable to CHO cells under these conditions and cell viability studies showed concentration- and cell-line-dependent cytotoxicity. Dynamic widefield microscopy studies revealed that in cancer cell lines, emission enhancement from the Ru(II) component occurred within further 4 hours following the initial uptake, the origin of which is thought to reflect the NP endosomal escape. Although RuBDP NPs can offer a ratiometric response to changes in late-endosomal oxygen levels, future studies will focus on exploiting the modifications to the particle exterior to achieve efficient endosomal escape and targeting of specific cellular organelles. Overall, the data indicate that these probes are suitable for non-invasive, dynamic, and quantitative measurements of oxygen in cellulo using a plate-reader assay or confocal microscopy (xyλ) and thus may be a useful tool for monitoring oxygen for mechanistic insights into cancer biology and diagnostics. Future studies will focus on promoting the targeting of key organelles and enhancing the oxygen response from sensors.Abbreviations
| CHO | Chinese hamster ovarian cells |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DLS | Dynamic light scattering |
| dpp | 4,7-Diphenyl-1,10-phenanthroline |
| FRET | Förster resonance energy transfer |
| GFP | Green fluorescent protein |
| LE | Late endosomes |
| MLCT | Metal-to-ligand charge-transfer |
| NPs | Nanoparticles |
| ox | Oxalate |
| phen | 1,10-Phenanthroline |
| SEM | Scanning electron microscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KSG and TEK gratefully acknowledge the Irish Research Council Scholarship: IRC Government of Ireland Postgraduate Scholarship GOIPG/2016/702. CSB, AH and TEK gratefully acknowledge the Government of Ireland Postdoctoral Fellowship, GOIPD/2018/495. This study is also based on the work supported by the Science Foundation Ireland under grant numbers 19/FFP/6428 and 12/RC/2276_P2. This publication has emanated from the research supported in part by a grant from the Science Foundation Ireland (SFI) and the European Regional Development Fund (ERDF) under grant number 13/RC/2073.Notes and references
- G. L. Semenza, Science, 2007, 318, 62–64 CrossRef CAS PubMed.
- T. C. O’Riordan, A. V. Zhdanov, G. V. Ponomarev and D. B. Papkovsky, Anal. Chem., 2007, 79, 9414–9419 CrossRef.
- G. L. Semenza, Biochem. J., 2007, 405, 1–9 CrossRef CAS PubMed.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed.
- Press release: The Nobel Prize in Physiology or Medicine 2019. NobelPrize.org. Nobel Media AB 2020. Sun. 04 Jan 2020. https://www.nobelprize.org/prizes/medicine/2019/press-release/.
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- X. Zheng, X. Wang, H. Mao, W. Wu, B. Liu and X. Jiang, Nat. Commun., 2015, 6, 5834 CrossRef CAS PubMed.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed.
- H. Kurokawa, H. Ito, M. Inoue, K. Tabata, Y. Sato, K. Yamagata, S. Kizaka-Kondoh, T. Kadonosono, S. Yano, M. Inoue and T. Kamachi, Sci. Rep., 2015, 5, 10657 CrossRef CAS PubMed.
- V. I. Shcheslavskiy, A. Neubauer, R. Bukowiecki, F. Dinter and W. Becker, Appl. Phys. Lett., 2016, 108, 091111 CrossRef.
- S. M. Borisov, R. Pommer, J. Svec, S. Peters, V. Novakova and I. Klimant, J. Mater. Chem. C, 2018, 6, 8999–9009 RSC.
- D. B. Papkovsky, Methods in Enzymology, Elsevier, 2004, vol. 381, pp. 715–735 Search PubMed.
- R. I. Dmitriev and D. B. Papkovsky, Cell. Mol. Life Sci., 2012, 69, 2025–2039 CrossRef CAS.
- A. Juris, S. Campagna, V. Balzani, G. Gremaud and A. Von Zelewsky, Inorg. Chem., 1988, 27, 3652–3655 CrossRef CAS.
- V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 280, pp. 1–36 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- Y.-Q. Fang, N. J. Taylor, G. S. Hanan, F. Loiseau, R. Passalacqua, S. Campagna, H. Nierengarten and A. V. Dorsselaer, J. Am. Chem. Soc., 2002, 124, 7912–7913 CrossRef CAS PubMed.
- S. M. Borisov, G. Nuss and I. Klimant, Anal. Chem., 2008, 80, 9435–9442 CrossRef CAS PubMed.
- M. Abrahamsson, M. Jäger, T. Österman, L. Eriksson, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 12616–12617 CrossRef CAS PubMed.
- D. Hara, Y. Umehara, A. Son, W. Asahi, S. Misu, R. Kurihara, T. Kondo and K. Tanabe, ChemBioChem, 2018, 19, 956–962 CrossRef CAS.
- L. Cosgrave, M. Devocelle, R. J. Forster and T. E. Keyes, Chem. Commun., 2010, 46, 103–105 RSC.
- C. S. Burke, A. Byrne and T. E. Keyes, Angew. Chem., Int. Ed., 2018, 57, 12420–12424 CrossRef CAS PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 RSC.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658 RSC.
- R. I. Dmitriev, H. M. Ropiak, G. V. Ponomarev, D. V. Yashunsky and D. B. Papkovsky, Bioconjugate Chem., 2011, 22, 2507–2518 CrossRef CAS PubMed.
- B. D. MacCraith, C. M. McDonagh, G. O’Keeffe, E. T. Keyes, J. G. Vos, B. O’Kelly and J. F. McGilp, Analyst, 1993, 118, 385–388 RSC.
- S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Iida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 4490–4498 CrossRef CAS PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300–15309 CrossRef CAS.
- S. Tobita, T. Yoshihara, R. Mukai, Y. Shimoda, I. Takahashi and H. Akiyama, Photochem. Photobiol. Sci., 2018, 17, 846 CrossRef PubMed.
- D. Wencel, C. Dolan, M. Barczak, T. E. Keyes and C. McDonagh, Nanotechnology, 2013, 24, 365705 CrossRef CAS PubMed.
- D. Hara, Y. Umehara, A. Son, W. Asahi, S. Misu, R. Kurihara, T. Kondo and K. Tanabe, ChemBioChem, 2018, 19, 956–962 CrossRef CAS.
- R. I. Dmitriev, S. M. Borisov, H. Düssmann, S. Sun, B. J. Müller, J. Prehn, V. P. Baklaushev, I. Klimant and D. B. Papkovsky, ACS Nano, 2015, 9, 5275–5288 CrossRef CAS PubMed.
- Z. Jiang, X. Yu, S. Zhai and Y. Hao, Sensors, 2017, 17, 548 CrossRef.
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885 RSC.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed.
- D. Hara, H. Komatsu, A. Son, S. Nishimoto and K. Tanabe, Bioconjugate Chem., 2015, 26, 645–649 CrossRef CAS PubMed.
- H. Bian, X. Song, N. Li, H. Man and Y. Xiao, J. Mater. Chem. B, 2018, 6, 1699–1705 RSC.
- K. Y. Zhang, P. Gao, G. Sun, T. Zhang, X. Li, S. Liu, Q. Zhao, K. K.-W. Lo and W. Huang, J. Am. Chem. Soc., 2018, 140, 7827–7834 CrossRef CAS.
- T. Yoshihara, S. Murayama and S. Tobita, Sensors, 2015, 15, 13503–13521 CrossRef CAS PubMed.
- Y.-E. L. Koo, Y. Cao, R. Kopelman, S. M. Koo, M. Brasuel and M. A. Philbert, Anal. Chem., 2004, 76, 2498–2505 CrossRef CAS PubMed.
- X. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. M. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901 RSC.
- A. V. Kondrashina, R. I. Dmitriev, S. M. Borisov, I. Klimant, I. O’Brien, Y. M. Nolan, A. V. Zhdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931–4939 CrossRef CAS.
- R. I. Dmitriev, S. M. Borisov, H. Düssmann, S. Sun, B. J. Müller, J. Prehn, V. P. Baklaushev, I. Klimant and D. B. Papkovsky, ACS Nano, 2015, 9, 5275–5288 CrossRef CAS PubMed.
- Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Chem. Sci., 2015, 6, 1825–1831 RSC.
- Q. Zhao, T. Pan, G. Xiang, Z. Mei, J. Jiang, G. Li, X. Zou, M. Chen, D. Sun, S. Jiang and Y. Tian, Sens. Actuators, B, 2018, 273, 242–252 CrossRef CAS.
- L. Zang, H. Zhao, J. Hua, F. Qin, Y. Zheng, Z. Zhang and W. Cao, Sens. Actuators, B, 2016, 231, 539–546 CrossRef CAS.
- P. J. Cywinski, A. J. Moro, S. E. Stanca, C. Biskup and G. J. Mohr, Sens. Actuators, B, 2009, 135, 472–477 CrossRef CAS.
- X.-H. Wang, H.-S. Peng, H. Ding, F.-T. You, S.-H. Huang, F. Teng, B. Dong and H.-W. Song, J. Mater. Chem., 2012, 22, 16066 RSC.
- D. Lambrechts, M. Roeffaers, G. Kerckhofs, S. J. Roberts, J. Hofkens, T. Van de Putte, H. Van Oosterwyck and J. Schrooten, Biomaterials, 2013, 34, 922–929 CrossRef CAS PubMed.
- T. Mayr, S. M. Borisov, T. Abel, B. Enko, K. Waich, G. Mistlberger and I. Klimant, Anal. Chem., 2009, 81, 6541–6545 CrossRef CAS.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed.
- H. Shi, X. Ma, Q. Zhao, B. Liu, Q. Qu, Z. An, Y. Zhao and W. Huang, Adv. Funct. Mater., 2014, 24, 4823–4830 CrossRef CAS.
- A. Martin, A. Byrne, C. Dolan, R. J. Forster and T. E. Keyes, Chem. Commun., 2015, 51, 15839–15841 RSC.
- S. M. King, S. Claire, R. I. Teixeira, A. N. Dosumu, A. J. Carrod, H. Dehghani, M. J. Hannon, A. D. Ward, R. Bicknell, S. W. Botchway, N. J. Hodges and Z. Pikramenou, J. Am. Chem. Soc., 2018, 140, 10242–10249 CrossRef CAS PubMed.
- Y. Cao, Y.-E. Lee Koo and R. Kopelman, Analyst, 2004, 129, 745 RSC.
- C. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741–2745 CrossRef CAS PubMed.
- X. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. M. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901 RSC.
- J. Napp, T. Behnke, L. Fischer, C. Würth, M. Wottawa, D. M. Katschinski, F. Alves, U. Resch-Genger and M. Schäferling, Anal. Chem., 2011, 83, 9039–9046 CrossRef CAS PubMed.
- N. W. Choi, S. S. Verbridge, R. M. Williams, J. Chen, J.-Y. Kim, R. Schmehl, C. E. Farnum, W. R. Zipfel, C. Fischbach and A. D. Stroock, Biomaterials, 2012, 33, 2710–2722 CrossRef CAS PubMed.
- A. V. Kondrashina, R. I. Dmitriev, S. M. Borisov, I. Klimant, I. O’Brien, Y. M. Nolan, A. V. Zhdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931–4939 CrossRef CAS.
- R. I. Dmitriev, S. M. Borisov, H. Düssmann, S. Sun, B. J. Müller, J. Prehn, V. P. Baklaushev, I. Klimant and D. B. Papkovsky, ACS Nano, 2015, 9, 5275–5288 CrossRef CAS.
- Y.-E. Koo Lee, E. E. Ulbrich, G. Kim, H. Hah, C. Strollo, W. Fan, R. Gurjar, S. Koo and R. Kopelman, Anal. Chem., 2010, 82, 8446–8455 CrossRef CAS.
- A. Byrne, J. Jacobs, C. S. Burke, A. Martin, A. Heise and T. E. Keyes, Analyst, 2017, 142, 3400–3406 RSC.
- G. Bœuf, G. V. Roullin, J. Moreau, L. Van Gulick, N. Zambrano Pineda, C. Terryn, D. Ploton, M. C. Andry, F. Chuburu, S. Dukic, M. Molinari and G. Lemercier, ChemPlusChem, 2014, 79, 171–180 CrossRef.
- W. Sun, S. Li, B. Häupler, J. Liu, S. Jin, W. Steffen, U. S. Schubert, H.-J. Butt, X.-J. Liang and S. Wu, Adv. Mater., 2017, 29, 1603702 CrossRef PubMed.
- J. Shen, H.-C. Kim, J. Wolfram, C. Mu, W. Zhang, H. Liu, Y. Xie, J. Mai, H. Zhang, Z. Li, M. Guevara, Z.-W. Mao and H. Shen, Nano Lett., 2017, 17, 2913–2920 CrossRef CAS PubMed.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS.
- Y. Ellahioui, M. Patra, C. Mari, R. Kaabi, J. Karges, G. Gasser and S. Gómez-Ruiz, Dalton Trans., 2019, 48, 5940–5951 RSC.
- N. Soliman, L. K. McKenzie, J. Karges, E. Bertrand, M. Tharaud, M. Jakubaszek, V. Guérineau, B. Goud, M. Hollenstein, G. Gasser and C. M. Thomas, Chem. Sci., 2020, 11, 2657–2663 RSC.
- A. Martin, R. D. Moriarty, C. Long, R. J. Forster and T. E. Keyes, Asian J. Org. Chem., 2013, 2, 763–778 CrossRef CAS.
- C. S. Burke and T. E. Keyes, RSC Adv., 2016, 6, 40869–40877 RSC.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204 RSC.
- T. Gunnlaugsson, J. P. Leonard, K. Sénéchal and A. J. Harte, J. Am. Chem. Soc., 2003, 125, 12062–12063 CrossRef CAS.
- J. N. Demas, E. W. Harris and R. P. McBride, J. Am. Chem. Soc., 1977, 99, 3547–3551 CrossRef CAS.
- K. Adamson, C. Dolan, N. Moran, R. J. Forster and T. E. Keyes, Bioconjugate Chem., 2014, 25, 928–944 CrossRef CAS PubMed.
- G. A. Crosby and R. J. Watts, J. Am. Chem. Soc., 1971, 93, 3184–3188 CrossRef CAS.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 337–342 CrossRef CAS.
- S. Draxler, M. E. Lippitsch, I. Klimant, H. Kraus and O. S. Wolfbeis, J. Phys. Chem., 1995, 99, 3162–3167 CrossRef CAS.
- C. Zhou, L. Ma, J. Ping, L. Guo, J. Qin, M. Yuan, Z. Geng, F. You and H. Peng, Anal. Bioanal. Chem., 2020, 412, 2579–2587 CrossRef CAS PubMed.
- R. M. Bukowski, R. Ciriminna, M. Pagliaro and F. V. Bright, Anal. Chem., 2005, 77, 2670–2672 CrossRef CAS PubMed.
- J. Jacobs, N. Gathergood and A. Heise, Macromol. Rapid Commun., 2013, 34, 1325–1329 CrossRef CAS PubMed.
- J. Jacobs, A. Byrne, N. Gathergood, T. E. Keyes, J. P. A. Heuts and A. Heise, Macromolecules, 2014, 47, 7303–7310 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
- Y. Michiels, P. Puyvelde and B. Sels, Appl. Sci., 2017, 7, 665 CrossRef.
- C.-W. Lai, Y.-H. Wang, C.-H. Lai, M.-J. Yang, C.-Y. Chen, P.-T. Chou, C.-S. Chan, Y. Chi, Y.-C. Chen and J.-K. Hsiao, Small, 2008, 4, 218–224 CrossRef CAS PubMed.
- S. P.-Y. Li, C. T.-S. Lau, M.-W. Louie, Y.-W. Lam, S. H. Cheng and K. K.-W. Lo, Biomaterials, 2013, 34, 7519–7532 CrossRef CAS PubMed.
- P. Ceroni, A. Y. Lebedev, E. Marchi, M. Yuan, T. V. Esipova, G. Bergamini, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Photochem. Photobiol. Sci., 2011, 10, 1056 CrossRef CAS PubMed.
- S. Hirohara, M. Obata, H. Alitomo, K. Sharyo, T. Ando, S. Yano and M. Tanihara, Bioconjugate Chem., 2009, 20, 944–952 CrossRef CAS PubMed.
- A. Ruggi, F. W. B. van Leeuwen and A. H. Velders, Coord. Chem. Rev., 2011, 255, 2542–2554 CrossRef CAS.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS.
- A. K. Oyelere, P. C. Chen, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Bioconjugate Chem., 2007, 18, 1490–1497 CrossRef CAS PubMed.
- B. Kang, M. A. Mackey and M. A. El-Sayed, J. Am. Chem. Soc., 2010, 132, 1517–1519 CrossRef CAS.
- N. S. Chandel and P. T. Schumacker, J. Appl. Physiol., 2000, 88, 1880–1889 CrossRef CAS PubMed.
- C. T. Taylor, Biochem. J., 2008, 409, 19–26 CrossRef CAS PubMed.
- H. Bian, X. Song, N. Li, H. Man and Y. Xiao, J. Mater. Chem. B, 2018, 6, 1699–1705 RSC.
- P. V. Escribá, Trends Mol. Med., 2006, 12, 34–43 CrossRef.
- P. V. Escribá, J. M. González-Ros, F. M. Goñi, P. K. J. Kinnunen, L. Vigh, L. Sánchez-Magraner, A. M. Fernández, X. Busquets, I. Horváth and G. Barceló-Coblijn, J. Cell. Mol. Med., 2008, 12, 829–875 CrossRef.
- D. C. Wallace, Nat. Rev. Cancer, 2012, 12, 685–698 CrossRef CAS PubMed.
- R. J. DeBerardinis and N. S. Chandel, Sci. Adv., 2016, 2, e1600200 CrossRef.
- I. Mellman and Y. Yarden, Cold Spring Harbor Perspect. Biol., 2013, 5, a016949–a016949 CrossRef.
- N. Bernardes and A. Fialho, IJMS, 2018, 19, 3871 CrossRef PubMed.
- M. Sok, M. Šentjurc, M. Schara, J. Stare and T. Rott, Ann. Thorac. Surg., 2002, 73, 1567–1571 CrossRef PubMed.
- R. F. A. Zwaal, P. Comfurius and E. M. Bevers, CMLS, Cell. Mol. Life Sci., 2005, 62, 971–988 CrossRef CAS PubMed.
- S. Riedl, B. Rinner, M. Asslaber, H. Schaider, S. Walzer, A. Novak, K. Lohner and D. Zweytick, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2638–2645 CrossRef CAS PubMed.
- A. Fercher, S. M. Borisov, A. V. Zhdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499–5508 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- J. Gruenberg and H. Stenmark, Nat. Rev. Mol. Cell Biol., 2004, 5, 317–323 CrossRef CAS PubMed.
- J. P. Luzio, P. R. Pryor and N. A. Bright, Nat. Rev. Mol. Cell Biol., 2007, 8, 622–632 CrossRef CAS.
- J. M. Kelly, A. B. Tossi, D. J. McConnell and C. OhUigin, Nucl. Acids Res., 1985, 13, 6017–6034 CrossRef CAS PubMed.
- M. B. Fleisher, K. C. Waterman, N. J. Turro and J. K. Barton, Inorg. Chem., 1986, 25, 3549–3551 CrossRef CAS.
- Y. Liu, R. Hammitt, D. A. Lutterman, L. E. Joyce, R. P. Thummel and C. Turro, Inorg. Chem., 2009, 48, 375–385 CrossRef CAS PubMed.
- R. D. Srivastava, A. F. Mcmillan and I. J. Harris, Can. J. Chem. Eng., 1968, 46, 181–184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR analysis, mass spectrometry, photophysical data, lifetime decays, phototoxicity, and uptake studies. See DOI: 10.1039/d1cb00102g |
| This journal is © The Royal Society of Chemistry 2021 |