
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mechanism-inspired UDP-N-acetylglucosamine pyrophosphorylase inhibitor†
Olawale G.
Raimi‡
 a,
Ramon
Hurtado-Guerrero‡§
a,
Vladimir
Borodkin
a,
Andrew
Ferenbach
a,
Michael D.
Urbaniak¶
b,
Michael A. J.
Ferguson
b and
Daan M. F.
van Aalten
*a
a,
Ramon
Hurtado-Guerrero‡§
a,
Vladimir
Borodkin
a,
Andrew
Ferenbach
a,
Michael D.
Urbaniak¶
b,
Michael A. J.
Ferguson
b and
Daan M. F.
van Aalten
*a
aDivision of Gene Regulation and Expression, School of Life Sciences, University of Dundee, Dow Street, DD1 5EH Dundee, UK. E-mail: dmfvanaalten@dundee.ac.uk
bWellcome Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, DD1 5EH Dundee, UK
First published on 24th March 2020
Abstract
UDP-N-acetylglucosamine pyrophosphorylase (UAP1) catalyses the last step in eukaryotic biosynthesis of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), converting UTP and GlcNAc-1P to the sugar nucleotide. Gene disruption studies have shown that this gene is essential in eukaryotes and a possible antifungal target, yet no inhibitors of fungal UAP1 have so far been reported. Here we describe the crystal structures of substrate/product complexes of UAP1 from Aspergillus fumigatus that together provide snapshots of catalysis. A structure with UDP-GlcNAc, pyrophosphate and Mg2+ provides the first Michaelis complex trapped for this class of enzyme, revealing the structural basis of the previously reported Mg2+ dependence and direct observation of pyrophosphorolysis. We also show that a highly conserved lysine mimics the role of a second metal observed in structures of bacterial orthologues. A mechanism-inspired UTP α,β-methylenebisphosphonate analogue (meUTP) was designed and synthesized and was shown to be a micromolar inhibitor of the enzyme. The mechanistic insights and inhibitor described here will facilitate future studies towards the discovery of small molecule inhibitors of this currently unexploited potential antifungal drug target.
Introduction
Yeast and fungi are encapsulated by a cell wall that is important for morphogenesis and provides chemical/mechanical protection from the environment. Fungal cell walls are composed mainly of glucans, chitin and mannoproteins.1 Chitin is a β(1,4)-linked homopolymer of N-acetylglucosamine residues, accounting for only 1–2% of the cell mass in yeast,2,3 whereas the cell wall of filamentous fungi contains higher amounts of chitin, up to 10–20% of the dry weight.4 Chitin is synthesized from UDP-GlcNAc, which is also a sugar donor for the synthesis of glycoproteins, glycosaminoglycans and glycosylphosphatidylinositol (GPI) anchors in eukaryotes as well as molecules such as lipopolysaccharide in prokaryotes. In eukaryotes, UDP-GlcNAc is synthesized from fructose-6-phosphate (Fru-6P) and L-glutamine by a cascade of four enzymatic reactions (Fig. 1); (i) conversion of Fru-6P to GlcN-6P, (ii) acetylation of GlcN-6P to GlcNAc-6P, (iii) isomerization of GlcNAc-6P to GlcNAc-1P, and (iv) the uridylation of GlcNAc-1P to the sugar nucleotide UDP-GlcNAc. The enzymes involved in this pathway are (i) GlcN-6P synthase (GFA1), (ii) GlcN-6P acetyltransferase (GNA1), (iii) phosphoacetylglucosamine mutase (AGM1) and (iv) UDP-GlcNAc pyrophosphorylase (UAP1) (Fig. 1), respectively.5 Knock out studies of any of these genes have produced lethal phenotypes in a range of eukaryotes,6–8 with the exception of the knockout of GFA1, which produces GlcN/GlcNAc auxotrophic mutants,9 and the enzymes are considered to be potential drug targets. One species-specific allosteric inhibitor has been described for T. brucei UAP1,10 but no mechanism-based inhibitors have so far been reported for any of the last three enzymes in the pathway.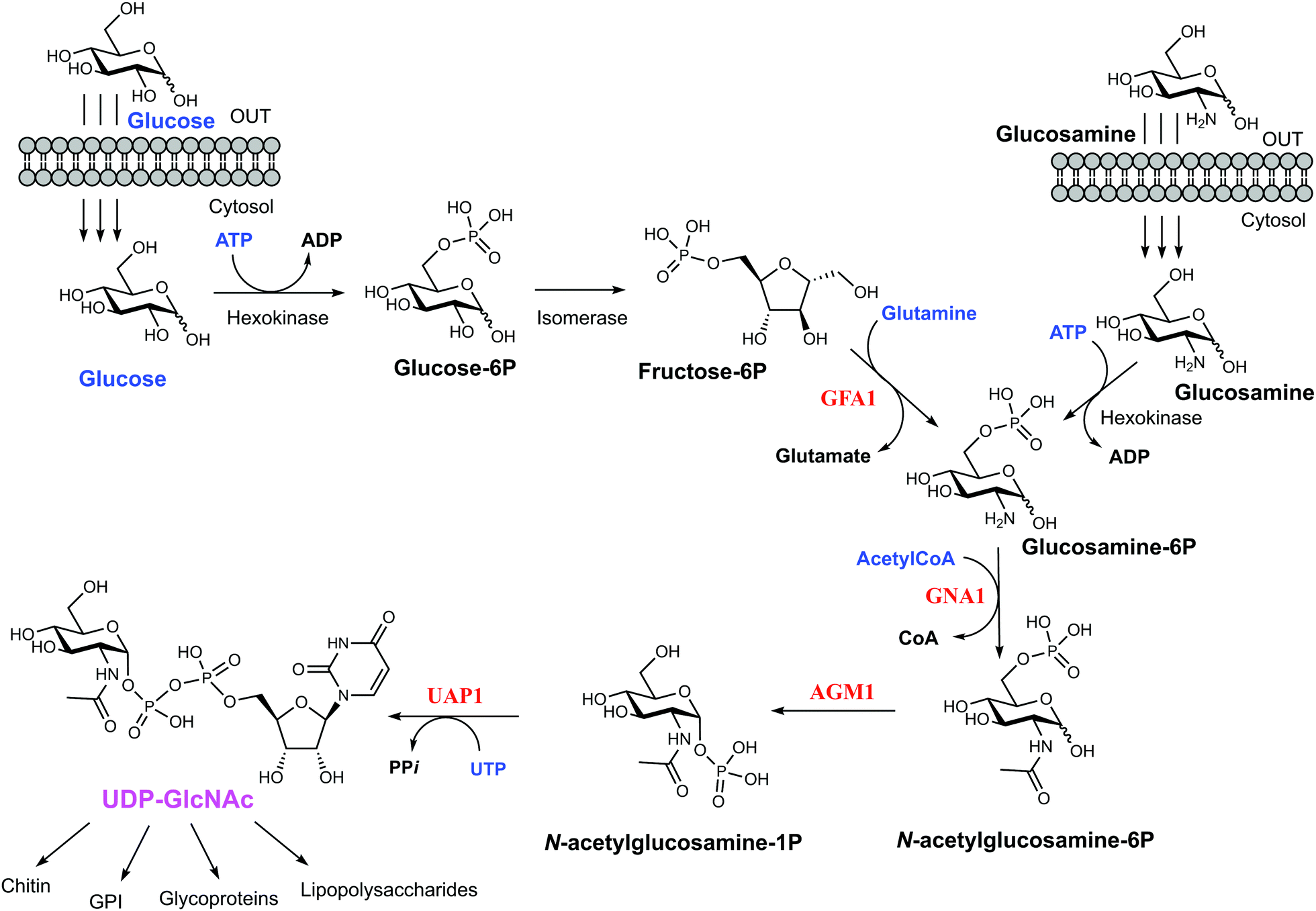 | ||
| Fig. 1 UDP-GlcNAc biosynthetic pathway. The four main enzymes of the pathway are coloured in red. Metabolites feeding the pathway are in blue and the final product of the pathway is coloured in pink. The various fates of UDP-GlcNAc are linked by the branching arrows. | ||
UDP-GlcNAc pyrophosphorylase (E.C.2.7.7.23) is a member of the large family of nucleotide diphosphate sugar pyrophosphorylases.7 Eukaryotic UDP-GlcNAc pyrophosphorylase catalyzes the synthesis of UDP-GlcNAc from UTP and GlcNAc-1P and also the reverse pyrophosphorolysis of UDP-GlcNAc (UTP + GlcNAc-1P ⇔ UDP-GlcNAc + PPi). Eukaryotic UAP1s have been categorized into mammalian UAP1 and non-mammalian UAP1.11 The crystal structures of the two isoforms (AGX1 and AGX2) of human UAP1 (hUAP1) were the first UAP1 structures to be reported.12 UAP1s consist of an N-terminal domain, a central pyrophosphorylase domain and a C-terminal domain. The functions of the N and C-terminal domains are not yet known.11,12 Non-mammalian UAP1s possess a primary structure similar to that of mammalian UAP1s, but lack an “insertion loop” of more than 10 amino acids residues11 as visualised in the crystal structures of Candida albicans and Aspergillus fumigatus UAP1s (CaUAP1 and AfUAP1 respectively) (Fig. S1, ESI†).11,13 Together, these human and C. albicans UAP1 structures, solved in complex with substrates/products, suggest an SN2 reaction mechanism in which phosphate oxygen of GlcNAc-1P attacks the UTP α-phosphate, forming UDP-GlcNAc and releasing inorganic pyrophosphate (PPi). Although it has been shown that divalent cations facilitate the reactions catalysed by UAP1, their mechanistic roles are not clear, and the available structures have been determined in the absence of any such ions.
Aspergillus fumigatus is the major cause of Aspergillosis, an often invasive and potentially life-threatening infection, found most frequently in immunocompromised patients.14,15 As a step towards chemically validating A. fumigatus UDP-N-acetylglucosamine pyrophosphorylase (AfUAP1) as a potential drug target, we report the crystal structure of AfUAP1 in complex with the two reaction products, UDP-GlcNAc and pyrophosphate, and the cofactor magnesium ion, representing the first Michaelis complex for this enzyme family. Together with additional substrate/product complexes of this enzyme, this provides a range of snapshots of the reaction mechanism, revealing key conformational changes and essential participation of the magnesium ion and Lys437 in catalysis. A mechanism-inspired α,β-methylenebisphosphonate UTP analogue (meUTP) is the first example of a eukaryotic pyrophosphorylase inhibitor, providing a basis for the future design of small molecule inhibitor for this class of enzymes.
Experimental
Recombinant AfUAP1 production and structure determination
AfUAP1 was expressed as described recently.13 The protein (20 mg mL−1) was co-crystallized with UTP, GlcNAc-1P and UDP-GlcNAc respectively by incubating the protein with 5 mM of the ligands at 4 °C for 2 h before setting up crystal trays. Crystals grew after three days in a condition containing 0.2 M sodium formate and 20% PEG3350. To obtain the UAP1–UDP-GlcNAc–PPi–Mg2+ ion complex, an AfUAP1–UDP-GlcNAc crystal was soaked for 10 seconds in a cryo-protectant (mother liquor + 10% glycerol) containing 10 mM pyrophosphate and 10 mM magnesium. Prior to data collection, crystals were cryo-protected in mother liquor containing 10% glycerol. Data were collected at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France) and processed with the HKL suite.16 The AfUAP1–UDP-GlcNAc structure and other complexes described here were solved by molecular replacement with MOLREP17 using the PDB entry 4BMA as the template.13 This gave a solution with two molecules in the asymmetric unit. The resulting electron density map was of good quality, allowing WarpNtrace18 to build a model containing most residues. REFMAC19 was used for further refinement, iterated with model building using COOT,20 producing a final models with the statistics shown in Table 1. Models for ligands were not included until their conformations were fully defined by unbiased |Fo| − |Fc|, φcalc electron density maps. Ligand structures and topologies were generated by PRODRG.21 WHATIF22 was used to calculate hydrogen bonds and PyMOL was used to generate figures.23| UDP-GlcNAc + PPi, Mg2+ | GlcNAc-1P | UTP | |
|---|---|---|---|
| Values in parenthesis are for the highest resolution shell. All measured data were included in the refinement. | |||
| Resolution (Å) | 20.0 (1.81–1.75) | 20.0 (2.36–2.28) | 20.0 (2.49–2.40) |
| Space group | P212121 | P212121 | P212121 |
| Unit cell | |||
| a (Å) | 55.2 | 55.7 | 55.6 |
| b (Å) | 139.8 | 138.1 | 140.0 |
| c (Å) | 144.6 | 145.1 | 146.3 |
| α = β = γ (°) | 90 | 90 | 90 |
| No. of reflections | 441![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 409 409 |
157074 |
177172 |
| No. of unique reflections | 113044 |
49491 |
46020 |
| I/σ (I) | 16.4 (2.4) | 11.2 (2.4) | 13.7 (3.2) |
| Completeness (%) | 99.1 | 95.5 | 99.2 |
| Redundancy | 3.9 (3.8) | 3.2 (3.0) | 3.8 (3.0) |
| R merge (%) | 5.8 (56.8) | 11.6 (39.2) | 8.6 (32.5) |
| RMSD from ideal geometry | |||
| Bond distance (Å) | 0.008 | 0.009 | 0.010 |
| Bond angle (°) | 1.08 | 1.0 | 1.23 |
| R work (%) | 21.0 | 20.6 | 22.7 |
| R free (%) | 24.0 | 25.9 | 27.7 |
| No. of residues | 925 | 905 | 928 |
| No. of water mol. | 559 | 231 | 120 |
| B-factors (Å2) | |||
| Overall | 23.8 | 48.6 | 34.8 |
| Protein | 23.4 | 48.8 | 34.8 |
| Ligand | 16.7 | 42.5 | 43.4 |
| Solvent | 29.3 | 44.5 | 29.2 |
| PDB ID | 6G9V | 6TN3 | 6G9W |
Enzyme kinetics and inhibition
AfUAP1 steady-state kinetics was studied under linear conditions with not more than 10% of the substrate converted to products. The activity of the enzyme in the forward direction was determined as described previously.13 The enzymes were used at 20 nM with the exception of K148M mutant that was used at 185 nM. The absorbance intensity data were analyzed with nonlinear regression analysis using GRAFIT 524 with the default equations for the first-order reaction rates and Michaelis–Menten steady-state kinetics. To investigate AfUAP1 inhibition by meUTP the forward reaction (formation of UDP-GlcNAc) was assayed using high performance anion exchange chromatography (HPAEC) based assay. The reaction mixtures (20 µM UTP, 50 µM GlcNAc-1P, 1 mM DTT, 5 nM enzyme, 50 mM Tris–HCl pH 8.0, 10 mM MgCl2 and 2% glycerol, total volume 100 µL) with and without varying concentrations of the inhibitor (0–1000 µM) were incubated for 30 min and the reactions were terminated by the addition of 10 µL of 0.1 M NaOH (∼10 mM final concentration) prior to analysis on a CarboPac-PA-1 column (Dionex) equilibrated with a 80:20 mixture of 1 mM NaOH and a 1:1 mixture of 1 mM NaOH and 1 M sodium acetate as described previously.25 The eluent was monitored at 260 nm and peaks were identified by comparison to standards. The IC50 was calculated using Graphpad Prism software, CA, USA (http://www.graphpad.com).
Synthesis of an α,β-methylenebisphosphonate UTP (meUTP)
HRMS (positive) m/z 501.1478; expected for C17H31N2O11P2 [M + H]+ 501.1403.
1H NMR (500 MHz, CDCl3) δ 10.20 (2 × d, 1H), 7.43 (d, J = 2.8, 0.5H), 7.41 (d, J = 2.7, 0.5H), 5.73 (d, J = 2.3, 0.5H), 5.69 (d, J = 2.3, 0.5H), 5.67 (d, J = 5.8, 0.5H), 5.65 (d, J = 5.8, 0.5H), 4.94–4.91 (m, 1H), 4.86 (dd, J = 6.4, 3.7, 0.5H), 4.82 (dd, J = 6.5, 3.5, 0.5H), 4.34–4.22 (m, 3H), 3.76–3.74 (m, 4.5H), 3.73–3.72 (m, 4.5H), 2.50 (dt, J = 21.2, 1.2, 2H), 1.49 (s, 3H), 1.27 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 163.89, 150.36, 142.41, 142.33, 114.29, 102.49, 93.64, 93.30, 85.27, 85.22, 84.11, 80.50, 80.41, 77.50, 65.88, 65.82, 65.77, 53.42, 53.33, 53.27, 53.21, 52.44, 52.39, 26.97, 25.13, 23.66 (dt, J = 137.6, 8.2).
31P NMR (202 MHz, CDCl3) δ 21.81 (d, J = 6.4), 21.76 (d, J = 6.4), 21.70 (d, J = 6.4), 21.41 (d, J = 6.4).
HRMS (positive) m/z 858.1673; expected for C21H35N2O13P2 585.1614 [M + H]+ 585.1614.
1H NMR (500 MHz, CDCl3) δ 10.10 (2 × d, 1H), 7.74 (d, J = 8.2, 0.5H), 7.67 (d, J = 8.2, 0.5H), 6.13 (dd, J = 6.6, 1.3, 0.5H), 5.77 (dd, J = 8.1, 1.9, 0.5H), 5.73 (dd, J = 8.1, 1.9, 0.5H), 5.44 (dd, J = 5.9, 3.7, 0.5H), 5.37 (dd, J = 5.9, 3.5, 0.5H), 5.30 (t, J = 6.2, 0.5H), 5.25 (t, J = 6.4, 0.5H), 4.45–4.41 (m, 0.5H), 4.39–4.32 (m, 1H), 4.24–4.20 (m, 1H), 3.82–3.74 (m, 9H), 2.62–2.46 (m, 4H), 1.14 (bs, 3H), 1.13 (bs, 3H), 1.10–1.07 (m, 6H).
13C NMR (126 MHz, CDCl3) δ 175.79, 175.69, 175.51, 163.39, 163.36, 150.69, 150.65, 140.07, 139.99, 103.50, 103.36, 86.13, 85.89, 81.21, 81.15, 81.04, 80.98, 77.37, 72.38, 72.31, 69.91, 69.87, 65.37, 65.34, 53.33 (m), 33.65, 33.54, 23.93 (dt, J = 137.3, 14.2) 18.82, 18.71, 18.63.
31P NMR (202 MHz, CDCl3) δ 22.44 (d, J = 7.3), 21.87 (d, J = 6.6), 21.46 (d, J = 7.3), 21.41 (d, J = 6.7).
:1) (1 mL). The residue was co-evaporated with toluene (2 × 1 mL) to remove traces of water. The residue was dissolved in anhydrous MeOH (1 mL) and treated with a 25% stock solution of NaOMe (0.05 mL) at room temperature (RT) for 3 h. The reaction was quenched with Dowex50W8-Et3NH+. The resin was filtered off and the filtrate was concentrated. The residue was dissolved in water and freeze-dried to give 0.072 g (0.1 mmol) of the target compound as a fluffy white solid.
HRMS (negative) m/z 401.0149; expected for C10H15N2O11P2 [M − H]− 401.0151.
1H NMR (500 MHz, MeOD) δ 7.76 (d, J = 8.1, 1H), 5.73 (d, J = 4.6, 1H), 5.71 (d, J = 8.1, 1H), 4.14 (dt, J = 9.8, 5.2, 2H), 4.05–4.00 (m, 1H), 3.98–3.87 (m, 2H), 2.96 (q, J = 7.3, 12H), 1.97 (dd, J = 27.1, 11.5, 2H), 1.03 (t, J = 7.3, 18H).
13C NMR (126 MHz, MeOD) δ 166.20, 151.74, 141.81, 102.46, 88.37, 83.31 (d, J = 7.6), 73.69, 69.54, 63.28 (d, J = 4.3), 46.57, 27.19 (t, J = 125.0), 8.
31P NMR (202 MHz, MeOD) δ 16.43, 14.2.
HRMS (negative) m/z 480.9857; expected for C10H16N2O14P3 [M − H]− 480.9814.
1H NMR (500 MHz, D2O) δ 7.89 (d, J = 8.1, 1H), 5.85–5.81 (m, 2H), 4.29–4.25 (m, 2H), 4.13 (d, J = 1.9, 1H), 4.10–4.01 (m, 2H), 2.29–2.18 (dt, J = 21, 2, 2H).
31P NMR (202 MHz, D2O) δ 17.80 (d, J = 9.3), 6.18 (dd, J = 23.6, 9.3), −9.25 (d, J = 23.6).
Results and discussion
AfUAP1 undergoes large conformational changes during catalysis
To explore UAP1 catalytic mechanism and provide a structural framework for the design and optimization of inhibitors, crystal structures of AfUAP1 in complex with substrates/products were determined. Initially, a high-resolution dataset of a Michaelis complex of the enzyme at 1.75 Å was trapped using crystals grown in the presence of UDP-GlcNAc and subsequently soaked with PPi and Mg2+ (Table 1 and Fig. 2; RMSD of 1.6 Å on Cα atoms compared to the “apo-like” structure described earlier with PDB 4BMA13). One of the two protein molecules in the asymmetric unit showed an empty active site, and is considered here as the “apo-like” form (Fig. 2). To understand the catalytic mechanism, two further complexes were also determined – a substrate complex with GlcNAc-1P and a substrate complex with UTP (Table 1 and Fig. 2).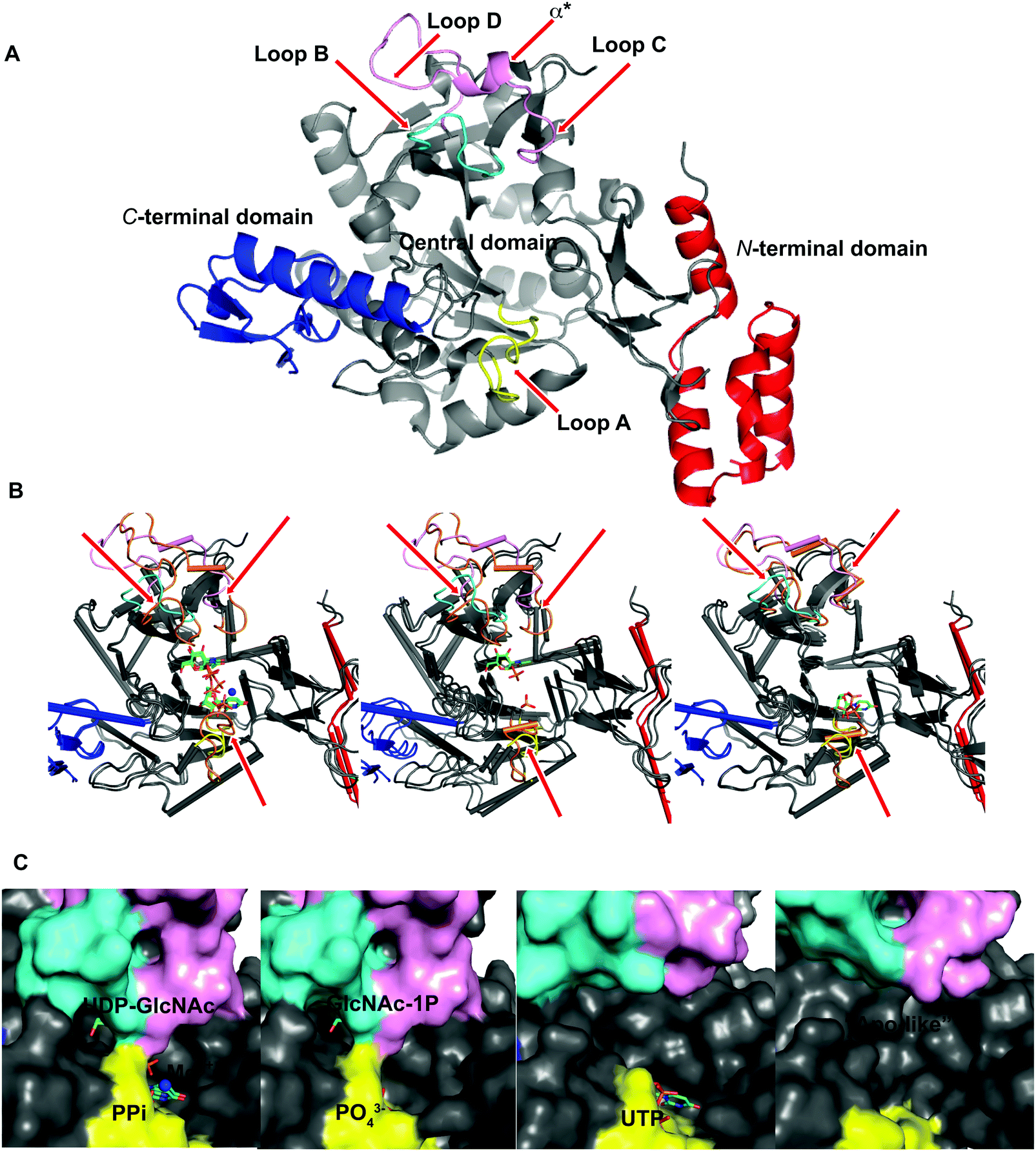 | ||
| Fig. 2 Ligand-induced conformational changes of AfUAP1. (A) Cartoon representation of AfUAP1, the N-terminal domain is coloured red, the C-terminal domain is coloured blue and the pyrophosphorylase domain is coloured in grey. Loops A, B, and C–D together with α* are coloured in yellow, cyan and pink, respectively. The flexible regions are indicated by red arrows. (B) Cartoon representation of the superposed “apo-like” structure with the different complexes showing a large area around the active site. While the colours for the mobile regions are maintained for the “apo-like” structure, the same loops and α* are coloured in orange in the different complexes. (C) The surface representation shows conformational changes around the active site in the various AfUAP1 complexes. The substrates and the products are shown as sticks, the Mg2+ ion is shown as a blue sphere. | ||
The overall structure of AfUAP1 is similar to that previously described for eukaryotic UAP1s CaUAP111 and HsUAP112 (Fig. 3A and B). AfUAP1 and CaUAP1 share 52% sequence identity (Fig. S1, ESI†) and superposition of the overall structures of these two proteins reveals an RMSD of 1.1 Å (on all Cα atoms), indicating a similar overall structure. AfUAP1 possesses a three-domain fold with the central catalytic domain flanked by two domains of unknown function (Fig. 2A). While the N-terminal domain of AfUAP1 consists entirely of four α-helices (Fig. 2A), the central domain consists of seventeen β-strands and twelve helices adopting the Rossmann fold. The C-terminal domain consists of four short β-strands and a long helix that connects to the central domain (Fig. 2A). AfUAP1 has a large active site cleft, with its wall being formed by the central domain and the C-terminal domains (Fig. 2). Ligand binding at the bottom of the active site induces a number of significant conformational changes compared to the “apo-like” structure. The binding of GlcNAc-1P to AfUAP1 is stabilized by hydrogen bonds with Asn249, Gly316, Tyr330, Glu329, Asn355 and Lys437. Interestingly, a PO43− molecule which occupies the same position as the UTP γ-phosphate interacts with R141 and T140 (Fig. 2B, C and 4) and induce a shift of loops A, B, C, D and α* on the central domain, closing the active site (Fig. 2A and B) (RMSD vs. “apo-like” structure = 2.1 Å on all Cα atoms). Similar interactions were seen in the CaUAP1–GlcNAc-1P complex with equivalent residues (Tyr310, Asn227, Glu309, Gly294, Asn335, and Lys421) except for the absence of the PO43− molecule, which prevents the ordering of R116 (R141 in AfUAP1) (Fig. 3C and D).11 We hypothesise that in the absence of this PO43− molecule, the binding of GlcNAc-1P alone would only cause partial closure of the active site. In contrast, UTP appears to bind AfUAP1 with no significant conformational changes (RMSD vs. “apo-like” structure = 1.1 Å on all Cα atoms), interacting with Gly136, Gln138, Gly139, Thr140, Lys148, Arg141, Gln222, Gly248, Cys277 and Lys383 (Fig. 2B, C and 4). However, binding of UDP-GlcNAc, magnesium and PPi to the enzyme causes a considerable change to the overall structure of the protein (Fig. 2B, C and 3) (RMSD vs. “apo-like” structure = 2.1 Å on all Cα atoms). UDP-GlcNAc binds to the enzyme with the GlcNAc and uridine moieties of the molecule occupying positions equivalent to GlcNAc-1P (max. atomic positional shift = 0.6 Å) and UTP (max. atomic positional shift = 1.0 Å), also establishing similar hydrogen bonding interactions (Fig. 2B, C and 4). Further hydrogen bonds are established between α and β phosphates of UDP-GlcNAc and Arg141 and Lys437 (Fig. 4). The PPi molecule occupies a position approximately equivalent to the β and γ-phosphates of UTP (max. positional shift on phosphorus atoms = 1.5 Å), and is tethered by interactions with positively charged residues, such as Arg141, Lys437 and Lys148 (Fig. 4). A comparison of AfUAP1–UDP-GlcNAc–Mg+2–PPi and CaUAP1–UDP-GlcNAc complexes reveals that UDP-GlcNAc occupies the same position and makes identical interactions with equivalent residues in both enzymes (Fig. 3C and D).11 Interestingly a SO42− ion occupies the same position as one of the phosphates of the PPi seen in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ complex (Fig. 3D and 4). Taken together, it appears that to accommodate substrates and release products, AfUAP1 undergoes large conformational changes during catalysis.
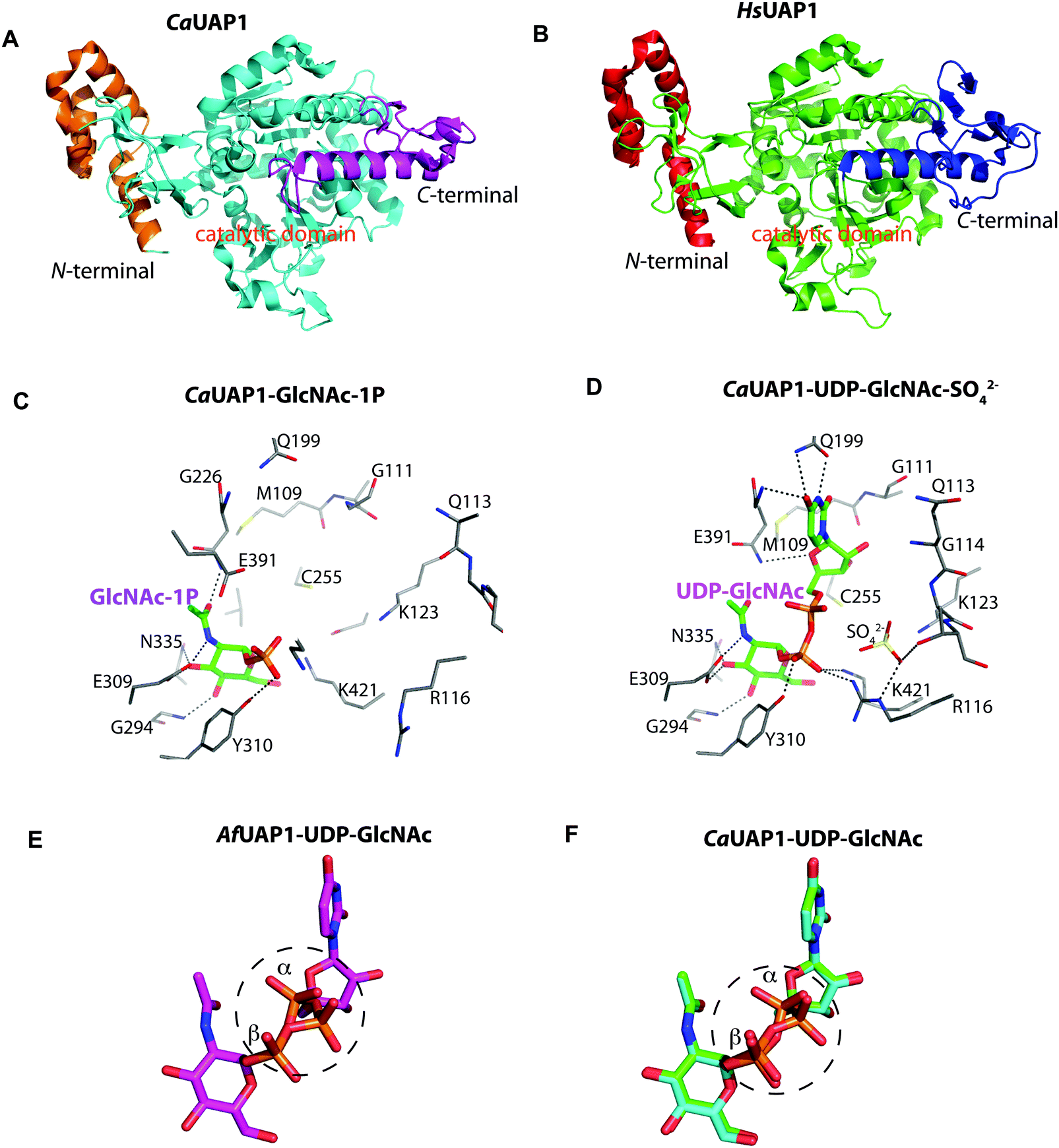 | ||
| Fig. 3 Comparison of AfUAP1 with other UAP1s. (A) Overall structure of CaUAP1. Coloured in orange, cyan and purple are the N-terminal, catalytic and C-terminal domains, respectively. (B) Overall structure of HsUAP1. Coloured in red, green and blue are the N-terminal, catalytic and C-terminal domains, respectively. (C) Active site of CaUAP1 with GlcNAc-1P bound. Active site residues are shown in sticks. Hydrogen bond interactions are shown as black dashes. (D) Active site of CaUAP1 with UDP-GlcNAc–SO42− bound. Active site residues are shown in sticks. Hydrogen bond interactions are shown as black dashes. SO42− is shown as sticks. (E) Stick representation of UDP-GlcNAc in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ ternary complex, showing the observed dual conformation around the α-phosphate. (F) Superposition of UDP-GlcNAc from the CaUAP1–UDP-GlcNAc product complex (PDB: 2yqs, blue) with UDP-GlcNAc from the CaUAP1–UDP-GlcNAc reaction-completed product complex (PDB: 2yqj, green). This superposition recapitulates the dual conformation observed in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ complex. | ||
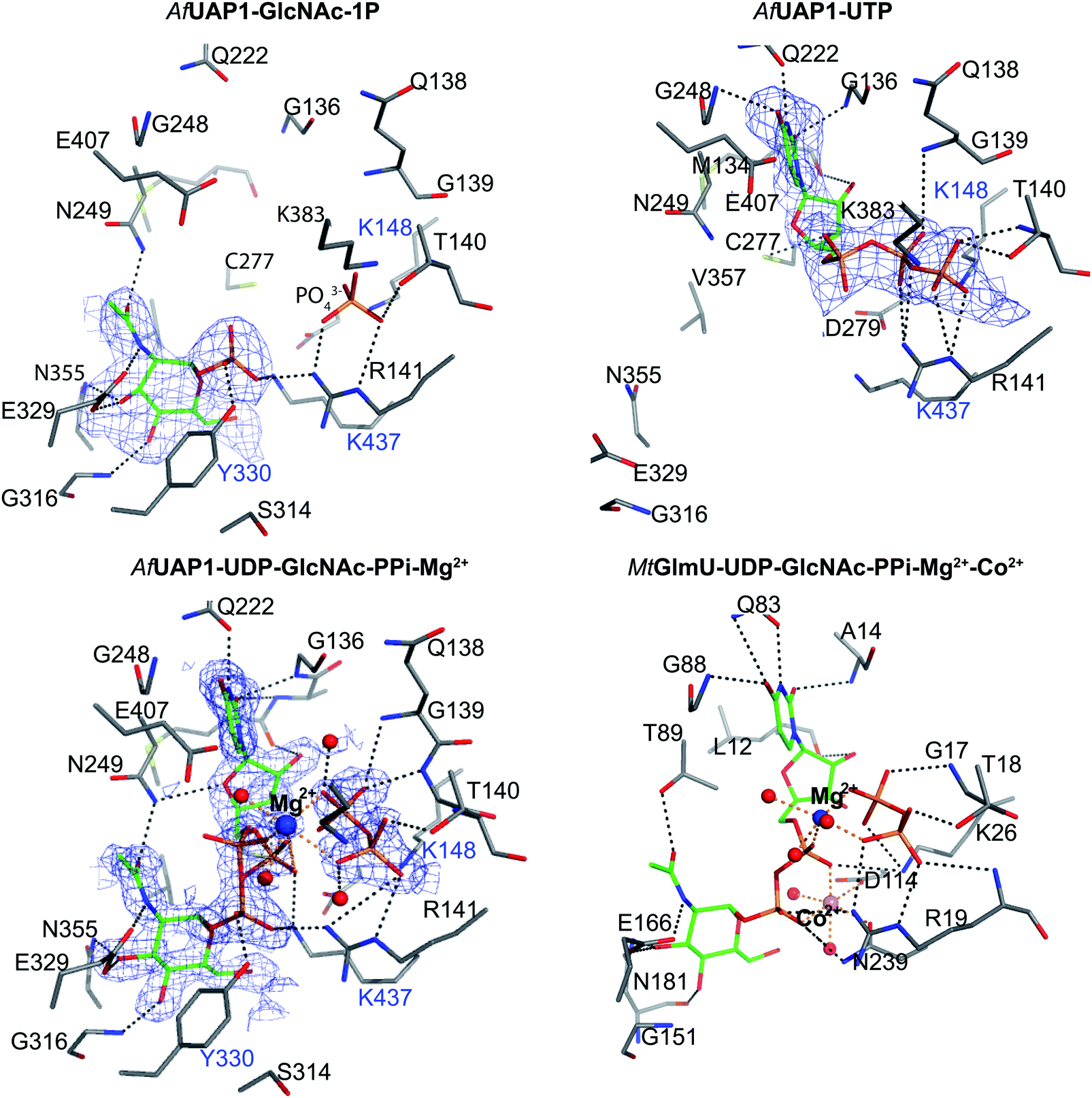 | ||
| Fig. 4 Active site of AfUAP1 complexes and comparison with the MtGlmU ternary complex. Amino acids and ligands are shown as sticks with grey and green carbon atoms respectively. Water molecules, Mg2+ and Co2+ atoms are shown as spheres in red, blue and pink respectively. Hydrogen bonds and metal coordination are represented with dotted lines coloured black and orange respectively. The unbiased |Fo| − |Fc|φcalc electron density maps for the ligands is shown in blue, contoured from 2.2 to 2.5σ depending on the ligand. The blue characters indicate the residues mutated in the mutagenesis assay. | ||
The Mg2+ ion aligns the reactants for catalysis
Several reports have shown that UAP1 requires divalent cations, such as Mg2+, for full activity.7,11,12 The UDP-GlcNAc–PPi–Mg2+ Michaelis complex of the reverse reaction trapped here reveals the key role the metal plays in coordinating the reactants/products (Fig. 4 and 5). The magnesium ion with an occupancy of 1.0, is pentacoordinated by the phosphate groups of pyrophosphate, UDP-GlcNAc and two water molecules in a tetragonal bipyramidal arrangement (distances ranging between 2.0 and 2.4 Å) while the 6th ligand is absent (presumably a disordered water molecule), providing the necessary geometry to place the attacking oxygen of pyrophosphate within 2.2 Å distance (angle of attack 166°) from the UDP-GlcNAc α-phosphate (Fig. 5A), compatible with a direct displacement mechanism (Fig. 4). Interestingly, the high-resolution electron density map also reveals an alternative conformation for the UDP-GlcNAc α-phosphate that flips 1.7 Å resulting in an “off conformation”, and placed 3.5 Å from the PPi oxyanion (Fig. 4 and 5). It is possible that this “off conformation” resembles the product of the forward synthesis preceding a conformation that is the Michaelis complex of the reverse pyrophosphorolysis reaction. Interestingly, superposition of two separate CaUAP1–UDP-GlcNAc complexes (PDB entries 2yqs and 2yqj11) reveals a similar dual conformation as observed in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ complex described here (Fig. 3E and F). The role of the metal is firstly to correctly align the UTP with GlcNAc-1P, and UDP-GlcNAc with PPi, and secondly, to facilitate close proximity of the negatively charged phosphate groups. Similarly, Pelissier et al.27 also proposed that Mg2+ prevents electrostatic repulsion between phosphate groups of the substrates Man-1P and GTP of the thermophilic bacterium Thermotoga maritima pyrophosphorylase. Thus, the Mg2+ ion aligns the reactants for catalysis.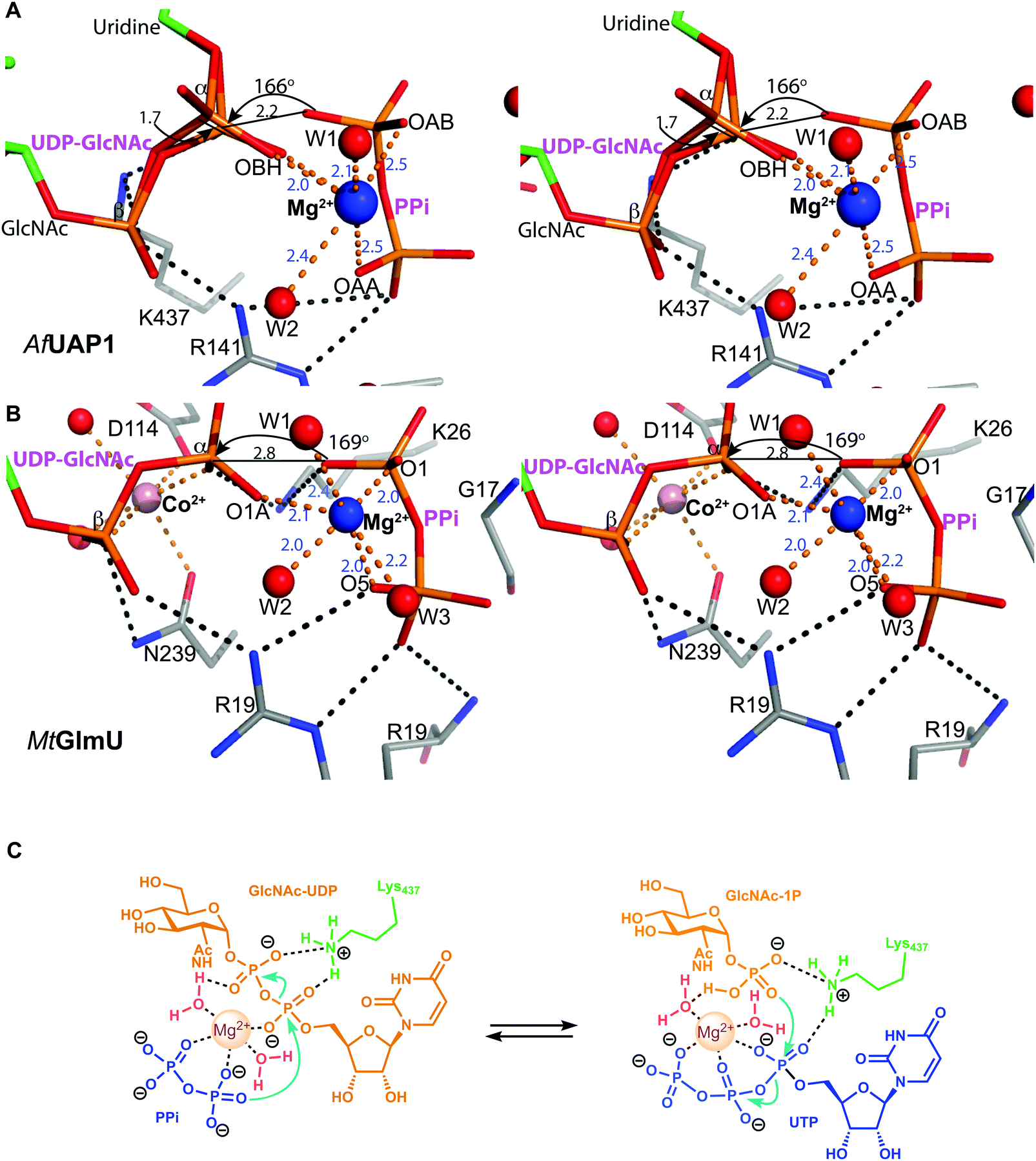 | ||
| Fig. 5 Magnesium coordination and AfUAP1 mechanism. (A) Magnesium coordination in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ complex active site. A view of the active site of the AfUAP1–UDP-GlcNAc–PPi–Mg2+ and MtGlmU–UDP-GlcNAc–PPi–Mg2+–Co2+ complexes. The attack distance for pyrophosphorolysis is 2.2 and 2.8 Å and the attack angle is 166° and 169° for UAP1 and GlmU, respectively. The distance between the α-phosphates in both UDP-GlcNAc adopting the dual conformation is 1.7 Å. The Mg2+ is tetragonal bipyramidally coordinated (albeit with a disordered 6th ligand in AfUAP1) as suggested by coordination distances and angles. (B) Magnesium coordination in the MtGlmU–UDP-GlcNAc–PPi–Mg2+–Co2+ complex active site. (C) Schematic diagram of the proposed forward and reverse reaction mechanisms. The left panel indicates chemical bond formation/cleavage (cyan arrows) in the AfUAP1–PPi–UDP-GlcNAc ternary complex shown in Fig. 5A leading to the formation of GlcNAc-1P and UTP. The right panel using the same ternary complex structure as a template indicates chemical bond formation/cleavage in the reverse reaction leading to the formation of UDP-GlcNAc and PPi from GlcNAc-1P and UTP is proposed. | ||
A conserved lysine occupies the position of a second metal ion in GlmU
The structure suggests that the reverse (pyrophosphorolysis) reaction may be facilitated by an increase in electrophilicity of the α-phosphorus induced by a salt bridge interaction between a lysine that is conserved in eukaryotes (Lys437 in AfUAP1, Fig. S1, ESI†) and the α phosphate of UDP-GlcNAc (Fig. 5). Similarly, Lys437 may facilitate the forward reaction by inducing an increase of the electrophilic character of the UTP α-phosphorus (Fig. 5).Recently the structures of a ternary complex of a bacterial UAP from M. tuberculosis named MtGlmU, and other classes of nucleotidyltransferases,28,29 revealed an unusual catalytic mechanism dependent on two-metal-ions30 (Fig. 4 and 5B). GlmUs are bacterial enzymes with two domains catalysing two independent reactions. While the C-terminal domain catalyses an acetyltransfer reaction from acetyl-CoA to glucosamine-1P (GlcN-1P) to form N-acetylglucosamine-1P (GlcNAc-1P), the N-terminal domain possesses uridylation activity converting GlcNAc-1P to UDP-GlcNAc in the presence of UTP.30 In the MtGlmU structure, UDP-GlcNAc and PPi interact through hydrogen bonds with Ala14, Gly17, Thr18, Gln83, Gly88, Thr89, Gly151, Glu166 and Asn181, and salt bridges with Lys26 and Arg19 (Fig. 4). The magnesium atom in both the AfUAP1 and MtGlmU ternary complexes occupy similar positions but they differ in how they are coordinated (Fig. 5). The metal ion in AfUAP1 is pentacoordinated, whereas the magnesium atom in GlmU is hexacoordinated by the phosphate groups of PPi and UDP-GlcNAc, and three water molecules (Fig. 5). The average bond angle of coordination of magnesium in the AfUAP1–UDP-GlcNAc–PPi–Mg2+ complex is 89 ± 10° while in the MtGlmU–UDP-GlcNAc–PPi–Mg2+–Co2+ complex it is 90 ± 17°. A comparison of the angles of magnesium coordination in AfUAP1 and MtGlmU complexes is shown in Table 2. With respect to the reaction mechanism, the pyrophosphate is within 2.8 Å (with an angle of attack of 169°) from the UDP-GlcNAc α-phosphate and therefore at a distance not as optimal as that found in the Michaelis complex of AfUAP1, suggesting that a positional shift of UDP-GlcNAc α-phosphate might be required in order to facilitate catalysis (Fig. 5A).
| AfUAP1–UDP-GlcNAc–PPi–Mg2+ | MtGlmu–UDP-GlcNAc–PPi–Mg2+–Co2+ | ||
|---|---|---|---|
| Atoms | Angle (°) | Atoms | Angle (°) |
| W1 = water 1, W2 = water 2 and W3 = water 3 as indicated in Fig. 5. nd = not detectable due to absence of W3 in the AfUAP1 structure. | |||
| W1Mg2+W2 | 100.1 | W1Mg2+W2 | 80.7 |
| W1Mg2+W3 | nd | W1Mg2+W3 | 103.3 |
| W1Mg2+OBH | 87.9 | W1Mg2+O1A | 88.7 |
| W1Mg2+OAB | 105.2 | W1Mg2+O1 | 107.9 |
| W3Mg2+OAA | nd | W3Mg2+O5 | 69.6 |
| W3Mg2+W2 | nd | W3Mg2+W2 | 69.7 |
| OAAMg2+OAB | 79.7 | O5Mg2+O1 | 80.0 |
| OABMg2+OBH | 82.8 | O1Mg2+O1A | 93.9 |
| W3Mg2+OAB | nd | W3Mg2+O1 | 116.1 |
| W2Mg2+OAA | 76.0 | W2Mg2+O5 | 92.1 |
| W2Mg2+OBH | 85.0 | W2Mg2+O1A | 76.4 |
| OAAMg2+OBH | 96.1 | O1AMg2+O5 | 94.2 |
Interestingly, the crystal structure reveals the presence of a second metal in the MtGlmU identified as cobalt, although experiments in solution confirm that GlmU requires two magnesium ions for catalysis.30 Furthermore, this second metal binding site is found in a position occupied by Lys437 in AfUAP1 (Fig. 4 and 5B). In the C. albicans UAP1 crystal structure, Lys421 occupies a similar position and was also predicted to play the role of the second metal ion.11 In Leishmania major UDP–glucose pyrophosphorylase31 and a bacterial GDP–mannose pyrophosphorylase,27 a lysine was also observed to occupy the position of the second Mg2+ seen in C. glutamicum UGP.29 It is possible that bacterial GlmUs and eukaryotic UAP1s follow a similar catalytic mechanism but differ in the number of metal atoms and consequently in how the electrophilic character of α-phosphorous is enhanced. In eukaryotes a single magnesium atom is essential for the reaction while the position of the second magnesium atom present in bacteria is occupied by a basic amino acid. Thus, a conserved lysine in AfUAP1 occupies the position of a second metal ion in GlmU.
Lys437 is required for AfUAP1 catalysis
Several of the amino acid residues (e.g. equivalents of Thr140, Arg141 and Lys148 in AfUAP1, Fig. 4) shown to bind the phosphates of nucleotides/phosphosugars in complexes with other UAP1 enzymes have been mutated previously and shown to be important for substrate binding/catalysis.7 However, to probe the role of Lys437 in catalysis, we carried out site-directed mutagenesis of this basic residue and two other amino acids involved in stabilization of substrates/products.7,11,12 The mutants have the same expression levels as the wild type enzyme as confirmed by SDS-PAGE (Fig. S2A, ESI†). As mentioned earlier, Lys437 makes direct contacts with the phosphate group of GlcNAc-1P (Fig. 4) and with the α and β-phosphate of UDP-GlcNAc (Fig. 4). Tyr330 establishes a hydrogen bond with the phosphate group of GlcNAc-1P, and Lys148 is involved in UTP binding, contacting the phosphate groups of UTP and pyrophosphate (Fig. 4) through direct and indirect hydrogen bonds. In agreement with the structural data, the K148M and Y330F mutants affect the Km values of UTP (10-fold increase) and GlcNAc-1P (6-fold increase), respectively (Table 3). The K437A mutant shows an increase in the Km values of both substrates (32 and 10-fold increase in the Kms of GlcNAc-1P and UTP respectively), which may be the result of disrupting the interaction of Lys437 with the phosphate groups of GlcNAc-1P and UDP-GlcNAc (Fig. 4). To probe the effect of this residue further we made K437R and K437M mutants. The K437R mutant shows a slight increase in Km by 5- and 4-fold for GlcNAc-1P and UTP, respectively while the K437M mutant completely abolishes enzyme activity (Table 3 and Fig. S2B, ESI†). The increase in activity by the K437R mutant compared to the K437A could be as a result of the arginine mimicking the positive charge present on Lys437, which is essential for an optimal catalytic efficiency. However, the activity of K437R is 2.9-fold lower than the wild type enzyme likely due to the bulkier arginine side chain not being positioned optimally with respect to the α-phosphate group of either UTP or UDP-GlcNAc. Overall, all the four mutants reduce kcat (Table 3). Out of the five mutants tested, K437A has the largest effect on catalytic efficiencies with a 433 and 117-fold reduction towards GlcNAc-1P and UTP, respectively (Table 3) and K437M completely inactivates the enzyme. Therefore, our data suggest that these residues play key roles in aligning the substrates for UAP1-catalyzed UDP-GlcNAc synthesis/pyrophosphorolysis, in particular supporting the role of Lys437 as being essential for catalysis in line with the proposed catalytic mechanism (Fig. 5C).| GlcNAc-1P | UTP | GlcNAc-1P | UTP | |||
|---|---|---|---|---|---|---|
| K m (µM) | K m (µM) | V max (µM s−1) | k cat (s−1) | k cat/Km (s−1 µM−1) | k cat/Km (s−1 µM−1) | |
| nd: not detectable. | ||||||
| WT | 34 ± 3 | 21 ± 2 | 0.044 ± 0.001 | 2.2 ± 0.3 | 0.065 | 0.10 |
| K148M | 44 ± 13 | 200 ± 31 | 0.0074 ± 0.0007 | 0.4 ± 0.01 | 0.0091 | 0.002 |
| Y330F | 200 ± 72 | 25 ± 5 | 0.016 ± 0.001 | 0.8 ± 0.1 | 0.004 | 0.032 |
| K437A | 1100 ± 900 | 200 ± 62 | 0.0033 ± 0.0004 | 0.17 ± 0.02 | 0.00015 | 0.00085 |
| K437R | 155 ± 29 | 88 ± 12 | 0.027 ± 0.003 | 1.3 ± 0.1 | 0.008 | 0.015 |
| K437M | nd | nd | nd | nd | nd | nd |
Design and synthesis of meUTP as a mechanism-inspired inhibitor
We attempted to use the AfUAP1 structural/mechanistic insights generated here to design a mechanism-inspired substrate analogue inhibitor. The AfUAP1–UDP-GlcNAc–PPi Michaelis complex (Fig. 4) and the proposed reaction mechanism (Fig. 5) reveal the direct attack of the phosphate group of GlcNAc-1P on UTP α,β-phosphodiester bond leading to the formation of the products UDP-GlcNAc and PPi. This suggests that an α,β-methylenebisphosphonate analogue of UTP (meUTP) in which the scissile α,β-pyrophosphate bond is replaced with a methylenebisphosphonate could inhibit this forward reaction (Fig. 6A). Synthesis of this analogue was initiated with the preparation of a fully protected UDP methylenebisphosphonate analogue 2 (Scheme 1) using a variant of Mitsunobu esterification of 2′,3′-O-isopropylideneuridine with methylenebisphosphonic acid trimethylester as a key step (Scheme 1).26 In these conditions, the target compound was obtained in 90% yield on a gram scale after conventional chromatography. This novel and highly effective method offers valuable advantages over the reported procedure for direct synthesis of nucleoside 5′-methylenebisphosphonates, which requires large excess of methylenebisphosphonic acid tetrachloride32 to be used to minimize formation of symmetrical and/or cyclic by-products. According to the initial synthetic plan, compound 2 was converted into the 2′,3′-di-O-isobutyrate derivative 3 in a two-step protective group exchange (Scheme 1). Unfortunately, all attempts to perform selective mono demethylation at the terminal phosphorous atom of the methylene bisphosphonate moiety in 3 to ensure regioselective formation of the triphosphate bond were unsuccessful. After some experimentation, conventional phosphorylation of the fully deprotected α,β-methylenebisphosphonate UDP analogue 4 was proven to be the easiest way to reach the target molecule. To this end, stepwise removal of the protecting groups from 3 gave rise to 4 in near quantitative yield. Finally, phosphorylation of 4 with N,N′-carbonyldiimidazole and tributylammonium phosphate in DMF for 16 h furnished the desired compound 5 or meUTP (Fig. 6A) in a modest 15% yield after ion-exchange chromatography. The structure of compound 5 was ultimately confirmed by 31P NMR, with the spectrum showing the characteristic pattern of three different phosphorous signals as doublet at δ 17.8 (J = 9.3), doublet of doublets at δ 6.18 (J = 23.6, 9.3), and doublet at δ −9.25 (J = 23.6) corresponding to γ, β, and α phosphorous atoms only compatible with the linear phosphate-methylenebisphosphonate structural motif. The HPLC profile of the purified compound 5 (Fig. S3A, ESI†) reveals the content of the main substance no less than 98% along with a contamination (2%) corresponding to meUDP.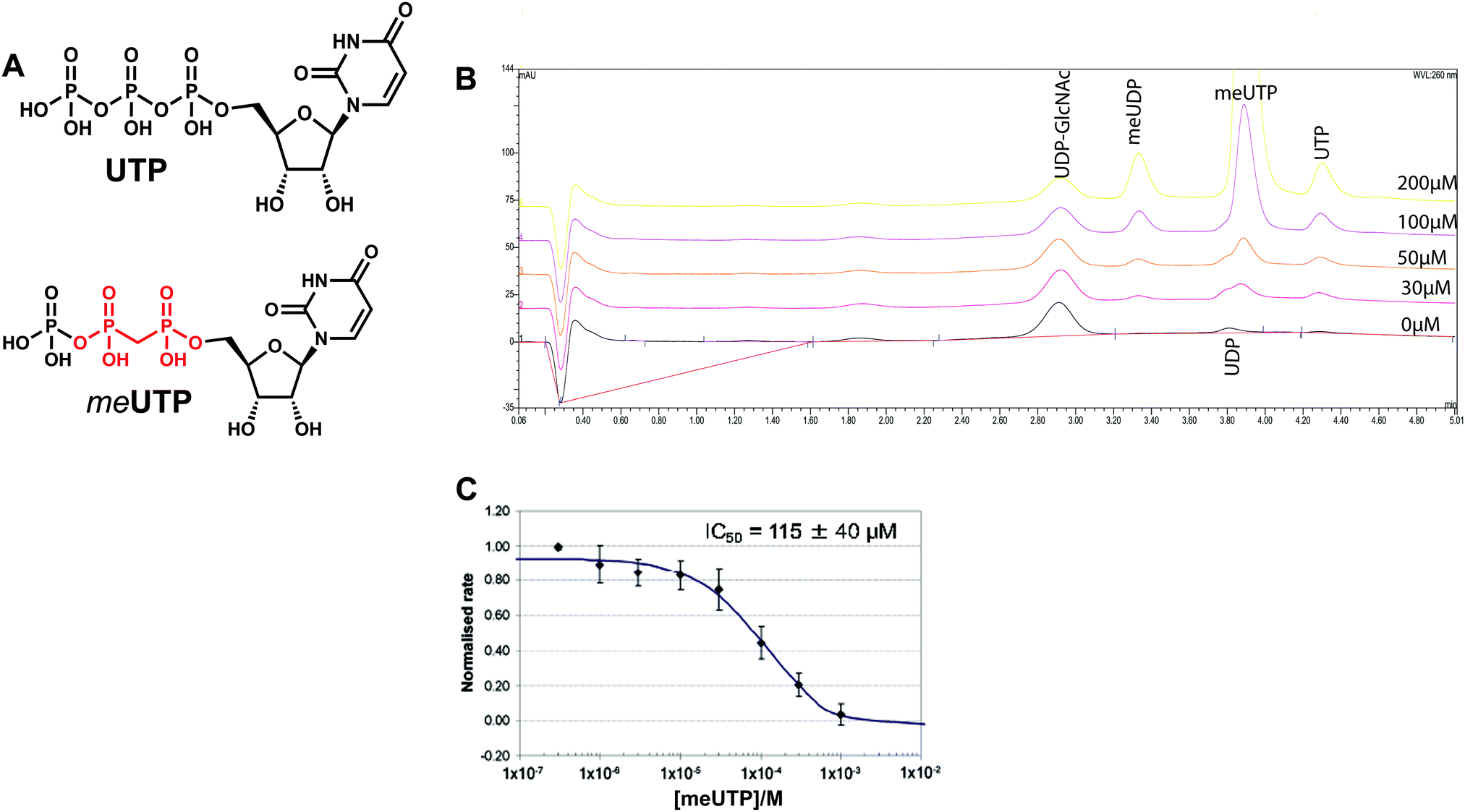 | ||
| Fig. 6 Kinetic characterization of meUTP (compound 5). (A) Chemical structures of UTP and meUTP. The α,β-methylenebisphosphanate is indicated in red. (B) HPAEC chromatogram of AfUAP1 inhibition by meUTP, where [UTP] ≈ Km and [GlcNAc-1-P] is in excess. The inhibitor concentration is indicated on each curve (0–200 µM). The reaction mixtures (20 µM UTP, 50 µM GlcNAc-1P, 1 mM DTT, 5 nM enzyme, 50 mM Tris–HCl pH 8.0, 10 mM MgCl2 and 2% glycerol, total volume 100 µL) with and without varying concentrations of the inhibitor were incubated for 30 min and the reactions were terminated by the addition of 10 µL of 0.1 M NaOH (∼10 mM final concentration) prior to analysis on a CarboPac-PA-1 column (Dionex) equilibrated with a 80:20 mixture of 1 mM NaOH and a 1:1 mixture of 1 mM NaOH and 1 M sodium acetate. The eluent was monitored at 260 nm and peaks were identified by comparison to standards. (C) IC50 curves of meUTP with fixed [UTP] and [GlcNAc-1P] and varying concentration of inhibitor. | ||
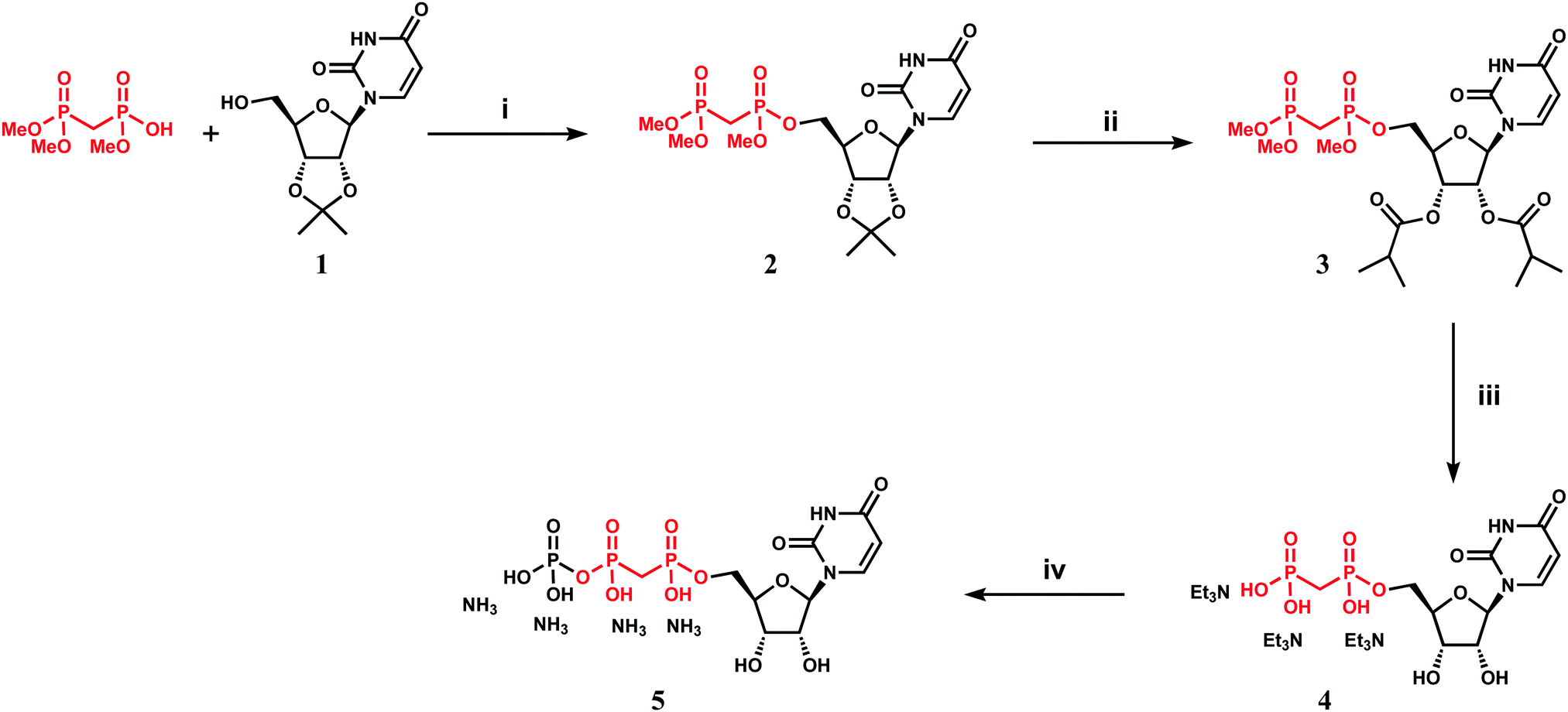 | ||
| Scheme 1 Synthesis of α,β-methylenebisphosphanate UTP analogue (meUTP). Reagents and conditions: (i) DIAD, PPh3, trimethyl methylenediphosphonate, THF, 2 h, reflux, 92%; (ii) (a) TFA–H2O (9:1), DCM, 2 h, RT, 85%, (b) (i-PrCO)2O, Py, DMAP, 5 h, RT, 87%; (iii) (a) TMSBr, MeCN, 1 h, RT, (b) MeOH, MeONa, then Dowex50W8-Et3NH+, quant; (iv) Im2CO, DMF, then (Bu3NH)3PO4, 16 h, RT, ion-exchange chromatography, 15%. | ||
Initial experiments showed that AfUAP1 was unable to utilize the meUTP as substrate. Therefore, potential inhibition by this compound was studied by an HPAEC assay, allowing us to follow the conversion of UTP to UDP-GlcNAc. Using this approach, it was observed that at increasing concentrations of the meUTP, less UTP is consumed while the amount of UDP-GlcNAc produced decreases, suggesting that meUTP is an AfUAP1 inhibitor (Fig. 6B). We have noted that while the concentration of the inhibitor increases, a peak at Rt = 3.33 min corresponding to meUDP impurity also increases steadily. At 200 µmol concentration of the inhibitor the estimated concentration of meUDP reaches about 10–15 µmol, which was deduced from the comparison of the areas under the meUDP, UDP-GlcNAc, and UTP peaks. This estimation exceeds the calculated concentration of meUDP impurity based on 2% contamination of the meUTP 2 to 3 folds. It is not clear if this is a result of enzymatic conversion of meUTP or whether the meUDP level increases due to partial hydrolysis of the inhibitor in mildly basic conditions of the reaction quench/sample processing. Additionally, a peak at Rt = 3.80 min was identified as UDP and probably originates from the similar decay of UTP during processing of the enzymatic reaction. To rule out whether meUDP is the source of the observed enzyme inhibition, we showed that AfUAP1 is not inhibited by 500 µM meUDP (Fig. S3B and C, ESI†), eliminating this possibility.
The data show that the α,β-methylenebisphosphonate UTP analogue 5 (meUTP) inhibits AfUAP1 activity with an IC50 of 115 ± 40 µM (Fig. 6C), representing one of the few examples of a rationally designed inhibitor of this class of enzyme. Although this compound is likely to be an aspecific inhibitor of UAP1s and other UTP-dependent enzymes, further structural modifications guided by small differences between the human and the fungal enzymes13 may lead to development of selective inhibitors of fungal UAP1. The possibility of inhibition of specific UAP1s, despite high sequence conservation in this family, has recently been demonstrated with a selective UTP-competitive inhibitor of T. brucei UAP1 (TbUAP1).10 This inhibitor was shown to compete with UTP, binding with a KD of 9.3 µM (IC50 of 37 ± 4 µM), making it more potent than the meUTP.10 In addition, the TbUAP1 inhibitor was selective towards this parasite enzyme because it binds to a unique allosteric site on TbUAP1 that is not present in either the human or the fungal UAP1.
Concluding remarks
The structural data described here provide detailed snapshots of the enzyme in complex with substrates/products, revealing for the first time the key role that the magnesium ion plays in stabilizing the Michaelis complex of pyrophosphorolysis, aligning the reactants for a direct attack by the pyrophosphate group, in parallel with significant conformational changes in the enzyme. We also show that eukaryotic UAP1s depend solely on one magnesium ion unlike the bacterial GlmUs that require two magnesium ions for catalysis. Conserved amino acids interacting with the negative charges on the phosphates were shown to be important for full catalytic activity and in particular we demonstrated that eukaryotic UAP1s use a conserved lysine (Lys437 in AfUAP1) as a key electron withdrawer to increase the electrophilic character of the α-phosphorous of either UTP or UDP-GlcNAc in order to promote the forward or reverse catalytic reaction respectively. Furthermore, we have reported for the first time that an α,β-methylenebisphosphonate UTP analogue (meUTP) inhibits AfUAP1 with an IC50 of 115 µM. This and its derivatives may also be a useful reagent for the study of the broader family of sugar nucleotide pyrophosphorylases. For example, it could be used in kinetic protein crystallography experiments that could help to trap kinetic states of pyrophosphorylases, and to determine the outcome of inhibiting these enzymes in appropriate cell models. Together, the novel mechanistic insights and the substrate-based inhibitor reported here will aid the future exploitation of this genetically validated enzyme as a potential target for the discovery of novel antifungal compounds.Author contribution
D. M. F. v. A., conceived the idea. R. H.-G. and O. G. R. designed the crystallization constructs and solved the crystal structures. R. H.-G. and O. G. R. purified the enzymes, crystallized the complexes, and refined the crystal structures. O. G. R. performed the kinetic experiments. V. B., synthesized the meUTP, A. F. cloned the K437R mutant, M. D. U. performed the HPAEC assay, M. A. J. F., R. H.-G., O. G. R. and D. M. F. v. A. wrote the manuscript. All authors read and approved the final manuscript.Accession codes
The coordinates of all the structures have been deposited to the PDB with the accession codes 6G9V, 6TN3 and 6G9W.Conflicts of interest
There are no conflict of interest to declare.Acknowledgements
The authors would like to thank the European Synchrotron Radiation facility (ESRF), Grenoble, for beamline time. This work was funded by MRC Programme Grant M004139 to DvA.References
- A. F. Ram, et al., The cell wall stress response in Aspergillus niger involves increased expression of the glutamine: fructose-6-phosphate amidotransferase-encoding gene (gfaA) and increased deposition of chitin in the cell wall, Microbiology, 2004, 150(Pt 10), 3315–3326 CrossRef CAS PubMed.
- F. M. Klis, Review: cell wall assembly in yeast, Yeast, 1994, 10(7), 851–869 CrossRef CAS PubMed.
- F. M. Klis, P. Mol, K. Hellingwerf and S. Brul, Dynamics of cell wall structure in Saccharomyces cerevisiae, FEMS Microbiol. Rev., 2002, 26(3), 239–256 CrossRef CAS PubMed.
- M. Bernard and J. P. Latge, Aspergillus fumigatus cell wall: composition and biosynthesis, Med. Mycol., 2001, 39(suppl. 1), 9–17 CrossRef CAS PubMed.
- Y. Nishitani, et al., Crystal structures of N-acetylglucosamine-phosphate mutase, a member of the alpha-D-phosphohexomutase superfamily, and its substrate and product complexes, J. Biol. Chem., 2006, 281(28), 19740–19747 CrossRef CAS PubMed.
- S. Milewski, I. Gabriel and J. Olchowy, Enzymes of UDP-GlcNAc biosynthesis in yeast, Yeast, 2006, 23(1), 1–14 CrossRef CAS PubMed.
- T. Mio, T. Yabe, M. Arisawa and H. Yamada-Okabe, The eukaryotic UDP-N-acetylglucosamine pyrophosphorylases. Gene cloning, protein expression, and catalytic mechanism, J. Biol. Chem., 1998, 273(23), 14392–14397 CrossRef CAS PubMed.
- T. Mio, T. Yamada-Okabe, M. Arisawa and H. Yamada-Okabe, Saccharomyces cerevisiae GNA1, an essential gene encoding a novel acetyltransferase involved in UDP-N-acetylglucosamine synthesis, J. Biol. Chem., 1999, 274(1), 424–429 CrossRef CAS PubMed.
- T. Naderer, E. Wee and M. J. McConville, Role of hexosamine biosynthesis in Leishmania growth and virulence, Mol. Microbiol., 2008, 69(4), 858–869 CrossRef CAS PubMed.
- M. D. Urbaniak, et al., A novel allosteric inhibitor of the uridine diphosphate N-acetylglucosamine pyrophosphorylase from Trypanosoma brucei, ACS Chem. Biol., 2013, 8(9), 1981–1987 CrossRef CAS PubMed.
- D. Maruyama, et al., Crystal structure of uridine-diphospho-N-acetylglucosamine pyrophosphorylase from Candida albicans and catalytic reaction mechanism, J. Biol. Chem., 2007, 282(23), 17221–17230 CrossRef CAS PubMed.
- C. Peneff, et al., Crystal structures of two human pyrophosphorylase isoforms in complexes with UDPGlc(Gal)NAc: role of the alternatively spliced insert in the enzyme oligomeric assembly and active site architecture, EMBO J., 2001, 20(22), 6191–6202 CrossRef CAS PubMed.
- W. Fang, et al., Genetic and structural validation of Aspergillus fumigatus UDP-N-acetylglucosamine pyrophosphorylase as an antifungal target, Mol. Microbiol., 2013, 89(3), 479–493 CrossRef CAS PubMed.
- R. Hurtado-Guerrero, et al., Structural and kinetic differences between human and Aspergillus fumigatus D-glucosamine-6-phosphate N-acetyltransferase, Biochem. J., 2008, 415(2), 217–223 CrossRef CAS PubMed.
- D. A. Stevens, et al., Practice guidelines for diseases caused by Aspergillus. Infectious Diseases Society of America, Clin. Infect. Dis., 2000, 30(4), 696–709 CrossRef CAS PubMed.
- Z. Otwinowski and A. Minor, Processing of X-ray diffraction data collected in oscillation mode, Methods in Enzymology, Macromolecular Crystallography, 1997, vol. 276, pp. 307–326 Search PubMed.
- A. Vagin and A. Teplyakov, MOLREP: an automated program for molecular replacement, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- A. Perrakis, R. Morris and V. S. Lamzin, Automated protein model building combined with iterative structure refinement, Nat. Struct. Biol., 1999, 6(5), 458–463 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53(Pt 3), 240–255 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60(Pt 12 Pt 1), 2126–2132 CrossRef PubMed.
- A. W. Schuettelkopf and D. M. F. van Aalten, PRODRG: a tool for high-throughput crystallography of protein–ligand complexes, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- G. Vriend, WHAT IF: a molecular modeling and drug design program, J. Mol. Graphics, 1990, 8(1), 52–56 CrossRef CAS PubMed.
- W. L. DeLano, Use of PYMOL as a communication tool for molecular science, Abstr. Pap. Am. Chem. Soc., 2004, 228, 030-CHED Search PubMed.
- R. J. Leatherbarrow, Grafit version 5, Erithacus Software Ltd., Horley, UK, 2001 Search PubMed.
- N. Tomiya, E. Ailor, S. M. Lawerence, M. J. Bettenbaugh and Y. C. Lee, Determination of nucleotides and sugar nucleotides involved in protein glycosylation by high-performance anion-exchange chromatography: sugar nucleotide contents in cultured insect cells and mammalian cells, Anal. Biochem., 2002, 293, 129–137 CrossRef PubMed.
- V. S. Borodkin, M. A. J. Ferguson and A. V. Nikolaev, Synthesis of potential bisubstrate inhibitors of Leishmania elongating α-D-mannosyl phosphate transferase, Tetrahedron Lett., 2004, 45, 857–862 CrossRef CAS.
- M. C. Pelissier, S. A. Lesley, P. Kuhn and Y. Bourne, Structural insights into the catalytic mechanism of bacterial guanosine-diphospho-D-mannose pyrophosphorylase and its regulation by divalent ions, J. Biol. Chem., 2010, 285(35), 27468–27476 CrossRef CAS PubMed.
- J. Lohofener, et al., The Activation Mechanism of 2′-5′-Oligoadenylate Synthetase Gives New Insights Into OAS/cGAS Triggers of Innate Immunity, Structure, 2015, 23(5), 851–862 CrossRef PubMed.
- J. B. Thoden and H. M. Holden, Active site geometry of glucose-1-phosphate uridylyltransferase, Protein Sci., 2007, 16(7), 1379–1388 CrossRef CAS PubMed.
- P. K. Jagtap, S. K. Verma, N. Vithani, V. S. Bais and B. Prakash, Crystal structures identify an atypical two-metal-ion mechanism for uridyltransfer in GlmU: its significance to sugar nucleotidyl transferases, J. Mol. Biol., 2013, 425(10), 1745–1759 CrossRef CAS PubMed.
- J. I. C. Fuehring, J. T. Routier, F. H. Lamerz, A.-C. Baruch, P. Gerardy-Schahn and R. Fedorov, Catalytic Mechanism and Allosteric Regulation of UDP-Glucose Pyrophosphorylase from Leishmania major, ACS Catal., 2013, 3(12), 2976–2985 CrossRef CAS.
- M. Kalek, J. Jemielity, J. Stepinski, R. Stolarski and E. Darzynkiewicz, A direct method for the synthesis of nucleoside 5′-methylenebis(phosphonate)s from nucleosides, Tetrahedron Lett., 2005, 46, 2417–2421 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1. Multiple sequence alignment of UAP1s with MtGlmU. Multiple sequence alignment of AfUAP1, the human AGX1 and AGX2 isoforms, C. albicans UAP1 (CaUAP1) and MtGlmU. Secondary structure elements from the AfUAP1 structure are shown with α-helices and β-strands in red. The key highly conserved lysine in eukaryotes (Lys437 in AfUAP1) is highlighted in cyan and indicated with a green inverted triangle. The insertion loop is underlined and AfUAP1 loops are indicated with a line and labelled appropriately as well as helix α*. Fig. S2. Role of K437 in catalysis. (A) SDS-PAGE of all mutants. 1. WT, 2. K437A, 3. Y330F, 4. K148M, 5. K437R and 6. K437M. All proteins were overexpressed and in the soluble fraction. (B) Activity of K437A, K437R and K437M compared to the wild type. Experiment was done in triplicates and result expressed as mean ± SEM. The Biomol Green assay was performed as described in the materials and methods using equal enzyme concentration of 20 nM enzyme, 300 µM UTP and 250 µM GlcNAc-1P terminating after 20 min. Fig. S3. (A) HPLC chromatogram of meUTP. The major component (meUTP) has a retention time of 6.94. The residual peak of meUDP has a retention time of 3.56 min. The major compound comprises 98% of the mixture. (B) AfUAP1 activity in the absence of meUDP. To investigate the effect of meUDP an HPLC assay was performed (Waters Xselect 150 × 4.6 mm column, flow rate 1 mL min−1. Buffer A 100 mM phosphate/2.5 mM Bu4HSO4 in water; Buffer B 100 mM phosphate/2.5 mM Bu4HSO4 in water–MeCN 1:1). The assay was performed using 5 nM enzyme, 25 µM UTP and 50 µM GlcNAc-1P, for 10 min. The chromatogram shows the production of UDP-GlcNAc (Rt = 4.5 min). It also shows a peak corresponding to UDP (Rt = 5.55 min) which is thought to be a result of gradual hydrolysis of UTP. The remaining UTP elutes at Rt = 9.5 min. (C) Effect of meUDP on AfUAP1 activity. The experiment was done as described for B in the presence of 500 µM meUDP. The chromatogram shows meUDP elutes at Rt = 3.56 min; concentration far above the scale. Production of UDP-GlcNAc (Rt = 4.5 min) remains unaffected. It also shows a peak corresponding to UDP (Rt = 5.55 min) which is taken to be a result of gradual hydrolysis of UTP. The remaining UTP elutes at Rt = 9.44 min. See DOI: 10.1039/c9cb00017h |
| ‡ These authors contributed equally to this work. |
| § Current address: Institute of Biocomputation and Physics of Complex Systems (BIFI), University of Zaragoza, BIFI-IQFR (CSIC) Joint Unit, Mariano Esquillor s/n, Campus Rio Ebro, Edificio I+D; Fundacion ARAID, Spain. |
| ¶ Current address: Faculty of Health and Medicine, Lancaster University, Lancaster LA1 4YG, UK. |
| This journal is © The Royal Society of Chemistry 2020 |