
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting the RNA demethylase FTO for cancer therapy
Lin-Lin
Zhou
 ab,
Hongjiao
Xu
ab,
Yue
Huang
ac and
Cai-Guang
Yang
*abc
ab,
Hongjiao
Xu
ab,
Yue
Huang
ac and
Cai-Guang
Yang
*abc
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: yangcg@simm.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, China
cSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 26th July 2021
Abstract
N 6-Methyladenosine (m6A) is the most prevalent internal modification on mRNA and represents a new layer of gene expression in eukaryotes. The field of m6A-encoded epitranscriptomics was rejuvenated with the discovery of fat mass and obesity-associated protein (FTO) as the first m6A demethylase responsible for RNA modification in cells. Increasing evidence has revealed that FTO is significantly involved in physiological processes, and its dysregulation is implicated in various human diseases. Considering this functional significance, developing small-molecule modulators of the FTO protein represents a novel direction for biology research. However, such modulators remain in the early stages of development. Here, our review mainly focuses on the progress of FTO inhibitor development to date. We summarize screening methods used to identify FTO modulators, techniques used to assess the biological effects of these modulators, strategies used to achieve selective inhibition of FTO rather than its homologues, and the results of investigations of FTO modulator modes of action and anticancer efficacy. Thus, this review aims to facilitate novel chemical entity discovery, probe FTO biology, and promote the validation of FTO as a clinical drug target for cancer treatment.
 Lin-Lin Zhou | Lin-Lin Zhou is a graduate student at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. She studies chemical intervention on untargeted proteins for anticancer therapy in the Yang lab. She received an BS degree in Pharmacy at China Pharmaceutical University in 2018. |
 Hongjiao Xu | Hongjiao Xu studied the biological function of FTO with FTO inhibitors as chemical tools in the Yang lab, and received her PhD degree at Shanghai Institute of Materia Medica and the University of Chinese Academy of Sciences in 2021. She received her BS degree at Shandong University in 2016. |
 Yue Huang | Yue Huang graduated from Nanjing Normal University in 2012 and received her PhD degree in 2017 from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, under the supervision of Professor Cai-Guang Yang. After the postdoctoral training at Shanghai Institute of Materia Medica, she was appointed as an assistant professor in 2019, and promoted to an associate professor in 2020. Her research interest lies in the development of chemical probes of RNA demethylase FTO and the application in drug discovery. |
 Cai-Guang Yang | Cai-Guang Yang is a professor at Shanghai Institute of Materia Medica. His research interests span many traditional and emerging disciplines of chemistry and biology with noteworthy focuses on the use of medicinal chemistry, structural biology and cell biology to target the untargeted proteins for drug discovery. He received his BS degree from Huazhong University of Science and Technology in 1997, and then studied total synthesis of natural products at Shanghai Institute of Organic Chemistry and earned his PhD in 2002. His postdoctoral training focused on the mechanism of protein/DNA interaction in Chemistry Department at the University of Chicago until 2008. |
1. Introduction
Extensive progress has been made in understanding both the physiological and pathological significance of DNA methylation and histone modifications since the field of epigenetics was introduced in 1942.1–3 However, although N6-methyladenosine (m6A) on mRNA was first reported in 1974, and more than 170 types of RNA modifications have been identified to date,4–6 the biology of RNA modifications remained largely underexplored until the identification of the first RNA demethylase, fat mass and obesity-associated protein (FTO), in 2011.7 The discovery of m6A on mRNA as the cellular substrate of FTO demethylase has revealed the reversible and dynamic features of RNA methylation.8 Along with the development of the MeRIP–Seq method for m6A profiling,9,10 epitranscriptomics has emerged as a new research field, and a race to develop drugs that can target the “epitranscriptome code” has followed.11Emerging investigations of FTO functions have clearly demonstrated the essential role of FTO in both physiological and pathological processes. Dysregulation of FTO demethylation has been identified as a driving factor in various diseases, including cancers, metabolic diseases, and neuropsychiatric disorders.12–14 Therefore, efforts have been made to develop small-molecule inhibitors of FTO to probe m6A biology and therapeutically target m6A modifications, which has been partially reviewed previously.15–18 Our review will focus on potential strategies for achieving selective inhibition of FTO over other AlkB homologues, summarize screening methods, and underline the mode of action and potency of FTO inhibitors discovered to date in detail. Thus, this review aims to promote the development of novel chemical entities as chemical tools to probe FTO biology and investigate the druggability of FTO demethylase for anticancer.
2. Biology of the FTO demethylase and its association with cancer
Reversible m6A methylation is the best-characterized modification of mRNA, which is strictly regulated through a functional network of writer (METTL3/METTL14 complex and METTL16), eraser (FTO and ALKBH5) and reader proteins (YTH domain-containing proteins, etc.) (Fig. 1A).19 The m6A modification of mRNA plays a regulatory role in diverse physiological processes as well as cancer progression by controlling gene expression.20,21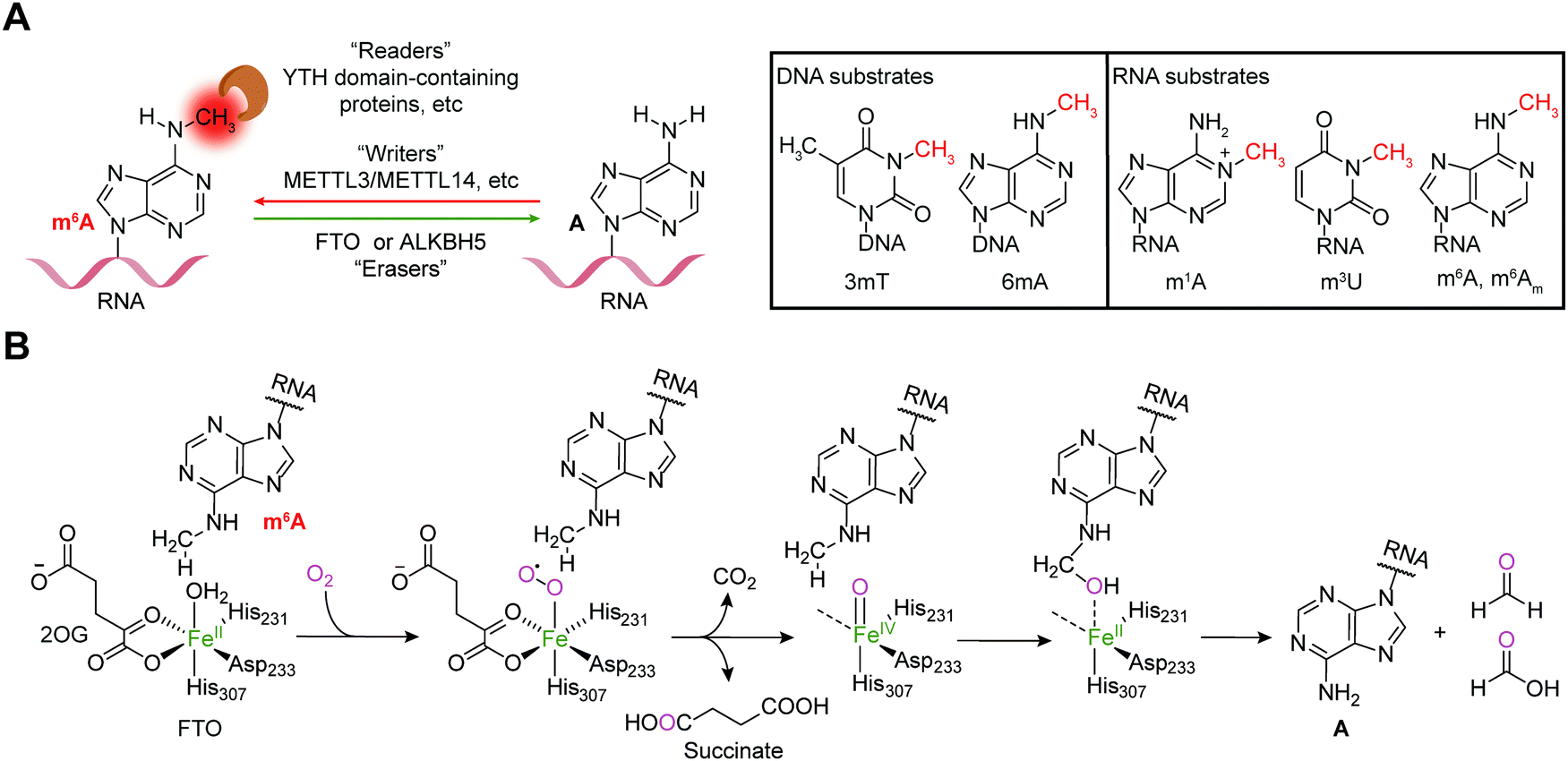 | ||
| Fig. 1 Dynamic and reversible m6A modification. (A) m6A is regulated by writers (METTL3/METTL14 and METTL16) and erasers (FTO and ALKBH5) and recognized by readers (YTH domain-containing proteins, etc.); the substrates that can be demethylated by FTO are shown. (B) The catalytic mechanism of m6A demethylation by FTO. | ||
The FTO gene was initially identified to be involved in obesity and type 2 diabetes in three independent genome-wide association studies (GWAS) (Fig. 2),22–24 which encodes the FTO protein that belongs to the Fe2+ and 2-oxoglutarate (2OG)-dependent AlkB dioxygenase family.25 FTO was initially reported to catalyse the oxidative demethylation of N3-methylthymine (3mT) in single-stranded DNA (ssDNA) and N3-methyluridine (m3U) in single-stranded RNA (ssRNA) in vitro with low efficiency (Fig. 1A).25,26 Later, in 2011, m6A in nuclear RNA was established as the major substrate of FTO both in vitro and in living cells.7 Mechanistically, FTO use cofactors 2OG and Fe2+ to catalyse the removal of m6A (Fig. 1B), and it generates N6-hydroxymethyladenosine and N6-formyladenosine in a stepwise manner.27 Notably, recent studies have revealed multiple substrates of FTO in addition to m6A on mRNA, including m6A on small nuclear RNAs (snRNAs), cap N6,2′-O-dimethyladenosine (m6Am) on messenger RNAs (mRNAs) and snRNAs, and N1-methyladenosine (1mA in DNA and m1A on RNA) on transfer RNAs (tRNAs) (Fig. 1A).28,29 Although the precise mechanism used by FTO to selectively recognize and demethylate the methylated nucleic acids remains largely underexplored,30–32 these studies of the m6A demethylase FTO have revealed the reversibility and dynamics of RNA methylations.
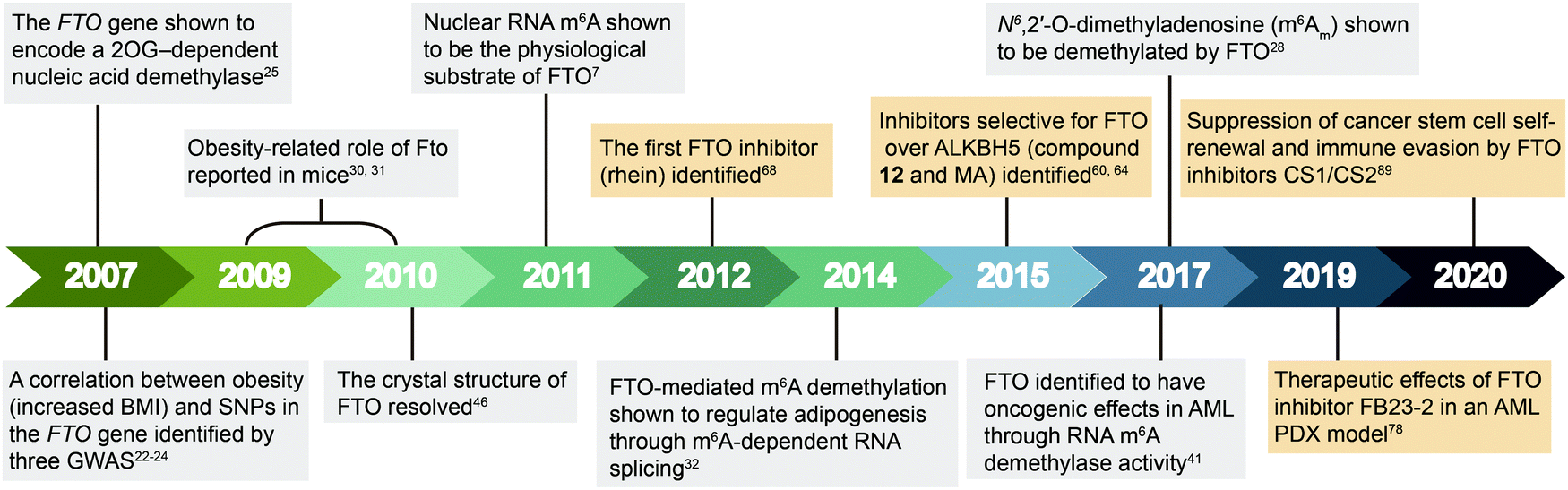 | ||
| Fig. 2 Timeline of key discoveries related to FTO. Studies of FTO biology (grey boxes) and inhibitors (wheat-coloured boxes) are indicated. | ||
FTO exhibits complicated biological functions in physiological processes, and dysregulation of FTO has been connected to various human diseases. Genetic manipulation in mouse models has verified the obesity-related function of FTO: Fto-deficiency leads to reduced adipose tissue and a lean phenotype, while overexpression of Fto increases body and fat mass.33,34 FTO-facilitated m6A demethylation in adipocytes was found to regulate mRNA splicing and affect preadipocyte differentiation, which provided the first evidence of the effects of FTO-dependent m6A regulation on mRNA splicing.35 The Fto gene is widely expressed in both foetal and adult tissues, with the highest expression in the brain,25 and it has been reported that the Fto gene regulates dopaminergic signalling to control neuronal activity in dopaminergic neurons as well as the behavioural response through demethylation of a set of mRNAs in vivo.36 FTO also participates in postnatal neurodevelopment and adult neurogenesis in vivo.37 Therefore, it is not surprising that FTO has been indicated to be involved in neuropsychiatric diseases such as Alzheimer's disease, Parkinson's disease, epilepsy, anxiety and depression.13
Abnormal expression of FTO demethylase promote tumorigenesis, progression and chemoresistance in several cancers.14,38–44 Significantly high expression of FTO has been observed in cancers compared with adjacent tissues.14 Estrogen-induced FTO accumulation in nucleus through mTOR signalling pathway enhanced the proliferation of endometrial cancer cells.45 Increased FTO expression level in gastric cancer tissues also correlates with tumour stages and predicts poor prognosis in patients, and forced expression of FTO increased the proliferation, migration and invasion in gastric cancer cell lines.46 Li et al. provided the first evidence that FTO plays an oncogenic role through downregulating the expression of downstream targets RARA and ASB2 in a m6A-dependent manner in acute myeloid leukaemia (AML).47 Overexpressed FTO facilitated glycolysis of breast cancer cells through PI3K/AKT signalling pathway.48 Yang et al. reported that FTO is upregulated in human-derived melanoma samples and cell lines, and increased FTO has oncogenic effects by modulating the expression of critical protumorigenic cell-intrinsic genes such as PD-1, CXCR4 and SOX10 via m6A modification.41 FTO is also overexpressed in VHL-deficient clear cell renal cell carcinoma (ccRCC) tumours compared to adjacent tissues. Genetic inhibition of FTO impairs the growth and survival of VHL-deficient cancer cells.49 In non-small-cell lung cancer, upregulation of FTO facilitates tumour progression by regulating the expression of USP7 and MZF1.39,40 In addition, high expression of FTO causes resistance to chemotherapy and radiotherapy. For example, increased FTO protein enhances the anti-PD-1 resistance in melanoma,41 inhibits all-trans-retinoic acid-induced AML cell differentiation,47 contributes to acquired resistance to tyrosine kinase inhibitors in leukaemia cells,50 and causes resistance to chemo-radiotherapy by targeting β-catenin in cervical squamous cell carcinoma.51 On the contrary, down-regulated FTO has been also observed in some cases,52,53 for example, expression of FTO is significantly lower compared with normal control and knockdown of FTO enhances the proliferation and migration in bladder cancer cells; FTO in ovarian cancer stem cells hindered the self-renewal and inhibited tumorigenesis in vivo. In summary, both up- and down-regulated FTO demethylase have been suggested to drive tumorigenesis and progression, which indicates the necessity to identify precise biomarkers in the development of FTO inhibitors as anticancer therapy.
FTO is functionally important in physiological processes, and its dysregulation has been associated with various human diseases. Therefore, developing small-molecule modulators to exploit its biological function and understanding the pathogenic mechanisms in-depth, as well as performing target validation for the treatment of human diseases, especially cancer, are warranted.
3. Structural insights into FTO
Determinations of the FTO structure have revealed the mechanisms of substrate recognition and catalytic demethylation, which has facilitated the structure-based design of small-molecule inhibitors. Initially, the crystal structure of FTO was determined in the presence of the 3mT mononucleotide (PDB: 3LFM), and later, it was determined with bound N6-methyldeoxyadenosin (6mA)-modified ssDNA (PDB: 5ZMD).54,55 FTO is composed of two domains, a catalytic N-terminal domain (NTD, residues 32–326, yellow) and a C-terminal domain (CTD, residues 327–498, grey), a newly identified fold with no significant homology (Fig. 3). The NTD contains a double-stranded β-helix fold termed the jelly-roll motif (corresponding to residues 201–322); this motif is highly conserved in the AlkB family as a catalytic domain in which a metal ion is accommodated by the conserved residues H231, D233 and H307, and the bidentate ligand N-oxalylglycine (NOG, an inert 2OG analogue) is mainly stabilized through hydrogen bonds formed with the side chains of N205, Y295, R316, S318, and R322 (Fig. 3). Furthermore, Zhang et al. determined the structure of FTO bound to 6mA-modified ssDNA, which revealed the mechanism underlying the preference of FTO for the nucleobase N6-methyladenine over other reported substrates.55 6mA is extensively stabilized in the substrate binding site, in which the purine ring is stacked by multiple residues through hydrophobic interactions (with residues Y108, L109, V228, and H231) and hydrogen binding (between the N1, N6, and N7 positions of 6mA and the R96 and E234 residues of FTO), the methyl group localizes well in a hydrophobic pocket (formed by R96, Y106, Y108, L203, and R322), and the deoxyribose ring is stabilized mainly through hydrophobic interactions (with residues I85, V228, S229, W230, and H231) (Fig. 3). The structure of FTO bound to 6mA-modified ssDNA indicates that the N6-methyladenine instead of the ribose ring determines the substrate specificity of FTO.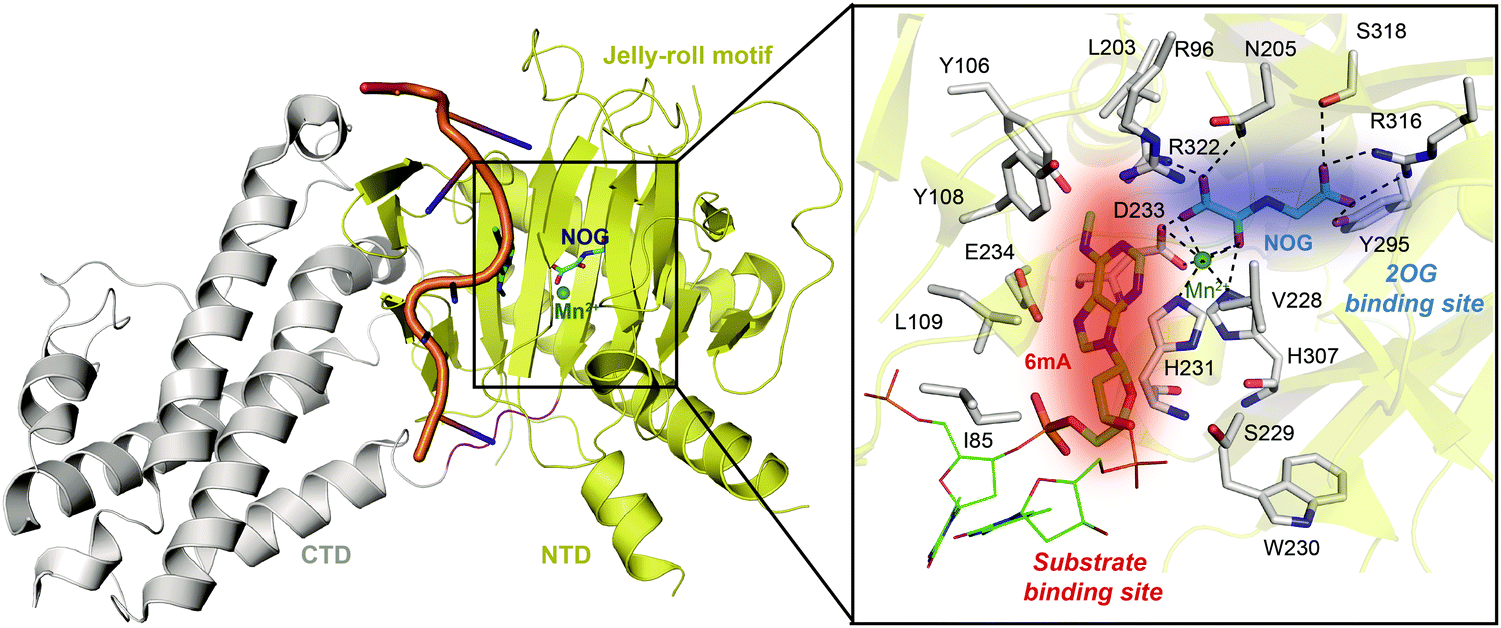 | ||
| Fig. 3 Overall structure of FTO bound to 6mA-modified ssDNA (PDB: 5ZMD).55 The CTD is shown in grey, the NTD is shown in yellow, and the modified ssDNA is shown in orange. The black box shows a magnified view of the connections FTO makes to accommodate NOG (cyan) and Mn2+ (green sphere). The side chains of key residues interacting with 6mA (green) and cofactors within the jelly-roll motif are labelled and shown as grey sticks. The substrate binding site and 2OG binding site are indicated in red and blue, respectively. | ||
The CTD is mainly composed of α-helixes; in the CTD, one three-helix bundle interacts extensively with the NTD, which likely plays an indispensable role in maintaining the catalytic activity of FTO.54 The exact mechanism remains largely unknown, however. Recently, it has been suggested that SFPQ, an adaptor protein facilitating RNA-processing enzymes, interacts with the CTD of FTO to mediate selective demethylation on m6A-containing RNAs.56
To further investigate the structural features of FTO homologues, a structure-based sequence alignment of FTO (PDB: 5ZMD),55 ALKBH2 (PDB: 3RZL),31 ALKBH3 (PDB: 2IUW),57 ALKBH5 (PDB: 4NJ4)58 and AlkB (4RFR)59 was performed with the web server mTM-align and ESPript (Fig. 4A).60–62 The alignment revealed the conserved jelly-roll motif for the coordinating cofactors Fe2+ and 2OG (Fig. 4B). The alignment also indicated that one of the most notable structural differences between FTO and other AlkB family proteins is a long loop covering one side of the jelly-roll motif (L1, residues 213–224, violet box in Fig. 4A and magenta in Fig. 4C). This unique loop exhibits steric hindrance for double-stranded DNA (dsDNA)/RNA and interacts with the base of the RNA substrate, thus playing an important role in substrate recognition.55 The other RNA demethylase, ALKBH5, also possesses an α-helix to block dsDNA/RNA access (β4–5, residues 230–243, orange box in Fig. 4A and orange in Fig. 4C).58 The structures of AlkB, ALKBH2, and ALKBH3 lack a similar loop motif, however.
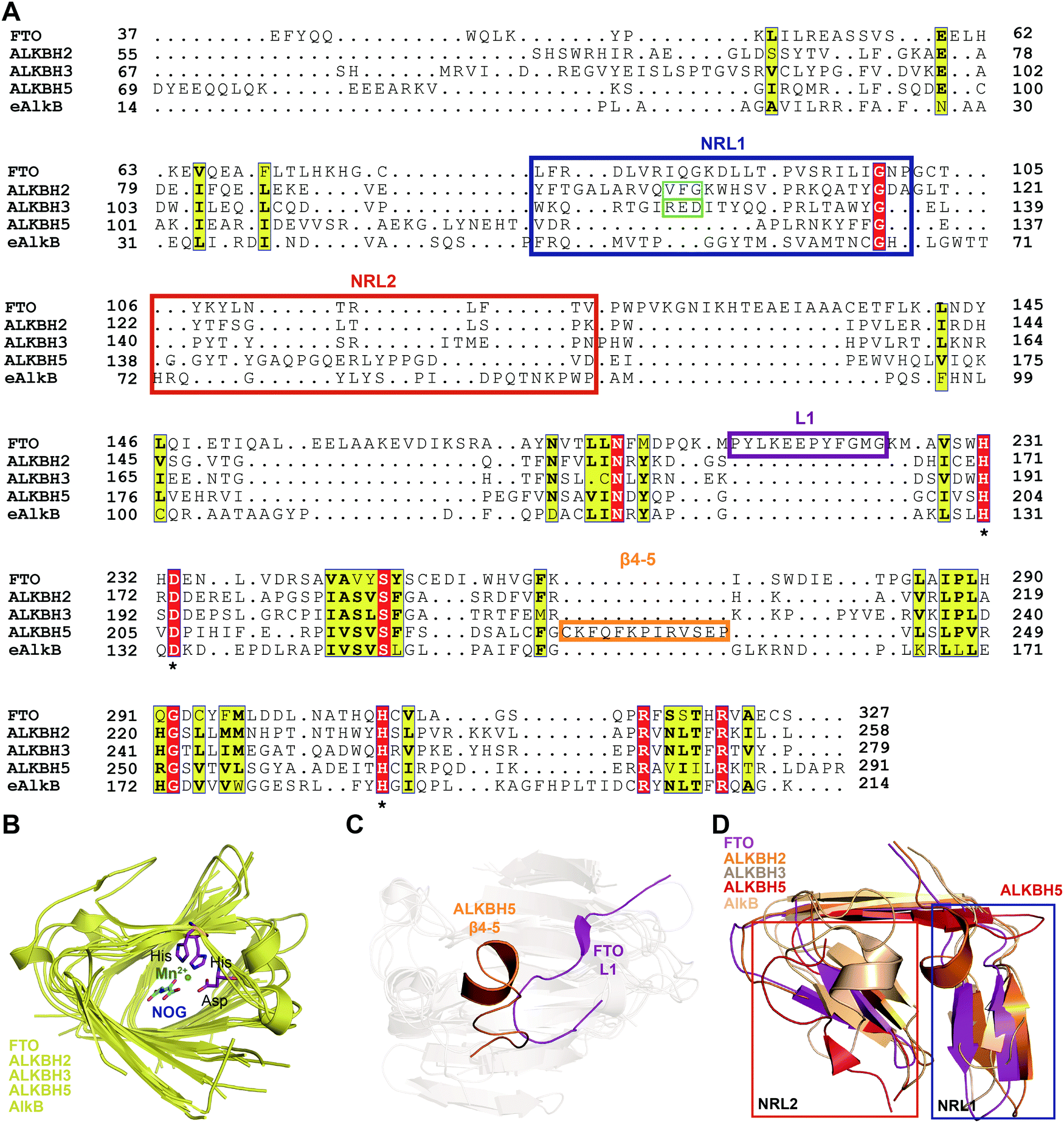 | ||
| Fig. 4 Structure-based sequence alignment and structural analysis. (A) Structure-based sequence alignment of FTO (PDB: 5ZMD), ALKBH2 (PDB: 2RZL), ALKBH3 (PDB: 2IUW), ALKBH5 (PDB: 4NJ4) and AlkB (PDB: 4RFR), performed with the web servers mTM-align (https://yanglab.nankai.edu.cn/mTM-align/) and ESPript 3.0 (http://espript.ibcp.fr). The highly conserved residues H231, D233 and H307 that enable binding of Fe2+ are labelled with ‘*’ under the sequences. Boxes indicate the residues that form NRL1 (blue), NRL2 (red), the insertion loop L1 of FTO (violet), and β4–5 of ALKBH5 (orange); the residues that exert the substrate specificity of ALKBH2 (cyan) and ALKBH3 (green) are also shown. (B) Structural alignment of the conserved jelly-roll motif shared by AlkB family proteins. The conserved residues are shown as magenta sticks using those of FTO as a representative, and Mn2+ is shown as a green sphere. (C) The non-conserved loops among AlkB family proteins. L1 of FTO is highlighted in magenta, and β4–5 of ALKBH5 is highlighted in red. (D) NRL comparisons between AlkB family proteins. The NRL1 conformation of ALKBH5 (red) exhibits extreme differences that leave a large space over the active site pocket. The alignment in Fig. 4B–D was conducted using FTO (PDB: 5ZMD), ALKBH2 (PDB: 3RZL), ALKBH3 (PDB: 2IUW), ALKBH5 (PDB: 4NJ4) and AlkB (PDB: 2FD8).63 | ||
The other structural difference is in the motif termed the nucleotide recognition lid (NRL), which was first proposed in the structure of Escherichia coli AlkB.63,64 The NRL motif (consisting of NRL1 and NRL2) covers the active site and exhibits conformational flexibility to help recognize and bind nucleotide substrates.63 This structure is not conserved across the AlkB family proteins, however. The diverse composition of amino acids in the NRL motif results in various conformations and distinctive interactions with different substrates, which partially determine the substrate specificity (blue and red boxes, respectively, in Fig. 4A and D). Although not identical in terms of amino acid sequence, AlkB, ALKBH2, ALKBH3 and FTO show similar structures over the active site. However, the NRL motif of ALKBH5 possess a distinct spatial structure that leaves a large open space over the active site (Fig. 4D).58 These differences in the exposure of the active site may confer substrate selectivity to AlkB family proteins. As m6A demethylases, FTO and ALKBH5 are likely to use this structural difference to partly determine substrate specificity. These structural features provide insight into the mechanisms underlying substrate preference and recognition and indicate potential strategies for developing selective small-molecule modulators.
4. Strategies to achieve selective inhibition of FTO
Given the essential roles of FTO in biology and pathology, great efforts have been devoted to developing small-molecule inhibitors as functional probes and potential lead compounds for therapeutic purposes. However, the development of selective inhibitors targeting FTO remains a great challenge due to the highly conserved jelly-roll motif shared by AlkB family members. Initially, small-molecule inhibitors mainly targeted cofactor binding sites, for example, 2OG competitors and iron chelators (green box in Fig. 5A), most of which also non-selectively inhibit other Fe2+- and 2OG-dependent dioxygenases. However, the composition and conformation of the nucleotide binding site as well as the adjacent regions are critical for specific recognition and binding of different methylated substrates (Fig. 5A). For instance, the determined structural complexes of rhein bound to AlkB and FTO show disparate occupancy within the active site (Fig. 5B), indicating that the structural differences between AlkB demethylases might offer an opportunity to achieve selective inhibition.59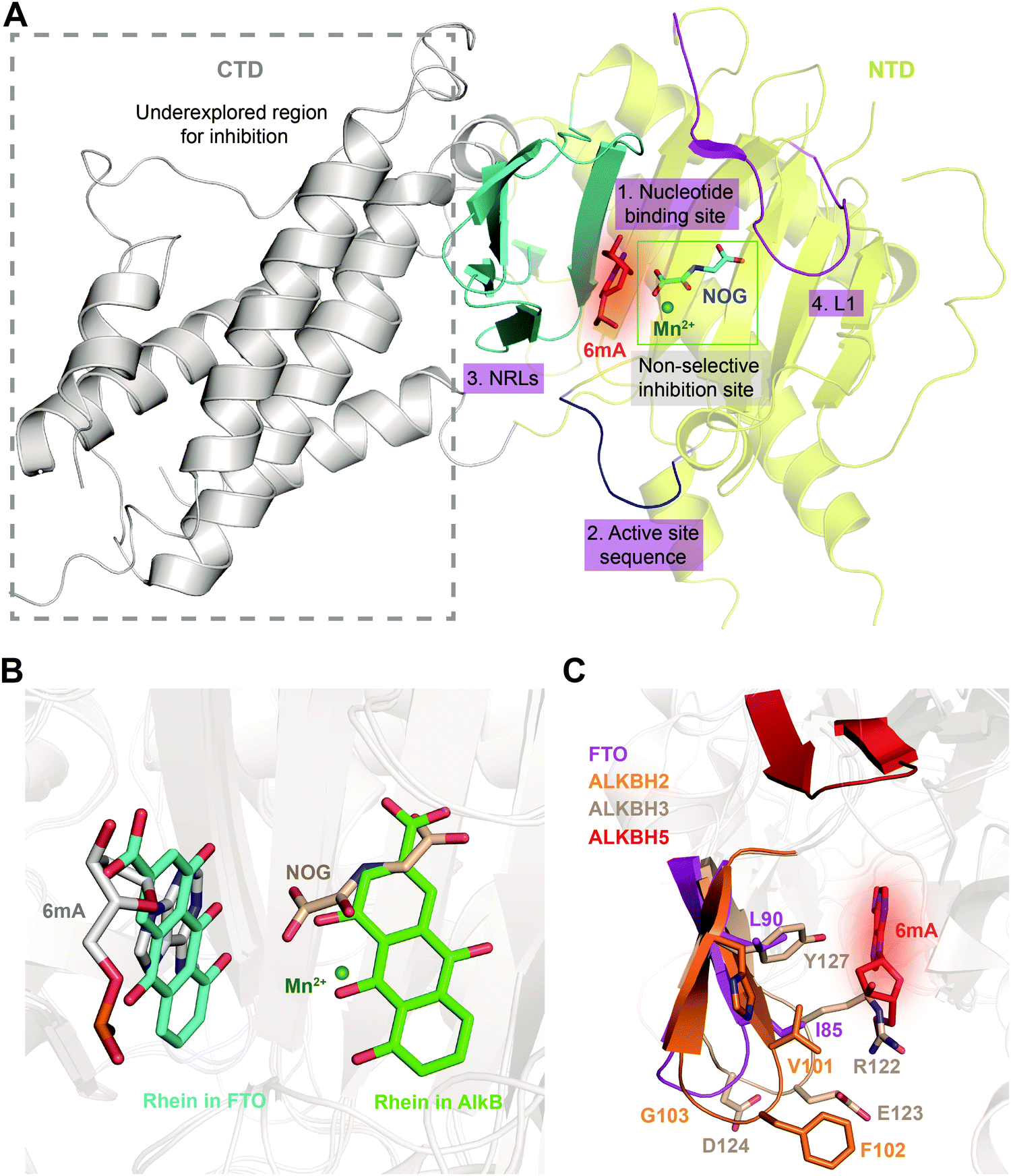 | ||
| Fig. 5 Strategies for achieving selective inhibition of FTO. (A) Structural features of FTO that can be exploited for the development of selective inhibitors. Inhibitors targeting the CTD have been underexplored. The CTD is shown in grey. The non-selective inhibition site (Mn2+ in green and NOG in cyan) is in the green box. The nucleotide binding site has a red background with 6mA shown as a red stick. The active site loop is shown in blue. The NRLs are shown in cyan, and L1 is coloured magenta. (B) Rhein inhibits FTO and AlkB through different mechanisms. Rhein occupies the 2OG binding site in AlkB and the substrate binding site in FTO. Structural alignment was performed using the FTO/rhein structure (PDB: 4IE7), AlkB/rhein structure (PDB: 4RFR) and FTO/6mA-ssDNA with a 6mA-modified nucleotide structure (PDB: 5ZMD). Rhein in FTO is represented by a cyan-coloured stick while that in AlkB is represented by a green-coloured stick. 6mA is represented by a grey stick, Mn2+ is shown as a green sphere, and NOG is represented by a wheat-coloured stick. (C) Structural comparison of NRL1 motifs. Different amino acid compositions of the loop endow ALKBH2 (orange) and ALKBH3 (wheat) with different substrate preferences. The amino acids of FTO NRL1 (magenta stick) are distinct between ALKBH2 and ALKBH3. The NRL1 region of ALKBH5 is relatively short compared with those of the other AlkB family proteins. The 6mA nucleotide is shown as a red stick. | ||
Intriguingly, the structural differences inside the nucleotide binding site could provide opportunities for achieving selective inhibition of FTO. As the structures indicate, several key residues (Y108, H231, L109, V228, R96 and E234) contribute to selective interactions between FTO and the methylated nucleotide (number 1, red in Fig. 5A).65 These residues in FTO are either substituted or absent in the other AlkB family members, which results in loss or weakened interaction between 3mT/6mA and the residues in FTO and therefore causes the low (even no) affinity of other AlkB demethylases for 3mT/6mA.65 In addition, the structure of FTO bound to 6mA-modified ssDNA shows that the hydrogen bonds formed by R96 and E234 of FTO are the most critical contributors to specific substrate base recognition; for example, the N6 and N7 atoms of 6mA form strong hydrogen bonds with the side chain of E234, while hydrogen bonds formed between the O4 atom of 3mT and E234 are weaker. Therefore, E234 in FTO has been revealed to participate in nucleobase selection and to recognize structures inside the catalytic pocket.55 The structural elements inside the nucleotide binding site of FTO make it possible to use a chemical moiety mimicking the 3mT/6mA interactions to achieve selective modulation of FTO. Because of the continuous pocket of the substrate and the 2OG binding sites, it is also possible to achieve selective inhibition by occupying both sites.
Additionally, a loop region within the active site (residues 235–240, number 2, blue cartoon in Fig. 5A) in FTO that is located in the C-terminally to the conserved HxD motif displays divergent amino acid composition across AlkB family demethylases and contributes to substrate specificity.30,66 Sequence alignment showed that this active site loop utilizes amino acids with neutral side chains to interact with neutrally charged 6mA/m6A and negative side chains to interact with positively charged 1mA/m1A substrates. A change of the amino acids in the loop region of FTO and ALKBH5 to those of AlkB or ALKBH2 results in increased 1mA-targeted demethylation and decreased 6mA-targeted demthylation.66 Crystallographic and kinetic studies of AlkB have illustrated that the distinctive active site loop (residues 135–139 in AlkB) plays a role in discriminating between 1mA and 6mA. These data suggested that this region is involved in substrate recognition and demethylase specificity among AlkB family proteins. Thus, exploiting the different sequences in the active site loops to aid the design of small molecules that can selectively modulate these homologous demethylases is reasonable.66
Furthermore, it might also be promising to rationally design inhibitors with selectivity by taking advantage of the different structural features outside the conserved jelly-roll motif. Except for ALKBH7, all structurally characterized AlkB family members (AlkB, ALKBH1, ALKBH2, ALKBH3, ALKBH5, ALKBH8 and FTO) possess NRLs, but these NRLs are divergent in composition and conformation. The V101–F102–G103 residues in NRL1 of ALKBH2 form a hydrophobic motif that is important for damage searching and repair of ssDNA and dsDNA (cyan box in Fig. 4 and 5C).30,31 In contrast, ALKBH3 utilizes charged, hydrophilic and polar residues (R122–E123–D124) and forms a bulky salt bridge to prevent the binding of a dsDNA substrate (green box in Fig. 4 and 5C).67 These sequence divergences determine the substrate specificity of ALKBH2 and ALKBH3. There are two parts of the NRL motif in FTO, residues 77–102 and 103–116 (number 3, cyan in Fig. 5A). The loop structure in NRL1 of FTO is shorter than that of other AlkB family members.55 Additionally, NRL1 in FTO features small and hydrophobic residues (I85 and L90), while these are replaced either by a hydrophilic residue (H106) in ALKBH2 or by bulky residues (R122 and Y127) in ALKBH3 (Fig. 5C).68 Compared with those of FTO, ALKBH2 and ALKBH3, the NRLs in ALKBH5 are distinctive, with one short part of NRL1 (residues 124–137) and another part of NRL2 (residues 138–161) exhibiting much flexibility (Fig. 5C). Collectively, the characteristics of NRLs at least partially determine the protein's substrate specificity. The structural differences in NRLs make it feasible to develop selective small-molecule inhibitors that interact with these regions. In addition, the insertion loop L1 of FTO is unique in amino acid composition and conformation, providing additional opportunity for selective inhibition beyond the active site (number 4, magenta in Fig. 5A).
Interestingly, the CTD of FTO helps stabilize the conformation of the NTD and is catalytically indispensable (grey in Fig. 5A). Mutations of F114D and C392D in the CTD, which would be expected to disrupt the NTD–CTD interaction, compromise the demethylase activity,54 and this result raises the possibility of allosterically regulating FTO activity by inducing a conformational change of the CTD. Such a change would affect the interactions between the CTD and NTD and eventually impair the demethylation activity. Since the CTD of FTO is unique, it is possible to selectively manipulate FTO activity by targeting this domain or its interaction with the NTD.69
5. Methods of screening FTO inhibitors
Given that the processes of FTO demethylation mainly consist of substrate recognition, substrate binding and catalysis, methods for identifying FTO inhibitors can be grouped into two categories: (1) methods detecting the changes induced by protein–ligand binding and (2) methods directly detecting demethylation-inhibiting activity. Biochemical and biophysical assays measure the changes in catalytic activity or the binding properties of FTO when compounds modulate or bind with it. In addition, cell-based assays generate a functional read-out relevant to FTO demethylation activity in living cells, which ultimately provides small-molecule modulators with intracellular functions (Table 1).| Protein-based assays | Cell-based assays | |||
|---|---|---|---|---|
| Binding detection | Demethylation activity determination | Demethylase activity determination | ||
| Antibody-dependent | Enzyme-dependent | Fluorescence- and radioactivity-dependent | ||
| Virtual screening | Dot blot | Dpn II | Fluorometric RNA probe | Fluorescent m6A-switchable probe |
| FP | INB | MazF | Radio-labelled RNA oligonucleotides | |
| DSF | HPLC-MS | |||
| Thermal shift | TLC | |||
| Capillary electrophoresis | Co-products release | |||
5.1 Binding-based assays for in vitro screening
Detecting FTO–ligand binding directly or measuring biophysical changes in target proteins caused by ligand binding are widely applied approaches for target-based hit identification. Computer-aided high-throughput virtual screening is typically the first choice when the structure of a target of interest is available. Several FTO inhibitors have been discovered through virtual screenings and further evaluation and structural optimization.Most experiment-based assays for identifying potential FTO inhibitors depend on biophysical methods, which either measure the binding affinity between the target protein and small-molecule ligands or monitor the disruption of FTO binding to methylated substrates in vitro. Frequently used assays include fluorescence intensity, capillary electrophoresis, fluorescence polarization (FP), differential scanning fluorimetry (DSF), and thermal shift assays. For instance, the FP assay measures the changes in the fluorescein signal induced by ligand binding and enables the identification of substrate-competitive inhibitors, while it might miss potential inhibitors that perturb the demethylation activity without abrogating substrate binding (e.g., allosteric inhibitors). In addition, DSF assays can be used for high-throughput screening of FTO ligands. Other biophysical assays for detecting interaction, such as isothermal titration calorimetry, surface plasmon resonance, nuclear magnetic resonance, and mass spectrometry, are also widely used; however, the throughput is relatively low for screening.
In addition, modifying the “old” ideas to generate “new” methods has led to the development of subfamily selective probes. By combining DSF with multiprotein dynamic combinatorial chemistry, Das et al. developed an assay that simultaneously measures the melting temperatures of ALKBH3, ALKBH5, and FTO in dynamic chemistry libraries, which enabled the identification of selective inhibitors for FTO (with IC50 = 2.6 μM) and ALKBH3.70
Overall, screening assays detecting FTO ligand binding indeed promote the development of FTO inhibitors; however, they also identify many false positives that do not disturb demethylation in vitro. Additionally, some potential inhibitors can also be missed because of the inherent specific limitations of each assay.
5.2 In vitro demethylation-based assays
Binding-based biophysical strategies are robust for high-throughput screening, but the inhibition of enzymatic activity needs further validation. Therefore, assays directly determining FTO demethylation activity have received more interest. Based on the read-out dependency, in vitro demethylation-based assays can be classified into three categories: antibody-dependent assays, enzyme-dependent assays, and fluorescence- or radioactivity-based assays (Table 1).Antibodies specifically recognizing the N6-methylation on RNAs (i.e., both m6A and m6Am) have been used for detection of RNA methylation. The dot blot assay and immuno-northern blotting (INB) are commonly used to semiquantify the methylation states of RNA substrates (chemically synthesized oligonucleotides and RNA samples isolated from cells).71 These methods are suitable for cellular samples. However, antibody-based assays are mainly performed to evaluate potential FTO modulators rather than to carry out large-scale screening to our knowledge.
Assays with read-outs dependent on enzymes are the most prevalent methods used to determine FTO catalytic activity. Methyl-sensitive restriction enzymes (e.g., DpnII and MazF) cleave the modified substrate (designed and chemically synthesized RNA oligonucleotides) once the methyl group is removed. Therefore, the methylation states are reflected by the subsequent determination of the digestion products (gel electrophoresis) or the released fluorescence (FRET) or by using nanomaterial for fluorescence detection.72–75 In addition, some assays (HPLC-MS/MS and 2D thin-layer chromatography) digest the original RNA samples or radiolabelled RNA samples into single nucleotides and then quantify the nucleosides.7 These methods determine the FTO inhibitory effect in cells with isolated RNAs and are able to discriminate the two similar modifications m6A and m6Am. However, most of the enzyme-dependent assays (except the FRET-based assay) are low-throughput and not effective for large-scale screening.73
Assays using fluorescence and radioactivity as read-outs have also been proposed. Svensen and Jaffrey developed a fluorometric RNA substrate that showed increased fluorescence intensity after demethylation by FTO.76 This strategy provides a high-throughput screening approach based on the demethylase activity of FTO. Cheong et al. also developed a fluorescent m6A-switchable probe that shows fluorescence upon demethylation by FTO both in vitro and in living cells.77 Notably, this m6A probe is sensitively and selectively demethylated by FTO instead of by ALKBH5. This strategy has successfully identified known FTO inhibitors with diverse chemical scaffolds and is also suitable for high-throughput screening. Similarly, Yang et al. developed a m6A-switchable probe that could be measured through the DSF technique.78 In addition, demethylation of radiolabelled m6A by ALKBH5 can be measured directly by determining radioactivity, which is likely also possible for FTO.79
5.3 Cell-based assays for monitoring FTO activity
Assays to determine the inhibition of FTO demethylation in living cells can be used to deliver active cellular inhibitors. Cell-based assays are direct and powerful because they consider cellular target engagement in the early stage of inhibitor screening. The fluorescent m6A-switchable probe developed by Cheong et al. can be delivered into cells and achieve real-time imaging and single-cell flow cytometry analysis of FTO activity.77 By monitoring demethylase activity directly in living cells, the m6A probe makes it possible to determine the cellular activity of FTO inhibitors.77 However, cell-based high-throughput screening for FTO modulator discovery has not been achieved.6. Small-molecule inhibitors of FTO
Because of the efforts to develop FTO inhibitors, numerous compounds have been identified (Table 2). The current known FTO inhibitors were mainly discovered through virtual screening and rational design, and some inhibitors have achieved selectivity among AlkB family proteins. The active site of FTO provides two binding pockets (the 2OG binding site and the substrate binding site) for chemical intervention. Several inhibitors have shown anticancer effects, which provides proof-of-concept for the FTO demethylase as a potential anticancer target in drug discovery. The representative FTO inhibitors can be grouped into 2OG-competitive, substrate-competitive, dual 2OG- and substrate-competitive and mechanism-unverified inhibitors according to their mode of action (Fig. 6).| Mode of action | Inhibitor | Discovery method | PDB code | Enzymatic activity (method, substrate) | Anticancer potency | Ref. |
|---|---|---|---|---|---|---|
| N/A, not available. | ||||||
| 2OG-competitive | 2,4-PDCA | DSF-based assay | 4IE0 | 8.3 μM (UPLC, 3mT nucleoside) | N/A | 80 |
| IOX1 | DSF-based assay | 4IE4 | 3.3 μM (UPLC, 3mT nucleoside) | N/A | 80 | |
| R-2HG | N/A | 133.3 μM (dot blot, ssRNA) | Inhibited survival of sensitive AML cells; inhibited AML progression and prolonged survival in xeno-transplanted mice (sensitive cells) | 81 | ||
| Substrate-competitive | Rhein | Virtual screening | 4IE7 | 30.0 μM (HPLC, ssRNA) | >20 μM | 72 and 80 |
| MA | FP assay | 4QKN | 8.6 μM (HPLC, ssRNA) | Inhibited growth and self-renewal of GSC lines (MA2); suppressed tumour progression and prolonged lifespan of GSC-grafted mice (MA2) | 68 | |
| MA2 | ||||||
| FB23 | Rational design | 6AKW | 0.06 μM | Inhibited proliferation of AML cells (FB23-2); inhibited progression of AML cells in xeno-transplanted mice (FB23-2) | 82 | |
| FB23-2 | 2.6 μM (HPLC, ssRNA) | |||||
| Dac51 | Rational design | 7CKK | 0.4 μM (HPLC, ssRNA) | Inhibited tumour growth and prolonged survival of melanoma mice models in combination with immunotherapy | 83 | |
| FL1 | Structural similarity screening | 4ZS3 | 6.6 μM (LC/MS, ssRNA) | N/A | 84 | |
| FTO-4 | Rational design; molecular docking | N/A | 3.4 μM (fluorescence, m6A7-Broccoli RNA) | Impaired self-renewal and inhibited neurosphere formation of GSCs | 85 | |
| N-CDPCB | Rational design | 5DAB | 5.0 μM (LC/MS, ssRNA) | >20 μM | 86 | |
| CHTB | Rational design | 5F8P | 39.2 μM (LC/MS, ssRNA) | N/A | 87 | |
| Radicicol | Virtual screening; similarity search | N/A | 16.0 μM (LC/MS, ssRNA) | N/A | 90 | |
| Substrate- and 2OG-competitive | 12 | Rational design | 4CXW | 0.6 μM (HPLC, ssRNA) | >20 μM (an ethyl ester derivatives of 12) | 65 |
| Entacapone | Virtual screening | 6AK4 | 3.5 μM (LC/MS, ssDNA) | N/A | 88 | |
| IOX3 | DSF-based assay | 4IE6 | 2.8 μM (UPLC, 3mT nucleoside) | N/A | 80,89 | |
| Inhibitors with unverified mechanisms | MO-I-500 | N/A | 8.7 μM (HPLC, ssDNA) | Inhibited survival and colony formation of metabolic challenge-resistant SUM149 cells | 91 | |
| Nafamostat mesilate | N/A | 13.8 μM (LC/MS, ssRNA) | N/A | 92 | ||
| Clausine E | N/A | 27.8 μM (LC/MS, ssRNA) | N/A | 93 | ||
| Diacerein | Single-QD-based FRET nanosensor | N/A | 1.5 μM (single-QD-based FRET nanosensor, ssDNA) | N/A | 75 | |
| CS1 (Bisantrene) | Virtual screening | N/A | 142.6 nM (cell-free m6A demethylase assay) | Inhibited proliferation of AML cells and sensitized AML cells to T cell cytotoxicity; delayed progression and improved survival in PDX AML models and synergy with T cell treatment | 94 | |
| CS2 (Brequinar) | Virtual screening | N/A | 712.8 nM (cell-free m6A demethylase assay) | 94 | ||
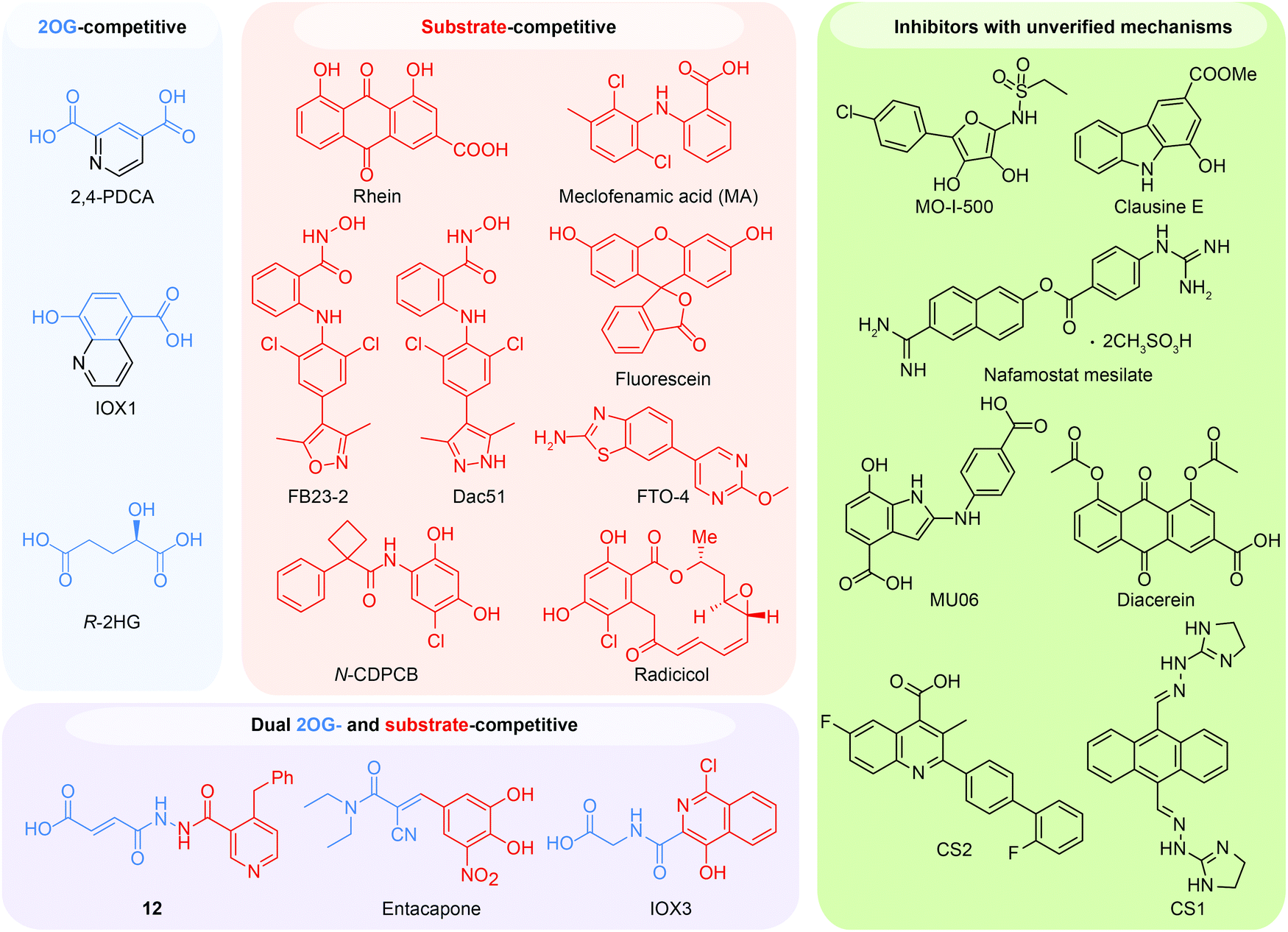 | ||
| Fig. 6 Representative FTO inhibitors classified into four groups based on their mode of action. The 2OG binding site and the substrate binding site can be competitively occupied, and representative molecules are shown. A strategy to simultaneously bind both the 2OG binding site and the substrate binding pocket has also successfully produced dual competitive inhibitors. The structures coloured blue and red occupy the 2OG and substrate binding site respectively. The chemical structures of other unclassified FTO inhibitors with unverified mechanisms are also listed. | ||
6.1 2OG-competitive inhibitors
In principle, the catalytic oxidative demethylation of 2OG-dependent dioxygenases can be inhibited by 2OG and its analogues (Fig. 7A).95 Therefore, FTO can also be competitively inhibited by non-reactive 2OG analogues by directly occupying the 2OG binding site. Several 2OG analogues have been reported to show inhibitory effects on FTO demethylation, for instance, the pan 2OG dioxygenase inhibitors NOG, pyridine-2,4-dicarboxylate (2,4-PDCA) and IOX1 (Fig. 6).80 Although these inhibitors cannot selectively target FTO, their co-crystal structures provide the first step towards potent and selective FTO inhibitor development (Fig. 7B).80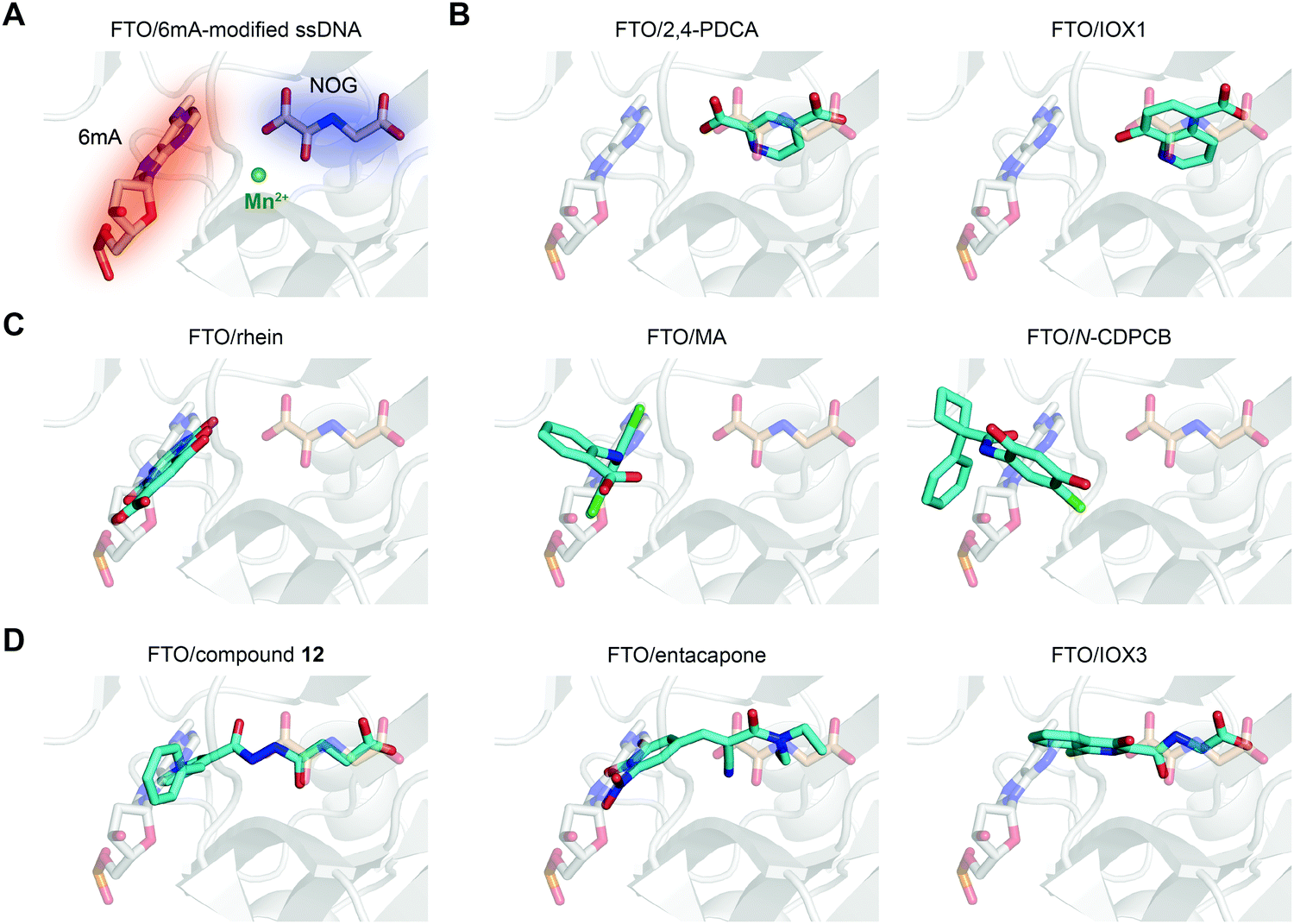 | ||
| Fig. 7 The binding poses of representative FTO inhibitors. (A) The 6mA substrate (grey stick) and NOG (wheat stick) form two binding sites for chemical intervention (PDB: 5ZMD). Mn2+ is shown as a green sphere. (B) Representative 2OG-competitive FTO inhibitors 2,4-PDCA (PDB: 4IE0) and IOX1 (PDB: 4IE4). (C) Representative substrate-competitive FTO inhibitors rhein (PDB: 4IE7), MA (PDB: 4QKN) and N-CDPCB (PDB: 5DAB). (D) Representative binding poses of both 2OG- and substrate-competitive FTO inhibitors compound 12 (PDB: 4CXW), entacapone (PDB: 6AK4) and IOX3 (PDB: 4IE6). All inhibitors are coloured cyan. | ||
The R enantiomer of 2-hydroxyglutarate (R-2HG) is produced from 2OG by a neomorphic function of IDH1 and IDH2 mutants.96 Although R-2HG is a broad-spectrum inhibitor as an analogue of 2OG, FTO has been validated as the main target of R-2HG in sensitive leukemia cells. R-2HG inhibits FTO catalytic activity with an IC50 of 133.3 μM in vitro.81 Biochemical and cell-based experiments, such as the cellular thermal shift assay (CETSA) and drug affinity responsive target stability (DARTS), have well characterized the target engagement of R-2HG in cells. R-2HG significantly suppressed the survival and proliferation of sensitive leukemia cells and glioma cells by inducing cell cycle arrest and apoptosis. Treatment with R-2HG in mice xeno-transplanted with sensitive cells reduced the aggressive leukemic symptoms, impaired AML progression and extended survival. Mechanistically, R-2HG-induced FTO inhibition upregulates the m6A modification of MYC and CEBPA transcripts, reduces mRNA stability, and downregulates protein levels to achieve antitumour effects. Additionally, R-2HG also attenuates cancer metabolism through suppression of PFKP and LDHB-related aerobic glycolysis.97
6.2 Substrate-competitive inhibitors
Most inhibitors of the RNA demethylase FTO are substrate-competitive inhibitors (Fig. 6 and 7A). In 2012, Chen et al. identified the first FTO inhibitor by performing virtual screening on the drug-like SPECS database.72 During the search for potential compounds capable of occupying the substrate binding site, the natural product rhein was identified to inhibit FTO demethylation in vitro with an IC50 of 30 μM. Instead of chelating Fe2+ or mimicking 2OG, rhein acts as a reversible inhibitor competitively occupying the substrate binding site in FTO, and this mechanism was further verified by the crystal structure of rhein in complex with FTO (PDB: 4IE7, Fig. 7C left).80 In addition, treatment with rhein can elevate the levels of m6A on mRNA in cells, which indicates that cellular RNA methylation could be affected by the presence of small-molecule compounds. However, rhein cannot achieve selective inhibition among AlkB family members or other 2OG-dependent dioxygenases (e.g., the JMJD2 histone demethylase).To selectively inhibit the FTO demethylase rather than another RNA demethylase, ALKBH5, Huang et al. performed an FP assay to screen compounds that selectively disrupt FTO binding to methylated ssDNA over ALKBH5 binding.68 In a screen of a small-molecule library containing more than 900 small-molecule drugs in-house, meclofenamic acid (MA), a non-steroidal anti-inflammatory drug, showed selective and competitive disruption of FTO binding to substrate over ALKBH5 binding to substrate. Further enzymatic inhibition assays showed that MA selectively inhibits FTO demethylation with an IC50 of 8.6 μM, while no inhibition was detected for ALKBH5, ALKBH2 or ALKBH3. Biochemical and crystallographic experiments (PDB: 4QKN, Fig. 7C middle) revealed that MA exerts inhibitory effects by competing for substrate binding with FTO instead of being a 2OG mimic or Fe2+ chelator. Notably, the hydrophobic interactions between FTO and MA partly come from NRL1, and this motif is absent in ALKBH5, which likely accounts for the selectivity of MA among the two m6A demethylases. Although ALKBH2 and ALKBH3 possess the NRL1 region, the presence of hydrophilic and bulky residues hinders their interaction with and binding to MA (Fig. 5C). MA2, an ethyl ester derivative of MA, significantly increases the level of m6A on mRNA in cells in an FTO-dependent manner instead of targeting ALKBH5.68 In addition, MA2-induced m6A upregulation inhibits the growth and self-renewal of glioblastoma stem cells (GSCs) and reverses the aggressive phenotypes caused by knockdown of METTL3 or METTL14.98In vivo, MA2 decreases the tumour size in GSC-initiated tumorigenesis and extends survival in mice models. MA2-mediated FTO inhibition also inhibits the development of cervical cancer in vivo,99 downregulates the PI3K/Akt/mTOR signalling pathway in gastrointestinal cancer cells,100 and inhibits the migration and invasion of HER2-positive breast cancer cells.101
Based on these structural insights into the selective binding and inhibition of MA towards FTO, Huang et al. further employed structure-guided design to improve the inhibitory potency on FTO demethylation.82 By retaining the benzyl carboxylic acid to maintain the selectivity for FTO over ALKBH5 and using a bulky ligand in dichloride-substituted benzene to extend to a deeper pocket, FB23 was designed; FB23 shows an IC50 of 0.06 μM, a more than 100-fold increase over that of MA.82 The co-crystal structure of FB23 bound to FTO (PDB: 6AKW) was determined to show that FB23 utilizes a similar interaction as MA to bind to FTO but has extra hydrogen bonds between the nitrogen or oxygen in the five-membered heterocyclic ring and E234 of FTO, which might contribute to the enhanced inhibitory effect towards FTO (Fig. 8). Because of the poor permeability of FB23 in cells, further optimization based on the bioisosterism principle was applied to modify the carboxylic acid of FB23, which led to the discovery of the benzohydroxamic acid FB23-2, which has an IC50 value of 2.6 μM. DARTS assays and knockout experiments showed that FB23-2 binds FTO in cell lysates and exhibits antiproliferative effects through FTO inhibition. FB23-2 displays selectivity for FTO over other AlkB family members, epigenetic targets, kinases, oncogenic proteases, and COX-1/2 (the primary targets of MA) in vitro. FB23-2 efficiently inhibited the proliferation of a panel of AML cell lines and patient-derived primary AML cells. Of note, FB23-2 significantly suppressed leukemia progression, induced AML cell differentiation, and prolonged survival in both xeno-transplantion and patient-derived xeno-transplantation (PDX) AML models, suggesting that FTO is a druggable target and that targeting FTO with small-molecule inhibitors holds potential to treat AML.102
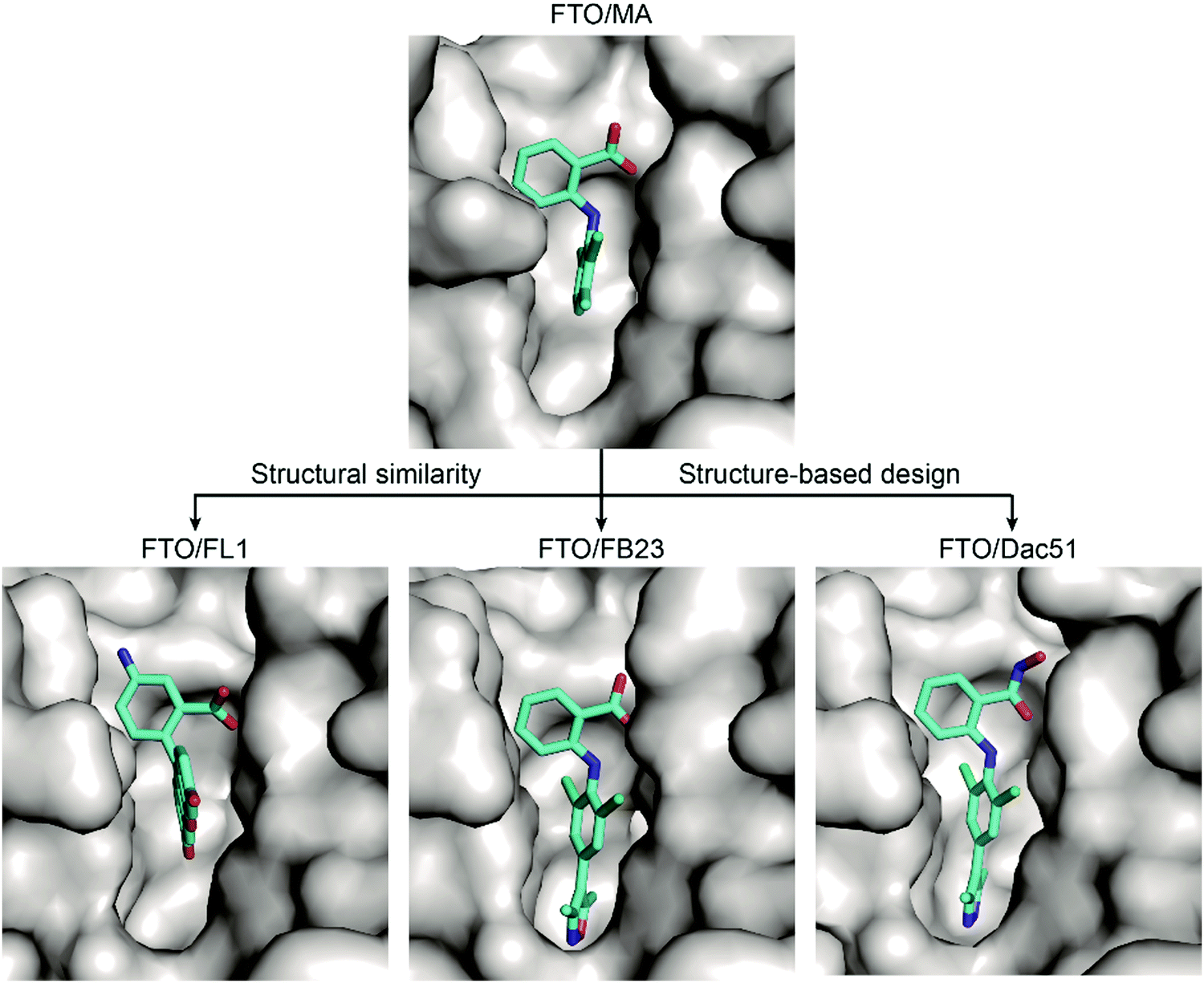 | ||
| Fig. 8 Binding mechanisms of MA and further inhibitors discovered through structural similarity searches (the fluorescein derivative FL1) and structure-based rational design (FB23 and Dac51). The inhibitors are coloured cyan; the surface of FTO protein is coloured grey. | ||
Through optimization of FB23, Dac51 was developed to inhibit FTO demethylation with an improved IC50 value of 0.4 μM.83 Structural insights of the FTO/Dac51 complex (PDB: 7CKK) revealed a similar binding mode as FB23 (Fig. 8). The analysis of CETSA and m6A abundance verified the target engagement of Dac51 in cells. Dac51-mediated FTO inhibition downregulated the bZIP family transcription factors and glycolysis-related genes in B16-OVA and LLC cell lines and patient-derived organoids. In vivo, Dac51 inhibited the tumour growth in mice xeno-transplanted with B16-OVA cells in a T cell-dependent manner. Notably, combining Dac51 with immune checkpoint blockade agent remarkably inhibited tumour growth and prolonged the overall survival, without recurrence via memory T cell responses.
Inspired by the structural similarity between MA and fluorescein derivatives, Wang et al. attempted to discover potential FTO inhibitors with the ability to monitor the location of cellular FTO simultaneously (Fig. 8).84 Fluorescein and its derivatives (FL1–8) were validated as FTO inhibitors and showed selective inhibitory activity towards FTO over ALKBH5 with IC50 values between 1.0 μM and 7.0 μM. The derivatives FL6 and FL8 also significantly increased the m6A level on mRNA in cells. As revealed in structural complexes of FL1 (PDB: 4ZS3) and FL4 (PDB: 4ZS2) bound to FTO, fluorescein inhibitors adopt a similar binding mode as MA: they partially occupy the substrate binding site to competitively inhibit FTO. In addition, selectivity is also achieved through hydrophobic interactions provided by the NRL1 motif in FTO. By introducing the photoreactive group diazirine into FL2, the product FL2-DZ was created; FL2-DZ is able to selectively label recombinant FTO over ALKBH2/3/5 in vitro and has the bioimaging capacity to label endogenous FTO in situ while maintaining inhibitory activity towards FTO.84
Based on the MA binding site of FTO, Huff et al. performed structure-based drug design and molecular docking experiments and identified FTO-2 and FTO-4 as selective competitive FTO inhibitors over ALKBH5 with IC50 values of 2.2 μM and 3.4 μM (Fig. 6).85 The selectivity among other AlkB family proteins and dioxygenases has not been disclosed, however. FTO-4 elevated both m6A and m6Am levels in GSCs. Treatment with FTO-4 impaired the self-renewal of patient-derived GSC lines and inhibited neurosphere formation.
Through rational design, N-CDPCB was developed to inhibit FTO; N-CDPCB has an IC50 of 5.0 μM, and it induces increased cellular m6A levels on mRNA (Fig. 6).86N-CDPCB was revealed to extensively interact with NRL1 as well as the L1 loop of FTO (PDB: 5DAB, Fig. 7C right), occupying a novel binding site.86 Wang et al. conducted virtual screening and similarity searches based on N-CDPCB.90 Radicicol was discovered to bind and inhibit FTO demethylation with an IC50 of 16.04 μM, and it occupies a similar site as N-CDPCB. Similarly, structure-based design also helped to identify CHTB as an FTO inhibitor with an IC50 value of 39.24 μM.87 CHTB occupies a similar binding site as MA and can increase the m6A level in cells. However, the selectivity of N-CDPCB and CHTB is largely unknown.
6.3 Dual 2OG- and substrate-competitive inhibitors
Given the continuous pockets of cofactor and substrate binding sites in dioxygenases (Fig. 7A), a 2OG-tethering strategy was proposed for the development of more potent inhibitors.65 This method links analogues of both 2OG and nucleotides to occupy the entire pocket used for FTO demethylation. Through exploitation of the structural differences within the nucleotide binding sites among AlkB subfamilies and modelling studies to identify potential side chains mimicking substrate interaction, Toh et al. identified compound 12 (IC50 = 0.6 μM) as an inhibitor showing selectivity for FTO over other AlkB subfamily members as well as other human 2OG dioxygenases (PHD2 and JMJD2A) in vitro.65 Compound 12 was designed to link fumarate hydrazide (a 2OG analogue) and 4-benzylpyridine (a nucleotide competitor) through a chemical linker. As predicted, the determined structure (PDB: 4CXW, Fig. 7D left) revealed that the fumarate hydrazide well occupies the 2OG binding site and interacts with the metal ion, and the 4-benzyl pyridine side chain occupies the position of the nucleotide substrate and exhibits similar interactions with FTO as 3mT. Furthermore, the ethyl ester derivative of compound 12 also shows dose-dependent cellular inhibition of mRNA m6A demethylation. The discovery of compound 12 as well as MA provides potential functional tools for determining FTO biological significance and its individual role in m6A regulation irrespective of ALKBH5.65In a structure-based virtual screening with FDA-approved drugs, entacapone was reported to inhibit FTO demethylase activity with an IC50 value of 3.5 μM.88 The crystal structure of FTO bound with entacapone (PDB: 6AK4, Fig. 7D middle) revealed that the small-molecule inhibitor occupies both the substrate and 2OG binding sites in FTO. Both structural complex and SAR exploration have revealed that the meta-position hydroxyl group forms hydrogen bonds with R322 and Y106, which is critical for inhibitory activity. The nitrile chelates Zn2+, and the carbonyl group forms a hydrogen bond with Q205. No inhibitory effect of entacapone towards the ALKBH5 and TET1 demethylases was found. Investigations of the molecular mechanism and involved signalling pathways showed that the glucose metabolism-associated genes glucose 6-phosphatase and phosphoenolpyruvate carboxykinase 1 are downregulated through the transcription factor FOXO1 in entacapone-treated Hep-G2 cells. In addition, entacapone administration reduces body weight and lowers fasting blood glucose concentrations in diet-induced obese mice, suggesting a potential effect of FTO inhibitors on obesity.88
IOX3 inhibited FTO demethylation in vitro with an IC50 of 2.8 μM. Inspection of the co-crystal structure (PDB: 4IE6, Fig. 7D right) shows that IOX3 occupies both the 2OG and substrate binding sites.80 This compound is a non-selective inhibitor that has been well characterized to inhibit PHD1–3 in cells and animals.95,103
6.4 Inhibitors with unverified mechanisms
MO-I-500 was identified in the course of searching for anticonvulsant compounds targeting prolyl-4-hydroxylase; however, this compound was proven to inhibit FTO demethylation in vitro with an IC50 of 8.7 μM.91 MO-I-500 is a 2OG and ascorbic acid mimic as well as an ion chelator; however, it shows a certain degree of specificity for FTO over some 2OG-dependent dioxygenases, such as JMJD2A. Unfortunately, the selectivity among AlkB proteins is unknown. MO-I-500 at 2 μM significantly inhibits the survival and colony formation of metabolic challenge-resistant SUM149 cells in glutamine-deficient medium.104Several FTO binders have been developed using structure-based design (Fig. 6); however, the mechanisms remain mostly unverified. For example, nafamostat mesilate and clausine E bind and inhibit demethylase activity in vitro;92,93 MU06 was designed through a scaffold hopping approach, and further evaluation using molecular docking simulations indicated direct binding with FTO;105 in a single-quantum-dot (QD)-based FRET nanosensor assay, diacerein, which has a similar structure to rhein, showed an inhibitory effect on FTO with an IC50 of 1.5 μM.75
Most recently, Su et al. conducted a structure-based virtual screening combined with assessment of anti-leukemic efficacy in human cell lines and ultimately identified CS1 and CS2 (also named bisantrene and brequinar, respectively) as FTO inhibitors.94 The docking models revealed that these two small molecules inhibit demethylase activity by preventing the m6A-modified substrate from entering the catalytic pocket. Although CS1 and CS2 have been suggested to bind to FTO through NMR, DARTS, and CETSA assays, and the essential amino acids for binding were also investigated in the mutagenesis assays, the complex structure of CS1 or CS2 bound to FTO is currently unavailable. CS1 and CS2 significantly inhibited the cell viability of AML cell lines, with the cellular sensitivity dependent on FTO protein levels.94 In PDX AML models, CS1 and CS2 delayed leukemia progression, mitigated leukemia burden and prolonged survival.
Discussion and conclusion
With the discovery of m6A demethylation by FTO in living cells, the existence of dynamic and reversible RNA methylation has been realized. And such processes reveal a new form of post-transcriptional regulation of gene expression at the RNA level termed epitranscriptomics. As the most abundant internal modification of mRNA, m6A regulates gene expression by modulating mRNA metabolism in several stages, which is critical in physiological processes.20 The important role of m6A modification in the initiation and progression of human diseases especially cancer has also been increasingly discovered.106 Thus, modulating RNA methylation has become a potential therapeutic strategy just as modulating epigenetic factors such as histone modifications and DNA methylation has. It has been established that m6A is deposited by methyltransferases, removed by FTO and ALKBH5, and selectively recognized by several reader proteins. These findings reveal that RNA methylation occurs in a complex regulatory network, setting challenges for precise modulation, either global or targeted, of m6A modifications.To date, almost all m6A regulatory proteins have been demonstrated to be closely involved in diverse types of cancer.38,106 However, the chemical manipulation of m6A mainly focuses on targeting RNA demethylases. An activator of the METTL3/14/WTAP complex and inhibitors of METTL3 were identified in vitro.107,108 Most recently, STM2457, a potent and selective METT3 inhibitor has been developed and showed pharmacological effects on AML in mice model.109 Bedi et al. performed computational screening of fragments and found thirty m6A mimics with the potential to bind to reader proteins.110 There are also few selective modulators for another m6A demethylase, ALKBH5. For example, Li et al. identified the ALKBH5 inhibitor ALK-04 (the structure remains unreleased) through in silico screening, which significantly enhances the efficacy of anti-PD-1 therapy.111 However, no any activator for RNA demethylases has been identified. We reason that it is critical to exploit not only RNA demethylases but also other m6A regulatory proteins to develop chemical probes to deeply understand the function of m6A modification and even to develop drugs for clinical use.
The abnormal expression of FTO is increasingly implicated in various diseases, especially cancer, making it of vital importance to develop FTO modulators for potential therapeutic applications. To our knowledge, FTO acts as an oncogene in promoting cancer initiation and progression in some cancers. Encouragingly, several FTO inhibitors perturbing demethylase activity have exhibited significant therapeutic effects in different cancers, validating the potential of identifying drugs targeting epitranscriptomic RNA methylation in drug discovery.
The antitumour effects of FTO inhibitors have expanded from leukemia to solid tumours,83,94 though further exploration is needed for elucidation of biological effects of FTO inhibitors, either alone or in combination. Due to the heterogeneity property of cancer, identification of precise biomarkers is very important for individual treatment, improved outcome and potential toxicity prediction.112 Currently, lack of predictive biomarker partly hinders the applicability of FTO inhibitors in different cancers. It has been revealed that the expression level of FTO is upregulated in some cancers while downregulated in others, which indicate the significance to define the context-specific role of FTO demethylation in different types of cancer. However, the biological function of FTO and the molecular mechanisms underlying the role of FTO in different cancers have yet to be well understood. Therefore, further investigation of FTO tumour biology, development of high-quality chemical probes, and preclinical studies will help the target validation of FTO.
Most of the reported FTO inhibitors are cytotoxic, while noncytotoxic inhibitors modulating the immune system also represents a promising strategy for cancer treatment especially in immunotherapy. To this end, the FTO inhibitor Dac51 exhibited minimal effects on cell viability under the tested concentrations, but it reprogramed the glycolytic metabolism and rejuvenated T cell response, thus improving efficacy of anti-PD-L1 blockade in vivo.83 In addition, the ALKBH5 inhibitor ALK-04 showed no cytotoxicity while it remarkably synergized with anti-PD-1 therapy in vivo.111 Taken together, the development of small-molecule inhibitors for RNA demethylases could open new avenues in the field of immunotherapy for treating cancer.
Multiple FTO inhibitors have been developed so far; however, most of them show non-selective inhibitory effects towards FTO. Fortunately, potential strategies to achieve selectivity have been proposed, some of which have been successfully applied in identifying inhibitors that are selective for FTO over other AlkB family proteins and 2OG-dependent dioxygenases. Structure-based rational design also takes advantage of the structural features of FTO to facilitate drug discovery. Though selective FTO inhibitors are important for investigating the specific FTO-regulated signaling pathway, small molecules targeting both m6A demethylases might exhibit more therapeutic effects because of dramatically increased m6A methylation levels either in a global context or on a certain target. However, the biological output of dual inhibition on RNA demethylases in caners was rarely investigated. It was established that each demethylase regulates specific set of target transcripts, while overlapped targets of FTO and ALKBH5 cannot be excluded. In this circumstance, pan inhibitors of RNA demethylases would hold the potential to treat cancer more efficiently.
Most of the FTO inhibitors occupy the catalytic pocket to achieve inhibition, and developing allosteric modulators also represents a new challenge and chance for chemical modulation of FTO function. In addition, using small molecules to disturb the protein–protein interactions of FTO or the protein–nucleotide interactions is also a promising strategy for achieving selective inhibition. It remains a challenge to realize transcript and m6A modification site-specific regulation through small molecules. In addition to inhibitors, the development of activators of FTO is also appealing and requires new methods for compound screening and evaluation. These FTO modulators would open a new avenue for chemical intervention on FTO activity, and likely make FTO a promising anticancer target.
Assays for FTO modulator (inhibitor, activator, and binder) discovery have progressed in recent years. Both biochemical and biophysical assays detecting enzymatic activities and binding are commonly used methods. However, most of the assays possess a low throughput and lack sensitivity, making them less adaptable to high-throughput screening. Novel screening methods with high throughput, especially enzymatic activity-based measurements, are urgently needed to facilitate more efficient discovery of new potent FTO modulators. In addition, very few assays can be applied to measure the demethylase activity of FTO in living cells. Strategies to detect the cellular FTO activity during high-throughput screening remain to be identified.
Considering the complicated function of m6A on mRNAs and ncRNAs, it is notable that altering m6A methylation by targeting demethylases may cause distinct global m6A distribution, and this outcome has been confirmed by several m6A-seq results (most of the sequencing focused on m6A on mRNAs). Altered m6A patterns may affect gene expression in several ways, conveying substantially different information and affecting a series of biological processes. Therefore, the clinical usage of m6A-modulating agents for treatment should be based on a comprehensive and deep understanding of RNA m6A biology and the individual role of the selected target in m6A regulation, and attention should be paid to the toxicity of potential drug candidates. Moreover, considering that m6A on mRNA is not the only substrate of FTO, the roles of the other substrates, including m1A on tRNA, m6A on snRNAs and cap m6Am on mRNAs and snRNAs, which have been overlooked in recent studies, also need to be explored extensively to fully understand the biological function of FTO. The discovery of diverse high-quality chemical probes for FTO would speed up investigations of epitranscriptomics.
In conclusion, selective and potent FTO modulators will soon be identified with advances in rational design and the development of novel methods for high-throughput screening and in silico assistance. These small-molecule compounds will profoundly facilitate investigations of FTO biological functions and RNA methylation biology and, most importantly, the discovery of new drugs that regulate RNA methylation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We apologize to colleagues whose work could not be cited due to space constraints. Our research has been supported by the National Natural Science Foundation of China (21907101 to Y. H. and 21725801 to C. Y.) and the Science and Technology Commission of Shanghai Municipality (18YF1428500 to Y. H. and 18431907100 to C. Y.).References
- S. R. Bhaumik, E. Smith and A. Shilatifard, Nat. Struct. Mol. Biol., 2007, 14, 1008–1016 CrossRef CAS PubMed.
- G. Cavalli and E. Heard, Nature, 2019, 571, 489–499 CrossRef CAS PubMed.
- H. P. Mohammad, O. Barbash and C. L. Creasy, Nat. Med., 2019, 25, 403–418 CrossRef CAS PubMed.
- R. P. Perry and D. E. Kelley, Cell, 1974, 1, 37–42 CrossRef CAS.
- R. Desrosiers, K. Friderici and F. Rottman, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3971–3975 CrossRef CAS PubMed.
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, T. K. Wirecki, V. de Crécy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303–D307 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- B. S. Zhao, I. A. Roundtree and C. He, Nat. Rev. Mol. Cell Biol., 2016, 18, 31–42 CrossRef PubMed.
- Kate D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, Christopher E. Mason and Samie R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- C&EN Global Enterprise, 2019, vol. 97, pp. 34–39.
- R. J. Loos and G. S. Yeo, Nat. Rev. Endocrinol., 2014, 10, 51–61 CrossRef CAS PubMed.
- P. K. Annapoorna, H. Iyer, T. Parnaik, H. Narasimhan, A. Bhattacharya and A. Kumar, Neuroscience, 2019, 418, 15–24 CrossRef CAS PubMed.
- J. Y. Wang, L. J. Chen and P. Qiang, Onco Targets Ther, 2020, 13, 12845–12856 CrossRef CAS.
- L. J. Xie, L. Liu and L. Cheng, Biochemistry, 2020, 59, 230–239 CrossRef CAS PubMed.
- J. Gu, J. Xu, Q. You and X. Guo, Eur. J. Med. Chem., 2020, 196, 112325 CrossRef CAS PubMed.
- L.-L. Zhou and C.-G. Yang, Biochemistry, 2020, 59, 125–127 CrossRef CAS PubMed.
- G.-Q. Lai, L.-L. Zhou and C.-G. Yang, Chin. J. Chem., 2020, 38, 420–421 CrossRef CAS.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- H. Shi, J. Wei and C. He, Mol. Cell, 2019, 74, 640–650 CrossRef CAS.
- P. C. He and C. He, EMBO J., 2021, 40, e105977 CrossRef CAS PubMed.
- C. Dina, D. Meyre, S. Gallina, E. Durand, A. Korner, P. Jacobson, L. M. Carlsson, W. Kiess, V. Vatin, C. Lecoeur, J. Delplanque, E. Vaillant, F. Pattou, J. Ruiz, J. Weill, C. Levy-Marchal, F. Horber, N. Potoczna, S. Hercberg, C. Le Stunff, P. Bougneres, P. Kovacs, M. Marre, B. Balkau, S. Cauchi, J. C. Chevre and P. Froguel, Nat. Genet., 2007, 39, 724–726 CrossRef CAS PubMed.
- T. M. Frayling, N. J. Timpson, M. N. Weedon, E. Zeggini, R. M. Freathy, C. M. Lindgren, J. R. Perry, K. S. Elliott, H. Lango, N. W. Rayner, B. Shields, L. W. Harries, J. C. Barrett, S. Ellard, C. J. Groves, B. Knight, A. M. Patch, A. R. Ness, S. Ebrahim, D. A. Lawlor, S. M. Ring, Y. Ben-Shlomo, M. R. Jarvelin, U. Sovio, A. J. Bennett, D. Melzer, L. Ferrucci, R. J. Loos, I. Barroso, N. J. Wareham, F. Karpe, K. R. Owen, L. R. Cardon, M. Walker, G. A. Hitman, C. N. Palmer, A. S. Doney, A. D. Morris, G. D. Smith, A. T. Hattersley and M. I. McCarthy, Science, 2007, 316, 889–894 CrossRef CAS PubMed.
- A. Scuteri, S. Sanna, W. M. Chen, M. Uda, G. Albai, J. Strait, S. Najjar, R. Nagaraja, M. Orru, G. Usala, M. Dei, S. Lai, A. Maschio, F. Busonero, A. Mulas, G. B. Ehret, A. A. Fink, A. B. Weder, R. S. Cooper, P. Galan, A. Chakravarti, D. Schlessinger, A. Cao, E. Lakatta and G. R. Abecasis, PLoS Genet., 2007, 3, e115 CrossRef PubMed.
- T. Gerken, C. A. Girard, Y. C. Tung, C. J. Webby, V. Saudek, K. S. Hewitson, G. S. Yeo, M. A. McDonough, S. Cunliffe, L. A. McNeill, J. Galvanovskis, P. Rorsman, P. Robins, X. Prieur, A. P. Coll, M. Ma, Z. Jovanovic, I. S. Farooqi, B. Sedgwick, I. Barroso, T. Lindahl, C. P. Ponting, F. M. Ashcroft, S. O'Rahilly and C. J. Schofield, Science, 2007, 318, 1469–1472 CrossRef CAS PubMed.
- G. Jia, C.-G. Yang, S. Yang, X. Jian, C. Yi, Z. Zhou and C. He, FEBS Lett., 2008, 582, 3313–3319 CrossRef CAS PubMed.
- Y. Fu, G. Jia, X. Pang, R. N. Wang, X. Wang, C. J. Li, S. Smemo, Q. Dai, K. A. Bailey, M. A. Nobrega, K. L. Han, Q. Cui and C. He, Nat. Commun., 2013, 4, 1798 CrossRef PubMed.
- J. Mauer, X. Luo, A. Blanjoie, X. Jiao, A. V. Grozhik, D. P. Patil, B. Linder, B. F. Pickering, J.-J. Vasseur, Q. Chen, S. S. Gross, O. Elemento, F. Debart, M. Kiledjian and S. R. Jaffrey, Nature, 2017, 541, 371–375 CrossRef CAS PubMed.
- J. Wei, F. Liu, Z. Lu, Q. Fei, Y. Ai, P. C. He, H. Shi, X. Cui, R. Su, A. Klungland, G. Jia, J. Chen and C. He, Mol. Cell, 2018, 71, 973–985.e975 CrossRef CAS PubMed.
- B. Chen, H. Liu, X. Sun and C. G. Yang, Mol. BioSyst., 2010, 6, 2143–2149 RSC.
- C. Yi, B. Chen, B. Qi, W. Zhang, G. Jia, L. Zhang, C. J. Li, A. R. Dinner, C. G. Yang and C. He, Nat. Struct. Mol. Biol., 2012, 19, 671–676 CrossRef CAS.
- B. Chen, J. Gan and C. Yang, Sci. China: Chem., 2014, 57, 307–313 CrossRef CAS.
- J. Fischer, L. Koch, C. Emmerling, J. Vierkotten, T. Peters, J. C. Bruning and U. Ruther, Nature, 2009, 458, 894–898 CrossRef CAS PubMed.
- C. Church, L. Moir, F. McMurray, C. Girard, G. T. Banks, L. Teboul, S. Wells, J. C. Bruning, P. M. Nolan, F. M. Ashcroft and R. D. Cox, Nat. Genet., 2010, 42, 1086–1092 CrossRef CAS PubMed.
- X. Zhao, Y. Yang, B.-F. Sun, Y. Shi, X. Yang, W. Xiao, Y.-J. Hao, X.-L. Ping, Y.-S. Chen, W.-J. Wang, K.-X. Jin, X. Wang, C.-M. Huang, Y. Fu, X.-M. Ge, S.-H. Song, H. S. Jeong, H. Yanagisawa, Y. Niu, G.-F. Jia, W. Wu, W.-M. Tong, A. Okamoto, C. He, J. M. Rendtlew Danielsen, X.-J. Wang and Y.-G. Yang, Cell Res., 2014, 24, 1403–1419 CrossRef CAS PubMed.
- M. E. Hess, S. Hess, K. D. Meyer, L. A. Verhagen, L. Koch, H. S. Bronneke, M. O. Dietrich, S. D. Jordan, Y. Saletore, O. Elemento, B. F. Belgardt, T. Franz, T. L. Horvath, U. Ruther, S. R. Jaffrey, P. Kloppenburg and J. C. Bruning, Nat. Neurosci., 2013, 16, 1042–1048 CrossRef CAS PubMed.
- L. Li, L. Zang, F. Zhang, J. Chen, H. Shen, L. Shu, F. Liang, C. Feng, D. Chen, H. Tao, T. Xu, Z. Li, Y. Kang, H. Wu, L. Tang, P. Zhang, P. Jin, Q. Shu and X. Li, Hum. Mol. Genet., 2017, 26, 2398–2411 CrossRef CAS PubMed.
- H. Huang, H. Weng and J. Chen, Cancer Cell, 2020, 37, 270–288 CrossRef CAS PubMed.
- J. Liu, D. Ren, Z. Du, H. Wang, H. Zhang and Y. Jin, Biochem. Biophys. Res. Commun., 2018, 502, 456–464 CrossRef CAS PubMed.
- J. Li, Y. Han, H. Zhang, Z. Qian, W. Jia, Y. Gao, H. Zheng and B. Li, Biochem. Biophys. Res. Commun., 2019, 512, 479–485 CrossRef CAS PubMed.
- S. Yang, J. Wei, Y. H. Cui, G. Park, P. Shah, Y. Deng, A. E. Aplin, Z. Lu, S. Hwang, C. He and Y. Y. He, Nat. Commun., 2019, 10, 2782 CrossRef.
- Y. Niu, Z. Lin, A. Wan, H. Chen, H. Liang, L. Sun, Y. Wang, X. Li, X.-F. Xiong, B. Wei, X. Wu and G. Wan, Mol. Cancer, 2019, 18, 46 CrossRef PubMed.
- X. Tang, S. Liu, D. Chen, Z. Zhao and J. Zhou, Oncol. Lett., 2019, 17, 2473–2478 CAS.
- D. Zou, L. Dong, C. Li, Z. Yin, S. Rao and Q. Zhou, Cancer Cell Int, 2019, 19, 321 CrossRef CAS PubMed.
- Y. Zhu, J. Shen, L. Gao and Y. Feng, Oncol. Rep., 2016, 35, 2391–2397 CrossRef CAS PubMed.
- D. Xu, W. Shao, Y. Jiang, X. Wang, Y. Liu and X. Liu, Oncol. Rep., 2017, 38, 2285–2292 CrossRef CAS PubMed.
- Z. Li, H. Weng, R. Su, X. Weng, Z. Zuo, C. Li, H. Huang, S. Nachtergaele, L. Dong, C. Hu, X. Qin, L. Tang, Y. Wang, G. M. Hong, H. Huang, X. Wang, P. Chen, S. Gurbuxani, S. Arnovitz, Y. Li, S. Li, J. Strong, M. B. Neilly, R. A. Larson, X. Jiang, P. Zhang, J. Jin, C. He and J. Chen, Cancer Cell, 2017, 31, 127–141 CrossRef PubMed.
- Y. Liu, R. Wang, L. Zhang, J. Li, K. Lou and B. Shi, Oncol. Lett., 2017, 13, 4685–4690 CrossRef CAS PubMed.
- Y. Xiao, K. N. Thakkar, H. Zhao, J. Broughton, Y. Li, J. A. Seoane, A. N. Diep, T. J. Metzner, R. von Eyben, D. L. Dill, J. D. Brooks, C. Curtis, J. T. Leppert, J. Ye, D. M. Peehl, A. J. Giaccia, S. Sinha and E. B. Rankin, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21441–21449 CrossRef CAS PubMed.
- F. Yan, A. Al-Kali, Z. Zhang, J. Liu, J. Pang, N. Zhao, C. He, M. R. Litzow and S. Liu, Cell Res., 2018, 28, 1062–1076 CrossRef CAS PubMed.
- S. Zhou, Z. L. Bai, D. Xia, Z. J. Zhao, R. Zhao, Y. Y. Wang and H. Zhe, Mol. Carcinog., 2018, 57, 590–597 CrossRef CAS PubMed.
- L. Wen, X. Pan, Y. Yu and B. Yang, BMC Urol., 2020, 20, 39 CrossRef CAS PubMed.
- H. Huang, Y. Wang, M. Kandpal, G. Zhao, H. Cardenas, Y. Ji, A. Chaparala, E. J. Tanner, J. Chen, R. V. Davuluri and D. Matei, Cancer Res., 2020, 80, 3200–3214 CrossRef CAS.
- Z. Han, T. Niu, J. Chang, X. Lei, M. Zhao, Q. Wang, W. Cheng, J. Wang, Y. Feng and J. Chai, Nature, 2010, 464, 1205–1209 CrossRef CAS PubMed.
- X. Zhang, L. H. Wei, Y. Wang, Y. Xiao, J. Liu, W. Zhang, N. Yan, G. Amu, X. Tang, L. Zhang and G. Jia, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2919–2924 CrossRef CAS PubMed.
- H. Song, Y. Wang, R. Wang, X. Zhang, Y. Liu, G. Jia and P. R. Chen, Cell Chem. Biol., 2020, 27, 283–291.e286 CrossRef CAS PubMed.
- O. Sundheim, C. B. Vågbø, M. Bjørås, M. M. L. Sousa, V. Talstad, P. A. Aas, F. Drabløs, H. E. Krokan, J. A. Tainer and G. Slupphaug, EMBO J., 2006, 25, 3389–3397 CrossRef CAS PubMed.
- W. Aik, J. S. Scotti, H. Choi, L. Gong, M. Demetriades, C. J. Schofield and M. A. McDonough, Nucleic Acids Res., 2014, 42, 4741–4754 CrossRef CAS PubMed.
- Q. Li, Y. Huang, X. Liu, J. Gan, H. Chen and C. G. Yang, J. Biol. Chem., 2016, 291, 11083–11093 CrossRef CAS PubMed.
- R. Dong, S. Pan, Z. Peng, Y. Zhang and J. Yang, Nucleic Acids Res., 2018, 46, W380–W386 CrossRef CAS PubMed.
- R. Dong, Z. Peng, Y. Zhang and J. Yang, Bioinformatics, 2018, 34, 1719–1725 CrossRef CAS PubMed.
- X. Robert and P. Gouet, Nucleic Acids Res., 2014, 42, W320–W324 CrossRef CAS PubMed.
- B. Yu, W. C. Edstrom, J. Benach, Y. Hamuro, P. C. Weber, B. R. Gibney and J. F. Hunt, Nature, 2006, 439, 879–884 CrossRef CAS PubMed.
- C. G. Yang, C. Yi, E. M. Duguid, C. T. Sullivan, X. Jian, P. A. Rice and C. He, Nature, 2008, 452, 961–965 CrossRef CAS PubMed.
- J. D. W. Toh, L. Sun, L. Z. M. Lau, J. Tan, J. J. A. Low, C. W. Q. Tang, E. J. Y. Cheong, M. J. H. Tan, Y. Chen, W. Hong, Y. G. Gao and E. C. Y. Woon, Chem. Sci., 2015, 6, 112–122 RSC.
- C. Zhu and C. Yi, Angew. Chem., Int. Ed., 2014, 53, 3659–3662 CrossRef CAS PubMed.
- V. T. Monsen, O. Sundheim, P. A. Aas, M. P. Westbye, M. M. Sousa, G. Slupphaug and H. E. Krokan, Nucleic Acids Res., 2010, 38, 6447–6455 CrossRef CAS PubMed.
- Y. Huang, J. Yan, Q. Li, J. Li, S. Gong, H. Zhou, J. Gan, H. Jiang, G.-F. Jia, C. Luo and C.-G. Yang, Nucleic Acids Res., 2015, 43, 373–384 CrossRef CAS PubMed.
- Z. Han, N. Huang, T. Niu and J. Chai, Protein Cell, 2010, 1, 616–620 CrossRef CAS PubMed.
- M. Das, T. Yang, J. Dong, F. Prasetya, Y. Xie, K. H. Q. Wong, A. Cheong and E. C. Y. Woon, Chem. – Asian J., 2018, 13, 2854–2867 CrossRef CAS.
- E. Mishima, D. Jinno, Y. Akiyama, K. Itoh, S. Nankumo, H. Shima, K. Kikuchi, Y. Takeuchi, A. Elkordy, T. Suzuki, K. Niizuma, S. Ito, Y. Tomioka and T. Abe, PLoS One, 2015, 10, e0143756 CrossRef PubMed.
- B. Chen, F. Ye, L. Yu, G. Jia, X. Huang, X. Zhang, S. Peng, K. Chen, M. Wang, S. Gong, R. Zhang, J. Yin, H. Li, Y. Yang, H. Liu, J. Zhang, H. Zhang, A. Zhang, H. Jiang, C. Luo and C.-G. Yang, J. Am. Chem. Soc., 2012, 134, 17963–17971 CrossRef CAS.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- S. H. Yim, H. J. Cha, S. J. Park, Y. Yim, J. S. Woo and D. H. Min, Chem. Commun., 2020, 56, 4716–4719 RSC.
- Y. Zhang, Q. N. Li, K. Zhou, Q. Xu and C. Y. Zhang, Anal. Chem., 2020, 92, 13936–13944 CrossRef CAS PubMed.
- N. Svensen and S. R. Jaffrey, Cell Chem. Biol., 2016, 23, 415–425 CrossRef PubMed.
- A. Cheong, J. J. A. Low, A. Lim, P. M. Yen and E. C. Y. Woon, Chem. Sci., 2018, 9, 7174–7185 RSC.
- T. Yang, A. Cheong, X. Mai, S. Zou and E. C. Woon, Chem. Commun., 2016, 52, 6181–6184 RSC.
- F. Li, S. Kennedy, T. Hajian, E. Gibson, A. Seitova, C. Xu, C. H. Arrowsmith and M. Vedadi, J. Biomol. Screen., 2016, 21, 290–297 CrossRef CAS PubMed.
- W. Aik, M. Demetriades, M. K. K. Hamdan, E. A. L. Bagg, K. K. Yeoh, C. Lejeune, Z. Zhang, M. A. McDonough and C. J. Schofield, J. Med. Chem., 2013, 56, 3680–3688 CrossRef CAS PubMed.
- R. Su, L. Dong, C. Li, S. Nachtergaele, M. Wunderlich, Y. Qing, X. Deng, Y. Wang, X. Weng, C. Hu, M. Yu, J. Skibbe, Q. Dai, D. Zou, T. Wu, K. Yu, H. Weng, H. Huang, K. Ferchen, X. Qin, B. Zhang, J. Qi, A. T. Sasaki, D. R. Plas, J. E. Bradner, M. Wei, G. Marcucci, X. Jiang, J. C. Mulloy, J. Jin, C. He and J. Chen, Cell, 2018, 172, 90–105.e123 CrossRef CAS PubMed.
- Y. Huang, R. Su, Y. Sheng, L. Dong, Z. Dong, H. Xu, T. Ni, Z. S. Zhang, T. Zhang, C. Li, L. Han, Z. Zhu, F. Lian, J. Wei, Q. Deng, Y. Wang, M. Wunderlich, Z. Gao, G. Pan, D. Zhong, H. Zhou, N. Zhang, J. Gan, H. Jiang, J. C. Mulloy, Z. Qian, J. Chen and C.-G. Yang, Cancer Cell, 2019, 35, 677–691.e610 CrossRef CAS PubMed.
- Y. Liu, G. Liang, H. Xu, W. Dong, Z. Dong, Z. Qiu, Z. Zhang, F. Li, Y. Huang, Y. Li, J. Wu, S. Yin, Y. Zhang, P. Guo, J. Liu, J. J. Xi, P. Jiang, D. Han, C. G. Yang and M. M. Xu, Cell Metab., 2021, 33, 1221–1233.e1211 CrossRef CAS PubMed.
- T. Wang, T. Hong, Y. Huang, H. Su, F. Wu, Y. Chen, L. Wei, W. Huang, X. Hua, Y. Xia, J. Xu, J. Gan, B. Yuan, Y. Feng, X. Zhang, C.-G. Yang and X. Zhou, J. Am. Chem. Soc., 2015, 137, 13736–13739 CrossRef CAS.
- S. Huff, S. K. Tiwari, G. M. Gonzalez, Y. Wang and T. M. Rana, ACS Chem. Biol., 2021, 16, 324–333 CrossRef CAS PubMed.
- W. He, B. Zhou, W. Liu, M. Zhang, Z. Shen, Z. Han, Q. Jiang, Q. Yang, C. Song, R. Wang, T. Niu, S. Han, L. Zhang, J. Wu, F. Guo, R. Zhao, W. Yu, J. Chai and J. Chang, J. Med. Chem., 2015, 58, 7341–7348 CrossRef CAS.
- Y. Qiao, B. Zhou, M. Zhang, W. Liu, Z. Han, C. Song, W. Yu, Q. Yang, R. Wang, S. Wang, S. Shi, R. Zhao, J. Chai and J. Chang, Biochemistry, 2016, 55, 1516–1522 CrossRef CAS PubMed.
- S. Peng, W. Xiao, D. Ju, B. Sun, N. Hou, Q. Liu, Y. Wang, H. Zhao, C. Gao, S. Zhang, R. Cao, P. Li, H. Huang, Y. Ma, Y. Wang, W. Lai, Z. Ma, W. Zhang, S. Huang, H. Wang, Z. Zhang, L. Zhao, T. Cai, Y. L. Zhao, F. Wang, Y. Nie, G. Zhi, Y. G. Yang, E. E. Zhang and N. Huang, Sci. Transl. Med., 2019, 11, eaau7116 CrossRef PubMed.
- F. McMurray, M. Demetriades, W. Aik, M. Merkestein, H. Kramer, D. S. Andrew, C. L. Scudamore, T. A. Hough, S. Wells, F. M. Ashcroft, M. A. McDonough, C. J. Schofield and R. D. Cox, PLoS One, 2015, 10, e0121829 CrossRef PubMed.
- R. Wang, Z. Han, B. Liu, B. Zhou, N. Wang, Q. Jiang, Y. Qiao, C. Song, J. Chai and J. Chang, Mol. Pharm., 2018, 15, 4092–4098 CrossRef CAS PubMed.
- G. Zheng, T. Cox, L. Tribbey, G. Z. Wang, P. Iacoban, M. E. Booher, G. J. Gabriel, L. Zhou, N. Bae, J. Rowles, C. He and M. J. Olsen, ACS Chem. Neurosci., 2014, 5, 658–665 CrossRef CAS PubMed.
- X. Han, N. Wang, J. Li, Y. Wang, R. Wang and J. Chang, Chem. – Biol. Interact., 2019, 297, 80–84 CrossRef CAS PubMed.
- Y. Wang, J. Li, X. Han, N. Wang, C. Song, R. Wang and J. Chang, J. Mol. Recognit., 2019, 32, e2800 CrossRef PubMed.
- R. Su, L. Dong, Y. Li, M. Gao, L. Han, M. Wunderlich, X. Deng, H. Li, Y. Huang, L. Gao, C. Li, Z. Zhao, S. Robinson, B. Tan, Y. Qing, X. Qin, E. Prince, J. Xie, H. Qin, W. Li, C. Shen, J. Sun, P. Kulkarni, H. Weng, H. Huang, Z. Chen, B. Zhang, X. Wu, M. J. Olsen, M. Müschen, G. Marcucci, R. Salgia, L. Li, A. T. Fathi, Z. Li, J. C. Mulloy, M. Wei, D. Horne and J. Chen, Cancer Cell, 2020, 38, 79–96.e11 CrossRef CAS PubMed.
- N. R. Rose, M. A. McDonough, O. N. King, A. Kawamura and C. J. Schofield, Chem. Soc. Rev., 2011, 40, 4364–4397 RSC.
- M. S. Waitkus, B. H. Diplas and H. Yan, Cancer Cell, 2018, 34, 186–195 CrossRef CAS PubMed.
- Y. Qing, L. Dong, L. Gao, C. Li, Y. Li, L. Han, E. Prince, B. Tan, X. Deng, C. Wetzel, C. Shen, M. Gao, Z. Chen, W. Li, B. Zhang, D. Braas, J. Ten Hoeve, G. J. Sanchez, H. Chen, L. N. Chan, C. W. Chen, D. Ann, L. Jiang, M. Müschen, G. Marcucci, D. R. Plas, Z. Li, R. Su and J. Chen, Mol. Cell, 2021, 81, 922–939.e929 CrossRef CAS PubMed.
- Q. Cui, H. Shi, P. Ye, L. Li, Q. Qu, G. Sun, G. Sun, Z. Lu, Y. Huang, C. G. Yang, A. D. Riggs, C. He and Y. Shi, Cell Rep., 2017, 18, 2622–2634 CrossRef CAS PubMed.
- X. Wang, Z. Li, B. Kong, C. Song, J. Cong, J. Hou and S. Wang, Oncotarget, 2017, 8, 98918–98930 CrossRef PubMed.
- Q. Zhao, Y. Zhao, W. Hu, Y. Zhang, X. Wu, J. Lu, M. Li, W. Li, W. Wu, J. Wang, F. Du, H. Ji, X. Yang, Z. Xu, L. Wan, Q. Wen, X. Li, C. H. Cho, C. Zou, J. Shen and Z. Xiao, Theranostics, 2020, 10, 9528–9543 CrossRef CAS PubMed.
- Y. Xu, S. Ye, N. Zhang, S. Zheng, H. Liu, K. Zhou, L. Wang, Y. Cao, P. Sun and T. Wang, Cancer Commun., 2020, 40, 484–500 CrossRef.
- I. Van Der Werf and C. Jamieson, Cancer Cell, 2019, 35, 540–541 CrossRef CAS PubMed.
- L. Yan, V. J. Colandrea and J. J. Hale, Expert Opin. Ther. Pat., 2010, 20, 1219–1245 CrossRef CAS PubMed.
- B. Singh, H. E. Kinne, R. D. Milligan, L. J. Washburn, M. Olsen and A. Lucci, PLoS One, 2016, 11, e0159072 CrossRef PubMed.
- M. Padariya and U. Kalathiya, Comput. Biol. Chem., 2016, 64, 414–425 CrossRef CAS PubMed.
- I. Barbieri and T. Kouzarides, Nat. Rev. Cancer, 2020, 20, 303–322 CrossRef CAS.
- S. Selberg, D. Blokhina, M. Aatonen, P. Koivisto, A. Siltanen, E. Mervaala, E. Kankuri and M. Karelson, Cell Rep., 2019, 26, 3762–3771.e3765 CrossRef CAS PubMed.
- R. K. Bedi, D. Huang, S. A. Eberle, L. Wiedmer, P. Śledź and A. Caflisch, ChemMedChem, 2020, 15, 744–748 CrossRef CAS PubMed.
- E. Yankova, W. Blackaby, M. Albertella, J. Rak, E. De Braekeleer, G. Tsagkogeorga, E. S. Pilka, D. Aspris, D. Leggate, A. G. Hendrick, N. A. Webster, B. Andrews, R. Fosbeary, P. Guest, N. Irigoyen, M. Eleftheriou, M. Gozdecka, J. M. L. Dias, A. J. Bannister, B. Vick, I. Jeremias, G. S. Vassiliou, O. Rausch, K. Tzelepis and T. Kouzarides, Nature, 2021, 593, 597–601 CrossRef CAS PubMed.
- R. K. Bedi, D. Huang, L. Wiedmer, Y. Li, A. Dolbois, J. A. Wojdyla, M. E. Sharpe, A. Caflisch and P. Sledz, ACS Chem. Biol., 2020, 15, 618–625 CrossRef CAS PubMed.
- N. Li, Y. Kang, L. Wang, S. Huff, R. Tang, H. Hui, K. Agrawal, G. M. Gonzalez, Y. Wang, S. P. Patel and T. M. Rana, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20159–20170 CrossRef CAS PubMed.
- L. Wu and X. Qu, Chem. Soc. Rev., 2015, 44, 2963–2997 RSC.
| This journal is © The Royal Society of Chemistry 2021 |