
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-cleaving ribozymes: substrate specificity and synthetic biology applications
Huan
Peng†
a,
Brandon
Latifi†
 b,
Sabine
Müller
c,
Andrej
Lupták
*b and
Irene A.
Chen
*a
b,
Sabine
Müller
c,
Andrej
Lupták
*b and
Irene A.
Chen
*a
aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095, USA. E-mail: ireneachen@ucla.edu
bDepartment of Pharmaceutical Sciences, University of California, Irvine, CA 92697, USA. E-mail: aluptak@uci.edu
cInstitute for Biochemistry, University Greifswald, 17487 Greifswald, Germany
First published on 2nd July 2021
Abstract
Various self-cleaving ribozymes appearing in nature catalyze the sequence-specific intramolecular cleavage of RNA and can be engineered to catalyze cleavage of appropriate substrates in an intermolecular fashion, thus acting as true catalysts. The mechanisms of the small, self-cleaving ribozymes have been extensively studied and reviewed previously. Self-cleaving ribozymes can possess high catalytic activity and high substrate specificity; however, substrate specificity is also engineerable within the constraints of the ribozyme structure. While these ribozymes share a common fundamental catalytic mechanism, each ribozyme family has a unique overall architecture and active site organization, indicating that several distinct structures yield this chemical activity. The multitude of catalytic structures, combined with some flexibility in substrate specificity within each family, suggests that such catalytic RNAs, taken together, could access a wide variety of substrates. Here, we give an overview of 10 classes of self-cleaving ribozymes and capture what is understood about their substrate specificity and synthetic applications. Evolution of these ribozymes in an RNA world might be characterized by the emergence of a new ribozyme family followed by rapid adaptation or diversification for specific substrates.
Introduction
Functional ribonucleic acid (RNA) is a molecule of special interest due to its dual ability to store information and catalyze chemical reactions. Ribozymes are a class of RNAs that typically catalyze self-referential activities (e.g., self-cleavage, self-splicing, ligation, template-directed polymerization) and are the foundation of a branch of synthetic biology aimed at creating a simple cell with primitive metabolism.1 Self-cleaving ribozymes are some of the most well-studied ribozymes and are often used as model RNAs for testing new methods or reaction conditions. These have been discovered as naturally occurring transcripts (e.g., as viral/viroid replication intermediates), through in vitro selection from a pool of random or genomic sequences, or, increasingly, through bioinformatic searches. There are now more than eleven characterized families of small, self-cleaving ribozymes. Here, we review what is known about the sequence specificity of the naturally-occurring families of these ribozymes, and indicate how some of the ribozymes could have synthetic biology applications.Ribozyme specificity is an important consideration for possible applications, such as mRNA inactivation. In particular, high sequence specificity of the ribozymes for their substrates, i.e., the ability to distinguish between target and closely related alternative sequences, is desirable for such applications. Conversely, substrate promiscuity is the basis for the potential evolvability of ribozymes to process new substrates. It has been previously suggested that early ribozymes might have been characterized by low activity and low specificity, allowing diversifying evolution toward multiple functions in parallel.2–4 Substrate specificity (or promiscuity) would therefore be a critical aspect influencing the ability of an RNA world to expand in complexity during early evolution.
The families of small self-cleaving ribozymes include the hepatitis delta virus (HDV)-like, glucosamine-6-phosphate synthase (glmS), hatchet, hammerhead, hairpin, twister, twister sister, pistol, Neurospora Varkud satellite (VS); in addition to recently discovered mammalian SINE and hovlinc ribozymes. In general, these ribozymes act in cis (i.e., on the same strand), and therefore execute a single catalytic turnover. It is usually possible to engineer the RNA into separate catalytic and substrate strands such that the catalytic strand acts in trans. In those cases, the ribozyme can often perform multiple turnovers.
Although the different ribozymes have distinct primary, secondary, and tertiary structures, as well as self-scission sites, they accelerate the same transesterification reaction. The chemical mechanism of the cleavage reaction is achieved through general acid–base catalysis (Fig. 1). The ribozymes need to be correctly folded to bring functional nucleotides to the vicinity of the cleavage site and adopt a conformation that positions the cleavage site in the required in-line geometry. In most cases, the ribozymes use nucleobases from their structure or exogenous cofactors as the general base or acid. The general base deprotonates the 2′-hydroxyl group, thus enhancing nucleophilicity of the oxygen, which attacks the adjacent phosphorous. As a result, a trigonal bipyramidal transition state (or intermediate, if stabilized) is formed. Protonation of the 5′-oxygen in the pentavalent phosphorane by a general acid leads to departure of the leaving group and formation of the characteristic products: a 2′,3′-cyclic phosphate and a free 5′-OH group.4 Mechanisms of these ribozymes have been extensively reviewed recently and are not covered in depth here.5–8 In this review, we provide a brief introduction to the mechanism of each ribozyme in order to contextualize the accompanying description of substrate specificity. We also discuss how gaining more insight into these ribozymes can allow for synthetic biology applications to be realized, such as engineering of aptazymes to control gene expression through cleavage of the 5′ and 3′ untranslated regions (UTRs) of mRNAs.
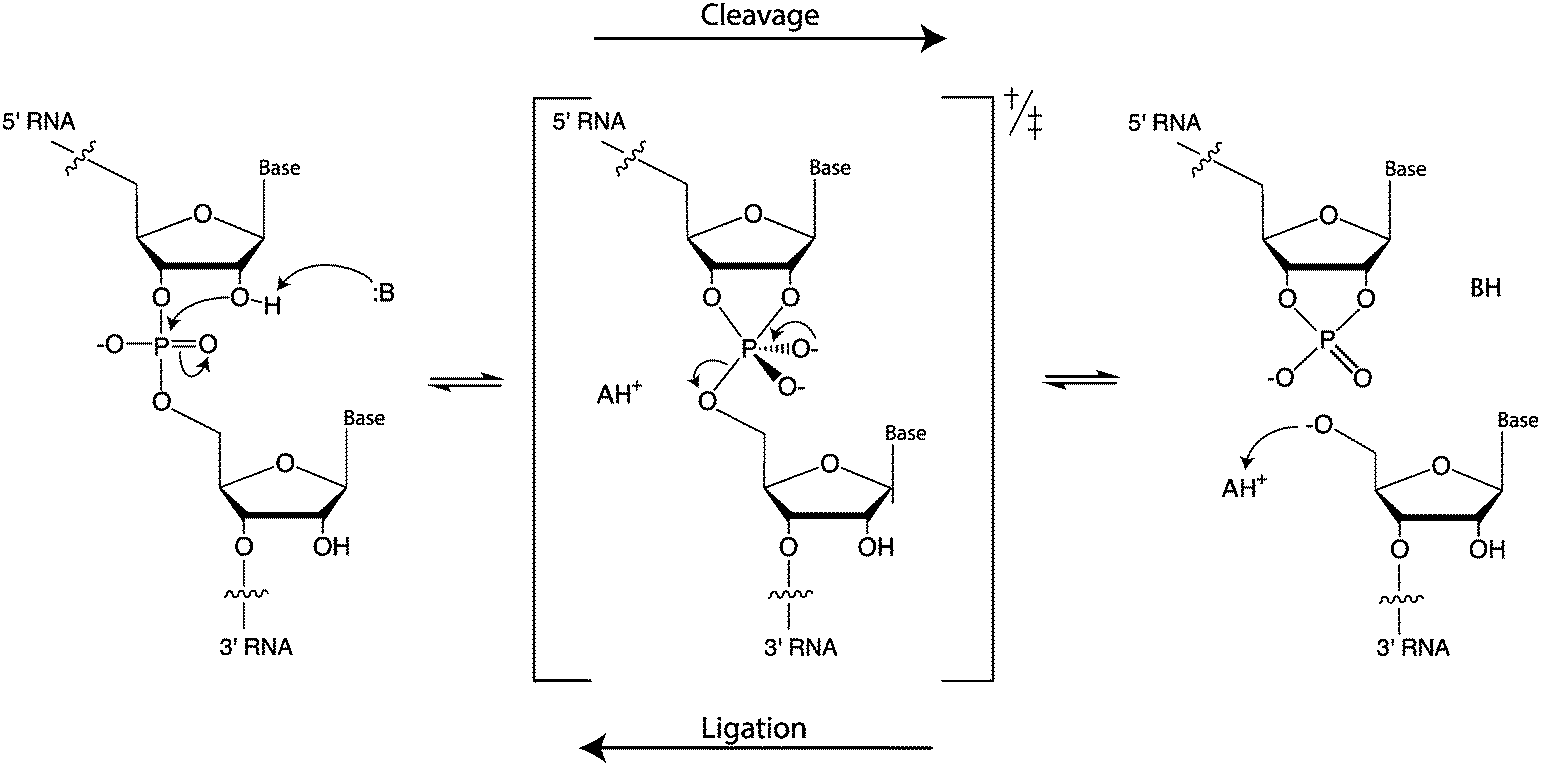 | ||
| Fig. 1 Mechanism of RNA self-scission and ligation by general acid–base catalysis. | ||
Hepatitis delta virus (HDV)-like ribozymes
HDV is an RNA satellite of the hepatitis B virus (HBV) that relies on host cellular machinery for rolling-circle replication of its circular, single-stranded, 1700-nucleotide (nt) RNA genome.13 This family of ribozymes is widespread in genomes of eukaryotes, including mammals,14–16 with several examples mapping to bacterial genomes.17,18 The cis-acting HDV ribozyme consists of five helical regions (P1 through P4 and P 1.1), which form two coaxial stacks: P1 and P1.1 stack on P4 while P2 stacks on P3 (Table 1).19–21 In the trans-acting ribozyme, the substrate is typically derived from the 5′ strand of P1 and the remainder of the sequence forms the ribozyme.| 2° Structure | Active site | Substrate strand requirement |
|---|---|---|

|
Cleavage site: peripheral | Upstream: single nucleotide |
| General acid: C75 in J2/4 | Downstream: at least 6 base pairs, starting with a G + 1 | |
| General base: hydrated divalent metal ion | ||
| Ligation: no | ||
| Ref. 9 and 10 | ||
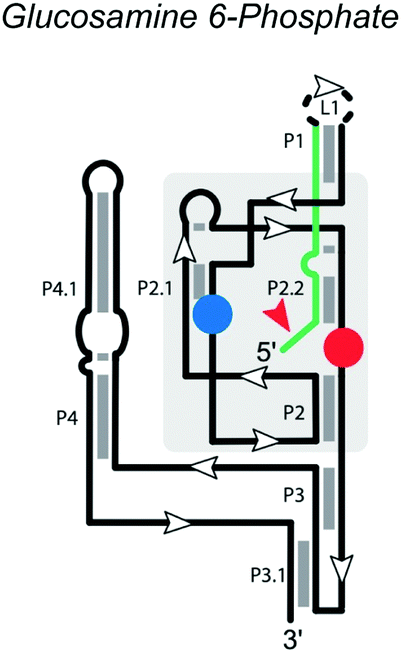
|
Cleavage site: peripheral | Upstream: single nucleotide |
| General acid: GlcN6P | Downstream: base pairing | |
| General base: G40 in P2.1 | ||
| Ligation: no | ||
| Ref. 11 | ||
|
Cleavage site: peripheral | Upstream: single nucleotide |
| General acid: G30 or Mg2+-H2O coordinated G30 located in base pair and stacks on P2 | Downstream: base pairing | |
| General base: potentially G31 located in base pair and stacks underneath P3 | Notes: G30 and G31 mutations poorly tolerated. | |
| Ligation: no | ||
| Ref. 12 | ||
A hydroxide from the hydration shell of a divalent metal ion, such as Mg2+ or Ca2+, and C75, which maps to the beginning of the J4/2 strand, serve as the general base and general acid, respectively. The crystal structure of the self-cleaved ribozyme suggested that the N3 of C75 can hydrogen bond with the leaving group,19 donating a proton to the 5′-oxyanion of G1.22 Although Mg2+ is not absolutely required for the reaction, changing the pH from 6 to 8 either enhances or reduces the reaction rate, in the presence or absence of Mg2+, respectively,23 consistent with a hydrated Mg2+ acting as the general base when present, accepting the proton from the 2′-OH nucleophile. The nucleophile appears to exist in the protonated form in the ground state, but becomes negatively charged at the beginning of the reaction.24 This appears to be supported by the active site Mg2+ ion.25 The pKa of the general acid C75 is shifted to facilitate protonation of the leaving group.26,27 In the HDV and HDV-like ribozymes (i.e., other non-HDV, self-cleaving ribozymes with structural and biochemical similarities to the HDV ribozyme28), self-scission is supported by a number of divalent cations.29
Two forms of a trans-acting HDV ribozyme have been generated from the cis-acting ribozyme sequences. In the first form, the ribozyme was interrupted in the loop of P4, yielding a large piece containing the substrate strand and a relatively small strand containing the catalytic cytosine. The two pieces interact via P4 and P2 to cleave the 5′-fragment of P1.26,30,31 Because the active site is split between the two parts, the construct is useful for mechanistic studies of the ribozyme, but not for applications involving cleaving target RNAs in trans.
In the second form, the 5′ segment of P1 is separated from P2 in J1/2, forming the substrate strand, which associates by base pairing in P1 to the larger catalytic domain.32 In this situation, a circularly permuted trans-acting ribozyme can be generated by closing the end of P2 with a short loop.33 Interchanging 1–4 nucleotide pairs between the substrate strand and the catalytic strand maintains cleavage activity.34–37 The peripheral terminus of the stem formed by the substrate and ribozyme strands appears to be more tolerant of a distortion of the P1 stem caused by mismatches compared to internal positions. Nucleotides in the middle of the substrate strand are of critical importance to substrate recognition and cleavage activity, with the wobble pair (G–U) at the cleavage site being essential. Simultaneous alteration of two nucleotides in the middle of the P1 stem eliminates cleavage activity in both trans- and cis-acting HDV ribozymes, possibly by disrupting formation of the metal-ion binding site.38 The trans-acting ribozyme can cleave a substrate with a minimum of six nucleotides adjacent to the cleavage site in the 3′ direction. Substrates with a single mismatch show low cleavage rates, likely due to the poor binding between the substrate and the ribozyme.
The 5′ region of the cleavage site (positions upstream of site −1) also affects substrate specificity.39 A systematic study of mutations at positions −1 to −4 of the substrate40,41 revealed that cleavage is somewhat sensitive to these positions. The consensus cleavable substrate from −1 to −4 was found to be −1HRHY−4 (H = U, C, or A; R = A or G; Y = C or U). For position −1, the order of reactivity was A > C > U, with G being a poorly-cleavable substrate, while for position −2, the order was A > G ≫ U, with C being a low-cleavage substrate. The reactivity of the nucleotide at position −2 also depended on the base at position −1. Positions −1 to −4 appear to be important for formation of the P1.1 pseudoknot, which is essential for cleavage activity.42,43 Base-pairing between the single-stranded 5′ end of the substrate and the J1/4 junction of the ribozyme inhibits folding of the P1.1 stem, causing a significant decrease of cleavage. Thus, some substrate sequences may inhibit activity by causing misfolding of the ribozyme-substrate complex.
Longer substrates have also been studied, illustrating a trade-off between substrate affinity and sequence specificity, which are improved by additional base-pairing interactions formed in the J1/2 region. However, longer base-paired products result in very slow off-rates, greatly decreasing turnover.44 Overall, at least 6 nucleotides are invariant to make an active HDV-like ribozyme, but only one of these (G1) resides in the substrate strand. The multitude of natural HDV-like ribozymes provides a good starting point for designing ribozymes with variable sequence specificity and size, particularly for trans-cleavage.28 The substrate helix (P1) must be at least 6 bp long,29 but can be extended to an arbitrarily long substrate through interactions in a P1.2 helix.29,45
Glucosamine-6-phosphate synthase (glmS) ribozyme
The glmS ribozyme, found in many Gram-positive bacteria, regulates the expression of the glmS gene, which encodes glutamine-fructose-6-phosphate transaminase.46 It is a conserved motif located in the 5′ untranslated region of the glmS gene and the only self-cleaving ribozyme family known to use the metabolite glucosamine 6-phosphate (GlcN6P) as a cofactor for catalysis. The secondary structure (Table 1) of the glmS ribozyme contains three near-parallel coaxial stacks, with P1 stacking on P3.1, P4 stacking on P4.1, and P2.1. The core structure of the glmS ribozyme is a double pseudoknot involving both P2.1 and P2.2. P1 and P2 contain the necessary conserved base-pairs for the catalytic center, located in the pseudoknots of P2.1 and P2.2. P2.2 forms part of the binding site for the GlcN6P cofactor and also serves as the substrate strand. Folding of P2.2 enables the ribozyme to adopt the active conformation.47 Although P3 and P4 are not necessary for catalytic activity, they provide structural stability and enhance cleavage ability,46,48 especially at low Mg2+ concentrations.49Unlike many riboswitches, ligand binding of the glmS ribozyme does not cause a major conformational change.50 GlcN6P is buried in the binding pocket with its sugar ring stacked on the substrate-strand G1 nucleobase and its phosphate, forming a hydrogen bond with N1 of G1 and coordinating a Mg2+ ion. The GlcN6P cofactor appears to have multiple roles, including protonation of the 5′-O leaving group, alignment of the active site, activation of the 2′-O nucleophile through disruption of an inhibitory interaction, and charge stabilization of the nonbridging oxygen atoms during the reaction.50–52 The glmS ribozyme thus utilizes an overdetermined set of competing hydrogen bond donors in the active site to ensure potent activation and regulation by the cofactor GlcN6P. In the active site, G40 of the Thermoanaerobacter tengcongesis glmS ribozyme (shown in Table 1) or guanosine G33 of the B. anthracis glmS ribozyme acts as the general base by hydrogen bonding with the A-1 2′-OH nucleophile.50,53,54
Replacement of the C2-amine of GlcN6P by a hydroxyl group in Glc6P incurs a ∼104-fold reduction of activity, similar to the activity without GlcN6P,46,55 although the importance of GlcN6P varies somewhat in this family of ribozymes.55 Interestingly, G33 modulates the pKa of both the phosphate and the amine of GlcN6P.56 In some cases, a different amine may functionally substitute for GlcN6P, but the rate enhancement of GlcN6P is approximately 5 orders of magnitude higher than that by Tris at the same concentration (10 mM). Similar results were observed when using glucosamine or serinol to replace GlcN6P. On the other hand, glucose, ethanol, methylamine and ammonium ions cannot activate the self-cleavage reaction, suggesting that adjacent amine and hydroxyl groups are necessary to support the activity of the glmS ribozyme.55
A trans-acting core version of the B. cereus glmS ribozyme has been created by removing the closing loop of helix P1.57 Compared with the cis-acting full-length glmS ribozyme, the apparent GlcN6P and Mg2+ affinity of the trans-acting ribozyme was ∼24-fold and ∼10-fold weaker, respectively. Nevertheless, like the cis-acting glmS ribozyme, this trans-acting variant shows a very slow cleavage rate in the absence of GlcN6P and is activated by ∼5000-fold with 10 mM GlcN6P. Addition of comparable amounts of GlcN, a ligand analog lacking the 6-phosphate moiety, activates the trans-acting glmS weakly (by ∼30-fold), and Glc6P, lacking the 2-amino group, does not significantly activate the ribozyme. The trans-acting glmS ribozyme is fully folded in solution before binding GlcN6P, supporting a direct role of this cofactor in the catalytic mechanism rather than folding. The cleavage site is located at the 5′ end of the ribozyme, with only one nucleotide required upstream of the cut site. Downstream, only base-pairing is required within the P2.2 and P1 regions.
Hatchet ribozyme
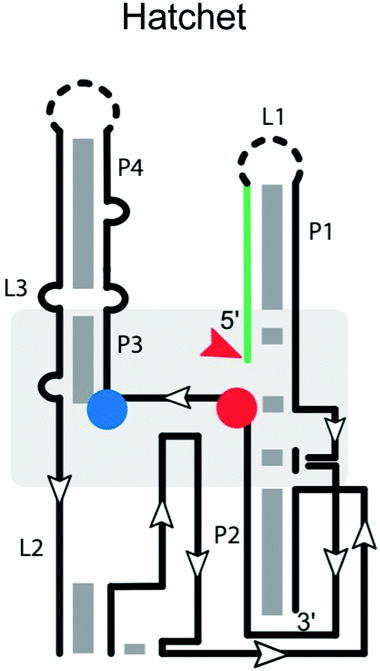
Many ribozymes have been found by bioinformatic searches for conserved RNA structures.5 This applies to the GlmS, pistol, twister, twister sister, and hatchet ribozymes. The hatchet ribozyme58 has a secondary structure comprising four stems (P1, P2, P3 and P4), with the first two linked by three highly conserved residues and the latter three bridged by two internal loops (L2 and L3) (Table 1).59 Loop L2 harbors most of the highly conserved residues. The crystal structure of the product state59 shows a compact pseudo-symmetrical RNA, with each ‘half’ stabilized by long-range interactions involving highly conserved nucleotides in close proximity to the scissile phosphate. The cleavage site of the hatchet ribozyme is located at the 5′ end of stem P1—a distal location like that in the HDV-like ribozyme. Modeling of C(−1) into the active site pocket suggested that N7 of G31 may act as the general base to deprotonate the 2′-OH of C(−1) for nucleophilic attack.59 A hatchet variant with 7-deazaguanosine in position 31 is completely inactivated. Additionally, Mg2+ is essential for self-cleavage,17 and important for ribozyme folding,60 but not necessarily for active site chemistry. Further work on these ribozymes will be necessary to reveal key determinants of substrate specificity.The hammerhead ribozyme
The hammerhead ribozymes are a widespread class of self-cleaving RNAs that were originally found in plant viroids and satellites, catalyzing specific cleavage reactions during processing of rolling-circle transcripts.68 The cis-acting hammerhead ribozyme consists of a universally conserved junction sequence and three helices (stem I, II and III;69Table 2).69 The general base that activates the nucleophile is the N1 of G12 from stem II, which forms an H-bond to the nucleophilic 2′-OH of C17.70,71 Additional studies indicate that a divalent metal cation activates G12 and helps organize the active site.69 G8 is stabilized in place by Watson–Crick base-pairing to C3.72 However, a more recent study indicates that a Mn2+-bound water, rather than the 2′-OH of G8, serves as the general acid in the cleavage reaction.73| 2° Structure | Active site | Substrate strand requirement |
|---|---|---|
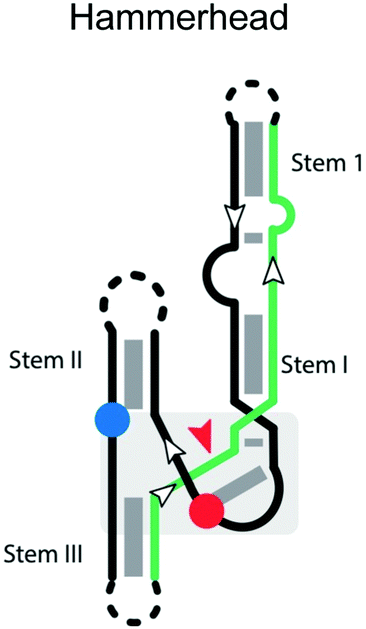
|
Cleavage site: internal | Upstream: stem III base pairing |
| General acid: invariant G8 in ribozyme strand of loop within catalytic core | Downstream: stem I base pairing | |
| General base: invariant G12 in between stem II & stem III | Notes: cleavage is between CN | |
| Ligation: yes | *All terminal loops can be manipulated into a trans version | |
| Ref. 61 | ||
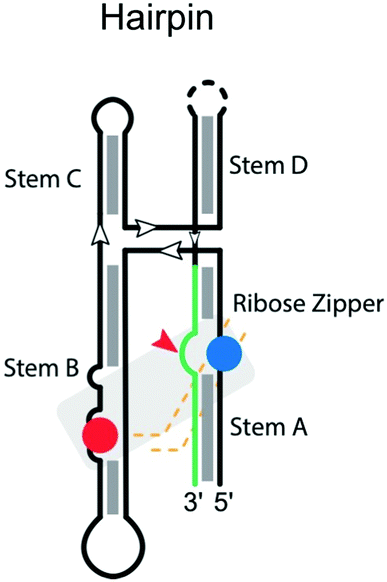
|
Cleavage site: internal | Upstream: base pairing within stem A and D |
| General acid: A38 in stem B | Downstream: base pairing following G + 1 in stem A | |
| General base: G8 in stem A | Notes: H-bond substitutions for G + 1 destroy activity. A38 cannot be mutated | |
| Ligation: yes | ||
| Ref. 62 | ||
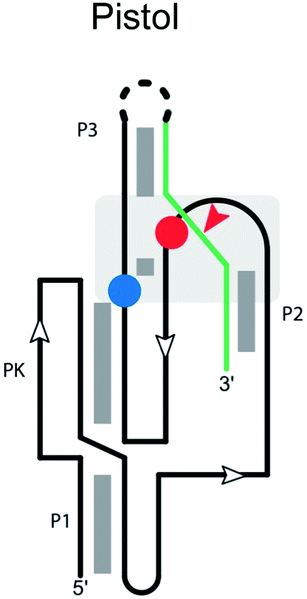
|
Cleavage site: internal | Upstream: base pairing |
| General acid: A32 on the ribozyme strand between P2 and pseudoknot (PK) | Downstream: base pairing | |
| General base: G40 located on the ribozyme strand between pseudoknot (PK) and P3 | ||
| Ligation: yes63 | ||
| Ref. 64 | ||
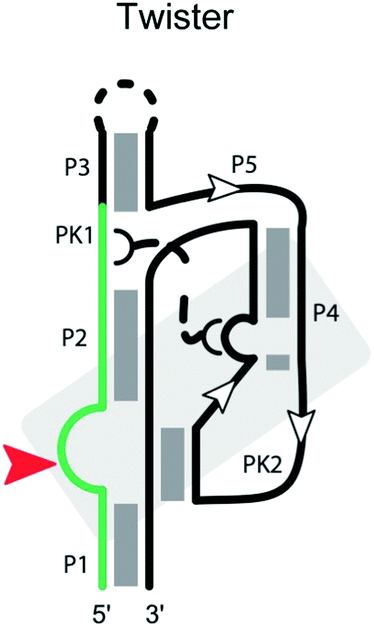
|
Cleavage site: internal | Upstream: base pairing |
| General acid: A10? or A1?65 | Downstream: base pairing, starting with A | |
| General base: A63? G62?66 G3365 | Notes: core of twister is stabilized by stacking and two pseudoknots | |
| Ligation: ? | ||
| Ref. 66 | ||
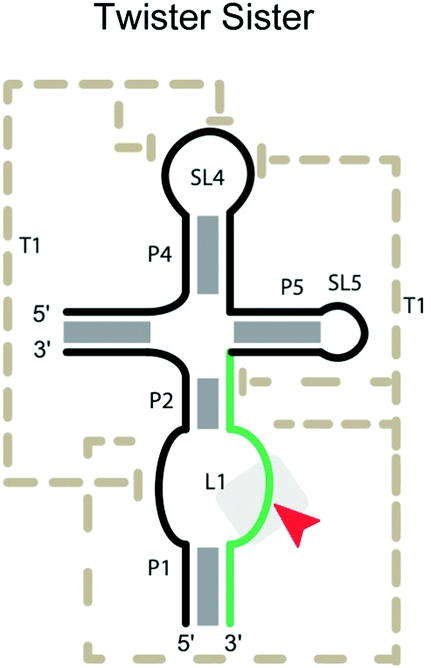
|
Cleavage site: internal | Upstream: base pairing |
| General acid: ? | Downstream: base pairing | |
| General base: ? | Notes: mutation of A63 and G5 cannot be mutated | |
| Ligation: ? | ||
| Ref. 60 and 67 | ||
G12 and G8 are critical residues that should be preserved in the catalytic strand, as well as for proper folding and positioning of the scissile phosphate in the required in-line geometry of the catalytic core. Mutation of G12 to any other nucleobases or substitution of G12 by analogs perturbing the pKa of N1 were found to significantly decrease the reaction rate.74,75 The 2′-OH of G8 is similarly essential,76,77 and mutation of G8 to C decreases the reaction rate by 105-fold.72,75 Interestingly, the reaction rate of the G8C mutant can be partially restored by the compensatory mutation C3G,72 suggesting some flexibility in the identity of these important residues.
A major determinant of sequence specificity is the stable base-pairing of the substrate strand to the catalytic strand. The sequence specificity of the hammerhead ribozyme was reported to be NUH,78 where N is any nucleotide, and H is any nucleotide except G. Eckstein and coworkers re-investigated the sequence specificity of the hammerhead ribozyme reaction and rewrote the NUH rule to be NHH.79,80 In the trans-acting hammerhead ribozyme, cleavage specificity is largely determined by the different binding affinities between the matched and mismatched substrates,81 which is influenced by the length and base composition of the helices formed upon ribozyme-substrate binding. Therefore, for high specificity, these helices should have the minimum length necessary to ensure binding, because overly long recognition helices reduce the specificity of cleavage and the bound form can have a substantial lifetime even with a suboptimal match. Adding more nucleotides to the recognition sequence ultimately reduces substrate specificity,82 because the cleavage rate of both matched and mismatched substrate is still much faster than the substrate dissociation rate. Thus, optimal substrate specificity can be achieved by increasing the substrate dissociation rate (reducing the binding affinity) to be close to the cleavage rate. The position of mismatches is also important, with mismatches in the innermost base pairs of stem I and III having the greatest effect on cleavage rate, and distal mismatches having less effect.83 Overall, while the influence of bases involved in catalysis may be more difficult to predict, sequence specificity caused by substrate association can be understood in this framework and is therefore amenable to engineering.
The hairpin ribozyme
Like the hammerhead ribozyme, the hairpin ribozyme is found in RNA satellites of plant viruses like the tobacco ringspot,68 chicory yellow mottle,84 and arabis mosaic virus.85 The hairpin ribozyme performs a reversible self-cleavage reaction in processing multimeric RNA during rolling-circle replication of the viral genome. There are four helical stems anchoring a four-way junction in the secondary structure of the hairpin ribozyme. The catalytic center involves a complex ribose zipper formed by A10 and G11 and A24 and C25 (Table 2).86 Residues G8 and A38 on the ribozyme strand, located within stems B and A, respectively, serve as the general base and general acid, respectively.87,88 The microscopic pKa of the N1 imino group of A38 matches the macroscopic pKa measured for the cleavage reaction for the most active, four-way helical junction, form of the ribozyme, consistent with its proposed role as general acid.89 Interestingly, crowded environments also pose a critical influence on the catalysis of the hairpin ribozyme, stabilizing the active ‘docked’ conformation and the transition state, leading to increased catalytic activity.90Hairpin ribozyme mutants were extensively studied using in vitro selection to screen for active variants,91,92 identifying key catalytic residues in both loops (positions 8–10 and 21–25). These studies demonstrated a very strong preference for G at the +1 position, but the base identity at positions −1, +2, and +3 was less constrained. Accordingly, the consensus sequence at the cleavage site was defined as N*GUY.93 However, later studies demonstrated that also G*GUN, G*GGR, and U*GUA substrates were cleaved, although with about five-fold lower activity.94 To improve activity on poorly processed substrates, hairpin ribozyme variants were engineered for G*GUA95 and A*GCU sites,96 thus further extending substrate specificity. These two latter studies clearly demonstrated that, as implicated before,97 compensatory base changes of the ribozyme strand can rescue nucleobase substitutions in the substrate, thus recovering not only cleavage but also ligation activity. For example, mutation of C + 3 to A in the substrate can be tolerated by substitution of A7 by U in the ribozyme strand, compensating for the lost interaction between wild-type C + 3 and A7.95 Similarly, mutation of U + 2 to C in the substrate is compensated by replacement of A8 with G in the ribozyme strand.96 Thus, substrate specificity can be expanded by appropriate engineering of the hairpin ribozyme.
Pistol ribozyme
The secondary structure of the pistol ribozyme is composed of one pseudoknot and three helical stems (P1, P2 and P3; Table 2), which are connected by three loops (loops 1, 2 and 3), with the P1 stem and pseudoknot forming a stacked structure.98 In the trans construct,99 stems P1 and P2 each comprise five Watson–Crick base pairs, and stem P2 contains an additional U–U base pair between U29 on the ribozyme and U1 on the substrate strand. The U–U base pair has two potential roles, helping to adjust the orientation of the scissile phosphate for the cleavage reaction and/or stabilizing U1 after the cleavage reaction. Three highly conserved adenosine residues in loop 1 form an A-minor interaction with stem P1 and orient stem P2 in the active site. Loops 2 and 3 flank the pseudoknot and form the active site. The scissile phosphate on the substrate strand is located on a sharp turn between stems P2 and P3.The N1 of G40 appears to act as the general base, activating the 2′-OH nucleophile to initiate cleavage.99 To do this, the N1 of G40 needs to be deprotonated by ionization or tautomerization. A32 is also in proximity to the active site and it has been suggested to act as the general acid by neutralizing the charge of the 5′-O during the reaction. Instead, because disruption of the inner-sphere coordination of the active-site Mg2+ cation to N7 of G33 significantly reduces the cleavage rate, it has been proposed that the hydrated Mg2+ acts as a general acid. A crystal structure of the ribozyme100 suggested that this cation plays a major role in catalysis. A second Mg2+ stabilizing the 2′,3′-cyclic phosphate in the product complex further supports the cleavage process.101
The ribozyme displays full activity with an A32G mutation, consistent with interactions with the ribose ring being more critical than the base. Overall, while data on specificity are limited, residues affecting positions 32 and 40, and the U–U wobble base pair in stem P2, are expected to influence substrate specificity.
Twister ribozyme
The twister ribozyme is a small catalytic RNA motif, present in many species of bacteria and eukaryotes.102 It comprises five stems (P1, P2, P3, P4 and P5; Table 2) and internal loops stabilized by two pseudoknots.66,102–106 Cleavage of the twister ribozyme is folding-dependent. In particular, the central pseudoknot displays reversible opening and closing dynamics at physiological Mg2+ concentration, and proper orientation of the substrate is required to achieve the cleavage-competent conformation.107 A Mg2+ ion in the env22 twister ribozyme appears to coordinate the non-bridging phosphate oxygen at the U–A cleavage site,103,106 potentially stabilizing the transition state into an intermediate. Consistent with these influences, ribozyme activity strongly depends on the environmental conditions including pH, Mg2+ concentration and temperature.102,103,105A highly conserved guanine at the cleavage site appears to be critical for the ribozyme activity. In the env22 twister ribozyme structure,103 as well as the env9 and O. sativa twister ribozyme structures,66,105 the guanine is thought to act as the general base, whereas an adenine at the cleavage site is suggested to play the role of a general acid.106 The locations of other key residues, such as the conserved U at the cleavage site, differ among the ribozyme structures, possibly reflecting conformational changes in the transition state of the twister ribozyme.
At least ten strongly conserved nucleotides are located at the active site,66 which contains the scissile phosphate between a conserved A and U adjoining stem P1. Mutants at these conserved positions display largely reduced activity. Although the twister ribozyme has not yet been as closely examined as other self-cleaving ribozymes, we expect the substrate specificity of the twister ribozyme to be largely determined by base-pairing between the ribozyme strand and the substrate strand, as well as the conserved nucleotides and tertiary interactions in the cleavage center, including interactions with the Mg2+ ion.
Twister sister ribozyme
The twister sister ribozymes are another family discovered through bioinformatics, and have similar sequence and secondary structure to the twister ribozymes.17 However, they lack the double pseudoknot interaction found in the twister ribozymes. In the pre-catalytic structure of the four-way version (Table 2),67 Mg2+ cations mediate long-range interactions, which bring 11 conserved, but spatially separated loop nucleotides close to the core of the ribozyme. C62 and A63, which flank the cleavage site on the internal loop between stems P1 and P2, are splayed apart, unlike in the stack found in the three-way junctional version. Instead, the scissile phosphate is anchored in place by a network of hydrogen bonding, including the N1H of G5, the inner sphere water of a hydrated Mg2+ and a non-bridging phosphate oxygen.67Mutation of G5 and A63 can completely inactivate the four-way junction ribozyme. The unusual number of coordinated Mg2+ ions suggests that, in addition to mutations affecting the active site and base-pairing between ribozyme and substrate strands, mutations disturbing the binding of Mg2+ influence catalytic efficiency. A caveat of the current understanding is that the structural information is derived from complexes that may be far from the catalytically active conformation. Understanding the substrate specificity of this ribozyme is less developed compared to the ribozymes discussed earlier.
Neurospora Varkud satellite (VS) ribozyme
The Varkud satellite (VS) RNA is a transcript found in some strains of Neurospora.112 It is the largest nucleolytic ribozyme known (∼150 nucleotides) and functions in replication of the single-stranded RNA satellite. Its secondary structure contains seven helices (1–7) (Table 3) forming three three-way junctions: 2-3-6, 3-4-5, and 1-7-2.109 The cleavage site of the VS ribozyme is in the inner loop of stem 1.113 Therefore, part of stem I acts as the substrate strand in the cis-acting VS ribozyme. The sequence in the junction domain is critically important for the coaxial stacking of stems.113 The catalytic center of the ribozyme is located within stem 6 and 1. A stable kissing loop interaction is formed between the GUC in stem loop 1 and the GAC in stem loop 5, docking the loops together114 to form the active site and bring the cleavage site into the catalytic center. Additionally, the kissing loop interaction leads to an intramolecular secondary structure rearrangement in stem 1, which is important for the activity of the VS ribozyme,114 but requires surmounting a major thermodynamic barrier.115 A756 and G638 are adjacent to the scissile phosphate,116 and are believed to play the role of general acid and general base, respectively.42,101 The crystal structure of the VS ribozyme from Neurospora intermedia indicates that the ribozyme actually exists as an intertwined dimer formed by the exchange of substrate helices,109 creating two active sites in trans around cleavage site nucleotides G620 and A621. The proposed general base G638 forms a cross-strand stack with G620, which contains the 2′-O nucleophile. The imino group of G638 is within hydrogen-bonding distance of the scissile phosphate, suggesting that G638 stabilizes the in-line conformation and facilitates catalysis directly via hydrogen bonding or proton transfer. In addition, Mg2+ in the active site appears to interact critically with the scissile phosphate and also activates G638.117

|
Cleavage site: internal | Upstream: base pairing, ending with a loop |
| General acid: A756 located in loop between stem 6B and 6C | Downstream: base pairing | |
| General base: G638 located in loop between stem 1A & stem 1B | Notes: active sites are assembled by interactions between internal loops found within two separate helices (G638 & A756 loops) | |
| Ligation: yes108 | ||
| Ref. 109 and 110 | ||
|
Cleavage site: internal | Upstream: base pairing |
| General acid: unknown | Downstream: residues involved in Pseudoknot | |
| General base: Unknown | Notes: disruption of base pairing in S4 helix and pseudoknot abolishes cleavage activity. | |
| Ligation: unknown | ||
| Ref. 111 | ||
Formation of the kissing loop and the preservation of the function of A756 and G638 are important considerations for substrate specificity of the cis-acting VS ribozyme. Mutation of A756 to G or G638 to A causes a significant decrease (103–104-fold) in the rate of cleavage.42,118 Introducing low pKa analogs, such as purine or 8-aza-adenosine at A756 interferes with ribozyme activity, but activity can be rescued by adjusting the pH.119 This is consistent with the protonated form at A756 being important, e.g., for donating a proton to the leaving group or stabilizing the transition state.
A trans-acting VS ribozyme containing 144 nucleotides (helices 2–6) can be created by disconnecting the stem-loop of 1 from the rest of the RNA.110 A minimal substrate requires one base upstream of the cleavage site and 19 bases downstream of the cleavage site to form a stable stem-loop hairpin. These interactions are supplemented by tertiary contacts between the ribozyme and substrate, such as the GUC/GAC kissing-loop interaction.120 The ribozyme appears to be amenable to extensive engineering for substrate specificity. For example, activity can be maintained while varying the number of base pairs in stem 1, as long as a compensating number of base pairs are added to stem 5.121 Also, the 1/5 kissing-loop interaction can be replaced by other kissing-loop interactions while maintaining efficient substrate cleavage.122 These features illustrate how substrate specificity depends on the unusual structure of this ribozyme.
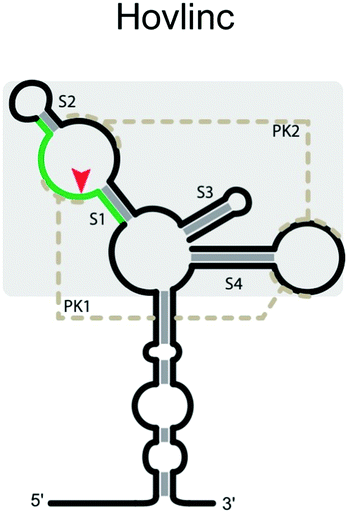
Hovlinc ribozyme
A new type of self-cleaving ribozyme (Hovlinc) has recently emerged through an experimental genome-wide search aimed at uncovering naturally-occurring ribozymes in humans.111,123 While there is limited information about this ribozyme's mechanism of cleavage, RNA structure analysis suggests that the catalytic core consists of two stem-loops with two pseudoknots (Table 3). The ribozyme exhibited a dependence on Mg2+ and pH and showed the highest activity in Mn2+ with the reactivity changing depending on the cation present (Mn2+ > Mg2+ > Ca2+). Given that the ribozyme appears to have emerged relatively recently and that its activity is linearly dependent on pH and divalent metal ions, it is likely that the ribozyme acts primarily through positioning of the scissile phosphate for a near-optimal geometry for scission and through coordination of a hydrated metal ion.124 Thus, its active site may be simpler than active sites of other ribozymes. Additional studies will be required to further understand the structure and the mechanism of cleavage of this ribozyme.Utilization and considerations for using self-cleaving ribozymes for synthetic biology applications
Deeper understanding and exploration of self-cleaving ribozymes brought a newer field of interest to the spotlight. Aptazymes are a class of RNAs that have the unique capability to contain the specificity of an aptamer domain, while also encompassing the catalytic potential of a ribozyme. Aptazymes form an emerging field of study in the RNA world because of their exploitation to control gene expression by utilizing a ligand dependent switch that ultimately leads to inducing cleavage and degradation of their own mRNAs.46,125 However, while much effort has been put into understanding the design aspects of creating aptazymes,126,127 no naturally occurring aptazymes have been discovered (with the exception of potential allosteric regulation of a bacterial HDV-like ribozyme by GlcN6P).18 Some of the ribozymes described above have been extensively studied in order to engineer and design aptazymes for dynamic control of gene expression.127,128 Throughout studies in constructing synthetic aptazymes for that purpose, only four classes of self-cleaving ribozymes (HDV, hammerhead, pistol and twister) have been shown to produce working aptazymes.128–131Researchers have been taking advantage of either the 5′ or 3′ untranslated regions (UTR) within mRNA to control gene expression. In bacteria, this has been shown by either repression due to rho-independent transcriptional termination or by translation initiation by occluding the Shine–Dalgarno sequence.132,133 It has also been suggested that RNAs bearing 5′-OH groups have been able to avoid degradation by RNaseE because they are poorer substrates compared to 5′ phosphorylated RNAs.134,135 This idea has been used in designing aptazymes for the purposes of stabilizing downstream transcripts upon cleavage by ligand-dependent ribozymes.136
While most of the literature for bacterial UTRs has focused on the 5′ region, in eukaryotic systems more information about manipulating the 3′ region is coming to light.8,137 Several eukaryotic examples have suggested downregulation of mRNA expression in the 3′ UTR, one being the theophylline-binding aptamer with a hammerhead ribozyme. In the presence of theophylline, the aptamer changes conformation and activates the hammerhead to self-cleave, preventing expression of β-lactamase, leading to decreased antibiotic resistance138 (Fig. 2B). In another approach, an aptazyme was engineered by replacing the P4–L4 stem-loop of an HDV ribozyme with an aptamer for guanine, which, upon binding, stabilizes the catalytically competent form of the ribozyme, and result in down-regulation of EGFP. The aptazyme was placed in the 3′ UTR of the mRNA, and in response to guanine, causes HDV to cleave its 5′ end from the transcript130 (Fig. 2A). Another group constructed an artificial library containing pistol-based aptazymes and utilized a deep sequencing approach to analyze over 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 aptazyme mutants. The guanine aptamer was fused in tandem vs the conventional placement within a ribozyme, which opens up a new potential aptazyme architecture and consideration for engineering aptazymes137,138 (Fig. 2C). While this method was viable, a new, rapid identification method was developed for high throughput screening for conditionally cleaved ribozymes in cellular systems through barcode amplification. This not only uncovered tet-hammerhead and guanine-HDV on and off switches, but also a tet-twister based aptazyme.139
000 aptazyme mutants. The guanine aptamer was fused in tandem vs the conventional placement within a ribozyme, which opens up a new potential aptazyme architecture and consideration for engineering aptazymes137,138 (Fig. 2C). While this method was viable, a new, rapid identification method was developed for high throughput screening for conditionally cleaved ribozymes in cellular systems through barcode amplification. This not only uncovered tet-hammerhead and guanine-HDV on and off switches, but also a tet-twister based aptazyme.139
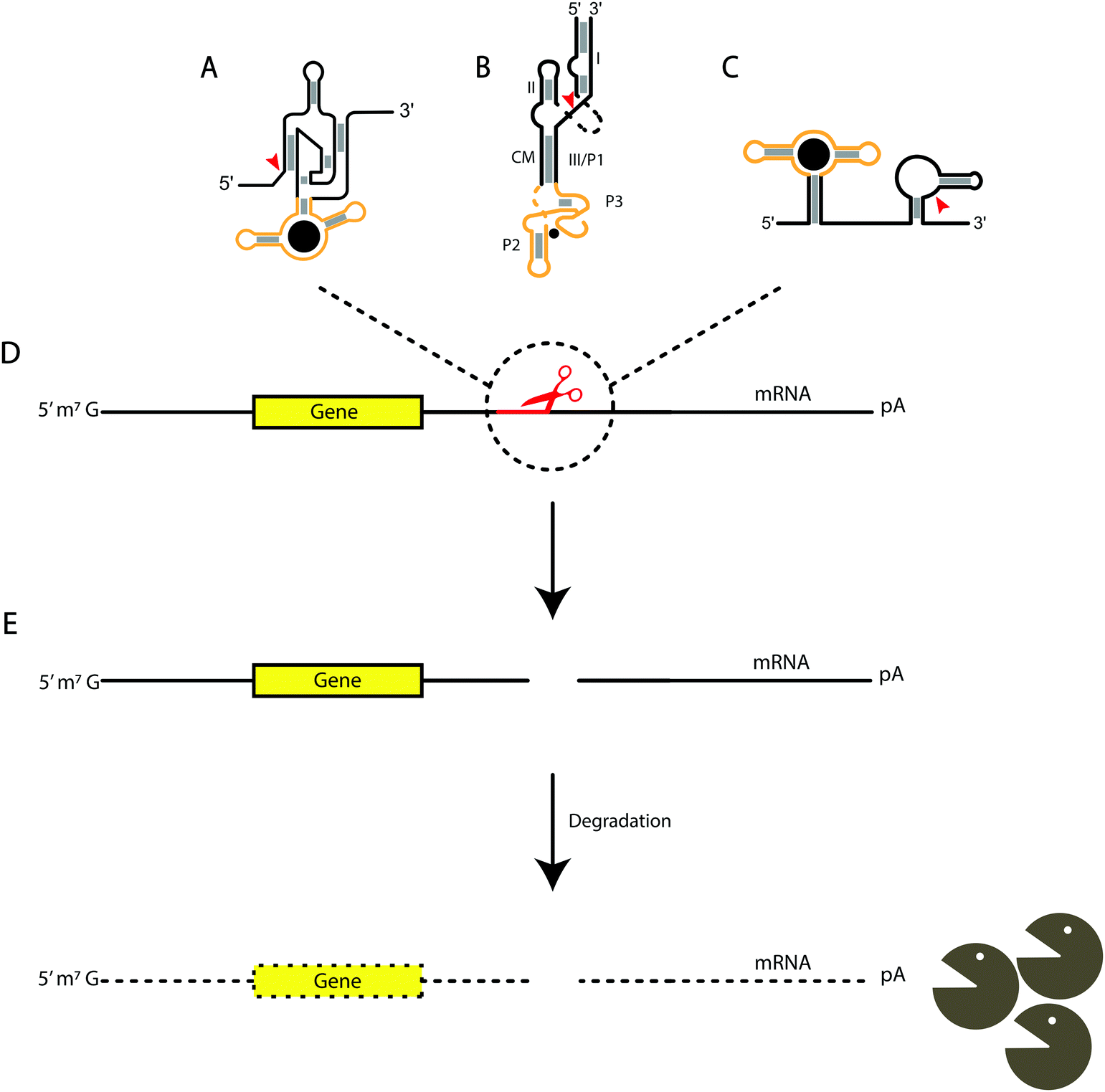 | ||
| Fig. 2 Aptazyme-based cleavage for mRNA degradation. The yellow regions indicate the aptamer domain, while the black areas indicate the associated ribozyme. The gray boxes indicate base-pairing and the red arrows are the sites of cleavage for each ribozyme. (A) HDV-based, (B) hammerhead-based, and (C) pistol-based aptazymes. (D) Aptazymes inserted in the 3′ untranslated regions of mRNA and cleaving after binding their respective ligands. (E) Cleavage of the ribozyme portion of the aptazyme leads to degradation of the mRNA. | ||
Overall, there is much that needs to be understood about the mechanisms and applications for utilizing self-cleaving ribozymes and aptazymes. This includes the requirements for designing aptazymes, which contain not only the aptamer domain and ribozyme selected, but also the communication module between the aptamer and ribozyme domains.140 These modules play an important role, as it has been shown that connector stability can impact the activity of the ribozyme. Multiple groups have mutagenized and in vitro selected a library of potential aptazymes, which were subsequently analyzed for the best combination of the communication module, ribozyme, and aptamer to allow for increased cleavage under their desired conditions.130,141–143 Other concerns include the consideration that too much ribozyme could lead to high levels of background cleavage, or an excessively stable aptamer domain may not give a sufficient ligand-dependent response when looking at a response range for downregulation of gene expression. Utilizing all the discussed properties of these catalytic RNAs can allow for other applications in controlling gene expression.
Conclusion
The discovery of ribozymes is considered as one of the strongest pieces of evidence for the RNA World theory, which states that early life forms relied primarily on RNA for genetic information storage and biochemical function (including catalysis), without major participation in cellular processes by DNA or proteins. The specificity of ribozymes for their substrates is a key factor in the behavior of a collective of RNAs. Highly specific ribozymes may be desirable in some cases (e.g., self-aminoacylating ribozymes that would be precursors to the tRNA/synthetase system), but highly promiscuous ribozymes might be desired in other cases (e.g., an RNA polymerase ‘copying’ ribozyme). In addition, network-level behaviors can emerge from the peculiarities of substrate specificities (e.g., as mediated by internal guide sequences144). The important conceptual role of ribozyme specificity was highlighted by Levy and Ellington,145 who surmised that, absent other mechanisms of compartmentalization, cooperating ribozymes in the RNA World would need to recognize each other through sequence ‘tags’ to avoid parasitization by non-catalytic RNAs. Such target-specific polymerases have been recently demonstrated.146In this review, we have discussed substrate specificity of small nucleolytic ribozymes, which is closely tied to their mechanism and structure. In general, substrate specificity is influenced strongly by base-pairing interactions with the substrate strand, and at least in some cases, can be rationally engineered with knowledge of base-pair stabilities. We have also indicated key structural features that affect substrate recognition, as well as requirements for the activity of the ribozymes. Mapping out the key nucleotides involved in the self-cleaving mechanism and structural requirements that assemble the catalytically competent conformations (kissing loops, ribose zippers, tertiary interactions, etc.) will allow more efficient engineering of new ribozyme variants and facilitate further analysis of trans-cleaving versions of these ribozymes. Substrate nucleotides that affect the catalytic mechanism (e.g., Mg2+ binding, proper folding) add to the sequence-specificity of the ribozyme. Ribozymes in which sequence specificity of the substrate strand has been extensively investigated reveal that the base-pairing between the substrate and ribozyme strands plays an important role. Typically, central residues are conserved while mutations are tolerated at more distal sites, and specificity is maximized when the correct substrate binds strongly, but not strongly enough that mismatches would be tolerated (except when such redundancy is desired).
It is noteworthy that the site of cleavage for most of these ribozymes occurs between two base-paired regions (Table 2). Cleavage then results in products which each have lower affinity for the ribozyme than the substrate. While this is advantageous for multiple-turnover reactions, sequence requirements on both sides of the cleavage site constrain potential substrates and applications. On the other hand, some ribozymes (e.g., HDV, hatchet; Table 1) cleave peripherally of the specified substrate sequence, and the products thus have similar affinity to the ribozyme as the substrates. Although this decreases substrate turnover, the placement of most sequence determinants on one side of the cleavage site is a useful property when designing constructs to synthesize RNAs with a defined 3′ end.
The increasing number of these ribozyme motifs discovered demonstrates that, with respect to the small endonucleolytic ribozymes, the same biochemical goal can be reached with a variety of RNA folds. A given ribozyme can be adapted to different substrates, as long as those contain essential sequence elements required for catalysis. If these requirements are not fulfilled, another ribozyme having different requirements for its substrate sequence may be able to process the substrate in question. For example, HDV ribozymes have been widely used to process guide RNAs in CRISPR-based gene editing applications. However, there are guide sequences that may base-pair with critical segments of the HDV ribozyme, leading to poor self-scission. In such cases one of the many other examples of the HDV-like ribozymes can be used, or the processing can be achieved by the hatchet or the metabolite-dependent GlmS ribozyme, which also cleave peripherally (Table 1), allowing the ribozyme to be wholly independent of the guide RNA sequence and structure. Similarly, an RNA world may have had the potential to process numerous substrates while simultaneously maintaining sequence-specific catalysis. Understanding the determinants of specificity in these ribozymes and others may aid synthetic biology efforts to re-create an RNA World.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the Simons Foundation Collaboration on the Origins of Life [290356FY18], the NIH New Innovator Program [DP2GM123457], and NSF [1935087] to I. A. C. and by NSF-CBET 1804220 and NASA 20-EXO20-0110 to A. L.Notes and references
- A. Pressman, C. Blanco and I. A. Chen, Curr. Biol., 2015, 25, R953–R963 CrossRef CAS PubMed.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed.
- R. A. Jensen, Annu. Rev. Microbiol., 1976, 30, 409–425 CrossRef CAS PubMed.
- E. Janzen, C. Blanco, H. Peng, J. Kenchel and I. A. Chen, Chem. Rev., 2020, 120, 4879–4897 CrossRef CAS PubMed.
- C. E. Weinberg, Z. Weinberg and C. Hammann, Nucleic Acids Res., 2019, 47, 9480–9494 CrossRef CAS PubMed.
- D. Lilley, F1000Research, 2019, 8, 1462 CAS.
- D. D. Seith, J. L. Bingaman, A. J. Veenis, A. C. Button and P. C. Bevilacqua, ACS Catal., 2018, 8, 314–327 CrossRef CAS PubMed.
- A. Ren, R. Micura and D. J. Patel, Curr. Opin. Chem. Biol., 2017, 41, 71–83 CrossRef CAS PubMed.
- C. Reymond, J.-D. Beaudoin and J.-P. Perreault, Cell. Mol. Life Sci., 2009, 66, 3937 CrossRef CAS PubMed.
- N. Riccitelli and A. Lupták, in Progress in Molecular Biology and Translational Science, ed. G. A. Soukup, Academic Press, 2013, vol. 120, pp. 123–171 Search PubMed.
- A. R. Ferré-D'Amaré, Q. Rev. Biophys., 2010, 43, 423–447 CrossRef PubMed.
- L. Zheng, C. Falschlunger, K. Huang, E. Mairhofer, S. Yuan, J. Wang, D. J. Patel, R. Micura and A. Ren, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10783–10791 CrossRef CAS PubMed.
- J. M. Taylor, Intervirology, 1999, 42, 173–178 CrossRef CAS PubMed.
- K. Salehi-Ashtiani, A. Lupták, A. Litovchick and J. W. Szostak, Science, 2006, 313, 1788–1792 CrossRef CAS PubMed.
- C.-H. T. Webb, N. J. Riccitelli, D. J. Ruminski and A. Lupták, Science, 2009, 326, 953 CrossRef CAS PubMed.
- D. J. Ruminski, C.-H. T. Webb, N. J. Riccitelli and A. Lupták, J. Biol. Chem., 2011, 286, 41286–41295 CrossRef CAS PubMed.
- Z. Weinberg, P. B. Kim, T. H. Chen, S. Li, K. A. Harris, C. E. Lünse and R. R. Breaker, Nat. Chem. Biol., 2015, 11, 606–610 CrossRef CAS PubMed.
- L. F. M. Passalacqua, R. M. Jimenez, J. Y. Fong and A. Lupták, Biochemistry, 2017, 56, 6006–6014 CrossRef CAS PubMed.
- A. R. Ferré-D'Amaré, K. Zhou and J. A. Doudna, Nature, 1998, 395, 567–574 CrossRef PubMed.
- A. Ke, K. Zhou, F. Ding, J. H. D. Cate and J. A. Doudna, Nature, 2004, 429, 201–205 CrossRef CAS PubMed.
- J. Chen, A. Ganguly, Z. Miswan, S. Hammes-Schiffer, P. C. Bevilacqua and B. L. Golden, Biochemistry, 2013, 52, 557–567 CrossRef CAS PubMed.
- S. R. Das and J. A. Piccirilli, Nat. Chem. Biol., 2005, 1, 45–52 CrossRef CAS PubMed.
- S. Nakano, D. M. Chadalavada and P. C. Bevilacqua, Science, 2000, 287, 1493–1497 CrossRef CAS PubMed.
- J. Lu, S. C. Koo, B. P. Weissman, M. E. Harris, N.-S. Li and J. A. Piccirilli, Biochemistry, 2018, 57, 3465–3472 CrossRef CAS PubMed.
- Y. Chen, N. V. Eldho, T. K. Dayie and P. R. Carey, Biochemistry, 2010, 49, 3427–3435 CrossRef CAS PubMed.
- A. Lupták, A. R. Ferré-D'Amaré, K. Zhou, K. W. Zilm and J. A. Doudna, J. Am. Chem. Soc., 2001, 123, 8447–8452 CrossRef PubMed.
- B. Gong, J.-H. Chen, E. Chase, D. M. Chadalavada, R. Yajima, B. L. Golden, P. C. Bevilacqua and P. R. Carey, J. Am. Chem. Soc., 2007, 129, 13335–13342 CrossRef CAS PubMed.
- C.-H. T. Webb and A. Lupták, RNA Biol., 2011, 8, 719–727 CrossRef CAS PubMed.
- N. J. Riccitelli, E. Delwart and A. Lupták, Biochemistry, 2014, 53, 1616–1626 CrossRef CAS PubMed.
- A. T. Perrotta and M. D. Been, Nucleic Acids Res., 1993, 21, 3959–3965 CrossRef CAS PubMed.
- A. D. Branch and H. D. Robertson, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10163–10167 CrossRef CAS PubMed.
- A. T. Perrotta and M. D. Been, Biochemistry, 1992, 31, 16–21 CrossRef CAS PubMed.
- M. Puttaraju, A. T. Perrotta and M. D. Been, Nucleic Acids Res., 1993, 21, 4253–4258 CrossRef CAS PubMed.
- M. D. Been, A. T. Perrotta and S. P. Rosenstein, Biochemistry, 1992, 31, 11843–11852 CrossRef CAS PubMed.
- H. N. Wu, Y. J. Wang, C. F. Hung, H. J. Lee and M. M. C. Lai, J. Mol. Biol., 1992, 223, 233–245 CrossRef CAS PubMed.
- P. K. Kumar, Y. A. Suh, K. Taira and S. Nishikawa, FASEB J., 1993, 7, 124–129 CrossRef CAS PubMed.
- A. T. Perrotta and M. D. Been, Nature, 1991, 350, 434–436 CrossRef CAS PubMed.
- S. Ananvoranich and J. P. Perreault, J. Biol. Chem., 1998, 273, 13182–13188 CrossRef CAS PubMed.
- G. Roy, S. Ananvoranich and J. P. Perreault, Nucleic Acids Res., 1999, 27, 942–948 CrossRef CAS.
- P. Deschenes, D. A. Lafontaine, S. Charland and J. P. Perreault, Antisense Nucleic Acid Drug Dev., 2000, 10, 53–61 CrossRef CAS PubMed.
- P. Deschênes, J. Ouellet, J. Perreault and J. P. Perreault, Nucleic Acids Res., 2003, 31, 2087–2096 CrossRef PubMed.
- T. J. Wilson, A. C. McLeod and D. M. J. Lilley, EMBO J., 2007, 26, 2489–2500 CrossRef CAS PubMed.
- D. J. Ruminski, C.-H. T. Webb, N. J. Riccitelli and A. Lupták, J. Biol. Chem., 2011, 286, 41286–41295 CrossRef CAS PubMed.
- C.-H. T. Webb and A. Lupták, Biochemistry, 2018, 57, 1440–1450 CrossRef CAS PubMed.
- C.-H. T. Webb, D. Nguyen, M. Myszka and A. Lupták, Sci. Rep., 2016, 6, 28179 CrossRef CAS PubMed.
- W. C. Winkler, A. Nahvi, A. Roth, J. A. Collins and R. R. Breaker, Nature, 2004, 428, 281–286 CrossRef CAS PubMed.
- A. Savinov and S. M. Block, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11976–11981 CrossRef CAS PubMed.
- A. Roth, A. Nahvi, M. Lee, I. Jona and R. R. Breaker, RNA, 2006, 12, 607–619 CrossRef CAS PubMed.
- C. M. Hull, A. Anmangandla and P. C. Bevilacqua, ACS Chem. Biol., 2016, 11, 1118–1127 CrossRef CAS PubMed.
- D. J. Klein and A. R. Ferré-D'Amaré, Science, 2006, 313, 1752–1756 CrossRef CAS PubMed.
- J. L. Bingaman, S. Zhang, D. R. Stevens, N. H. Yennawar, S. Hammes-Schiffer and P. C. Bevilacqua, Nat. Chem. Biol., 2017, 13, 439–445 CrossRef CAS PubMed.
- J. L. Bingaman, I. Y. Gonzalez, B. Wang and P. C. Bevilacqua, Biochemistry, 2017, 56, 4313–4317 CrossRef CAS PubMed.
- J. C. Cochrane, S. V. Lipchock and S. A. Strobel, Chem. Biol., 2007, 14, 97–105 CrossRef CAS PubMed.
- J. H. Davis, B. F. Dunican and S. A. Strobel, Biochemistry, 2011, 50, 7236–7242 CrossRef CAS PubMed.
- T. J. McCarthy, M. A. Plog, S. A. Floy, J. A. Jansen, J. K. Soukup and G. A. Soukup, Chem. Biol., 2005, 12, 1221–1226 CrossRef CAS.
- B. Gong, D. J. Klein, A. R. Ferré-D’Amaré and P. R. Carey, J. Am. Chem. Soc., 2011, 133, 14188–14191 CrossRef CAS PubMed.
- R. A. Tinsley, J. R. W. Furchak and N. G. Walter, RNA, 2007, 13, 468–477 CrossRef CAS PubMed.
- S. Li, C. E. Lünse, K. A. Harris and R. R. Breaker, RNA, 2015, 21, 1845–1851 CrossRef CAS PubMed.
- L. Zheng, C. Falschlunger, K. Huang, E. Mairhofer, S. Yuan, J. Wang, D. J. Patel, R. Micura and A. Ren, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10783–10791 CrossRef CAS PubMed.
- C. Gasser, J. Gebetsberger, M. Gebetsberger and R. Micura, Nucleic Acids Res., 2018, 46, 6983–6995 CrossRef CAS PubMed.
- R. Przybilski and C. Hammann, ChemBioChem, 2006, 7, 1641–1644 CrossRef CAS PubMed.
- P. B. Rupert and A. R. Ferré-D'Amaré, Nature, 2001, 410, 780–786 CrossRef CAS PubMed.
- Y. Lihanova and C. E. Weinberg, RNA Biol., 2021, 1–9, DOI:10.1080/15476286.2021.1874706.
- T. J. Wilson, Y. Liu, N.-S. Li, Q. Dai, J. A. Piccirilli and D. M. J. Lilley, J. Am. Chem. Soc., 2019, 141, 7865–7875 CrossRef CAS PubMed.
- C. S. Gaines and D. M. York, J. Am. Chem. Soc., 2016, 138, 3058–3065 CrossRef CAS PubMed.
- D. Eiler, J. Wang and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13028–13033 CrossRef CAS PubMed.
- L. Zheng, E. Mairhofer, M. Teplova, Y. Zhang, J. Ma, D. J. Patel, R. Micura and A. Ren, Nat. Commun., 2017, 8, 1180 CrossRef PubMed.
- G. A. Prody, J. T. Bakos, J. M. Buzayan, I. R. Schneider and G. Bruening, Science, 1986, 231, 1577–1580 CrossRef CAS PubMed.
- A. Mir, J. Chen, K. Robinson, E. Lendy, J. Goodman, D. Neau and B. L. Golden, Biochemistry, 2015, 54, 6369–6381 CrossRef CAS PubMed.
- M. Anderson, E. P. Schultz, M. Martick and W. G. Scott, J. Mol. Biol., 2013, 425, 3790–3798 CrossRef CAS PubMed.
- Y.-I. Chi, M. Martick, M. Lares, R. Kim, W. G. Scott and S.-H. Kim, PLoS Biol., 2008, 6, e234 CrossRef PubMed.
- M. Martick and W. G. Scott, Cell, 2006, 126, 309–320 CrossRef CAS PubMed.
- A. Mir and B. L. Golden, Biochemistry, 2016, 55, 633–636 CrossRef CAS PubMed.
- J. Han and J. M. Burke, Biochemistry, 2005, 44, 7864–7870 CrossRef CAS PubMed.
- D. E. Ruffner, G. D. Stormo and O. C. Uhlenbeck, Biochemistry, 1990, 29, 10695–10702 CrossRef CAS PubMed.
- D. M. Williams, W. A. Pieken and F. Eckstein, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 918–921 CrossRef CAS PubMed.
- D. J. Fu and L. W. McLaughlin, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3985–3989 CrossRef CAS PubMed.
- K. R. Birikh, P. A. Heaton and F. Eckstein, Eur. J. Biochem., 1997, 245, 1–16 CrossRef CAS PubMed.
- A. R. Kore, N. K. Vaish, U. Kutzke and F. Eckstein, Nucleic Acids Res., 1998, 26, 4116–4120 CrossRef CAS PubMed.
- J. Ludwig, M. Blaschke and B. S. Sproat, Nucleic Acids Res., 1998, 26, 2279–2285 CrossRef CAS PubMed.
- S. T. Crooke and B. Lebleu, Antisense research and applications, CRC Press, Boca Raton, 1993 Search PubMed.
- D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6921–6925 CrossRef CAS PubMed.
- M. Werner and O. C. Uhlenbeck, Nucleic Acids Res., 1995, 23, 2092–2096 CrossRef CAS PubMed.
- L. Rubino, M. E. Tousignant, G. Steger and J. M. Kaper, J. Gen. Virol., 1990, 71, 1897–1903 CrossRef CAS PubMed.
- J. M. Kaper, M. E. Tousignant and G. Steger, Biochem. Biophys. Res. Commun., 1988, 154, 318–325 CrossRef CAS PubMed.
- M. L. Hamm, J. A. Piccirilli, A. Karpeisky, L. Beigelman, B. S. Ross, M. Manoharan and M. J. Gait, Biochemistry, 2000, 39, 6410–6421 CrossRef PubMed.
- P. B. Rupert, A. P. Massey, S. T. Sigurdsson and A. R. Ferré-D'Amaré, Science, 2002, 298, 1421–1424 CrossRef CAS PubMed.
- S. Kath-Schorr, T. J. Wilson, N.-S. Li, J. Lu, J. A. Piccirilli and D. M. J. Lilley, J. Am. Chem. Soc., 2012, 134, 16717–16724 CrossRef CAS PubMed.
- J. A. Liberman, M. Guo, J. L. Jenkins, J. Krucinska, Y. Chen, P. R. Carey and J. E. Wedekind, J. Am. Chem. Soc., 2012, 134, 16933–16936 CrossRef CAS PubMed.
- B. P. Paudel and D. Rueda, J. Am. Chem. Soc., 2014, 136, 16700–16703 CrossRef CAS PubMed.
- A. Berzal-Herranz, S. Joseph and J. M. Burke, Genes Dev., 1992, 6, 129–134 CrossRef CAS PubMed.
- A. Berzal-Herranz, S. Joseph, B. M. Chowrira, S. E. Butcher and J. M. Burke, EMBO J., 1993, 12, 2567–2573 CrossRef CAS PubMed.
- S. Joseph, A. Berzal-Herranz, B. M. Chowrira, S. E. Butcher and J. M. Burke, Genes Dev., 1993, 7, 130–138 CrossRef CAS PubMed.
- M. Pérez-Ruiz, A. Barroso-delJesus and A. Berzal-Herranz, J. Biol. Chem., 1999, 274, 29376–29380 CrossRef PubMed.
- I. Drude, A. Strahl, D. Galla, O. Müller and S. Müller, FEBS J., 2011, 278, 622–633 CrossRef CAS PubMed.
- D. Balke, I. Zieten, A. Strahl, O. Müller and S. Müller, ChemMedChem, 2014, 9, 2128–2137 CrossRef CAS PubMed.
- R. Pinard, D. Lambert, N. G. Walter, J. E. Heckman, F. Major and J. M. Burke, Biochemistry, 1999, 38, 16035–16039 CrossRef CAS PubMed.
- A. Ren, N. Vušurović, J. Gebetsberger, P. Gao, M. Juen, C. Kreutz, R. Micura and D. J. Patel, Nat. Chem. Biol., 2016, 12, 702–708 CrossRef CAS PubMed.
- L. A. Nguyen, J. Wang and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1021–1026 CrossRef CAS PubMed.
- M. Teplova, C. Falschlunger, O. Krasheninina, M. Egger, A. Ren, D. J. Patel and R. Micura, Angew. Chem., Int. Ed., 2020, 59, 2837–2843 CrossRef CAS PubMed.
- T. J. Wilson, N.-S. Li, J. Lu, J. K. Frederiksen, J. A. Piccirilli and D. M. J. Lilley, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11751–11756 CrossRef CAS PubMed.
- A. Roth, Z. Weinberg, A. G. Y. Chen, P. B. Kim, T. D. Ames and R. R. Breaker, Nat. Chem. Biol., 2014, 10, 56–60 CrossRef CAS PubMed.
- A. Ren, M. Košutić, K. R. Rajashankar, M. Frener, T. Santner, E. Westhof, R. Micura and D. J. Patel, Nat. Commun., 2014, 5, 5534 CrossRef CAS PubMed.
- Z. Weinberg, J. X. Wang, J. Bogue, J. Yang, K. Corbino, R. H. Moy and R. R. Breaker, Genome Biol., 2010, 11, R31 CrossRef PubMed.
- Y. Liu, T. J. Wilson, S. A. McPhee and D. M. J. Lilley, Nat. Chem. Biol., 2014, 10, 739–744 CrossRef CAS PubMed.
- M. Košutić, S. Neuner, A. Ren, S. Flür, C. Wunderlich, E. Mairhofer, N. Vušurović, J. Seikowski, K. Breuker, C. Höbartner, D. J. Patel, C. Kreutz and R. Micura, Angew. Chem., Int. Ed., 2015, 54, 15128–15133 CrossRef PubMed.
- N. Vušurović, R. B. Altman, D. S. Terry, R. Micura and S. C. Blanchard, J. Am. Chem. Soc., 2017, 139, 8186–8193 CrossRef PubMed.
- F. D. Jones, S. P. Ryder and S. A. Strobel, Nucleic Acids Res., 2001, 29, 5115–5120 CrossRef CAS PubMed.
- N. B. Suslov, S. DasGupta, H. Huang, J. R. Fuller, D. M. J. Lilley, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2015, 11, 840–846 CrossRef CAS PubMed.
- H. C. Guo and R. A. Collins, EMBO J., 1995, 14, 368–376 CrossRef CAS PubMed.
- Y. Chen, F. Qi, F. Gao, H. Cao, D. Xu, K. Salehi-Ashtiani and P. Kapranov, Nat. Chem. Biol., 2021, 17, 601–607 CrossRef CAS PubMed.
- R. A. Collins, Biochem. Soc. Trans., 2002, 30, 1122–1126 CrossRef CAS PubMed.
- D. A. Lafontaine, D. G. Norman and D. M. J. Lilley, EMBO J., 2001, 20, 1415–1424 CrossRef CAS PubMed.
- A. A. Andersen and R. A. Collins, Mol. Cell, 2000, 5, 469–478 CrossRef CAS PubMed.
- P. Bouchard and P. Legault, RNA, 2014, 20, 1451–1464 CrossRef CAS PubMed.
- J. Lipfert, J. Ouellet, D. G. Norman, S. Doniach and D. M. J. Lilley, Structure, 2008, 16, 1357–1367 CrossRef CAS PubMed.
- A. Ganguly, B. P. Weissman, T. J. Giese, N.-S. Li, S. Hoshika, S. Rao, S. A. Benner, J. A. Piccirilli and D. M. York, Nat. Chem., 2020, 12, 193–201 CrossRef CAS PubMed.
- D. A. Lafontaine, D. G. Norman and D. M. J. Lilley, EMBO J., 2002, 21, 2461–2471 CrossRef CAS PubMed.
- F. D. Jones and S. A. Strobel, Biochemistry, 2003, 42, 4265–4276 CrossRef CAS PubMed.
- T. Rastogi, T. L. Beattie, J. E. Olive and R. A. Collins, EMBO J., 1996, 15, 2820–2825 CrossRef CAS PubMed.
- J. Lacroix-Labonté, N. Girard, S. Lemieux and P. Legault, Nucleic Acids Res., 2011, 40, 2284–2293 CrossRef PubMed.
- J. Lacroix-Labonté, N. Girard, P. Dagenais and P. Legault, Nucleic Acids Res., 2016, 44, 6924–6934 CrossRef PubMed.
- C. C. Chen and A. Lupták, Nat. Chem. Biol., 2021, 17, 507–508 CrossRef CAS PubMed.
- Y. Li and R. R. Breaker, J. Am. Chem. Soc., 1999, 121, 5364–5372 CrossRef CAS.
- A. Ogawa and M. Maeda, ChemBioChem, 2008, 9, 206–209 CrossRef CAS PubMed.
- J. Tang and R. R. Breaker, Chem. Biol., 1997, 4, 453–459 CrossRef CAS PubMed.
- G. Zhong, H. Wang, C. C. Bailey, G. Gao and M. Farzan, eLife, 2016, 5, e18858 CrossRef PubMed.
- Y. Yokobayashi, Curr. Opin. Chem. Biol., 2019, 52, 72–78 CrossRef CAS PubMed.
- S. Kobori, K. Takahashi and Y. Yokobayashi, ACS Synth. Biol., 2017, 6, 1283–1288 CrossRef CAS PubMed.
- Y. Nomura, L. Zhou, A. Miu and Y. Yokobayashi, ACS Synth. Biol., 2013, 2, 684–689 CrossRef CAS PubMed.
- H. Cheng, Y. Zhang, H. Wang, N. Sun, M. Liu, H. Chen and R. Pei, Mol. BioSyst., 2016, 12, 3370–3376 RSC.
- C. Hammann and E. Westhof, Genome Biol., 2007, 8, 210 CrossRef PubMed.
- R. R. Breaker, Mol. Cell, 2011, 43, 867–879 CrossRef CAS PubMed.
- A. Deana, H. Celesnik and J. G. Belasco, Nature, 2008, 451, 355–358 CrossRef CAS PubMed.
- H. Celesnik, A. Deana and J. G. Belasco, Mol. Cell, 2007, 27, 79–90 CrossRef CAS PubMed.
- J. M. Carothers, J. A. Goler, D. Juminaga and J. D. Keasling, Science, 2011, 36498, 1716–1719 CrossRef PubMed.
- M. Felletti, A. Bieber and J. S. Hartig, RNA Biol., 2017, 14, 1522–1533 CrossRef PubMed.
- X. Feng, L. Liu, X. Duan and S. Wang, Chem. Commun., 2011, 47, 173–175 RSC.
- B. Strobel, M. Spöring, H. Klein, D. Blazevic, W. Rust, S. Sayols, J. S. Hartig and S. Kreuz, Nat. Commun., 2020, 11, 714 CrossRef CAS PubMed.
- X. Chen and A. Ellington, PLoS Comput. Biol., 2009, 5, e1000620 CrossRef PubMed.
- M. Wieland and J. S. Hartig, Angew. Chem., Int. Ed., 2008, 47, 2604–2607 CrossRef CAS PubMed.
- A. Wittmann and B. Suess, Mol. BioSyst., 2011, 7, 2419–2427 RSC.
- B. Klauser, C. Rehm, D. Summerer and J. S. Hartig, Methods Enzymol., 2015, 550, 301–320 CAS.
- N. Vaidya, M. L. Manapat, I. A. Chen, R. Xulvi-Brunet, E. J. Hayden and N. Lehman, Nature, 2012, 491, 72–77 CrossRef CAS PubMed.
- M. Levy and A. D. Ellington, Nat. Struct. Biol., 2001, 8, 580–582 CrossRef CAS PubMed.
- R. Cojocaru and P. J. Unrau, Science, 2021, 371, 1225–1232 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |