
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous Rh and Rh/Ag bimetallic nanoparticle catalysts immobilized on chiral polymers†
Hyemin
Min
,
Hiroyuki
Miyamura
,
Tomohiro
Yasukawa
and
Shū
Kobayashi
 *
*
Department of Chemistry, School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shu_kobayashi@chem.s.u-tokyo.ac.jp
First published on 27th June 2019
Abstract
The development of heterogeneous chiral catalysts has lagged far behind that of homogeneous chiral catalysts in spite of their advantages, such as environmental friendliness for a sustainable society. We describe herein novel heterogeneous chiral Rh and Rh/Ag bimetallic nanoparticle catalysts consisting of polystyrene-based polymers with chiral diene moieties. The catalysts enable high-to-excellent yields and enantioselectivities to be obtained in asymmetric 1,4-addition reactions of arylboronic acids with α,β-unsaturated carbonyl compounds such as ketones, esters, and amides, and in other asymmetric reactions. The catalysts could be readily recovered by simple filtration and reused; they could also be applied to continuous-flow synthesis. We also discuss the nature of possible reaction species based on XPS analysis.
Introduction
Catalytic asymmetric C–C bond-forming reactions provide among the most efficient protocols for preparing optically active compounds. While much progress has been made with homogeneous chiral complexes as catalysts, the development of heterogeneous chiral catalysts has lagged far behind in spite of their advantages, such as environmental friendliness for a sustainable society.1 A general method for the preparation of heterogeneous chiral catalysts involves formation of metal–ligand complexes from supported/immobilized chiral ligands and metal sources.2–5 However, these metal–ligand complexes are often unstable, and metal leaching and decomposition of ligands sometimes occur.5,6 The use of chiral metal nanoparticles (NPs) can address these issues because of their high activity/selectivity as well as their robustness.7–9 Indeed, some metal NP catalysts immobilized on achiral supports, such as metal oxides and activated carbon, with externally added chiral modifiers have been reported for some asymmetric reactions.10,11 In these cases, unfortunately, collapse of the chiral reaction environments can easily occur and recovery of chiral ligands is difficult. To overcome these problems, a few catalysts in which metal NPs were immobilized on chiral supports were reported. Glorius and coworkers synthesized Pd NPs supported on Fe3O4 that contained chiral N-heterocyclic carbene moieties, and this catalyst was applied to asymmetric allylation of 4-nitrobenzaldehyde with allyltributyltin.12 Toste, Somorjai, and coworkers reported Au NPs in a chiral self-assembled monolayer as an efficient catalyst for intermolecular cyclopropane synthesis.13 In both cases, however, only moderate enantioselectivities were observed, and the development of heterogeneous chiral NP catalysts with chiral supports for asymmetric C–C bond-forming reactions that show high enantioselectivity is still desired.14Since our initial study in 2012, we have reported on asymmetric 1,4-addition reactions of arylboronic acids to α,β-unsaturated carbonyl compounds, which constitute one of the most useful methods for the preparation of chiral aryl-substituted compounds15–17 using Rh/Ag bimetallic NP catalysts with externally added binap or chiral diene ligands. Excellent yields and outstanding enantioselectivities were observed, especially with amide-substituted chiral diene ligands, without metal leaching.18–23 Recovery and reuse of these catalysts was possible because of their robustness; however, in every reaction for the recovery and reuse, the chiral ligand had to be externally added. Moreover, in a continuous-flow reaction, the chiral ligand must flow together with substrates.
Herein, we describe novel heterogeneous chiral Rh and Rh/Ag bimetallic NP catalysts consisting of polystyrene-based polymers with chiral diene moieties (Scheme 1). The catalysts successfully promoted asymmetric 1,4-addition reactions of arylboronic acid with α,β-unsaturated carbonyl compounds and other asymmetric reactions. The catalysts could be readily recovered by simple filtration and reused, and further applied to continuous-flow synthesis.
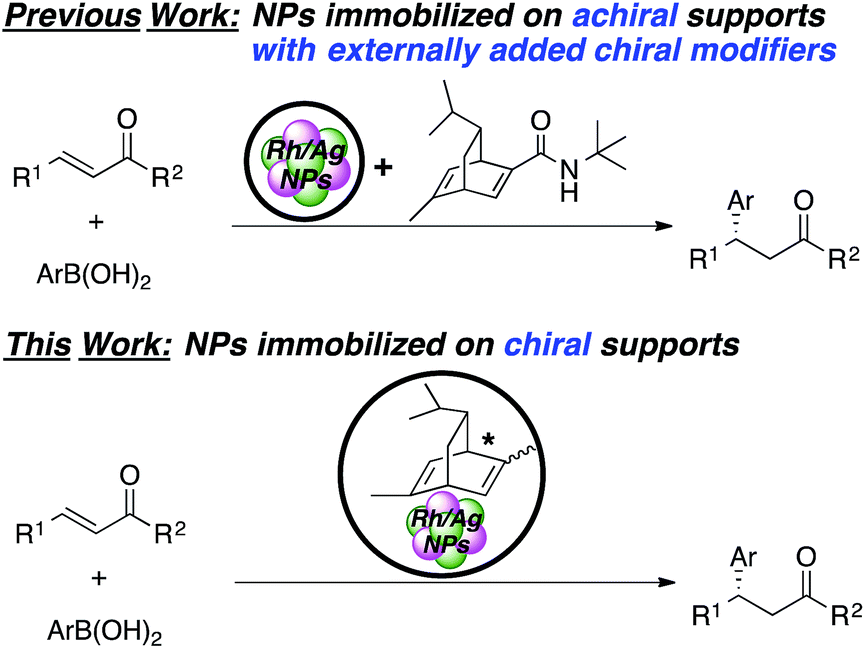 | ||
| Scheme 1 Concept scheme for this work. | ||
Results and discussion
Preparation of polymers and catalysts
Chiral polymer 1 was prepared by copolymerization of styrene, trimethoxysilylstyrene (a cross-linking moiety),24,25 and a chiral diene monomer, according to Scheme 2(a). By using copolymer 1 and carbon black (CB) as a second support,26,27 Rh and Rh/Ag bimetallic NP catalysts were prepared as shown in Scheme 2(b). Rh or Rh/Ag bimetallic NPs were formed from reduction with NaBH4, and then cross-linking was conducted by heating under basic conditions in a sol–gel process. A second reduction with NaBH4 was conducted to increase the activity of the catalyst in some cases.18 STEM and EDX mapping experiments showed that NPs of 4 nm average size were formed and, in the case of Rh/Ag, an alloyed conformation was confirmed, while the ratios of Rh and Ag were random, depending on NPs (see the ESI, Fig. S1–S5†). In this chiral ligand-immobilized organic–inorganic hybrid CB Rh NP catalyst (LIHBCB-Rh (2)), metal NPs were stabilized by multiple weak interactions from abundant π-electrons of the benzene rings, and these interactions meant that active but stable metal NP catalysts could be realized.28 Actual loadings of Rh, determined by Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analyses, were close to the target loadings.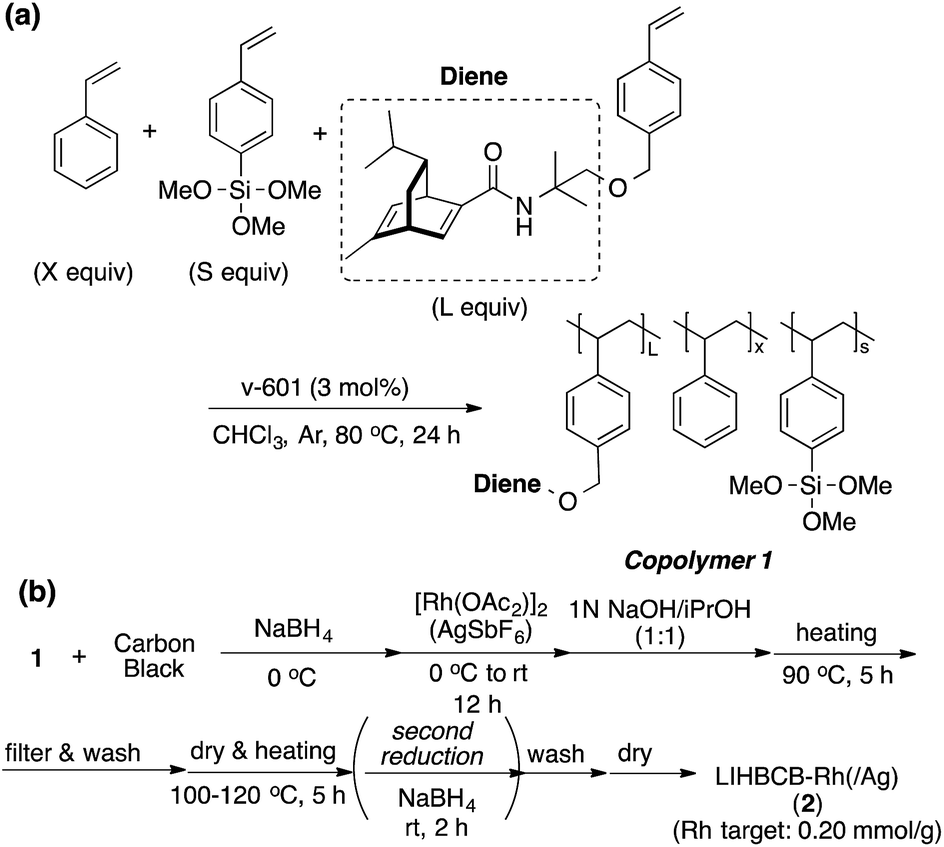 | ||
| Scheme 2 Preparation of chiral polymer and catalyst. | ||
First, we prepared catalysts 2a, 2b, and 2c containing different amounts of the chiral diene ligand moiety while maintaining the amount of the cross-linking moiety (trimethoxysilyl group). The ratios of the monomer moieties were determined by 1H NMR analysis, and almost the same amount of the ligand moiety as the target amount was immobilized. Actual Rh loadings were also close to the target loadings of Rh (0.2 mmol g−1). The activity of the LIHBCB-Rh catalysts (2a, 2b, and 2c) was estimated in the 1,4-addition of phenylboronic acid (4a) to 2-cyclohexenone (3a) (Table 1). When around 10% (mole fraction) of the ligand monomer moiety (L) was immobilized (2a), an excellent enantioselectivity was obtained, but with a yield of 79% (Table 1, entry 1). When the ratio of L in the polymer was increased to 20% (2b), the yield was improved and high ee was maintained (entry 2); the same result was observed with a further increased ratio of L (2c) (entry 3). Thus, an excess amount of L against Rh was required to achieve a high yield. On the other hand, excellent enantioselectivities were observed in all cases. Based on these results, the polymer that was used in catalyst 2b was used for further optimizations.
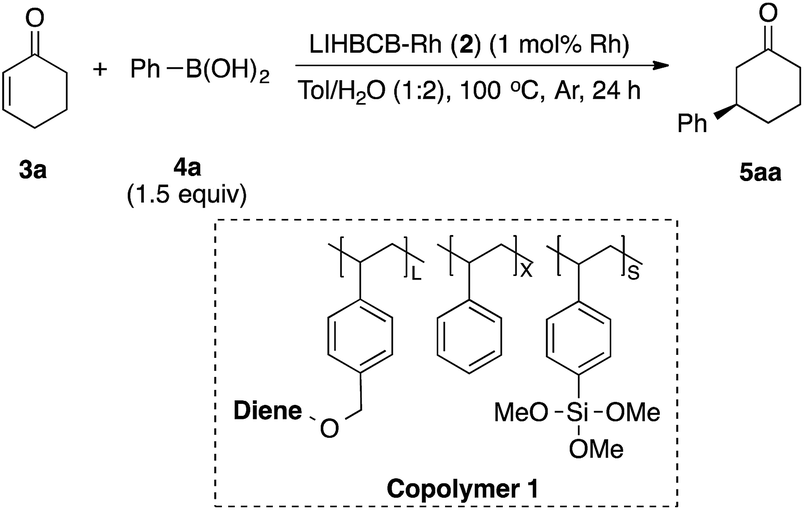
Optimization of catalyst and reaction conditions with α,β-unsaturated acyclic ketone substrate
LIHBCB-Rh (2b) showed good activity in the reaction with α,β-unsaturated cyclic ketone substrate 3a. We then applied this catalyst to the α,β-unsaturated acyclic ketone substrate benzalacetone (3b) (Table 2).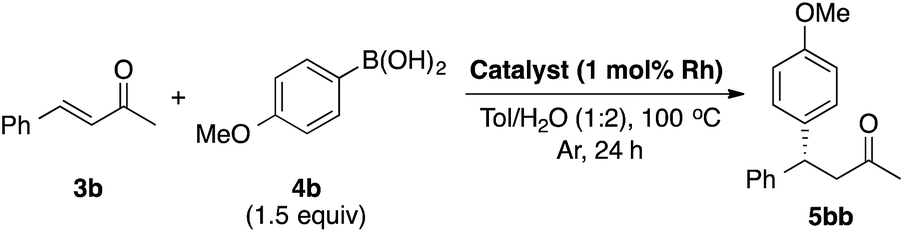
In the reaction between benzalacetone (3b) and 4-methoxyphenylboronic acid (4b) under the same conditions, the desired product was obtained in 52% yield with excellent enantioselectivity (Table 2, entry 1). Although no improvement of the yield was observed using 2 mol% catalyst (entry 2), a slight increase of the yield was obtained using 2 equivalents of 4b (entry 3). In our previous report, Rh/Ag bimetallic NP catalysts showed higher activity than Rh NP catalysts because of better dispersion of Rh NPs.18 Considering this result, we prepared chiral diene-immobilized hybrid Rh–Ag bimetallic NP catalysts with different ratios of Rh and Ag; LIHBCB-Rh/Ag (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (2d), LIHBCB-Rh/Ag (2:1) (2e), and LIHBCB-Rh/Ag (1:2) (2f). Among them, 2f showed the highest reactivity while maintaining excellent enantioselectivity (entries 4, 5, and 6); when 2 equivalents of 4b were used, high yield and excellent enantioselectivity were obtained (entry 7).
:1) (2d), LIHBCB-Rh/Ag (2:1) (2e), and LIHBCB-Rh/Ag (1:2) (2f). Among them, 2f showed the highest reactivity while maintaining excellent enantioselectivity (entries 4, 5, and 6); when 2 equivalents of 4b were used, high yield and excellent enantioselectivity were obtained (entry 7).
Substrate scope for 1,4-addition of boronic acids to α,β-unsaturated carbonyl compounds
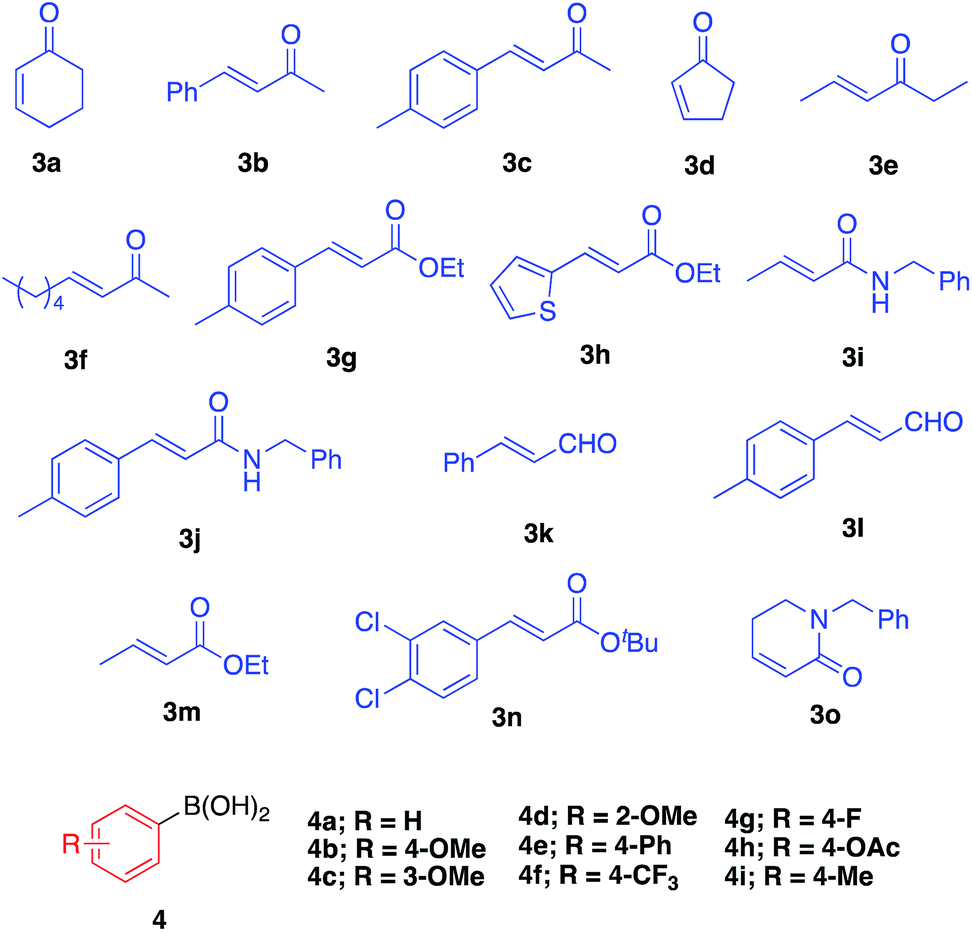
We then surveyed the substrate scope of this asymmetric 1,4-addition reaction (Table 3). First, the reaction of 3a with a range of arylboronic acids including various substituents was examined. With phenylboronic acid (4a), excellent yield and enantioselectivity were obtained using Rh 2b (entry 1). Rh/Ag bimetallic catalyst 2f also worked as well as catalyst 2b (entry 2). When para-, meta-, and ortho-methoxyphenylboronic acids 4b–d were used, excellent results were obtained in all cases (entries 3–5). With a phenyl group at the para-position of phenylboronic acid (4e), satisfactory results were obtained (entry 6). Next, the reactions with arylboronic acids having electron-withdrawing groups such as trifluoromethyl (4f), fluoro (4g), and acetoxy groups (4h) were examined. It was found that these substrates were less active than arylboronic acids having electron-donating groups; however, high yields with excellent enantioselectivities were obtained by using a slight excess of the arylboronic acids (entries 7–9). For further less-active substrates such as 2-cyclopentenone (3d) and acyclic α,β-unsaturated ketones (3b, 3c, 3e and 3f), high yields with excellent enantioselectivities were observed by using Rh/Ag 2f (entries 10–14) instead of Rh 2b. We then optimized less reactive unsaturated carbonyl compounds.29 Rh/Ag 2f also showed good activity toward α,β-unsaturated esters 3g and 3h, although 1 equivalent of K2CO3 was needed as an additive (entries 15–18). Interestingly, high yield and excellent enantioselectivity were obtained with α,β-unsaturated amides 3i and 3j without any additive (entries 19 and 20). We also tried cinnamaldehyde (3k) and 4-methyl substituted cinnamaldehyde (3l) as substrate, and it showed good yields and enantioselectivities with various arylboronic acid (entries 21–23). To confirm further utility of this catalyst, the preparation of natural product and drug precursors was examined. Ester 5mi can be used as a precursor of various anticancer and antimicrobial materials,30,31 and it was successfully prepared by asymmetric 1,4-addition of 4-methoxyphenylboronic acid (4i) to α,β-unsaturated ester 3m in 87% yield with 94% enantioselectivity (entry 24). Ester 5na, a precursor of (−)-indatraline, was also obtained in good yield with high enantioselectivity (entry 25).32 Amide substrate 3o reacted with 4-fluorophenylboronic acid (4g) to afford 5og, a precursor of (−)-paroxetine, in good yield with excellent enantioselectivity (entry 26).21,33|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 | 4 | Conditionsa | Yield (%) | Eeb (%) |
| a Conditions A: LIHBCB-Rh (2b, 1 mol% Rh), ArB(OH)2 (1.5 equiv.); B: LIHBCB-Rh/Ag (2f, 1 mol% Rh), ArB(OH)2 (2 equiv.); C: LIHBCB-Rh (2b, 1 mol% Rh), ArB(OH)2 (2 equiv.); D: LIHBCB-Rh/Ag (2d, 1 mol% Rh), ArB(OH)2 (2 equiv.). b Determined by HPLC analysis. c 1 equiv. of K2CO3 was used. d 3 mol% of 2f was used. e 2 mol% of 2f was used. f 3 equiv. of 4a were used. | |||||
| 1 | 3a | 4a | A | 98 | 98 |
| 2 | 3a | 4a | B | 95 | 98 |
| 3 | 3a | 4b | A | 94 | 97 |
| 4 | 3a | 4c | A | 96 | 97 |
| 5 | 3a | 4d | A | 95 | 97 |
| 6 | 3a | 4e | A | 89 | 98 |
| 7 | 3a | 4f | C | 91 | 98 |
| 8 | 3a | 4g | C | 89 | 98 |
| 9 | 3a | 4h | C | 92 | 98 |
| 10 | 3b | 4b | B | 88 | 96 |
| 11 | 3c | 4a | B | 93 | 91 |
| 12 | 3d | 4a | B | 94 | 97 |
| 13 | 3e | 4a | B | 90 | 96 |
| 14 | 3f | 4a | B | 87 | 97 |
| 15c | 3g | 4a | B | Quant. | 97 |
| 16c | 3g | 4c | B | 91 | 98 |
| 17c | 3h | 4c | B | 63 | 94 |
| 18c,d | 3h | 4c | B | 73 | 94 |
| 19e | 3i | 4a | B | 90 | 98 |
| 20e | 3j | 4a | B | 90 | 98 |
| 21 | 3k | 4b | D | 64 | 87 |
| 22 | 3k | 4g | D | 70 | 93 |
| 23 | 3l | 4a | D | 71 | 93 |
| 24 | 3m | 4i | B | 87 | 94 |
| 25f | 3n | 4a | B | 85 | 93 |
| 26 | 3o | 4g | B | 74 | 99 |
|
|||||
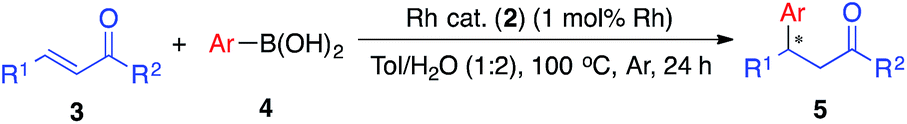
Recovery and reuse of the catalyst
We next conducted catalyst recovery and reuse experiments. The heterogeneous catalyst could be recovered by simple filtration, and the recovered catalyst was used for the next run after washing with toluene/H2O and drying in vacuo. Both monometallic Rh catalyst (2b, for 5aa) and Rh/Ag bimetallic catalyst (2f, for 5ia) were tested, and could be recovered and reused 3–4 times without significant decrease in activity (Table 4).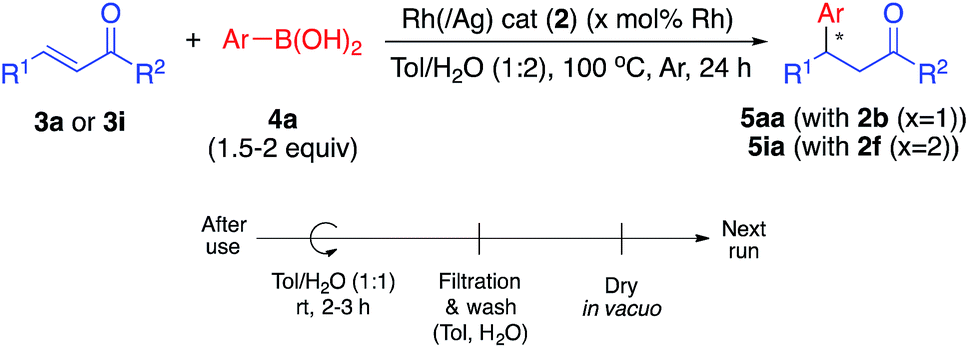
In the 4th or 5th run, the yields decreased, but the enantioselectivities remained high. We carefully analyzed the reused catalysts; however, no significant aggregation or damage was observed by STEM analysis (see ESI, Fig. S6 and S7†). We consider that the decreased yields might arise from adsorbed contaminants generated during the reactions (byproducts or co-products). We check the reaction profile in the recovery/reuse experiments until 3rd run, and gradually decreased reaction rates in the successive runs were observed, while yields were reached >90% after 24 h reaction time in each run (see ESI, Fig. S18†). It was probably derived from decrease in available reaction sites due to remained contaminants. Washing the catalyst in basic media (0.5–1 M NaOH (aq.)) or acidic media (0.5 M HCl (aq.)) was conducted for removal; however, no further improvement was obtained. Another possibility for the reduced yield is decomposition of the diene ligand during the reaction. To test this, we conducted a number of control experiments (Table 5).
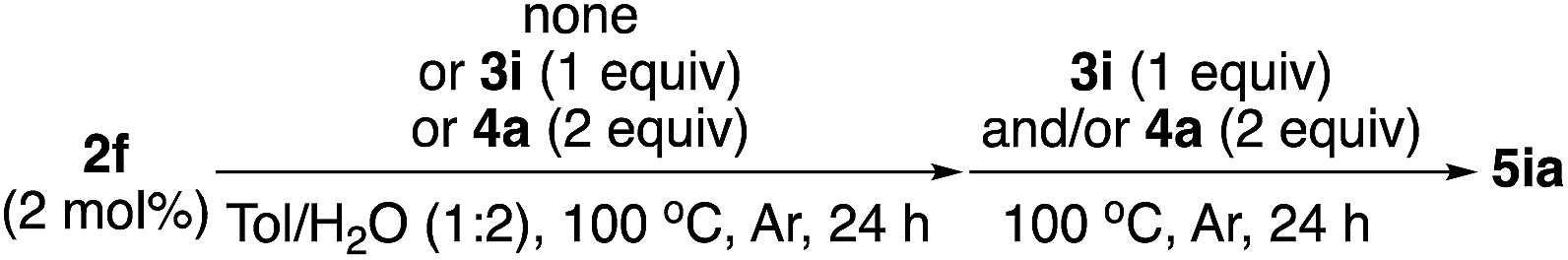
Catalyst 2f was stirred at 100 °C in the presence of the solvent for 96 h, which was the same as the total reaction time until the 4th run in recovery/reuse experiments, and then both substrates 3i and 4a were added. In this case, excellent yield and ee of the desired product (5ia) were obtained, and the possibility of ligand degradation through thermal decomposition could be excluded (Table 5, entry 1). To examine the effect of substrates in the reaction medium, catalyst 2f was heated at 100 °C in the presence of 3i or 4a for 96 h, followed by the addition of other substrates. In the case of heating at 100 °C with amide 3i, an excellent result was still obtained (entry 2). We also conducted the heating treatment with phenylboronic acid 4a; in this case, the desired product (5ia) was not obtained, probably because of the hydrolysis of 4a to benzene during the first 96 h (entry 3). On the other hand, when a new portion of 4a was added after the same heating treatment with 3i, a slight decrease in yield was obtained; however, the desired product 5ia was obtained with high enantioselectivity. It may be possible that decomposed (or co-products of) arylboronic acid generated in the asymmetric 1,4-addition reaction somehow deactivated the catalysts in the recovery and reuse experiments.
We also analyzed the amount of adsorbed hydrogen gas (H2) in HBCB-Rh/Ag (1:2) (6), prepared without a ligand component in the polymer, fresh 2f, and recovered 2f after the 5th run (Table 6). Whereas catalyst 6 adsorbed 0.2413 cm3 g−1 H2, fresh 2f adsorbed only 0.0783 cm3 g−1 H2, even though the size of NPs is smaller than 6 (13 nm (6) vs. 4 nm (2f)). This may be explained by considering that certain amounts of ligand coordinate to the surface of metal NPs competing with H2 in 2f. Interestingly, in the case of recovered 2f after the 5th run, the amount of adsorbed H2 decreased further. As a control, we also examined the H2 adsorption of an immobilized ligand (containing carbon black, without metal NPs); no adsorption of H2 was observed, so H2 would be adsorbed on metal NPs. Although these results are indirect, they suggested that contaminants not removed by washing gradually coordinated to the surface of the metal NPs and accumulated during the recovery and reuse of the catalyst.
| Catalyst | Amount of adsorbed hydrogen gas (cm3 g−1) |
|---|---|
| HBCB cat. (6) | 0.2413 |
| Fresh 2f | 0.0783 |
| Deactivated 2f (after 5th run) | 0.0206 |
| Immobilized ligand + CB | 0 |
Mechanistic insight
There are broadly three categories of possible catalytically active species in NP catalyst systems, as shown in Scheme 3:34,35 (a) a reaction occurs on the surface of metal NPs coordinated with immobilized ligands; (b) a detached metal complexes or a small metal clusters coordinated with immobilized ligands catalyzes the reaction as an active species, which returns back to NPs after the reaction, and NPs act as a reservoir for these small clusters; and (c) a leached homogeneous metal complex from nanoparticles works as an active species. Hot filtration tests were conducted to determine whether the reaction proceeded through mechanism (c) by leached homogeneous complex (Fig. 1). The reaction profile was monitored by using a filtrate obtained in the middle of a reaction. We conducted this test for both cyclic (3a) and acyclic (3e) ketone substrates, and no increase in yield was observed after removal of the catalyst in either of the experiments; therefore, it was confirmed that this reaction was catalyzed in a heterogeneous manner. ICP analysis of the reaction medium revealed that trace amounts of Rh and Ag were detected after the reaction of 3e (0.111% Rh, 0.035% Ag (Rh detection limit: 0.045%, Ag detection limit: 0.007%)). Based on the results of the hot filtration tests, we conclude that leached Rh or Ag did not catalyze the reactions, and that metal species in the catalysts that were not fully immobilized/fixed were washed out during the reactions.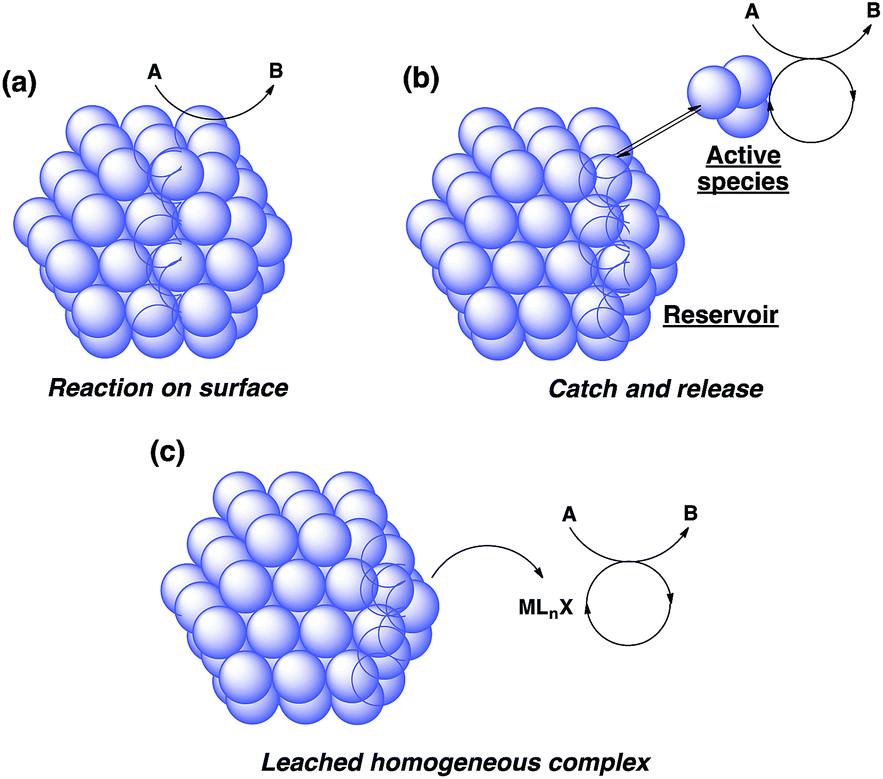 | ||
| Scheme 3 Possible mechanisms of heterogeneous metal nanoparticle catalysts. | ||
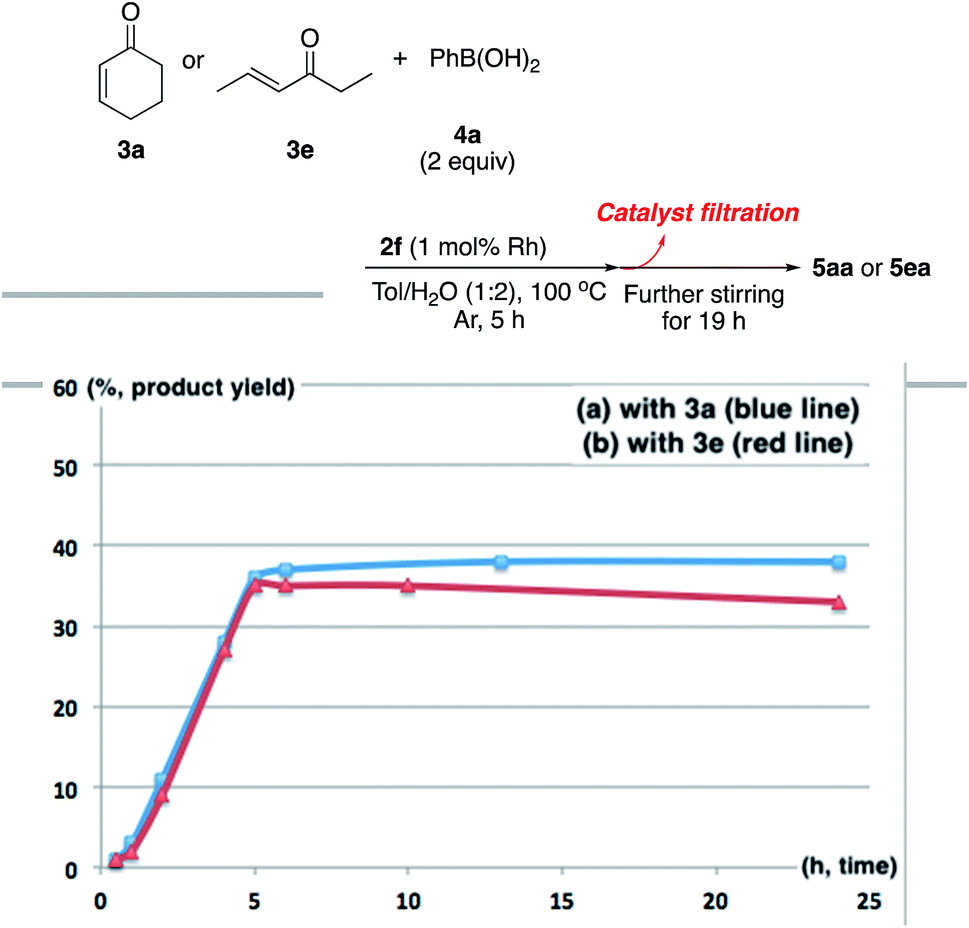 | ||
| Fig. 1 Hot filtration test. | ||
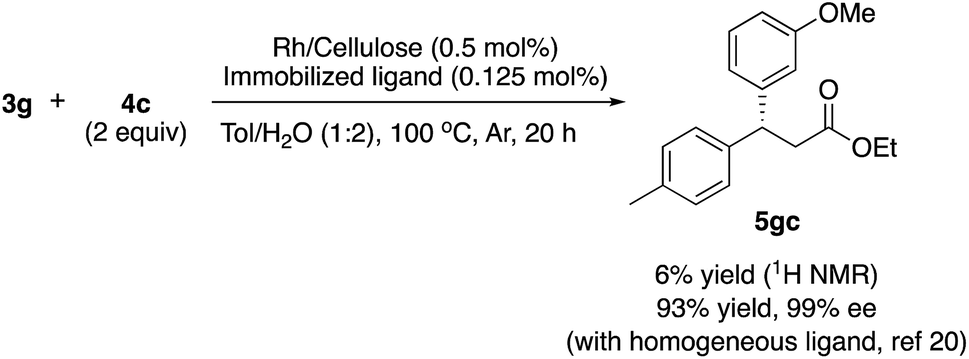
To confirm the importance of physical proximity between metal NPs and chiral diene ligands, we prepared an immobilized ligand on carbon black (see ESI, Scheme S3†), and the reaction was conducted together with a Rh/cellulose catalyst, which was an easily prepared and highly active catalyst (Scheme 4).20 As a result, only a trace amount of the product (5gc) was obtained. It confirms that this reaction proceeds by ligand acceleration and the reaction hardly occurs on the non-coordinated metal NPs. Also, physical proximity is an important factor required to make highly active species. To gather further information on the reaction mechanism and the active species, we conducted XPS analysis of several states of catalyst 2f: (A) fresh, (B) recovered during the reaction (1 h), (C) after 1st run, and (D) after 5th run. HBCB-Rh/Ag (1:2) catalyst (6) was also analyzed: (E) fresh and (F) after the reaction using 2.5 mol% of externally added amide-substituted chiral diene (Table 7).
 | ||
| Scheme 4 Reaction with the mixture of Rh NPs and chiral ligand on different supports. | ||
| State | 2f | |||
|---|---|---|---|---|
| (A) | (B) | (C) | (D) | |
| a For 2f: (A) fresh, (B) recovered during the reaction (1 h), (C) after 1st run, (D) after 5th run; for 6: (E) fresh, (F) after the reaction using 2.5 mol% of externally added amide-substituted chiral diene. | ||||
| Rh (eV) (3p3/2) | 497.0 | 497.4 | 496.9 | 496.9 |
| State | 6 | Rh(OH)–diene (7) | |
|---|---|---|---|
| (E) | (F) | ||
| Rh (eV) (3p3/2) | 497.2 | 498.2 | 497.3 |
In all data, Rh binding energy (BE) showed more distinct differences compared with that of Ag (see ESI, Table S1†), and based on these results, we concluded the following. First, lower Rh BE was obtained in fresh 2f (A) compared with that of 6 (E), and we supposed that this difference in Rh BE came from coordination of the ligand to Rh NPs. Second, in the case of 2f, almost the same Rh BE was observed in the fresh catalyst (A) and the recovered catalysts after the reactions (C and D), while Rh BE of 6 was changed after the reaction using an externally added chiral diene (E and F). Moreover, a significant amount of metal leaching was observed in the reaction with 6 and the externally added chiral diene ligand (see ESI, Scheme S5†). Although it is unclear whether this change in Rh BE of 6 is due to metal leaching or changed states of Rh NPs, it was assumed that more stable Rh species was generated and maintained in the presence of the immobilized chiral diene ligand. Lastly, a change in Rh BE was observed when the catalyst was recovered during the reaction (B). For comparison, we prepared Rh(OH)–diene complex (7) adsorbed on carbon (see ESI, Scheme S4†), and confirmed that the Rh BE of 7 was similar to that in (B). We consider that the active species of this reaction might be a species which has a similar electronic state to hydroxyl-rhodium species; however, it is not defined whether monometallic complex or oligomeric nanoclusters formed at this stage. Based on the experiments described above, it is feasible that detached Rh species from NPs coordinate to the chiral diene ligand immobilized on the polymer support, may be active for this asymmetric 1,4-addition reaction. Since the XPS analysis indicted the same Rh BE after use, the active species presumably returns to the NPs after the reaction. From these results, catch-and-release mechanism (b) may be more likely than reaction-on-surface mechanism (a), but we cannot exclude the possibility of mechanism (a).36
Asymmetric arylation of nitroolefin and imine compounds
We next examined the ability of the current heterogeneous chiral Rh/Ag bimetallic NP catalyst (2d) to mediate other asymmetric reactions; to this end, we examined the asymmetric arylation of nitroolefins and the asymmetric arylation of imines (Table 8).|
|
||||
|---|---|---|---|---|
| Entry | 8 | 4 | Yield (%) | Eeb (%) |
|
a Solvent ratio: Tol/H2O (5:1) for nitroolefin (entries 1–6), Tol/H2O (8:1) for imine (entries 7–12).
b Determined by HPLC analysis.
c 2 mol% 2d, 3 equiv. of 4k were used.
|
||||
| 1 | 8a | 4g | 84 | 88 |
| 2 | 8a | 4j | Quant. | 92 |
| 3a | 8a | 4k | 69 | 98 |
| 4 | 8b | 4c | 93 | 88 |
| 5 | 8c | 4c | 98 | 86 |
| 6 | 8d | 4c | 63 | 82 |
| 7 | 9a | 4a | 50 | 98 |
| 8 | 9a | 4g | 53 | 99 |
| 9 | 9b | 4a | 61 | 98 |
| 10 | 9c | 4a | 51 | 97 |
| 11 | 9d | 4a | 59 | 98 |
| 12 | 9e | 4a | 70 | 97 |
|
||||
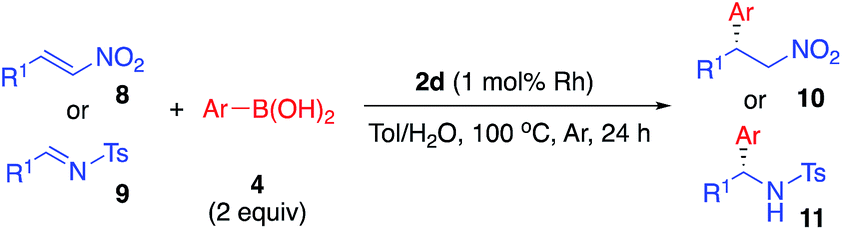
In the reaction of nitrostyrene (8a) with phenylboronic acid bearing electron-withdrawing groups (4g and 4j), the desired 1,4-addition products were obtained in high-to-excellent yields with high enantioselectivities (Table 8, entries 1 and 2). With 2-methylphenylboronic acid, which has a sterically-hindered structure, a good yield and excellent enantioselectivity were obtained, although increased amounts of both catalyst and boronic acid were needed (entry 3). Other nitroolefins that contain an electron-donating group or a heteroaromatic ring were also reacted, and the desired products were obtained in excellent yields with high enantioselectivities (entries 4 and 5). Notably, even the aliphatic nitroolefin 8d could form the desired 1,4-addition product in good yield with high enantioselectivity (entry 6). On the other hand, in the reactions of imine substrates, it was found that yields were a little lower because of the relatively rapid hydrolysis of imines; however, excellent enantioselectivities were obtained. 4-Methyl-substituted tosyl imine (9a) reacted with phenylboronic acid (4a) and 4-fluorophenylboronic acid (4g) to afford the desired 1,2-addition products in 50% yield with 98% ee (entry 7) and in 53% yield with 99% ee (entry 8), respectively. In the reaction of 4-methoxy-substituted tosyl imine (9b), the yield was slightly increased and the excellent enantioselectivity was maintained (entry 9). The desired amine products were also obtained with good yields and excellent enantioselectivities from 2- or 1-naphthyl (9c or 9d) and 2-thienyl substituted imine (9e) substrates (entries 10–12). It is noted that chiral Rh/Ag bimetallic NP catalyst 2d worked well for both asymmetric arylation of nitroolefins and asymmetric arylation of imines. Given that nitro groups can be readily reduced to the corresponding amines, two types of optically active amine derivatives can be prepared using these methods.
Application to flow system
Finally, this catalyst system was applied to a continuous-flow system (Scheme 5). Flow reactions have advantages over batch reactions in terms of environmental friendliness, efficiency, and safety.37 Among several flow reactions, those using heterogeneous catalysts are very efficient because a product can be obtained simply by passing starting materials through a column filled with the catalyst and, at the same time, the pro duct and the catalyst can be separated.38 In spite of the potential utility of this system, applicable heterogeneous catalysts, particularly heterogeneous chiral catalysts, are limited.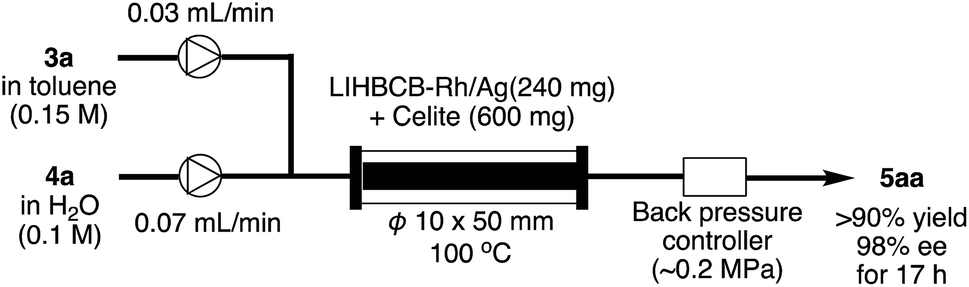 | ||
| Scheme 5 Application to flow system. | ||
Rh/Ag 2f was mixed with Celite and the mixture was packed in a column. α,β-Unsaturated ketone 3a in toluene and an aqueous solution of phenylboronic acid 4a were flowed into the column using separate pumps. To prevent evaporation of the solvents in the column, back-pressure was maintained. As a result, the desired product 5aa was obtained in >90% yield with 98% ee for 17 h (productivity (STY): 348 g dL−1).
Conclusions
We have developed heterogeneous chiral Rh and Rh/Ag bimetallic NP catalysts immobilized on a chiral diene-containing polymer. The catalysts showed high-to-excellent yields and enantioselectivities in asymmetric 1,4-addition reactions of arylboronic acids with α,β-unsaturated carbonyl compounds such as ketones, esters, and amides. The catalysts could be recovered and reused without external addition of other portions of the chiral diene ligand while maintaining their activities. We also presented a reaction mechanism and discussed the nature of the active species involved in the heterogeneous asymmetric catalysis. Furthermore, the catalyst could be applied to other asymmetric reactions, asymmetric arylation of nitroolefins and imines, and a continuous-flow system. To our knowledge, this is the first example of a catalyst in which a chiral ligand is immobilized together with metal NPs on the same support that shows both high yields and enantioselectivities in asymmetric C–C bond-formation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Mr Kohei Nishino for his contribution to the initial stage of this work. This work was supported in part by a Grant-in-Aid for Scientific Research from JSPS, The University of Tokyo, and MEXT (Japan), JST. We also thank Mr Noriaki Kuramitsu (The University of Tokyo) and Mr Tei Maki (JEOL, The University of Tokyo) for STEM and EDX analysis.Notes and references
- K. Ding and Y. Uozumi, Handbook of Asymmetric Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- B. Pugin and H. U. Blaser, Top. Catal., 2010, 53, 953–962 CrossRef CAS.
- T. Sawano, P. F. Ji, A. R. McIsaac, Z. K. Lin, C. W. Abney and W. B. Lin, Chem. Sci., 2015, 6, 7163–7168 RSC.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- D. Astruc, Nanoparticles and Catalysis, Wiley-VCH Verlag GmbH & Co.: KGaA, 2008 Search PubMed.
- H. Cong and J. A. Porco, ACS Catal., 2012, 2, 65–70 CrossRef CAS PubMed.
- Y. Orito, S. Imai, S. Niwa and G. H. Nguyen, J. Synth. Org. Chem., Jpn., 1979, 37, 173–174 CrossRef CAS.
- T. Yasukawa, H. Miyamura and S. Kobayashi, Chem. Soc. Rev., 2014, 43, 1450–1461 RSC.
- K. V. S. Ranganath, A. H. Schafer and F. Glorius, ChemCatChem, 2011, 3, 1889–1891 CrossRef CAS.
- E. Gross, J. H. Liu, S. Alayoglu, M. A. Marcus, S. C. Fakra, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 3881–3886 CrossRef CAS PubMed.
- In 2016, Hua et al. reported asymmetric oxidation reactions using bimetallic nanoclusters supported on chiral-substituted poly-N-vinylpyrrolidinones with good to excellent enantioselectivities. B. Hao, M. J. Gunaratna, M. Zhang, S. Weerasekara, S. N. Seiwald, V. T. Nguyen, A. Meier and D. H. Hua, J. Am. Chem. Soc., 2016, 138, 16839–16848 CrossRef CAS PubMed.
- T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed.
- P. Tian, H. Q. Dong and G. Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS.
- K. Okamoto, T. Hayashi and V. H. Rawal, Org. Lett., 2008, 10, 4387–4389 CrossRef CAS PubMed.
- T. Yasukawa, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 16963–16966 CrossRef CAS PubMed.
- T. Yasukawa, A. Suzuki, H. Miyamura, K. Nishino and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 6616–6623 CrossRef CAS PubMed.
- T. Yasukawa, H. Miyamura and S. Kobayashi, Chem. Sci., 2015, 6, 6224–6229 RSC.
- T. Yasukawa, Y. Saito, H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2016, 55, 8058–8061 CrossRef CAS PubMed.
- T. Yasukawa, T. Kuremoto, H. Miyamura and S. Kobayashi, Org. Lett., 2016, 18, 2716–2718 CrossRef CAS PubMed.
- H. Miyamura, K. Nishino, T. Yasukawa and S. Kobayashi, Chem. Sci., 2017, 8, 8362–8372 RSC.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 239–243 CrossRef CAS PubMed.
- H. Miyamura, K. Maehata and S. Kobayashi, Chem. Commun., 2010, 46, 8052–8054 RSC.
- W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 3095–3101 CrossRef CAS PubMed.
- H. Miyamura, A. Sonoyama, D. Hyrapetyan and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 10559–10563 CrossRef CAS PubMed.
- S. Kobayashi and H. Miyamura, Aldrichimica Acta, 2013, 46, 3–19 CAS.
- We monitored profiles of the reactions with α,β-unsaturated ketone, ester, amide and aldehyde substrates under the same conditions. As a result, slower reaction rate (ester, aldehyde) or lower final yield (amide) was observed, and we optimized reaction conditions for each substrates (see ESI, Table S2 and Fig. S17†).
- S. Afewerki, P. Breistein, K. Pirttila, L. Deiana, P. Dziedzic, I. Ibrahem and A. Cordova, Chem.–Eur. J., 2011, 17, 8784–8788 CrossRef CAS PubMed.
- J. Q. Li, X. Quan and P. G. Andersson, Chem.–Eur. J., 2012, 18, 10609–10616 CrossRef CAS PubMed.
- W. T. Wei, J. Y. Yeh, T. S. Kuo and H. L. Wu, Chem.–Eur. J., 2011, 17, 11405–11409 CrossRef CAS PubMed.
- M. S. Yu, I. Lantos, Z. Q. Peng, J. Yu and T. Cacchio, Tetrahedron Lett., 2000, 41, 5647–5651 CrossRef CAS.
- T. Yasukawa, H. Miyamura and S. Kobayashi, ACS Catal., 2016, 6, 7979–7988 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
- Preparation of small nanoclusters on same polymer support was tried by lowering the target loading, but still colonies of nanoparticles were formed (see ESI, Fig. S11†).
- S. Kobayashi, Chem.–Asian J., 2016, 11, 425–436 CrossRef CAS PubMed.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02670c |
| This journal is © The Royal Society of Chemistry 2019 |