
Design of Au@Ag/BiOCl–OV photocatalyst and its application in selective alcohol oxidation driven by plasmonic carriers using O2 as the oxidant†
 *d
*d
Abstract
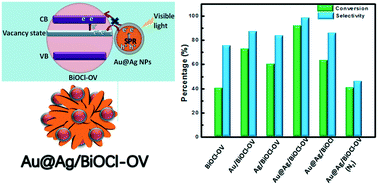
The hot holes produced by plasmonic noble metals have a mild oxidizing ability and provide an ideal alternative for photocatalytic selective oxidation. In this work, BiOCl with oxygen vacancies (BiOCl–OV) was photosensitized to the visible spectrum using plasmonic Au@Ag nanoparticles (NPs) to study photocatalytic selective alcohol oxidation using O2 as the oxidant. In the Au@Ag/BiOCl–OV system, the Au@Ag NPs consisting of a plasmonic Au core and covered by a very thin Ag shell were distributed on BiOCl spheres with the assistance of 3-aminopropyl-triethoxysilane (APTES). The combination of Au@Ag NPs and BiOCl–OV into one system endowed it with remarkable advantages in photocatalytic selective oxidation. Firstly, strong O2-adsorption capacity was achieved on the BiOCl–OV surface due to the presence of oxygen vacancies that serve as active sites. Secondly, the electron–hole generation capability is enhanced due to the extended optical absorption properties and amplified electromagnetic (EM) field effect that originated from the surface plasmon resonance (SPR) coupling effect of Au and Ag. Thirdly, the enhanced separation efficiency of hot electrons and holes was achieved through the trapping of hot electrons by the oxygen vacancies. The aforesaid, resulted in the remarkable enhancement of Au@Ag/BiOCl–OV as a photocatalyst in the selective oxidation of benzyl alcohol to benzaldehyde. We believe that the present strategy based on the plasmonic effect would be a significant contribution in the design and preparation of efficient photocatalysts for the selective oxidation.
 Please wait while we load your content...
Please wait while we load your content...

