
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA perspective on oxide-supported single-atom catalysts
Junyi
Zhou
 ,
Zhen
Xu
,
Meijia
Xu
,
Xiong
Zhou
* and
Kai
Wu
*
,
Zhen
Xu
,
Meijia
Xu
,
Xiong
Zhou
* and
Kai
Wu
*
BNLMS, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: kaiwu@pku.edu.cn; xiongzhou@pku.edu.cn
First published on 14th July 2020
Abstract
Single-atom catalysts (SACs) can not only maximize the metal atom utilization efficiency, but also show drastically improved catalytic performance for various important catalytic processes. Insights into the working principles of SACs provide rational guidance to design and prepare advanced catalysts. Many factors have been claimed to affect the performance of SACs, which makes it very challenging to clarify the correlation between the catalytic performance and physicochemical characteristics of SACs. Oxide-supported SACs are one of the most extensively explored systems. In this minireview, some latest developments on the determining factors of the stability, activity and selectivity of SACs on oxide supports are overviewed. Discussed also are the reaction mechanisms for different systems and methods that are employed to correlate the properties with the catalyst structures at the atomic level. In particular, a recently proposed surface free energy approach is introduced to fabricate well-defined modelled SACs that may help address some key issues in the development of SACs in the future.
1 Introduction
In recent years, single-atom catalysts (SACs) or alternatively single site catalysts (SSCs) with isolated metal atoms dispersed on solid supports have drawn tremendous attention and accordingly very rapid progress has been achieved.1–6 People have been fascinated by the unique properties that SACs have exhibited in specific reactions, including minimum loading of precious metal atoms to reach maximum catalytic efficiency. The concept of the ‘SAC’ was initially proposed by Zhang and co-workers in their pioneering work in 2011.7 They successfully synthesized a SAC that consists of isolated Pt atoms anchored to the surface of iron oxide. Ever since, a dramatically increased number of SACs with a promising catalytic performance have been designed and fabricated, together with careful structural characterization for the dispersion of the metallic species. However, it's also well known that aimlessly and endlessly downsizing the metal particles does not necessarily guarantee a better catalytic performance.8–15 The so-called “size effect” is too simplified to explain various phenomena that have been experimentally observed in different catalytic reactions by all sorts of SACs. In addition, not only does the single atom matter in reactions, but the environment including the oxide support and the metal–support interaction also plays a critical role in the catalytic performance. Historically, the “ensemble effect” has long been proposed and applied in designing novel heterogeneous catalysts because many supported metal catalysts require specific reaction sites that are formed by either metal atoms or metal–support contacts. Thus, it is meaningful to find concrete intrinsic principles to elucidate this ensemble effect. Despite the rapid progress and valuable insights, the intrinsic principles of SACs still remain elusive, which in return spurs further explorations in SACs.In terms of the configuration of the single atoms supported on oxides, there exist some general considerations without subtle inspections of specific SAC cases and their structural details.16–24 In general, the surface free energy of a bulk metal increases significantly as its size decreases. To obtain thermally stable single metal atoms on oxides, one approach is to strengthen the metal–oxide interaction so that the metal atoms could highly disperse rather than aggregate into clusters. Meanwhile, ultrathin oxide films may serve as a new group of structural support materials whose surface free energy and adhesion to the added metals can be fine-tuned by the film thickness, providing another approach to stabilize the single atoms. Therefore, adoption of either a strong metal interaction or an ultrathin oxide film becomes the pre-requisite for the development of these SACs.
Furthermore, the oxide support may act as an oxygen reservoir that stores and releases oxygen, and takes part in redox reactions via a Mars–van Krevelen (MvK) mechanism.25 The absence of neighbouring metal atoms in SACs obviously generates a maximum metal–oxide contact, leading to a fascinating metal–support interface effect that promotes redox reactions. The charge state of a metal particle essentially remains close to neutrality because the transferred electrons at the interface are shared by abundant atoms in the particle, while the partially transferred charge is localized at the single atoms, resulting in a crucial charge effect on the performance of SACs. Low coordination is often regarded as a common characteristic of SACs, though it is not necessary that single atoms may be highly coordinated to the oxide support, and attributed to be responsible for their high activity. Moreover, owing to their limited number of available metal sites, SACs are conceived to have limitations for complex reactions, which can be alternatively applied to improve the selectivity of SACs under certain circumstances.
Quite a few reviews on SACs have been published in the past decades and provided excellent overviews of this rapidly growing research field.26–43 These reviews have already summarized synthetic strategies, structural characterization, and/or catalytic evaluations for specific reactions. In this minireview, we instead try to provide a fundamental understanding of the intrinsic principles of SACs in their catalytic performance with some very recent typical case studies in the past years. Given that stability, activity and selectivity are three main assessment indexes for catalytic performance, the following section will be framed in this sequence, aiming at illuminating the connotations that make SACs unique and different from supported metal nanoparticles. In addition, we also provide our perspective on the surface free energy approach recently developed by our group and our understanding of reaction mechanisms on oxide-supported SACs.
2 Stability improvement
One of the key issues in the development of SACs is how to stabilize the single metal atoms under practical reaction conditions. In common situations, metal atoms on oxides are too facile to diffuse and aggregate into nanoparticles to lower their surface free energy or chemical potential, which spontaneously triggers the Ostwald ripening process at elevated temperatures. Many efforts have been devoted to retaining the atomic dispersion state of the unstable metal atoms. Herein, we summarize some main aspects to describe the development of the stability of SACs.2.1 Metal–oxide interaction
Conventionally, SACs are routinely prepared by wet chemistry which includes impregnation, co-precipitation and deposition–precipitation. In this way, metal atoms may be stabilized by the strong metal–oxide interaction via either their incorporation into solid lattices or replacement of metal cations in oxides. Physical deposition is another powerful methodology to prepare highly dispersed SACs under high-vacuum conditions for fundamental research. Regardless of preparation methods, the anchoring position of single atoms in/on oxides can be divided into three types: type a, location on the oxide surfaces; type b, replacement of the surface metal cations; and type c, incorporation into the oxide lattices.Interestingly, Jakub et al. have reported a study of the Ir1/Fe3O4(001) (where Ir1 stands for a single Ir atom) model catalyst that contains all three types of SACs mentioned above and provided a direct estimation of their stability.44 As shown in Fig. 1, Ir single atoms are obtained by deposition onto the Fe3O4(001) surface at 300 K (Fig. 1A). The Ir single atoms are experimentally observed to be located in between two neighbouring lattice oxygens with a 2-fold coordination. The adsorption energy per Ir atom is 5.25 eV according to DFT calculations. These Ir single atoms are unstable and can be transformed into a 5-fold coordination geometry upon thermal treatment at 623 K (Fig. 1B). They displace the surface iron cations with an increase in their adsorption energy up to 6.22 eV. When the model catalyst is further warmed up to 723 K (Fig. 1C), the Ir atoms are incorporated into the subsurface layer to become 6-fold coordinated. The adsorption energy further increases to 7.14 eV, and the Ir single atoms eventually aggregate into metallic Ir clusters as the sample temperature is elevated to 900 K.
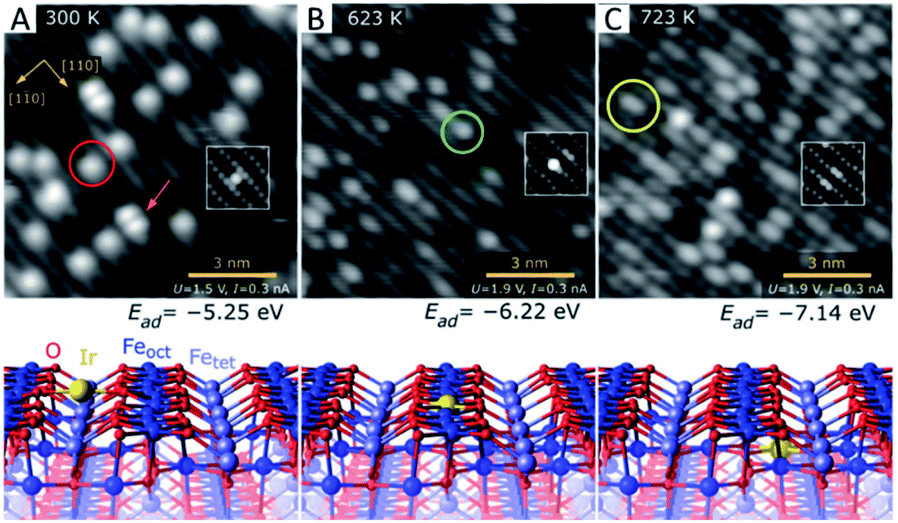 | ||
| Fig. 1 Local structure of Ir1/Fe3O4(001) determined by room-temperature STM (up) and the corresponding DFT model (down). (A) 2-fold coordinated Ir1 atom on Fe3O4(001) prepared at 300 K. (B) 5-fold coordinated Ir atom incorporated within the Fe3O4(001) surface after thermal annealing at 623 K. (C) 6-fold coordinated Ir adatom incorporated into the subsurface layer of Fe3O4(001) after thermal annealing at 723 K. Reproduced with permission.44 Copyright 2019, Wiley-VCH. | ||
Obviously, highly coordinated single atoms incorporated into the subsurface (type c) have the highest stability. In such a situation, the catalytic performance is, however, largely affected because the active metal atoms are no longer in direct contact with the reactants. It is therefore necessary to retain the atomic dispersion state of the active and unstable metal atoms in order to balance their high catalytic activity. Great efforts have been made to create plenty of real cases where the balance can be indeed achieved and the existence of the metal–oxide interaction is confirmed as well.
Meier et al.45 have proven that the Fe3O4(001) surface can stabilize ordered arrays of metal adatoms (Ag and Cu) with a high thermal stability up to 550 K. The metal adatoms do occupy the bulk-continuation cation sites above the surface and become two-fold coordinated to the surface oxygen atoms (type a). Zhang et al.46 have reported a highly stable and atomically dispersed Pt catalyst supported on mesoporous Al2O3 that survives CO oxidation between 100 °C and 400 °C for one month. The Pt atoms are firmly anchored to the inner surface of the support, occupying the unsaturated pentahedral Al3+ centres and coordinating with four oxygen anions (type b). Kim et al.87 have successfully synthesized Pt SACs on antimony-doped tin oxide (Pt1/ATO) by incipient wet impregnation and demonstrated that the single Pt atoms survive repeated cyclic voltammetry for 1800 cycles where the Pt1/ATO serves as the anode catalyst. The stability stems from the replacement of the Sb atoms with Pt in the ATO support (type b).
2.2 Protecting functional groups or gaseous adsorbents
Except the oxide itself, other extrinsic functional groups or gaseous adsorbates have been introduced to stabilize SACs. Thang et al.47 have explored the nature of the stability of single Pt atoms on anatase TiO2. The Pt atoms bind to two excess oxygen atoms on the surface (PtO2), which arise from the formation of surface hydroxyl groups and provide a solid anchor to the oxide support. They also suggest that the stability of atomically dispersed Ru atoms on ZrO2 originates from the interaction of the Ru atoms with one OH group of the surface with elimination of H2 by condensation, leaving the RuO species strongly bound to the zirconia surface.48 Millet et al.49 have prepared Ni SACs on MgO via co-precipitation. The catalysts stay stable between 200 °C and 300 °C over 100 h on stream without sintering. The stability relates to the formation of hydrogenated carbonate species on the surface during the reactions.Interestingly, Duan et al.50 have synthesized a series of high Pt1 density Pt1/Fe2O3 SACs (Fig. 2A) by a facile wet chemical method and investigated their stability in different gas environments. In oxidative gases like O2, the Pt single atoms are stable at high temperatures (Fig. 2B). In the presence of reductive gases, either H2 (Fig. 2C) or CO (Fig. 2D), however, facilitates the movement of the Pt atoms. It is observed that the strong interaction between CO and Pt weakens the binding of Pt to the support, or the dissociated H2 molecules dislodge the surface oxygen-anchored Pt atoms, resulting in the formation of Pt clusters. Other reports have also indicated that reductive gases or functional groups can induce the movement of single metal atoms such as Pt51,52 and Pd53,54 in a CO atmosphere, leading to the destabilization of the single metal atoms. Similar to the destabilization effect of CO, adsorption of H2 or H2O can also induce sintering.50,55 The H2 or H2O molecules are supposed to dissociatively adsorb on the metal atoms and then spill over to the metal–support interface to create –OH species. The produced –OH species further diffuse onto the oxide surface to consume lattice oxygen, resulting in the bond (M1–On–) weakening.
 | ||
| Fig. 2 (A) HAADF-STEM images of the as-prepared 1.66 wt% Pt1/Fe2O3 SAC and the samples treated in (B) O2, (C) H2, and (D) CO at 250 °C for 2 h. Reproduced with permission.50 Copyright 2018, IOP Publishing Ltd. | ||
However, the destabilization effect of reductive gases does not work universally for all metal–oxide SAC systems. Jakub et al.44 have reported that, instead of accelerated sintering, CO adsorption can stabilize Ir adatoms to prevent their incorporation into the substrate. The formed IrCO species remains on the site until 610 K at which CO desorption takes place. In contrast, bare Ir single atoms incorporate into the substrate even below 100 K. CO adsorption makes the Ir atoms bond more strongly to the surface and the Ir–O bond length becomes shortened.
2.3 Surface free energy
Another approach to improve the stability of SACs is to mediate the surface free energy of the oxide support by the preparation of an ultrathin oxide film on a bulk metal substrate. In this method, the ultrathin oxide film is first grown on a metal single crystal surface. The electronic effect from the oxide–metal interface leads to a drastic surface property change of the grown thin oxide film, especially an increase in the surface free energy of the oxide film. Then the surface free energy or chemical potential of the supported single metal atoms becomes comparable to or even lower than that of the supported thin oxide film. As such, the single metal atoms are prevented from aggregating into nanoparticles. Distinguished from the strategy of the metal–oxide interaction, the SAC stability via the surface free energy approach is together regulated by the underlying oxide film and metal support. Zhou et al.56 have successfully applied this methodology to prepare supported single-atom model catalysts, dispersed Au supported on an ultrathin CuO film grown on Cu(110) (Fig. 3A). The Au single atoms are stable upon warming up to 400 K, while thermal annealing at 500 K leads to a distinct decrease in the number of Au single atoms, leaving enlarged Au islands rather than particles at the surface. The Au atom can be precisely determined to be sitting at the mid-point of two neighbouring O anions along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction, and positioning inside the rectangle formed by four topmost neighbouring Cu cations. Chen et al.57 have also obtained K single atoms with this method. They have employed a Pt(111) single crystal instead as the substrate to grow monolayered ceria islands, and then deposited K atoms on the ceria/Pt(111) sample (Fig. 3B). These K single atoms are stable above room temperature. Therrien et al.58,59 have used a Cu2O monolayer grown on the Cu(111) support to prepare Pt single atoms that are afterwards applied to catalyse CO oxidation and water activation. It can be seen that such a free energy approach to preparing single atoms is simple, straightforward and highly reproducible. In addition, the oxide film remains intact and the local ensemble environment of the metal atoms and their surrounding cations in the oxide is well defined, which is suitable to be characterized by scanning tunnelling microscopy at the atomic level.
0] direction, and positioning inside the rectangle formed by four topmost neighbouring Cu cations. Chen et al.57 have also obtained K single atoms with this method. They have employed a Pt(111) single crystal instead as the substrate to grow monolayered ceria islands, and then deposited K atoms on the ceria/Pt(111) sample (Fig. 3B). These K single atoms are stable above room temperature. Therrien et al.58,59 have used a Cu2O monolayer grown on the Cu(111) support to prepare Pt single atoms that are afterwards applied to catalyse CO oxidation and water activation. It can be seen that such a free energy approach to preparing single atoms is simple, straightforward and highly reproducible. In addition, the oxide film remains intact and the local ensemble environment of the metal atoms and their surrounding cations in the oxide is well defined, which is suitable to be characterized by scanning tunnelling microscopy at the atomic level.
 | ||
| Fig. 3 (A) Au single atoms stabilized by monolayered CuO on a Cu(110) single crystal. Reproduced with permission.56 Copyright 2018, American Chemical Society. (B) K single atoms stabilized by monolayered ceria on a Pt(111) single crystal. Reproduced with permission.57 Copyright 2019, American Institute of Physics. | ||
3 Activity tunability
Numerous pieces of evidence have indicated that SACs normally show quite different catalytic performance compared to their nanoparticle counterparts. Such a difference has a tremendous impact on the activities of these entities, either a remarkable enhancement46,60–63 or complete annihilation12,64–66 for a specific catalytic process. Low coordination is also often regarded as a basic element for the high activity of SACs, although it is too simple to explain the various performances in reactions. As described in the Introduction section, charge and interface effects become more obvious for SACs than for their particle counterparts. Besides, it needs to be noted that not all single atoms perform well in given reactions. We hereby also present some cases in which nanoparticle catalysts catalytically outperform SACs.3.1 Charge effect
Strong binding of a single metal atom to an oxide support usually leads to a charge transfer due to different electron affinities. The charge effect has been confirmed to be critical for catalytic performance in many cases, while the spectacular coordination environments of SACs on oxide supports can even generate a more sensitive charge effect than the metal particle catalysts do.Recently Zhou et al.56 have reported the vital role of the charge effect of Au adatoms in tuning their catalytic activity for CO oxidation. Au single atoms supported by a CuO monolayer are successfully prepared on a single crystalline Cu(110) substrate. The strong Au–O interaction gives rise to negatively charged Au atoms, as evidenced by X-ray photoemission spectroscopy. Once the Au1/CuO sample is exposed to CO at room temperature (RT), O vacancies appear next to the Au single-atoms (Fig. 4A). These O vacancies are created by the reaction of CO with the O anions in the CuO lattice adjacent to the single Au atoms, which lead to neutralization of the negatively charged Au. The neutralized Au atoms turn out to be inactive for further CO oxidation (Fig. 4B). It is observed that the activity for CO oxidation is enhanced by the presence of negatively charged Au atoms through two possible ways. One is to weaken the Cu–O bond by electron withdrawal from the neighbouring oxygen atoms. The other is to activate the adsorbed CO molecules by enhancement of back-donation from the Au d-electrons to the anti-bonding π* orbital of the adsorbed CO molecules.
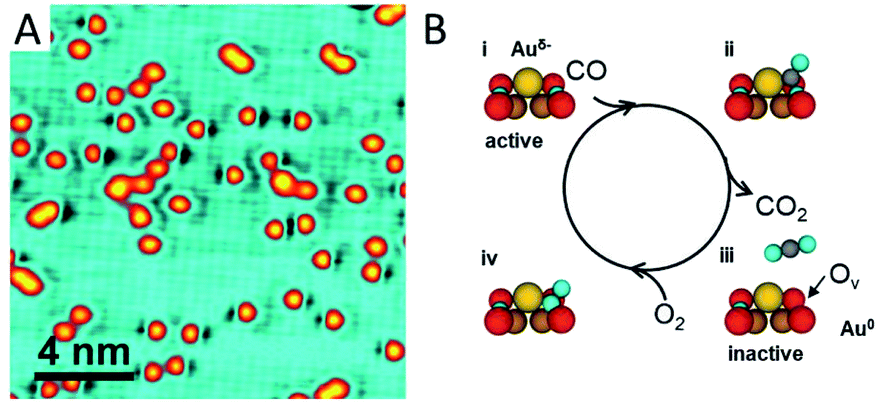 | ||
| Fig. 4 CO oxidation catalysed by Au single atoms on the CuO monolayer. (A) STM image of the Au single atoms exposed to CO at RT. (B) Schematic illustration of the CO oxidation process activated by the Au single atoms on the CuO monolayer. Reproduced with permission.56 Copyright 2018, American Chemical Society. | ||
Fu et al.67 have shown that doping the surface of TiO2 with Pt single atoms enhances the formation of surface oxygen vacancies which are active redox centres for the selective cleavage of the C–O bonds via the reverse Mars–van Krevelen mechanism. They have revealed that the cationic redox Pt atoms on the TiO2 surface are more active than metallic Pt particle sites for the C–O bond activation. Kwon et al.62 have reported that Rh SACs can enable the conversion of methane to value-added products (methane or methanol) by using H2O2 or O2 as oxidants below 300 °C, whereas Rh nanoparticles merely produce CO2. The Rh single atoms dispersed on the ZrO2 surface are oxidative in nature. It's also proven that the cationic Rh single atoms play a critical role in the stabilization of on-surface yielded CH3 intermediates and prevention of their subsequent over-oxidation.
3.2 Interface effect
Since the interfaces between metal atoms and oxide supports are believed to be the location of the active sites in heterogeneous catalysis,68 the interface effect may play a pivotal or even decisive role in shaping the catalytic performance of SACs. Lattice oxygen atoms at the interface in a metal/oxide system are conceived to be more reactive and selective than O2 molecules in oxidation reactions.69 Due to the metal–support interaction, single metal atoms frequently change the formation feasibility of O vacancies on oxide supports.For example, Chen et al.70 were able to prepare Pt/FeOx catalysts with the Pt entity exclusively in single atom or nanoparticle states. The catalytic activity of the Pt/FeOx catalyst has been experimentally identified to increase with the size decrease of the Pt entity. The supported Pt single atoms, however, turn out to be among the most active for the water–gas shift (WGS) reaction (Fig. 5A). Such high activity originates from the facilitation of the Pt adatoms to generate interfacial O vacancies on FeOx. The produced O vacancies are able to dissociate H2O into H2, and the lattice oxygen on the FeOx surface can react with CO, which implies that a “redox” process takes place (Fig. 5C). In contrast, CO adsorbs more strongly on the Pt nanoparticles (Pt–NP) and subsequently reacts with the OH species generated by the activation of H2O to form the formate intermediate, leading to a different pathway and lower activity for the WGS (Fig. 5B). Similar results are also observed in other SAC systems such as Rh/TiO2 (ref. 60) and Pd/FeOx,71 where single metal atoms accelerate the formation of the O vacancies and show a higher activity towards the WGS than their counterparts, the metal nanoparticles, on the same catalytic support. Besides, SACs of the same active metal atoms sometimes exhibit varied catalytic activities upon dispersion on different oxides. For instance, the CO oxidation activity of Pt single atoms increases in sequence when the Pt adatoms are dispersed on γ-Al2O3, Fe2O3 and ZnO.72 This interface effect between the catalytic activity and employed support can actually provide a novel strategy to regulate the SAC performance via support selection. The selectivity can be also enhanced by support selection through the oxide supporting effect.20–22,49
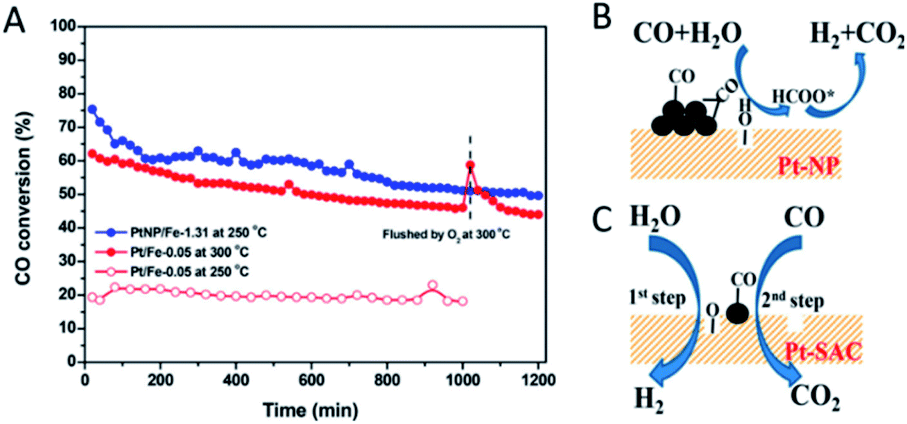 | ||
| Fig. 5 (A) CO conversions as a function of time on stream during the WGS over Pt–SAC (Pt/Fe-0.05) at 250 and 300 °C, and Pt–NP (PtNP/Fe-1.31) at 250 °C, respectively. (B and C) Schematic illustration of the WGS activated by (B) Pt–NP and (C) Pt–SAC on FeOx supports. Reproduced with permission.70 Copyright 2018, American Chemical Society. | ||
3.3 Counter examples
It should be noticed that SACs are not guaranteed to be more active than their nanoparticle counterparts. Many reported cases have indicated that some SACs have failed in certain catalytic reactions. Understanding the science behind the failure should help design high-performance SACs.One reason is the varied adsorption capability of the reactants. Ding et al.73 have demonstrated that only Pt nanoparticles show activity for CO oxidation and water–gas shift at low temperature in the coexistence of Pt single atoms and nanoparticles in many conventional catalysts. The lack of catalytic activity of Pt single atoms is partly attributed to their strong binding to the CO molecules (Fig. 6A). Zhou et al.13 have reported another system of the Pt species on CuO films that the Pt nanoclusters have an optimized size for CO oxidation in the sub-nanometre regime, i.e. 0.7–1.1 nm. In contrast, Pt single atoms possess no reactivity for CO oxidation due to the early and complete desorption of CO before its oxidation on the model catalysts commences (Fig. 6B). Additionally, sub-nanometric Ir clusters have been reported to register a higher activity for CO oxidation than Ir single atoms with or without the presence of H2.14 This arises from the promotion effect of the Ir clusters on the CO adsorption and OH formation. On the one hand, sub-nanometric Ir clusters promote CO adsorption. On the other hand, H2 can be feasibly activated and react with O species to form OH species. A subsequent reaction between the adsorbed CO and OH species possesses a lower activation energy, making the production of CO2 rather feasible. On the contrary, Ir single atoms have a weaker CO adsorption capability and cannot activate the H2 molecule so that the Ir single atoms exhibit a much lower activity for CO oxidation.
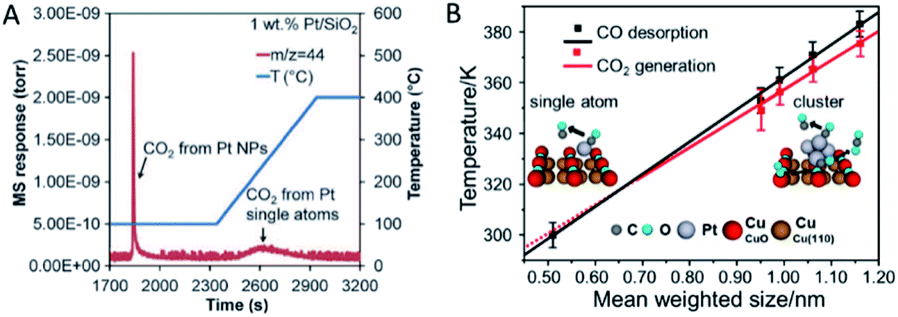 | ||
| Fig. 6 (A) CO2 signal monitored by the mass spectrum during the temperature-programmed oxidation process of the pre-adsorbed CO on Pt/SiO2. Reproduced with permission.73 Copyright 2015, American Association for the Advancement of Science. (B) Plots of the CO desorption and CO2 generation temperatures versus the mean size of the Pt nanoclusters. Reproduced with permission.13 Copyright 2016, American Chemical Society. | ||
Another reason for the failure of SACs is the limitation of active sites. Complex reactions normally involve the activation, adsorption and migration of reactants and possible intermediates. SACs fail to provide continuously packed metal sites that can work cooperatively to complete these multiple steps. Zuo et al.12 have reported that Ni single atoms are inactive for the complex dry reforming of the methane reaction, while Ni clusters show a higher performance for this reaction. This difference comes from the incapability of Ni single atoms to dissociate CO2 and CH4, while Ni clusters are able to activate both reactants and hence enable the production of CO, H2, and H2O.
4 Selectivity enhancement
4.1 Site limitation
Due to the lack of abundant metal sites, SACs have been reported to show poor performance for complex reactions and they may adopt a reaction mechanism completely different from that of their nanoparticle counterparts. This is often envisioned as a main drawback for SACs. However, such a disadvantage can be utilized to actually improve the selectivity of SACs under certain circumstances.For instance, Millet et al.49 have successfully prepared a Ni SAC by using a solid solution approach inside the MgO structure (NixMg1−xO). By controlling the reaction temperature, they are able to tune the formation of Ni clusters. Below 300 °C, the Ni SAC displays an astonishing selectivity for CO2 hydrogenation towards CO production, avoiding over-reduction in the reverse WGS reaction. An isolated Ni atom is capable of weakening the bond strength of CO2 to a low-coordinated surface sites and promote H2 dissociation. To obtain hydrogenation products such as CH4 or MeOH, it is necessary to utilize the Ni clusters that are formed above 300 °C (Fig. 7A). Guo et al.15 have demonstrated an atom-economical direct CH4 conversion process, enabled by lattice-confined single iron sites embedded within a silica matrix, which can activate CH4 and generate methyl radicals. The absence of adjacent iron sites prevents catalytic C–C coupling, further oligomerization, and hence coke deposition (Fig. 7B). Xie et al.74 have reported that Pt1/CeO2 catalysts exhibit a catalytic performance superior to their nanoparticle counterparts, achieving 14.4% of methane conversion at 975 °C and 74.6% selectivity toward C2 products (ethane, ethylene, and acetylene). Comparative studies of Pt1/CeO2 catalysts of different loadings and the nanoparticle counterparts have revealed that Pt single atoms are the active sites for the selective conversion of methane into C2 hydrocarbons.
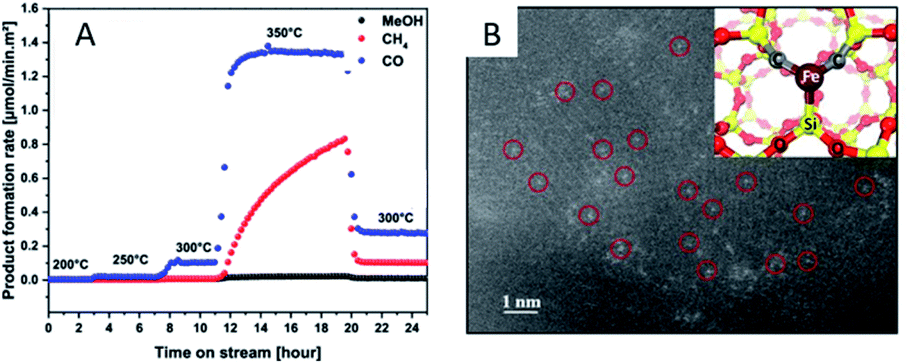 | ||
| Fig. 7 (A) Product formation rate on the NixMg1−xO samples at different temperatures. Reproduced with permission.49 Copyright 2019, American Chemical Society. (B) STEM-HAADF image of the catalyst after the reaction, with the inset showing the computational model of the single iron atom bonded to two C atoms and one Si within the silica matrix. Reproduced with permission15 Copyright 2014, American Association for the Advancement of Science. | ||
5 Summary and perspectives
This minireview overviews some recent advancements in exploring the intrinsic principles of oxide-supported SACs in catalytic performance, including stability, activity and selectivity. A consensus has been reached that some SACs possess distinctive and even unique properties against their nanoparticle counterparts. Such a difference in catalytic performance could not be linearly scaled by the simple size effect. SACs have their own merits in heterogeneous catalysis, including remarkable enhancement or complete annihilation for catalytic activity, simplification of reaction pathways, generation of new products, and cost-effective preparation of catalysts containing precious metals. Therefore, SACs do provide a new opportunity for catalyst design and development. The catalytic performance of SACs is largely determined by governing principles including metal–oxide interaction, surface free energy, charge effect, interface effect, site limitation and so on. These intrinsic properties may be traded off to improve the catalytic performance of SACs. Comprehensive consideration is required to develop a practical catalyst that meets the main criteria: high stability, excellent catalytic activity and high selectivity toward target products.In the foreseeable future, more and more SACs with unique properties are anticipated to appear due to the remarkable advancements in nanoscience and technology. Until today, most SACs have been exploited for relatively simple catalytic reactions. Multi-electron complex catalytic reactions have already appeared on the new horizon because more and more bi-metallically alloyed SACs have been reported in the literature that are beyond the scope of this minireview.29 At the present stage, more SACs should be tried and tested to build up a practical database that may be analysed with artificial intelligence to uncover the unknown principles of heterogeneous catalytic processes. In addition, it should be pointed out that heterogeneous catalysts undergo rigorous dynamic changes in both chemical states and structures under reaction conditions that instantly vary the reaction mechanisms in their ever-changing reaction atmospheres. The majority of our present knowledge about heterogeneous catalysis is built upon “static” catalytic models and pictures. This can lead to contradictory and frustrating conclusions.75 To tackle these challenging issues, detailed and systematic observations, characterization studies and analyses at the atomic level are highly needed to establish the exact connections between various structures of reactants and catalysts with specific catalytic characteristics.76,77 Such an atomic-level understanding will provide valuable guidance in designing and preparing advanced catalysts.24,78–86 Moreover, in situ and operando studies concerning the dynamic changes during the catalytic reactions are required to further reveal the mechanism involved in catalysis. Compared with SACs made by wet chemistry that are indeed closer to practical applications, single atoms fabricated by physical deposition on ultrathin oxide films (surface free energy approach) provide better-defined models for fundamental studies, especially for local and dynamic characterization studies, of practical catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the NSFC (21821004 and 21932001) and MOST (2017YFA0204702), China.References
- X. Zhang, Z. Sun, B. Wang, Y. Tang, N. Luan, Y. Li and F. F. Tao, J. Am. Chem. Soc., 2018, 140, 954–962 CrossRef CAS PubMed.
- Z. Chen, V. Evgeniya, M. Sharon, F. Edvin, M. A. Ortuño, L. Núria, S. M. Collins, P. A. Midgley, R. Sylvia, V. Gianvito and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- Y. Peng, Z. Geng, S. Zhao, L. Wang, H. Li, X. Wang, X. Zheng, J. Zhu, Z. Li, R. Si and J. Zeng, Nano Lett., 2018, 18, 3785–3791 CrossRef CAS PubMed.
- E. Vorobyeva, E. Fako, Z. Chen, S. M. Collins, D. Johnstone, P. A. Midgley, R. Hauert, O. V. Safonova, G. Vilé and N. López, Angew. Chem., Int. Ed., 2019, 58, 8724–8729 CrossRef CAS PubMed.
- R. Lin, D. Albani, E. Fako, S. K. Kaiser, O. V. Safonova, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 58, 514–519 Search PubMed.
- S. Wei, A. Li, J. C. Liu, L. Zhi, W. Chen, G. Yue, Q. Zhang, C. Weng-Chon, W. Yu, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Hakkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- M. J. Berr, F. F. Schweinberger, M. Döblinger, K. E. Sanwald, C. Wolff, J. Breimeier, A. S. Crampton, C. J. Ridge, M. Tschurl, U. Heiz, F. Jackel and F. Jochen, Nano Lett., 2012, 12, 5903–5906 CrossRef CAS PubMed.
- J. N. Kuhn, W. Huang, C.-K. Tsung, Y. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026–14027 CrossRef CAS PubMed.
- J. Wang, H. Tan, S. Yu and K. Zhou, ACS Catal., 2015, 5, 2873–2881 CrossRef CAS.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- X. Zhou, W. Yang, Q. Chen, Z. Geng, X. Shao, J. Li, Y. Wang, D. Dai, W. Chen, G. Xu, X. Yang and K. Wu, J. Phys. Chem. C, 2016, 120, 1709–1715 CrossRef CAS.
- J. Lin, Y. Chen, Y. Zhou, L. Li, B. Qiao, A. Wang, J. Liu, X. Wang and T. Zhang, AIChE J., 2017, 63, 4003–4012 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- B. B. Sarma, J. Kim, J. Amsler, G. Agostini, C. Weidenthaler, N. Pfander, R. Arenal, P. Concepcion, P. N. Plessow, F. Studt and G. Prieto, Angew. Chem., 2020, 132, 5806–5815 CrossRef PubMed.
- Y. Wang, H. Arandiyan, J. Scott, K.-F. Aguey-Zinsou and R. Amal, ACS Appl. Energy Mater., 2018, 1, 6781–6789 CrossRef CAS.
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lü, F. Gao, X. Xie, C. Du, H. Yi, D. Kang and X. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- M. Babucci, C. Fang, J. E. Perezaguilar, A. S. Hoffman, A. Boubnov, E. Guan, S. R. Bare, B. C. Gates and A. Uzun, Chem. Sci., 2019, 10, 2623–2632 RSC.
- E. Guan, L. M. Debefve, M. Vasiliu, S. Zhang, D. A. Dixon and B. C. Gates, ACS Catal., 2019, 9, 9545–9553 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- Y. Chen, Z. Huang, M. Zhou, Z. Ma, J. Chen and X. Tang, Environ. Sci. Technol., 2017, 51, 2304–2311 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and M. Ding, Angew. Chem., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- Z. Huang, Z. Xu, J. Zhou, H. Chen, W. Rong, Y. Lin, X. Wen, H. Zhu and K. Wu, J. Phys. Chem. C, 2019, 123, 17823–17828 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- H. Schwarz, Catal. Sci. Technol., 2017, 7, 4302–4314 RSC.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS PubMed.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- J. Liu, Chin. J. Catal., 2017, 38, 1460–1472 CrossRef CAS.
- Z. Li, D. Wang, Y. Wu and Y. Li, Natl. Sci. Rev., 2018, 5, 673–689 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- M. J. Hülsey, J. Zhang and N. Yan, Adv. Mater., 2018, 30, 1802304 CrossRef PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- Y. Guo, R. Lang and B. Qiao, Catalysts, 2019, 9, 135 CrossRef.
- G. S. Parkinson, Catal. Lett., 2019, 149, 1137–1146 CrossRef CAS PubMed.
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC.
- Z. Jakub, J. Hulva, M. Meier, R. Bliem, F. Kraushofer, M. Setvin, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Angew. Chem., Int. Ed., 2019, 58, 13961–13968 CrossRef CAS PubMed.
- M. Meier, Z. Jakub, J. Balajka, J. Hulva, R. Bliem, P. K. Thakur, T.-L. Lee, C. Franchini, M. Schmid, U. Diebold, F. Allegretti, D. A. Duncan and G. S. Parkinson, Nanoscale, 2018, 10, 2226–2230 RSC.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104–114 CrossRef CAS.
- H. V. Thang, S. Tosoni, L. Fang, P. Bruijnincx and G. Pacchioni, ChemCatChem, 2018, 10, 2634–2645 CrossRef CAS.
- M.-M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- S. Duan, R. Wang and J. Liu, Nanotechnology, 2018, 29, 204002 CrossRef PubMed.
- R. Bliem, J. E. V. D. Hoeven Je, J. Hulva, J. Pavelec, O. Gamba, P. E. de Jongh, M. Schmid, P. Blaha, U. Diebold and G. S. Parkinson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8921–8926 CrossRef CAS PubMed.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724–728 CrossRef CAS PubMed.
- G. Spezzati, Y. Su, J. P. Hofmann, A. D. Benavidez, A. T. DeLaRiva, J. McCabe, A. K. Datye and E. J. M. Hensen, ACS Catal., 2017, 7, 6887–6891 CrossRef CAS PubMed.
- D. B. Rasmussen, T. V. W. Janssens, B. Temel, T. Bligaard, B. Hinnemann, S. Helveg and J. Sehested, J. Catal., 2012, 293, 205–214 CrossRef CAS.
- X. Zhou, Q. Shen, K. Yuan, W. Yang, Q. Chen, Z. Geng, J. Zhang, X. Shao, W. Chen, G. Xu, X. Yang and K. Wu, J. Am. Chem. Soc., 2018, 140, 554–557 CrossRef CAS PubMed.
- H. Chen, W. Rong, Z. Huang, Z. Peng, Z. Xu, J. Zhou, B. Di, X. Zhou and K. Wu, J. Chem. Phys., 2019, 151, 184703 CrossRef PubMed.
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J. S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS.
- A. J. Therrien, G. Kyle, A. J. R. Hensley, A. C. Schilling, R. T. Hannagan, M. D. Marcinkowski, P. Alex, F. R. Lucci, E. C. H. Sykes and M. E. Jean-Sabin, J. Catal., 2018, 364, 166–173 CrossRef CAS.
- H. Guan, J. Lin, B. Qiao, S. Miao, A.-Q. Wang, X. Wang and T. Zhang, AIChE J., 2017, 63, 2081–2088 CrossRef CAS.
- C. Yang, L. Jian, L. Lin, B. Qiao, J. Liu, Y. Su and X. Wang, ACS Catal., 2018, 8, 859–868 CrossRef.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- S. Zhang, Y. Tang, L. Nguyen, Y. F. Zhao, Z. Wu, T. W. Goh, J. J. Liu, Y. Li, T. Zhu, W. Huang, A. I. Frenkel, J. Li and F. F. Tao, ACS Catal., 2018, 8, 110–121 CrossRef CAS.
- C. Dessal, L. Martínez, C. Maheu, T. Len, F. Morfin, J. L. Rousset, E. Puzenat, P. Afanasiev, M. Aouine, L. Soler, J. Llorca and L. Piccolo, J. Catal., 2019, 375, 155–163 CrossRef CAS.
- C. K. Narula, L. F. Allard, M. Moses-Debusk, G. M. Stocks and Z. Wu, Sci. Rep., 2017, 7, 560 CrossRef PubMed.
- J. Fu, J. Lym, W. Zheng, K. Alexopoulos, A. V. Mironenko, N. Li, J. A. Boscoboinik, D. Su, R. T. Weber and D. G. Vlachos, Nat. Catal., 2020, 3, 446–453 CrossRef CAS.
- G. Zhao, Y. Fan, Z. Chen, Q. Liu, Y. Ji, Z. Yi, Z. Niu, J. Mao, X. Bao and P. Hu, Nat. Commun., 2017, 8, 14039 CrossRef PubMed.
- T. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Zhang, J. Huang, H. Sun, M. Haruta, A. Wang, B. Qiao, J. Li and T. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed.
- Y. Chen, J. Lin, L. Li, B. Qiao, J. Liu, Y. Su and X. Wang, ACS Catal., 2018, 8, 859–868 CrossRef CAS.
- X. Sun, J. Lin, Y. Zhou, L. Lin, Y. Su, X. Wang and T. Zhang, AIChE J., 2017, 63, 4022–4031 CrossRef CAS.
- Y. Lou and J. Liu, Ind. Eng. Chem. Res., 2017, 56, 6916–6925 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- S. C. Ammal and A. Heyden, ACS Catal., 2017, 7, 301–309 CrossRef CAS.
- H. Chen, H. Zhu, Z. Huang, W. Rong and K. Wu, Adv. Mater., 2019, 31, 1902080 CrossRef CAS PubMed.
- L. Xing, Z. Peng, W. Li and K. Wu, Acc. Chem. Res., 2019, 52, 1048–1058 CrossRef CAS PubMed.
- F. Dvořak, M. F. Camellone, A. Tovt, N. Tran, F. R. Negreiros, M. Vorokhta, T. Skala, I. Matolinova, J. Myslivecek, V. Matolin and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- J. Ftouni, A. Muñoz-Murillo, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, L. Lu, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Catal., 2016, 6, 5462–5472 CrossRef CAS.
- L. Liu, U. Diaz, R. Arenal, G. Agostini, P. Concepcion and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- H. Fei, J. Dong, M. J. Arellanojimenez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- J. Fu, J. Lym, W. Zheng, K. Alexopoulos, A. V. Mironenko, N. Li, J. A. Boscoboinik, D. Su, R. T. Weber and D. G. Vlachos, Nat. Catal., 2020, 3, 446–453 CrossRef CAS.
- C. Kuo, Y. Lu, L. Kovarik, M. H. Engelhard and A. M. Karim, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- Z. Lu, X. Liu, B. Zhang, Z. Gan, S. Tang, L. Ma, T. Wu, G. J. Nelson, Y. Qin, C. H. Turner and Yu Lei, J. Catal., 2019, 377, 419–428 CrossRef CAS.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS PubMed.
- J. Kim, C. W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |