
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electronic structure, surface properties, and in situ N2O decomposition of mechanochemically synthesised LaMnO3†
Rachel H.
Blackmore
 ab,
Maria Elena
Rivas
c,
George F.
Tierney
ab,
Khaled M. H.
Mohammed
bd,
Donato
Decarolis
ae,
Shusaku
Hayama
f,
Federica
Venturini
f,
Georg
Held
f,
Rosa
Arrigo
fg,
Monica
Amboage
f,
Pip
Hellier
ae,
Evan
Lynch
ab,
Mahrez
Amri
c,
Marianna
Casavola
b,
Tugce
Eralp Erden
c,
Paul
Collier
c and
Peter P.
Wells
*abf
ab,
Maria Elena
Rivas
c,
George F.
Tierney
ab,
Khaled M. H.
Mohammed
bd,
Donato
Decarolis
ae,
Shusaku
Hayama
f,
Federica
Venturini
f,
Georg
Held
f,
Rosa
Arrigo
fg,
Monica
Amboage
f,
Pip
Hellier
ae,
Evan
Lynch
ab,
Mahrez
Amri
c,
Marianna
Casavola
b,
Tugce
Eralp Erden
c,
Paul
Collier
c and
Peter P.
Wells
*abf
aUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratories, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0FA, UK
bSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: P.P.Wells@soton.ac.uk
cJohnson Matthey Technology Centre, Blounts Court Road, Sonning Common, Reading, RG4 9NH, UK
dDepartment of Chemistry, Faculty of Science, Sohag University, Sohag, P. O. Box 82524, Egypt
eSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
fDiamond Light Source Ltd., Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
gSchool of Science, Engineering and Environment, University of Salford, Manchester M5 4WT, UK
First published on 30th June 2020
Abstract
The use of mechanochemistry to prepare catalytic materials is of significant interest; it offers an environmentally beneficial, solvent-free, route and produces highly complex structures of mixed amorphous and crystalline phases. This study reports on the effect of milling atmosphere, either air or argon, on mechanochemically prepared LaMnO3 and the catalytic performance towards N2O decomposition (deN2O). In this work, high energy resolution fluorescence detection (HERFD), X-ray absorption near edge structure (XANES), X-ray emission, and X-ray photoelectron spectroscopy (XPS) have been used to probe the electronic structural properties of the mechanochemically prepared materials. Moreover, in situ studies using near ambient pressure (NAP)-XPS, to follow the materials during catalysis, and high pressure energy dispersive EXAFS studies, to mimic the preparation conditions, have also been performed. The studies show that there are clear differences between the air and argon milled samples, with the most pronounced changes observed using NAP-XPS. The XPS results find increased levels of active adsorbed oxygen species, linked to the presence of surface oxide vacancies, for the sample prepared in argon. Furthermore, the argon milled LaMnO3 shows improved catalytic activity towards deN2O at lower temperatures compared to the air milled and sol–gel synthesised LaMnO3. Assessing this improved catalytic behaviour during deN2O of argon milled LaMnO3 by in situ NAP-XPS suggests increased interaction of N2O at room temperature within the O 1s region. This study further demonstrates the complexity of mechanochemically prepared materials and through careful choice of characterisation methods how their properties can be understood.
1. Introduction
Developing catalytic processes that are both economically viable and sustainable represents a significant challenge. To meet this ambition, the development of new technologies can no longer rely on iterative trial and error approaches and must instead be design-led using sophisticated characterisation methods.1 Synchrotron techniques, such as X-ray absorption spectroscopy (XAS), have multiple benefits, which aid the in-depth understanding of chemically important yet complex systems.2–4Previously, we have reported the advantages of XAS for studying the preparation of metal oxides prepared through mechanochemistry; this solvent-free synthesis route produces less waste than traditional methods and prepares materials with enhanced catalytic properties.5 Our recent study focussed on the mechanochemical synthesis of LaMnO3 from its single oxide precursors. During this process there was a significant amount of amorphous material produced at different stages of milling. Traditional characterisation methods, such as X-ray diffraction (XRD), provided an incomplete understanding of the underlying chemistry. Using XAS, which provides structural insights on all length scales, allowed us to learn more about the principal chemical steps within the milling process. Furthermore, these studies also increased the understanding of how these materials promote the catalytic decomposition of the environmental pollutant N2O (deN2O).
However, even after performing conventional XAS at the Mn K-edge information such as the local charge density and electronic configuration still eluded us. In this work we discuss the use of further advanced characterisation techniques, such as high energy resolution fluorescence detection (HERFD) to improve the XANES spectral resolution,6,7 and X-ray emission spectroscopy (XES) to attain information on electronic structure and oxidation state.8,9 Furthermore, we endeavoured to mimic the environmental conditions experienced during planetary ball milling by performing an in situ XAS study at high pressure in a diamond anvil cell (DAC). This was performed in conjunction with in situ vibrational ball milling by XRD. These new structural insights have then been coupled with in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) to further understand how ball milled materials have improved catalytic activity at lower temperatures during deN2O.
By combining these local structural and electronic techniques, along with surface sensitive characterisation, it can be a powerful tool for understanding how structural properties, induced by the syntheitic route, can affect catalysis. Herein, we report an in-depth analysis during the mechanochemically synthesised LaMnO3 by XES and HERFD, not previously reported. Furthermore, we can now link these structural properties of the final material to the enhancement of catalytic activity for the in situ deN2O combined with NAP-XPS.
2. Experimental
a. LaMnO3 sample preparation
High energy planetary ball milling was performed at room temperature in a 4-station Fritsch Pulverisette 5 Planetary Ball Mill. ZrO2 vessels were prepared with precursors Mn2O3 and La2O3 at the correct proportions to synthesise stoichiometric LaMnO3, along with 5 mm ZrO2 spheres at a sphere![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :powder ratio of 10:1. Milling was then conducted up to 4 hours at 400 rpm in sessions of 20 min, in both atmospheric and inert environments. For the sol–gel synthesis stoichiometric amounts of Mn(NO3)2·4H2O, and La(NO3)3·6H2O were dissolved in deionised water and added to a gel prepared by mixing equimolecular amounts of citric acid (99.5%) and ethylene glycol (99.5%) as a polydentate ligand. The excess water was slowly removed on a hot plate until a viscous liquid was obtained. Subsequently, the final slurry was slowly heated in air (1 °C min−1) from room temperature up to 700 °C and kept at this temperature for 4 h. These conditions are essential to obtain a crystalline material.
:powder ratio of 10:1. Milling was then conducted up to 4 hours at 400 rpm in sessions of 20 min, in both atmospheric and inert environments. For the sol–gel synthesis stoichiometric amounts of Mn(NO3)2·4H2O, and La(NO3)3·6H2O were dissolved in deionised water and added to a gel prepared by mixing equimolecular amounts of citric acid (99.5%) and ethylene glycol (99.5%) as a polydentate ligand. The excess water was slowly removed on a hot plate until a viscous liquid was obtained. Subsequently, the final slurry was slowly heated in air (1 °C min−1) from room temperature up to 700 °C and kept at this temperature for 4 h. These conditions are essential to obtain a crystalline material.
b. Characterisation
XES/HERFD measurements were performed on the scanning branch of the I20 beamline at the Diamond Light Source, Didcot, UK. Measurements were taken using a Si(111) four-bounce monochromator using a high-resolution X-ray emission spectrometer.10 For this experiment, the spectrometer was equipped with three Ge(440) analysers to allow the Kβ spectra to be measured.10 Mn K-edge XES spectra were fitted using three Voigt functions which represent the Kβ1,3, Kβx and Kβ′ peaks.11 Prior to the fitting, spectra were normalized, and linear background was subtracted. During the fitting, peak splittings were kept constant at theoretical values;12 (i) splitting between Kβ1,3 and Kβ′ peaks ≈ 14–16 eV and (ii) Kβx was lower than main, Kβ1,3, peak by ∼3 eV. The same fitting procedures were applied on spectra taken for Mn standard materials for comparison purposes. HERFD were measured by scanning the incident energy and detecting the fluorescence intensity at the maximum of the Mn Kβ1,3 emission line with time resolution of ∼20 min per spectrum with 3 spectra collected. Data processing, background submission and normalisation was performed using Athena package. In situ XRD ball milling was performed at the ID15A beamline at the European Synchrotron Radiation Facility using a modified MM200 Retsch mill reported by Halasz et al.13,14 The set-up comprised of a PMMA milling jar containing one 5 mm WC milling media with Mn2O3 and La2O3 precursor powders. The mill operated at 30 Hz for ∼19 h. The in situ XRD was collected at 69 keV, λ = 0.17971 Å with a beam size of 300 × 300 μm2. A Dectris Pilatus X area detector was used with an exposure of 5s and a readout time of 120 s. The incident wavelength and detector distance (430 mm) were calibrated using a NIST CeO2. The Raw data frames were integrated using the internal ESRF Matlab.Eva 5.0.0.22/PDF-4+ 2020 software package was used for phase identification and data plotting. In situ high pressure EDE-XAFS were performed at I20-EDE, the energy dispersive beamline at the Diamond Light Source, Didcot, UK. Energy-dispersive EXAFS was performed at the Mn K-edge (6539 eV) in transmission mode using a Si(111) polychromator. A diamond anvil cell (DAC) was fitted with 0.5 mm thick diamonds in order to reduce the diamonds absorption and enable XAS measurements at the Mn K-edge. The single-crystal diamond mini-anvils were mounted on fully perforated diamonds. Pressure was measured by the ruby fluorescence method.15 The dispersive X-ray beam was focussed on the sample in the DAC with a spot size of 0.05 mm diameter. Diamond single crystal reflections were deglitched following a similar procedure to Hong et al.16 Spectra were recorded every 2 GPa of pressure. XRD was collected on a Bruker AXS D8 diffractometer with Cu Kα radiation over a range of 2θ = 10–130° with 0.044° step size (Johnson Matthey, Sonning Common, UK). Phase identification was performed using Bruker-AXS Diffrac Eva V4.2 with Rietveld refinement performed using Bruker-AXS Topas 4.2. XAS measurements were performed at the B18 Beamline, Diamond Light Source. XAS measurements at the Mn K-edge (6539 eV) and La L3-edge (5483 eV) were performed in transmission mode using QEXAFS setup with fast scanning Si(111) double crystal monochromator. An appropriate foil was placed between It and Iref and measured concurrently with XAS spectra acquisition. At the Mn K-edge XAS spectra was acquired with a time resolution of 20 min per spectrum (kmax = 14) averaged over 3 scans. At the La L3-edge XAS spectra were acquired with a time resolution of 5 min per spectrum (kmax = 10) averaged over 3 scans. Data processing was background submission and normalisation was performed using Athena and Artemis software package.17,18 XPS was carried out with a Thermo Escalab 250. The radiation used was monochromatised aluminium Kα radiation with a 650 μm spot size. Charge compensation was provided by the in-lens electron flood gun at a 2 eV setting and the “401” unit for “zero energy” argon ions. Sensitivity factors after Scofield used in quantification performed by Johnson Matthey, Sonning Common, UK.c. Catalytic activity (deN2O)
DeN2O was carried out in a fixed-bed quartz reactor (Hiden CATLAB) with the temperature range of 100–800 °C, using a ramp rate of 10 °C min−1. The reaction was performed at 30 mL min−1 flow of 0.5% N2O in He over 400 mg of catalyst with sieve fraction 125–250 μm to result in a GHSV = 18000 h−1. The exhaust gases composition was measured using a Hiden QGA mass spectrometer for He (m/z = 4), N2 (m/z = 28), O2 (m/z = 32), N2O(m/z = 46).
d. In situ NAP-XPS deN2O
The catalytic reaction was performed on 3 samples; mechanochemically prepared LaMnO3, milled under air and argon gas atmospheres for 4 h, and compared to sol–gel synthesised LaMnO3.NAP-XPS was performed at the beamline B07-C (VerSoX) at the Diamond Light Source. Spectra were recorded at the La 3d, Mn 2p, O 1s, C 1s and Mn 3s levels at 834.5–855 eV, 641–655 eV, 529–532 eV, 284–289 eV and 82–89 eV binding energies, respectively, during the deN2O. Samples were pressed into 8 mm pellets containing 50 mg of LaMnO3. The reaction was performed under 0.5% N2O in He, with scans performed at room temperature in vacuum, room temperature with 10 mbar of N2O and then subsequently at 400 °C and 600 °C (still under 10 mbar of N2O). The measurements were performed at specific incident energies for each XPS region to ensure the same kinetic energy/depth of the emitted electron (∼350 eV). To achieve a variable depth profile the Mn 3s region was recorded with an incident energy of 1200 eV. Each XPS region was aligned to a reference C 1s peak at 284.8 eV19 to compensate for charging effects. Peak positions and FWHM determined after using Shirley background subtraction on CasaXPS, with all residual standard deviations of the curve fitting reported to be under 1.
3. Results and discussion
3.1 Understanding the mechanochemical synthesis of LaMnO3
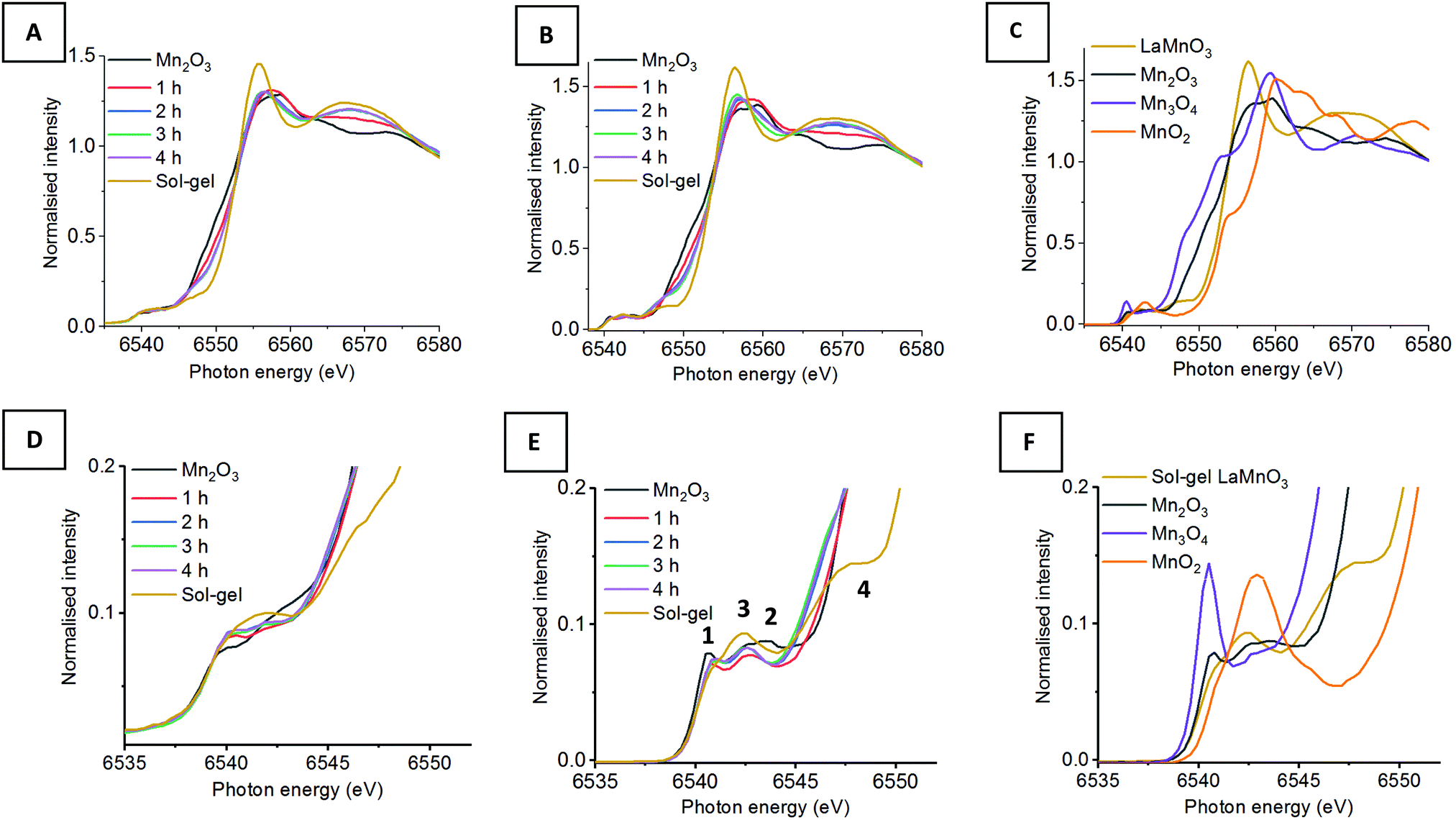 | ||
| Fig. 1 (A) XAS Mn K-edge XANES compared to (B) HERFD-XANES of time-slices during the mechanochemial synthesis if LaMnO3 compared to (C) reference Mn(II), (III), (IV) oxides and sol–gel synthesised LaMnO3 with (D–F) their respective highlighted pre-edge region. | ||
Fig. 1 shows the XANES and HERFD-XANES data, with expanded pre-edge regions for the milled materials and the Mn oxide reference compounds. Assessing the Mn K-edge HERFD-XANES, the level of spectral resolution has vastly improved compared to the conventional XANES data. This increased resolution is more clearly pronounced in the pre-edge region, which shows more defined transitions that are well separated from the main edge. The HERFD-XANES spectra of the reference Mn oxides (Mn2O3, Mn3O4, MnO2 and LaMnO3) and their interpretation allow for an increased understanding of the ball milled intermediates and final material.
For the Kβ1,3 HERFD-XANES data the main edge results from an allowed 1s → 4p transition. The position of this main edge transition is dependent on the oxidation state of the system; an increased formal charge on the Mn results in the transition shifting to higher energy. In the pre-edge region, there are multiple transitions that give rise to the complex structure observed. The 1s → 3d transitions, that are normally forbidden by dipole selection rules, in this region can be quadrupolar or dipolar in nature, dependent on the local mixing of 3d/4p wavefunctions and the presence of non-local excitations; these occur with 3d states of neighbouring metal sites, through an oxygen-mediated intersite hybridisation, e.g. Mn(4p)–O(2p)–Mn′(3d).21 The size and shape of the pre-edge peaks is therefore highly dependent on the geometry of central absorbing atom.7
Assessing the pre-edge region for Mn2O3 two clear peaks can be observed at 6540.8 eV and 6543.0 eV. These different features arise as a consequence of the structure of Mn2O3 that contains two different Mn(III) coordination sites. Both sites are 6 coordinate, with respect to oxygen, however, one site adopts a more regular centrosymmetric Oh geometry, whereas the other displays a large degree of Jahn–Teller distortion. The work by Farges showed that the lower energy peak can be assigned to the centrosymmetric Mn site and the higher energy peak to the Jahn–Teller distorted Mn site.22 This can further be applied to the pre-edge features for the mixed valent Mn3O4, which contains both Mn(II) and Mn(III). The intense peak at 6540.5 eV results from the tetrahedrally coordinated Mn(II), allowing for 3d–4p local hybridisation, resulting in an intense dipole transition. A low intensity peak is also observed at 6542.6 eV, originating from Mn(III). It should be noted that the intensity of these two peaks, at 6540.5 eV and 6542.6 eV, do not relate to the proportion of Mn(II) and Mn(III). Work by Radu et al. shows the Mn(II) peak as a single feature, with the Mn(III) feature extending below the Mn(II) peak due to the multiplet effect of the final state (1s13d5).23 For MnO2, the Mn(IV) cation is positioned in an octahedral site, however, the pre-edge for Mn(IV) is extremely complex and even HERFD-XANES struggles to resolve the features.22 Previous work shows that the broad feature observed at 6542.9 eV is a combination of 1s → 3d quadrupole transitions and non-local excitations.22
LaMnO3, synthesised here by the Pechini method,24 has two peaks in the pre-edge region observed at 6542.3 eV and (a shoulder at) 6547.7 eV. The peak at 6542.3 eV has been demonstrated to be two overlapping peaks from both a 1s→ 3d quadrupole transition and a 1s → 3d4p non-local transition. This non-local transition occurs from the 3d states of neighbouring metal sites, Mn(4p)–O(2p)–Mn′(3d), through the vertex linked octahedral units.25 The shoulder observed at 6547.8 eV is often overlooked within the Mn K-edge XANES of LaMnO3. However, work by Ignatov et al. showed they could not effectively model this shoulder within LaMnO3 XANES without considering the contribution of d and f orbitals, suggesting this shoulder arises due to Mn 4p and La 5d hybridisation.26
Using this analysis of reference Mn oxides we can now effectively assess the changes within the pre-edge region of Mn during the evolution of LaMnO3 by mechanochemical synthesis. After 1 h of milling there are already clear differences in the HERFD-XANES spectrum compared to Mn2O3. The key differences are a decrease in intensity of the transitions at (1) 6540.8 and (2) 6543.0 eV. These changes provide important information about the chemical steps occurring during the milling process. The decrease in intensity of the transition at (2) indicates that the extended crystallites of Mn2O3 have been significantly disrupted; the transition is a non-local excitation to a neighbouring site and its reduced intensity is clear evidence for less extensive Mn–O–Mn interactions. The decrease in the transition at (1) indicates that there has been alterations to the local geometry of Mn sites. On increasing milling time to 2 h, there is then an increase in intensity of the transitions associated with LaMnO3 at (3) 6542.3 eV, indicating that there are changes to the local structure indicative of the formation of a perovskite unit. There are no further discernible changes in the spectra for milling times of 2–4 h. Comparing the final ball milled, LaMnO3, to the sol–gel reference, clear differences are observed, most noticeably lower intensity transitions at (3) and (4) at 6547.7 eV.
The peak at (3) is assigned to a 1s → 3d4p non-local hybridised transition to neighbouring Mn sites, the strength of this transition is dictated by the metal–oxygen bond length and the metal–oxygen–metal bond angle.27 The strongest transition, and ideal hybridisation, is achieved by a short bond length and a linear bond angle.25 In our previous study, the Mn–O bond lengths calculated by EXAFS for both the sol–gel and mechanochemically prepared LaMnO3 have comparable distances within the error range of the technique. Moreover, the final ball-milled LaMnO3 is a complex mixture of both amorphous and crystalline content, consistent with multiple metal–oxygen–metal bond angles. The crystalline LaMnO3 has a more linear metal–oxygen–metal bond angle and it has a higher intensity peak for this transition at (3). Furthermore, the absence of the shoulder at (4) for the ball milled materials suggests the lack of Mn4p La5d hybridisation.26 In our previous work, we reported that the EXAFS fitting model for the ball milled material required two Mn–La scattering paths at 3.24(1) Å and 3.37(1) Å, which is significantly different to that observed for the crystalline sol–gel sample. These changes in Mn–La coordination have disrupted the Mn 4p La 5d hybridisation and affect the transition at (4).
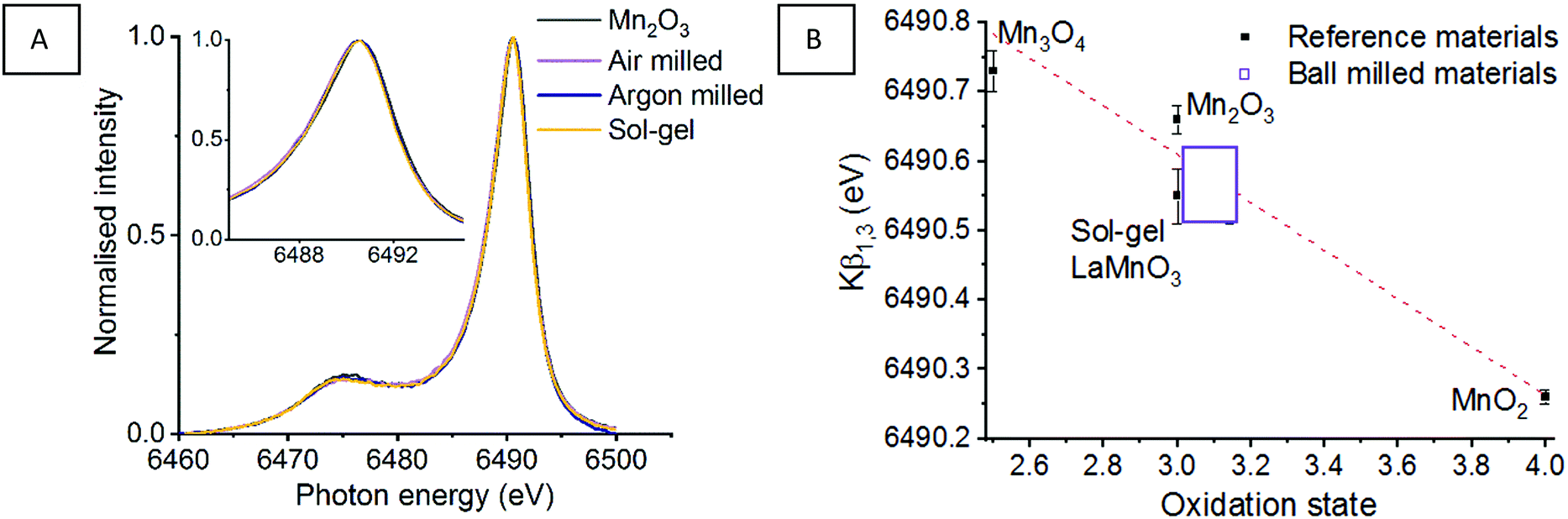
Fig. 2 shows the Mn Kβ mainlines, consisting of a strong Mn Kβ1,3 peak and with a Mn Kβ′ satellite at a lower energy, for the 3p → 1s transition of reference Mn oxides and a sol–gel synthesised LaMnO3. The splitting of these peaks originate from the strong coupling interaction between the 3p hole and the total spin of the 3d electrons.28,29 Any energy shifts of the Mn Kβ1,3 peak is expected to result from a combination of effective nuclear charge (Zeff) and spin state. Work by Beckwith et al. showed that for Mn complexes the Zeff and 3d spin effects oppose one another.30 Therefore, an increase in oxidation state corresponds to an decrease in total spin, which tends to shift the Mn Kβ1,3 to a lower energy.21
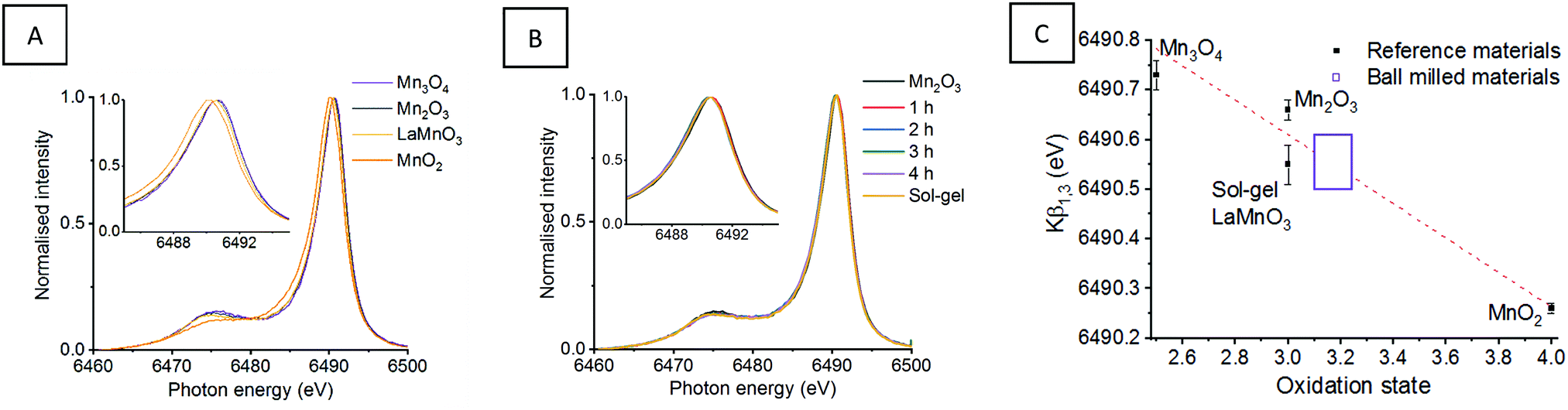 | ||
| Fig. 2 Mn Kβ XES mainlines for (A) Mn reference oxides and sol–gel synthesised LaMnO3 with (B) ‘time-slices’ during the mechanochemical synthesis of LaMnO3 (C) presents the Kβ1,3 max as a function of oxidation state, with a line of best fit calculated from the reference materials and used to calculate the oxidation state of the ball milled materials.11 | ||
This is strongly reflected in the Mn reference materials reported here, which have fitted Mn Kβ1,3 peaks for MnO2, Mn2O3 and M3O4 species at 6490.26, 6490.66 and 6490.73 eV, respectively (Table 1) (Fig. S1, ESI†). When considering the reference sol–gel synthesised LaMnO3, the fitted Mn Kβ1,3 peak position is reported at 6490.55 eV. This indicates that the Mn(III) oxidation state shows a small variation in the Mn Kβ1,3 peak position.
| Compound | Kβ1,3 max (eV) | Oxidation state |
|---|---|---|
| MnO2 | 6490.26 | IV |
| Mn2O3 | 6490.66 | III |
| Mn3O4 | 6490.73 | II/III |
| Sol–gel LaMnO3 | 6490.55 | III |
| 1 h of milling | 6490.57 | 3.0–3.3 |
| 2 h of milling | 6490.53 | 3.0–3.3 |
| 3 h of milling | 6490.54 | 3.0–3.3 |
| 4 h of milling | 6490.56 | 3.0–3.3 |
Assessing the position of the fitted Mn Kβ1,3 peak for reference Mn oxide materials as a function of oxidation state allows us to extract oxidation state information (Fig. 2C). The positions of the Mn Kβ1,3 emission lines suggests that the milled materials have an average oxidation state higher than the Mn(III) oxidation state expected. This is consistent with our previous XPS analysis which identified Mn(IV) at the surface of the mechanically prepared LaMnO3. However, there is an associated error in determining the positions of deconvoluted peaks. When this is taking into consideration, alongside the variance Mn(III) reference materials (Mn2O3 and sol–gel LaMnO3), all that can be reliably confirmed is that the net oxidation state for all milled samples is very similar to that of the sol–gel prepared LaMnO3.
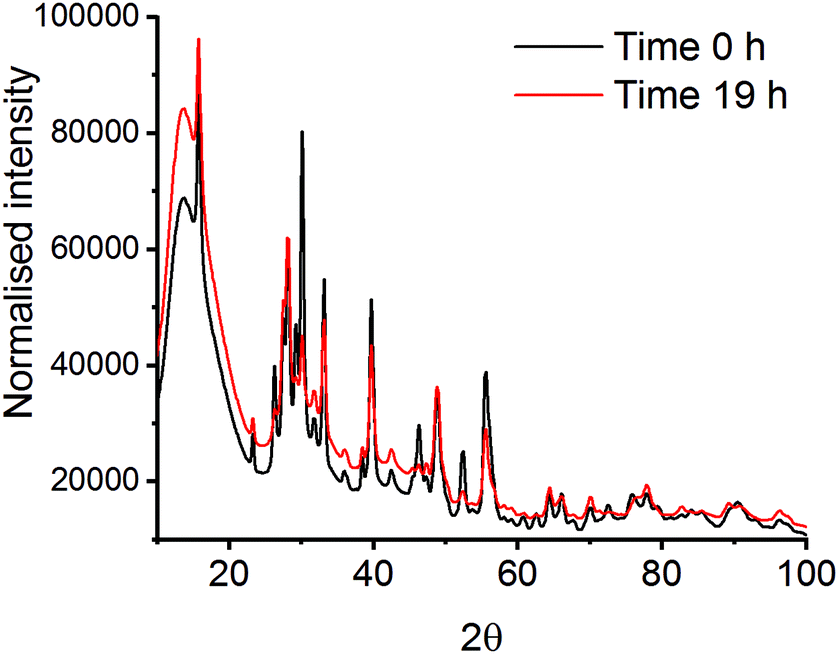 | ||
| Fig. 3 XRD patterns at time 0 h and time 19 h after milling La2O3 and Mn2O3 with a PMMA vibrational mill. | ||
Mechanochemistry results in extremely localised high temperature and pressure spots on the powdered materials due to collisions between milling media and the milling jar.31,32 High thermal synthesise (1300–1500 K), known as the ceramic method, is commonly known as a traditional preparation method for perovskites and are already well documented in literature.33,34 Though high pressures have been applied to already synthesised mixed metal oxides, there is limited information on using pressure alone to synthesise the LaMnO3 phase.35,36 In order to mimic the forces transferred to the powdered materials through the mechanochemical action of the mill, in situ high pressure studies using a diamond anvil cell (DAC) have been monitored by energy-dispersive-EXAFS (EDE) at the Mn K-edge. Systematic studies were performed on homogeneous mixtures of La2O3 and Mn2O3 up to pressures of 30 GPa at room temperature. Even at the highest pressure achieved with the perforated DAC, on mixtures that had been ball milled for 30 min, no significant changes in the EDE spectra were observed (Fig. 4). This indicates that either higher pressures or a combination of increased pressure and temperature are needed to mimic the conditions that drive mechanochemical transformations. Ultimately, this information combined with the results from the in situ vibrational mill, confirms that the mechanochemical synthesis of these metal oxide systems are extremely complicated to monitor in situ.
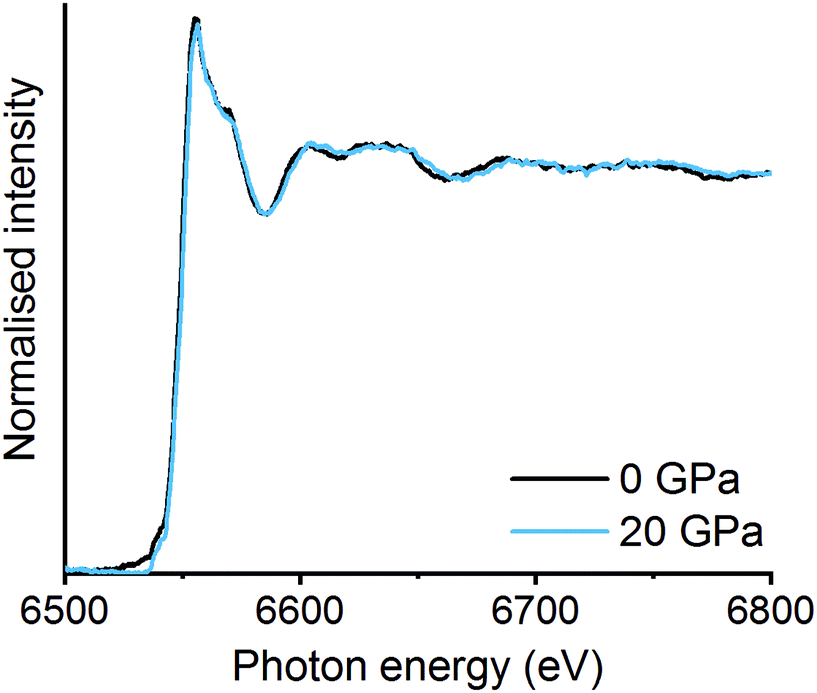 | ||
| Fig. 4 In situ high pressure EDE spectra at 0 GPa and 20 GPa on La2O3 and Mn2O3 mixture after high energy ball milling for 30 min. | ||
3.2 Comparison of milling environment
The La oxide precursor is significantly hydroscopic and readily forms a hydroxide on exposure to air. To understand the effect this change of precursor has on the final milled material, we have conducted the milling procedure in an inert atmosphere of argon.37 The mechanochemical production of LaMnO3 under argon was performed for 4 h, with the final material then subsequently exposed to air.The XRD data was analysed using Rietveld refinement38 to confirm the fraction of crystalline phases in the final milled material (Fig. 5). The data confirmed that for the mechanochemically prepared LaMnO3 under inert conditions a milling time of 4 h was not sufficient to achieve complete conversion to perovskite LaMnO3, with 77% crystalline perovskite calculated (100% was achieved when milling in air). The residual 23% comprises unreacted crystalline precursor phases. Here, the excess moisture/oxygen present in the milled material prepared in air is clearly beneficial in forming a crystalline final product.
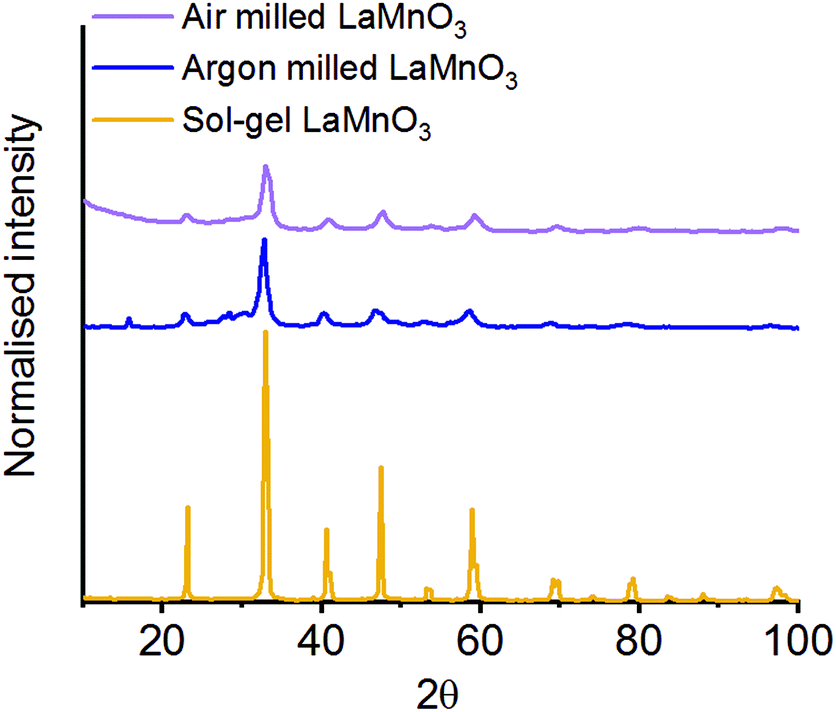 | ||
| Fig. 5 XRD patterns LaMnO3 mechanochemically synthesised 4 h under atmospheric and inert (argon) conditions compared to the sol–gel synthesised perovskite. | ||
To assess the crystalline and amorphous content of the argon milled LaMnO3 EXAFS analysis was also performed (Fig. 6) (Table S1, ESI†). The argon milled LaMnO3 EXAFS data is well modelled, using a single Mn–O and two Mn–La scattering paths. The two Mn–La scattering paths were calculated to be 3.13 Å and 3.30 Å, similar to that found for the air milled LaMnO3; the shorter and longer distance La–Mn scattering paths arise due to amorphous and crystalline content, respectively. In general the EXAFS data of the air and argon milled samples are similar, however, there are differences in the Fourier transform resulting from changes in the amorphous/crystalline LaMn phases present. This model also exhibits a degree of oxygen deficiency, as with the air milled sample. To further investigate this, HERFD-XANES and Mn Kβ XES studies have been performed.
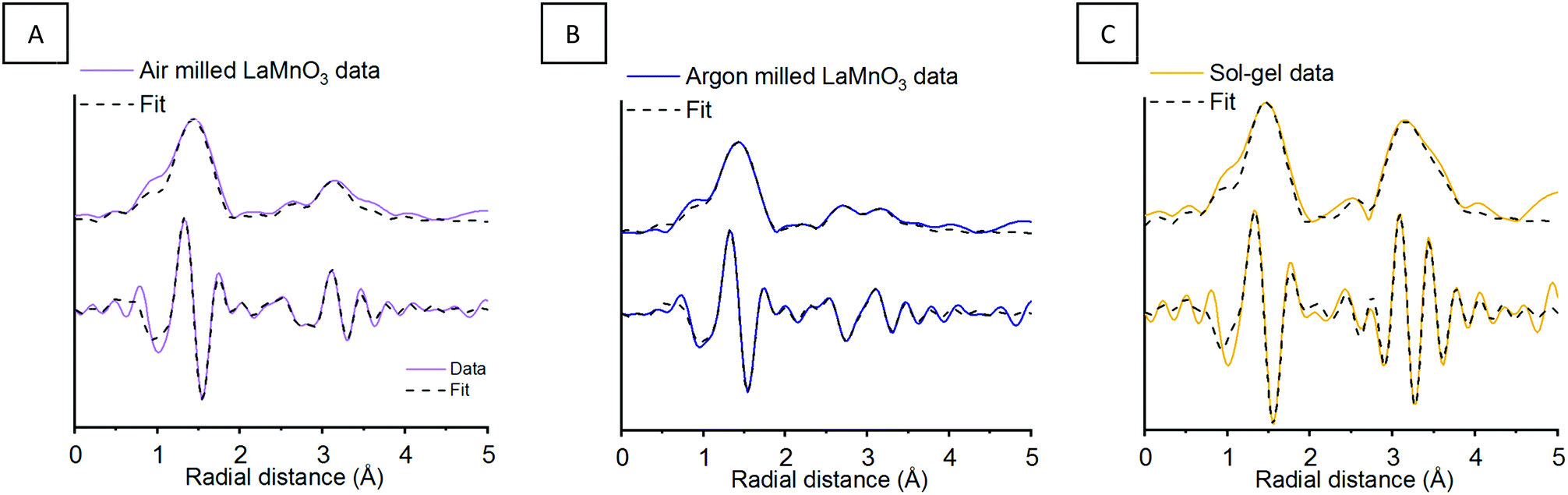 | ||
| Fig. 6 Mn K-edge EXAFS data after 4 h of milling in (A) atmospheric and (B) inert milling conditions showing the magnitude and imaginary components of the k2-weighted FT data and fits compared to (C) sol–gel synthesised LaMnO3. | ||
As seen in Section 1.1, broad unresolved features are observed within the traditional XANES, with HERFD-XANES showing a much greater resolution (Fig. 1). Fig. 7 shows the XANES and HERFD-XANES data, with expanded pre-edge regions for LaMnO3 catalysts synthesised by mechanochemistry (in air and argon) and sol–gel, compared to the Mn2O3 precursor. The argon milled sample clearly shows an unshifted peak (1) at 6540.8 eV, which arises as a consequence of unreacted Mn2O3. The absence of peak (2) and the presence of peak (3) at 6542.6 eV suggest the formation of perovskite-like structures; peak (3) resulting from a transition to the non-local neighbouring Mn sites within perovskites. The decrease in intensity of peak (3) is indicative of a greater proportion of structural disorder. Furthermore, the argon milled material lacks the shoulder (4) at 6547.8 eV, suggesting an absence of Mn 4p La 5d hybridisation.26 In the EXAFS model the argon milled LaMnO3 lacks the longer scattering path of 3.70 Å present in the sol–gel prepared material (Table S1, ESI†). This further suggests that the changes in the Mn–La coordination have disrupted the Mn 4p La 5d hybridisation and have affected the transition at 6547.8 eV.
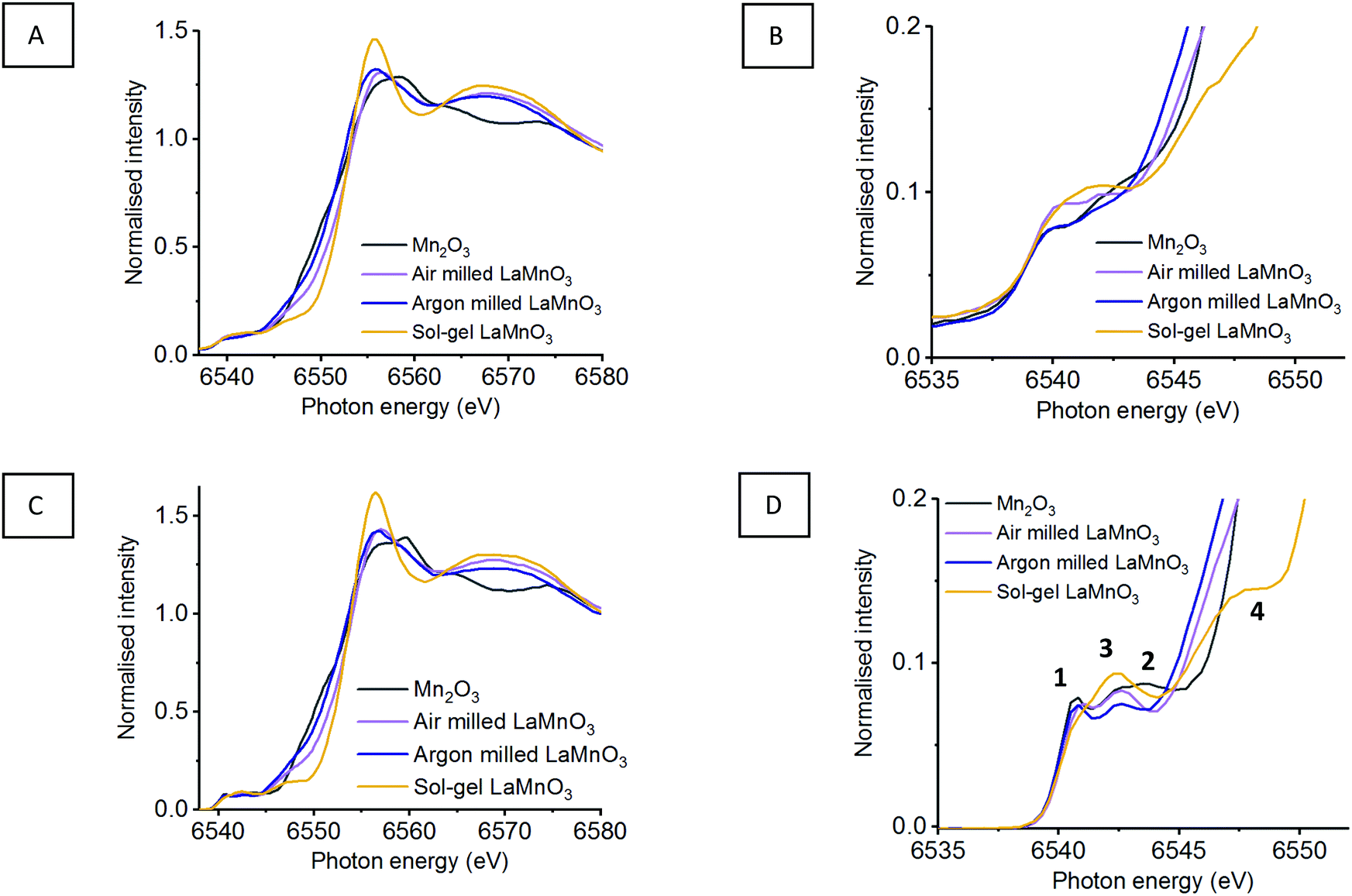 | ||
| Fig. 7 (A) Mn K-edge XANES with (B) highlighted pre-edge region compared to (C) HERFD-XANES with (D) highlighted pre-edge region of mechanochemial synthesis if LaMnO3 within air and argon environments compared to sol–gel LaMnO3 and precursor Mn2O3. | ||
The observed shift in the main edge for the argon catalyst could suggest a reduction in Mn charge. However, the edge position at the Mn K-edge is not a suitable measure for oxidation state as geometry and symmetry effects also dominate the XANES and therefore Kβ1,3 emission spectroscopy has been performed in order to achieve more reliable information.
Using the fitting analysis stated in Section 3.1.2, the Mn Kβ mainlines have now been assessed for the argon milled LaMnO3, compared to the air milled and sol–gel prepared perovskite (Fig. 8) (Fig. S3, ESI†). Both air and argon milled LaMnO3 have fitted Mn Kβ1,3 peak positions within error of one another, which is consistent with the sol–gel LaMnO3, as stated earlier.
 | ||
| Fig. 8 (A) Mn Kβ XES mainlines of time-slices through the mechanochemical synthesis of LaMnO3 from precursors La2O3 and Mn2O3 along with (B) which presents the Kβ1,3 max as a function of oxidation state, with a line of best fit calculated from the reference materials and used to calculate the oxidation state of the ball milled materials.11 | ||
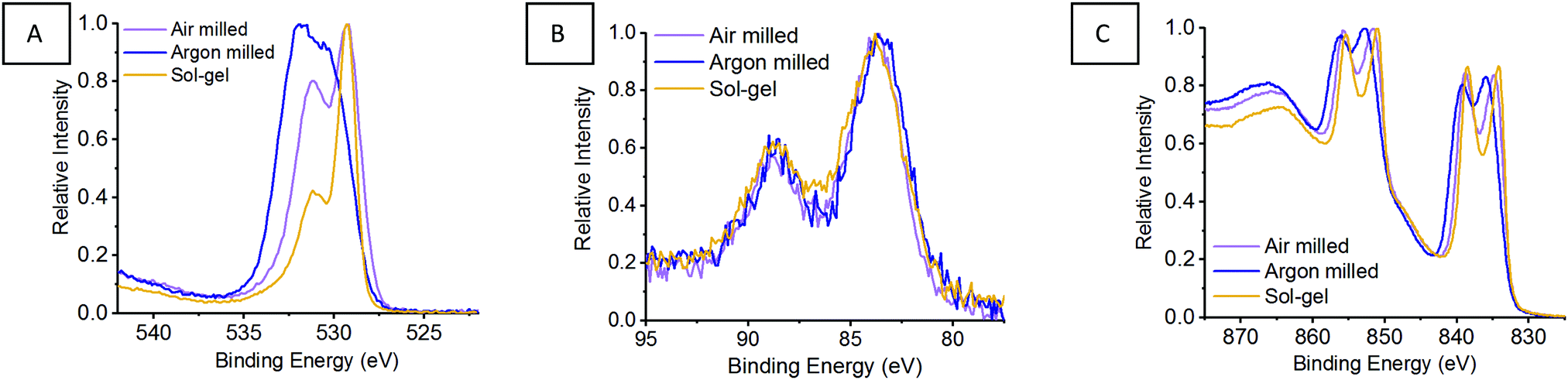
Previously, we reported the importance of assessing the surface of the LaMnO3 catalysts to identify possible active sites for catalysis by X-ray photoelectron spectroscopy (XPS).5 The atomic percent ratios of La, Mn and O species at the surface of LaMnO3 show the argon milled to have higher ratio of La to Mn on the surface compared to the air milled and sol–gel synthesised samples (Table S2, ESI†). Fig. 9A shows the O 1s XPS signals of mechanochemically and sol–gel synthesised LaMnO3. The argon milled sample follows a vastly different spectral profile compared to that of both the air milled and sol–gel synthesised LaMnO3. Previously, we reported using curve fitting to assess the change in adsorbed species on the catalyst surface.5
 | ||
| Fig. 9 XPS in the (A) O 1s, (B) Mn 3s and (C) La 3d region for LaMnO3 synthesised by sol–gel and mechanochemistry with air and argon environments. | ||
Using curve fitting here, three features were deconvoluted within the O 1s XPS region at 529.3–529.6 eV, 531.1–531.7 eV and 533.4–535.0 eV. The lowest energy binding peak at 529.3–529.6 eV is assigned to lattice-type oxygens in the perovskite (O22−), with the peak at 531.1–531.7 eV assigned to adsorbed species, such as O2−, O− or OH−.39–41 The peak at ∼533.5 eV corresponds to adventitious carbon, with analogous C–O peaks confirmed within the C 1s XPS region at 286.2 eV and 288.6 eV.39–41
Commonly it is assumed that active oxygen species adsorb on the surface to compensate for lattice oxygen vacancies.41 Firstly, the Brunauer–Emmett–Teller (BET) surface areas were calculated to be within range of one another; ∼5 m2 g−1 for the ball milled samples and ∼8 m2 g−1 for the sol–gel LaMnO3. Therefore, comparing the adsorbed oxygen to lattice oxygen can give an indication of the changes to adsorption centres, independent of surface area.42–44 The argon milled sample shows a greater intensity for the higher energy binding peak, indicating more adsorbed species on the surface compared to the other LaMnO3 samples. The relative intensity of the adsorbed oxygen feature decreases in the order; argon milled >4 h milled ≈ sol–gel preparation.
The XPS Mn 3s region spectra (Fig. 9B) indicate a mixed Mn oxidation state with peak splitting at 4.8–5.0 eV suggesting the presence of Mn(IV) and Mn(III) species (Table S3, ESI†). Analysis in the La 3d region (Fig. 9C) indicate a 3+ oxidation state for all samples, however, the argon milled LaMnO3 presents a different multiplet splitting (Table S3, ESI†). A reduced multiplet splitting of 3.7 eV is indicative of La(OH)3 species, consistent with the higher surface ratio of La (Table S2, ESI†), and the higher percentage of adsorbed species, –OH, within the O 1s region. This XPS analysis signifies the importance of surface sensitive techniques, as well as bulk structure analysis, XAS and XES, in order to understand how different synthetic routes effect the structural and catalytic properties of these LaMnO3 catalysts.
3.3 Catalytic testing
N2O decomposition (deN2O) has been performed on the differently synthesised LaMnO3 catalysts, with the light-off temperature curves detailing the percentage conversion of N2O to N2 (Fig. 10). Good reproducibility of this performance is shown in Fig. S4 (ESI†). Air milled and the sol–gel synthesised LaMnO3 show similar activity, which begins ∼350 °C with 100% conversion achieved by 550 °C. The argon milled LaMnO3, however, shows activity starting below 300 °C. This indicates that the catalytic activity cannot be explained by analysis of the bulk structures alone, as both ball milled catalysts show similarities in structure by HERFD-XANES and XES. The surface analysis of the O 1s region by XPS, however, indicates changes at the surface for the argon milled LaMnO3. With the desorption of oxygen well understood to be the rate controlling step within the mechanism of deN2O this supports our previous work, that proposes the higher amount of surface adsorption centres is linked to a higher conversion of N2O to N2 at lower temperatures.5,45,46 Furthermore, this argon milled catalyst shows a change in the light-off curve at 550 °C, which has previously been correlated to an increase in crystalline perovskite phase for other ball milled species during deN2O. Here, the high reaction temperatures have resulted in annealing of the ball milled perovskite, changing the catalyst structure and thus the catalytic activity (Fig. S5, ESI†). However, the argon milled catalyst still shows to be largely effective at lower temperatures, before the phase transformation takes place.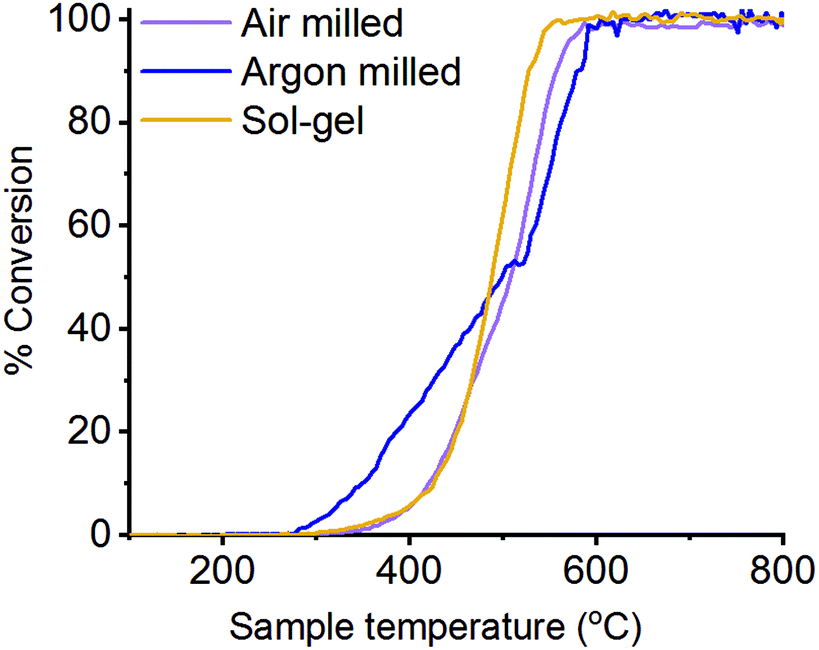 | ||
| Fig. 10 Light-off curve of the percentage of deN2O to N2 over LaMnO3 catalysts synthesised by mechanochemistry and sol–gel synthesised with 0.5% N2O/He at 30 mL min−1 with a pre-treatment of He at 30 mL min−1. | ||
Furthermore, with XPS analysis showing the argon milled catalyst to have a higher proportion of La at the surface, it raises a very interesting feature as traditionally it is thought that the Mn is the active species for catalysis in this system.47 Due to La electronic configuration it does not possess redox capabilities and is most stable in a 3+ oxidation state. However, differences do occur in the multiplet splitting within the XPS La 3d region, with the argon milled catalyst having a higher percentage of adsorbed oxygen species. In order to understand the role of La during deN2O and how the other XPS regions are affected during the reaction in situ near ambient pressure (NAP)-XPS will now be discussed.
3.4 In situ NAP-XPS deN2O
In the previous sections, we were able to link the catalytic activity towards deN2O to the surface properties of the LaMnO3 catalysts. It is critical to understand how the deN2O process effects the surface properties and stability of our LaMnO3 catalysts during N2O adsorption, dissociation and desorption of N2. Our previous lab-based XPS was performed under vacuum with a fixed incident beam. This ex situ analysis of the surface of the catalysts often presents vastly different properties compared to that under reaction conditions.48 Furthermore, controlling the incident energy allows for the same specific penetration depth of the surface for each XPS region.49 In this section, NAP-XPS has been performed under working conditions during deN2O with spectra recorded at the La 3d, Mn 2p, and O 1s regions at 834.5–855 eV, 641–655 eV and 529–532 eV binding energies, respectively, for the differently synthesised perovskite catalysts (Fig. S6–S8, ESI†). To achieve a comparison depth profile the Mn 3s region was performed with a greater incident energy. With the argon milled LaMnO3 catalyst showing promising activity at lower temperatures (Fig. 10) we now the detail the surface properties during in situ deN2O compared to the other LaMnO3 catalysts.Initially, the Mn XPS regions (Mn 2p and Mn 3s) have been assessed for the argon milled LaMnO3 with scans performed at RT under a N2O atmosphere (Fig. 11A) and then after heating at 400 °C and 600 °C (Fig. S9, ESI†). Within the Mn 2p region two main peaks are observed at ∼641.5 eV and ∼653 eV, originating from spin–orbit coupling, assigned as Mn 2p3/2 and Mn 2p1/2 respectively.50 These peak positions give an indication of the Mn oxidation state at the surface. Due to the broad and asymmetric nature of the peaks, deconvolution of both Mn 2p3/2 and Mn 2p1/2 peaks by curve fitting resulted in a total of four peaks for the region, suggesting mixed valency. The deconvoluted peak positions at RT in N2O for Mn 2p3/2 were recorded at 641.5 and 643.2 eV and for Mn 2p1/2 at 653.3 and 655.6 eV representing Mn(III) and Mn(IV), respectively (Table 2).51–53 On subsequent heating to 400 °C and 600 °C the peak positions are observed to remain ±0.2 eV within each other. Assessing the area of the deconvoluted peaks within Mn 2p3/2 region can give an indication of the relative abundance of Mn(III) and Mn(IV) contributions (Table 2).40 Calculating a ratio of the areas for each measurement results in consistent 1:1.3 ± 0.1 ratio of Mn(IV):Mn(III) during in situ deN2O for the argon milled catalyst.
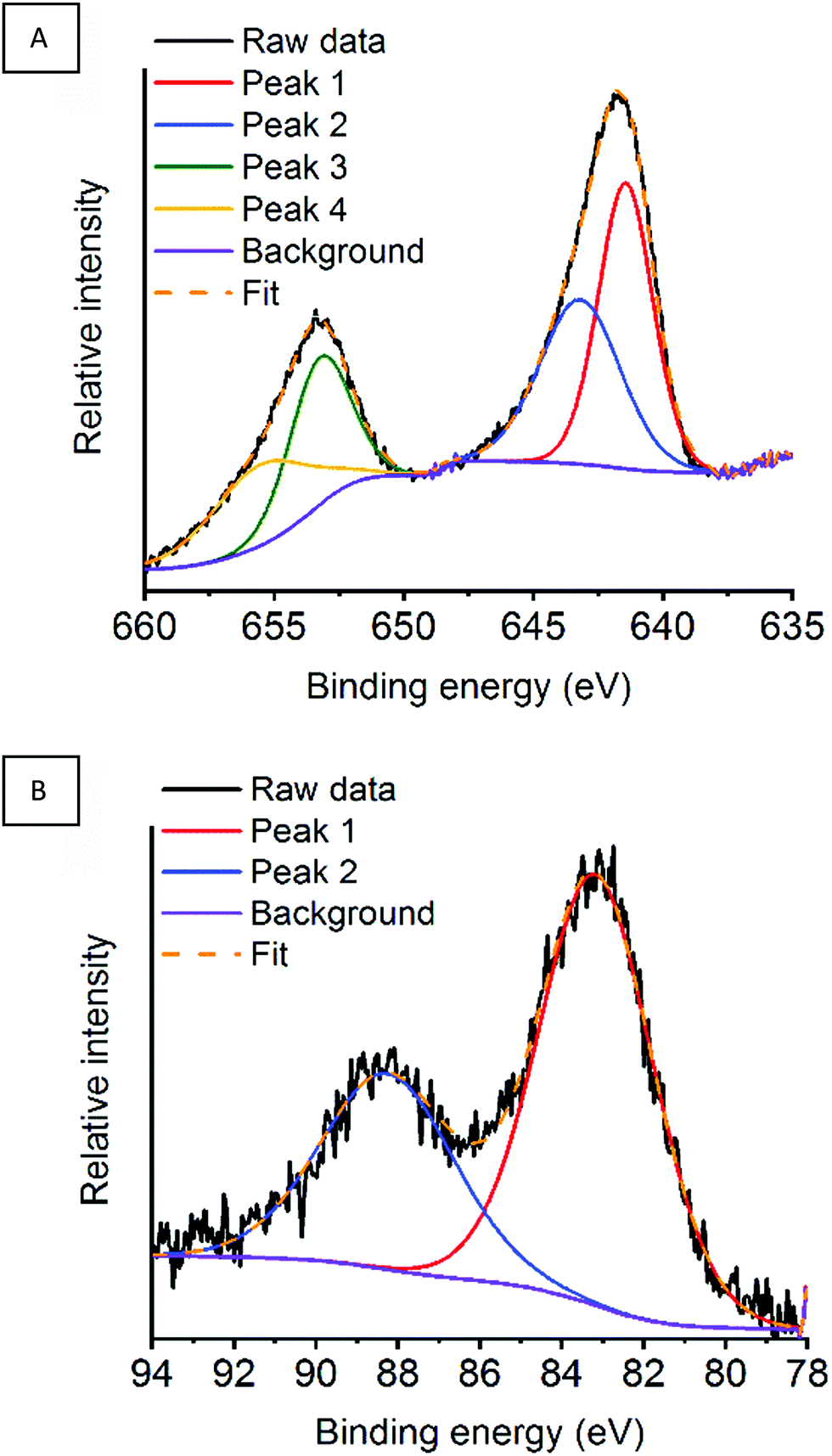 | ||
| Fig. 11 (A) Mn 2p region and (B) Mn 3s region for the argon milled catalyst at RT under a N2O atmosphere. | ||
| RT vacuum | RT in N2O | 400 °C in N2O | 600 °C in N2O | ||
|---|---|---|---|---|---|
| Mn 2p | Peak 1 | — | 641.5, 46205 |
641.3, 52777 |
641.1, 63001 |
| Peak 2 | — | 643.2, 39338 |
643.3, 40166 |
643.3, 44761 |
|
| Peak 3 | — | 653.3, 26307 |
653.0, 24094 |
652.6, 20800 |
|
| Peak 4 | — | 655.6, 23371 |
655.1, 18093 |
654.3, 22707 |
|
| Spin coupling | — | 11.8 | 11.7 | 11.5 | |
| Ratio Mn(IV):Mn(III) |
— | 1.1 | 1.3 | 1.4 | |
| Mn 3s | Peak 1 | 83.12 | 83.18 | 83.55 | 83.36 |
| Peak 2 | 88.18 | 88.26 | 88.83 | 88.72 | |
| Satellite | — | — | 93.38 | 92.01 | |
| Splitting | 5.1 | 5.1 | 5.3 | 5.4 | |
| La 3d | Peak 1 | 834.19 | 834.52 | 833.9 | 834.0 |
| Peak 2 | 838.13 | 838.32 | 838.2 | 838.5 | |
| Peak 3 | 836.46 | 836.79 | 836.0 | 836.2 | |
| Peak 4 | 850.95 | 851.29 | 850.8 | 850.9 | |
| Peak 5 | 854.91 | 855.26 | 855.1 | 855.3 | |
| Peak 6 | 853.09 | 853.68 | 852.9 | 853.1 | |
| La 3d5/2 splitting | 3.9 | 3.8 | 4.3 | 4.4 | |
| Spin coupling | 16.8 | 16.8 | 16.9 | 16.8 | |
| O 1s | Peak 1 | 529.3, 4867 | 529.3, 26741 |
529.3, 46686 |
529.7, 37254 |
| Peak 2 | 531.3, 6909 | 531.1, 51708 |
531.5, 44746 |
530.9, 28693 |
|
| Peak 3 | 533.4, 1940 | 533.0, 16494 |
534.0, 1472 | 532.2, 6739 | |
| Peak 4 | — | 534.6, 6911 | — | — |
Comparison of the argon milled catalyst to the air milled and sol–gel prepared LaMnO3 shows that there are distinct variations of the initial Mn(IV)/Mn(III) peak positions and area contributions (Tables S4 and S5) (Fig. S10 and S11) (ESI†). However, on heating to 400 °C and 600 °C during deN2O the Mn(IV)/Mn(III) ratios for all catalysts remain within ±0.2 of one another, with similar deconvoluted peak positions recorded. This is indicative of surface reactions occurring during catalysis that change the net oxidation state.
The Mn 3s region provides complimentary information and was recorded at an increased kinetic energy of ∼770 eV, for enhanced depth profiling. The splitting of the Mn 3s peak (Fig. 11B) at ∼83.5 and ∼88.5 eV (Table 2) is a final state effect due to the parallel and anti-parallel coupling between the spins of the remaining 3s electron and the 3d electrons.54 On increasing oxidation state, i.e. a decrease of d electrons, the separation between these two peaks is known to decrease.55 For the argon milled catalyst a Mn 3s peak splitting value of 5.1 eV is observed at room temperature in vacuum. This suggests, a mixed Mn(IV)/Mn(III) valency, with literature reporting a peak splitting of 5.4 eV for Mn2O3 and 4.4 eV for MnO2.52,53 No change is observed for the Mn 3s peak splitting on the exposure to N2O gas. However, on increasing temperature, this splitting increases to 5.4 eV, which is indicative of the Mn(III) oxidation state.53 This increased depth-profiling observes the diffusion of sub-surface oxygen towards the exterior of the particle, which is responsible for the observed change in oxidation state.56,57 The lattice oxygen that has migrated towards the surface is then available to participate in the deN2O process.58,59 With the mixed Mn(III) and Mn(IV) valences still observed at 600 °C at a lower depth penetration (Mn 2p region) it suggests this is a highly surface sensitive reaction.
This behaviour, of the splitting energy increase due to elevated temperatures, within the Mn 3s region was also found for the other LaMnO3 catalysts (Tables S4 and S5) (Fig. S10 and S11) (ESI†). Understanding the Mn 2p and Mn 3s XPS regions highlights that, though the differently synthesised LaMnO3 catalysts have varying bulk properties, at the surface there are similarities in how the Mn environment acts under working conditions.
The La 3d XPS region (Fig. 12 and Fig. S12, ESI†) shows the typical doublet splitting, with the lower energy doublet assigned to La 3d5/2 and the higher, La 3d3/2.60 The peak splitting of the La 3d5/2 doublet has been used to understand the nature of the La species.61 The asymmetric nature of the peaks within the La 3d region required three peaks to deconvolute each doublet (Fig. 12 and Fig. S12, ESI†).62,63 For the argon milled catalyst, all the values of La 3d5/2 peak splitting during deN2O are typical of La(III) compounds, which is further confirmed by the spin–orbit coupling values of 16.8–16.9 eV for all species (Table 2).41 Initially, at RT under vacuum, the La 3d5/2 peak splitting of 3.9 eV is indicative of La(OH)3 (Table 2). This is further confirmed by the presence of La(OH)3 diffraction peaks in Fig. 5. Increasing the temperature during deN2O results in an increase in the peak splitting value, suggesting the formation of an oxide species at the surface.64
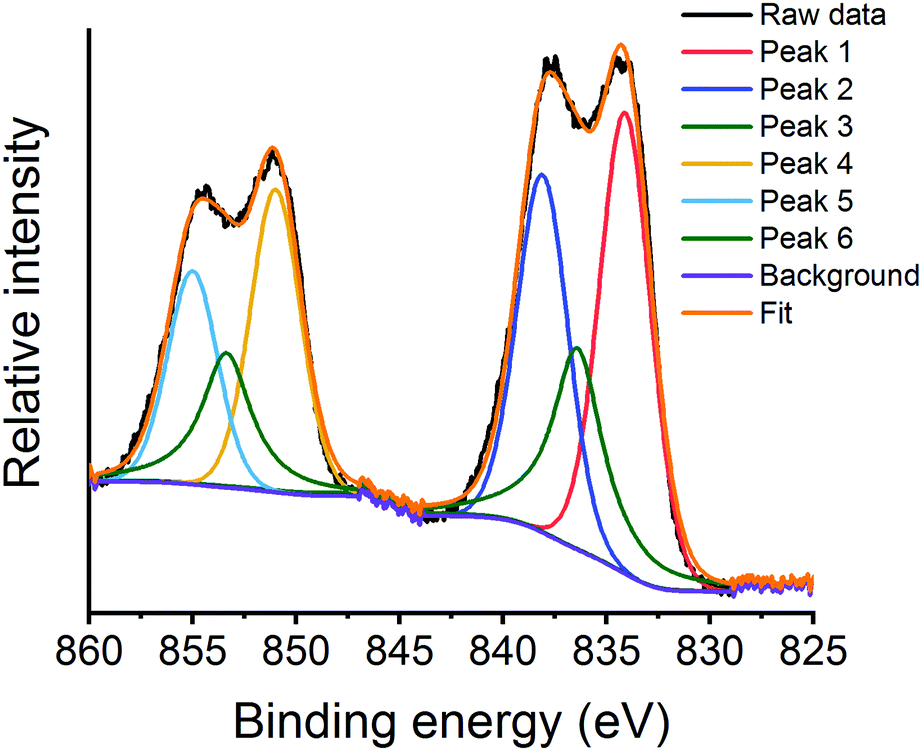 | ||
| Fig. 12 La 3d XPS region for the argon milled catalyst showing the deconvoluted peaks at RT under a N2O atmosphere. | ||
Both the air milled and sol–gel LaMnO3 start with a higher La 3d5/2 splitting value, of 4.1 eV and 4.2 eV respectively, compared to that of the argon milled catalyst, indicating a more oxide surface initially (Tables S4 and S5) (Fig. S13 and S14) (ESI†).65 The ball milled samples record a decrease in the La 3d splitting on exposure to N2O (∼0.1 eV). This could be indicative that the adsorbed N2O is interacting with surface La species.
Fig. 13 shows the deconvoluted fitted peaks for the O 1s region in argon milled LaMnO3 at RT under a N2O atmosphere and then at 600 °C (full data set Fig. S15, ESI†). On deconvolution of the O 1s region three oxygen species can be identified (Table 2). In Table 2, peak 1 is assigned to lattice-type oxygens (O22−), peak 2 to adsorbed species such as O2−, O− or OH− and peak 3 and 4 is associated with adsorbed adventitious carbon or molecular water.39–41
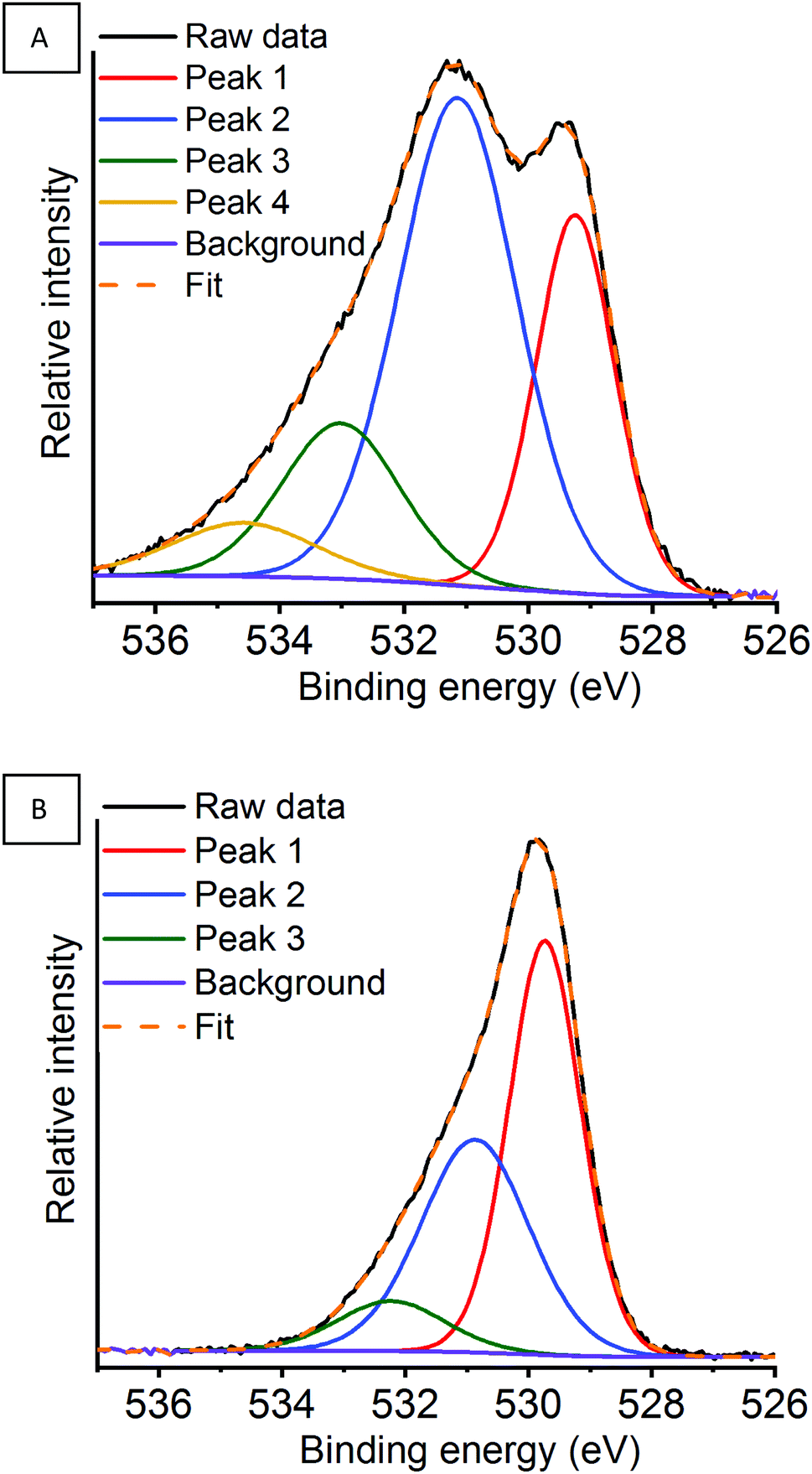 | ||
| Fig. 13 O 1s region for the argon milled catalyst showing the deconvoluted peaks at (A) RT and (B) 600 °C under a N2O atmosphere. | ||
By monitoring in situ deN2O by NAP-XPS clear changes can be observed within the O 1s region for the argon milled catalyst, which have not been previously reported. Whilst remaining at RT, on the introduction of N2O an increase in the proportion of adsorbed species can be observed, along with the presence of a higher binding energy peak 4 at 534.6 eV (Fig. S15A, ESI†). This is indicative that N2O is adsorbed on the surface at room temperature, in agreement with La 3d XPS data. A change in the area of peak 1, arising due to lattice-type oxygens, is observed on exposure to N2O. This could indicate a possible rearrangement of the surface structure due to N2O interaction. On increasing the temperature to 400 °C the surface adsorbed species decrease. Further increase of the temperature to 600 °C shows a significant change in the peak shape, along with a 0.5 eV energy increase in the binding energy of peak 1. Here, the change in relative oxygen species abundance and peak positions could relate to either the hydrothermal removal of adsorbed oxygen species from the surface, as seen in the La 3d region (Table 2), or changes due to the Mn enivronment.66,67 The Mn 2p and Mn 3s XPS data confirm that under these conditions the surface Mn species are relatively unchanged, whilst at greater depths oxygen is transferred towards the surface. Considering, that the O 1s data is acquired at the same depth penetration as the Mn 2p, it infers that the changes observed are correlated to changes in La speciation.
Both the air milled and sol–gel prepared LaMnO3 also recorded a higher proportion of adsorbed species at the surface on exposure to N2O (Tables S4 and S5) (Fig. S16 and S17) (ESI†). Significantly, the air milled and sol–gel catalysts do not have peak 4, fitted at 534.6 eV for the argon milled LaMnO3, present within the O 1s region. This suggests that there are distinct surface sites for the argon milled LaMnO3 that are involved in the deN2O process at lower temperatures, increasing the catalytic activity (Fig. 10).58,59 Furthermore, the air milled catalyst also suggests a rearrangement in surface structure due to exposure of N2O, as a reduction in peak 1, corresponding to lattice-type oxygen, is observed.
The deN2O catalytic testing observed an abrupt change to the light off profiles for the ball milled prepared samples that have been assigned to a phase transformation (Fig. 10). This behaviour mirrors what is found in the O 1s XPS data; between 400 °C and 600 °C there are significant changes within the O 1s profile for the balled milled samples. Conversely, the sol–gel prepared LaMnO3 shows a similar peak profile within the O 1s region at both 400 °C and 600 °C, suggesting a relatively stable oxygen environment.
Although the role of Mn towards the catalytic activity is clearly important, in this work the Mn 2p region shows minimal changes during deN2O.63 However, there are significant changes, within the La 3d and O 1s regions that have provided additional value in understanding the catalytic activity of these LaMnO3 systems. Furthermore, variations within the in situ deN2O NAP-XPS regions compared to the ex situ XPS reported in Section 3.2 show the importance in tuning the incident beam in order to achieve equivalent data for each XPS region at the same penetration depth.
4. Conclusions
Here we have successfully provided in-depth insights into understanding the electronic and geometric changes during the mechanochemical synthesis of LaMnO3. By performing ex situ HERFD measurements on ‘time-slices’ during milling we successfully identified the reduction of precursor Mn2O3 features within the pre-edge region and the evolution of peaks assigned to LaMnO3. However, significant alterations are observed within the ball milled perovskite which indicate disruption to the Mn–La coordination, specifically to the lack of Mn 4p La 5d hybridisation, in comparison to the sol–gel prepared LaMnO3. XES performed at the MnKβ1,3 emission line suggest a ‘bulk’ Mn(III) oxidation state throughout the mechanochemical synthesis.Efforts to perform in situ milling and to replicate the conditions experienced during milling by in situ high pressure experiments on La2O3 and Mn2O3 precursors were unable to induce structural changes. This confirms that the mechanochemical synthesis of these metal oxide systems are extremely complicated to monitor in situ.
Investigating how the milling atmosphere effects the formation of the perovskite phase, shows that an argon environment decreases the proportion of crystalline LaMnO3 after 4 h of milling, compared to an air milling atmosphere. However, performing deN2O shows this argon milled catalyst to have an improved performance at lower temperatures compared to the air milled and sol–gel LaMnO3. With the bulk structural analysis of the ball milled materials reporting similar structures it indicated that the catalytic activity of deN2O is strongly correlated to the proportion of oxygen vacancies recorded at the surface from ex situ XPS.
By performing in situ NAP-XPS deN2O we were able to further the understanding of this improved catalytic activity whilst studying the different LaMnO3 catalysts under working conditions. Within the O 1s region the argon milled catalyst showed a higher proportion of adsorbed species on exposure to N2O at RT, indicating an increase interaction with this species. It also highlighted that all catalysts remained with a mixed Mn(III)/Mn(IV) valency, even at elevated temperatures of 600 °C whilst working under catalytic conditions.
This work, therefore, demonstrates how the use of further advanced characterisation, such as HERFD, XES and NAP-XPS, can provide an in-depth understanding of the electronic, structural and surface properties of LaMnO3 induced by the mechanochemical synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Diamond Light Source for provision of beamtimes (experiments SP20129, SP20200 and SP22063) along with the UK Catalysis Hub for provision of beamtime SP15151-8. The staff on B18, I20-Scanning, I20-EDE and B07-C at Diamond Light Source are thanked. Particularly Dr Diego Gianolio is thanked for his assistance in collecting data on B18 and Dave Grinter, Pilar Ferrer-Escorihuela and Rosa Arrigo for establishing the B07-C beamline. Also, to the ID15A beamline and their staff at the European Synchrotron Radiation Facility and particularly to the Ruđer Bošković Institute for the use of their in situ milling set-up (experiment CH-5331). The RCaH are acknowledged for use of facilities and staff support. Johnson Matthey is acknowledged for their provision of precursor materials and milling equipment. The Johnson Matthey advanced analytical department are also thanked for their help and support throughout the project. The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium (portfolio grants EP//K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/I019693/1). The University of Southampton and EPSRC are thanked for the iCASE studentship of RHB. PW and KM wish to acknowledge the STFC for funding the position of KM (ST/R002754/1). PW and MC wish to acknowledge the EPSRC for funding the position of MC (EP/R011710/1). All data supporting this study are openly available from the University of Southampton repository at DOI: 10.5258/SOTON/D1342.References
- J. F. Jenck, F. Agterberg and M. J. Droescher, Green Chem., 2004, 6, 544 RSC.
- D. C. Koningsberger, B. L. Mojet, G. E. Van Dorssen and D. E. Ramaker, Top. Catal., 2000, 10, 143–155 CrossRef CAS.
- M. Newville, Fundamentals of XAFS, 2004.
- J. Evans, X-Ray Absorption Spectroscopy for the Chemical and Materials Sciences, John Wiley & Sons Ltd, 2017 Search PubMed.
- R. H. Blackmore, M. E. Rivas and T. E. Erden, Dalton Trans., 2020, 49, 232–240 RSC.
- S. Lafuerza, J. García, G. Subías, J. Blasco and P. Glatzel, Phys. Rev. B, 2016, 93, 31–33 CrossRef.
- F. De Groot, G. Vankó and P. Glatzel, J. Phys.: Condens. Matter, 2009, 21, 104207 CrossRef PubMed.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- P. Glatzel, T. C. Weng, K. Kvashnina, J. Swarbrick, M. Sikora, E. Gallo, N. Smolentsev and R. A. Mori, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 17–25 CrossRef CAS.
- S. Hayama, G. Duller, J. P. Sutter, M. Amboage, R. Boada, A. Freeman, L. Keenan, B. Nutter, L. Cahill, P. Leicester, B. Kemp, N. Rubies and S. Diaz-Moreno, J. Synchrotron Radiat., 2018, 25, 1556–1564 CrossRef CAS PubMed.
- S. Limandri, S. Ceppi, G. Tirao, G. Stutz, C. G. Sánchez and J. A. Riveros, Chem. Phys., 2010, 367, 93–98 CrossRef CAS.
- T. A. Tyson, Q. Qian, C.-C. Kao, J.-P. Rueff, F. M. F. De Groot, M. Croft, S.-W. Cheong, M. Greenblatt and M. A. Subramanian, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 4665–4674 CrossRef CAS.
- I. Halasz, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. C. Nightingale, R. E. Dinnebier and T. Friščić, Nat. Protoc., 2013, 8, 1718–1729 CrossRef PubMed.
- T. Frišči, I. Halasz, P. J. Beldon and A. M. Belenguer, Nat. Chem., 2012, 5, 66–73 CrossRef PubMed.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
- X. Hong, M. Newville, V. B. Prakapenka, M. L. Rivers and S. R. Sutton, Rev. Sci. Instrum., 2009, 80, 073908 CrossRef PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. Bauer, Phys. Chem. Chem. Phys., 2014, 16, 13827–13837 RSC.
- D. Rybicki, M. Sikora, J. Przewoznik, C. Kapusta and J. F. Mitchell, Phys. Rev. B, 2018, 97, 1–8 CrossRef.
- F. Farges, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–14 CrossRef.
- D. C. Radu, P. Glatzel, W. M. Heijboer, J. H. Bitter, B. M. Weckhuysen and F. M. F. de Groot, Mn and Fe Ions and Oxo clusters in ZSM-5: Pushing the limits of X-ray Spectroscopy, Elsevier B.V., 2007, vol. 170 Search PubMed.
- L. A. Isupova, G. M. Alikina, S. V. Tsybulya, A. N. Salanov, N. N. Boldyreva, E. S. Rusina, I. A. Ovsyannikova, V. A. Rogov, R. V. Bunina and V. A. Sadykov, Catal. Today, 2002, 75, 305–315 CrossRef CAS.
- V. Cuartero, S. Lafuerza, M. Rovezzi, J. García, J. Blasco, G. Subías and E. Jiménez, Phys. Rev. B, 2016, 94, 1–10 CrossRef.
- A. Y. Ignatov, N. Ali and S. Khalid, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 1–16 CrossRef.
- F. M. F. De Groot, S. Huotari, R. J. Cava, T. Lorenz and M. Reuther, 2008, arXiv:0802.2744.
- R. Baran, L. Valentin, J. M. Krafft, T. Grzybek, P. Glatzel and S. Dzwigaj, Phys. Chem. Chem. Phys., 2017, 19, 13553–13561 RSC.
- S. D. Gamblin and D. S. Urch, J. Electron Spectrosc. Relat. Phenom., 2001, 113, 179–192 CrossRef CAS.
- M. A. Beckwith, M. Roemelt, C. Duboc, T. Weng, U. Bergmann, P. Glatzel, F. Neese and S. Debeer, Inorg. Chem., 2011, 8397–8409 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- P. Baláž, M. Achimovičová and M. Baláž, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- M. a Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef CAS PubMed.
- P. Granger, V. I. Parvulescu, S. Kaliaguine and W. Prellier, Perovskites and Related Mixed Oxides Concepts and Applications, Wiley-VCH, 2015 Search PubMed.
- Y. Syono and S. Akimoto, J. Phys. Soc. Jpn., 1969, 26, 993–999 CrossRef CAS.
- J. A. M. Van Roosmalen, P. van Vlaanderen and E. H. P. Cordfunke, J. Solid State Chem., 1995, 114, 516–523 CrossRef CAS.
- Y. Zhao, Materials, 2012, 5, 1413–1438 CrossRef CAS.
- L. B. Mccusker, R. B. Von Dreele, D. E. Cox, D. Louër and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- K. Jirátová, J. Mikulová, J. Klempa, T. Grygar, Z. Bastl and F. Kovanda, Appl. Catal., A, 2009, 361, 106–116 CrossRef.
- V. P. Santos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 353–363 CrossRef CAS.
- A. Machocki, T. Ioannides, B. Stasinska, W. Gac, G. Avgouropoulos, D. Delimaris, W. Grzegorczyk and S. Pasieczna, J. Catal., 2004, 227, 282–296 CrossRef CAS.
- S. K. Gupta, M. Sahu, P. S. Ghosh, D. Tyagi, M. K. Saxena and R. M. Kadam, Dalton Trans., 2015, 44, 18957–18969 RSC.
- Y. Yang, S. Zhang, S. Wang, K. Zhang, H. Wang, J. Huang, S. Deng, B. Wang, Y. Wang and G. Yu, Environ. Sci. Technol., 2015, 49, 4473–4480 CrossRef CAS PubMed.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- N. Gunasekaran, S. Rajadurai and J. J. Carberry, Catal. Lett., 1995, 35, 373–382 CrossRef CAS.
- N. Russo, D. Mescia, D. Fino, G. Saracco and V. Specchia, Ind. Eng. Chem. Res., 2007, 46, 4226–4231 CrossRef CAS.
- H. Najjar and H. Batis, Catal. Rev., 2016, 58, 371–438 CrossRef CAS.
- C. Escudero, P. Jiang, E. Pach, F. Borondics, M. W. West, A. Tuxen, M. Chintapalli, S. Carenco, J. Guo and M. Salmeron, J. Synchrotron Radiat., 2013, 20, 504–508 CrossRef CAS PubMed.
- N. H. Turner, Anal. Chem., 1986, 58, 153–165 CrossRef.
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586–19601 CrossRef CAS.
- A. J. Nelson, J. G. Reynolds and J. W. Roos, J. Vac. Sci. Technol., A, 2000, 18, 1072–1076 CrossRef CAS.
- M. A. Stranick, Surf. Sci. Spectra, 1999, 6, 31–38 CrossRef CAS.
- M. A. Stranick, Surf. Sci. Spectra, 1999, 6, 39–46 CrossRef CAS.
- D. J. Lam, B. W. Veal and D. E. Ellis, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 5730–5739 CrossRef CAS.
- V. R. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. A. Ovechkina, E. Z. Kurmaev, N. I. Lobachevskaya, Y. M. Mukovskii, J. Mitchell and D. L. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 1–4 CrossRef.
- A. Staykov, H. Téllez, T. Akbay, J. Druce, T. Ishihara and J. Kilner, Chem. Mater., 2015, 27, 8273–8281 CrossRef CAS.
- H. Najjar, J. F. Lamonier, O. Mentré, J. M. Giraudon and H. Batis, Appl. Catal., B, 2011, 106, 149–159 CAS.
- T. A. Egerton, F. S. Stone and J. C. Vickerman, J. Catal., 1974, 33, 307–315 CrossRef CAS.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- R. Dudric, A. Vladescu, V. Rednic, M. Neumann, I. G. Deac and R. Tetean, J. Mol. Struct., 2014, 1073, 66–77 CrossRef CAS.
- T. S. Kharlamova, A. S. Matveev, A. V. Ishchenko, A. N. Salanov, S. V. Koshcheev, A. I. Boronin and V. A. Sadykov, Kinet. Catal., 2014, 55, 361–371 CrossRef CAS.
- D. F. Mullica, C. K. C. Lok, H. O. Perkins and V. Young, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 4039–4042 CrossRef CAS PubMed.
- M. F. Sunding, K. Hadidi, S. Diplas, O. M. Løvvik, T. E. Norby and A. E. Gunnæs, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 399–409 CrossRef CAS.
- Y. Zhang-Steenwinkel, J. Beckers and A. Bliek, Appl. Catal., A, 2002, 235, 79–92 CrossRef CAS.
- M. E. Rivas, C. E. Hori, J. L. G. Fierro, M. R. Goldwasser and A. Griboval-Constant, J. Power Sources, 2008, 184, 265–275 CrossRef CAS.
- J. G. Kang, Y. Il Kim, D. Won Cho and Y. Sohn, Mater. Sci. Semicond. Process., 2015, 40, 737–743 CrossRef CAS.
- P. Fleming, R. A. Farrell, J. D. Holmes and M. A. Morris, J. Am. Ceram. Soc., 2010, 93, 1187–1194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00793e |
| This journal is © the Owner Societies 2020 |