
Crystal engineering of coordination-polymer-based iodine adsorbents using a π-electron-rich polycarboxylate aryl ether ligand†
Junling
Chen
 ab,
Bo
Li
b,
Zhenzhen
Shi
b,
Cheng
He
a,
Chunying
Duan
a,
Tiexin
Zhang
*a and
Li-Ya
Wang
*bc
ab,
Bo
Li
b,
Zhenzhen
Shi
b,
Cheng
He
a,
Chunying
Duan
a,
Tiexin
Zhang
*a and
Li-Ya
Wang
*bc
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: zhangtiexin@dlut.edu.cn
bCollege of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang, 473061, P. R. China. E-mail: wly@nynu.edu.cn
cCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450001, P. R. China
First published on 7th September 2020
Abstract
Efficient capture and storage of radioactive iodine isotopes are important in nuclear waste treatment and environmental protection. Coordination polymers (CPs) are a family of newly emerging potential iodine adsorbents. However, the exact structure–activity relationship between various structural CP hosts and their noncovalent adsorption effects toward iodine guest molecules has not been elucidated. Herein, a crystal engineering strategy was employed to systematically study the effects of porosity and arrangement of noncovalent interaction sites of CP hosts on the iodine uptake performance. Three isomeric CPs—{[Zn3(μ3-OH)(L)(H2O)3]·2H2O}n (1), {[Zn3(μ3-OH)(L)(H2O)]·2DMF·3H2O}n (2), and {[Zn3(μ3-OH)(L)(DMF)2]·H2O}n (3)—having different structures were synthesized from a π-electron-rich polycarboxylate ligand, H5L [5,5′-((5-carboxy-1,3-phenylene)bis(oxy))diisophthalic acid], containing potential sites for charge-transfer interaction and halogen bonding. CP 2, with a medium channel size and suitable arrangement of noncovalent interaction sites, exhibited higher I2 adsorption capacity than the other two CPs. The synergistic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯I and charge-transfer π⋯I interactions between the I2 guest and the carboxylate moiety and the benzene ring of CP 2, respectively, were validated by X-ray diffraction analysis, electrochemical impedance spectroscopy, and solid-state fluorescence spectroscopy.
O⋯I and charge-transfer π⋯I interactions between the I2 guest and the carboxylate moiety and the benzene ring of CP 2, respectively, were validated by X-ray diffraction analysis, electrochemical impedance spectroscopy, and solid-state fluorescence spectroscopy.
1. Introduction
Various isotopes of iodine, including I129, I125, I123, and I131, have been extensively utilized in a wide range of applications such as in monitoring contaminants, imaging, cancer treatment, and catalysis and as animal feed and tags for proteins.1–7 However, these isotopes are harmful to human health as they can be absorbed by the thyroid gland when inhaled, causing thyroid cancer, leukemia, and other diseases.8–10 Because of this health concern, effective techniques must be developed for the controllable recovery and release of I2. So far, a few types of porous materials have been developed for capturing I2, such as chalcogenide aerogels, functionalized clays, and silver-based porous zeolitic materials.11–14 However, because of the non-uniform distribution of the functional sites for iodine adsorption, further modification and redesign of the porous materials on the basis of structure–activity relationships are challenging.Crystalline porous coordination polymers (CPs), which are self-assembled structures of metal ions and organic ligands, have attracted considerable attention as platforms for host–guest chemistry. These materials have promising applications in many fields such as drug delivery,15 sensing,16 gas adsorption/separation,17,18 ion exchange,19 catalysis,20–22 and water harvesting.23 Because of the tailorable porosity and the uniform arrangement of noncovalent sorption sites within the frameworks of CPs,24 CPs serve as effective candidates for capturing I2. Thus, CPs have attracted increasing attention in the recent years.25–31 It was found that the adsorption of I2 by crystalline porous materials, including CPs, was based on non-covalent interactions like halogen bonding interactions between I2 and Lewis basic sites32–35 or charge-transfer interactions between the electron-poor I2 and π-electron rich aromatics.36–41 Thus, a combination of halogen bond and CT interactions between the guest I2 molecules and the host frameworks imparts a comprehensive tuning effect on the iodine capture process. However, it is still unclear as to how the synergistic effect of the framework porosities and the multiple noncovalent interactions affects the iodine uptake performances. Moreover, the highly directional nature of halogen bonds and charge-transfer interactions makes it challenging to appropriately arrange the Lewis basic42 and electron-rich aromatic sites within the pores of CP-based adsorbents for the analysis of the structure–activity relationships.
In this study, a flexible polycarboxylate aryl ether ligand, H5L (H5L = 5,5′-((5-carboxy-1,3-phenylene)bis(oxy))diisophthalic acid), along with zinc salts was chosen for the crystal engineering and tuning of the iodine uptake ability of CP-based adsorbents. The abundant π-electron-rich aryl moieties and Lewis basic carboxylates available, following the deprotonation/complexation process, provide potential binding sites for the capture of iodine. The flexible aryl ether hinges and the multiple carboxylic coordination groups of the H5L ligand were believed to be able to tolerate the formation of variable topology and porosity of the CP candidates in crystal engineering.43 Three trinuclear isomers, namely, {[Zn3(μ3-OH)(L)(H2O)3]·2H2O}n (1), {[Zn3(μ3-OH)(L)(H2O)]·2DMF·3H2O}n (2), and {[Zn3(μ3-OH)(L)(DMF)2]·H2O}n (3), having different porosities and arrangements of noncovalent interaction sites were obtained through crystal engineering under the tuned solvothermal conditions. The structures of these isomers were characterized by single-crystal X-ray diffraction and powder X-ray diffraction (PXRD) analyses in combination with thermogravimetric analysis (TGA). The differential I2 adsorption of CPs 1–3 was studied through infrared (IR) spectroscopy and single-crystal X-ray diffraction and solid-state fluorescence analyses to understand their structure–activity relationships (Scheme 1).44–46
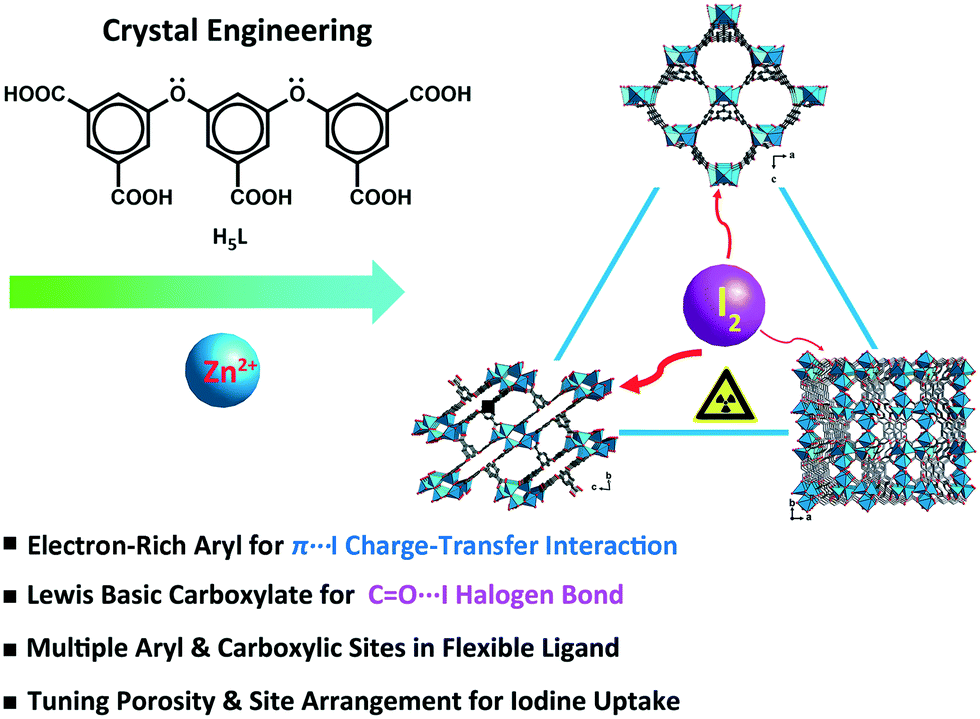 | ||
| Scheme 1 Schematic representation of the crystal engineering of π-electron-rich polycarboxylate coordination polymers for tuning the iodine uptake. | ||
2. Experimental section
2.1. X-ray crystal structures
The crystallographic data for CPs 1–3 and the iodine adsorbed CP I2@2 were evaluated by using a CCD X-ray diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved by the direct method and the non-hydrogen atoms were refined anisotropically by least-squares on F2 using the SHELXTL program package. The crystallographic data for 1–3 and I2@2 are summarized in Table 1. Selected bond lengths and angles are given in Table S1.†| CPs | 1 | 2 | 3 | I2@2a |
|---|---|---|---|---|
| a The residual electron densities were flattened by using the SQUEEZE option of PLATON. b R 1 = ∑‖Fo| − |Fc‖/∑|Fo|; wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP], in which P = (Fo2 + 2Fc2)/3. | ||||
| Chemical formula | C23H18O17Zn3 | C23H12O14Zn3 | C29H26N2O16Zn3 | C23H12I0.52O14Zn3 |
| CCDC No. | 1911376 | 1883449 | 1588500 | 2007800 |
| Formula weight | 762.48 | 708.44 | 854.63 | 775.06 |
| T (K) | 293 | 293 | 293 | 100 |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Triclinic |
| Space group | Pmn21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
| a (Å) | 17.6198(4) | 10.1200(5) | 12.8756(6) | 10.2531(2) |
| b (Å) | 11.9264(3) | 12.3807(6) | 17.0258(7) | 12.2578(3) |
| c (Å) | 14.1457(4) | 15.6793(8) | 14.8329(8) | 15.7790(4) |
| α (deg) | 90 | 79.300(1) | 90 | 101.524(2) |
| β (deg) | 90 | 87.854(2) | 101.033(5) | 92.146(2) |
| γ (deg) | 90 | 75.715(1) | 90 | 102.223(2) |
| V (Å3) | 2972.59(13) | 1870.59(16) | 3191.5(3) | 1892.49(8) |
| Z | 2 | 2 | 4 | 2 |
| D c (g cm−3) | 0.852 | 1.258 | 1.779 | 1.360 |
| μ (mm−1) | 1.239 | 1.957 | 2.317 | 2.364 |
| F(000) | 764 | 704 | 1728 | 760 |
| θ min, θmax/deg | 3.349, 28.473 | 2.354, 25.000 | 3.339, 25.000 | 1.740, 24.998 |
| Total, unique data | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 558, 6885 558, 6885 |
46142, 6571 |
16990, 5593 |
18683, 6648 |
| R int | 0.0436 | 0.0850 | 0.0294 | 0.0316 |
| GOF | 1.001 | 1.043 | 1.040 | 1.078 |
| R 1, wR2 [I > 2σ(I)]b | 0.0381, 0.0870 | 0.0443, 0.1028 | 0.0343, 0.0796 | 0.0408, 0.1174 |
| R 1, wR2 (all data) | 0.0528, 0.0928 | 0.0628, 0.1084 | 0.0456, 0.0837 | 0.0503, 0.1217 |
| Largest diff. peak and hole (e Å−3) | −0.277, 0.373 | −1.287, 1.594 | −0.454, 0.535 | −0.808, 1.439 |
2.2. Synthesis of {[Zn3(μ3-OH)(L)(H2O)3]·2H2O}n (1)
H5L (0.05 mmol, 24.1 mg) and Zn(NO3)2·6H2O (0.1 mmol, 29.7 mg) were dissolved in a solvent mixture of 4 mL diethylformamide (DEF) and 2 mL H2O (2:1, v:v). The solution was autoclaved at 100 °C for 3 days, and then slowly cooled to room temperature (r.t.), giving rise to rectangular block-shaped colorless crystals in 51% yield (based on Zn(II)). Elemental analysis (%), calcd for C23H20O18Zn3: C, 35.39; H, 2.58. Found: C, 35.43; H, 2.61. FT-IR (KBr, cm−1): 3424 (m), 1649 (s), 1568 (s), 1457 (m), 1391 (s), 1132 (w), 1016 (w), 777 (m), 726 (w), 671 (w). It should be noted that small amounts of unknown microcrystals were randomly found in the reaction mixture, which could be easily separated from the desired product (CP 1) by washing with the DEF solvent.
2.3. Synthesis of {[Zn3(μ3-OH)(L)(H2O)]·2DMF·3H2O}n (2)
The synthesis protocol of CP 2 was similar to that of CP 1, except that DEF in the mixed solvent was replaced with dimethylformamide (DMF). The block-shaped colorless crystals were obtained in 49% yield (based on Zn(II)). Elemental analysis (%), calcd for C29H32O19N2Zn3: C, 38.32; H, 3.55; N, 3.08. Found: C, 38.28; H, 3.57; N, 3.11. FT-IR (KBr, cm−1): 3368 (m), 1624 (s), 1573 (s), 1380 (s), 1249 (m), 1137 (m), 1016 (m), 782 (s), 726 (m), 665 (w).2.4. Synthesis of {[Zn3(μ3-OH)(L)(DMF)2]·H2O}n (3)
The synthesis process of CP 3 was analogous to that of CP 2, except that the volume ratio of DMF and H2O in the mixed solvent was changed to 1:5 (1.0 mL:5.0 mL). The block-shaped colorless crystals were obtained in 43% yield (based on Zn(II)). Elemental analysis (%), calcd for C29H26N2O16Zn3: C, 40.75; H, 3.07; N, 3.28. Found: C, 40.79; H, 3.05; N, 3.31. FT-IR (KBr, cm−1): 3454 (m), 1665 (s), 1624 (s), 1568 (s), 1457 (s), 1380 (s), 1249 (m), 1137 (m), 1016 (m), 889 (w), 782 (s), 731 (m), 665 (w).
3. Results and discussion
3.1. Crystal structure of {[Zn3(μ3-OH)(L)(H2O)3]·2H2O}n (1)
Single-crystal X-ray analysis revealed that CP 1 crystallized in the orthorhombic system, having the space group Pmn21. Each asymmetric unit consisted of one and a half crystallographically independent Zn(II) atoms, half of an L5− ligand, one μ3-OH, and two and a half water molecules. One of the water molecules was free in the channel (squeezed by PLATON47) and the other one and a half water molecules were attached to the Zn(II) atoms. Zn1 was coordinated by three carboxylic oxygen atoms from three different L5− ligands and one μ3-OH oxygen atom. The structure displayed a slightly distorted tetrahedral coordination geometry. Zn2 forms a distorted/twisted octahedral geometry with two oxygen atoms from two different L5− ligands, one μ3-OH oxygen atom and three oxygen atoms from three different lattice water molecules. Additionally, Zn1, Zn2, and Zn1#3 shared the μ3-OH oxygen atom to form trinuclear nodes [Zn3(μ3-OH)(H2O)3(COO)5], wherein, the distances of the Zn–O bonds ranged from 1.94 to 2.19 Å (Fig. 1a).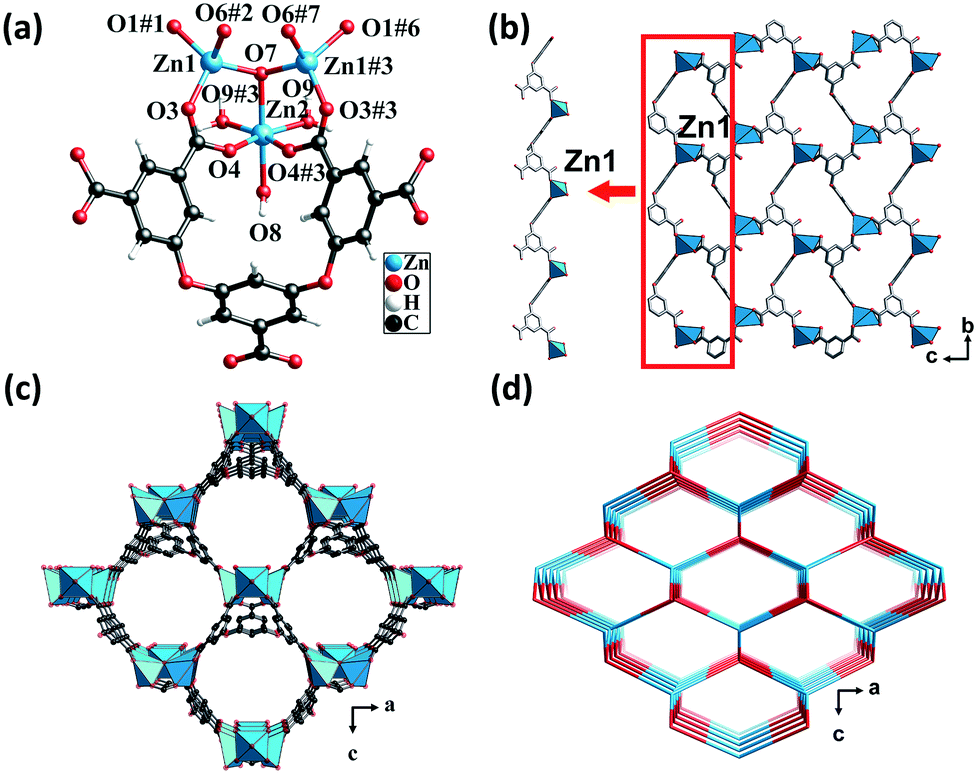 | ||
| Fig. 1 (a) Coordination environment of the Zn(II) center in CP 1. Color code: Sky blue – Zn; red – O; gray – C. (b) The 1D infinite chain and 2D layered structure formed by the Zn(II) atoms and L5− ligands in CP 1. (c) Perspective view of the 3D open framework in the b direction. (d) The topological network with the point symbol (66). | ||
The fully deprotonated carboxylic groups of the L5− ligand linked the Zn1 atoms via the μ2-η1:η1 and μ1-η1:η1 bridging modes to form 1D infinite chains (Scheme S1†). Subsequently, the 1D chains linked to each other to form a 2D layer (Fig. 1b), which in turn was further fused by Zn1, Zn2, and H5L ligands into a 3D structure (Fig. 1c). As calculated by the PLATON program, the solvent-accessible void volume of CP 1 was 64.5%. To gain a better understanding of the framework, both the trinuclear nodes and the L5− ligands were simplified as 4-connected nodes, so that CP 1 could be regarded as a uninodal net with a topological point symbol of (66) (Fig. 1d). This 3D framework comprised large honeycomb cavities (10.99 Å × 13.13 Å), and the π-electron-rich benzene rings and Lewis basic carboxylate moieties of the ligands were evenly distributed in the inner walls of the pore. These might act as the interaction sites for the iodine guest molecule encapsulation.
3.2. Crystal structure of {[Zn3(μ3-OH)(L)(H2O)]·2DMF·3H2O}n (2)
Single-crystal X-ray diffraction analysis revealed that CP 2 crystallized in the triclinic crystal system with the P space group. Each asymmetric unit consisted of three crystallographically unique Zn(II) atoms, one fully deprotonated L5− ligand, one μ3-OH, one coordinated water molecule, two DMF molecules, and three lattice water molecules (squeezed by PLATON). All the Zn(II) atoms formed a distorted triangular bipyramidal geometry. The Zn1 atom was coordinated by three oxygen atoms from three different L5− ligands, one μ3-OH group, and one μ2-O from a water molecule. The Zn3 atom was coordinated by two oxygen atoms from two different ligands, one oxygen atom from the μ3-OH group and one μ2-O from a water molecule. The Zn2 atom was coordinated by three oxygen atoms from three different ligands and one oxygen atom from the μ3-OH group. The separations of Zn–O bonds varied from 1.90 to 2.51 Å. Meanwhile, the Zn(II) atoms shared the μ3-OH oxygen atoms and μ2-H2O molecules to form hexanuclear nodes [Zn6(μ3-OH)2(μ2-OH)2(COO)10], in which the distances of Zn1⋯Zn2, Zn1⋯Zn3, and Zn2⋯Zn3 were 3.19 Å, 3.43 Å, and 3.48 Å, respectively (Fig. 2a).
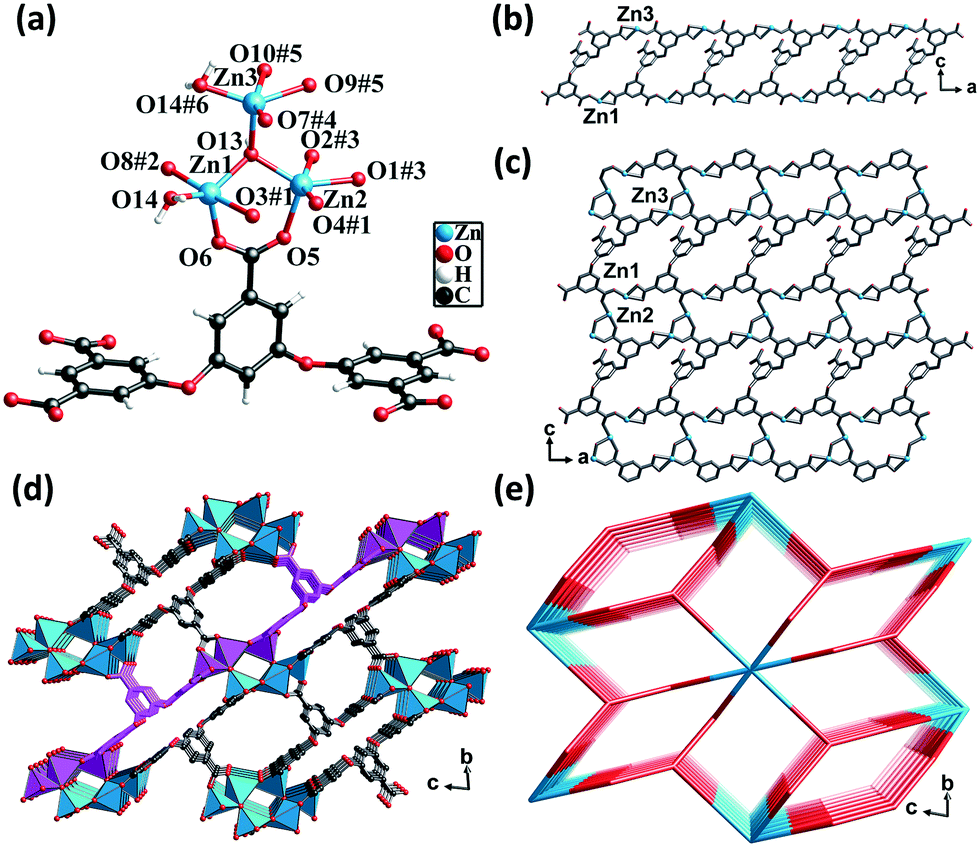 | ||
| Fig. 2 (a) Coordination environment of the Zn(II) center in CP 2. Color code: Sky blue – Zn; red – O; gray – C. (b) The 1D double chain in CP 2. (c) 2D layered structure in CP 2. (d) Perspective view of the 3D framework in the a direction. (e) The topological network with the point symbol (410)2(428·616·8). | ||
The carboxylic groups of the fully deprotonated L5− ligand followed the μ2-η1:η1 and μ1-η1:η1 bridging modes (Scheme S1†). The Zn1 and Zn3 atoms linked the L5− ligands to form 1D double chains along the b direction (Fig. 2b). The chains were in turn connected by Zn2 atoms to give rise to 2D layers (Fig. 2c). The adjacent 2D layers were further interconnected into a 3D structure featuring a six-nuclear Zn cluster node (Fig. 2d). The void space of the unit cell of CP 2 calculated by PLATON, after removing the guest solvent molecules, was found to be 45.3%. From the perspective of topology, CP 2 could be simplified as a 2-nodal net with a point symbol of (410)2(428·616·8), wherein the hexanuclear clusters could be rationalized as a 10-connected node and the L5− ligands could be considered as a 5-connected linker (Fig. 2e).
The framework of CP 2 comprised three types of rectangular channels—a long narrow cavity with dimensions of 3.7 Å × 14 Å and two larger cavities with dimensions of 6.8 Å × 8.2 Å and 6.8 Å × 9.2 Å. The π-electron-rich benzene ring and Lewis basic carboxylate sites were uniformly distributed in the inner wall. The reduced cross-sections in CP 2, in comparison with that in CP 1, ensures a more compact arrangement of the π-electron rich benzene ring and the Lewis basic carboxylate moieties in the channels of CP 2. This might provide more opportunities for the synergy between the two types of noncovalent interaction sites during the adsorption of iodine molecules.
3.3. Crystal structure of {[Zn3(μ3-OH)(L)(DMF)2]·H2O}n (3)
Single-crystal structural analysis revealed that CP 3 crystallized in the monoclinic system with the P21/c space group. Each asymmetric unit of CP 3 contained three crystallographically unique Zn(II) atoms, one completely deprotonated L5− ligand, one μ3-OH group, two DMF molecules, and one lattice water molecule. Zn1 and Zn3 atoms were coordinated by three O atoms from three different carboxylate groups and one O atom from the hydroxyl group, thus displaying a trigonal pyramidal geometry. The Zn2 atom was coordinated by three O atoms from three different carboxylate groups, one O atom from the hydroxyl group, and two O atoms from two different DMF molecules, giving rise to an octahedral geometry. The Zn–O distances varied from 1.93 to 2.17 Å, which were similar to those found in the other Zn(II) CPs (Fig. 3a).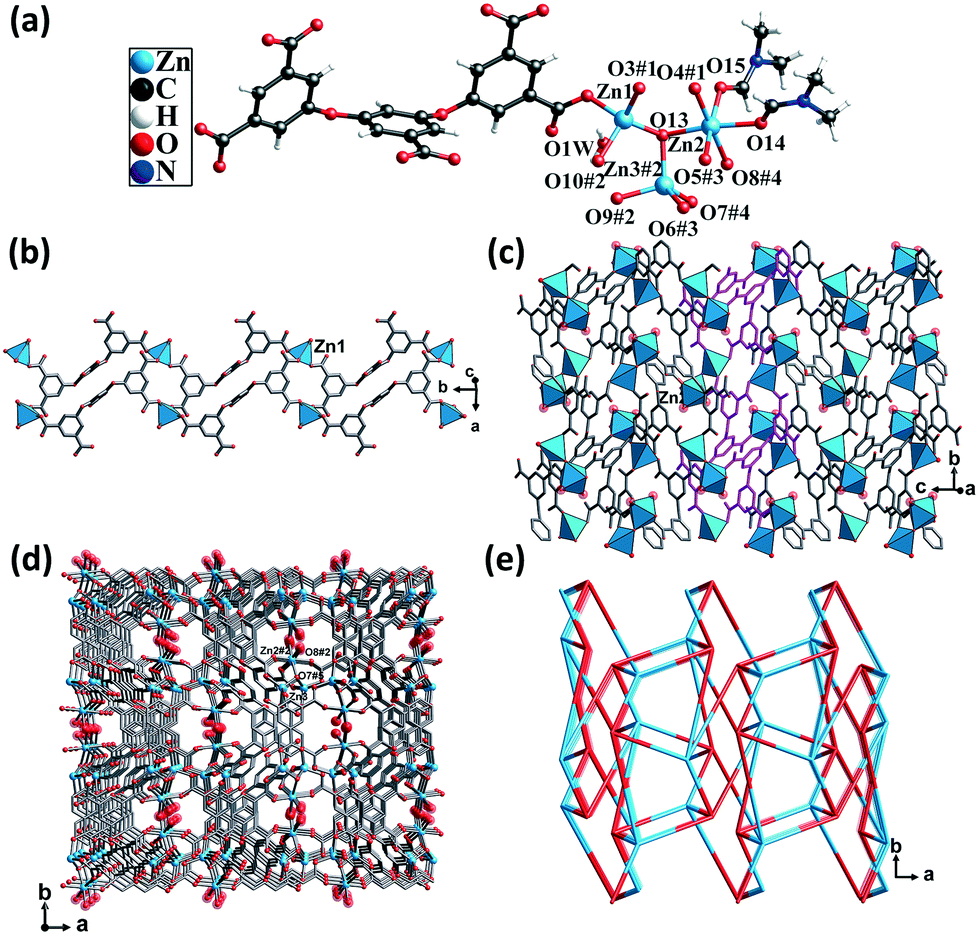 | ||
| Fig. 3 (a) Coordination environment of the Zn(II) atoms in CP 3. Color code: Sky blue – Zn; red – O; gray – C. (b) The 1D chain formed by the Zn1 atoms and L5− ligands. (c) The 2D layer formed by the Zn2 atoms and 1D chains. (d) 3D framework. (e) The topological network with the point symbol (45·65). | ||
The carboxylic groups of the fully deprotonated L5− ligand followed the μ2-η1:η1 and μ1-η1:η0 bridging modes (Scheme S1†). Each of the Zn1 atoms were linked to L5− ligands, giving rise to 1D double chains along the b axis (Fig. 3b). Such infinite double chains were bridged through the Zn2 atoms, forming a 2D layered structure (Fig. 3c). These layers were in turn extended through Zn–O bonds, resulting in a 3D framework (Fig. 3d). The PLATON calculations suggested that the solvent-accessible void space volume was about 34.4% after excluding DMF and free water molecules. In the topological view, the structure of CP 3 can be simplified as a uninodal network with a topological point symbol of (45·65) (Fig. 3e).
This structure consisted of channels that were much smaller than those of both CPs 1 and 2. The π-electron and carboxyl interaction sites were more densely dispersed. It is believed that the limited pore sizes and the cramped arrangement of the two types of interaction sites might limit the mass diffusion of the guest molecules.
3.4. Effect of solvents on the structure by crystal engineering
Based on the above results, it can be surmised that the mixed amide-type solvents and water play a vital role in the self-assembly process. The variability of these structures indicated that the porosity could be tuned by adjusting the amide-type solvents and/or the ratio of water. On keeping the ratios of water constant in the mixed solvents, it was observed that DEF having a larger molecular size exhibited an obvious template effect and facilitated the formation of larger pores in CP 1. In contrast, narrower pore diameters were induced in CP 2 by the solvent DMF having a smaller size. In the presence of higher ratios of water in the mixed solvents, the hydrophobic π-electron-rich aryl ether moieties tended to stack more densely, giving rise to narrower channels and a more compact arrangement of the potential noncovalent interaction sites in CP 3 as compared to CP 2. Under the comprehensive solvent effects, the porosity of CP 1 was found to be the highest, followed by CP 2 and CP 3. Additionally, the flexible aryl ether backbone of the ligand tolerated these structural variations and the multiple carboxylate coordination groups allowed the diversified coordination modes to adapt to the varied frameworks in CPs 1–3.3.5. PXRD, TGA, and fluorescence studies
PXRD patterns and TGA results of CPs 1–3 were evaluated to verify their phase purity and thermal stability (Fig. 4). The PXRD patterns of CPs 1–3 matched well with the corresponding simulated patterns from the single-crystal structure analysis, confirming the phase purity of the crystalline samples. CP 1 exhibited an initial weight loss of 11.8% (calc. 11.5%) below 160 °C, which is attributed to the release of the water molecules. Subsequently, the TGA curve decreased slowly from 186 °C, which can be ascribed to the degradation of the framework. CP 2 displayed a primary weight loss of 23.4% (calc. 24.0%) from r.t. to 320 °C, which denoted the removal of three water molecules and two DMF molecules squeezed by the SQUEEZE routine in PLATON. Subsequently, the entire framework began to degrade. CP 3 displayed a primary weight loss of 20.4% (calc. 19.2%) from r.t. to 300 °C, which corresponds to the release of one water molecule and two DMF molecules. Following this, a rapid weight loss indicated the collapse of the structure.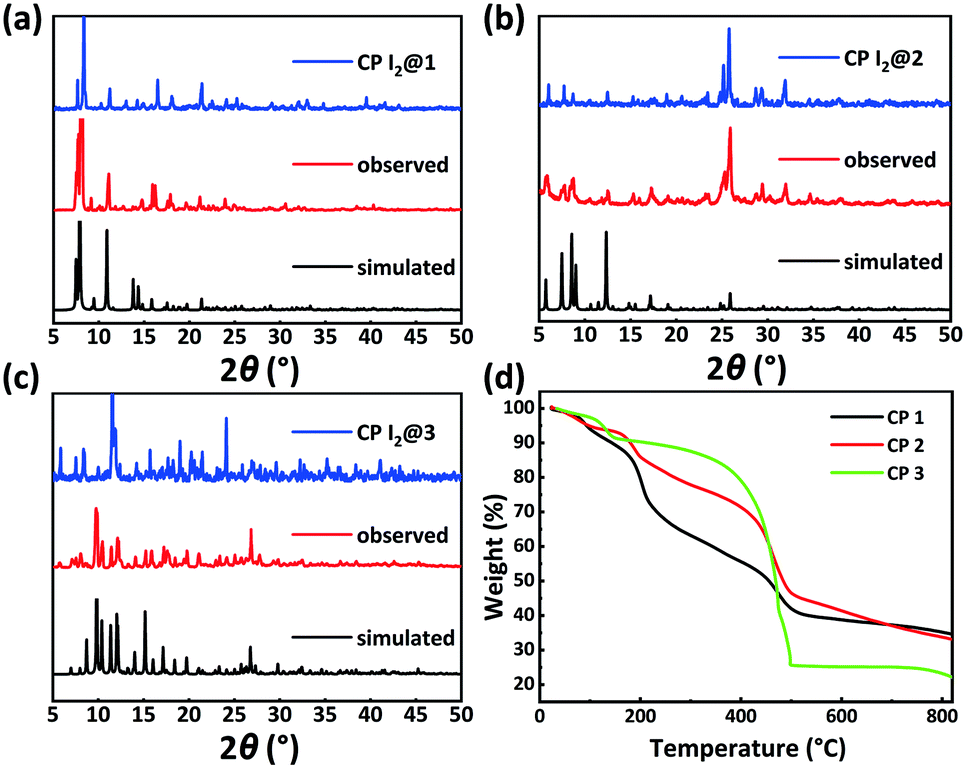 | ||
| Fig. 4 PXRD patterns of CPs: (a) 1, (b) 2, and (c) 3 and (d) TGA diagrams of CPs 1–3. | ||
Solid-state fluorescence spectra of the H5L ligand and CPs 1–3 were measured at r.t. Under irradiation at 326 nm, CPs 1, 2, and 3 exhibited emission bands centered at 452, 445, and 420 nm, respectively, which could be assigned to the π → π* or n → π* transitions of the ligand.48 In comparison to the emission maximum of CP 1, the blue-shifted maxima and decreased intensities of the fluorescence peaks of CPs 2 and 3 might be due to the varying degrees of inter-ligand interactions. These observation points toward the increasingly dense distributions of Lewis basic carboxylates and π-electron-rich aryl ether moieties in CPs 2 and 3 (Fig. 5c and S1†).34–36
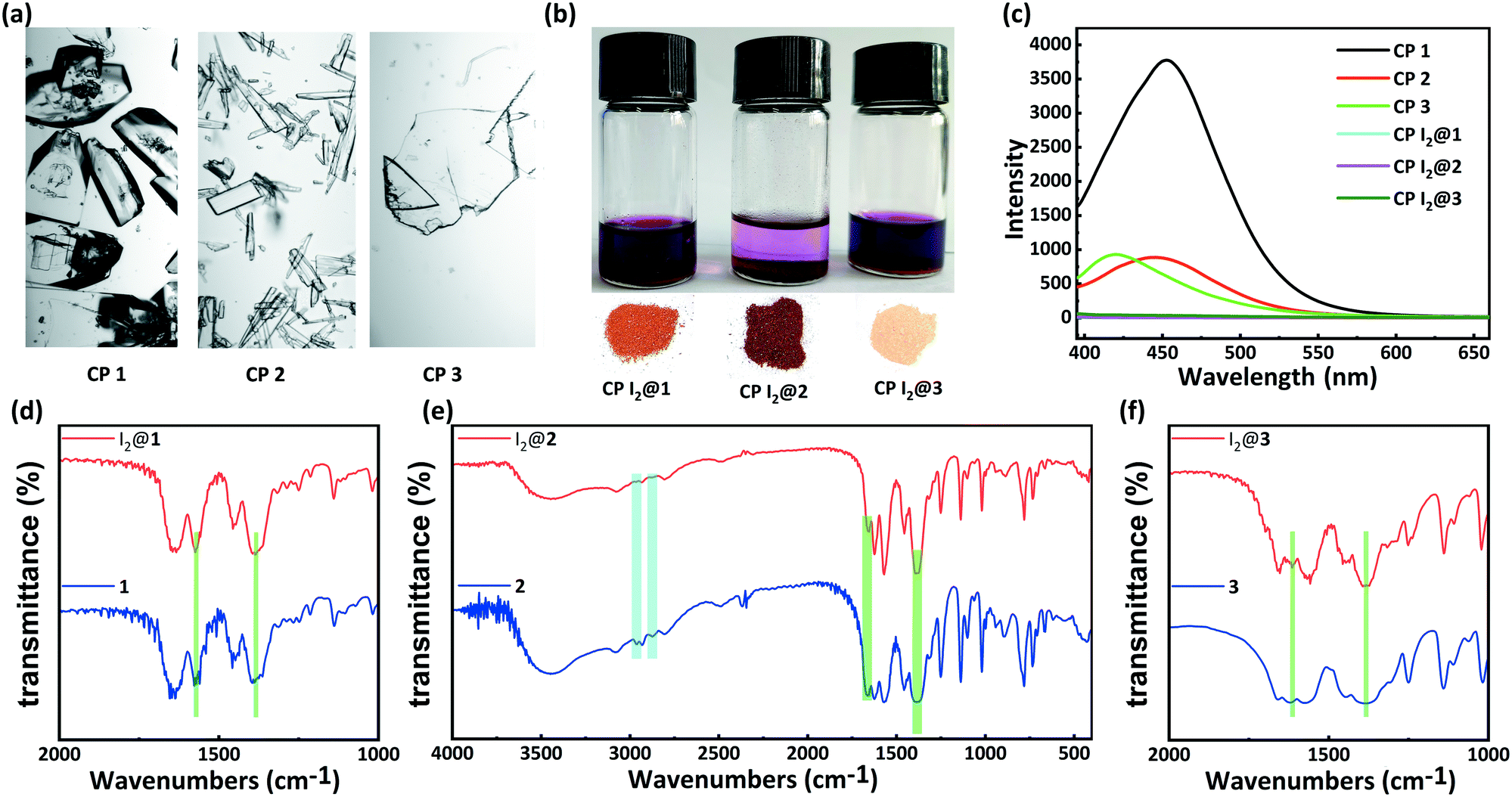 | ||
| Fig. 5 (a) Photographic images of CPs 1 (left), 2 (middle), and 3 (right). (b) Photographs of I2 adsorption in solutions and crystalline I2 inclusion compounds of CPs 1 (left), 2 (middle), 3 (right). (c) Solid luminescence spectra of CPs 1–3 before and after I2 adsorption in cyclohexane solution of I2 for 48 h. IR spectra of CPs (d) 1, (e) 2, (f) 3 before and after I2 adsorption in cyclohexane solution of I2 for 48 h. | ||
3.6. I2 adsorption studies
Inspired by the special structural features (tunable porosity and enriched aryl ether moieties as potential interaction sites) of CPs 1–3, the adsorption of I2 was studied using the soaking method. Iodine in cyclohexane exhibited an adsorption peak at 523 nm as measured by UV-vis spectroscopy. When 50 mg of CPs 1–3 crystals was exposed to a cyclohexane solution containing 0.01 M I2 at r.t. for 48 h, the crystals changed from the initial colorless state to pale brown and then to black. And the adsorption amount qt of CP 2 was ca. 152.1 mg g−1 (Table S2†). Meanwhile, the solution faded from dark red to pale red. The morphology and crystallinity were retained (Fig. 5a and b). The I2 adsorption capacities of CPs 1–3 were monitored by using UV-vis spectroscopy. The removal efficiency of iodine was defined using the following equation:where Ci and Ce are the initial and equilibrium concentrations in cyclohexane (mg mL−1). Notably, the removal efficiency of CP 2 (99.4%) substantially surpassed that of CP 1 (66.3%) and CP 3 (35.6%). The PXRD patterns (Fig. 4a–c) of CPs 1–3 confirmed that the structural frameworks maintained their crystalline integrity after the adsorption of I2. Additionally, the luminescence of CPs 1–3 was investigated after soaking the crystals in cyclohexane solutions of I2 for 48 h (Fig. 5c). It was found that the luminescence of CPs 1–3 was quenched completely after the loading of I2, which might indicate the possible existence of CT interactions between the CP hosts and the I2 guest molecules.49
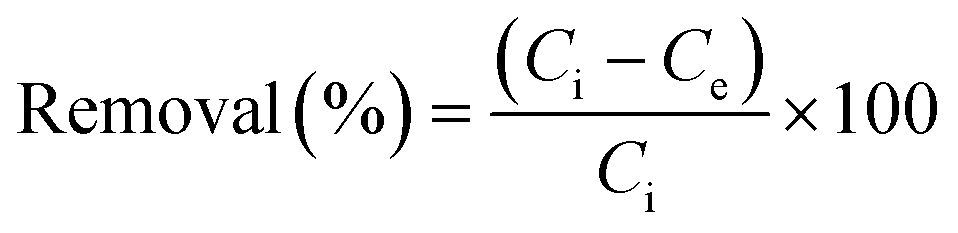
Moreover, the IR spectra of CPs 1–3 were measured to study the interactions between the CP scaffolds and I2. The IR spectrum of CP 2 after the adsorption of I2 clearly indicates that the intensities of the typical aromatic C–H stretching peaks (2967 cm−1 and 2880 cm−1) and the asymmetric vibration peak of the ester moiety (1657 cm−1) decreased remarkably. In contrast, the intensity of the symmetric vibration peak of the carboxylate moiety (1384 cm−1) increased (Fig. 5e). The iodine-encapsulated CPs 1 and 3 also exhibited varied intensities of carboxylate peaks. In particular, the augmented and sharpened symmetric vibrations at 1384 cm−1 reflected the interactions between the carboxylate moieties and the encapsulated iodine molecules. However, no variations were found in the C–H stretching peaks of the phenyl moieties in the iodine-encapsulated CPs 1 and 3, implying that the interactions between the aromatic scaffolds and the encapsulated I2 were relatively weak (Fig. 5d and f). These results demonstrated that the carboxylate⋯iodine interactions were favorable in all the CPs regardless of the different sizes of their pores. However, the coexistence of carboxylate⋯iodine and aromatic⋯iodine interactions gave rise to a higher iodine uptake ability. This essential synergy between the two types of noncovalent interactions seemed to be correlated to the crystal structures of the CP hosts.
We were able to obtain the incubated I2@2 crystals after immersing CP 2 in a cyclohexane solution of I2 (0.01 M) for 48 h. Single-crystal X-ray diffraction of I2@2 revealed that the I2 molecules were “held” in the channels via weak interactions (Fig. 6a) by multiple CO⋯I (ca. 3.58, 3.60, and 3.78 Å) and π⋯I (ca. 4.15 Å) interactions (Fig. 6b), and the theoretical total site occupancy of I2 was 0.52 per unit cell. Electrochemical impedance spectroscopy of CP 2 and the encapsulated structure I2@2 was conducted to study the effect of host–guest interactions. The lower charge-transfer resistance (Rct) value of I2@2 than that of CP 2 validated the CT interaction between the π-electron-rich aryl moieties of the ligands and the guest molecule I2 (Fig. 6c).50–53
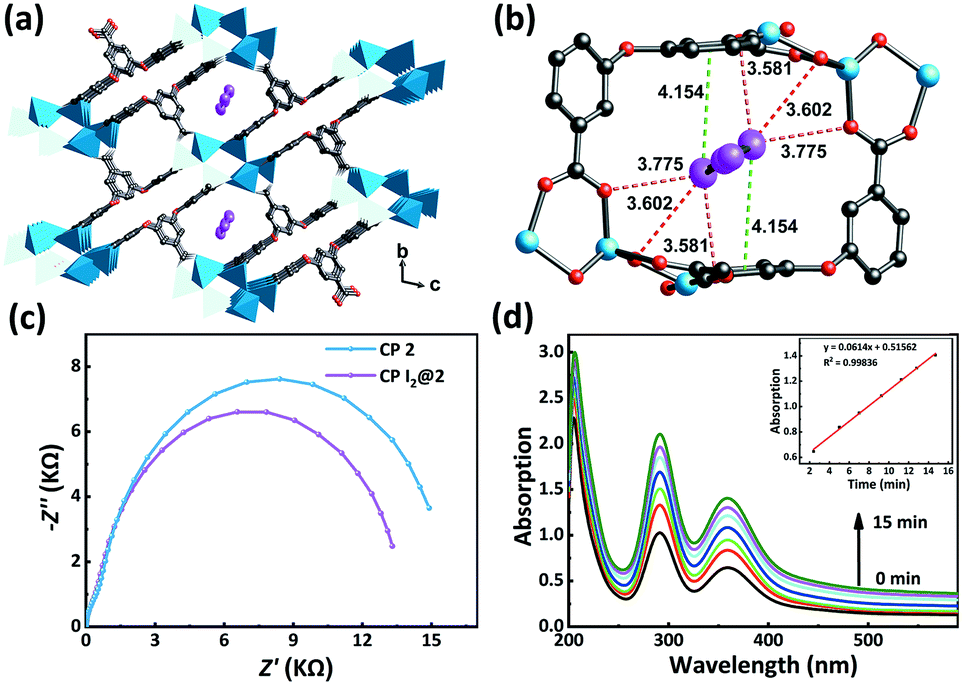 | ||
| Fig. 6 (a) I2 guest molecules are encapsulated in the channels in CP I2@2. (b) The linkages between I2 and the framework through CO⋯I and π⋯I interactions (green dashed line – π⋯I interaction; red dashed line – CO⋯I interaction). (c) Electrochemical impedance spectra of CP 2 and CP I2@2. (d) The I2 release process of CP I2@2. The inset of figure is the kinetic study of I2 release over time. | ||
Next, to gain better visualization and understanding of the desorption process, we have analyzed the case of CP 2, which had the highest adsorption efficiency. When the encapsulated I2@2 sample was dipped in fresh ethanol, the color of the crystals faded, while the color of the ethanol solution deepened gradually. This observation indicated that the highly polar solvent interfered with the noncovalent interactions between the framework and I2 molecules. In other words, I2 could be extracted from the pores using the ethanol solvent. Subsequently, the desorption process of I2 from I2@2 was further recorded by UV-vis spectroscopy. A total of 3 mg of I2@2 crystals was immersed in fresh ethanol (3 mL) at r.t. for monitoring the release of I2. It was observed that the concentration of I2 in the solution increased linearly with time, indicating that the I2 release process followed zero-order kinetics, and the desorption behavior was controlled by the host–guest interactions (Fig. 6d).
Finally, on the basis of all the above experimental observations, it was speculated that the mechanism of I2 adsorption depends on the intermolecular π⋯I interactions between the benzene rings of H5L and I2 as well as the CO⋯I halogen bond between the carboxylic coordination group and I2. These interactions enabled controllable entrance and accommodation and release of I2 in the channels of CPs 1–3. In addition, the structural characteristics of the CPs and their I2 adsorption capacity connote that although the uptake of I2 in CPs 1–3 was correlated with porosity, it was not proportional to the void space. The pore size of CP 1 was large enough to accommodate I2. However, considering the ca. 120° intersection angles of the CP 1 channels and the highly directional nature of CO⋯I and π⋯I interactions, it might not be easy for the carboxylate and the aryl moieties on the inner walls of the channels to allow two types of noncovalent interactions that converge to the same I2 molecule. Conversely, the small pore sizes of CP 3 did not facilitate the ingress of iodine molecules, leading to a relatively low adsorption capacity. In comparison, the suitable channel size of CP 2 and the appropriate spatial distribution of the carboxylates and the electron-rich aromatic rings allowed the synergy of CO⋯I and π⋯I interactions to ensure more durable I2 inclusion and a higher uptake amount. Notably, the empirical rule revealed here applies to the structure–activity relationship analysis for the iodine adsorption of related CPs based on similar polycarboxylate aryl ether ligands and zinc nodes. In the work of Su and Lan,54 the microporous channels surrounded by π-electron-rich walls of polycarboxylate aryl ether ligands endowed the zinc-based CP IFMC-69 with remarkably higher iodine adsorption ability compared to the isomeric IFMC-68 containing much larger cavities.
4. Conclusions
Three isomeric CPs were obtained based on a π-electron-rich polycarboxylate ligand by tuning the solvothermal reaction conditions. The synergistic effects of halogen bonds and CT interactions were systematically investigated in CPs with varied structures. Compared to CPs 1 and 3, CP 2, which possessed the most appropriate channel size and arrangement of the noncovalent interaction sites, exhibited the optimal iodine adsorption capacity. This was achieved through the synergy of CO⋯I and π⋯I interactions between the encapsulated iodine molecules and the Lewis basic carboxylate and π-electron-rich benzene moieties, respectively. This work showcased a structure–activity relationship-oriented design of CP-based iodine adsorbents and the crystal engineering strategy for screening the performance of CP candidates in a comparative study manner. This study can facilitate the future development of value-added host–guest systems based on the CP-derived porous materials with well-defined noncovalent interaction functional sites.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge financial support from the National Basic Research Program (973 Program) (No. 2014CB660804), the National Natural Science Foundation of China (No. 21971031, 21671114, and 21401112), the Scientific Research and Service Platform Fund of Henan Province (No. 2016151), the Fund of Scientific and Technological Innovation Team of Water Ecological Security for the Water Source Region of Mid-line of South-to-North Diversion Project of Henan Province, and Nanyang Normal University.Notes and references
- X. Yi, K. Yang, C. Liang, X. Zhong, P. Ning, G. Song, D. Wang, C. Ge, C. Chen, Z. Chai and Z. Liu, Adv. Funct. Mater., 2015, 25, 4689–4699 CrossRef CAS.
- P. M. Gignac, N. J. Kley, J. A. Clarke, M. W. Colbert, A. C. Morhardt, D. Cerio, I. N. Cost, P. G. Cox, J. D. Daza, C. M. Early, M. S. Echols, R. M. Henkelman, A. N. Herdina, C. M. Holliday, Z. Li, K. Mahlow, S. Merchant, J. Müller, C. P. Orsbon, D. J. Paluh, M. L. Thies, H. P. Tsai and L. M. Witmer, J. Anat., 2016, 228, 889–909 CrossRef CAS.
- X. Ge, J. Yu, Z. Wang, Y. Xu, C. Pan, L. Jiang, Y. Yang, K. Yuan and W. Liu, BMC Cancer, 2018, 18, 1120 CrossRef CAS.
- F. Schöne, K. Spörl and M. Leiterer, J. Trace Elem. Med. Biol., 2017, 39, 202–209 CrossRef.
- O. L. van der Reijden, M. B. Zimmermann and V. Galetti, Best Pract. Res., Clin. Endocrinol. Metab., 2017, 31, 385–395 CrossRef CAS.
- C. Martínez, A. E. Bosnidou, S. Allmendinger and K. Muñiz, Chem. – Eur. J., 2016, 22, 9929–9932 CrossRef.
- T. Duhamel, C. J. Stein, C. Martínez, M. Reiher and K. Muñiz, ACS Catal., 2018, 8, 3918–3925 CrossRef CAS.
- T. Müller and D. Winter, Mol. Cell. Proteomics, 2017, 16, 1173–1187 CrossRef.
- A. Oluwasanjo, R. Pathak, A. Ukaigwe and O. Alese, Cancer Causes Control, 2016, 27, 143–146 CrossRef.
- G. H. Seo, Y. Y. Cho, J. H. Chung and S. W. Kim, Thyroid, 2015, 25, 927–934 CrossRef.
- K. S. Subrahmanyam, D. Sarma, C. D. Malliakas, K. Polychronopoulou, B. J. Riley, D. A. Pierce, J. Chun and M. G. Kanatzidis, Chem. Mater., 2015, 27, 2619–2626 CrossRef CAS.
- B. J. Riley, J. Chun, J. V. Ryan, J. Matyáš, X. S. Li, D. W. Matson, S. K. Sundaram, D. M. Strachan and J. D. Vienna, RSC Adv., 2011, 1, 1704 RSC.
- A. Miller, J. Kruichak, M. Mills and Y. Wang, J. Environ. Radioact., 2015, 147, 108–114 CrossRef CAS.
- Z. Tauanov and V. J. Inglezakis, Sci. Total Environ., 2019, 682, 259–270 CrossRef CAS.
- B. E. Souza, L. Donà, K. Titov, P. Bruzzese, Z. Zeng, Y. Zhang, A. S. Babal, A. F. Möslein, M. D. Frogley, M. Wolna, G. Cinque, B. Civalleri and J.-C. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 5147–5156 CrossRef CAS.
- Z. Hu, W. P. Lustig, J. Zhang, C. Zheng, H. Wang, S. J. Teat, Q. Gong, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2015, 137, 16209–16215 CrossRef CAS.
- M. Xu, S.-S. Yang and Z.-Y. Gu, Chem. – Eur. J., 2018, 24, 15131–15142 CrossRef CAS.
- Z. Chang, R.-B. Lin, Y. Ye, C. Duan and B. Chen, J. Mater. Chem. A, 2019, 7, 25567–25572 RSC.
- D. Chen, W. Shi and P. Cheng, Chem. Commun., 2015, 51, 370–372 RSC.
- D. Shi, L. Zeng, Z. Ming, C. He, C. Meng and C. Duan, RSC Adv., 2016, 6, 51936–51940 RSC.
- T. Zhang, Y. Jin, Y. Shi, M. Li, J. Li and C. Duan, Coord. Chem. Rev., 2019, 380, 201–229 CrossRef CAS.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- X. Feng, Y. Feng, N. Guo, Y. Sun, T. Zhang, L. Ma and L. Wang, Inorg. Chem., 2017, 56, 1713–1721 CrossRef CAS.
- B. Guo, F. Li, C. Wang, L. Zhang and D. Sun, J. Mater. Chem. A, 2019, 7, 13173–13179 RSC.
- M. Jia, J. Li, S. Che, L. Kan, G. Li and Y. Liu, Inorg. Chem. Front., 2019, 6, 1261–1266 RSC.
- J.-H. Liu, Y.-J. Qi, D. Zhao, H.-H. Li and S.-T. Zheng, Inorg. Chem., 2019, 58, 516–523 CrossRef CAS.
- Y. Tang, H. Huang, J. Li, W. Xue and C. Zhong, J. Mater. Chem. A, 2019, 7, 18324–18329 RSC.
- W. Xie, D. Cui, S.-R. Zhang, Y.-H. Xu and D.-L. Jiang, Mater. Horiz., 2019, 6, 1571–1595 RSC.
- A. Beheshti, E. S. Mousavi Fard, M. Kubicki, P. Mayer, C. T. Abrahams and S. E. Razatofighi, CrystEngComm, 2019, 21, 251–262 RSC.
- Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2011, 47, 7185–7187 RSC.
- R.-X. Yao, X. Cui, X.-X. Jia, F.-Q. Zhang and X.-M. Zhang, Inorg. Chem., 2016, 55, 9270–9275 CrossRef CAS.
- A. Peuronen, A. Valkonen, M. Kortelainen, K. Rissanen and M. Lahtinen, Cryst. Growth Des., 2012, 12, 4157–4169 CrossRef CAS.
- L.-M. Zhao, W.-T. Zhang, K.-Y. Song, Q.-Q. Wu, Y. Li, H.-H. Li and Z.-R. Chen, CrystEngComm, 2018, 20, 2245–2252 RSC.
- P. Shen, W.-W. He, D.-Y. Du, H.-L. Jiang, S.-L. Li, Z.-L. Lang, Z.-M. Su, Q. Fu and Y.-Q. Lan, Chem. Sci., 2014, 5, 1368–1374 RSC.
- X. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitórica-Yrezábal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudić, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS.
- M.-H. Zeng, Q.-X. Wang, Y.-X. Tan, S. Hu, H.-X. Zhao, L.-S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS.
- D. Banerjee, X. Chen, S. S. Lobanov, A. M. Plonka, X. Chan, J. A. Daly, T. Kim, P. K. Thallapally and J. B. Parise, ACS Appl. Mater. Interfaces, 2018, 10, 10622–10626 CrossRef CAS.
- N. Zhang, L.-X. Sun, Y.-H. Xing and F.-Y. Bai, Cryst. Growth Des., 2019, 19, 5686–5695 CrossRef CAS.
- D. Wang, D. Zhang, S.-D. Han, J. Pan, Z.-Z. Xue, J.-H. Li and G.-M. Wang, Dalton Trans., 2019, 48, 602–608 RSC.
- L. Esrafili, A. A. Tehrani, A. Morsali, L. Carlucci and D. M. Proserpio, Inorg. Chim. Acta, 2019, 484, 386–393 CrossRef CAS.
- S.-L. Wang, F.-L. Hu, J.-Y. Zhou, Y. Zhou, Q. Huang and J.-P. Lang, Cryst. Growth Des., 2015, 15, 4087–4097 CrossRef CAS.
- D. Shi, C. He, B. Qi, C. Chen, J. Niu and C. Duan, Chem. Sci., 2015, 6, 1035–1042 RSC.
- L. Liu, Z. Yao, Y. Ye, Q. Lin, S. Chen, Z. Zhang and S. Xiang, Cryst. Growth Des., 2018, 18, 3724–3728 CrossRef CAS.
- K. P. Rao, M. Higuchi, J. Duan and S. Kitagawa, Cryst. Growth Des., 2013, 13, 981–985 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- J. Cui, Y. Li, Z. Guo and H. Zheng, Chem. Commun., 2013, 49, 555–557 RSC.
- X.-L. Hu, F.-H. Liu, H.-N. Wang, C. Qin, C.-Y. Sun, Z.-M. Su and F.-C. Liu, J. Mater. Chem. A, 2014, 2, 14827–14834 RSC.
- Y.-H. Luo, X.-Y. Yu, J.-J. Yang and H. Zhang, CrystEngComm, 2014, 16, 47–50 RSC.
- Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, S. K. Kwak, J.-W. Yoo and R. S. Ruoff, J. Am. Chem. Soc., 2019, 141, 16884–16893 CrossRef CAS.
- X. Zhang, I. da Silva, R. Fazzi, A. M. Sheveleva, X. Han, B. F. Spencer, S. A. Sapchenko, F. Tuna, E. J. L. McInnes, M. Li, S. Yang and M. Schröder, Inorg. Chem., 2019, 58, 14145–14150 CrossRef CAS.
- K. Naskar, A. Dey, S. Maity, M. K. Bhunia, P. P. Ray and C. Sinha, Cryst. Growth Des., 2019, 19, 2206–2218 CrossRef CAS.
- P. Shen, W.-W. He, D.-Y. Du, H.-L. Jiang, S.-L. Li, Z.-L. Lang, Z.-M. Su, Q. Fu and Y.-Q. Lan, Chem. Sci., 2014, 5, 1368–1374 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Coordination modes of H5L ligands and selected bond lengths and angles for 1–3 and I2@2. CCDC 1588500, 1883449, 1911376 and 2007800. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01004a |
| This journal is © The Royal Society of Chemistry 2020 |