
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic visualization of type II peptidyl carrier protein recognition in pyoluteorin biosynthesis†
Joshua C.
Corpuz
 a,
Larissa M.
Podust
b,
Tony D.
Davis
a,
Matt J.
Jaremko
a and
Michael D.
Burkart
*a
a,
Larissa M.
Podust
b,
Tony D.
Davis
a,
Matt J.
Jaremko
a and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California-San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
bSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California-San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0755, USA
First published on 24th March 2020
Abstract
Using a covalent chemical probe and X-ray crystallography coupled to nuclear magnetic resonance data, we elucidated the dynamic molecular basis of protein recognition between the carrier protein and adenylation domain in pyoluteorin biosynthesis. These findings reveal a unique binding mode, which contrasts previously solved carrier protein and partner protein interfaces.
The type II non-ribosomal peptide synthetase (NRPS) system consists of stand-alone enzymes that commonly participate in hybrid pathways along with fatty acid synthase (FAS) and polyketide synthase (PKS) enzymes.1 The stand-alone architecture of type II NRPS proteins makes them promising candidates for metabolic engineering, as they commonly serve to install unique chemical functionality into growing metabolites. These hybrid pathways afford complex natural products that include antibiotic, antitumor, and antifungal agents.2 Examples of functionalized natural products include those that utilize dehydrogenated prolines as a pharmacophore, including prodigiosin, pyoluteorin, and chlorizidine A, all of which require type II NRPS proteins to functionalize and incorporate a pyrrole into the natural product (Fig. 1).3–5
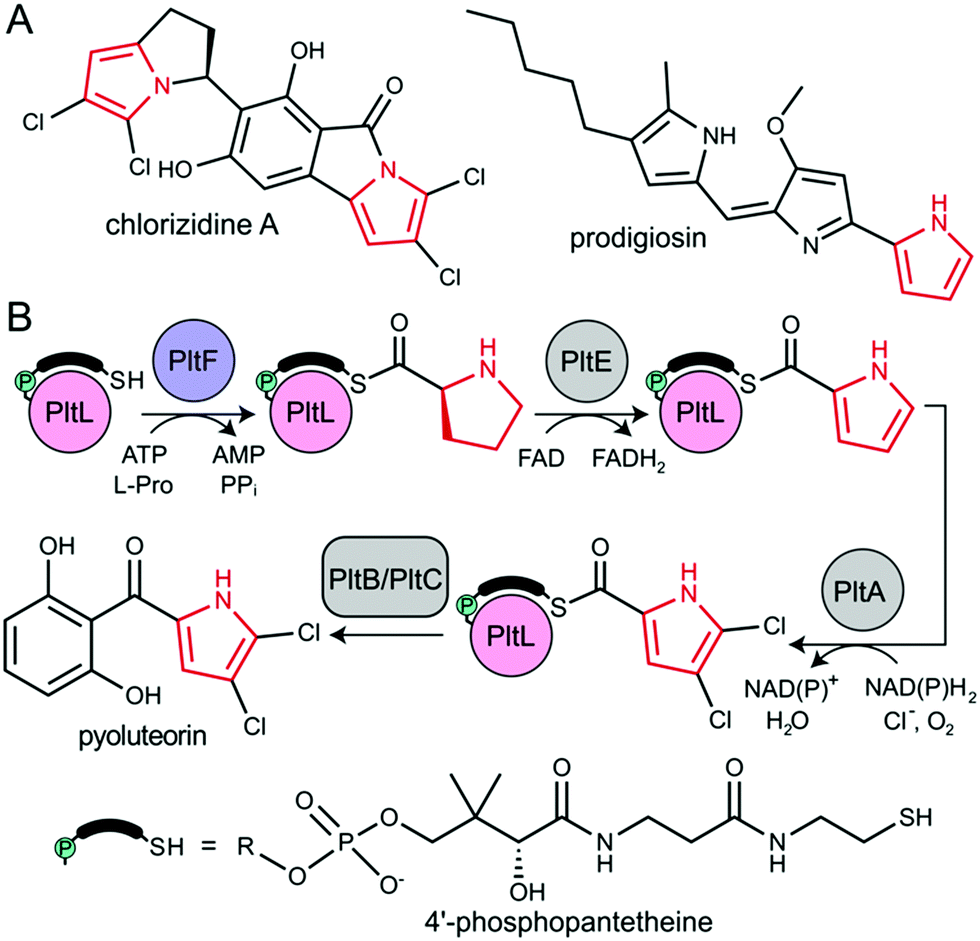 | ||
| Fig. 1 (A) Natural product examples that incorporate pyrrole (red) via type II NRPS proteins. (B) Pyrrole functionalization and installation in pyoluteorin via type II NRPS proteins. PltB and PltC is a type I PKS. | ||
All NRPS systems include an adenylation (A) and peptidyl carrier protein (PCP) domain to activate and load substrates. The A domain activates a specific amino acid substrate and subsequently installs the substrate onto the thiol of the 4′-phosphopantetheine (PPant) arm of the PCP (Fig. 2A).6 The PCP is a 10 kDa protein that consists of a 4-helix bundle.7 The PPant modification is attached onto an invariant serine via a phosphopantetheinyl transferase to activate the PCP to the holo-form. The PCP can transport acyl or aminoacyl cargo to a variety of enzymatic domains for functionalization and incorporation of the substrate into the nascent natural product.
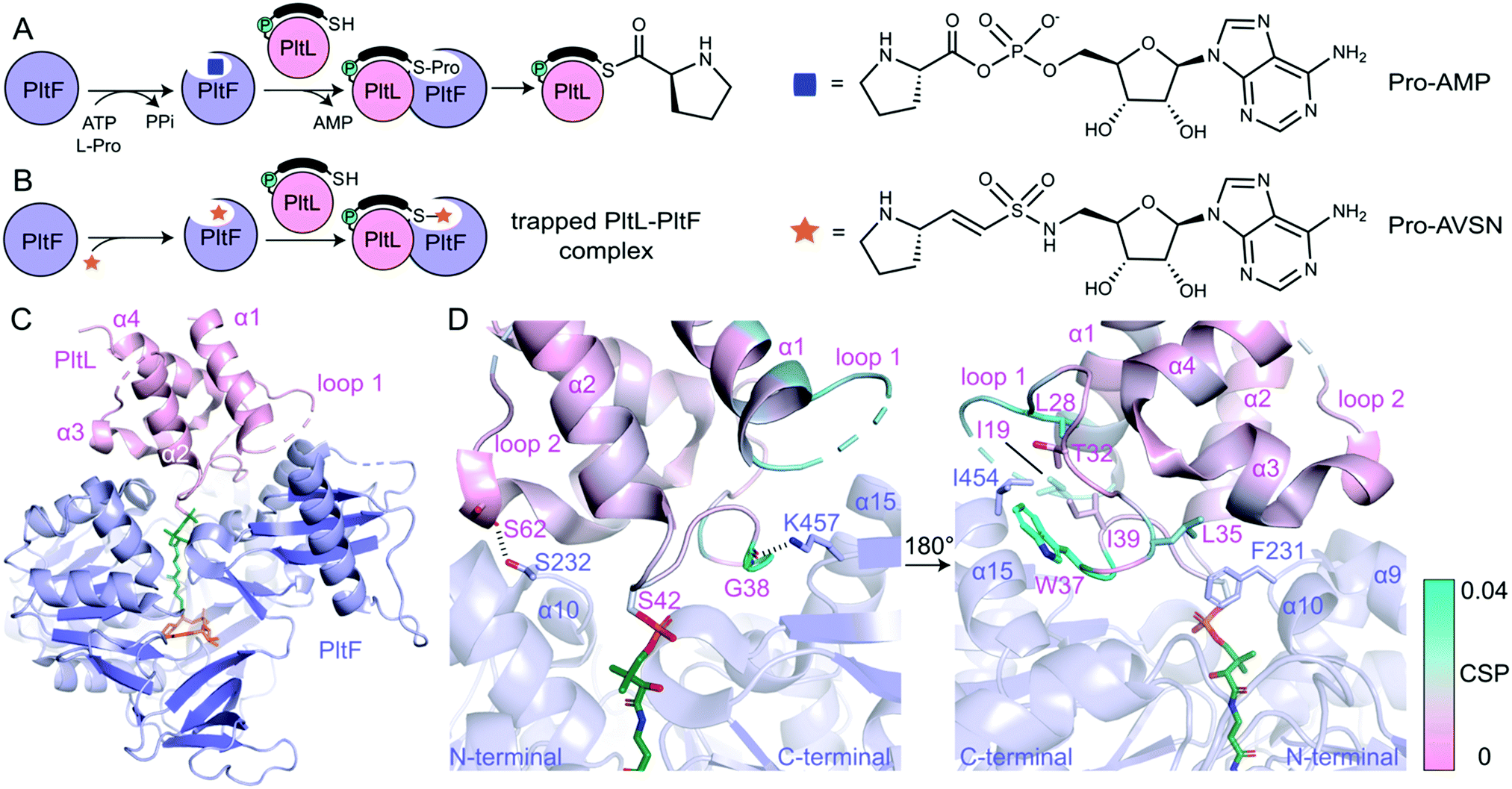 | ||
| Fig. 2 The PltL–PltF crystal structure. (A) Adenylation and thiolation of L-Pro by PltF. (B) Trapping of the PltL–PltF complex with Pro-AVSN (orange star). (C) 2.15 Å resolution crystal structure of the PltL–PltF complex. PltL (pink) is trapped to PltF (purple) via Pro-AVSN (orange). PPant is shown in green. (D) Close-up of the PltL–PltF interface. The left panel displays hydrogen bonding interactions. The right panel, which is rotated 180° from the left, displays hydrophobic interactions. The CSPs (cyan) from a recent NMR titration of PltL with PltF are mapped onto the bound PltL. | ||
During pyoluteorin biosynthesis in Pseudomonas fluorescens, the type II NRPS PCP, holo-PltL, is loaded with L-Pro by the A domain, PltF (Fig. 1). While protecting its substrate in a hydrophobic cleft,8 prolyl-PltL transports the L-Pro for dehydrogenation and dichlorination before being off-loaded onto a type I PKS for the incorporation of the dichloropyrrolyl substrate into pyoluteorin.9
PltL has been shown to exhibit specificity towards PltF and no interactivity towards homologous A domains.10,11 This suggested the requirement of a specific protein–protein interaction motif for A domain activity. Studies have attempted to control the partner protein specificity of PltL; solution-phase nuclear magnetic resonance (NMR) titration experiments revealed a region of PltL, loop 1 (residues 19–41), that was postulated to form the protein–protein interface with PltF.10 Mutagenesis of residues in this region disrupted activity, however, these studies could not resolve the PCP-A domain interface clearly enough to accurately manipulate PltL specificity.
In order to determine the basis of molecular recognition between PltL and PltF, we set out to structurally characterize the PltL–PltF complex with X-ray crystallography. Carrier proteins (CPs) and partner proteins often form weak transient interactions and high disassociation may impede co-crystallization.12 To stabilize the interaction between PltL and PltF, a substrate mimic of the proline adenosine monophosphate (Pro-AMP) intermediate was deemed necessary. Based on a covalent inhibitor motif developed by the Aldrich and Tan groups,13–18 the proline adenosine vinylsulfonamide (Pro-AVSN) was synthesized (see ESI†) and employed to trap PltL with PltF (Fig. 2B). Incubation of holo-PltL, PltF and Pro-AVSN enabled crystallization and optimization led to a crystal that diffracted to 2.15 Å. Molecular replacement was carried out using the A domain, DltA (PDB ID 3E7W), in the thiolation state19 and with PltL (PDB ID 2N5H) (Fig. S2, ESI†).8
The PltL–PltF crystal structure contains PltL trapped with PltF in the thiolation state via addition to Pro-AVSN (Fig. 2C). Successful trapping of the complex is shown by electron density of the PPant extended into the active site of PltF, with its terminal thiol covalently linked to the sulfonamide β-carbon of Pro-AVSN (Fig. S3, ESI†). The putative K486 responsible for adenylation is 25 Å away from the active site, consistent with the domain alternation hypothesis proposed by Gulick and coworkers (Fig. S4A, ESI†).20 The N-terminal domain (NTD) contains an AMP binding pocket and L-Pro binding pocket that is conserved amongst A domains that process L-Pro (Fig. S4A and S5, ESI†).21 The bound PltL maintains the conserved four-α helix bundle, with the PPant attached onto Ser42 at the top of helix 2 (Fig. S4B, ESI†). Loop 1 residues 20–25 had poor electron density, so no atomic model was built for this region.
The PltL–PltF crystal structure reveals a protein–protein interface that is mediated by interactions between the loop 1 region of PltL with PltF helix 15 and the turn between helix 9 and 10 (Fig. 2D and 3B). A portion of PltL helix 2 and loop 2 also contribute to the interface. The backbone carbonyl of PltL Ser62 and Gly38 form hydrogen bonds (H-bonds) with PltF Ser232 and Lys457, respectively (Fig. 2D). The phosphate of the PPant arm also forms an electrostatic interaction with PltF Arg404. PltF Ile454 is positioned inside a hydrophobic pocket created by PltL loop 1 residues Ile19, Leu28, Trp37, and Ile39 (Fig. 2D and Fig. S6B, ESI†). The indole ring of PltL Trp37 is buried along PltF helix 15 (Fig. S6A, ESI†). PltF Phe231 is observed in a hydrophobic cleft between PltL helices 2, 3 and loop 1 residue Leu35 (Fig. 2D and 3C).
Previously reported NMR titration studies of holo-15N-PltL with PltF revealed significant chemical shift perturbations (CSPs) in PltL loop 1 residues (Fig. S7, ESI†), implicating their participation in, or response to, the binding event.10 Of those residues, Ile19, Leu28, Leu35, Trp37, and Gly38 were seen at the protein–protein interface in the crystal structure (Fig. 2D). The remaining residues with high CSPs are likely due to changes in the secondary structural interactions between helix 1 and loop 1 (Fig. S8, ESI†).
To gain insight into the highest relative CSPs found in PltL Trp37 and Gly38, the solution NMR structure of holo-PltL was aligned and superposed to PltL in the bound state with PltF (Fig. 3B). Comparison of the different states reveal the lack of the loop 1 hydrophobic pocket in the unbound PltL that contacts PltF Ile454 (Fig. 3A and Fig. S8, ESI†). The superposition shows the pocket formation via a 5.5 Å displacement of the indole ring of PltL Trp37 by PltF Ile454. Adjacent to Trp37 is Gly38, which forms a H-bond via its backbone carbonyl with PltF Lys457 that may stabilize the displacement. The relevance of the hydrophobic pocket formation and H-bonding was demonstrated by mutagenesis of Ile454Ala and Lys457Ala, respectively, where individual mutations both stunted aminoacylation activity (Fig. 3D). Furthermore, previous molecular dynamic simulations revealed the relative flexibility of PltL loop 1, which supports loop 1 reorganization upon binding.10 These data provide evidence that specific H-bonding and hydrophobic interactions allowed by the conformational flexibility of PltL loop 1 is responsible for its recognition towards PltF.
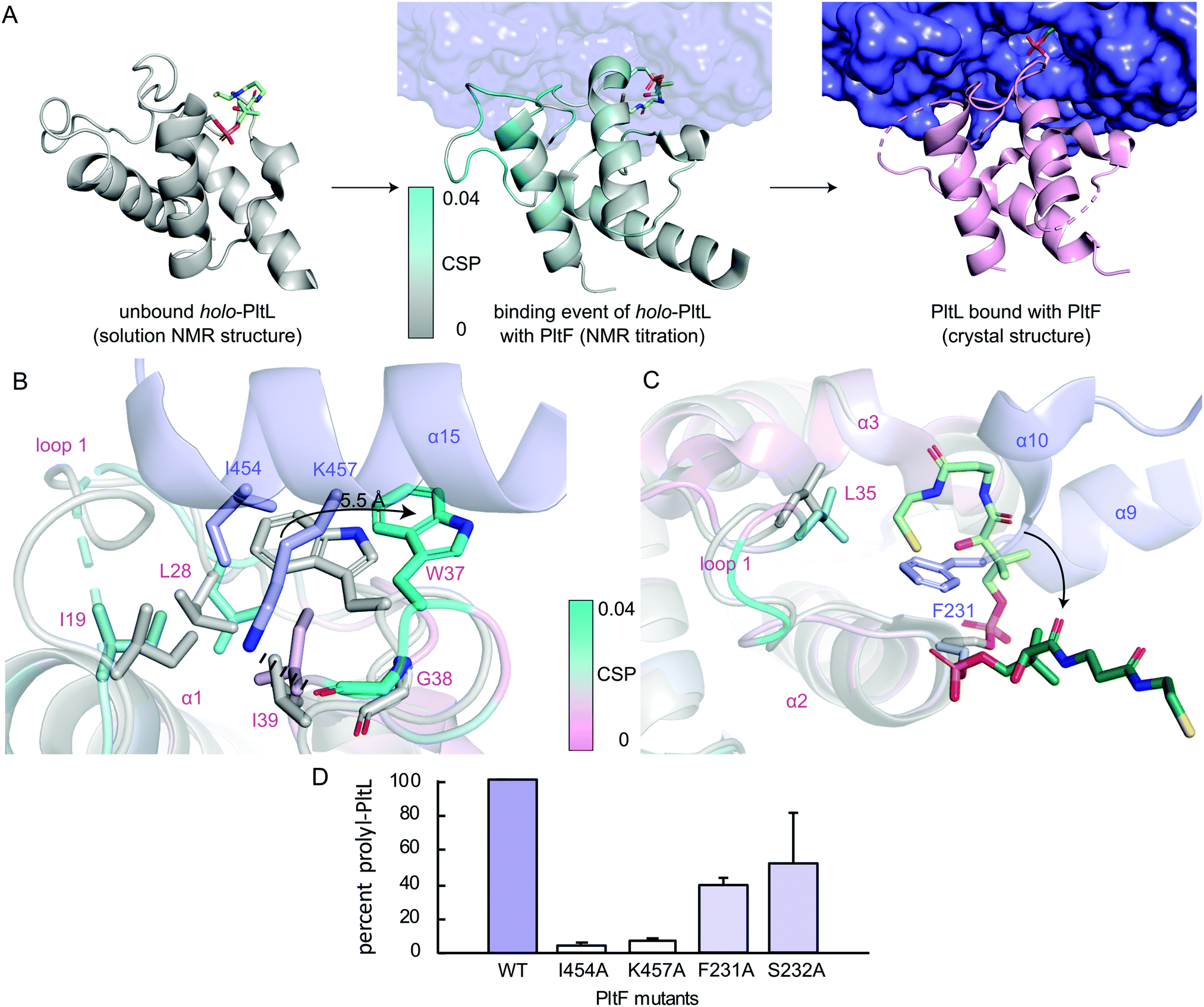 | ||
| Fig. 3 Visualization of the PltL–PltF binding event. (A) Linking of the solution NMR structure of holo-PltL (gray, PDB ID 2N5H), the CSPs (cyan) from the NMR titration, and the PltL–PltF (pink, purple) complex. (B) Superposition of the bound (pink, cyan) and unbound (gray) PltL with PltF Ile454 (purple) and (C) Phe231 (purple). Pro-AVSN was omitted for clarity. (D) PltF mutant aminoacylation assay with PltL. | ||
Superposition of bound and unbound PltL also reveals PltF Phe231 residing between helices 2 and 3 of PltL, which occludes the hydrophobic cleft that protects the substrate (Fig. 3C).8 This suggests that the hydrophobic cleft is not only involved in substrate protection but also molecular recognition. Mutagenesis of PltF Phe231Ala and Ser232Ala results in decreased PltL aminoacylation, which supports its relevance in PltL–PltF recognition (Fig. 3D). Recent structural analysis of the type II PCP of pentabromopseudilin biosynthesis, Bmp1, in complex with the oxidase, Bmp2, yields similar conclusions.22
Next, we compared the PltL–PltF interface against other PCP-A domain interfaces from the crystal structures of LgrA, EntF, EntE–EntB, and PA1221.14,15,23,24 The most outstanding difference is the location of the interface interactions. The PCP interfaces of LgrA, EntF, PA1221, and EntE–EntB involve the loop 1 and, to a larger extent, helix 2 of the PCP (Fig. 4). In contrast, the PltL–PltF structure reveals that only the first residue of PltL helix 2, Met43, is partially buried at the interface. The loop 1 region of PltL is instead the main contributor to the interface with PltF. This interface independent of helix 2 is surprising considering the established prevalence of helix 2 at the interface of CP-dependent pathways, such as the E. coli type II FAS, where the CP–enzyme interface primarily relies on helix 2 for binding (Fig. S11, ESI†).25–27
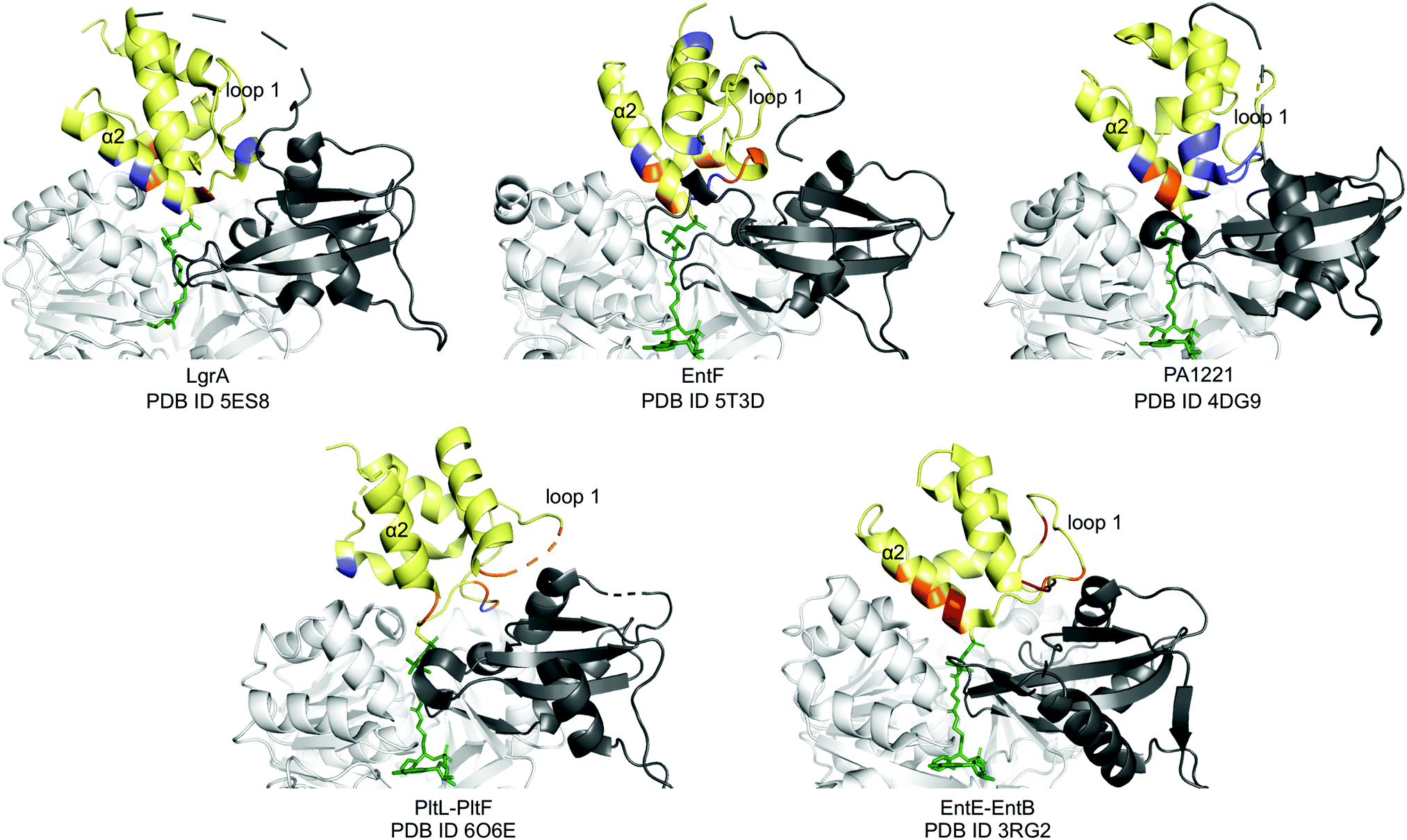 | ||
| Fig. 4 PCP-A domain interface comparison. Yellow, PCP; dark grey, A domain C-terminal domain; white, A domain N-terminal domain; blue, H-bonding; red, salt bridge; orange, hydrophobic. | ||
While PCP-A domain interfaces consist of hydrophobic interactions, each pathway differs in number of H-bonds and salt bridges for partner protein recognition (Fig. 4). The PltL–PltF structure reveals two H-bonds at the interface, whereas EntE–EntB employs multiple salt bridges. In contrast, the LgrA, EntF, and PA1221 structures contain four or more H-bonds. The type of interaction in addition to the location of each interaction presents a challenge towards the combinatorial biosynthesis of CP-dependent pathways.
Conclusions
The current structural investigation enhances our understanding of the molecular basis of PltL–PltF interactions. While our previous NMR titrations identified potential interface residues on PltL, mutagenesis studies did not afford noncognate activity. Here, combining NMR titrations, chemical trapping, X-ray crystallography, and mutagenesis assays allows us to more precisely identify the interface to understand specificity and predict functional mutations.This structure of the trapped PltL–PltF complex, complemented by prior NMR studies, has revealed new insights into a distinct mechanism of recognition used by A domains and PCPs. Resolving structural features of the protein–protein interface revealed the differences in the type, location, and dynamics of interfacial interactions that govern CP and partner protein recognition. These differences may serve a fundamental reason behind the limited success of prior combinatorial biosynthetic efforts in NRPSs. With our discoveries on the type II PCP and A domain from pyoluteorin biosynthesis, a layer of complexity has been revealed that will inform the future combinatorial biosynthetic efforts and engineering of CP-dependent pathways.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jefferey Mindrebo and Dr Ashay Patel for providing feedback on the manuscript, Dr Yongxuan Su for MS analyses, Dr Anthony Mrse and Dr Xuimei Huang for NMR assistance, and George Meigs, Dr James Holton, and Jane Tanamachi for X-ray data collection assistance. JCC was funded by the NIH Molecular Biophysics Training Grant T32GM008326. Funding was provided by NIH R01GM095970 to MDB and T32CA009523 to MJJ. TDD is a San Diego IRACDA postdoctoral fellow funded by NIH K12GM068524 and NIH K99GM129454. LMP was supported by the UC San Diego start-up funds. Beamline 8.3.1 of the Advanced Light Source, a DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the NIH, NIGMS, grant P30GM124169-01.References
- M. J. Jaremko, T. D. Davis, J. C. Corpuz and M. D. Burkart, Type II non-ribosomal peptide synthetase proteins: structure, mechanism, and protein–protein interactions, Nat. Prod. Rep., 2020 10.1039/C9NP00047J.
- D. J. Newman and G. M. Cragg, Natural Products as Sources of New Drugs from 1981 to 2014, J. Nat. Prod., 2016, 79(3), 629–661 CrossRef CAS PubMed.
- N. R. Williamson, H. T. Simonsen, R. A. A. Ahmed, G. Goldet, H. Slater and L. Woodley, et al., Biosynthesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel 2-methyl-3-n-amyl-pyrrole (MAP) assembly pathway, definition of the terminal condensing enzyme, and implications for undecylprodigiosin biosynthesis in Streptomyces: The biosynthetic pathway of prodigiosin in Serratia, Mol. Microbiol., 2005, 56(4), 971–989 CrossRef CAS PubMed.
- M. G. Thomas, M. D. Burkart and C. T. Walsh, Conversion of L-Proline to Pyrrolyl-2-Carboxyl-S-PCP during Undecylprodigiosin and Pyoluteorin Biosynthesis, Chem. Biol., 2002, 9(2), 171–184 CrossRef CAS PubMed.
- X. Álvarez-Micó, D. D. Rocha, L. A. Guimarães, A. Ambrose, E. Chapman and L. V. Costa-Lotufo, et al., The Hybrid Pyrroloisoindolone-Dehydropyrrolizine Alkaloid (−)-Chlorizidine A Targets Proteins within the Glycolytic Pathway, ChemBioChem, 2015, 16(14), 2002–2006 CrossRef PubMed.
- G. H. Hur, C. R. Vickery and M. D. Burkart, Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology, Nat. Prod. Rep., 2012, 29(10), 1074 RSC.
- A. C. Mercer and M. D. Burkart, The ubiquitous carrier protein—a window to metabolite biosynthesis, Nat. Prod. Rep., 2007, 24(4), 750 RSC.
- M. J. Jaremko, D. J. Lee, S. J. Opella and M. D. Burkart, Structure and Substrate Sequestration in the Pyoluteorin Type II Peptidyl Carrier Protein PltL, J. Am. Chem. Soc., 2015, 137(36), 11546–11549 CrossRef CAS PubMed.
- H. Gross and J. E. Loper, Genomics of secondary metabolite production by Pseudomonas spp, Nat. Prod. Rep., 2009, 26(11), 1408 RSC.
- M. J. Jaremko, D. J. Lee, A. Patel, V. Winslow, S. J. Opella and J. A. McCammon, et al., Manipulating Protein–Protein Interactions in Nonribosomal Peptide Synthetase Type II Peptidyl Carrier Proteins, Biochemistry, 2017, 56(40), 5269–5273 CrossRef CAS PubMed.
- J. Beld, K. Finzel and M. D. Burkart, Versatility of Acyl-Acyl Carrier Protein Synthetases, Chem. Biol., 2014, 21(10), 1293–1299 CrossRef CAS PubMed.
- A. M. Gulick and C. C. Aldrich, Trapping interactions between catalytic domains and carrier proteins of modular biosynthetic enzymes with chemical probes, Nat. Prod. Rep., 2018, 35(11), 1156–1184 RSC.
- C. Qiao, D. J. Wilson, E. M. Bennett and C. C. Aldrich, A Mechanism-Based Aryl Carrier Protein/Thiolation Domain Affinity Probe, J. Am. Chem. Soc., 2007, 129(20), 6350–6351 CrossRef CAS PubMed.
- C. A. Mitchell, C. Shi, C. C. Aldrich and A. M. Gulick, Structure of PA1221, a Nonribosomal Peptide Synthetase Containing Adenylation and Peptidyl Carrier Protein Domains, Biochemistry, 2012, 51(15), 3252–3263 CrossRef CAS PubMed.
- J. A. Sundlov, C. Shi, D. J. Wilson, C. C. Aldrich and A. M. Gulick, Structural and Functional Investigation of the Intermolecular Interaction between NRPS Adenylation and Carrier Protein Domains, Chem. Biol., 2012, 19(2), 188–198 CrossRef CAS PubMed.
- X. Lu, H. Zhang, P. J. Tonge and D. S. Tan, Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis, Bioorg. Med. Chem. Lett., 2008, 18(22), 5963–5966 CrossRef CAS PubMed.
- X. Lu, S. K. Olsen, A. D. Capili, J. S. Cisar, C. D. Lima and D. S. Tan, Designed Semisynthetic Protein Inhibitors of Ub/Ubl E1 Activating Enzymes, J. Am. Chem. Soc., 2010, 132(6), 1748–1749 CrossRef CAS PubMed.
- C. Ji, I. Sharma, D. Pratihar, L. L. Hudson, D. Maura and T. Guney, et al., Designed Small-Molecule Inhibitors of the Anthranilyl-CoA Synthetase PqsA Block Quinolone Biosynthesis in Pseudomonas aeruginosa, ACS Chem. Biol., 2016, 11(11), 3061–3067 CrossRef CAS PubMed.
- H. Yonus, P. Neumann, S. Zimmermann, J. J. May, M. A. Marahiel and M. T. Stubbs, Crystal Structure of DltA: Implications for the Reaction Mechanism of Non-Ribosomal Peptide Synthetase Adenylation Domains, J. Biol. Chem., 2008, 283(47), 32484–32491 CrossRef CAS PubMed.
- A. M. Gulick, Conformational Dynamics in the Acyl-CoA Synthetases, Adenylation Domains of Non-ribosomal Peptide Synthetases, and Firefly Luciferase, ACS Chem. Biol., 2009, 4(10), 811–827 CrossRef CAS PubMed.
- S. Schmelz and J. H. Naismith, Adenylate-forming enzymes, Curr. Opin. Struct. Biol., 2009, 19(6), 666–671 CrossRef CAS PubMed.
- H. R. Thapa, J. M. Robbins, B. S. Moore and V. Agarwal, Insights into Thiotemplated Pyrrole Biosynthesis Gained from the Crystal Structure of Flavin-Dependent Oxidase in Complex with Carrier Protein, Biochemistry, 2019, 58(7), 918–929 CrossRef CAS PubMed.
- J. M. Reimer, M. N. Aloise, P. M. Harrison and T. Martin Schmeing, Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase, Nature, 2016, 529(7585), 239–242 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov and C. Leigh Allen, et al., Structures of two distinct conformations of holo-non-ribosomal peptide synthetases, Nature, 2016, 529(7585), 235–238 CrossRef CAS PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. L. Markwick, J. Bruegger and G. Caldara-Festin, et al., Trapping the dynamic acyl carrier protein in fatty acid biosynthesis, Nature, 2014, 505(7483), 427–431 CrossRef CAS PubMed.
- T. Izoré and M. J. Cryle, The many faces and important roles of protein–protein interactions during non-ribosomal peptide synthesis, Nat. Prod. Rep., 2018, 35(11), 1120–1139 RSC.
- J. Crosby and M. P. Crump, The structural role of the carrier protein – active controller or passive carrier, Nat. Prod. Rep., 2012, 29(10), 1111 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cb00015a |
| This journal is © The Royal Society of Chemistry 2020 |