
Calix[8]arene-constructed stable polyoxo-titanium clusters for efficient CO2 photoreduction†
Ning
Li‡
a,
Jing-Jing
Liu‡
b,
Jia-Wei
Sun
b,
Bao-Xia
Dong
*a,
Long-Zhang
Dong
 b,
Su-Juan
Yao
b,
Zhifeng
Xin
c,
Shun-Li
Li
b and
Ya-Qian
Lan
*b
b,
Su-Juan
Yao
b,
Zhifeng
Xin
c,
Shun-Li
Li
b and
Ya-Qian
Lan
*b
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, P. R. China. E-mail: bxdong@yzu.edu.cn
bJiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, No. 1, Wenyuan Road, NanJing 210023, China. E-mail: yqlan@njnu.edu.cn
cInstitute of Molecular Engineering and Applied Chemistry, Anhui University of Technology, Ma'anshan, Anhui 243002, PR China
First published on 20th July 2020
Abstract
It is still rare for coordination complexes to achieve CO2 reduction reaction in water. Demonstrated here is the self-assembly and CO2 photoreduction performance of two thermodynamically reversibly converted polyoxo-titanium clusters (PTCs), Ti4-C8A and Ti7-C8A, using calix[8]arene with high molecular degrees of freedom and variable conformations as an organic “functional armor”. Because of the rich coordination sites and bridging modes of calix[8]arene, the obvious electron transfer effect generated between the titanium–oxo core and calix[8]arene ligand extended the light absorption of the two PTCs from the traditional ultraviolet to the visible region. Moreover, the hydrophobic benzene groups in calix[8]arene endowed the two clusters with high structural and chemical stability in aqueous solutions with different pH values. On this foundation, these two stable and photosensitive clusters were used as heterogeneous molecular photocatalysts for efficient CO2 reduction in water (with triethanolamine as a sacrificial agent) and they exhibited very high CO2-to-HCOO− conversion activity and selectivity. Most importantly, this is the first report of molecular coordination complexes in water containing a sacrificial agent to perform heterogeneous CO2 reduction reaction.
Introduction
The large amount of CO2 emission from fuel combustion has created serious environmental burden and energy crisis. By imitating natural photosynthesis, solar energy-driven artificial CO2 reduction reaction (CO2RR) performed in water (H2O) is considered the most energy-efficient and eco-friendly means to achieving CO2 recycling, since it can reduce CO2 into value-added carbon-based chemicals under the action of a photocatalyst.1–4 Thus, as the crucial factor of CO2RR, the fundamental design requirements of photocatalyst structures are usually water-stability, photosensitivity, and recyclability. Although many stable and efficient nanostructured photocatalyst materials have been explored to perform CO2RR,1,4–9 in most cases the identification of catalytically active sites is limited by their complicated hybrid or composite structures. In this context, the development of efficient, photosensitive, and heterogeneous photocatalysts with well-defined structures is highly important.10–12In recent years, the exploration of photocatalysts for CO2RR has targeted structurally adjustable coordination complexes including metal–organic frameworks (MOFs) and metal–organic molecules/clusters (MOMs/MOCs),13–26 because their clear structure information can provide a visual platform for the study of catalytically active sites and reaction mechanisms. However, the poor water stability and photosensitivity of these crystalline materials make them mostly used as homogeneous or heterogeneous photocatalysts to perform CO2RR in toxic organic solvents containing an auxiliary photosensitizer.2,3,25,27 In this regard, effectively increasing the light absorption and H2O stability of crystalline coordination complexes is the most fundamental and significant design requirement for them to perform heterogeneous photocatalytic CO2RR in eco-friendly H2O. In recent years, some well-defined, conjugated organic polymer/covalent-organic framework/single-atom catalyst materials have been developed to reduce CO2 in pure H2O.28–33 However, coordination complexes that can achieve CO2RR in H2O containing/excluding a sacrificial agent are still very rare.16,34
Polyoxo-titanium clusters (PTCs), due to their intrinsic low-toxicity and photosensitivity, have been widely used in many photocatalytic applications.35,36 Importantly, these PTCs under the modification of specifically functionalized organic ligands can display high water and chemical stability, and their photosensitivity can also be enhanced by efficient charge transfer between the titanium (Ti)–oxo core and organic ligand.37,38 This means that they have great potential to be used as crystalline heterogeneous molecular photocatalysts for efficient CO2RR in H2O. Nevertheless, the most prominent obstacles for traditional PTCs applied in heterogeneous photocatalytic CO2RR are their narrow light responsive range (ultraviolet region) and insolubility in H2O and organic solvents.23,39–42 Consequently, the selection of suitable functionalized organic ligands to construct water-stable and light absorption-extended PTCs is the key to solving these problems.
In this work, we designed and synthesized two calix[8]arene (H8C8A)-modified PTCs, ellipse-shaped [Ti4O2(C8A)(OiPr)4(HOiPr)] (Ti4-C8A, HOiPr = isopropanol) and bowknot-shaped [Ti7O4(C8A)(CH3O)12] (Ti7-C8A), and their crystallographic structures can achieve thermodynamic interconversion under different solvent and temperature conditions (Scheme 1). Because of the hydrophobic benzene group of C8A,43 these two PTCs displayed high structural and chemical stability in aqueous solutions. Moreover, their light absorption ranges were extended to the visible light region through strong charge transfer between the titanium–oxo core and hydroxyl groups of the C8A ligand. In consideration of these advantages, two water-stable and photosensitive PTCs were further used as heterogeneous photocatalysts to perform CO2RR in H2O containing sacrificial triethanolamine (TEOA). We found that both could reduce CO2 to HCOOH with very high activity (253.88 ± 13.29 and 488.35 ± 21.41 μmol g−1 h−1) and selectivity (96.0% and 99.7%) under ultraviolet light, which is even comparable to the reported superior coordination complex photocatalysts for CO2-to-HCOO− conversion in organic solvents under visible light irradiation.13,15,16,21,44–46 Significantly, this is the first case of a crystalline molecular coordination system as heterogeneous photocatalysts to perform CO2RR in H2O in the presence of TEOA. This work provides a new strategy toward developing more stable and photosensitive PTCs applied in artificial CO2RR.
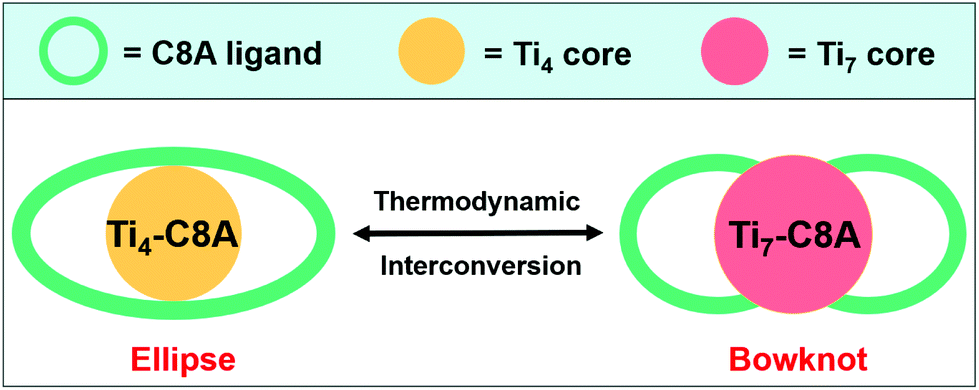 | ||
| Scheme 1 Simplified structures of ellipse-shaped Ti4-C8A and bowknot-shaped Ti7-C8A. Their crystallographic structures can undergo thermodynamic interconversion. | ||
Synthetic materials, procedures, and methods
All reagents and solvents employed in this work were commercially available and used without further purification. Infrared spectra using KBr pellets were measured on a Bruker Tensor 27 in a range of 4000–400 cm−1. Thermogravimetric (TG) analysis was performed on a Netzch STA449F3 analyzer at a heating rate of 10 °C min−1 from ambient temperature to 800 °C under a nitrogen gas atmosphere. Room-temperature powder X-ray diffraction (PXRD) spectra were recorded on a Rigaku D/Max 2500/PC diffractometer at 40 kV and 200 mA with a Cu-target tube and a graphite monochromator. UV-Vis absorption spectra were acquired on a Varian Cary 5000 UV-Vis spectrophotometer in a wavelength range of 250–800 nm. The photocatalytic liquid products were analyzed by ion chromatography (LC-2010 PLUS). Gas chromatography was performed on GC-7920A (Aulight Co.) equipped with a flame ionization detector (FID) with a methanizer and a thermal conductivity detector (TCD). 13CNMR spectra were measured using chromatography–mass spectrometry (7890A and 5975C, Agilent). Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) measurements were performed using Agilent 720.Synthesis of Ti4-C8A
A mixture of C8A (0.01 mmol, 8.5 mg) and phosphoric acid (10.0 mg) in isopropanol (5.0 mL) was ultrasonically dissolved, and then titanium isopropoxide (0.6 mL) was added into the above solution. After stirring for 10 min, this solution was transferred into a 15 mL Teflon-lined stainless steel autoclave for 72 h at 100 °C under autogenous pressure. After cooling down to room temperature, yellow crystals were collected by filtration and fully washed several times with isopropanol. Yield: ca. 89%.Synthesis of Ti7-C8A
The mixture of C8A (8.5 mg), phosphoric acid (30.0 mg) in isopropanol (2 mL), and CH3OH (3 mL) was ultrasonically dissolved. After stirring for 10 min, titanium isopropoxide (1.2 ml) was added into the resultant solution and stirred for another 10 min for dissolution. This solution was transferred into a 15 mL Teflon-lined stainless steel autoclave for 72 h at 120 °C under autogenous pressure. After cooling down to room temperature, reddish brown crystals were collected by filtration and fully washed several times with isopropanol. Yield: ca. 76%.Thermodynamic conversion of Ti4-C8A to Ti7-C8A
5.0 mg of Ti4-C8A crystals was added into 3 mL CH3OH and then transferred into a 15 mL Teflon-lined stainless steel autoclave for 72 h at 150 °C under autogenous pressure. After cooling down to room temperature, reddish brown crystals of Ti7-C8A were obtained and collected by filtration.Thermodynamic conversion of Ti7-C8A to Ti4-C8A
5.0 mg of Ti7-C8A crystals was dissolved in 3 mL isopropanol and then transferred into a 15 mL Teflon-lined stainless steel autoclave for 96 h at 100 °C under autogenous pressure. After cooling down to room temperature, yellow crystals of Ti4-C8A and a few co-crystals of Ti7-C8A were obtained and collected by filtration.X-ray crystallography
The single-crystal diffraction analysis of Ti4-C8A and Ti7-C8A were performed using a Bruker AXS smart Apex CCD diffractometer at 296 K. The X-ray generator was operated at 50 kV and 35 A using Mo Kα (λ = 0.71073 Å) radiation. The crystal structures were solved and refined by full matrix methods against F2 using the SHELXL-2014 program package and Olex-2 software. All non-hydrogen atoms were refined with anisotropic temperature parameters, and hydrogen positions were fixed at calculated positions and refined isotropically. The selected bond lengths and angles of the compounds are listed in Table S1b.† The crystal structures of the two clusters have been deposited, and CCDC numbers are 1999136 and 1999137† for Ti4-C8A and Ti7-C8A, respectively.Crystal structures
Single-crystal X-ray diffraction analysis reveals that Ti4-C8A crystallizes in a monoclinic system with the space group P21/n (Table S1a†) and is composed of four independent Ti atoms, one C8A ligand, two μ3-O atoms, and five isopropanol molecules. Within the cluster structure, five-coordinated Ti1, Ti2, and Ti3 atoms all exhibit a distorted tetragonal pyramid geometry, except six-coordinated Ti4 that exhibits a slightly distorted octahedral geometry (Fig. 1a). Ti2 and Ti3 atoms are linked by two μ3-O atoms, which are further connected with Ti1 and Ti4 atoms to constitute a tetranuclear titanium–oxo (Ti4O2) core. Then, the overall metal–oxo core of small size (ca. 5.27 × 2.16 Å) is firmly fixed into the cavity (ca. 8.17 × 4.99 Å) of the C8A ligand by coordinating its eight hydroxyl groups (Fig. 1b and c). Interestingly, when the Ti4-C8A crystals are soaked in methanol solution undergoing solvothermal reaction at 150 °C for 72 hours, they can be thermodynamically converted into another more stable neutral cluster, Ti7-C8A. The crystal structure of Ti7-C8A is different from that of Ti4-C8A, and the originally coordinated isopropanol groups in Ti4-C8A are all replaced and coordinated by more methoxy (OCH3) groups. Crystallographic structure analysis shows that Ti7-C8A crystallizes in the orthogonal Cmc21 space group with relatively high symmetry (Table S1a†). The asymmetric unit contains three and a half Ti atoms, one half of the C8A ligand, six OCH3 groups, one μ2-O atom, and one μ4-O atom. There are four independent Ti atoms within the cluster, and all of them display a slightly distorted octahedral geometry constructed by six O atoms from μ2-O, μ4-O, and μ3-OCH3 groups and/or the C8A ligand. The main difference is that the coordination environment of the Ti4 atom includes two terminal methoxy groups, which may be easily substituted by other small molecules in solution. Among these Ti atoms, the Ti3 atom is located at the intersection of the symmetric elements of glide and mirror planes, and thus it is the center of the metal–oxo core (Fig. S1†). The central Ti3 atom is connected by two sets of symmetric Ti1, Ti2, and Ti4 atoms through two μ2-O atoms, two μ4-O atoms, and two μ3-OCH3 groups to form a heptanuclear titanium–oxo (Ti7O16) core (Fig. 1d). The two sets of Ti1, Ti2, and Ti4 atoms on the periphery communicate with each other by six μ2-OCH3 groups. Analogously, the overall metal–oxo core is also captured by the cavity of the C8A ligand in which eight hydroxyl groups are coordinated to two Ti1 and two Ti2 atoms. However, the shared Ti3 atom and two Ti4 atoms are exposed above the cavity (ca. 8.17 × 5.50 Å) of the C8A ligand due to the larger size (ca. 11.12 × 5.98 Å) of the heptanuclear metal–oxo core (Fig. 1e and f). It should be noted that different morphologies of Ti7-C8A crystals can be obtained through slight changes in the synthetic protocol (Table 1 and Fig. 2a). Furthermore, the Ti7-C8A crystals can also be converted into Ti4-C8A by further soaking them in isopropanol at 100 °C for one week. More details about the supramolecular stacking, main bond lengths and bond angles of Ti4-C8A and Ti7-C8A are provided in Table S1b and Fig. S2, S3.†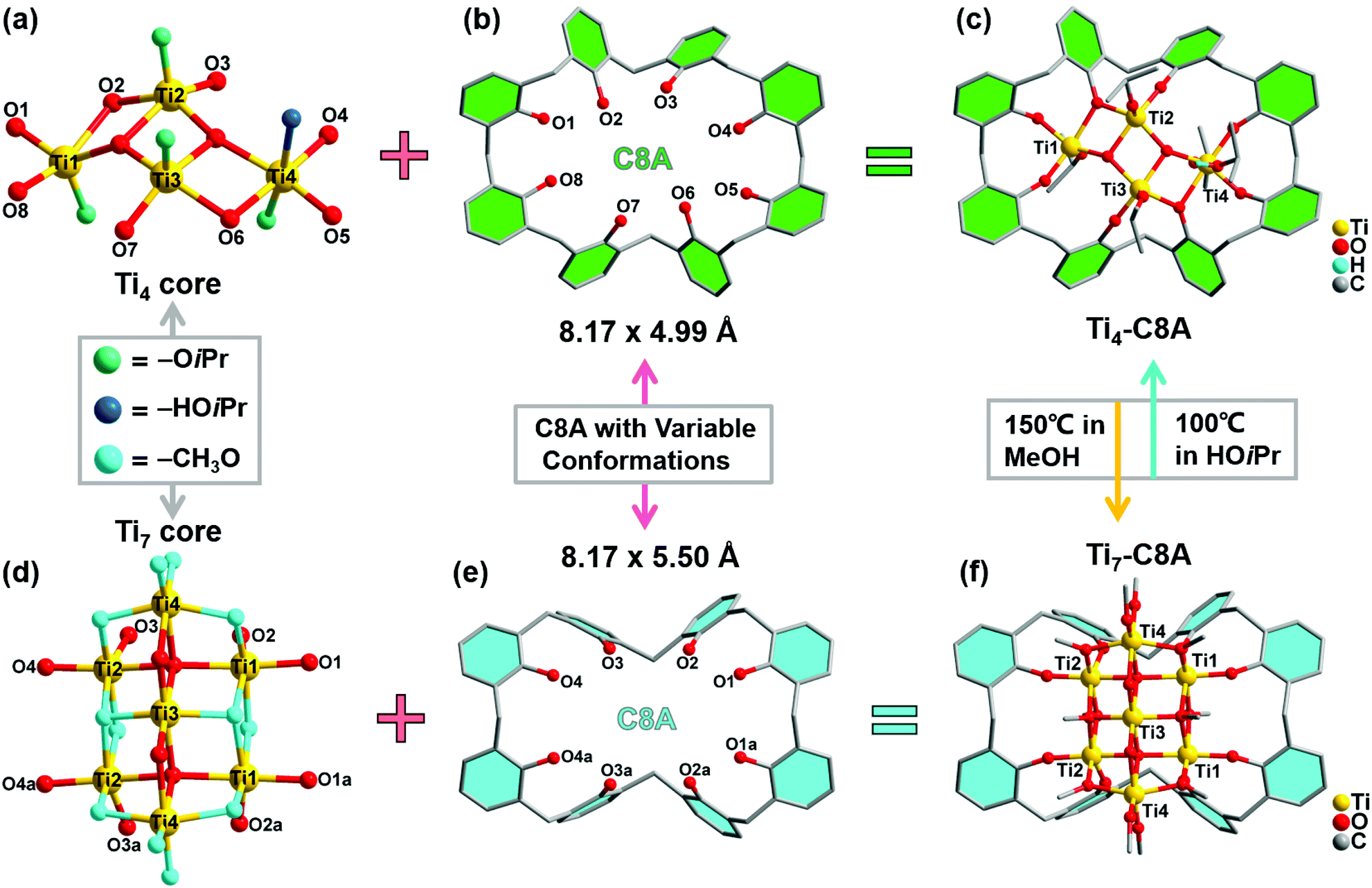 | ||
| Fig. 1 Schematic of the crystal structures of Ti4-C8A and Ti7-C8A. (a) The metal–oxo core, (b) C8A ligand (cavity of ca. 8.17 × 4.99 Å), and (c) overall cluster structure of Ti4-C8A. (d) The metal–oxo core, (e) C8A ligand (cavity of ca. 8.17 × 5.50 Å), and (f) overall cluster structure of Ti7-C8A. Except for the protonated –OiPr group (blue ball), all the hydrogen atoms are omitted for clarity. | ||
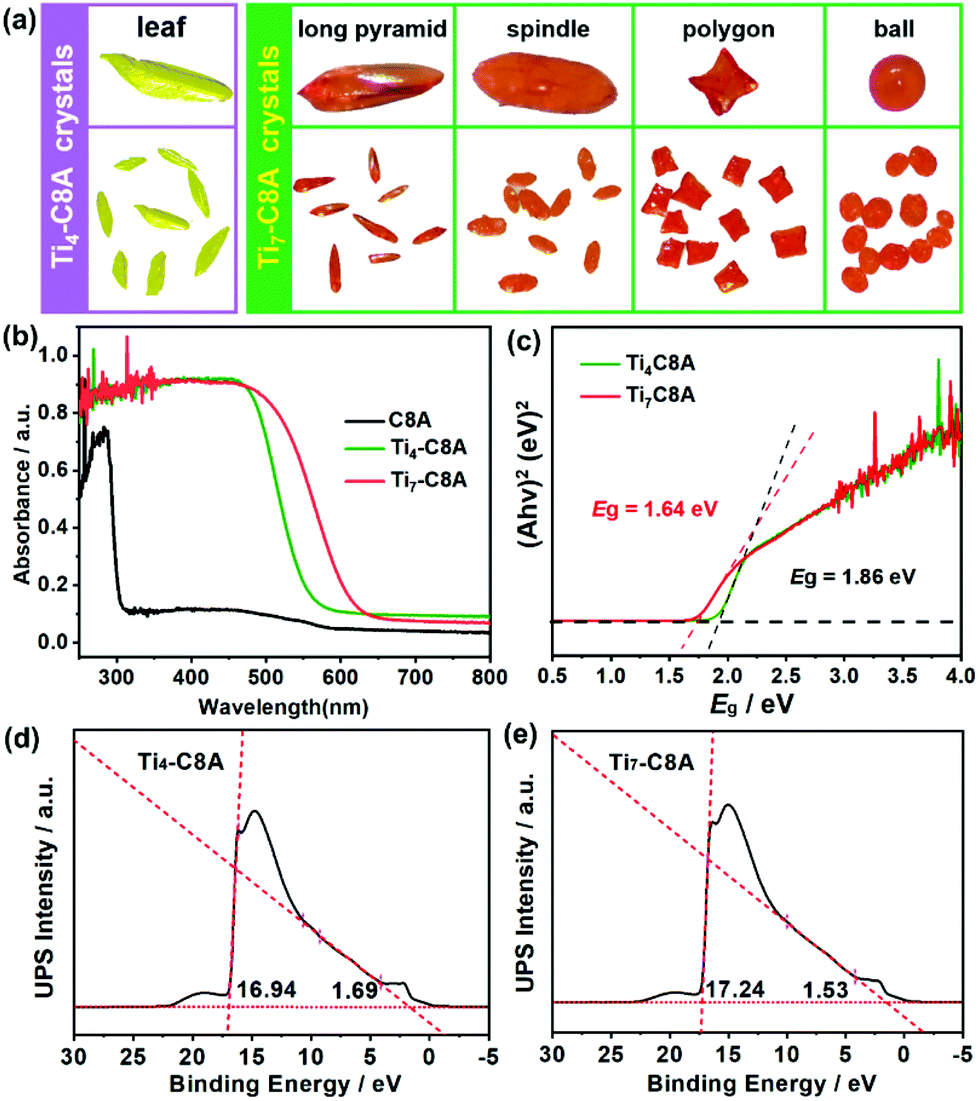 | ||
| Fig. 2 (a) Different morphologies of Ti4-C8A and Ti7-C8A crystals. (b) UV/vis spectra of the C8A ligand, Ti4-C8A, and Ti7-C8A. (c) Tauc plots of Ti4-C8A and Ti7-C8A. (d and e) Ultraviolet photoelectron spectroscopy (UPS) spectra of Ti4-C8A and Ti7-C8A. The intersections of the tangents with the baseline give the edges of the UPS spectra from which the UPS width is determined. UPS is used to determine the ionization potentials [equivalent to the HOMO energy] of Ti4-C8A and Ti7-C8A. Their HOMO levels are determined to be 5.97 (1.12 V vs. NHE) and 5.51 eV (0.66 V vs. NHE) by subtracting the width of the He I UPS spectra from the excitation energy (21.22 eV). The LUMO levels of Ti4-C8A and Ti7-C8A are thus calculated to be −0.74 V (vs. NHE) and −0.98 V (vs. NHE), respectively. | ||
| Ti(OiPr)4 | C8A ligand | H3PO4 | HOiPr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH :MeOH |
Crystal morphology |
|---|---|---|---|---|
| 4 mmol | 0.01 mmol | 30.0 mg | 1:4 (mL) |
Polygon |
| 4 mmol | 0.01 mmol | 30.0 mg | 0:5 (mL) |
Ball |
| 4 mmol | 0.01 mmol | 30.0 mg | 2:3 (mL) |
Spindle |
| 4 mmol | 0.01 mmol | 30.0 mg | 3:2 (mL) |
Long pyramid |
The phase purity and thermal stability of Ti4-C8A and Ti7-C8A were first confirmed by well-matched powder X-ray diffraction (PXRD) patterns and thermogravimetric analysis, respectively (Fig. S4–S6†). It should be noted that the hydrophobic benzene group modification of the C8A ligand endows these two PTCs with high structural and chemical stability in aqueous solutions with different pH values, which is essential and important for them to be used in water-dependent applications. Besides, it is well recognized that Ti-based nanostructured or crystalline materials generally exhibit intrinsic photoactivity and extensive photocatalytic applications, in which their light harvesting capability plays a key role in determining their photocatalytic performance. Therefore, ultraviolet-visible (UV-vis) absorption spectroscopy was performed to estimate the photosensitivity and optical bandgaps of Ti4-C8A and Ti7-C8A. In Fig. 2b, it can be observed that both the clusters coordinated with the C8A ligand exhibited better visible light collecting ability than the traditional UV-responsive PTCs and free C8A ligand, indicating that an obvious charge transfer occurred between the C8A ligand and titanium–oxo core. The optical bandgaps were thus calculated to be 1.86 (Ti4-C8A) and 1.64 eV (Ti7-C8A) from their Tauc plots (Fig. 2c), which means that these two water-stable PTCs also can display semiconductor-like properties. On this basis, the corresponding HOMO and LUMO energy levels of Ti4-C8A and Ti7-C8A were further determined by ultraviolet photoelectron spectroscopy (UPS) (Fig. 2d and e) and Mott–Schottky measurements (Fig. S7†), and they were converted to electrochemical energy potentials in volts vs. normal hydrogen electrode (NHE). It can be found that the Ti4-C8A and Ti7-C8A clusters have very negative LUMO levels; therefore, they are expected to serve as catalysts for photocatalytic reduction reactions.
CO2 photoreduction
Based on the abovementioned advantages, we conducted UV and visible light-driven photocatalytic CO2RR over Ti4-C8A and Ti7-C8A, with TEOA and H2O as the electron donor and reaction solvent (H2O/TEOA = 4/1), respectively. As we can see from Fig. 3a and b, the yield of HCOO− for these two PTC clusters shows a continuous growth with the extended irradiation time. After 17 hours, the HCOO− production of Ti4-C8A under UV light irradiation (200–400 nm) reaches up to 41.51 ± 1.87 μmol (488.35 ± 21.41 μmol g−1 h−1), which is nearly two times higher than that of Ti7-C8A (21.58 ± 1.13 μmol; 253.88 ± 13.29 μmol g−1 h−1). The photocatalytic results revealed that HCOO− is the only liquid product detected by ion chromatography (IC; Fig. S8†), while a small amount of competitive H2 and CO was observed by gas chromatography (GC; Fig. S9† and Fig. 3c). According to the reductive product distribution (Fig. 3c), the photocatalytic selectivity of CO2-to-HCOO− conversion for Ti4-C8A and Ti7-C8A was determined to be 96.0% and 99.7%, respectively. The higher photocatalytic activity of Ti4-C8A is probably related to its four potentially catalytically active sites with naked coordination space (five-coordinated Ti1, Ti2, and Ti3 atoms) or a coordinated isopropanol molecule (Ti4 atom), which are easily available sites for CO2 attack. For Ti7-C8A, however, only two Ti4 atoms coordinated with two terminal methoxy groups can function as catalytically active sites for CO2 reduction. Additionally, when an identical photocatalytic reaction was carried out under visible-light irradiation (420–800 nm), the HCOO− yield (8.25 ± 1.06 μmol; 97.06 ± 12.47 μmol g−1 h−1) of Ti4-C8A was slightly higher than that of Ti7-C8A (6.19 ± 1.32 μmol; 72.82 ± 15.53 μmol g−1 h−1). This is mainly because Ti7-C8A has a better visible-light absorption ability compared with Ti4-C8A. Moreover, no other gaseous or liquid reductive by-products were detected by IC and GC during the reaction process, which suggested that both the PTC clusters displayed a relatively higher selectivity (∼100%) for CO2-to-HCOO− conversion under visible light. From the structural viewpoint, we assume that the potentially active sites (Ti1, Ti2, Ti3, and Ti4 atoms) in Ti4-C8A are twice those (two Ti4 atoms) in Ti7-C8A. Consequently, we compared the catalytic activity of active sites in Ti4-C8A and Ti7-C8A using the turnover number (TON). As shown in Table S2,† each potential Ti4+ catalytic site (TONTi) in Ti7-C8A is more active than that in Ti4-C8A under UV or visible light irradiation, whereas the Ti4-C8A cluster has a higher overall TON. Therefore, the higher photocatalytic activity of Ti4-C8A compared with that of Ti7-C8A is mainly attributed to its more potentially active Ti4+ sites. To know the reasons behind the different photocatalytic activities of Ti4-C8A and Ti7-C8A, we first explored the charge separation efficiencies by their transient photocurrent responses to UV and visible light irradiation. It was revealed that Ti4-C8A had a slightly higher photocurrent than Ti7-C8A under UV or visible light irradiation (Fig. S10–S13†). However, the photocurrent is usually affected by the resistance of the material (both within a crystallite and at grain boundaries), conductivity between the ITO and the material, as well as the homogeneous distribution and connectivity among ITO, the material and Nafion used as a binder. All these factors affect the current that is measured. The photocurrent measured thus describes the complete electrode and not one particular component of the electrode. Our electrode consisting of ITO, Nafion, and Ti4-C8A shows a more efficient charge separation which does not mean that Ti4-C8A itself has a more effective charge separation than Ti7-C8A, because the fabricated electrodes were not used in photocatalysis. To further confirm the charge separation efficiency, we also conducted electrochemical impedance spectroscopy (EIS) measurements. As shown in Fig. S14,† the size of the Nyquist plot of Ti7-C8A is clearly smaller than that of Ti4-C8A, which indicates that the interfacial charge transfer process of Ti7-C8A is faster than that of Ti4-C8A. Therefore, Ti7-C8A does have a higher charge separation efficiency than Ti4-C8A. To further validate the exciton separation (or charge separation) efficiency of Ti4-C8A and Ti7-C8A, we also calculated the electron–hole Coulomb attraction energies (Ec) in the first excited state. In general, a small Ec results in high exciton separation efficiency. Density functional theory (DFT) and time-dependent DFT (TD-DFT) were employed in this work. The calculated Ec values are 3.42 and 2.16 eV for Ti4-C8A and Ti7-C8A, respectively (Fig. S15†). This result also suggested that Ti7-C8A has higher exciton separation efficiency than Ti4-C8A, which is consistent with our experimental data. The difference in the charge separation of these two clusters is mainly derived from their structural distinction, which results in different activities for photocatalytic CO2RR. Moreover, the apparent quantum efficiency (QE) for HCOO− evolution was measured using different monochromatic light (365/420/450/500 nm), and the corresponding results indicate that Ti4-C8A has higher QE than Ti7-C8A (see the QE Calculation section in the ESI†). It is worth noting that Ti4-C8A and Ti7-C8A not only showed high HCOOH production under UV light irradiation (Table S3†), but also they are the first crystalline coordination molecular system to perform heterogeneous photocatalytic CO2RR in H2O containing TEOA.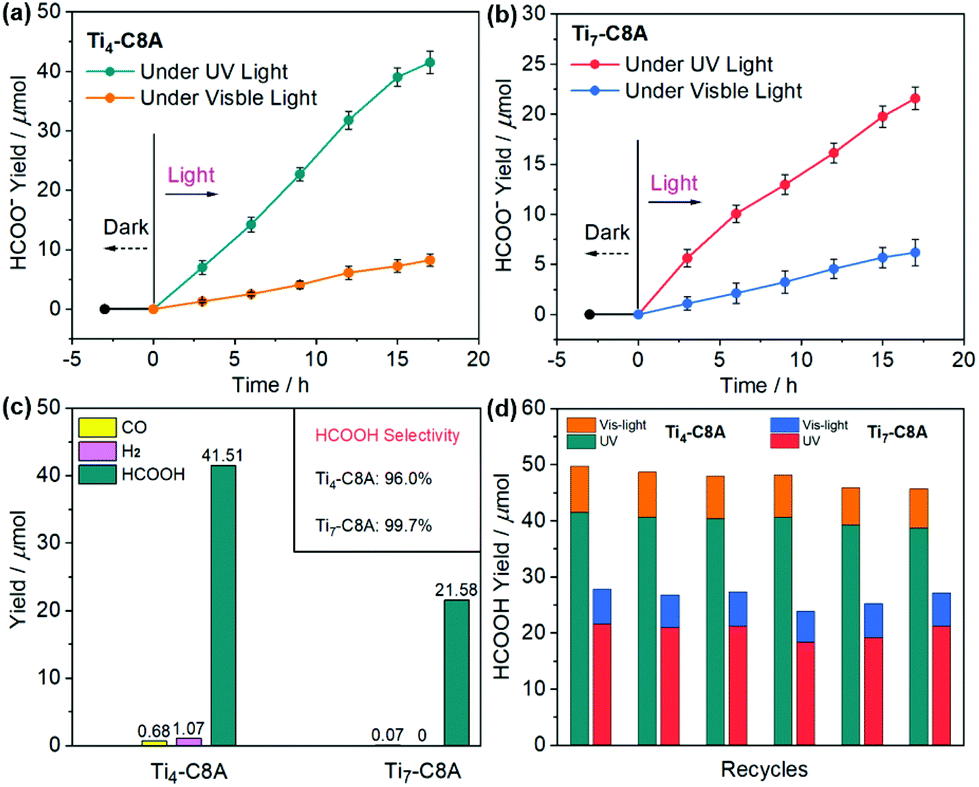 | ||
| Fig. 3 Photocatalytic characterization of Ti4-C8A and Ti7-C8A. (a and b) Amounts of HCOO− produced as a function of the irradiation time under UV and visible light irradiation. (c) Yield distribution of different photocatalytic reductive products and HCOO− selectivity for Ti4-C8A and Ti7-C8A under UV light irradiation. (d) The recycling experiments were performed by recovering and redispersing the used catalysts in a fresh H2O/TEOA solution for 17 hours of reaction, showing the photocatalytic durability of the catalyst. | ||
After the reaction, the photocatalyst solids were removed from the reaction solution. The Ti4+ residual ions in the resultant filtrate were evaluated to be 0.082% (Ti4-C8A) and 0.054% (Ti7-C8A) by inductively-coupled plasma analysis. At the same time, the UV-vis absorption spectra of the filtrates did not show any obvious signal (Fig. S16 and S17†), which excludes the influence of the decomposed active components from catalysts on the photocatalytic activity. Moreover, nearly unchanged solid IR spectra and PXRD patterns obtained before and after photocatalytic CO2RR also confirmed the heterogeneous catalysis nature of the Ti4-C8A and Ti7-C8A clusters (Fig. S18–S21†). A series of deletional control experiments were conducted to verify the photocatalytic activity of these two PTCs, in the absence of photocatalysts, CO2, TEOA, the C8A ligand or light irradiation. The results revealed that no detectable products were observed by IC and GC in the reaction system (Table S4†), corroborating the photocatalytic potential of these two PTC clusters for reducing CO2 into HCOO−. Subsequently, recycling experiments were performed for checking photocatalytic durability, in which Ti4-C8A and Ti7-C8A can maintain their initial activities for at least six cycles (Fig. 3d). The slightly reduced HCOO− yield was probably due to the mass loss of the photocatalysts in the recovery process. Since the catalysts had undergone centrifugation, washing, and redispersion processes, the quality was inevitably lost in the circulation process; therefore, it is relatively reasonable that the product yield slightly decreases. To further confirm the photocatalytic activities of Ti4-C8A and Ti7-C8A, an isotopic 13CO2 experiment under identical photocatalytic reaction conditions was performed to identify the carbon source origin of the produced HCOO−, and the products were identified by 13C NMR spectroscopy. As shown in Fig. S22–S24,† the 13C NMR spectrum gives very clear and strong signals corresponding to HCOO−, CO32−, and HCO3−, which are consistent with other previous important works.15,16,21 Also, these signals disappeared when using 12CO2 instead of 13CO2, except for the additional peaks corresponding to DMSO and TEOA. This fact unambiguously establishes that the produced HCOO− indeed originates from CO2. Therefore, Ti4-C8A and Ti7-C8A are indeed active for reducing CO2 to HCOO− under UV and visible-light irradiation.
Additionally, we further conducted electron spin-resonance spectroscopy (ESR) measurement to study the reaction mechanism behind CO2 photoreduction over Ti4-C8A and Ti7-C8A. As shown in Fig. S25,† the reaction systems including Ti4-C8A/Ti7-C8A and TEOA under a N2 atmosphere without light irradiation did not show any ESR signal. When the reaction systems were irradiated with a light source under identical conditions, a clear signal of Ti3+ (g = 1.948) could be observed from their ESR spectra. This means that the Ti4+ ions within the Ti4-C8A and Ti7-C8A clusters were reduced into Ti3+ ions by receiving photoexcited electrons transferred from the C8A ligand, while the corresponding photo-generated holes were quenched by TEOA. Moreover, the intensity of the Ti3+ signal increased with the extended irradiation time. When the reaction system was exposed to a CO2 atmosphere, the ESR signal of Ti3+ disappeared, indicating that the photogenerated Ti3+ is indeed involved in the CO2 reduction reaction. Besides, a sharp and narrow ESR signal (g = 2.002) of light-induced radical formation of the ligand also can be observed. The ESR results revealed that the Ti4+ ions within Ti4-C8A and Ti7-C8A were the photocatalytically active sites for the CO2-to-HCOOH reduction (Fig. S25†). Based on the above-mentioned experimental results and a deep understanding of the previously reported PTC-based photocatalysts (including Ti-MOFs and PTC clusters in our group) used for photocatalytic CO2RR,15,23 a proposed reaction mechanism for CO2 reduction is illustrated as shown in Fig. 4. Upon UV/visible-light irradiation, photo-excited electron–hole pairs are generated in the titanium–oxo cluster. Then, the Ti4+ ions within the cluster accept photo-generated electrons transferred from the C8A ligand and turn into Ti3+ ions, whereas the TEOA molecules in the reaction system act as electron donors to consume the produced photo-generated holes. Subsequently, the generated Ti3+ ions offer photo-excited electrons to the absorbed CO2 molecules for activation, and then go back to the Ti4+ ions. In this way, a complete CO2-to-HCOO− photosynthesis cycle can be achieved by Ti3+–Ti4+ intervalence charge transfer in Ti4-C8A and Ti7-C8A, in the presence of TEOA (as an electron and proton donor) and H2O.
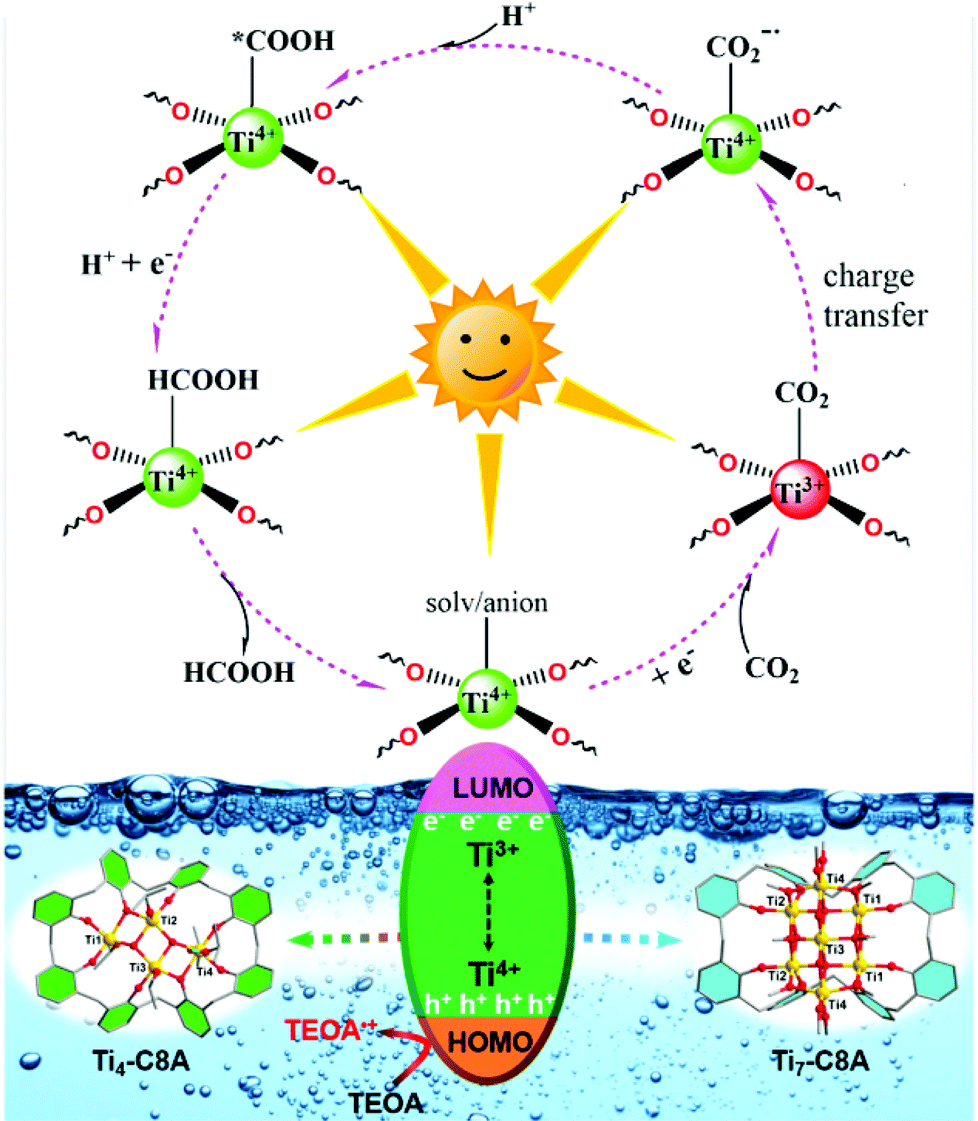 | ||
| Fig. 4 The proposed photocatalytic reaction mechanism for Ti4-C8A and Ti7-C8A. | ||
Conclusions
In conclusion, two water-stable and photosensitive PTCs were constructed by using the “functional armor” C8A with high molecular degrees of freedom and variable conformations, and they can be converted thermodynamically into each other under different solvothermal conditions. Thanks to the strong charge transfer generated from the intimate coordination effect between the titanium–oxo core and the C8A ligand, the light absorption range of these two PTCs is effectively extended from the traditional ultraviolet to the visible region. Moreover, the overall titanium–oxo core was surrounded by the hydrophobic benzene ring of C8A, resulting in high structural and chemical stability of the two PTCs in aqueous solutions. Based on these advantages, Ti4-C8A and Ti7-C8A were treated as heterogeneous photocatalysts to carry out CO2RR in H2O containing TEOA, and they finally exhibited very high photocatalytic activity and selectivity for CO2-to-HCOO− conversion. Significantly, this is the first time that a coordination molecular complex system performed heterogeneous photocatalytic CO2RR in H2O in the presence of TEOA. This work not only expanded the photocatalytic application of traditional PTCs, but also provided more insights into the design and preparation of heterogeneous molecular photocatalysts to achieve efficient CO2RR in H2O as the reaction solvent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation (No. 21622104, 21701085, and 20171169), Six Talent Peaks Project in Jiangsu Province (No. 2017-XNY-043), the projects funded by the High-Level Personnel Support Program of Yang-Zhou University, the Priority Academic Program Development of Jiangsu Higher Education Institutions, and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. XKYCX18_041).Notes and references
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- Q. Shi, J. Wu and S. Mu, J. Electroanal. Chem., 2018, 820, 1–8 CrossRef CAS.
- N.-T. Suen, Z.-R. Kong, C.-S. Hsu, H.-C. Chen, C.-W. Tung, Y.-R. Lu, C.-L. Dong, C.-C. Shen, J.-C. Chung and H. M. Chen, ACS Catal., 2019, 9, 5217–5222 CrossRef CAS.
- L. Rui, Z. Wang and Z. Kun, Adv. Mater., 2018, 30, e1705512 CrossRef.
- Y. Chen, D. Wang, X. Deng and Z. Li, Catal. Sci. Technol., 2017, 7, 4893–4904 RSC.
- H. Zhang, J. Nai, L. Yu and X. W. Lou, Joule, 2017, 1, 77–107 CrossRef CAS.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, Y.-S. Zhu, L.-Y. Liu, Y.-Y. Sun, X. Bu, J. Zhang and Q. Lin, Adv. Mater., 2018, 30, 1704388 CrossRef PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- N. Li, J. Liu, J. J. Liu, L. Z. Dong, Z. F. Xin, Y. L. Teng and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 5226–5231 CrossRef CAS PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- D. Chen, H. Xing, C. Wang and Z. Su, J. Mater. Chem. A, 2016, 4, 2657–2662 RSC.
- Y. Wang, N.-Y. Huang, J.-Q. Shen, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- Z.-H. Yan, M.-H. Du, J. Liu, S. Jin, C. Wang, G.-L. Zhuang, X.-J. Kong, L.-S. Long and L.-S. Zheng, Nat. Commun., 2018, 9, 3353 CrossRef PubMed.
- N. Li, J. Liu, J. J. Liu, L. Z. Dong, S. L. Li, B. X. Dong, Y. H. Kan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 17260–17264 CrossRef CAS PubMed.
- H. Zhang, Q. L. Hong, J. Li, F. Wang, X. Huang, S. Chen, W. Tu, D. Yu, R. Xu, T. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11752–11756 CrossRef CAS PubMed.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- J. Zhao, Q. Wang, C. Sun, T. Zheng, L. Yan, M. Li, K. Shao, X. Wang and Z. Su, J. Mater. Chem. A, 2017, 5, 12498–12505 RSC.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- Y. Yang, F. Li, J. Chen, J. Fan and Q. Xiang, ChemSusChem, 2020, 13, 1979–1985 CrossRef CAS PubMed.
- C. Dai, L. Zhong, X. Gong, L. Zeng, C. Xue, S. Li and B. Liu, Green Chem., 2019, 21, 6606–6610 RSC.
- L. Z. Dong, L. Zhang, J. Liu, Q. Huang, M. Lu, W. X. Ji and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 2659–2663 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2018, 47, 404–421 RSC.
- J.-X. Liu, M.-Y. Gao, W.-H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5160–5165 CrossRef CAS PubMed.
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. J. Teat, S. J. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC.
- W.-H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed.
- M.-Y. Gao, F. Wang, Z.-G. Gu, D.-X. Zhang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2556–2559 CrossRef CAS PubMed.
- G. Zhang, C. Liu, D.-L. Long, L. Cronin, C.-H. Tung and Y. Wang, J. Am. Chem. Soc., 2016, 138, 11097–11100 CrossRef CAS PubMed.
- Y. P. He, L. B. Yuan, G. H. Chen, Q. P. Lin, F. Wang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2017, 139, 16845–16851 CrossRef CAS PubMed.
- J. D. Ryan, K. J. Gagnon, S. J. Teat and R. D. McIntosh, Chem. Commun., 2016, 52, 9071–9073 RSC.
- Y. Lee, S. Kim, H. Fei, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 16549–16552 RSC.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- F. M. Wisser, M. Duguet, Q. Perrinet, A. C. Ghosh, M. Alves-Favaro, Y. Mohr, C. Lorentz, E. A. Quadrelli, R. Palkovits, D. Farrusseng, C. Mellot-Draznieks, V. de Waele and J. Canivet, Angew. Chem., Int. Ed., 2020, 59, 5116–5122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1999136 and 1999137. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc01497d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |